For an overview and description of Transfeminine Science, please visit our Home page.
Authors
The authors for Transfeminine Science include Aly, Sam, Lain, Mitzi, and Luna.
Aly
Aly (“she/her”) is a transgender woman from California. She has long had a passionate interest in transfeminine hormone therapy and pharmacology generally. Aly began her transition in the early 2010s and has studied the transgender medical literature since.
She has a bachelor’s degree with summa cum laude distinction from a University of California. Aly also completed some of the pre-medicine curriculum and worked for about a year in research labs at the university. Aside from medical areas, Aly has experience with computer programming and web development.
Aly is a volunteer editor on Wikipedia. She is a prolific medical editor at the online encyclopedia and has contributed a large amount of content in areas like sex hormone pharmacology and psychopharmacology over time. When Aly has included inline citations to Wikipedia on Transfeminine Science, they have usually been to content that she herself authored.
Sam (“she/they”) is a transgender woman who grew up in London, United Kingdom, before relocating north to the Yorkshire Dales National Park. She has kept up to date with the formal medical literature since she began to hormonally transition in 2018. Sam is currently employed working on the adoption of data-driven approaches, such as machine learning, to inform high-level business decision making. Her expertise includes biostatistics and computational modelling for predictive analysis.
Sam has contributed several articles to Transfeminine Science. Some of her other interests include hiking, astrophysics, and stargazing/astrophotography.
Lain (“they/them”) is non-binary and transfeminine. They completed a bachelor’s degree in applied mathematics and studied bioinformatics and computational biology at the Rochester Institute of Technology in New York. After withdrawing from their Ph.D. program, Lain migrated to the much warmer climes of the San Francisco Bay Area to work in tech.
As a person entrenched in nightlife community, specifically raving, Lain reformed and ran a nightlife harm reduction non-profit chapter for over 3 years, Bay Area DanceSafe. At DanceSafe they produced harm reductive literature on pertinent drugs and organized harm reduction booths at various festivals and raves. Through this direct outreach to the nightlife community, Lain led volunteers providing peer education on drugs, consent, sex education, and other pertinent topics as well as substance adulterant testing and harm reduction interventions. As a chapter head, they advocated for the normalization of harm reduction and the reform of drug policy working with peer organizations such as the Drug Policy Alliance (DPA), the Students for Sensible Drug Policy (SSDP), the Multidisciplinary Association for Psychedelic Studies (MAPS), and also by speaking at several events in the Bay Area.
As Lain began to medically transition, they began to passionately research endocrinology and transgender medicine and became involved with Transfeminine Science for a few years.
Mitzi (“she/they”) is a non-binary transfeminine person who lives in London, United Kingdom. She is prolifically active in DIY HRT communities, where she acts as an educator and advocates a harm reduction approach to self-medication. Mitzi frequently navigates situations that involve transgender homelessness, domestic violence, healthcare discrimination, and substance use both online and locally, and has held advisory roles for a variety of small grassroots organizations, including Trans Healthcare Network, Gender Construction Kit, and Bluelight.
Mitzi has a broad interest in medical academia, with a particular passion for endocrinology and psychopharmacology. She is an outspoken critic of her country’s transgender healthcare system, and has self-medicated for the duration of her own transition.
Transfeminine Science is no longer accepting contact inquiries. For questions and advice about hormone therapy, please refer instead to the online transgender community.
History
Transfeminine Science originated from the informational content on transfeminine hormone therapy that was created by transfeminine people in the transgender hormone therapy community on the social media website Reddit. Aly began creating content in August 2018, with other authors soon joining the effort. A couple years later, Aly developed and founded Transfeminine Science, which was launched in October 2020, and the existing content on Reddit was adapted and moved to the site. Transfeminine Science has served as a dedicated platform for the articles and for new content since it was started.
Disclaimer
The writers of Transfeminine Science are not medical professionals, academic researchers, or recognized experts in transgender health. Transfeminine Science articles emulate the format of scientific journal articles because this is an excellent format for scientific writing. However, it should be noted that Transfeminine Science itself is not a scientific journal and the content on this site has not been formally published nor scholarly peer-reviewed. Readers should not take the content on Transfeminine Science as authoritative. Instead, it should be considered as a supplementary resource to the information contained in transgender care guidelines and the medical literature in general.
Transfeminine Science is not specifically aimed at transfeminine people on do-it-yourself (DIY) hormone therapy. Rather, it is intended for transfeminine people on hormone therapy generally, as well as for medical providers and academics in transgender health. The authors of Transfeminine Science feel that wherever possible, decisions about medical care should be made in partnership with a health care professional. We recognize that this is not the case for many however, due to choice or circumstances, and we aim to help inform this important and undeserved community of individuals as well.
Recognition
Transfeminine Science has been cited, mentioned, and/or recognized in the published scientific literature by various academics. A partial list of these instances, with citations and excerpts, can be found here.
License
Transfeminine Science is copyright of Aly and the other authors of Transfeminine Science. We reserve all rights. We ask that readers please don’t reproduce content from Transfeminine Science unless given permission from Aly (e.g., for translation projects). If permission to reproduce content has been given, we ask that the content be appropriately attributed and linked back to the original page(s) on this site.
\ No newline at end of file
+About - Transfeminine Science
For an overview and description of Transfeminine Science, please visit our Home page.
Authors
The authors for Transfeminine Science include Aly, Sam, Lain, Mitzi, and Luna.
Aly
Aly (“she/her”) is a transgender woman from California. She has long had a passionate interest in transfeminine hormone therapy and pharmacology generally. Aly began her transition in the early 2010s and has studied the transgender medical literature since.
She has a bachelor’s degree with summa cum laude distinction from a University of California. Aly also completed some of the pre-medicine curriculum and worked for about a year in research labs at the university. Aside from medical areas, Aly has experience with computer programming and web development.
Aly is a volunteer editor on Wikipedia. She is a prolific medical editor at the online encyclopedia and has contributed a large amount of content in areas like sex hormone pharmacology and psychopharmacology over time. When Aly has included inline citations to Wikipedia on Transfeminine Science, they have usually been to content that she herself authored.
Sam (“she/they”) is a transgender woman who grew up in London, United Kingdom, before relocating north to the Yorkshire Dales National Park. She has kept up to date with the formal medical literature since she began to hormonally transition in 2018. Sam is currently employed working on the adoption of data-driven approaches, such as machine learning, to inform high-level business decision making. Her expertise includes biostatistics and computational modelling for predictive analysis.
Sam has contributed several articles to Transfeminine Science. Some of her other interests include hiking, astrophysics, and stargazing/astrophotography.
Lain (“they/them”) is non-binary and transfeminine. They completed a bachelor’s degree in applied mathematics and studied bioinformatics and computational biology at the Rochester Institute of Technology in New York. After withdrawing from their Ph.D. program, Lain migrated to the much warmer climes of the San Francisco Bay Area to work in tech.
As a person entrenched in nightlife community, specifically raving, Lain reformed and ran a nightlife harm reduction non-profit chapter for over 3 years, Bay Area DanceSafe. At DanceSafe they produced harm reductive literature on pertinent drugs and organized harm reduction booths at various festivals and raves. Through this direct outreach to the nightlife community, Lain led volunteers providing peer education on drugs, consent, sex education, and other pertinent topics as well as substance adulterant testing and harm reduction interventions. As a chapter head, they advocated for the normalization of harm reduction and the reform of drug policy working with peer organizations such as the Drug Policy Alliance (DPA), the Students for Sensible Drug Policy (SSDP), the Multidisciplinary Association for Psychedelic Studies (MAPS), and also by speaking at several events in the Bay Area.
As Lain began to medically transition, they began to passionately research endocrinology and transgender medicine and became involved with Transfeminine Science for a few years.
Mitzi (“she/they”) is a non-binary transfeminine person who lives in London, United Kingdom. She is prolifically active in DIY HRT communities, where she acts as an educator and advocates a harm reduction approach to self-medication. Mitzi frequently navigates situations that involve transgender homelessness, domestic violence, healthcare discrimination, and substance use both online and locally, and has held advisory roles for a variety of small grassroots organizations, including Trans Healthcare Network, Gender Construction Kit, and Bluelight.
Mitzi has a broad interest in medical academia, with a particular passion for endocrinology and psychopharmacology. She is an outspoken critic of her country’s transgender healthcare system, and has self-medicated for the duration of her own transition.
Transfeminine Science is no longer accepting contact inquiries. For questions and advice about hormone therapy, please refer instead to the online transgender community.
History
Transfeminine Science originated from the informational content on transfeminine hormone therapy that was created by transfeminine people in the transgender hormone therapy community on the social media website Reddit. Aly began creating content in August 2018, with other authors soon joining the effort. A couple years later, Aly developed and founded Transfeminine Science, which was launched in October 2020, and the existing content on Reddit was adapted and moved to the site. Transfeminine Science has served as a dedicated platform for the articles and for new content since it was started.
Disclaimer
The writers of Transfeminine Science are not medical professionals, academic researchers, or recognized experts in transgender health. Transfeminine Science articles emulate the format of scientific journal articles because this is an excellent format for scientific writing. However, it should be noted that Transfeminine Science itself is not a scientific journal and the content on this site has not been formally published nor scholarly peer-reviewed. Readers should not take the content on Transfeminine Science as authoritative. Instead, it should be considered as a supplementary resource to the information contained in transgender care guidelines and the medical literature in general.
Transfeminine Science is not specifically aimed at transfeminine people on do-it-yourself (DIY) hormone therapy. Rather, it is intended for transfeminine people on hormone therapy generally, as well as for medical providers and academics in transgender health. The authors of Transfeminine Science feel that wherever possible, decisions about medical care should be made in partnership with a health care professional. We recognize that this is not the case for many however, due to choice or circumstances, and we aim to help inform this important and undeserved community of individuals as well.
Recognition
Transfeminine Science has been cited, mentioned, and/or recognized in the published scientific literature by various academics. A partial list of these instances, with citations and excerpts, can be found here.
License
Transfeminine Science is copyright of Aly and the other authors of Transfeminine Science. We reserve all rights. We ask that readers please don’t reproduce content from Transfeminine Science unless given permission from Aly (e.g., for translation projects). If permission to reproduce content has been given, we ask that the content be appropriately attributed and linked back to the original page(s) on this site.
\ No newline at end of file
diff --git a/transfemscience.org/articles-by-author/aly/index.html b/transfemscience.org/articles-by-author/aly/index.html
index cf7852af..fdd6ee9a 100644
--- a/transfemscience.org/articles-by-author/aly/index.html
+++ b/transfemscience.org/articles-by-author/aly/index.html
@@ -1 +1 @@
-Articles by Aly - Transfeminine Science
\ No newline at end of file
diff --git a/transfemscience.org/articles-by-author/lain/index.html b/transfemscience.org/articles-by-author/lain/index.html
index a91a2017..b2202ee7 100644
--- a/transfemscience.org/articles-by-author/lain/index.html
+++ b/transfemscience.org/articles-by-author/lain/index.html
@@ -1 +1 @@
-Articles by Lain - Transfeminine Science
\ No newline at end of file
diff --git a/transfemscience.org/articles-by-author/mitzi/index.html b/transfemscience.org/articles-by-author/mitzi/index.html
index 972ae615..5f4e008c 100644
--- a/transfemscience.org/articles-by-author/mitzi/index.html
+++ b/transfemscience.org/articles-by-author/mitzi/index.html
@@ -1 +1 @@
-Articles by Mitzi - Transfeminine Science
\ No newline at end of file
diff --git a/transfemscience.org/articles-by-author/sam/index.html b/transfemscience.org/articles-by-author/sam/index.html
index 6047e85c..0fd094af 100644
--- a/transfemscience.org/articles-by-author/sam/index.html
+++ b/transfemscience.org/articles-by-author/sam/index.html
@@ -1 +1 @@
-Articles by Sam - Transfeminine Science
\ No newline at end of file
diff --git a/transfemscience.org/articles-by-date/index.html b/transfemscience.org/articles-by-date/index.html
index e26d0f8c..3210f8d0 100644
--- a/transfemscience.org/articles-by-date/index.html
+++ b/transfemscience.org/articles-by-date/index.html
@@ -1 +1 @@
-Articles by Date - Transfeminine Science
\ No newline at end of file
diff --git a/transfemscience.org/articles/aqueous-suspensions/index.html b/transfemscience.org/articles/aqueous-suspensions/index.html
index 822fa764..948d4471 100644
--- a/transfemscience.org/articles/aqueous-suspensions/index.html
+++ b/transfemscience.org/articles/aqueous-suspensions/index.html
@@ -1 +1 @@
-Injectable Aqueous Suspensions of Sex Hormones and How Depot Injectables Work - Transfeminine Science
Injectable Aqueous Suspensions of Sex Hormones and How Depot Injectables Work
By Aly | First published June 14, 2019 | Last modified August 2, 2022
Abstract / TL;DR
Depot injectable steroid preparations are formulated mainly as oil solutions and crystalline aqueous suspensions. Oil solutions of steroids are more well-known and widely used but aqueous suspensions also exist and continue to be employed in medicine. For example, depot medroxyprogesterone acetate and estradiol cypionate-containing combined injectable contraceptives are aqueous suspensions. Both oil solutions and aqueous suspensions produce a depot effect. However, they function in very different ways to produce this depot effect. Oil solutions form an oil depot at the site of injection which is slowly absorbed and from which the steroid slowly escapes, whereas aqueous suspensions deliver crystals of pure steroid which slowly dissolve and are absorbed. Aqueous suspensions are essentially like tiny pellet implants that can be injected instead of surgically inserted. Aqueous suspensions tend to have longer durations than oil solutions and can allow for much wider dosing intervals in comparison. The dissolution rates and durations of aqueous suspensions are highly dependent on crystal size. A limitation of aqueous suspensions is that they can cause pain and irritation at the site of injection in a way that oil solutions have not been associated with and this is likely responsible for their less widespread use. Aqueous suspensions of estradiol benzoate (Agofollin Depot) and progesterone (Agolutin Depot) remain limitedly and sporadically available and have much longer durations than the more common oil solutions of these steroids. These preparations may be potential options for use by transfeminine people.
Introduction
Depot injectable sex steroids are administered by intramuscular injection (into muscle) or subcutaneous injection (into fat) and are formulated in two main ways: 1) as oil solutions; and 2) as crystalline aqueous suspensions. What most transfeminine people are familiar with when it comes to injectable steroid preparations are oil solutions. This is how most common preparations like injectable estradiol valerate (Delestrogen, Progynon-Depot), estradiol cypionate (Depo-Estradiol), testosterone esters, progesterone, and hydroxyprogesterone caproate are formulated. However, injectable aqueous suspensions of sex steroids also exist and are used medically. The most major and well-known formulations include depot medroxyprogesterone acetate (Depo-Provera) and combined injectable contraceptives containing estradiol cypionate/medroxyprogesterone acetate (Cyclofem, Lunelle). Additionally, there exist more obscure preparations of aqueous suspensions such as estradiol benzoate (Agofollin Depot) and progesterone (Agolutin Depot) among others which are marketed in addition to the more common oil solutions of these steroids. These preparations may remain commercially available, including online from sites like EU Aibolit (Reddit). Many transfeminine people are unaware of what aqueous suspensions are or their medical availability. It is notable in this regard that aqueous suspensions are very different from oil solutions in their properties but can have much longer durations in comparison. This is potentially of therapeutic value especially in the case of otherwise shorter-acting injectables like progesterone. The purpose of this article is to shed light on injectable aqueous suspensions, describe how injectable oil solutions and aqueous suspensions work to achieve their depot effect and how they are different, and discuss how relevant aqueous suspensions can be obtained for medical use.
How Depot Injectables Work
Sex steroids are typically lipophilic (fat-soluble or “lipid-loving”) and hydrophobic (water-insoluble or “water-hating”). In other words, they dissolve in and are “attracted to” lipids (e.g., fats), and they are poorly soluble in and repelled by water. From chemistry, this is because sex steroids are very non-polar, whereas water is a quite polar molecule. As a result of their lipophilicity, sex steroids readily form clear homogenous solutions when mixed into oil—that is, they form oil solutions. In contrast, because sex steroids are hydrophobic, they do not easily form solutions when mixed into water—that is, they do not easily form aqueous solutions (like, e.g., salt and water). Instead of aqueous solutions, solid “clumps” or crystal particles of sex steroids can be mixed into and thereby suspended in water—that is, aqueous suspensions of sex steroid crystals can be made. Depot injectables are formulated as oil solutions or aqueous suspensions and these preparations have very different properties.
Injectable Oil Solutions
When an oil solution of a sex steroid is administered by intramuscular or subcutaneous injection, the solution is trapped within the tissue compartment it is injected into and remains there. As the tissue fluid is a water mixture, the oil solution stays together inside the tissue compartment and does not easily separate or distribute. This is because the lipophilic fats and sex steroids within the solution are attracted to each other and are repelled by water. Instead of rapidly dissolving, the fats and sex steroids at the edges of the oil solution are very slowly absorbed into the surrounding water. Once they have escaped the oil depot into the surrounding tissue fluid, they can be distributed into the bloodstream and then into other tissues to exert their biological effects. Eventually, the whole oil solution will be absorbed.
Oftentimes sex steroids that are used by intramuscular or subcutaneous injection are esterified with one or more lipophilic hydrocarbonesters. These esters include fatty acids like propanoic acid (propionate), pentanoic acid (valerate), hexanoic acid (caproate), heptanoic acid (enanthate), decanoic acid (decanote or decylate), and undecanoic acid (undecylate or undecanoate) as well as cyclic compounds like benzoic acid (benzoate), cyclopentylpropanoic acid (cypionate), and phenylpropanoic acid (phenylpropionate), among many others. Examples of these sex steroid esters include the well-known estradiol valerate, estradiol cypionate, estradiol benzoate, hydroxyprogesterone caproate, and numerous others. The attachment of a lipophilic ester (e.g., valeric acid) to a sex steroid (e.g., estradiol) will increase the lipophilicity of the sex steroid compared to merely injecting the unesterified sex steroid in an oil solution. The longer the carbon atom chain in the case of the simple fatty acid esters (e.g., propionate, valerate, enanthate, undecylate), the more lipophilic the resulting esterified sex steroid will be. As a result, the injected sex steroid ester will escape the oil tissue depot more slowly, lengthening the amount of time it takes for the sex steroid ester to be absorbed and therefore its duration in the body. The tables here and here show the lengthening duration of estradiol with longer or bulkier and and more lipophilic esters. Whereas an intramuscular injection of estradiol or progesterone in oil solution has a duration of only around 2 days, an intramuscular injection of an oil solution of estradiol undecylate, an ester of estradiol with a long fatty acid chain, has a duration measured in months. And an intramuscular injection of an oil solution of hydroxyprogesterone caproate, an ester of a derivative of progesterone that has a medium-length fatty acid chain, has a duration measured in weeks.
Most sex steroid esters themselves are biologically inactive. Once they have left the oil tissue depot, they are rapidly cleaved by esteraseenzymes into free unesterified steroid (e.g., estradiol, testosterone) and the previously connected ester moiety (e.g., valeric acid). Hence, most sex steroid esters are prodrugs and are otherwise identical to their parent sex steroids in their biological actions. In the case of esters of estradiol and testosterone, this means that they are bioidentical just like the unesterified steroids. Certain synthetic progesterone derivatives like hydroxyprogesterone caproate and medroxyprogesterone acetate are however not prodrugs and are not meaningfully cleaved into the unesterified parent compound. Instead, they have intrinsic hormonal activity of their own and act without bioactivation.
Injectable Aqueous Suspensions
Aqueous suspensions of sex steroids also form an injection-site depot and achieve a long-lasting depot effect when administered by subcutaneous or intramuscular injection. However, they work in a completely different way than oil solutions. Aqueous suspensions of sex steroids consist of tiny crystal particles of pure sex steroid that are suspended in water. These sex steroid particles are highly lipophilic and hydrophobic. When injected, the hydrophilic water vehicle is rapidly mixed into the fluid of the tissue compartment and absorbed by the body. But the hydrophobic sex steroid crystals are not, and instead float about in the fluid of the tissue compartment. As with oil solutions, the sex steroids at the edges of the crystals very slowly dissolve off the surface of the crystals into the surrounding water and are then distributed into the circulation and tissues. Eventually, the crystal will be fully absorbed into the body, but only after a long period of time. The rate of absorption of the particles is dependent on the properties of the particle crystal lattice and varies depending on the compound.
In the case of aqueous suspensions, the duration of the sex steroid is additionally highly dependent on particle size. These particle sizes have ranged from nanocrystalline to microcrystalline to macrocrystalline in their range. Almost always however it is microcrystalline particle sizes that have been used in injectable aqueous suspensions of sex steroids. (The present author has seen macrocrystalline preparations described a few times, specifically in research on combined injectable contraceptives (Garza-Flores, Del, & Perez-Palacios, 1992; Newton, d’Arcangues, & Hall, 1994; Sang, 1994), and is fairly sure that no such preparations have ever been marketed. On the other hand, nanocrystalline aqueous suspensions have been used with depot antipsychotics (Spanarello & Ferla, 2014; Correll et al., 2021).) Typically, there is a given particle size range for the formulation, such as 0.01 to 0.1 mm in diameter. The larger the particle sizes, the slower the absorption into the body, and the longer the duration of the preparation; the smaller the particle sizes, the faster the absorption, and the shorter the duration. When microcrystalline aqueous suspensions of sex steroids are manufactured nowadays, the particle sizes are defined and carefully controlled. Particle sizes influence the duration of injectable aqueous suspensions because they result in different surface areas from which sex steroid ester can escape particles. A single large particle has a smaller total surface area and hence dissolution rate than the same particle divided up into many smaller particles.
Particle sizes are manipulated during manufacturing via micronization—the process of decreasing the diameter of larger particles, such as via milling or grinding. Whereas more micronization improves the absorption and bioavailability of estradiol and progesterone with oral administration by increasing the surface area available for absorption into the body (Wiki; Wiki), less micronization decreases the rate of absorption of crystalline aqueous suspensions via depot injection and thereby extends the durations of these preparations by decreasing the total surface area for absorption.
There is a notable similarity of injectable aqueous suspensions of sex steroid to implantable sex steroid pellets, for instance of estradiol, testosterone, and progesterone (Wiki; Wiki; Wiki). Pellet implants are basically just pure crystalline sex steroid compressed into the shape of a small cylinder (Photo; Photo). They are inserted into subcutaneous fat in the body through a small incision using a large needle-like instrument called a trocar (Diagram). Once implanted, pellets slowly dissolve and absorb into the body over time, eventually disappearing completely. As they are nothing but pure crystalline hormone, there is no need for them to be removed or retrieved later on. In other words, implantation of a pellet is in a way the same thing as a subcutaneous injection of an aqueous suspension of sex steroid crystals—a single pellet is just one massive crystal instead of many tiny crystals suspended in water. And with very large crystals comes a very long duration—typically 6 months or more for each subcutaneous pellet of estradiol or testosterone (Kuhl, 2005; Wiki; Wiki). However, though injectable aqueous suspensions are typically much less prolonged than pellet implants, they have the advantages over pellets of being less expensive and not requiring a surgical incision. Due to the similarity between aqueous suspensions and pellet implants, aqueous suspensions have been described and marketed as “micropellets” in the past.
Medical Use of Injectable Aqueous Suspensions
Clinical Durations of Injectable Aqueous Suspensions
Studies that have compared sex steroids in injectable oil solutions versus injectable aqueous suspensions have generally found that the durations are considerably longer with aqueous suspensions than with oil solutions. Whereas injectable estradiol and estrone in oil solution have durations of only about 1 to 2 days, aqueous suspensions of these steroids have durations of 2 to 7 days (Table). Moreover, whereas injectable estradiol benzoate in oil solution has a duration of 4 to 6 days, injectable estradiol benzoate as a microcrystalline aqueous suspension has a duration of 2 to 3 weeks (Table). Such a duration is on par with the duration of longer-acting injectable estradiol esters like estradiol cypionate in oil solution and estradiol enanthate in oil solution. In addition, whereas injectable progesterone in oil solution has a duration of about 2 to 3 days, injectable aqueous suspensions of progesterone have a duration in the range of 1 to 2 weeks (Table). This is a duration that is comparable to that of injectable hydroxyprogesterone caproate in oil solution. The prolonged duration of injectable progesterone suspensions is particularly notable as progesterone cannot be esterified and hence injectable progesterone in oil solution cannot be prolonged except with structural modification (as in progestins like hydroxyprogesterone caproate and dihydroxyprogesterone acetophenide) (Wiki). The duration of injectable testosterone propionate in oil solution is 3 to 4 days, in a suspension of small crystals (0.04–0.1 mm) is 8 days, and as commercial-size crystals (0.05–0.2 mm) is 12 days (Sinkula, 1978). Testosterone isobutyrate as an aqueous suspension is said to have a duration of 2 to 3 weeks (Sinkula, 1978; Table). Some injectable aqueous suspensions can have extremely prolonged durations. Depot medroxyprogesterone acetate for instance has a duration of at least 3 months and as long as 6 to 9 months (Wiki). It greatly outlasts the related injectable progestogen norethisterone enanthate in oil solution (Noristerat) (Bassol & Garza-Flores, 1994; Paulen & Curtis, 2009).
Availability of Injectable Aqueous Suspensions
A list of injectable aqueous suspensions of sex steroids that are known to have been marketed can be found here. Most of them were introduced in the 1950s and many of them have been discontinued, but several of the preparations remain available today. These include the single-drug preparations estradiol benzoate (Agofollin Depot, Ovocyclin M), progesterone (Agolutin Depot), testosterone isobutyrate (Agovirin Depot), and medroxyprogesterone acetate (Depo-Provera, Depo-SubQ Provera 104) as well as the combination preparations estradiol benzoate/testosterone isobutyrate (Folivirin, Femandren M) and estradiol cypionate/medroxyprogesterone acetate (Cyclofem, Lunelle). Among the more notable injectable aqueous suspensions that are no longer marketed include estradiol (Aquadiol, Diogyn, Progynon Aqueous Suspension, Progynon Micropellets), estrone (Estrone Aqueous Suspension, Kestrone, Theelin Aqueous), and testosterone (Andronaq, Sterotate, Virosterone) as well as the combination preparation estradiol benzoate/progesterone (Sistocyclin).
Folivirin (estradiol benzoate/testosterone isobutyrate suspension): PDF; Google Translate
The company that manufactures these products, BB Pharma, is notably also the producer of the well-known Neofollin (injectable estradiol valerate in oil solution) in Europe.
Injection Procedure for Injectable Aqueous Suspensions
Aqueous suspensions require larger needle gauges (e.g., 20 or 21 gauge) than oil or aqueous solutions in order to allow the steroid crystals to pass through the needle lumen. The needle sizes may vary depending on the preparation and its crystal sizes. Injectable aqueous suspensions should be shaken adequately prior to injection as the crystals tend to aggregate. Aqueous suspensions are known to be more irritating and prone to causing pain and injection site reactions like redness and swelling than oil solutions, although this may vary depending on the preparation (Wiki; Wiki). This property is likely responsible for the discontinuation of many injectable suspensions and the greater popularity of injectable oil solutions. On the other hand, an advantage of aqueous suspensions over oil solutions is that in contrast them, there is no oil, and hence there is no risk of allergic reaction to the oil. As with injectable oil solutions, injectable aqueous suspensions are indicated for use specifically by intramuscular injection. Depot medroxyprogesterone acetate, which is available in both formulations for both intramuscular injection (Depo-Provera) and subcutaneous injection (Depo-SubQ Provera 104), is suggestive that aqueous suspensions have similar properties both by intramuscular and subcutaneous injection analogously to the case of injectable oil solutions (Wiki; Wiki). However, it should be noted that depot medroxyprogesterone acetate is formulated differently between these preparations.
Update: Pharmaceutical Suspensions Discontinued
Since this article was posted, Agofollin Depot (estradiol benzoate), Agolutin Depot (progesterone), and Folivirin (estradiol benzoate/testosterone isobutyrate) appear to have been discontinued by their manufacturer and seem to no longer be available from online pharmacies.
References
Bassol, S., & Garza-Flores, J. (1994). Review of ovulation return upon discontinuation of once-a-month injectable contraceptives. Contraception, 49(5), 441–453. [DOI:10.1016/0010-7824(94)90003-5]
Correll, C. U., Kim, E., Sliwa, J. K., Hamm, W., Gopal, S., Mathews, M., Venkatasubramanian, R., & Saklad, S. R. (2021). Pharmacokinetic Characteristics of Long-Acting Injectable Antipsychotics for Schizophrenia: An Overview. CNS Drugs, 35(1), 39–59. [DOI:10.1007/s40263-020-00779-5]
Garza-Flores, J., Cravioto, M. C., & Pérez-Palacios, G. (1992). Steroid injectable contraception: Current concepts and perspectives. In Sitruk-Ware, L. R., & Bardin, C. W. (Eds.). Contraception: Newer Pharmacological Agents, Devices, and Delivery Systems (pp. 41–70). New York: M. Dekker. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Newton, J. R., d’Arcangues, C., & Hall, P. E. (1994). A review of ‘once-a-month’ combined injectable contraceptives. Journal of Obstetrics and Gynaecology, 14(Suppl 1), S1–S34. [DOI:10.3109/01443619409027641]
Paulen, M. E., & Curtis, K. M. (2009). When can a woman have repeat progestogen-only injectables–depot medroxyprogesterone acetate or norethisterone enantate? Contraception, 80(4), 391–408. [DOI:10.1016/j.contraception.2009.03.023]
Sang, G. (1994). Pharmacodynamic effects of once-a-month combined injectable contraceptives. Contraception, 49(4), 361–385. [DOI:10.1016/0010-7824(94)90033-7]
Sinkula, A. A. (1978). Methods to Achieve Sustained Drug Delivery. The Chemical Approach. In Robinson, J. R. (Ed.). Sustained and Controlled Release Drug Delivery Systems (pp. 411–555). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books] [PDF]
Spanarello, S., & Ferla, T. (2014). The Pharmacokinetics of Long-Acting Antipsychotic Medications. Current Clinical Pharmacology, 9(3), 310–317. [DOI:10.2174/15748847113089990051]
\ No newline at end of file
+Injectable Aqueous Suspensions of Sex Hormones and How Depot Injectables Work - Transfeminine Science
Injectable Aqueous Suspensions of Sex Hormones and How Depot Injectables Work
By Aly | First published June 14, 2019 | Last modified August 2, 2022
Abstract / TL;DR
Depot injectable steroid preparations are formulated mainly as oil solutions and crystalline aqueous suspensions. Oil solutions of steroids are more well-known and widely used but aqueous suspensions also exist and continue to be employed in medicine. For example, depot medroxyprogesterone acetate and estradiol cypionate-containing combined injectable contraceptives are aqueous suspensions. Both oil solutions and aqueous suspensions produce a depot effect. However, they function in very different ways to produce this depot effect. Oil solutions form an oil depot at the site of injection which is slowly absorbed and from which the steroid slowly escapes, whereas aqueous suspensions deliver crystals of pure steroid which slowly dissolve and are absorbed. Aqueous suspensions are essentially like tiny pellet implants that can be injected instead of surgically inserted. Aqueous suspensions tend to have longer durations than oil solutions and can allow for much wider dosing intervals in comparison. The dissolution rates and durations of aqueous suspensions are highly dependent on crystal size. A limitation of aqueous suspensions is that they can cause pain and irritation at the site of injection in a way that oil solutions have not been associated with and this is likely responsible for their less widespread use. Aqueous suspensions of estradiol benzoate (Agofollin Depot) and progesterone (Agolutin Depot) remain limitedly and sporadically available and have much longer durations than the more common oil solutions of these steroids. These preparations may be potential options for use by transfeminine people.
Introduction
Depot injectable sex steroids are administered by intramuscular injection (into muscle) or subcutaneous injection (into fat) and are formulated in two main ways: 1) as oil solutions; and 2) as crystalline aqueous suspensions. What most transfeminine people are familiar with when it comes to injectable steroid preparations are oil solutions. This is how most common preparations like injectable estradiol valerate (Delestrogen, Progynon-Depot), estradiol cypionate (Depo-Estradiol), testosterone esters, progesterone, and hydroxyprogesterone caproate are formulated. However, injectable aqueous suspensions of sex steroids also exist and are used medically. The most major and well-known formulations include depot medroxyprogesterone acetate (Depo-Provera) and combined injectable contraceptives containing estradiol cypionate/medroxyprogesterone acetate (Cyclofem, Lunelle). Additionally, there exist more obscure preparations of aqueous suspensions such as estradiol benzoate (Agofollin Depot) and progesterone (Agolutin Depot) among others which are marketed in addition to the more common oil solutions of these steroids. These preparations may remain commercially available, including online from sites like EU Aibolit (Reddit). Many transfeminine people are unaware of what aqueous suspensions are or their medical availability. It is notable in this regard that aqueous suspensions are very different from oil solutions in their properties but can have much longer durations in comparison. This is potentially of therapeutic value especially in the case of otherwise shorter-acting injectables like progesterone. The purpose of this article is to shed light on injectable aqueous suspensions, describe how injectable oil solutions and aqueous suspensions work to achieve their depot effect and how they are different, and discuss how relevant aqueous suspensions can be obtained for medical use.
How Depot Injectables Work
Sex steroids are typically lipophilic (fat-soluble or “lipid-loving”) and hydrophobic (water-insoluble or “water-hating”). In other words, they dissolve in and are “attracted to” lipids (e.g., fats), and they are poorly soluble in and repelled by water. From chemistry, this is because sex steroids are very non-polar, whereas water is a quite polar molecule. As a result of their lipophilicity, sex steroids readily form clear homogenous solutions when mixed into oil—that is, they form oil solutions. In contrast, because sex steroids are hydrophobic, they do not easily form solutions when mixed into water—that is, they do not easily form aqueous solutions (like, e.g., salt and water). Instead of aqueous solutions, solid “clumps” or crystal particles of sex steroids can be mixed into and thereby suspended in water—that is, aqueous suspensions of sex steroid crystals can be made. Depot injectables are formulated as oil solutions or aqueous suspensions and these preparations have very different properties.
Injectable Oil Solutions
When an oil solution of a sex steroid is administered by intramuscular or subcutaneous injection, the solution is trapped within the tissue compartment it is injected into and remains there. As the tissue fluid is a water mixture, the oil solution stays together inside the tissue compartment and does not easily separate or distribute. This is because the lipophilic fats and sex steroids within the solution are attracted to each other and are repelled by water. Instead of rapidly dissolving, the fats and sex steroids at the edges of the oil solution are very slowly absorbed into the surrounding water. Once they have escaped the oil depot into the surrounding tissue fluid, they can be distributed into the bloodstream and then into other tissues to exert their biological effects. Eventually, the whole oil solution will be absorbed.
Oftentimes sex steroids that are used by intramuscular or subcutaneous injection are esterified with one or more lipophilic hydrocarbonesters. These esters include fatty acids like propanoic acid (propionate), pentanoic acid (valerate), hexanoic acid (caproate), heptanoic acid (enanthate), decanoic acid (decanote or decylate), and undecanoic acid (undecylate or undecanoate) as well as cyclic compounds like benzoic acid (benzoate), cyclopentylpropanoic acid (cypionate), and phenylpropanoic acid (phenylpropionate), among many others. Examples of these sex steroid esters include the well-known estradiol valerate, estradiol cypionate, estradiol benzoate, hydroxyprogesterone caproate, and numerous others. The attachment of a lipophilic ester (e.g., valeric acid) to a sex steroid (e.g., estradiol) will increase the lipophilicity of the sex steroid compared to merely injecting the unesterified sex steroid in an oil solution. The longer the carbon atom chain in the case of the simple fatty acid esters (e.g., propionate, valerate, enanthate, undecylate), the more lipophilic the resulting esterified sex steroid will be. As a result, the injected sex steroid ester will escape the oil tissue depot more slowly, lengthening the amount of time it takes for the sex steroid ester to be absorbed and therefore its duration in the body. The tables here and here show the lengthening duration of estradiol with longer or bulkier and and more lipophilic esters. Whereas an intramuscular injection of estradiol or progesterone in oil solution has a duration of only around 2 days, an intramuscular injection of an oil solution of estradiol undecylate, an ester of estradiol with a long fatty acid chain, has a duration measured in months. And an intramuscular injection of an oil solution of hydroxyprogesterone caproate, an ester of a derivative of progesterone that has a medium-length fatty acid chain, has a duration measured in weeks.
Most sex steroid esters themselves are biologically inactive. Once they have left the oil tissue depot, they are rapidly cleaved by esteraseenzymes into free unesterified steroid (e.g., estradiol, testosterone) and the previously connected ester moiety (e.g., valeric acid). Hence, most sex steroid esters are prodrugs and are otherwise identical to their parent sex steroids in their biological actions. In the case of esters of estradiol and testosterone, this means that they are bioidentical just like the unesterified steroids. Certain synthetic progesterone derivatives like hydroxyprogesterone caproate and medroxyprogesterone acetate are however not prodrugs and are not meaningfully cleaved into the unesterified parent compound. Instead, they have intrinsic hormonal activity of their own and act without bioactivation.
Injectable Aqueous Suspensions
Aqueous suspensions of sex steroids also form an injection-site depot and achieve a long-lasting depot effect when administered by subcutaneous or intramuscular injection. However, they work in a completely different way than oil solutions. Aqueous suspensions of sex steroids consist of tiny crystal particles of pure sex steroid that are suspended in water. These sex steroid particles are highly lipophilic and hydrophobic. When injected, the hydrophilic water vehicle is rapidly mixed into the fluid of the tissue compartment and absorbed by the body. But the hydrophobic sex steroid crystals are not, and instead float about in the fluid of the tissue compartment. As with oil solutions, the sex steroids at the edges of the crystals very slowly dissolve off the surface of the crystals into the surrounding water and are then distributed into the circulation and tissues. Eventually, the crystal will be fully absorbed into the body, but only after a long period of time. The rate of absorption of the particles is dependent on the properties of the particle crystal lattice and varies depending on the compound.
In the case of aqueous suspensions, the duration of the sex steroid is additionally highly dependent on particle size. These particle sizes have ranged from nanocrystalline to microcrystalline to macrocrystalline in their range. Almost always however it is microcrystalline particle sizes that have been used in injectable aqueous suspensions of sex steroids. (The present author has seen macrocrystalline preparations described a few times, specifically in research on combined injectable contraceptives (Garza-Flores, Del, & Perez-Palacios, 1992; Newton, d’Arcangues, & Hall, 1994; Sang, 1994), and is fairly sure that no such preparations have ever been marketed. On the other hand, nanocrystalline aqueous suspensions have been used with depot antipsychotics (Spanarello & Ferla, 2014; Correll et al., 2021).) Typically, there is a given particle size range for the formulation, such as 0.01 to 0.1 mm in diameter. The larger the particle sizes, the slower the absorption into the body, and the longer the duration of the preparation; the smaller the particle sizes, the faster the absorption, and the shorter the duration. When microcrystalline aqueous suspensions of sex steroids are manufactured nowadays, the particle sizes are defined and carefully controlled. Particle sizes influence the duration of injectable aqueous suspensions because they result in different surface areas from which sex steroid ester can escape particles. A single large particle has a smaller total surface area and hence dissolution rate than the same particle divided up into many smaller particles.
Particle sizes are manipulated during manufacturing via micronization—the process of decreasing the diameter of larger particles, such as via milling or grinding. Whereas more micronization improves the absorption and bioavailability of estradiol and progesterone with oral administration by increasing the surface area available for absorption into the body (Wiki; Wiki), less micronization decreases the rate of absorption of crystalline aqueous suspensions via depot injection and thereby extends the durations of these preparations by decreasing the total surface area for absorption.
There is a notable similarity of injectable aqueous suspensions of sex steroid to implantable sex steroid pellets, for instance of estradiol, testosterone, and progesterone (Wiki; Wiki; Wiki). Pellet implants are basically just pure crystalline sex steroid compressed into the shape of a small cylinder (Photo; Photo). They are inserted into subcutaneous fat in the body through a small incision using a large needle-like instrument called a trocar (Diagram). Once implanted, pellets slowly dissolve and absorb into the body over time, eventually disappearing completely. As they are nothing but pure crystalline hormone, there is no need for them to be removed or retrieved later on. In other words, implantation of a pellet is in a way the same thing as a subcutaneous injection of an aqueous suspension of sex steroid crystals—a single pellet is just one massive crystal instead of many tiny crystals suspended in water. And with very large crystals comes a very long duration—typically 6 months or more for each subcutaneous pellet of estradiol or testosterone (Kuhl, 2005; Wiki; Wiki). However, though injectable aqueous suspensions are typically much less prolonged than pellet implants, they have the advantages over pellets of being less expensive and not requiring a surgical incision. Due to the similarity between aqueous suspensions and pellet implants, aqueous suspensions have been described and marketed as “micropellets” in the past.
Medical Use of Injectable Aqueous Suspensions
Clinical Durations of Injectable Aqueous Suspensions
Studies that have compared sex steroids in injectable oil solutions versus injectable aqueous suspensions have generally found that the durations are considerably longer with aqueous suspensions than with oil solutions. Whereas injectable estradiol and estrone in oil solution have durations of only about 1 to 2 days, aqueous suspensions of these steroids have durations of 2 to 7 days (Table). Moreover, whereas injectable estradiol benzoate in oil solution has a duration of 4 to 6 days, injectable estradiol benzoate as a microcrystalline aqueous suspension has a duration of 2 to 3 weeks (Table). Such a duration is on par with the duration of longer-acting injectable estradiol esters like estradiol cypionate in oil solution and estradiol enanthate in oil solution. In addition, whereas injectable progesterone in oil solution has a duration of about 2 to 3 days, injectable aqueous suspensions of progesterone have a duration in the range of 1 to 2 weeks (Table). This is a duration that is comparable to that of injectable hydroxyprogesterone caproate in oil solution. The prolonged duration of injectable progesterone suspensions is particularly notable as progesterone cannot be esterified and hence injectable progesterone in oil solution cannot be prolonged except with structural modification (as in progestins like hydroxyprogesterone caproate and dihydroxyprogesterone acetophenide) (Wiki). The duration of injectable testosterone propionate in oil solution is 3 to 4 days, in a suspension of small crystals (0.04–0.1 mm) is 8 days, and as commercial-size crystals (0.05–0.2 mm) is 12 days (Sinkula, 1978). Testosterone isobutyrate as an aqueous suspension is said to have a duration of 2 to 3 weeks (Sinkula, 1978; Table). Some injectable aqueous suspensions can have extremely prolonged durations. Depot medroxyprogesterone acetate for instance has a duration of at least 3 months and as long as 6 to 9 months (Wiki). It greatly outlasts the related injectable progestogen norethisterone enanthate in oil solution (Noristerat) (Bassol & Garza-Flores, 1994; Paulen & Curtis, 2009).
Availability of Injectable Aqueous Suspensions
A list of injectable aqueous suspensions of sex steroids that are known to have been marketed can be found here. Most of them were introduced in the 1950s and many of them have been discontinued, but several of the preparations remain available today. These include the single-drug preparations estradiol benzoate (Agofollin Depot, Ovocyclin M), progesterone (Agolutin Depot), testosterone isobutyrate (Agovirin Depot), and medroxyprogesterone acetate (Depo-Provera, Depo-SubQ Provera 104) as well as the combination preparations estradiol benzoate/testosterone isobutyrate (Folivirin, Femandren M) and estradiol cypionate/medroxyprogesterone acetate (Cyclofem, Lunelle). Among the more notable injectable aqueous suspensions that are no longer marketed include estradiol (Aquadiol, Diogyn, Progynon Aqueous Suspension, Progynon Micropellets), estrone (Estrone Aqueous Suspension, Kestrone, Theelin Aqueous), and testosterone (Andronaq, Sterotate, Virosterone) as well as the combination preparation estradiol benzoate/progesterone (Sistocyclin).
Folivirin (estradiol benzoate/testosterone isobutyrate suspension): PDF; Google Translate
The company that manufactures these products, BB Pharma, is notably also the producer of the well-known Neofollin (injectable estradiol valerate in oil solution) in Europe.
Injection Procedure for Injectable Aqueous Suspensions
Aqueous suspensions require larger needle gauges (e.g., 20 or 21 gauge) than oil or aqueous solutions in order to allow the steroid crystals to pass through the needle lumen. The needle sizes may vary depending on the preparation and its crystal sizes. Injectable aqueous suspensions should be shaken adequately prior to injection as the crystals tend to aggregate. Aqueous suspensions are known to be more irritating and prone to causing pain and injection site reactions like redness and swelling than oil solutions, although this may vary depending on the preparation (Wiki; Wiki). This property is likely responsible for the discontinuation of many injectable suspensions and the greater popularity of injectable oil solutions. On the other hand, an advantage of aqueous suspensions over oil solutions is that in contrast them, there is no oil, and hence there is no risk of allergic reaction to the oil. As with injectable oil solutions, injectable aqueous suspensions are indicated for use specifically by intramuscular injection. Depot medroxyprogesterone acetate, which is available in both formulations for both intramuscular injection (Depo-Provera) and subcutaneous injection (Depo-SubQ Provera 104), is suggestive that aqueous suspensions have similar properties both by intramuscular and subcutaneous injection analogously to the case of injectable oil solutions (Wiki; Wiki). However, it should be noted that depot medroxyprogesterone acetate is formulated differently between these preparations.
Update: Pharmaceutical Suspensions Discontinued
Since this article was posted, Agofollin Depot (estradiol benzoate), Agolutin Depot (progesterone), and Folivirin (estradiol benzoate/testosterone isobutyrate) appear to have been discontinued by their manufacturer and seem to no longer be available from online pharmacies.
References
Bassol, S., & Garza-Flores, J. (1994). Review of ovulation return upon discontinuation of once-a-month injectable contraceptives. Contraception, 49(5), 441–453. [DOI:10.1016/0010-7824(94)90003-5]
Correll, C. U., Kim, E., Sliwa, J. K., Hamm, W., Gopal, S., Mathews, M., Venkatasubramanian, R., & Saklad, S. R. (2021). Pharmacokinetic Characteristics of Long-Acting Injectable Antipsychotics for Schizophrenia: An Overview. CNS Drugs, 35(1), 39–59. [DOI:10.1007/s40263-020-00779-5]
Garza-Flores, J., Cravioto, M. C., & Pérez-Palacios, G. (1992). Steroid injectable contraception: Current concepts and perspectives. In Sitruk-Ware, L. R., & Bardin, C. W. (Eds.). Contraception: Newer Pharmacological Agents, Devices, and Delivery Systems (pp. 41–70). New York: M. Dekker. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Newton, J. R., d’Arcangues, C., & Hall, P. E. (1994). A review of ‘once-a-month’ combined injectable contraceptives. Journal of Obstetrics and Gynaecology, 14(Suppl 1), S1–S34. [DOI:10.3109/01443619409027641]
Paulen, M. E., & Curtis, K. M. (2009). When can a woman have repeat progestogen-only injectables–depot medroxyprogesterone acetate or norethisterone enantate? Contraception, 80(4), 391–408. [DOI:10.1016/j.contraception.2009.03.023]
Sang, G. (1994). Pharmacodynamic effects of once-a-month combined injectable contraceptives. Contraception, 49(4), 361–385. [DOI:10.1016/0010-7824(94)90033-7]
Sinkula, A. A. (1978). Methods to Achieve Sustained Drug Delivery. The Chemical Approach. In Robinson, J. R. (Ed.). Sustained and Controlled Release Drug Delivery Systems (pp. 411–555). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books] [PDF]
Spanarello, S., & Ferla, T. (2014). The Pharmacokinetics of Long-Acting Antipsychotic Medications. Current Clinical Pharmacology, 9(3), 310–317. [DOI:10.2174/15748847113089990051]
\ No newline at end of file
diff --git a/transfemscience.org/articles/bica-adoption/index.html b/transfemscience.org/articles/bica-adoption/index.html
index 3f50a6ac..9dad84f5 100644
--- a/transfemscience.org/articles/bica-adoption/index.html
+++ b/transfemscience.org/articles/bica-adoption/index.html
@@ -1 +1 @@
-Bicalutamide and its Adoption by the Medical Community for Use in Transfeminine Hormone Therapy - Transfeminine Science
Bicalutamide and its Adoption by the Medical Community for Use in Transfeminine Hormone Therapy
By Aly | First published July 1, 2020 | Last modified August 23, 2025
Abstract / TL;DR
Bicalutamide is an antiandrogen which was introduced for the treatment of prostate cancer many years ago. Cost precluded its widespread use for other indications for many years. However, its cost has since come down and bicalutamide is now seeing significant adoption for use in transfeminine hormone therapy as well as for treatment of androgen-dependent conditions in other populations like cisgender women. Bicalutamide has risks of certain rare adverse effects like liver toxicity which have generated concerns about its safety and have limited its use in transfeminine people. However, while still significant, these risks are low with appropriate monitoring and clinical management. Prominent researchers in transgender medicine have recently shown openness to bicalutamide for potential use in transfeminine people and have written positively about it. Bicalutamide could eventually come to be regarded as acceptably safe for use in transfeminine hormone therapy, similarly to other antiandrogens with rare risks like spironolactone and cyproterone acetate. However, more studies and characterization of bicalutamide in transfeminine people will likely be needed before it could see wider adoption in transgender medicine.
History of Bicalutamide for Transfeminine People
Bicalutamide (Casodex) is a nonsteroidal antiandrogen and selectiveantagonist of the androgen receptor which was originally introduced for the treatment of prostate cancer in cisgender men in 1995. Prostate cancer is an androgen-dependent disease, so antiandrogens are effective in treating it. Bicalutamide has major advantages over other antiandrogens such as spironolactone (Aldactone) and cyproterone acetate (Androcur) in terms of antiandrogenicpotency, clinical effectiveness, pharmacological selectivity, and tolerability. It also has improved potency, pharmacokinetic properties, and tolerability, as well as far better safety, compared to the older nonsteroidal antiandrogens flutamide (Eulexin) and nilutamide (Anandron, Nilandron). However, use of bicalutamide as an antiandrogen in transfeminine hormone therapy is very recent. The employment of bicalutamide for transfeminine people was largely precluded for many years by the fact that bicalutamide had pharmaceutical patent protection and was very expensive. However, this changed with the availability of generic versions of bicalutamide starting in 2007 to 2009. In addition, newer and more effective antiandrogens like abiraterone acetate (Zytiga) in 2011 and enzalutamide (Xtandi) in 2012 were introduced and superseded bicalutamide as the standard-of-care antiandrogen for the treatment of prostate cancer. These developments have greatly reduced the cost of bicalutamide and it has gradually become much more affordable in the last decade.
Before 2015, there were only a few mentions in the literature of bicalutamide for transfeminine people and a handful of anecdotal reports online of transfeminine people using it. The earliest clear mention of bicalutamide in the literature in the context of transfeminine hormone therapy was by Louis Gooren in 2011 (Gooren, 2011). Gooren is a major longtime researcher in the field of transgender medicine and is one of the coauthors of the Endocrine Society’s transgender hormone therapy guidelines (Hembree et al., 2009; Hembree et al., 2017). He and his colleagues at the Center of Expertise on Gender Dysphoria of the Vrije Universiteit Medical Center (VUMC) in Amsterdam, Netherlands had conducted studies on nilutamide (Anandron, Nilandron) as an antiandrogen for transfeminine people in the late 1980s and early 1990s (de Voogt et al., 1987a; de Voogt et al., 1987b; Gooren et al., 1987; Johannes et al., 1987; Rao et al., 1988; Asscheman, Gooren, & Peereboom-Wynia, 1989; van Kemenade et al., 1989; Wiki). However, they seem to have abandoned it—probably due to its high incidence of lung toxicity and other off-target side effects. Nonetheless, Gooren began including nonsteroidal antiandrogens like flutamide and nilutamide in his publications as potential treatment options for transfeminine hormone therapy starting in the 1990s (Asscheman & Gooren, 1992; Gooren, 1999). Subsequently, flutamide was included in transgender health guidelines and other publications, though not necessarily favorably (e.g., Israel & Tarver, 1997; Levy, Crown, & Reid, 2003; Dahl et al., 2006a; Dahl et al., 2006b; Hembree et al., 2009; Moreno-Pérez et al., 2012). As a researcher interested in nonsteroidal antiandrogens for transfeminine people, bicalutamide—with its far better safety profile than flutamide and nilutamide—may have been appealing to Gooren. However, Gooren and his colleagues didn’t conduct clinical studies on bicalutamide for transfeminine people and never went beyond brief mention of it for such uses in their publications. Nor did any other academics.
Although there was little discussion or use of bicalutamide in transfeminine people prior to 2015, this started to change in mid-2015. At that time, the Wikipedia content for bicalutamide was greatly expanded, which made information about bicalutamide more accessible. In addition, certain transfeminine people, noting its advantages over existing options and its excellent potential for use in transfeminine hormone therapy, began advocating for use of bicalutamide in transfeminine people in online circles. A number of open-minded clinicians started adopting bicalutamide in transfeminine people around this time and thereafter as well. The first clinical study of bicalutamide in transfeminine people, which began in 2013, was published as an abstract in 2017 and as a full paper in 2019 (Neyman, Fuqua, & Eugster, 2017; Neyman, Fuqua, & Eugster, 2019). It was a small retrospective chart review of bicalutamide alone as a second-line puberty blocker in adolescent transgender girls for whom gonadotropin-releasing hormone analogues were denied by insurance. As of present, it remains the only published clinical data on bicalutamide in transfeminine people. It’s not exactly great data by any means, but it’s a study at least. The researchers who conducted the study had previously published on bicalutamide as a puberty blocker in boys with gonadotropin-independent precocious puberty (e.g., Lenz et al., 2010; Haddad & Eugster, 2012). While limited in its findings, Neyman, Fuqua, and Eugster (2019) helped to generate significant interest among clinicians and researchers in bicalutamide for use in transfeminine hormone therapy.
In any case, due to the recent nature of bicalutamide as an option for use in transfeminine hormone therapy, as well as the lack of studies and characterization of bicalutamide in transfeminine people and concerns about its safety (see next section), bicalutamide isn’t widely used in transfeminine people at this time. In fact, transgender hormone therapy guidelines largely don’t even mention it still. At present, the use of bicalutamide in transfeminine people is mostly limited to a number of more flexible clinicians and to people in the transgender do-it-yourself (DIY) hormone therapy community.
Concerns About Bicalutamide Limiting its Use
The transgender medical community has been reluctant to endorse the use of bicalutamide in transfeminine people to date. This is because of the lack of clinical studies and characterization of bicalutamide in transfeminine people, most importantly in terms of safety. There have been concerns about rare instances of liver failure that have occurred with bicalutamide in men with prostate cancer (Wiki). The reported cases of liver toxicity with bicalutamide have generally been sudden-onset and severe. Rare liver toxicity is an acceptable risk in men with prostate cancer because the risk–benefit ratio of bicalutamide therapy is very favorable, with the benefit of treating prostate cancer vastly outweighing the harm of the very rare instances of liver problems. But transfeminine people are typically young and healthy, and bicalutamide isn’t treating a terminal illness when it’s used in us. If a transfeminine person develops liver failure and dies because of bicalutamide, that’s unnecessary harm and a life needlessly lost. Accordingly, the University of California San Francisco (UCSF) transgender care guidelines warn against use of bicalutamide in transfeminine people currently due to potential liver risks (Deutsch, 2016). Aside from risks, there is also a lack of data to guide appropriate dosing of bicalutamide in transfeminine people at this time. A typical bicalutamide dosage of 50 mg/day is being used and recommended, but this has been arbitrarily chosen with little basis to support it.
To date, there are 10 published case reports of serious liver toxicity in association with bicalutamide (Table). All of these cases were in men with prostate cancer and all occurred within 6 months of initiation of bicalutamide therapy, with two of the cases resulting in death. While this is not a lot of cases and may seem reassuring, it must be noted that quantity of published case reports tends to vastly underestimate the true incidence of rare adverse reactions. As an example, there are around 50 published case reports of meningioma with cyproterone acetate (Table), but a recent large study by the French government found that there were more than 500 operated instances of meningioma in association with high-dose cyproterone acetate over an 8-year period in France alone (Aly, 2020). Accordingly, as of writing there are 40 reports of liver failure, including 25 consequent deaths, in association with bicalutamide in the U.S. FDA’s international MedWatch/FAERS database. (As well as 240 cases of interstitial lung disease associated with bicalutamide notably—relative to only 14 published case reports; Table.) Even with this database however, fewer than 10% of serious adverse reactions are estimated to be reported (Graham, Ahmad, & Piazza-Hepp, 2002). Hence, the true numbers may be much greater. These instances are merely co-occurrences, and causality in terms of bicalutamide and liver toxicity has not been established. But they are concerning nonetheless. There is additionally an unpublished case anecdote of death in a young transfeminine person associated with bicalutamide that’s been making its rounds through the transgender medical community. Per certain very credible people in the field of transgender medicine (e.g., Asa Radix and Zil Goldstein), she is said to have been a 20-year-old transgender girl in Texas taking bicalutamide with rapid-onset liver failure and no warning signs. This case has given clinicians and researchers who are aware of it reservations about the use of bicalutamide in hormone therapy for transfeminine people. Another case of liver failure and death in a transgender person over 60 years of age who was treated with bicalutamide has also been informally reported (QueerDoc).
In any case, the reported cases of serious liver toxicity with bicalutamide in transgender people have not been published nor properly confirmed. In addition, the absolute incidence of liver toxicity with bicalutamide is likely to be very low. For instance, the incidence of abnormal liver function tests (i.e., elevated liver enzymes on blood work) was only 3.4% with high-dose (150 mg/day) bicalutamide monotherapy relative to 1.9% for placebo (a 1.5% difference attributable to bicalutamide) at 3.0 years of follow-up in the Early Prostate Cancer (EPC) clinical programme, a series of three phase 3randomized controlled trials consisting of over 8,000 patients in which bicalutamide was evaluated for treatment of early prostate cancer (Anderson, 2003; Iversen et al., 2004; Wiki; Wiki). Moreover, there were no cases of serious liver toxicity or liver failure with bicalutamide in the initial clinical development programme of bicalutamide for advanced prostate cancer, in which almost 4,000 men were treated with bicalutamide (Blackledge, 1996; Kolvenbag & Blackledge, 1996; McLeod, 1997; Anderson, 2003; Iversen et al., 2004; Wiki). However, it should be noted that this was with careful monitoring of liver function in patients and with prompt discontinuation of bicalutamide upon detection of clinically concerning hepatic abnormalities. About 0.5 to 1.5% of men taking 50 to 150 mg/day bicalutamide in the major clinical programmes of bicalutamide for prostate cancer developed liver changes sufficiently marked that they required discontinuation (Blackledge, 1996; See et al., 2002; Wiki). Hence, regular liver monitoring is essential with bicalutamide to ensure that the possibility of severe liver toxicity is avoided.
Bicalutamide has a much lower risk of liver toxicity than its analogue flutamide (Kolvenbag & Blackledge, 1996; Schellhammer et al., 1997; Thole et al., 2004; Manso et al., 2006; Table). However, it retains a small risk of liver toxicity of its own—one with the potential for serious consequences. Hence, caution is warranted with its use, and careful liver monitoring is a necessity for anyone taking it.
Recent Developments and the Future
Bicalutamide is currently being adopted and characterized for use in the treatment androgen-dependent skin and hair conditions in cisgender women. For instance, a rigorous Italian phase 3 randomized controlled trial of bicalutamide for hirsutism was recently published (Moretti et al., 2018). Retrospective chart reviews of bicalutamide for scalp hair loss in cisgender women have also been published recently (Fernandez-Nieto et al., 2019; Ismail et al., 2020; Fernandez-Nieto et al., 2020; Moussa et al., 2021). The hair loss studies have observed low though significant rates of liver changes with bicalutamide.
Certain transgender medical researchers are showing interest in bicalutamide as well. Perhaps most notably, Wylie Hembree—the lead author of the Endocrine Society’s 2009 and 2017 transgender hormone therapy guidelines (Hembree et al., 2009; Hembree et al., 2017)—wrote positively about bicalutamide for transfeminine people in a recent review (Fishman, Paliou, Poretsky, & Hembree, 2019). He and his colleagues cited the recent phase 3 trial of bicalutamide for hirsutism in cisgender women and the study of bicalutamide as a puberty blocker in transgender girls in support of potential use of bicalutamide for transfeminine people. Guy T’Sjoen—another major researcher in transgender medicine and co-author of the Endocrine Society guidelines (Hembree et al., 2017; Mitchell, 2020)—seemed to show openness to bicalutamide with his colleagues in a recent review as well (Iwamoto et al., 2019). Researchers outside of the United States in particular may be more open to bicalutamide, owing to accumulating health concerns with cyproterone acetate—the most commonly used antiandrogen outside of the United States (Aly, 2020). John Randolph, a researcher at the University of Michigan, has also written positively about bicalutamide (Randolph, 2018), though he may have since changed his mind on it (Michigan Medicine, 2020). On the other hand, other authors have not been as welcoming of bicalutamide for transfeminine people (e.g., Hamidi & Davidge-Pitts, 2019; Cocchetti et al., 2020).
The small risks of bicalutamide with appropriate monitoring may prove to be acceptable to the transgender medical community. This would perhaps be analogous to the rare incidences of serious adverse effects with say spironolactone (e.g., hyperkalemia) or cyproterone acetate (e.g., benign brain tumors, blood clots, breast cancer, liver toxicity). It’s possible that bicalutamide may not ultimately be recommended as a first-line therapy due to its risks. However, it could still be allowed as a second-line option when other antiandrogens are less feasible or not possible due to being for instance inadequately effective, poorly tolerated, contraindicated, or unavailable. The transgender medical community isn’t there at this time though. More developments—namely studies and characterization of bicalutamide in actual transfeminine people—are likely to be needed before bicalutamide could become more accepted for use in transfeminine people or recommended in transgender hormone therapy guidelines.
Updates
Update 1: Thompson et al. (2021) [Fenway Health Guidelines]
In March 2021, the Fenway Health transgender health clinical practice guidelines were updated from the last version (October 2015) to the following latest edition (Aly, 2020):
Thompson, J., Hopwood, R. A., deNormand, S., & Cavanaugh, T. (2021). Medical Care of Trans and Gender Diverse Adults. Boston: Fenway Health. [URL] [PDF]
This update is notable as these guidelines included bicalutamide as an antiandrogen option for transfeminine people. While they did not recommend bicalutamide as a first-line agent due to its limited characterization in transfeminine people and its known small risk of liver toxicity, they were cautiously permissive of its use in transfeminine hormone therapy:
Bicalutamide can be used for [gender-affirming hormone therapy], but there are very few studies examining its use and the relative risk/benefit for this purpose. Because of reported cases of fulminant hepatitis, consensus is that its use in gender affirming hormonal regimen should be carefully considered, used only after alternative options have been trialed or offered, and an in-depth discussion of these potential risks have been had.
These are the first transgender care guidelines to allow the use of bicalutamide, and only the second guidelines to include bicalutamide. Previously, only the UCSF guidelines mentioned bicalutamide, but they were not permissive of its use in transfeminine people.
Tomson, A., McLachlan, C., Wattrus, C., Adams, K., Addinall, R., Bothma, R., Jankelowitz, L., Kotze, E., Luvuno, Z., Madlala, N., Matyila, S., Padavatan, A., Pillay, M., Rakumakoe, M. D., Tomson-Myburgh, M., Venter, W., & de Vries, E. (2021). Southern African HIV Clinicians’ Society gender-affirming healthcare guideline for South Africa. Southern African Journal of HIV Medicine, 22(1), a1299. [DOI:10.4102/sajhivmed.v22i1.1299] [PDF]
Surprisingly, these guidelines not only included bicalutamide but recommended it as the preferred antiandrogen over spironolactone and cyproterone acetate. The reason stated for this was “less risk of neurosteroid depletion (does not cross blood-brain-barrier readily).” However, this supposed effect isn’t a known concern with antiandrogens besides 5α-reductase inhibitors, and bicalutamide actually does appear to be centrally permeable in humans (Wiki). Also surprisingly, no mention of liver toxicity or liver enzyme monitoring with bicalutamide was made in these guidelines. Considering these apparent oversights and others, these guidelines’s recommendations should probably be interpreted with caution.
Update 3: Coleman et al. (2022) [WPATH SOC8 Guidelines]
Bicalutamide is an antiandrogen that has been used in the treatment of prostate cancer. It competitively binds to the androgen receptor to block the binding of androgens. Data on the use of bicalutamide in trans feminine populations is very sparse and safety data is lacking. One small study looked at the use of bicalutamide 50 mg daily as a puberty blocker in 23 trans feminine adolescents who could not obtain treatment with a GnRH analogue (Neyman et al., 2019). All adolescents experienced breast development which is also commonly seen in men with prostate cancer who are treated with bicalutamide. Although rare, fulminant hepatotoxicity resulting in death has been described with bicalutamide (O’Bryant et al., 2008). Given that bicalutamide has not been adequately studied in trans feminine populations, we do not recommend its routine use.
When selecting a medication, we advise using those which have been studied in multiple transgender populations (i.e., estrogen, cyproterone acetate, GnRH agonists) rather than medications with little to no peer-reviewed scientific studies (i.e., bicalutamide, rectal progesterone, etc.) (Angus et al., 2021; Butler et al., 2017; Efstathiou et al., 2019; Tosun et al., 2019).
As can be seen, the WPATH SOC8 did not recommend the routine use of bicalutamide in transfeminine people owing to the lack of studies of it in this population and its potential risks. As touched on in the present article, it is likely that more studies of bicalutamide in transfeminine people will be needed before bicalutamide could become endorsed by major transgender care guidelines.
Update 4: Jamie Reed 2023 Bicalutamide Liver Toxicity Case
In February 2023, Jamie Reed, a former case manager at the The Washington University Transgender Center at St. Louis Children’s Hospital in St. Louis, Missouri, published the op-ed “I Thought I Was Saving Trans Kids. Now I’m Blowing the Whistle.” in a conservative online news outlet called The Free Press. In this article, Reed expressed that she had become disillusioned with the medical care of transgender youth and layed out her grievances. In addition however, she briefly described an additional case of liver toxicity with bicalutamide in a transfeminine person that had allegedly occurred at her center. This individual was said to be 15 years of age and was given bicalutamide as a puberty blocker by Dr. Christopher Lewis, one of the co-founders of the center. She was said to have subsequently developed liver toxicity and was taken off of bicalutamide. In an electronic message to the center, her mother said that they were “lucky her family was not the type to sue”. This instance, and Reed’s op-ed in general, were subsequently widely reported on in conservative news media, for instance on Fox News and in the Daily Mail (Google). In addition to her op-ed, Reed provided a sworn affidavit to the office of Republican Missouri attorney general Andrew Bailey, who proceeded to launch an investigation of the clinic (Missouri Government, 2023a). The following further information was released in the affidavit:
One doctor at the Center, Dr. Chris Lewis, is giving patients a drug called Bicalutamide. The drug has a legitimate use for treating pancreatic cancer [sic], but it has a side effect of causing breasts to grow, and it can poison the liver. There are no clinical studies for using this drug for gender transitions, and there are no established standards of care for using this drug.
Because of these risks and the lack of scientific studies, other centers that do gender transitions will not use Bicalutamide. The adult center affiliated with Washington University will not use this medication for this reason. But the Center treating children does.
I know of at least one patient at the Center who was advised by the renal department to stop taking Bicalutamide because the child was experiencing liver damage. The child’s parent reported this to the Center through the patient’s online self-reporting medical chart (MyChart). The parent said they were not the type to sue, but “this could be a huge PR problem for you.”
While unpublished and unverified like the earlier reports of liver toxicity with bicalutamide in transfeminine people, this case represents yet another report, and is notably by far the best-documented one. No other clinical details on the case were provided, and it is unclear whether it involved serious liver toxicity, merely asymptomatic liver function test abnormalities, or a clinical situation somewhere in-between these extremes. In any case, it does seem clear that this instance is not likely to have a positive influence on the further adoption of bicalutamide in transfeminine hormone therapy.
Subsequent to the investigation of the clinic being launched, in April 2023, Missouri greatly restricted gender-affirming care for transgender youth and adults, with some of the most severe limits that have been enacted in the United States (Associated Press, 2023a; Missouri Government, 2023b). Bicalutamide and the liver toxicity instance were not further described with these developments. The new state law restricting gender-affirming care took effect August 28, 2023, and Washington University announced that it would stop prescribing puberty blockers and hormone therapy to transgender youth shortly thereafter (Associated Press, 2023b).
A New York Times article with additional information on the case was also subsequently published (Ghorayshi, 2023 [Excerpts]). It was noted that the adolescent had been on bicalutamide for 1 year and definitely experienced hepatotoxicity. However, she also had a complicated medical history, including being immunocompromised, having recently had COVID-19, and having taken another drug known to be associated with hepatotoxicity. As such, the hepatotoxicity cannot be definitively attributed to bicalutamide, but it simultaneously cannot be ruled out that bicalutamide was involved or causative.
Subsequent Burgener et al. (2023, 2024) Findings
Following the preceding case, Lewis and colleagues went on to publish a conference abstract and preprint of a study of bicalutamide in transfeminine youth and young adults in which they stated that it does not increase liver enzymes in this population (Burgener et al., 2023; Burgener et al., 2024). However, a closer look at their data show that bicalutamide did statistically significantly elevate certain liver parameters relative to other antiandrogens, namely rates of elevated aspartate aminotransferase (AST) (upper limit of normal 10.7% vs. 1.5%, P = 0.02) (Burgener et al., 2024). Likewise, rates of elevated alanine aminotransferase (ALT) appeared to trend in the direction of being increased, though this was not statistically significant (upper limit of normal 16.7% vs. 11.6%, P = 0.37) (Burgener et al., 2024). In any case, rates of clinically significant elevations in liver enzymes with bicalutamide, defined as greater than three times the upper limit of normal, were not significantly increased in the study.
On the basis of the relevant research in men with prostate cancer (Wiki), Lewis and colleagues’ study, with a bicalutamide-group sample size of only 84 transfeminine individuals, was clearly greatly underpowered for evaluating liver function changes. Per the Early Prostate Cancer trial of high-dose bicalutamide monotherapy in men with prostate cancer, elevated liver enzymes appear to occur with bicalutamide at a rate of only about 1.5% more than placebo, or roughly an additional 1 in every 66 people (Wiki). Based on power analysis, this would require a far larger sample size to have adequate statistical power and actually have a chance of achieving statistical significance.
As such, it seems to the present author premature to conclude that bicalutamide does not elevate liver enzymes in transfeminine people.
Lewis and colleagues didn’t mention in their study paper the transfeminine adolescent liver toxicity case reported by Jamie Reed that was said to have occurred at their clinic nor have they published a case report about this instance. Instead, only the following is stated:
One case report published in 2024 described a transgender female adolescent prescribed bicalutamide 50 mg daily who presented to a hospital with liver toxicity that resolved after stopping bicalutamide (Wilde et al., 2024). This appears to be the first documented case of bicalutamide-induced hepatoxicity in a transgender female.
While this case was, coincidentally, also a 17-year-old transfeminine adolescent (Wilde et al., 2024), this instance, per the medical histories and reporting authors/institutions, appears to be distinct from Dr. Lewis’s that was reported by Jamie Reed.
However, Lewis and colleagues did note the following in their paper, which plausibly might have been the Jamie Reed case:
There was one individual in whom bicalutamide was stopped after the follow-up period designated for the study. This individual developed ALT and AST >2x ULN after an episode of COVID and had a thorough hepatology evaluation. As ALT and AST were never > 3x ULN, it was not recommended that bicalutamide be stopped; however, ultimately a clinical decision was made to stop the medication and ALT and AST normalized.
Another concern with Lewis and colleagues’ paper pertains to the following statements:
Whereas bicalutamide doses for prostate cancer reach 150 mg daily, doses used in the care of AMAB transfeminine individuals are much lower (25-50 mg daily).
Bicalutamide doses used in prostate cancer are up to 150 mg daily. Due to these concerns of liver toxicity, bicalutamide has not been routinely used as an anti-androgen in AMAB transfeminine individuals, despite the much lower doses needed in this population (∼25-50 mg daily).
In actuality, bicalutamide is most widely used in prostate cancer, in the form of combined androgen blockade with surgical or medical castration, at a dosage of 50 mg/day, whereas the 150 mg/day dosage is used less commonly, in the form of monotherapy (Wiki). Moreover, only the 50 mg/day dosage is used in the United States, where monotherapy is not approved. Among the published case reports of hepatotoxicity with bicalutamide in men with prostate cancer, half have been at a dose of 50 mg/day and the other half have been at a dose of 80 to 150 mg/day (Wiki). The two instances of death due to hepatotoxicity with bicalutamide were both at 50 mg/day. There is currently no evidence that the hepatotoxicity of bicalutamide is dose-dependent across its clinically used dosage range (Wiki), although employment of the lowest effective dose in transfeminine people nonetheless seems prudent just in case. Hence, in contrast to Lewis and colleague’s claims, a bicalutamide dosage of 50 mg/day is not less than that generally used in prostate cancer, and clearly retains substantial hepatotoxic potential.
Update 5: New Bicalutamide Publications in 2022 Through 2025
Angus, L., Nolan, B., Zajac, J., & Cheung, A. (November 2022). Use of bicalutamide as an androgen receptor antagonist in transgender women. ESA/SRB/APEG/NZSE ASM 2022, November 13-16, Christchurch, Abstracts and Programme, 127–127 (abstract no. 280). [URL] [PDF] [Full Abstract Book]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (November 2023). Bicalutamide as an anti-androgen in trans people: a cross-sectional study. AusPATH 2023 Symposium. [URL] [PDF] [Slides] [Trans Health Research Blog Post]
Bambilla, A., Beal, C., & Vigil, P. (2023). Improving Access to Bicalutamide in Gender Affirming Medical Care. [Unpubished/pending publication] [QueerCME Blog Post]
Burgener, K., DeBosch, B., Lewis, C., Wallendorf, M., & Herrick, C. (May 2023). Assessment of Liver Function and Toxicity in Transgender Female Adolescents Prescribed Bicalutamide. Hormone Research in Paediatrics, 96(Suppl 3 [Abstracts of the 2023 Pediatric Endocrine Society (PES) Annual Meeting’ to Hormone Research in Paediatrics]), 377–378 (abstract no. 6232). [DOI:10.1159/000531602] [PDF]
Gómez-Aguilar, F., Martínez-Sánchez, L., Arias-Constantí, V., Muñoz-Santanach, D., & Sarquella-Brugada, G. (2023). QT prolongation and Torsade de Pointes in a 13-year-old transgender adolescent in treatment with bicalutamide and tacrolimus. Clinical Toxicology, 61(Suppl 1 [43rd International Congress of the European Association of Poisons Centres and Clinical Toxicologists (EAPCCT), 23–26 May 2023, Palma de Mallorca, Spain]), 81–82 (abstract no. 170). [DOI:10.1080/15563650.2023.2192024] [PDF] [Reactions Weekly]
Karakılıç Özturan, E., Öztürk, A. P., Baş, F., Erdoğdu, A. B., Kaptan, S., Kardelen Al, A. D., Poyrazoğlu, Ş., Yıldız, M., Direk, N., Yüksel, Ş., & Darendeliler, F. (2023). Endocrinological Approach to Adolescents with Gender Dysphoria: Experience of a Pediatric Endocrinology Department in a Tertiary Center in Turkey. Journal of Clinical Research in Pediatric Endocrinology, 15(3), 276–284. [DOI:10.4274/jcrpe.galenos.2023.2023-1-13]
Vierregger, K., Tetzlaff, M., Zimmerman, B., Dunn, N., Finney, N., Lewis, K., Slomoff, R., & Strutner, S. (May 2023). Bicalutamide Use as Antiandrogen in Trans Feminine Adults - A Safety Profile. National Transgender Health Summit (NTHS) 2023 Symposium. [Event Agenda PDF] [Symposium Session] [Symposium Abstracts/Program Book]
Vierregger, K., Tetzlaf, M., Zimmerman, B., Dunn, N., Finney, N., Lewis, K., Slomoff, R., & Strutner, S. (November 2023). Bicalutamide Use as Antiandrogen in Trans Feminine Adults - A Safety Profile. USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 96–96 (abstract no. SAT-B2-T4). [Symposium Schedule] [PDF] [Full Abstract Book]
Warus, J., Rincon, M. G., Salvetti, B., & Olson-Kennedy, J. (November 2023). Safety of Bicalutamide as Anti-Androgenic Therapy in Gender Affirming Care for Adolescents and Young Adults: A Retrospective Chart Review. USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 124–124 (abstract no. SUN-B1-T5). [Symposium Schedule] [PDF] [Full Abstract Book]
Wilde, B., Diamond, J. B., Laborda, T. J., Frank, L., O’Gorman, M. A., & Kocolas, I. (2023). Bicalutamide-Induced Hepatotoxicity in a Transgender Male-to-Female Adolescent. Journal of Adolescent Health, 74(1), 202–204. [DOI:10.1016/j.jadohealth.2023.08.024]
Burgener, K., DeBosch, B., Wang, J., Lewis, C., & Herrick, C. (2025). Bicalutamide does not raise transaminases clinically significantly compared to alternative anti-androgen regimens among transfeminine adolescents and young adults: a retrospective cohort study. International Journal of Transgender Health, 1–10. [DOI:10.1080/26895269.2025.2452184]
Fuqua, J. S., Shi, E., & Eugster, E. A. (2024). A retrospective review of the use of bicalutamide in transfeminine youth; a single center experience. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2023.2294321]
Shumer, D., & Roberts, S. A. (2024). Placing a Report of Bicalutamide-Induced Hepatotoxicity in the Context of Current Standards of Care for Transgender Adolescents. Journal of Adolescent Health, 74(1), 5–6. [DOI:10.1016/j.jadohealth.2023.10.010]
Angus, L. M., Hong, Q. V., Cheung, A. S., & Nolan, B. J. (2024). Effect of bicalutamide on serum total testosterone concentration in transgender adults: a case series. Therapeutic Advances in Endocrinology and Metabolism, 15. [DOI:10.1177/20420188241305022]
Update 6: Original Bicalutamide Liver and Lung Toxicity Analysis by Sam
A few years ago back in 2021, Transfeminine Science author Sam conducted an original analysis of the incidence of liver and lung toxicity with bicalutamide in the published clinical trial literature. This project was never finished or made publicly available. However, with bicalutamide being increasingly studied and adopted for use in transfeminine people, it seems quite valuable and relevant today. As such, we have opted to now publish Sam’s analysis in this section.
Sam’s analysis can be found in the provided document here. In terms of methodology, she searched PubMed for all clinical trials of bicalutamide, collated all of the relevant results into a table, and then calculated the incidences of serious liver toxicity and lung toxicity from those data. In clinical trials, adverse events are rated in terms of grades of severity, with a Grade 3 adverse event defined as “severe”, Grade 4 as “life-threatening”, and Grade 5 as “death” (Wiki).
Of 229 results, 33 trials were found to be relevant and were included. Most of the trials were in men with prostate cancer, but a few were in women with cancer and boys with precocious puberty. Sam found that of a total of 7,703 evaluable participants, there were 2 instances of serious liver toxicity and 2 instances of serious lung toxicity with bicalutamide. This resulted in the same incidence rate of 0.026% (95% CI: 0.003% to 0.094%) or approximately 1 in 3,846 individuals for both liver toxicity and lung toxicity. Combining these toxicities resulted in a total incidence of serious liver or serious lung toxicity with bicalutamide of 0.052% (95% CI: 0.014% to 0.133%) or approximately 1 in 1,923 individuals. All of the observed toxicity events were rated as Grade 3 or 4. It should be noted that clinical trials of bicalutamide typically employ careful laboratory monitoring and assessment of clinical adverse events as well as prompt medication discontinuation upon unfavorable laboratory changes.
While the confidence intervals (CIs) in Sam’s analysis were wide and hence the estimates are very rough, they provide an idea of the potential real-world risk of serious toxicity with bicalutamide in transfeminine people based on high-quality clinical data. Notably, they do not suffer from the problem of under-reporting of adverse events that occurs with published case reports, pharmacovigilance databases, and certain types of observational studies. However, limitations of Sam’s analysis include (1) toxicity incidence rates for non-bicalutamide-treated controls not being assessed and (2) most of the patients having cancer and being of older age, and hence the generalizability of the findings to healthy transfeminine people not being fully clear. In any case, I was surprised by how high the incidence rates were when I first saw her analysis, and I suspect that others may be as well.
References
Anderson, J. (2003). The role of antiandrogen monotherapy in the treatment of prostate cancer. BJU International, 91(5), 455–461. [DOI:10.1046/j.1464-410X.2003.04026.x]
Angus, L., Nolan, B., Zajac, J., & Cheung, A. (2022). Use of bicalutamide as an androgen receptor antagonist in transgender women. ESA/SRB/APEG/NZSE ASM 2022, November 13-16, Christchurch, Abstracts and Programme, 127–127 (abstract no. 280). [URL] [PDF] [Full Abstract Book]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2023). Bicalutamide as an anti-androgen in trans people: a cross-sectional study. AusPATH 2023 Symposium. [URL] [PDF] [Slides] [Trans Health Research Blog Post]
Angus, L. M., Hong, Q. V., Cheung, A. S., & Nolan, B. J. (2024). Effect of bicalutamide on serum total testosterone concentration in transgender adults: a case series. Therapeutic Advances in Endocrinology and Metabolism, 15. [DOI:10.1177/20420188241305022]
Arya, V. B., & Davies, J. H. (2019). Idiopathic gonadotropin-independent precocious puberty - is regular surveillance required? Journal of Pediatric Endocrinology & Metabolism: JPEM, 32(4), 403–407. [DOI:10.1515/jpem-2018-0419]
Asscheman, H., Gooren, L. J., & Peereboom-Wynia, J. D. (1989). Reduction in undesired sexual hair growth with anandron in male-to-female transsexuals—experiences with a novel androgen receptor blocker. Clinical and Experimental Dermatology, 14(5), 361–363. [DOI:10.1111/j.1365-2230.1989.tb02585.x]
Asscheman, H., & Gooren, L. J. (1992). Hormone Treatment in Transsexuals. In Bocking, W. O., Coleman, E. (Eds). Gender Dysphoria: Interdisciplinary Approaches in Clinical Management (pp. 39–54). Binghamton: Haworth Press. / Journal of Psychology & Human Sexuality, 5(4), 39–54. [Google Scholar] [Google Books] [DOI:10.1300/J056v05n04_03]
AstraZeneca. (2008). Clinical Review. NDA. 22-310/S-001. Casodex (bicalutamide) for Testotoxicosis. Food and Drug Administration/AstraZeneca Pharmaceuticals. [URL] [PDF]
Bahceci, M., Tuzcu, A., Canoruc, N., Tuzun, Y., Kidir, V., & Aslan, C. (2004). Serum C-reactive protein (CRP) levels and insulin resistance in non-obese women with polycystic ovarian syndrome, and effect of bicalutamide on hirsutism, CRP levels and insulin resistance. Hormone Research in Paediatrics, 62(6), 283–287. [DOI:10.1159/000081973]
Bailey, A. (2023 February 9). Missouri Attorney General Andrew Bailey Confirms Launch of Multi-Agency Investigation into St. Louis Transgender Center for Harming Hundreds of Children. Attorney General Andrew Bailey, Missouri Government. [URL] [Affadavit of Jamie Reed]
Bailey, A. (2023 April 13). Missouri Attorney General Andrew Bailey Promulgates Emergency Regulation Targeting Gender Transition Procedures for Minors. Attorney General Andrew Bailey, Missouri Government. [URL] [Emergency Regulation PDF]
Ballentine, S., & Hollingsworth, H. (2023 April 13). Missouri to limit gender-affirming care for minors, adults. Associated Press. [URL]
Bambilla, A., Beal, C., & Vigil, P. (2023). Improving Access to Bicalutamide in Gender Affirming Medical Care. [Unpubished/pending publication] [QueerCME Blog Post]
Blackledge, G. R. P. (1996). Clinical progress with a new antiandrogen, Casodex™ (bicalutamide). European Urology, 29(Suppl 2), 96–104. [DOI:10.1159/000473847]
Burgener, K., DeBosch, B., Lewis, C., Wallendorf, M., & Herrick, C. (2023). Assessment of Liver Function and Toxicity in Transgender Female Adolescents Prescribed Bicalutamide. Hormone Research in Paediatrics, 96(Suppl 3 [Abstracts of the 2023 Pediatric Endocrine Society (PES) Annual Meeting’ to Hormone Research in Paediatrics]), 377–378 (abstract no. 6232). [DOI:10.1159/000531602] [PDF]
Burgener, K., DeBosch, B., Wang, J., Lewis, C., & Herrick, C. (2025). Bicalutamide does not raise transaminases clinically significantly compared to alternative anti-androgen regimens among transfeminine adolescents and young adults: a retrospective cohort study. International Journal of Transgender Health, 1–10. [DOI:10.1080/26895269.2025.2452184]
Cocchetti, C., Ristori, J., Romani, A., Maggi, M., & Fisher, A. D. (2020). Hormonal Treatment Strategies Tailored to Non-Binary Transgender Individuals. Journal of Clinical Medicine, 9(6), 1609. [DOI:10.3390/jcm9061609]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Dahl, M., Feldman, J. L., Goldberg, J. M., & Jaberi, A. (2006). Physical aspects of transgender endocrine therapy. International Journal of Transgenderism, 9(3–4), 111–134. [DOI:10.1300/J485v09n03_06]
Dahl, M., Feldman, J. L., Goldberg, J., & Jaberi, A. (2006). Endocrine Therapy for Transgender Adults in British Columbia: Suggested Guidelines. Physical Aspects of Transgender Endocrine Therapy. Vancouver: Vancouver Coastal Health. [Google Scholar] [PDF]
de Voogt, H. J., Rao, B. R., Gooren, L. J. G., Geldof, A. A., & Bouman, F. G. (1987). Considerations About the Use of Anti-Androgenic Drugs in Male-to-Female Transsexuals. Tenth International Symposium on Gender Dysphoria, Diagnosis and Treatment of Transsexualism, Vrije Universiteit Amsterdam, The Netherlands, June 9–12, 1987. Harry Benjamin International Gender Dysphoria Association. [PDF]
de Voogt, H. J., Rao, B. R., Geldof, A. A., Gooren, L. J. G., & Bouman, F. G. (1987). Androgen action blockade does not result in reduction in size but changes histology of the normal human prostate. The Prostate, 11(4), 305–311. [DOI:10.1002/pros.2990110403]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Fernandez-Nieto, D., Saceda-Corralo, D., Rodrigues-Barata, R., Hermosa-Gelbard, A., Moreno-Arrones, O., Jimenez-Cauhe, J., Ortega-Quijano, D., & Vano-Galvan, S. (2019). Oral bicalutamide for female pattern hair loss: A pilot study. Dermatologic Therapy, 32(6), e13096. [DOI:10.1111/dth.13096]
Fernandez-Nieto, D., Saceda-Corralo, D., Jimenez-Cauhe, J., Moreno-Arrones, O. M., Rodrigues-Barata, R., Hermosa-Gelbard, A., & Vano-Galvan, S. (2020). Bicalutamide: A potential new oral antiandrogenic drug for female pattern hair loss. Journal of the American Academy of Dermatology, 83(5), e355–e356. [DOI:10.1016/j.jaad.2020.04.054]
Finkle, A., Zavertnik, S., Myers, S., Cormier, D., Heithaus, J., & Augustyn, M. (2020). Growing Up Fast: Managing Autism Spectrum Disorder and Precocious Puberty. Journal of Developmental and Behavioral Pediatrics: JDBP, 41(9), 740–742. [DOI:10.1097/DBP.0000000000000865]
Fishman, S. L., Paliou, M., Poretsky, L., & Hembree, W. C. (2019). Endocrine Care of Transgender Adults. In Poretsky, L., & Hembree, W. C. (Eds.). Transgender Medicine (pp. 143–163). Cham: Humana Press. [DOI:10.1007/978-3-030-05683-4_8]
Food and Drug Administration (FDA). (2017). CASODEX (bicalutamide) tablet, for oral use. [URL] [PDF]
Fuqua, J. S., Shi, E., & Eugster, E. A. (2024). A retrospective review of the use of bicalutamide in transfeminine youth; a single center experience. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2023.2294321]
Ghorayshi, A. (2023 August 29). How a Small Gender Clinic Landed in a Political Storm. The New York Times. [URL]
Gómez-Aguilar, F., Martínez-Sánchez, L., Arias-Constantí, V., Muñoz-Santanach, D., & Sarquella-Brugada, G. (2023). QT prolongation and Torsade de Pointes in a 13-year-old transgender adolescent in treatment with bicalutamide and tacrolimus. Clinical Toxicology, 61(Suppl 1 [43rd International Congress of the European Association of Poisons Centres and Clinical Toxicologists (EAPCCT), 23–26 May 2023, Palma de Mallorca, Spain]), 81–82 (abstract no. 170). [DOI:10.1080/15563650.2023.2192024] [PDF] [Reactions Weekly]
Gooren, L., Spinder, T., Spijkstra, J. J., Van Kessel, H., Smals, A., Rao, B. R., & Hoogslag, M. (1987). Sex steroids and pulsatile luteinizing hormone release in men. Studies in estrogen-treated agonadal subjects and eugonadal subjects treated with a novel nonsteroidal antiandrogen. The Journal of Clinical Endocrinology & Metabolism, 64(4), 763–770. [DOI:10.1210/jcem-64-4-763]
Gooren, L. J. G. (1999). Hormonal Sex Reassignment. International Journal of Transgenderism, 3(3), 1–7. [Google Scholar] [URL] [Alt]
Gooren, L. J. (2011). Care of transsexual persons. New England Journal of Medicine, 364(13), 1251–1257. [DOI:10.1056/NEJMcp1008161]
Graham, D. J., Ahmad, S. R., & Piazza‐Hepp, T. (2002). Spontaneous Reporting—USA. In Mann, R. D., & Andrews, E. B. (Eds.). Pharmacovigilance (pp. 219–227). Chichester: John Wiley & Sons. [DOI:10.1002/0470853093.ch17]
Gurnurkar, S., DiLillo, E., & Carakushansky, M. (2021). A Case of Familial Male-limited Precocious Puberty with a Novel Mutation. Journal of Clinical Research in Pediatric Endocrinology, 13(2), 239–244. [DOI:10.4274/jcrpe.galenos.2020.2020.0067]
Haddad, N. G., & Eugster, E. A. (2012). Peripheral Precocious Puberty: Interventions to Improve Growth. In Preedy, V. R. (Ed.). Handbook of Growth and Growth Monitoring in Health and Disease (pp. 1199–1212). New York: Springer. [DOI:10.1007/978-1-4419-1795-9_71]
Hamidi, O., & Davidge-Pitts, C. J. (2019). Transfeminine Hormone Therapy. Endocrinology and Metabolism Clinics, 48(2), 341–355. [DOI:10.1016/j.ecl.2019.02.001]
Hembree, W. C., Cohen-Kettenis, P., Delemarre-Van De Waal, H. A., Gooren, L. J., Meyer III, W. J., Spack, N. P., Tangpricha, V., & Montori, V. M. (2009). Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism, 94(9), 3132–3154. [DOI:10.1210/jc.2009-0345]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Ismail, F. F., Meah, N., de Carvalho, L. T., Bhoyrul, B., Wall, D., & Sinclair, R. (2020). Safety of oral bicalutamide in female pattern hair loss: A retrospective review of 316 patients. Journal of the American Academy of Dermatology, 83(5), 1478–1479. [DOI:10.1016/j.jaad.2020.03.034]
Israel, G. E., & Tarver, D. E. (1997). Transgender Care: Recommended Guidelines, Practical Information, and Personal Accounts (p. 66). Philadelphia: Temple University Press. [Google Scholar] [Google Books]
Iversen, P., Johansson, J. E., Lodding, P., Lukkarinen, O., Lundmo, P., Klarskov, P., Tammela, T. L., Tasdemir, I., Morris, T., Carroll, K., & Scandinavian Prostatic Cancer Group. (2004). Bicalutamide (150 mg) versus placebo as immediate therapy alone or as adjuvant to therapy with curative intent for early nonmetastatic prostate cancer: 5.3-year median followup from the Scandinavian Prostate Cancer Group Study Number 6. The Journal of Urology, 172(5 Part 1), 1871–1876. [DOI:10.1097/01.ju.0000139719.99825.54]
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 2042018819871166. [DOI:10.1177/2042018819871166]
Johannes, F. L. M., van Kemenade, M. A., Cohen-Kettenis, P. T., Cohen, L., & Gooren, L. J. G. (1987). Effects of RU 23.903 (Anandron) on Lateralization and Spatial Ability in Male-to-Female-Transsexuals. Tenth International Symposium on Gender Dysphoria, Diagnosis and Treatment of Transsexualism, Vrije Universiteit Amsterdam, The Netherlands, June 9–12, 1987. Harry Benjamin International Gender Dysphoria Association. [PDF]
Karakılıç Özturan, E., Öztürk, A. P., Baş, F., Erdoğdu, A. B., Kaptan, S., Kardelen Al, A. D., Poyrazoğlu, Ş., Yıldız, M., Direk, N., Yüksel, Ş., & Darendeliler, F. (2023). Endocrinological Approach to Adolescents with Gender Dysphoria: Experience of a Pediatric Endocrinology Department in a Tertiary Center in Turkey. Journal of Clinical Research in Pediatric Endocrinology, 15(3), 276–284. [DOI:10.4274/jcrpe.galenos.2023.2023-1-13]
Kolvenbag, G. J., & Blackledge, G. R. (1996). Worldwide activity and safety of bicalutamide: a summary review. Urology, 47(1), 70–79. [DOI:10.1016/S0090-4295(96)80012-4]
Kor, Y. (2018). Central precocious puberty in a case of late-diagnosed familial testotoxicosis and long-term treatment monitoring. Hormones, 17(2), 275–278. [DOI:10.1007/s42000-018-0029-1]
Kreher, N. C., Pescovitz, O. H., Delameter, P., Tiulpakov, A., & Hochberg, Z. E. (2006). Treatment of familial male-limited precocious puberty with bicalutamide and anastrozole. The Journal of Pediatrics, 149(3), 416–420. [DOI:10.1016/j.jpeds.2006.04.027]
Lenz, A. M., Shulman, D., Eugster, E. A., Rahhal, S., Fuqua, J. S., Pescovitz, O. H., & Lewis, K. A. (2010). Bicalutamide and third-generation aromatase inhibitors in testotoxicosis. Pediatrics, 126(3), e728–e733. [DOI:10.1542/peds.2010-0596]
Levy, A., Crown, A., & Reid, R. (2003). Endocrine intervention for transsexuals. Clinical Endocrinology, 59(4), 409–418. [DOI:10.1046/j.1365-2265.2003.01821.x]
Lewis, K. A., Lenz, A., Eugster, E. A., Fuqua, J. S., Pescovitz, O. H., Rahhal, S., & Shulman, D. (2009). Long term bicalutamide and 3rd generation aromatase inhibitor therapy in two boys with testotoxicosis. Hormone Research [in Paediatrics], 72(Suppl 3), 266–267 (PO2-189). [Google Scholar] [DOI:10.1159/000239668]
Manso, G., Thole, Z., Salgueiro, E., Revuelta, P., & Hidalgo, A. (2006). Spontaneous reporting of hepatotoxicity associated with antiandrogens: data from the Spanish pharmacovigilance system. Pharmacoepidemiology and Drug Safety, 15(4), 253–259. [DOI:10.1002/pds.1168]
McLeod, D. G. (1997). Tolerability of Nonsteroidal Antiandrogens in the Treatment of Advanced Prostate Cancer. The Oncologist, 2(1), 18–27. [DOI:10.1634/theoncologist.2-1-18]
Michigan Medicine. (2020). Feminizing Gender-Affirming Hormone Care: The Michigan Medicine Approach. Ann Arbor: Michigan Medicine/University of Michigan. [URL] [PDF]
Mitchell, F. (2020). Guy T’Sjoen: collaborating to improve transgender wellbeing. The Lancet Diabetes & Endocrinology, 8(7), 568–568. [DOI:10.1016/S2213-8587(20)30192-3]
Mitre, N., & Lteif, A. (2009). Treatment of familial male-limited precocious puberty (testotoxicosis) with anastrozole and bicalutamide in a boy with a novel mutation in the luteinizing hormone receptor. Journal of Pediatric Endocrinology and Metabolism, 22(12), 1163–1168. [DOI:10.1515/JPEM.2009.22.12.1163]
Moreno-Pérez, Ó., De Antonio, I. E., & Grupo de Identidad y Diferenciación Sexual de la SEEN (GIDSEEN. (2012). Clinical practice guidelines for assessment and treatment of transsexualism. SEEN Identity and Sexual Differentiation Group (GIDSEEN). Endocrinología y Nutrición (English Edition), 59(6), 367–382. [DOI:10.1016/j.endoen.2012.07.004]
Moretti, C. G., Guccione, L., Di Giacinto, P., Cannuccia, A., Meleca, C., Lanzolla, G., Andreadi, A., & Lauro, D. (2016). Efficacy and Safety of Myo-Inositol Supplementation in the Treatment of Obese Hirsute PCOS Women: Comparative Evaluation with OCP+Bicalutamide Therapy. Endocrine Reviews, 37(2 Suppl 1), ?–? (abstract no. SUN-153). [URL] [DOI:10.1093/edrv/37.supp.1]
Moretti, C., Guccione, L., Di Giacinto, P., Simonelli, I., Exacoustos, C., Toscano, V., Motta, C., De Leo, V., Petraglia, F., & Lenzi, A. (2018). Combined oral contraception and bicalutamide in polycystic ovary syndrome and severe hirsutism: a double-blind randomized controlled trial. The Journal of Clinical Endocrinology & Metabolism, 103(3), 824–838. [DOI:10.1210/jc.2017-01186]
Moussa, A., Kazmi, A., Bokhari, L., & Sinclair, R. D. (2022). Bicalutamide improves minoxidil-induced hypertrichosis in female pattern hair loss: A retrospective review of 35 patients. Journal of the American Academy of Dermatology, 87(2), 488–490. [DOI:10.1016/j.jaad.2021.10.048]
Muderris, I. I., Bayram, F., & Guven, M. (1999). Bicalutamide 25 mg/day in the treatment of hirsutism. Human Reproduction, 14(Suppl 3), 366–366 (R-196). [DOI:10.1093/humrep/14.Suppl_3.366-a]
Müderris, I. I., Bayram, F., Özçelik, B., & Güven, M. (2002). New alternative treatment in hirsutism: bicalutamide 25 mg/day. Gynecological Endocrinology, 16(1), 63–66. [DOI:10.1080/gye.16.1.63.66]
Müderris, İ. İ., & Öner, G. (2009). Hirsutizm Tedavisinde Flutamid ve Bikalutamid Kullanımı. [Flutamide and Bicalutamide Treatment in Hirsutism.] Türkiye Klinikleri Endokrinoloji-Özel Dergisi, 2(2), 110–112. [ISSN:1308-0954] [URL] [PDF] [Translation]
Nabhan, Z. M., & Eugster, E. A. (2019). Testotoxicosis with an episodic course: an unusual case within a series. AACE Clinical Case Reports, 5(1), e50–e53. [DOI:10.4158/ACCR-2018-0246]
Neyman, A., Fuqua, J. S., & Eugster, E. A. (2017). Bicalutamide as an Androgen Blocker with Secondary Effect of Promoting Feminization in Male to Female (MTF) Transgender Adolescents. Hormone Research in Paediatrics, 88(Suppl 1 [10th Individual Abstracts for International Meeting of Pediatric Endocrinology: Free Communication and Poster Sessions, Abstracts]), 477–477 (abstract no. P3-605). [DOI:10.1159/000481424] [PDF]
Neyman, A., Fuqua, J. S., & Eugster, E. A. (2019). Bicalutamide as an androgen blocker with secondary effect of promoting feminization in male-to-female transgender adolescents. Journal of Adolescent Health, 64(4), 544–546. [DOI:10.1016/j.jadohealth.2018.10.296]
Özcabı, B., Ceylaner, S., Özcan, R., Büyükünal, C., Ercan, O., Tüysüz, B., & Evliyaoğlu, O. (2015). Testotoxicosis: Report of Two Cases, One with a Novel Mutation in LHCGR Gene. Journal of Clinical Research in Pediatric Endocrinology, 7(3), 242–248. [DOI:10.4274/jcrpe.2067]
Randolph Jr, J. F. (2018). Gender-Affirming Hormone Therapy for Transgender Females. Clinical Obstetrics and Gynecology, 61(4), 705–721. [DOI:10.1097/GRF.0000000000000396] [URL]
Rao, B. R., De Voogt, H. J., Geldof, A. A., Gooren, L. J. G., & Bouman, F. G. (1988). Merits and considerations in the use of anti-androgen. Journal of Steroid Biochemistry, 31(4), 731–737. [DOI:10.1016/0022-4731(88)90024-6]
Reed, J. (2023 February 9). I Thought I Was Saving Trans Kids. Now I’m Blowing the Whistle. The Free Press. [URL]
Reiter, E. O., Mauras, N., McCormick, K., Kulshreshtha, B., Amrhein, J., De Luca, F., O’Brien, S., Armstrong, J., & Melezinkova, H. (2010). Bicalutamide plus anastrozole for the treatment of gonadotropin-independent precocious puberty in boys with testotoxicosis: a phase II, open-label pilot study (BATT). Journal of Pediatric Endocrinology & Metabolism: JPEM, 23(10), 999–1009. [DOI:10.1515/jpem.2010.161]
Salter, J., & Ballentine, S. (2023 September 12). Missouri clinics halt transgender care for minors in wake of new state law. Associated Press. [URL]
Schellhammer, P. F., Sharifi, R., Block, N. L., Soloway, M. S., Venner, P. M., Patterson, A. L., Sarosdy, M. F., Vogelzang, N. J., Schellenger, J. J., Kolvenbag, G. J., & Casodex Combination Study Group. (1997). Clinical benefits of bicalutamide compared with flutamide in combined androgen blockade for patients with advanced prostatic carcinoma: final report of a double-blind, randomized, multicenter trial. Urology, 50(3), 330–336. [DOI:10.1016/s0090-4295(97)00279-3]
Shumer, D., & Roberts, S. A. (2024). Placing a Report of Bicalutamide-Induced Hepatotoxicity in the Context of Current Standards of Care for Transgender Adolescents. Journal of Adolescent Health, 74(1), 5–6. [DOI:10.1016/j.jadohealth.2023.10.010]
See, W. A., Wirth, M. P., McLeod, D. G., Iversen, P., Klimberg, I., Gleason, D., Chodak, G., Montie, J., Tyrrell, C., Wallace, D. M., Delaere, K. P., Vaage, S., Tammela, T. L., Lukkarinen, O., Persson, B. E., Carroll, K., Kolvenbag, G. J., & Casodex Early Prostate Cancer Trialist Group. (2002). Bicalutamide as immediate therapy either alone or as adjuvant to standard care of patients with localized or locally advanced prostate cancer: first analysis of the early prostate cancer program. The Journal of Urology, 168(2), 429–435. [DOI:10.1016/S0022-5347(05)64652-6]
Stenger, P. J., Wagner, L., Egelhoff, J., & Rose, S. R. (2009). Bicalutamide and letrozole treatment of precocious puberty due to elevated levels of human chorionic gonadotropin (hCG). Hormone Research [in Paediatrics], 72(Suppl 3), 391–391 (PO3-149). [Google Scholar] [DOI:10.1159/000239668]
Tessaris, D., Matarazzo, P., Mussa, A., Tuli, G., Verna, F., Fiore, L., & Lala, R. (2012). Combined treatment with bicalutamide and anastrozole in a young boy with peripheral precocious puberty due to McCune-Albright Syndrome. Endocrine Journal, 59(2), 111–117. [DOI:10.1507/endocrj.ej11-0214]
Thole, Z., Manso, G., Salgueiro, E., Revuelta, P., & Hidalgo, A. (2004). Hepatotoxicity induced by antiandrogens: a review of the literature. Urologia Internationalis, 73(4), 289–295. [DOI:10.1159/000081585]
Thompson, J., Hopwood, R. A., deNormand, S., & Cavanaugh, T. (2021). Medical Care of Trans and Gender Diverse Adults. Boston: Fenway Health. [URL] [PDF]
Tomson, A., McLachlan, C., Wattrus, C., Adams, K., Addinall, R., Bothma, R., Jankelowitz, L., Kotze, E., Luvuno, Z., Madlala, N., Matyila, S., Padavatan, A., Pillay, M., Rakumakoe, M. D., Tomson-Myburgh, M., Venter, W., & de Vries, E. (2021). Southern African HIV Clinicians’ Society gender-affirming healthcare guideline for South Africa. Southern African Journal of HIV Medicine, 22(1), a1299. [DOI:10.4102/sajhivmed.v22i1.1299] [PDF]
van Kemenade, J. F., Cohen-Kettenis, P. T., Cohen, L., & Gooren, L. J. (1989). Effects of the pure antiandrogen RU 23.903 (anandron) on sexuality, aggression, and mood in male-to-female transsexuals. Archives of Sexual Behavior, 18(3), 217–228. [DOI:10.1007/BF01543196]
Vierregger, K., Tetzlaff, M., Zimmerman, B., Dunn, N., Finney, N., Lewis, K., Slomoff, R., & Strutner, S. (2023). Bicalutamide Use as Antiandrogen in Trans Feminine Adults - A Safety Profile. National Transgender Health Summit (NTHS) 2023 Symposium. [Event Agenda PDF] [Symposium Session] [Symposium Abstracts/Program Book]
Vierregger, K., Tetzlaf, M., Zimmerman, B., Dunn, N., Finney, N., Lewis, K., Slomoff, R., & Strutner, S. (2023). Bicalutamide Use as Antiandrogen in Trans Feminine Adults - A Safety Profile. USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 96–96 (abstract no. SAT-B2-T4). [Symposium Schedule] [PDF] [Full Abstract Book]
Warus, J., Rincon, M. G., Salvetti, B., & Olson-Kennedy, J. (2023). Safety of Bicalutamide as Anti-Androgenic Therapy in Gender Affirming Care for Adolescents and Young Adults: A Retrospective Chart Review. USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 124–124 (abstract no. SUN-B1-T5). [Symposium Schedule] [PDF] [Full Abstract Book]
Wilde, B., Diamond, J. B., Laborda, T. J., Frank, L., O’Gorman, M. A., & Kocolas, I. (2023). Bicalutamide-Induced Hepatotoxicity in a Transgender Male-to-Female Adolescent. Journal of Adolescent Health, 74(1), 202–204. [DOI:10.1016/j.jadohealth.2023.08.024]
\ No newline at end of file
+Bicalutamide and its Adoption by the Medical Community for Use in Transfeminine Hormone Therapy - Transfeminine Science
Bicalutamide and its Adoption by the Medical Community for Use in Transfeminine Hormone Therapy
By Aly | First published July 1, 2020 | Last modified August 23, 2025
Abstract / TL;DR
Bicalutamide is an antiandrogen which was introduced for the treatment of prostate cancer many years ago. Cost precluded its widespread use for other indications for many years. However, its cost has since come down and bicalutamide is now seeing significant adoption for use in transfeminine hormone therapy as well as for treatment of androgen-dependent conditions in other populations like cisgender women. Bicalutamide has risks of certain rare adverse effects like liver toxicity which have generated concerns about its safety and have limited its use in transfeminine people. However, while still significant, these risks are low with appropriate monitoring and clinical management. Prominent researchers in transgender medicine have recently shown openness to bicalutamide for potential use in transfeminine people and have written positively about it. Bicalutamide could eventually come to be regarded as acceptably safe for use in transfeminine hormone therapy, similarly to other antiandrogens with rare risks like spironolactone and cyproterone acetate. However, more studies and characterization of bicalutamide in transfeminine people will likely be needed before it could see wider adoption in transgender medicine.
History of Bicalutamide for Transfeminine People
Bicalutamide (Casodex) is a nonsteroidal antiandrogen and selectiveantagonist of the androgen receptor which was originally introduced for the treatment of prostate cancer in cisgender men in 1995. Prostate cancer is an androgen-dependent disease, so antiandrogens are effective in treating it. Bicalutamide has major advantages over other antiandrogens such as spironolactone (Aldactone) and cyproterone acetate (Androcur) in terms of antiandrogenicpotency, clinical effectiveness, pharmacological selectivity, and tolerability. It also has improved potency, pharmacokinetic properties, and tolerability, as well as far better safety, compared to the older nonsteroidal antiandrogens flutamide (Eulexin) and nilutamide (Anandron, Nilandron). However, use of bicalutamide as an antiandrogen in transfeminine hormone therapy is very recent. The employment of bicalutamide for transfeminine people was largely precluded for many years by the fact that bicalutamide had pharmaceutical patent protection and was very expensive. However, this changed with the availability of generic versions of bicalutamide starting in 2007 to 2009. In addition, newer and more effective antiandrogens like abiraterone acetate (Zytiga) in 2011 and enzalutamide (Xtandi) in 2012 were introduced and superseded bicalutamide as the standard-of-care antiandrogen for the treatment of prostate cancer. These developments have greatly reduced the cost of bicalutamide and it has gradually become much more affordable in the last decade.
Before 2015, there were only a few mentions in the literature of bicalutamide for transfeminine people and a handful of anecdotal reports online of transfeminine people using it. The earliest clear mention of bicalutamide in the literature in the context of transfeminine hormone therapy was by Louis Gooren in 2011 (Gooren, 2011). Gooren is a major longtime researcher in the field of transgender medicine and is one of the coauthors of the Endocrine Society’s transgender hormone therapy guidelines (Hembree et al., 2009; Hembree et al., 2017). He and his colleagues at the Center of Expertise on Gender Dysphoria of the Vrije Universiteit Medical Center (VUMC) in Amsterdam, Netherlands had conducted studies on nilutamide (Anandron, Nilandron) as an antiandrogen for transfeminine people in the late 1980s and early 1990s (de Voogt et al., 1987a; de Voogt et al., 1987b; Gooren et al., 1987; Johannes et al., 1987; Rao et al., 1988; Asscheman, Gooren, & Peereboom-Wynia, 1989; van Kemenade et al., 1989; Wiki). However, they seem to have abandoned it—probably due to its high incidence of lung toxicity and other off-target side effects. Nonetheless, Gooren began including nonsteroidal antiandrogens like flutamide and nilutamide in his publications as potential treatment options for transfeminine hormone therapy starting in the 1990s (Asscheman & Gooren, 1992; Gooren, 1999). Subsequently, flutamide was included in transgender health guidelines and other publications, though not necessarily favorably (e.g., Israel & Tarver, 1997; Levy, Crown, & Reid, 2003; Dahl et al., 2006a; Dahl et al., 2006b; Hembree et al., 2009; Moreno-Pérez et al., 2012). As a researcher interested in nonsteroidal antiandrogens for transfeminine people, bicalutamide—with its far better safety profile than flutamide and nilutamide—may have been appealing to Gooren. However, Gooren and his colleagues didn’t conduct clinical studies on bicalutamide for transfeminine people and never went beyond brief mention of it for such uses in their publications. Nor did any other academics.
Although there was little discussion or use of bicalutamide in transfeminine people prior to 2015, this started to change in mid-2015. At that time, the Wikipedia content for bicalutamide was greatly expanded, which made information about bicalutamide more accessible. In addition, certain transfeminine people, noting its advantages over existing options and its excellent potential for use in transfeminine hormone therapy, began advocating for use of bicalutamide in transfeminine people in online circles. A number of open-minded clinicians started adopting bicalutamide in transfeminine people around this time and thereafter as well. The first clinical study of bicalutamide in transfeminine people, which began in 2013, was published as an abstract in 2017 and as a full paper in 2019 (Neyman, Fuqua, & Eugster, 2017; Neyman, Fuqua, & Eugster, 2019). It was a small retrospective chart review of bicalutamide alone as a second-line puberty blocker in adolescent transgender girls for whom gonadotropin-releasing hormone analogues were denied by insurance. As of present, it remains the only published clinical data on bicalutamide in transfeminine people. It’s not exactly great data by any means, but it’s a study at least. The researchers who conducted the study had previously published on bicalutamide as a puberty blocker in boys with gonadotropin-independent precocious puberty (e.g., Lenz et al., 2010; Haddad & Eugster, 2012). While limited in its findings, Neyman, Fuqua, and Eugster (2019) helped to generate significant interest among clinicians and researchers in bicalutamide for use in transfeminine hormone therapy.
In any case, due to the recent nature of bicalutamide as an option for use in transfeminine hormone therapy, as well as the lack of studies and characterization of bicalutamide in transfeminine people and concerns about its safety (see next section), bicalutamide isn’t widely used in transfeminine people at this time. In fact, transgender hormone therapy guidelines largely don’t even mention it still. At present, the use of bicalutamide in transfeminine people is mostly limited to a number of more flexible clinicians and to people in the transgender do-it-yourself (DIY) hormone therapy community.
Concerns About Bicalutamide Limiting its Use
The transgender medical community has been reluctant to endorse the use of bicalutamide in transfeminine people to date. This is because of the lack of clinical studies and characterization of bicalutamide in transfeminine people, most importantly in terms of safety. There have been concerns about rare instances of liver failure that have occurred with bicalutamide in men with prostate cancer (Wiki). The reported cases of liver toxicity with bicalutamide have generally been sudden-onset and severe. Rare liver toxicity is an acceptable risk in men with prostate cancer because the risk–benefit ratio of bicalutamide therapy is very favorable, with the benefit of treating prostate cancer vastly outweighing the harm of the very rare instances of liver problems. But transfeminine people are typically young and healthy, and bicalutamide isn’t treating a terminal illness when it’s used in us. If a transfeminine person develops liver failure and dies because of bicalutamide, that’s unnecessary harm and a life needlessly lost. Accordingly, the University of California San Francisco (UCSF) transgender care guidelines warn against use of bicalutamide in transfeminine people currently due to potential liver risks (Deutsch, 2016). Aside from risks, there is also a lack of data to guide appropriate dosing of bicalutamide in transfeminine people at this time. A typical bicalutamide dosage of 50 mg/day is being used and recommended, but this has been arbitrarily chosen with little basis to support it.
To date, there are 10 published case reports of serious liver toxicity in association with bicalutamide (Table). All of these cases were in men with prostate cancer and all occurred within 6 months of initiation of bicalutamide therapy, with two of the cases resulting in death. While this is not a lot of cases and may seem reassuring, it must be noted that quantity of published case reports tends to vastly underestimate the true incidence of rare adverse reactions. As an example, there are around 50 published case reports of meningioma with cyproterone acetate (Table), but a recent large study by the French government found that there were more than 500 operated instances of meningioma in association with high-dose cyproterone acetate over an 8-year period in France alone (Aly, 2020). Accordingly, as of writing there are 40 reports of liver failure, including 25 consequent deaths, in association with bicalutamide in the U.S. FDA’s international MedWatch/FAERS database. (As well as 240 cases of interstitial lung disease associated with bicalutamide notably—relative to only 14 published case reports (Table).) Even with this database however, fewer than 10% of serious adverse reactions are estimated to be reported (Graham, Ahmad, & Piazza-Hepp, 2002). Hence, the true numbers may be much greater. These instances are merely co-occurrences, and causality in terms of bicalutamide and liver toxicity has not been established. But they are concerning nonetheless. There is additionally an unpublished case anecdote of death in a young transfeminine person associated with bicalutamide that’s been making its rounds through the transgender medical community. Per certain very credible people in the field of transgender medicine (e.g., Asa Radix and Zil Goldstein), she is said to have been a 20-year-old transgender girl in Texas taking bicalutamide with rapid-onset liver failure and no warning signs. This case has given clinicians and researchers who are aware of it reservations about the use of bicalutamide in hormone therapy for transfeminine people. Another case of liver failure and death in a transgender person over 60 years of age who was treated with bicalutamide has also been informally reported (QueerDoc).
In any case, the reported cases of serious liver toxicity with bicalutamide in transgender people have not been published nor properly confirmed. In addition, the absolute incidence of liver toxicity with bicalutamide is likely to be very low. For instance, the incidence of abnormal liver function tests (i.e., elevated liver enzymes on blood work) was only 3.4% with high-dose (150 mg/day) bicalutamide monotherapy relative to 1.9% for placebo (a 1.5% difference attributable to bicalutamide) at 3.0 years of follow-up in the Early Prostate Cancer (EPC) clinical programme, a series of three phase 3randomized controlled trials consisting of over 8,000 patients in which bicalutamide was evaluated for treatment of early prostate cancer (Anderson, 2003; Iversen et al., 2004; Wiki; Wiki). Moreover, there were no cases of serious liver toxicity or liver failure with bicalutamide in the initial clinical development programme of bicalutamide for advanced prostate cancer, in which almost 4,000 men were treated with bicalutamide (Blackledge, 1996; Kolvenbag & Blackledge, 1996; McLeod, 1997; Anderson, 2003; Iversen et al., 2004; Wiki). However, it should be noted that this was with careful monitoring of liver function in patients and with prompt discontinuation of bicalutamide upon detection of clinically concerning hepatic abnormalities. About 0.5 to 1.5% of men taking 50 to 150 mg/day bicalutamide in the major clinical programmes of bicalutamide for prostate cancer developed liver changes sufficiently marked that they required discontinuation (Blackledge, 1996; See et al., 2002; Wiki). Hence, regular liver monitoring is essential with bicalutamide to ensure that the possibility of severe liver toxicity is avoided.
Bicalutamide has a much lower risk of liver toxicity than its analogue flutamide (Kolvenbag & Blackledge, 1996; Schellhammer et al., 1997; Thole et al., 2004; Manso et al., 2006; Table). However, it retains a small risk of liver toxicity of its own—one with the potential for serious consequences. Hence, caution is warranted with its use, and careful liver monitoring is a necessity for anyone taking it.
Recent Developments and the Future
Bicalutamide is currently being adopted and characterized for use in the treatment androgen-dependent skin and hair conditions in cisgender women. For instance, a rigorous Italian phase 3 randomized controlled trial of bicalutamide for hirsutism was recently published (Moretti et al., 2018). Retrospective chart reviews of bicalutamide for scalp hair loss in cisgender women have also been published recently (Fernandez-Nieto et al., 2019; Ismail et al., 2020; Fernandez-Nieto et al., 2020; Moussa et al., 2021). The hair loss studies have observed low though significant rates of liver changes with bicalutamide.
Certain transgender medical researchers are showing interest in bicalutamide as well. Perhaps most notably, Wylie Hembree—the lead author of the Endocrine Society’s 2009 and 2017 transgender hormone therapy guidelines (Hembree et al., 2009; Hembree et al., 2017)—wrote positively about bicalutamide for transfeminine people in a recent review (Fishman, Paliou, Poretsky, & Hembree, 2019). He and his colleagues cited the recent phase 3 trial of bicalutamide for hirsutism in cisgender women and the study of bicalutamide as a puberty blocker in transgender girls in support of potential use of bicalutamide for transfeminine people. Guy T’Sjoen—another major researcher in transgender medicine and co-author of the Endocrine Society guidelines (Hembree et al., 2017; Mitchell, 2020)—seemed to show openness to bicalutamide with his colleagues in a recent review as well (Iwamoto et al., 2019). Researchers outside of the United States in particular may be more open to bicalutamide, owing to accumulating health concerns with cyproterone acetate—the most commonly used antiandrogen outside of the United States (Aly, 2020). John Randolph, a researcher at the University of Michigan, has also written positively about bicalutamide (Randolph, 2018), though he may have since changed his mind on it (Michigan Medicine, 2020). On the other hand, other authors have not been as welcoming of bicalutamide for transfeminine people (e.g., Hamidi & Davidge-Pitts, 2019; Cocchetti et al., 2020).
The small risks of bicalutamide with appropriate monitoring may prove to be acceptable to the transgender medical community. This would perhaps be analogous to the rare incidences of serious adverse effects with say spironolactone (e.g., hyperkalemia) or cyproterone acetate (e.g., benign brain tumors, blood clots, breast cancer, liver toxicity). It’s possible that bicalutamide may not ultimately be recommended as a first-line therapy due to its risks. However, it could still be allowed as a second-line option when other antiandrogens are less feasible or not possible due to being for instance inadequately effective, poorly tolerated, contraindicated, or unavailable. The transgender medical community isn’t there at this time though. More developments—namely studies and characterization of bicalutamide in actual transfeminine people—are likely to be needed before bicalutamide could become more accepted for use in transfeminine people or recommended in transgender hormone therapy guidelines.
Updates
Update 1: Thompson et al. (2021) [Fenway Health Guidelines]
In March 2021, the Fenway Health transgender health clinical practice guidelines were updated from the last version (October 2015) to the following latest edition (Aly, 2020):
Thompson, J., Hopwood, R. A., deNormand, S., & Cavanaugh, T. (2021). Medical Care of Trans and Gender Diverse Adults. Boston: Fenway Health. [URL] [PDF]
This update is notable as these guidelines included bicalutamide as an antiandrogen option for transfeminine people. While they did not recommend bicalutamide as a first-line agent due to its limited characterization in transfeminine people and its known small risk of liver toxicity, they were cautiously permissive of its use in transfeminine hormone therapy:
Bicalutamide can be used for [gender-affirming hormone therapy], but there are very few studies examining its use and the relative risk/benefit for this purpose. Because of reported cases of fulminant hepatitis, consensus is that its use in gender affirming hormonal regimen should be carefully considered, used only after alternative options have been trialed or offered, and an in-depth discussion of these potential risks have been had.
These are the first transgender care guidelines to allow the use of bicalutamide, and only the second guidelines to include bicalutamide. Previously, only the UCSF guidelines mentioned bicalutamide, but they were not permissive of its use in transfeminine people.
Tomson, A., McLachlan, C., Wattrus, C., Adams, K., Addinall, R., Bothma, R., Jankelowitz, L., Kotze, E., Luvuno, Z., Madlala, N., Matyila, S., Padavatan, A., Pillay, M., Rakumakoe, M. D., Tomson-Myburgh, M., Venter, W., & de Vries, E. (2021). Southern African HIV Clinicians’ Society gender-affirming healthcare guideline for South Africa. Southern African Journal of HIV Medicine, 22(1), a1299. [DOI:10.4102/sajhivmed.v22i1.1299] [PDF]
Surprisingly, these guidelines not only included bicalutamide but recommended it as the preferred antiandrogen over spironolactone and cyproterone acetate. The reason stated for this was “less risk of neurosteroid depletion (does not cross blood-brain-barrier readily).” However, this supposed effect isn’t a known concern with antiandrogens besides 5α-reductase inhibitors, and bicalutamide actually does appear to be centrally permeable in humans (Wiki). Also surprisingly, no mention of liver toxicity or liver enzyme monitoring with bicalutamide was made in these guidelines. Considering these apparent oversights and others, these guidelines’s recommendations should probably be interpreted with caution.
Update 3: Coleman et al. (2022) [WPATH SOC8 Guidelines]
Bicalutamide is an antiandrogen that has been used in the treatment of prostate cancer. It competitively binds to the androgen receptor to block the binding of androgens. Data on the use of bicalutamide in trans feminine populations is very sparse and safety data is lacking. One small study looked at the use of bicalutamide 50 mg daily as a puberty blocker in 23 trans feminine adolescents who could not obtain treatment with a GnRH analogue (Neyman et al., 2019). All adolescents experienced breast development which is also commonly seen in men with prostate cancer who are treated with bicalutamide. Although rare, fulminant hepatotoxicity resulting in death has been described with bicalutamide (O’Bryant et al., 2008). Given that bicalutamide has not been adequately studied in trans feminine populations, we do not recommend its routine use.
When selecting a medication, we advise using those which have been studied in multiple transgender populations (i.e., estrogen, cyproterone acetate, GnRH agonists) rather than medications with little to no peer-reviewed scientific studies (i.e., bicalutamide, rectal progesterone, etc.) (Angus et al., 2021; Butler et al., 2017; Efstathiou et al., 2019; Tosun et al., 2019).
As can be seen, the WPATH SOC8 did not recommend the routine use of bicalutamide in transfeminine people owing to the lack of studies of it in this population and its potential risks. As touched on in the present article, it is likely that more studies of bicalutamide in transfeminine people will be needed before bicalutamide could become endorsed by major transgender care guidelines.
Update 4: Jamie Reed 2023 Bicalutamide Liver Toxicity Case
In February 2023, Jamie Reed, a former case manager at the The Washington University Transgender Center at St. Louis Children’s Hospital in St. Louis, Missouri, published the op-ed “I Thought I Was Saving Trans Kids. Now I’m Blowing the Whistle.” in a conservative online news outlet called The Free Press. In this article, Reed expressed that she had become disillusioned with the medical care of transgender youth and layed out her grievances. In addition however, she briefly described an additional case of liver toxicity with bicalutamide in a transfeminine person that had allegedly occurred at her center. This individual was said to be 15 years of age and was given bicalutamide as a puberty blocker by Dr. Christopher Lewis, one of the co-founders of the center. She was said to have subsequently developed liver toxicity and was taken off of bicalutamide. In an electronic message to the center, her mother said that they were “lucky her family was not the type to sue”. This instance, and Reed’s op-ed in general, were subsequently widely reported on in conservative news media, for instance on Fox News and in the Daily Mail (Google). In addition to her op-ed, Reed provided a sworn affidavit to the office of Republican Missouri attorney general Andrew Bailey, who proceeded to launch an investigation of the clinic (Missouri Government, 2023a). The following further information was released in the affidavit:
One doctor at the Center, Dr. Chris Lewis, is giving patients a drug called Bicalutamide. The drug has a legitimate use for treating pancreatic cancer [sic], but it has a side effect of causing breasts to grow, and it can poison the liver. There are no clinical studies for using this drug for gender transitions, and there are no established standards of care for using this drug.
Because of these risks and the lack of scientific studies, other centers that do gender transitions will not use Bicalutamide. The adult center affiliated with Washington University will not use this medication for this reason. But the Center treating children does.
I know of at least one patient at the Center who was advised by the renal department to stop taking Bicalutamide because the child was experiencing liver damage. The child’s parent reported this to the Center through the patient’s online self-reporting medical chart (MyChart). The parent said they were not the type to sue, but “this could be a huge PR problem for you.”
While unpublished and unverified like the earlier reports of liver toxicity with bicalutamide in transfeminine people, this case represents yet another report, and is notably by far the best-documented one. No other clinical details on the case were provided, and it is unclear whether it involved serious liver toxicity, merely asymptomatic liver function test abnormalities, or a clinical situation somewhere in-between these extremes. In any case, it does seem clear that this instance is not likely to have a positive influence on the further adoption of bicalutamide in transfeminine hormone therapy.
Subsequent to the investigation of the clinic being launched, in April 2023, Missouri greatly restricted gender-affirming care for transgender youth and adults, with some of the most severe limits that have been enacted in the United States (Associated Press, 2023a; Missouri Government, 2023b). Bicalutamide and the liver toxicity instance were not further described with these developments. The new state law restricting gender-affirming care took effect August 28, 2023, and Washington University announced that it would stop prescribing puberty blockers and hormone therapy to transgender youth shortly thereafter (Associated Press, 2023b).
A New York Times article with additional information on the case was also subsequently published (Ghorayshi, 2023 [Excerpts]). It was noted that the adolescent had been on bicalutamide for 1 year and definitely experienced hepatotoxicity. However, she also had a complicated medical history, including being immunocompromised, having recently had COVID-19, and having taken another drug known to be associated with hepatotoxicity. As such, the hepatotoxicity cannot be definitively attributed to bicalutamide, but it simultaneously cannot be ruled out that bicalutamide was involved or causative.
Subsequent Burgener et al. (2023, 2024) Findings
Following the preceding case, Lewis and colleagues went on to publish a conference abstract and preprint of a study of bicalutamide in transfeminine youth and young adults in which they stated that it does not increase liver enzymes in this population (Burgener et al., 2023; Burgener et al., 2024). However, a closer look at their data show that bicalutamide did statistically significantly elevate certain liver parameters relative to other antiandrogens, namely rates of elevated aspartate aminotransferase (AST) (upper limit of normal 10.7% vs. 1.5%, P = 0.02) (Burgener et al., 2024). Likewise, rates of elevated alanine aminotransferase (ALT) appeared to trend in the direction of being increased, though this was not statistically significant (upper limit of normal 16.7% vs. 11.6%, P = 0.37) (Burgener et al., 2024). In any case, rates of clinically significant elevations in liver enzymes with bicalutamide, defined as greater than three times the upper limit of normal, were not significantly increased in the study.
On the basis of the relevant research in men with prostate cancer (Wiki), Lewis and colleagues’ study, with a bicalutamide-group sample size of only 84 transfeminine individuals, was clearly greatly underpowered for evaluating liver function changes. Per the Early Prostate Cancer trial of high-dose bicalutamide monotherapy in men with prostate cancer, elevated liver enzymes appear to occur with bicalutamide at a rate of only about 1.5% more than placebo, or roughly an additional 1 in every 66 people (Wiki). Based on power analysis, this would require a far larger sample size to have adequate statistical power and actually have a chance of achieving statistical significance.
As such, it seems to the present author premature to conclude that bicalutamide does not elevate liver enzymes in transfeminine people.
Lewis and colleagues didn’t mention in their study paper the transfeminine adolescent liver toxicity case reported by Jamie Reed that was said to have occurred at their clinic nor have they published a case report about this instance. Instead, only the following is stated:
One case report published in 2024 described a transgender female adolescent prescribed bicalutamide 50 mg daily who presented to a hospital with liver toxicity that resolved after stopping bicalutamide (Wilde et al., 2024). This appears to be the first documented case of bicalutamide-induced hepatoxicity in a transgender female.
While this case was, coincidentally, also a 17-year-old transfeminine adolescent (Wilde et al., 2024), this instance, per the medical histories and reporting authors/institutions, appears to be distinct from Dr. Lewis’s that was reported by Jamie Reed.
However, Lewis and colleagues did note the following in their paper, which plausibly might have been the Jamie Reed case:
There was one individual in whom bicalutamide was stopped after the follow-up period designated for the study. This individual developed ALT and AST >2x ULN after an episode of COVID and had a thorough hepatology evaluation. As ALT and AST were never > 3x ULN, it was not recommended that bicalutamide be stopped; however, ultimately a clinical decision was made to stop the medication and ALT and AST normalized.
Another concern with Lewis and colleagues’ paper pertains to the following statements:
Whereas bicalutamide doses for prostate cancer reach 150 mg daily, doses used in the care of AMAB transfeminine individuals are much lower (25-50 mg daily).
Bicalutamide doses used in prostate cancer are up to 150 mg daily. Due to these concerns of liver toxicity, bicalutamide has not been routinely used as an anti-androgen in AMAB transfeminine individuals, despite the much lower doses needed in this population (∼25-50 mg daily).
In actuality, bicalutamide is most widely used in prostate cancer, in the form of combined androgen blockade with surgical or medical castration, at a dosage of 50 mg/day, whereas the 150 mg/day dosage is used less commonly, in the form of monotherapy (Wiki). Moreover, only the 50 mg/day dosage is used in the United States, where monotherapy is not approved. Among the published case reports of hepatotoxicity with bicalutamide in men with prostate cancer, half have been at a dose of 50 mg/day and the other half have been at a dose of 80 to 150 mg/day (Wiki). The two instances of death due to hepatotoxicity with bicalutamide were both at 50 mg/day. There is currently no evidence that the hepatotoxicity of bicalutamide is dose-dependent across its clinically used dosage range (Wiki), although employment of the lowest effective dose in transfeminine people nonetheless seems prudent just in case. Hence, in contrast to Lewis and colleague’s claims, a bicalutamide dosage of 50 mg/day is not less than that generally used in prostate cancer, and clearly retains substantial hepatotoxic potential.
Update 5: New Bicalutamide Publications in 2022 Through 2025
Angus, L., Nolan, B., Zajac, J., & Cheung, A. (November 2022). Use of bicalutamide as an androgen receptor antagonist in transgender women. ESA/SRB/APEG/NZSE ASM 2022, November 13-16, Christchurch, Abstracts and Programme, 127–127 (abstract no. 280). [URL] [PDF] [Full Abstract Book]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (November 2023). Bicalutamide as an anti-androgen in trans people: a cross-sectional study. AusPATH 2023 Symposium. [URL] [PDF] [Slides] [Trans Health Research Blog Post]
Bambilla, A., Beal, C., & Vigil, P. (2023). Improving Access to Bicalutamide in Gender Affirming Medical Care. [Unpubished/pending publication] [QueerCME Blog Post]
Burgener, K., DeBosch, B., Lewis, C., Wallendorf, M., & Herrick, C. (May 2023). Assessment of Liver Function and Toxicity in Transgender Female Adolescents Prescribed Bicalutamide. Hormone Research in Paediatrics, 96(Suppl 3 [Abstracts of the 2023 Pediatric Endocrine Society (PES) Annual Meeting’ to Hormone Research in Paediatrics]), 377–378 (abstract no. 6232). [DOI:10.1159/000531602] [PDF]
Gómez-Aguilar, F., Martínez-Sánchez, L., Arias-Constantí, V., Muñoz-Santanach, D., & Sarquella-Brugada, G. (2023). QT prolongation and Torsade de Pointes in a 13-year-old transgender adolescent in treatment with bicalutamide and tacrolimus. Clinical Toxicology, 61(Suppl 1 [43rd International Congress of the European Association of Poisons Centres and Clinical Toxicologists (EAPCCT), 23–26 May 2023, Palma de Mallorca, Spain]), 81–82 (abstract no. 170). [DOI:10.1080/15563650.2023.2192024] [PDF] [Reactions Weekly]
Karakılıç Özturan, E., Öztürk, A. P., Baş, F., Erdoğdu, A. B., Kaptan, S., Kardelen Al, A. D., Poyrazoğlu, Ş., Yıldız, M., Direk, N., Yüksel, Ş., & Darendeliler, F. (2023). Endocrinological Approach to Adolescents with Gender Dysphoria: Experience of a Pediatric Endocrinology Department in a Tertiary Center in Turkey. Journal of Clinical Research in Pediatric Endocrinology, 15(3), 276–284. [DOI:10.4274/jcrpe.galenos.2023.2023-1-13]
Vierregger, K., Tetzlaff, M., Zimmerman, B., Dunn, N., Finney, N., Lewis, K., Slomoff, R., & Strutner, S. (May 2023). Bicalutamide Use as Antiandrogen in Trans Feminine Adults - A Safety Profile. National Transgender Health Summit (NTHS) 2023 Symposium. [Event Agenda PDF] [Symposium Session] [Symposium Abstracts/Program Book]
Vierregger, K., Tetzlaf, M., Zimmerman, B., Dunn, N., Finney, N., Lewis, K., Slomoff, R., & Strutner, S. (November 2023). Bicalutamide Use as Antiandrogen in Trans Feminine Adults - A Safety Profile. USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 96–96 (abstract no. SAT-B2-T4). [Symposium Schedule] [PDF] [Full Abstract Book]
Warus, J., Rincon, M. G., Salvetti, B., & Olson-Kennedy, J. (November 2023). Safety of Bicalutamide as Anti-Androgenic Therapy in Gender Affirming Care for Adolescents and Young Adults: A Retrospective Chart Review. USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 124–124 (abstract no. SUN-B1-T5). [Symposium Schedule] [PDF] [Full Abstract Book]
Wilde, B., Diamond, J. B., Laborda, T. J., Frank, L., O’Gorman, M. A., & Kocolas, I. (2023). Bicalutamide-Induced Hepatotoxicity in a Transgender Male-to-Female Adolescent. Journal of Adolescent Health, 74(1), 202–204. [DOI:10.1016/j.jadohealth.2023.08.024]
Burgener, K., DeBosch, B., Wang, J., Lewis, C., & Herrick, C. (2025). Bicalutamide does not raise transaminases clinically significantly compared to alternative anti-androgen regimens among transfeminine adolescents and young adults: a retrospective cohort study. International Journal of Transgender Health, 1–10. [DOI:10.1080/26895269.2025.2452184]
Fuqua, J. S., Shi, E., & Eugster, E. A. (2024). A retrospective review of the use of bicalutamide in transfeminine youth; a single center experience. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2023.2294321]
Shumer, D., & Roberts, S. A. (2024). Placing a Report of Bicalutamide-Induced Hepatotoxicity in the Context of Current Standards of Care for Transgender Adolescents. Journal of Adolescent Health, 74(1), 5–6. [DOI:10.1016/j.jadohealth.2023.10.010]
Angus, L. M., Hong, Q. V., Cheung, A. S., & Nolan, B. J. (2024). Effect of bicalutamide on serum total testosterone concentration in transgender adults: a case series. Therapeutic Advances in Endocrinology and Metabolism, 15. [DOI:10.1177/20420188241305022]
Update 6: Original Bicalutamide Liver and Lung Toxicity Analysis by Sam
A few years ago back in 2021, Transfeminine Science author Sam conducted an original analysis of the incidence of liver and lung toxicity with bicalutamide in the published clinical trial literature. This project was never finished or made publicly available. However, with bicalutamide being increasingly studied and adopted for use in transfeminine people, it seems quite valuable and relevant today. As such, we have opted to now publish Sam’s analysis in this section.
Sam’s analysis can be found in the provided document here. In terms of methodology, she searched PubMed for all clinical trials of bicalutamide, collated all of the relevant results into a table, and then calculated the incidences of serious liver toxicity and lung toxicity from those data. In clinical trials, adverse events are rated in terms of grades of severity, with a Grade 3 adverse event defined as “severe”, Grade 4 as “life-threatening”, and Grade 5 as “death” (Wiki).
Of 229 results, 33 trials were found to be relevant and were included. Most of the trials were in men with prostate cancer, but a few were in women with cancer and boys with precocious puberty. Sam found that of a total of 7,703 evaluable participants, there were 2 instances of serious liver toxicity and 2 instances of serious lung toxicity with bicalutamide. This resulted in the same incidence rate of 0.026% (95% CI: 0.003% to 0.094%) or approximately 1 in 3,846 individuals for both liver toxicity and lung toxicity. Combining these toxicities resulted in a total incidence of serious liver or serious lung toxicity with bicalutamide of 0.052% (95% CI: 0.014% to 0.133%) or approximately 1 in 1,923 individuals. All of the observed toxicity events were rated as Grade 3 or 4. It should be noted that clinical trials of bicalutamide typically employ careful laboratory monitoring and assessment of clinical adverse events as well as prompt medication discontinuation upon unfavorable laboratory changes.
While the confidence intervals (CIs) in Sam’s analysis were wide and hence the estimates are very rough, they provide an idea of the potential real-world risk of serious toxicity with bicalutamide in transfeminine people based on high-quality clinical data. Notably, they do not suffer from the problem of under-reporting of adverse events that occurs with published case reports, pharmacovigilance databases, and certain types of observational studies. However, limitations of Sam’s analysis include (1) toxicity incidence rates for non-bicalutamide-treated controls not being assessed and (2) most of the patients having cancer and being of older age, and hence the generalizability of the findings to healthy transfeminine people not being fully clear. In any case, I was surprised by how high the incidence rates were when I first saw her analysis, and I suspect that others may be as well.
References
Aly. (2020). Clinical Guidelines with Information on Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Aly. (2020). Recent Developments on Cyproterone Acetate and Meningioma Risk Out of France and Implications for Transfeminine People. Transfeminine Science. [URL]
Anderson, J. (2003). The role of antiandrogen monotherapy in the treatment of prostate cancer. BJU International, 91(5), 455–461. [DOI:10.1046/j.1464-410X.2003.04026.x]
Angus, L., Nolan, B., Zajac, J., & Cheung, A. (2022). Use of bicalutamide as an androgen receptor antagonist in transgender women. ESA/SRB/APEG/NZSE ASM 2022, November 13-16, Christchurch, Abstracts and Programme, 127–127 (abstract no. 280). [URL] [PDF] [Full Abstract Book]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2023). Bicalutamide as an anti-androgen in trans people: a cross-sectional study. AusPATH 2023 Symposium. [URL] [PDF] [Slides] [Trans Health Research Blog Post]
Angus, L. M., Hong, Q. V., Cheung, A. S., & Nolan, B. J. (2024). Effect of bicalutamide on serum total testosterone concentration in transgender adults: a case series. Therapeutic Advances in Endocrinology and Metabolism, 15. [DOI:10.1177/20420188241305022]
Arya, V. B., & Davies, J. H. (2019). Idiopathic gonadotropin-independent precocious puberty - is regular surveillance required? Journal of Pediatric Endocrinology & Metabolism: JPEM, 32(4), 403–407. [DOI:10.1515/jpem-2018-0419]
Asscheman, H., Gooren, L. J., & Peereboom-Wynia, J. D. (1989). Reduction in undesired sexual hair growth with anandron in male-to-female transsexuals—experiences with a novel androgen receptor blocker. Clinical and Experimental Dermatology, 14(5), 361–363. [DOI:10.1111/j.1365-2230.1989.tb02585.x]
Asscheman, H., & Gooren, L. J. (1992). Hormone Treatment in Transsexuals. In Bocking, W. O., Coleman, E. (Eds). Gender Dysphoria: Interdisciplinary Approaches in Clinical Management (pp. 39–54). Binghamton: Haworth Press. / Journal of Psychology & Human Sexuality, 5(4), 39–54. [Google Scholar] [Google Books] [DOI:10.1300/J056v05n04_03]
AstraZeneca. (2008). Clinical Review. NDA. 22-310/S-001. Casodex (bicalutamide) for Testotoxicosis. Food and Drug Administration/AstraZeneca Pharmaceuticals. [URL] [PDF]
Bahceci, M., Tuzcu, A., Canoruc, N., Tuzun, Y., Kidir, V., & Aslan, C. (2004). Serum C-reactive protein (CRP) levels and insulin resistance in non-obese women with polycystic ovarian syndrome, and effect of bicalutamide on hirsutism, CRP levels and insulin resistance. Hormone Research in Paediatrics, 62(6), 283–287. [DOI:10.1159/000081973]
Bailey, A. (2023 February 9). Missouri Attorney General Andrew Bailey Confirms Launch of Multi-Agency Investigation into St. Louis Transgender Center for Harming Hundreds of Children. Attorney General Andrew Bailey, Missouri Government. [URL] [Affadavit of Jamie Reed]
Bailey, A. (2023 April 13). Missouri Attorney General Andrew Bailey Promulgates Emergency Regulation Targeting Gender Transition Procedures for Minors. Attorney General Andrew Bailey, Missouri Government. [URL] [Emergency Regulation PDF]
Ballentine, S., & Hollingsworth, H. (2023 April 13). Missouri to limit gender-affirming care for minors, adults. Associated Press. [URL]
Bambilla, A., Beal, C., & Vigil, P. (2023). Improving Access to Bicalutamide in Gender Affirming Medical Care. [Unpubished/pending publication] [QueerCME Blog Post]
Blackledge, G. R. P. (1996). Clinical progress with a new antiandrogen, Casodex™ (bicalutamide). European Urology, 29(Suppl 2), 96–104. [DOI:10.1159/000473847]
Burgener, K., DeBosch, B., Lewis, C., Wallendorf, M., & Herrick, C. (2023). Assessment of Liver Function and Toxicity in Transgender Female Adolescents Prescribed Bicalutamide. Hormone Research in Paediatrics, 96(Suppl 3 [Abstracts of the 2023 Pediatric Endocrine Society (PES) Annual Meeting’ to Hormone Research in Paediatrics]), 377–378 (abstract no. 6232). [DOI:10.1159/000531602] [PDF]
Burgener, K., DeBosch, B., Wang, J., Lewis, C., & Herrick, C. (2025). Bicalutamide does not raise transaminases clinically significantly compared to alternative anti-androgen regimens among transfeminine adolescents and young adults: a retrospective cohort study. International Journal of Transgender Health, 1–10. [DOI:10.1080/26895269.2025.2452184]
Cocchetti, C., Ristori, J., Romani, A., Maggi, M., & Fisher, A. D. (2020). Hormonal Treatment Strategies Tailored to Non-Binary Transgender Individuals. Journal of Clinical Medicine, 9(6), 1609. [DOI:10.3390/jcm9061609]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Dahl, M., Feldman, J. L., Goldberg, J. M., & Jaberi, A. (2006). Physical aspects of transgender endocrine therapy. International Journal of Transgenderism, 9(3–4), 111–134. [DOI:10.1300/J485v09n03_06]
Dahl, M., Feldman, J. L., Goldberg, J., & Jaberi, A. (2006). Endocrine Therapy for Transgender Adults in British Columbia: Suggested Guidelines. Physical Aspects of Transgender Endocrine Therapy. Vancouver: Vancouver Coastal Health. [Google Scholar] [PDF]
de Voogt, H. J., Rao, B. R., Gooren, L. J. G., Geldof, A. A., & Bouman, F. G. (1987). Considerations About the Use of Anti-Androgenic Drugs in Male-to-Female Transsexuals. Tenth International Symposium on Gender Dysphoria, Diagnosis and Treatment of Transsexualism, Vrije Universiteit Amsterdam, The Netherlands, June 9–12, 1987. Harry Benjamin International Gender Dysphoria Association. [PDF]
de Voogt, H. J., Rao, B. R., Geldof, A. A., Gooren, L. J. G., & Bouman, F. G. (1987). Androgen action blockade does not result in reduction in size but changes histology of the normal human prostate. The Prostate, 11(4), 305–311. [DOI:10.1002/pros.2990110403]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Fernandez-Nieto, D., Saceda-Corralo, D., Rodrigues-Barata, R., Hermosa-Gelbard, A., Moreno-Arrones, O., Jimenez-Cauhe, J., Ortega-Quijano, D., & Vano-Galvan, S. (2019). Oral bicalutamide for female pattern hair loss: A pilot study. Dermatologic Therapy, 32(6), e13096. [DOI:10.1111/dth.13096]
Fernandez-Nieto, D., Saceda-Corralo, D., Jimenez-Cauhe, J., Moreno-Arrones, O. M., Rodrigues-Barata, R., Hermosa-Gelbard, A., & Vano-Galvan, S. (2020). Bicalutamide: A potential new oral antiandrogenic drug for female pattern hair loss. Journal of the American Academy of Dermatology, 83(5), e355–e356. [DOI:10.1016/j.jaad.2020.04.054]
Finkle, A., Zavertnik, S., Myers, S., Cormier, D., Heithaus, J., & Augustyn, M. (2020). Growing Up Fast: Managing Autism Spectrum Disorder and Precocious Puberty. Journal of Developmental and Behavioral Pediatrics: JDBP, 41(9), 740–742. [DOI:10.1097/DBP.0000000000000865]
Fishman, S. L., Paliou, M., Poretsky, L., & Hembree, W. C. (2019). Endocrine Care of Transgender Adults. In Poretsky, L., & Hembree, W. C. (Eds.). Transgender Medicine (pp. 143–163). Cham: Humana Press. [DOI:10.1007/978-3-030-05683-4_8]
Food and Drug Administration (FDA). (2017). CASODEX (bicalutamide) tablet, for oral use. [URL] [PDF]
Fuqua, J. S., Shi, E., & Eugster, E. A. (2024). A retrospective review of the use of bicalutamide in transfeminine youth; a single center experience. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2023.2294321]
Ghorayshi, A. (2023 August 29). How a Small Gender Clinic Landed in a Political Storm. The New York Times. [URL]
Gómez-Aguilar, F., Martínez-Sánchez, L., Arias-Constantí, V., Muñoz-Santanach, D., & Sarquella-Brugada, G. (2023). QT prolongation and Torsade de Pointes in a 13-year-old transgender adolescent in treatment with bicalutamide and tacrolimus. Clinical Toxicology, 61(Suppl 1 [43rd International Congress of the European Association of Poisons Centres and Clinical Toxicologists (EAPCCT), 23–26 May 2023, Palma de Mallorca, Spain]), 81–82 (abstract no. 170). [DOI:10.1080/15563650.2023.2192024] [PDF] [Reactions Weekly]
Gooren, L., Spinder, T., Spijkstra, J. J., Van Kessel, H., Smals, A., Rao, B. R., & Hoogslag, M. (1987). Sex steroids and pulsatile luteinizing hormone release in men. Studies in estrogen-treated agonadal subjects and eugonadal subjects treated with a novel nonsteroidal antiandrogen. The Journal of Clinical Endocrinology & Metabolism, 64(4), 763–770. [DOI:10.1210/jcem-64-4-763]
Gooren, L. J. G. (1999). Hormonal Sex Reassignment. International Journal of Transgenderism, 3(3), 1–7. [Google Scholar] [URL] [Alt]
Gooren, L. J. (2011). Care of transsexual persons. New England Journal of Medicine, 364(13), 1251–1257. [DOI:10.1056/NEJMcp1008161]
Graham, D. J., Ahmad, S. R., & Piazza‐Hepp, T. (2002). Spontaneous Reporting—USA. In Mann, R. D., & Andrews, E. B. (Eds.). Pharmacovigilance (pp. 219–227). Chichester: John Wiley & Sons. [DOI:10.1002/0470853093.ch17]
Gurnurkar, S., DiLillo, E., & Carakushansky, M. (2021). A Case of Familial Male-limited Precocious Puberty with a Novel Mutation. Journal of Clinical Research in Pediatric Endocrinology, 13(2), 239–244. [DOI:10.4274/jcrpe.galenos.2020.2020.0067]
Haddad, N. G., & Eugster, E. A. (2012). Peripheral Precocious Puberty: Interventions to Improve Growth. In Preedy, V. R. (Ed.). Handbook of Growth and Growth Monitoring in Health and Disease (pp. 1199–1212). New York: Springer. [DOI:10.1007/978-1-4419-1795-9_71]
Hamidi, O., & Davidge-Pitts, C. J. (2019). Transfeminine Hormone Therapy. Endocrinology and Metabolism Clinics, 48(2), 341–355. [DOI:10.1016/j.ecl.2019.02.001]
Hembree, W. C., Cohen-Kettenis, P., Delemarre-Van De Waal, H. A., Gooren, L. J., Meyer III, W. J., Spack, N. P., Tangpricha, V., & Montori, V. M. (2009). Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism, 94(9), 3132–3154. [DOI:10.1210/jc.2009-0345]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Ismail, F. F., Meah, N., de Carvalho, L. T., Bhoyrul, B., Wall, D., & Sinclair, R. (2020). Safety of oral bicalutamide in female pattern hair loss: A retrospective review of 316 patients. Journal of the American Academy of Dermatology, 83(5), 1478–1479. [DOI:10.1016/j.jaad.2020.03.034]
Israel, G. E., & Tarver, D. E. (1997). Transgender Care: Recommended Guidelines, Practical Information, and Personal Accounts (p. 66). Philadelphia: Temple University Press. [Google Scholar] [Google Books]
Iversen, P., Johansson, J. E., Lodding, P., Lukkarinen, O., Lundmo, P., Klarskov, P., Tammela, T. L., Tasdemir, I., Morris, T., Carroll, K., & Scandinavian Prostatic Cancer Group. (2004). Bicalutamide (150 mg) versus placebo as immediate therapy alone or as adjuvant to therapy with curative intent for early nonmetastatic prostate cancer: 5.3-year median followup from the Scandinavian Prostate Cancer Group Study Number 6. The Journal of Urology, 172(5 Part 1), 1871–1876. [DOI:10.1097/01.ju.0000139719.99825.54]
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 2042018819871166. [DOI:10.1177/2042018819871166]
Johannes, F. L. M., van Kemenade, M. A., Cohen-Kettenis, P. T., Cohen, L., & Gooren, L. J. G. (1987). Effects of RU 23.903 (Anandron) on Lateralization and Spatial Ability in Male-to-Female-Transsexuals. Tenth International Symposium on Gender Dysphoria, Diagnosis and Treatment of Transsexualism, Vrije Universiteit Amsterdam, The Netherlands, June 9–12, 1987. Harry Benjamin International Gender Dysphoria Association. [PDF]
Karakılıç Özturan, E., Öztürk, A. P., Baş, F., Erdoğdu, A. B., Kaptan, S., Kardelen Al, A. D., Poyrazoğlu, Ş., Yıldız, M., Direk, N., Yüksel, Ş., & Darendeliler, F. (2023). Endocrinological Approach to Adolescents with Gender Dysphoria: Experience of a Pediatric Endocrinology Department in a Tertiary Center in Turkey. Journal of Clinical Research in Pediatric Endocrinology, 15(3), 276–284. [DOI:10.4274/jcrpe.galenos.2023.2023-1-13]
Kolvenbag, G. J., & Blackledge, G. R. (1996). Worldwide activity and safety of bicalutamide: a summary review. Urology, 47(1), 70–79. [DOI:10.1016/S0090-4295(96)80012-4]
Kor, Y. (2018). Central precocious puberty in a case of late-diagnosed familial testotoxicosis and long-term treatment monitoring. Hormones, 17(2), 275–278. [DOI:10.1007/s42000-018-0029-1]
Kreher, N. C., Pescovitz, O. H., Delameter, P., Tiulpakov, A., & Hochberg, Z. E. (2006). Treatment of familial male-limited precocious puberty with bicalutamide and anastrozole. The Journal of Pediatrics, 149(3), 416–420. [DOI:10.1016/j.jpeds.2006.04.027]
Lenz, A. M., Shulman, D., Eugster, E. A., Rahhal, S., Fuqua, J. S., Pescovitz, O. H., & Lewis, K. A. (2010). Bicalutamide and third-generation aromatase inhibitors in testotoxicosis. Pediatrics, 126(3), e728–e733. [DOI:10.1542/peds.2010-0596]
Levy, A., Crown, A., & Reid, R. (2003). Endocrine intervention for transsexuals. Clinical Endocrinology, 59(4), 409–418. [DOI:10.1046/j.1365-2265.2003.01821.x]
Lewis, K. A., Lenz, A., Eugster, E. A., Fuqua, J. S., Pescovitz, O. H., Rahhal, S., & Shulman, D. (2009). Long term bicalutamide and 3rd generation aromatase inhibitor therapy in two boys with testotoxicosis. Hormone Research [in Paediatrics], 72(Suppl 3), 266–267 (PO2-189). [Google Scholar] [DOI:10.1159/000239668]
Manso, G., Thole, Z., Salgueiro, E., Revuelta, P., & Hidalgo, A. (2006). Spontaneous reporting of hepatotoxicity associated with antiandrogens: data from the Spanish pharmacovigilance system. Pharmacoepidemiology and Drug Safety, 15(4), 253–259. [DOI:10.1002/pds.1168]
McLeod, D. G. (1997). Tolerability of Nonsteroidal Antiandrogens in the Treatment of Advanced Prostate Cancer. The Oncologist, 2(1), 18–27. [DOI:10.1634/theoncologist.2-1-18]
Michigan Medicine. (2020). Feminizing Gender-Affirming Hormone Care: The Michigan Medicine Approach. Ann Arbor: Michigan Medicine/University of Michigan. [URL] [PDF]
Mitchell, F. (2020). Guy T’Sjoen: collaborating to improve transgender wellbeing. The Lancet Diabetes & Endocrinology, 8(7), 568–568. [DOI:10.1016/S2213-8587(20)30192-3]
Mitre, N., & Lteif, A. (2009). Treatment of familial male-limited precocious puberty (testotoxicosis) with anastrozole and bicalutamide in a boy with a novel mutation in the luteinizing hormone receptor. Journal of Pediatric Endocrinology and Metabolism, 22(12), 1163–1168. [DOI:10.1515/JPEM.2009.22.12.1163]
Moreno-Pérez, Ó., De Antonio, I. E., & Grupo de Identidad y Diferenciación Sexual de la SEEN (GIDSEEN. (2012). Clinical practice guidelines for assessment and treatment of transsexualism. SEEN Identity and Sexual Differentiation Group (GIDSEEN). Endocrinología y Nutrición (English Edition), 59(6), 367–382. [DOI:10.1016/j.endoen.2012.07.004]
Moretti, C. G., Guccione, L., Di Giacinto, P., Cannuccia, A., Meleca, C., Lanzolla, G., Andreadi, A., & Lauro, D. (2016). Efficacy and Safety of Myo-Inositol Supplementation in the Treatment of Obese Hirsute PCOS Women: Comparative Evaluation with OCP+Bicalutamide Therapy. Endocrine Reviews, 37(2 Suppl 1), ?–? (abstract no. SUN-153). [URL] [DOI:10.1093/edrv/37.supp.1]
Moretti, C., Guccione, L., Di Giacinto, P., Simonelli, I., Exacoustos, C., Toscano, V., Motta, C., De Leo, V., Petraglia, F., & Lenzi, A. (2018). Combined oral contraception and bicalutamide in polycystic ovary syndrome and severe hirsutism: a double-blind randomized controlled trial. The Journal of Clinical Endocrinology & Metabolism, 103(3), 824–838. [DOI:10.1210/jc.2017-01186]
Moussa, A., Kazmi, A., Bokhari, L., & Sinclair, R. D. (2022). Bicalutamide improves minoxidil-induced hypertrichosis in female pattern hair loss: A retrospective review of 35 patients. Journal of the American Academy of Dermatology, 87(2), 488–490. [DOI:10.1016/j.jaad.2021.10.048]
Muderris, I. I., Bayram, F., & Guven, M. (1999). Bicalutamide 25 mg/day in the treatment of hirsutism. Human Reproduction, 14(Suppl 3), 366–366 (R-196). [DOI:10.1093/humrep/14.Suppl_3.366-a]
Müderris, I. I., Bayram, F., Özçelik, B., & Güven, M. (2002). New alternative treatment in hirsutism: bicalutamide 25 mg/day. Gynecological Endocrinology, 16(1), 63–66. [DOI:10.1080/gye.16.1.63.66]
Müderris, İ. İ., & Öner, G. (2009). Hirsutizm Tedavisinde Flutamid ve Bikalutamid Kullanımı. [Flutamide and Bicalutamide Treatment in Hirsutism.] Türkiye Klinikleri Endokrinoloji-Özel Dergisi, 2(2), 110–112. [ISSN:1308-0954] [URL] [PDF] [Translation]
Nabhan, Z. M., & Eugster, E. A. (2019). Testotoxicosis with an episodic course: an unusual case within a series. AACE Clinical Case Reports, 5(1), e50–e53. [DOI:10.4158/ACCR-2018-0246]
Neyman, A., Fuqua, J. S., & Eugster, E. A. (2017). Bicalutamide as an Androgen Blocker with Secondary Effect of Promoting Feminization in Male to Female (MTF) Transgender Adolescents. Hormone Research in Paediatrics, 88(Suppl 1 [10th Individual Abstracts for International Meeting of Pediatric Endocrinology: Free Communication and Poster Sessions, Abstracts]), 477–477 (abstract no. P3-605). [DOI:10.1159/000481424] [PDF]
Neyman, A., Fuqua, J. S., & Eugster, E. A. (2019). Bicalutamide as an androgen blocker with secondary effect of promoting feminization in male-to-female transgender adolescents. Journal of Adolescent Health, 64(4), 544–546. [DOI:10.1016/j.jadohealth.2018.10.296]
Özcabı, B., Ceylaner, S., Özcan, R., Büyükünal, C., Ercan, O., Tüysüz, B., & Evliyaoğlu, O. (2015). Testotoxicosis: Report of Two Cases, One with a Novel Mutation in LHCGR Gene. Journal of Clinical Research in Pediatric Endocrinology, 7(3), 242–248. [DOI:10.4274/jcrpe.2067]
Randolph Jr, J. F. (2018). Gender-Affirming Hormone Therapy for Transgender Females. Clinical Obstetrics and Gynecology, 61(4), 705–721. [DOI:10.1097/GRF.0000000000000396] [URL]
Rao, B. R., De Voogt, H. J., Geldof, A. A., Gooren, L. J. G., & Bouman, F. G. (1988). Merits and considerations in the use of anti-androgen. Journal of Steroid Biochemistry, 31(4), 731–737. [DOI:10.1016/0022-4731(88)90024-6]
Reed, J. (2023 February 9). I Thought I Was Saving Trans Kids. Now I’m Blowing the Whistle. The Free Press. [URL]
Reiter, E. O., Mauras, N., McCormick, K., Kulshreshtha, B., Amrhein, J., De Luca, F., O’Brien, S., Armstrong, J., & Melezinkova, H. (2010). Bicalutamide plus anastrozole for the treatment of gonadotropin-independent precocious puberty in boys with testotoxicosis: a phase II, open-label pilot study (BATT). Journal of Pediatric Endocrinology & Metabolism: JPEM, 23(10), 999–1009. [DOI:10.1515/jpem.2010.161]
Salter, J., & Ballentine, S. (2023 September 12). Missouri clinics halt transgender care for minors in wake of new state law. Associated Press. [URL]
Schellhammer, P. F., Sharifi, R., Block, N. L., Soloway, M. S., Venner, P. M., Patterson, A. L., Sarosdy, M. F., Vogelzang, N. J., Schellenger, J. J., Kolvenbag, G. J., & Casodex Combination Study Group. (1997). Clinical benefits of bicalutamide compared with flutamide in combined androgen blockade for patients with advanced prostatic carcinoma: final report of a double-blind, randomized, multicenter trial. Urology, 50(3), 330–336. [DOI:10.1016/s0090-4295(97)00279-3]
Shumer, D., & Roberts, S. A. (2024). Placing a Report of Bicalutamide-Induced Hepatotoxicity in the Context of Current Standards of Care for Transgender Adolescents. Journal of Adolescent Health, 74(1), 5–6. [DOI:10.1016/j.jadohealth.2023.10.010]
See, W. A., Wirth, M. P., McLeod, D. G., Iversen, P., Klimberg, I., Gleason, D., Chodak, G., Montie, J., Tyrrell, C., Wallace, D. M., Delaere, K. P., Vaage, S., Tammela, T. L., Lukkarinen, O., Persson, B. E., Carroll, K., Kolvenbag, G. J., & Casodex Early Prostate Cancer Trialist Group. (2002). Bicalutamide as immediate therapy either alone or as adjuvant to standard care of patients with localized or locally advanced prostate cancer: first analysis of the early prostate cancer program. The Journal of Urology, 168(2), 429–435. [DOI:10.1016/S0022-5347(05)64652-6]
Stenger, P. J., Wagner, L., Egelhoff, J., & Rose, S. R. (2009). Bicalutamide and letrozole treatment of precocious puberty due to elevated levels of human chorionic gonadotropin (hCG). Hormone Research [in Paediatrics], 72(Suppl 3), 391–391 (PO3-149). [Google Scholar] [DOI:10.1159/000239668]
Tessaris, D., Matarazzo, P., Mussa, A., Tuli, G., Verna, F., Fiore, L., & Lala, R. (2012). Combined treatment with bicalutamide and anastrozole in a young boy with peripheral precocious puberty due to McCune-Albright Syndrome. Endocrine Journal, 59(2), 111–117. [DOI:10.1507/endocrj.ej11-0214]
Thole, Z., Manso, G., Salgueiro, E., Revuelta, P., & Hidalgo, A. (2004). Hepatotoxicity induced by antiandrogens: a review of the literature. Urologia Internationalis, 73(4), 289–295. [DOI:10.1159/000081585]
Thompson, J., Hopwood, R. A., deNormand, S., & Cavanaugh, T. (2021). Medical Care of Trans and Gender Diverse Adults. Boston: Fenway Health. [URL] [PDF]
Tomson, A., McLachlan, C., Wattrus, C., Adams, K., Addinall, R., Bothma, R., Jankelowitz, L., Kotze, E., Luvuno, Z., Madlala, N., Matyila, S., Padavatan, A., Pillay, M., Rakumakoe, M. D., Tomson-Myburgh, M., Venter, W., & de Vries, E. (2021). Southern African HIV Clinicians’ Society gender-affirming healthcare guideline for South Africa. Southern African Journal of HIV Medicine, 22(1), a1299. [DOI:10.4102/sajhivmed.v22i1.1299] [PDF]
van Kemenade, J. F., Cohen-Kettenis, P. T., Cohen, L., & Gooren, L. J. (1989). Effects of the pure antiandrogen RU 23.903 (anandron) on sexuality, aggression, and mood in male-to-female transsexuals. Archives of Sexual Behavior, 18(3), 217–228. [DOI:10.1007/BF01543196]
Vierregger, K., Tetzlaff, M., Zimmerman, B., Dunn, N., Finney, N., Lewis, K., Slomoff, R., & Strutner, S. (2023). Bicalutamide Use as Antiandrogen in Trans Feminine Adults - A Safety Profile. National Transgender Health Summit (NTHS) 2023 Symposium. [Event Agenda PDF] [Symposium Session] [Symposium Abstracts/Program Book]
Vierregger, K., Tetzlaf, M., Zimmerman, B., Dunn, N., Finney, N., Lewis, K., Slomoff, R., & Strutner, S. (2023). Bicalutamide Use as Antiandrogen in Trans Feminine Adults - A Safety Profile. USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 96–96 (abstract no. SAT-B2-T4). [Symposium Schedule] [PDF] [Full Abstract Book]
Warus, J., Rincon, M. G., Salvetti, B., & Olson-Kennedy, J. (2023). Safety of Bicalutamide as Anti-Androgenic Therapy in Gender Affirming Care for Adolescents and Young Adults: A Retrospective Chart Review. USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 124–124 (abstract no. SUN-B1-T5). [Symposium Schedule] [PDF] [Full Abstract Book]
Wilde, B., Diamond, J. B., Laborda, T. J., Frank, L., O’Gorman, M. A., & Kocolas, I. (2023). Bicalutamide-Induced Hepatotoxicity in a Transgender Male-to-Female Adolescent. Journal of Adolescent Health, 74(1), 202–204. [DOI:10.1016/j.jadohealth.2023.08.024]
\ No newline at end of file
diff --git a/transfemscience.org/articles/bone-shape-changes/index.html b/transfemscience.org/articles/bone-shape-changes/index.html
index 9ce94263..b22fd89b 100644
--- a/transfemscience.org/articles/bone-shape-changes/index.html
+++ b/transfemscience.org/articles/bone-shape-changes/index.html
@@ -1 +1 @@
-On Changes in Bone Shape in Transfeminine Individuals Under Gender-Affirming Hormone Therapy - Transfeminine Science
The studies here do not show significant hip bone growth in adult transfeminine individuals under hormone therapy. They do show, however significant feminizing body shape changes that seem to largely be explained by increased fat placement around the hips and butt.
Introduction
An area of interest that I’ve seen a number of times is how does gender-affirming hormone therapy (GAHT) affect bones, specifically in adult transgender individuals. Within the context of transfeminine individuals the interest is often related to whether or not GAHT can cause an increase in hip bone area and give a body shape that is more feminine or gynoid.
However, one way to quantitatively measure these changes is via DXA scans. DXA or dual-energy X-ray absorptiometry is a non-invasive way to measure the distribution of different soft tissue types like fat and muscle as well as bone area and density. It is most commonly used to study bone mineral density (BMD) which is often used as a measure of bone health and when assessing risk for things like fractures or osteoporosis.
To date there have been two studies that I am aware of that look at body composition changes beyond changes in fat/muscle composition or external factors like waist–hip ratio (WHR) which I will review here.
This study, a component of the European Network for the Investigation of Gender Incongruence (ENIGI) initiative (Kreukels et al., 2012) investigated a wide variety of body composition changes in a controlled cohort of 49 transfeminine individuals with a mean age of 33 (± 12) years with a minimum of 17 and a maximum of 67 years before starting GAHT with 1 and 2 year follow ups and compared them to a cohort of age matched cis men. The GAHT regimen used here was standardized at 4 mg estradiol valerate daily orally (or 100 μg transdermal estradiol if over 45) with 50 mg cyproterone acetate.
With respect to bone area while there was – somewhat surprisingly – a significant difference between the control men and transfeminine individuals prior to starting GAHT in various bone area measurements and BMD there was no statistically significant change in bone area for hips, radius (lower arm bone), femoral neck (the length between the femoral head which fits into the pelvis and the rest of the femur) or lumbar spine (lower spine bones).
However, despite this lack of change in total hip bone area – there were significant changes in WHR.
Figure 2: Changes in body composition after 1 and 2 years of hormone therapy (Van Caenegem et al., 2014).
You can see that while total hip bone area did not change significantly and neither did waist circumference – hip circumference and WHR ratio changed significantly – with WHR on average being what is considered a more feminine range.
This study is an earlier work by the same group out of Belgium. Unlike the previous one this one did not have a cohort that tracked before, and after beginning GAHT but rather compared individuals on GAHT with control cis men. Another area of difference is that everyone in this study had undergone gender confirmation surgery at least 3 years prior to being enrolled and had a mean age of 41 (± 7) years.
Figure 3: Bone composition differences between the group of transfeminine individuals and cis men (Lapauw et al., 2008).
These results again find no significant difference in total bone area of total hip or other specific areas looked at between the transfeminine individuals and the cis men. This study unfortunately did not also look at WHR or area specific body fat composition.
Discussion
Van Caenegem et al. (2014) paints a compelling picture of the changes experienced in transfeminine individuals under GAHT. While total bone area of the hip did not significantly change, WHR shrank due to an increase in hip circumference to a more gynoid distribution as has been reported a number of times in other places.
That this is due to fat mass is further bolstered by Klaver et al. (2017) which looked at body composition changes in 179 transfeminine individuals at regional areas and found that the “gynoid” area – or area around the hips experienced a 34% increase in fat percentage (see Figure 4 below).
Figure 4: Body composition changes in regions of DXA scans (Klaver et al., 2017).
While it is worth noting that the GAHT regimen given to these individuals is fairly homogeneous it should be pointed out that it did cause significant changes in body fat and WHR.
I believe this illustrates that the current evidence we have supports the idea that bone mass does not significantly change in adults under GAHT and that the primary contributor to changes in WHR and hip circumference are tissue focused – seemingly largely from increased fat deposition.
References
Klaver, M., de Blok, C. J., Wiepjes, C. M., Nota, N. M., Dekker, M. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/eje-17-0496]
Kreukels, B., Haraldsen, I., De Cuypere, G., Richter-Appelt, H., Gijs, L., & Cohen-Kettenis, P. (2012). A European network for the investigation of gender incongruence: The ENIGI initiative. European Psychiatry, 27(6), 445–450. [DOI:10.1016/j.eurpsy.2010.04.009]
Lapauw, B., Taes, Y., Simoens, S., Van Caenegem, E., Weyers, S., Goemaere, S., Toye, K., Kaufman, J., & T’Sjoen, G. G. (2008). Body composition, volumetric and areal bone parameters in male-to-female transsexual persons. Bone, 43(6), 1016–1021. [DOI:10.1016/j.bone.2008.09.001]
Van Caenegem, E., Wierckx, K., Taes, Y., Schreiner, T., Vandewalle, S., Toye, K., Kaufman, J., & T’Sjoen, G. (2014). Preservation of volumetric bone density and geometry in trans women during cross-sex hormonal therapy: a prospective observational study. Osteoporosis International, 26(1), 35–47. [DOI:10.1007/s00198-014-2805-3]
\ No newline at end of file
+On Changes in Bone Shape in Transfeminine Individuals Under Gender-Affirming Hormone Therapy - Transfeminine Science
The studies here do not show significant hip bone growth in adult transfeminine individuals under hormone therapy. They do show, however significant feminizing body shape changes that seem to largely be explained by increased fat placement around the hips and butt.
Introduction
An area of interest that I’ve seen a number of times is how does gender-affirming hormone therapy (GAHT) affect bones, specifically in adult transgender individuals. Within the context of transfeminine individuals the interest is often related to whether or not GAHT can cause an increase in hip bone area and give a body shape that is more feminine or gynoid.
However, one way to quantitatively measure these changes is via DXA scans. DXA or dual-energy X-ray absorptiometry is a non-invasive way to measure the distribution of different soft tissue types like fat and muscle as well as bone area and density. It is most commonly used to study bone mineral density (BMD) which is often used as a measure of bone health and when assessing risk for things like fractures or osteoporosis.
To date there have been two studies that I am aware of that look at body composition changes beyond changes in fat/muscle composition or external factors like waist–hip ratio (WHR) which I will review here.
This study, a component of the European Network for the Investigation of Gender Incongruence (ENIGI) initiative (Kreukels et al., 2012) investigated a wide variety of body composition changes in a controlled cohort of 49 transfeminine individuals with a mean age of 33 (± 12) years with a minimum of 17 and a maximum of 67 years before starting GAHT with 1 and 2 year follow ups and compared them to a cohort of age matched cis men. The GAHT regimen used here was standardized at 4 mg estradiol valerate daily orally (or 100 μg transdermal estradiol if over 45) with 50 mg cyproterone acetate.
With respect to bone area while there was – somewhat surprisingly – a significant difference between the control men and transfeminine individuals prior to starting GAHT in various bone area measurements and BMD there was no statistically significant change in bone area for hips, radius (lower arm bone), femoral neck (the length between the femoral head which fits into the pelvis and the rest of the femur) or lumbar spine (lower spine bones).
However, despite this lack of change in total hip bone area – there were significant changes in WHR.
Figure 2: Changes in body composition after 1 and 2 years of hormone therapy (Van Caenegem et al., 2014).
You can see that while total hip bone area did not change significantly and neither did waist circumference – hip circumference and WHR ratio changed significantly – with WHR on average being what is considered a more feminine range.
This study is an earlier work by the same group out of Belgium. Unlike the previous one this one did not have a cohort that tracked before, and after beginning GAHT but rather compared individuals on GAHT with control cis men. Another area of difference is that everyone in this study had undergone gender confirmation surgery at least 3 years prior to being enrolled and had a mean age of 41 (± 7) years.
Figure 3: Bone composition differences between the group of transfeminine individuals and cis men (Lapauw et al., 2008).
These results again find no significant difference in total bone area of total hip or other specific areas looked at between the transfeminine individuals and the cis men. This study unfortunately did not also look at WHR or area specific body fat composition.
Discussion
Van Caenegem et al. (2014) paints a compelling picture of the changes experienced in transfeminine individuals under GAHT. While total bone area of the hip did not significantly change, WHR shrank due to an increase in hip circumference to a more gynoid distribution as has been reported a number of times in other places.
That this is due to fat mass is further bolstered by Klaver et al. (2017) which looked at body composition changes in 179 transfeminine individuals at regional areas and found that the “gynoid” area – or area around the hips experienced a 34% increase in fat percentage (see Figure 4 below).
Figure 4: Body composition changes in regions of DXA scans (Klaver et al., 2017).
While it is worth noting that the GAHT regimen given to these individuals is fairly homogeneous it should be pointed out that it did cause significant changes in body fat and WHR.
I believe this illustrates that the current evidence we have supports the idea that bone mass does not significantly change in adults under GAHT and that the primary contributor to changes in WHR and hip circumference are tissue focused – seemingly largely from increased fat deposition.
References
Klaver, M., de Blok, C. J., Wiepjes, C. M., Nota, N. M., Dekker, M. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/eje-17-0496]
Kreukels, B., Haraldsen, I., De Cuypere, G., Richter-Appelt, H., Gijs, L., & Cohen-Kettenis, P. (2012). A European network for the investigation of gender incongruence: The ENIGI initiative. European Psychiatry, 27(6), 445–450. [DOI:10.1016/j.eurpsy.2010.04.009]
Lapauw, B., Taes, Y., Simoens, S., Van Caenegem, E., Weyers, S., Goemaere, S., Toye, K., Kaufman, J., & T’Sjoen, G. G. (2008). Body composition, volumetric and areal bone parameters in male-to-female transsexual persons. Bone, 43(6), 1016–1021. [DOI:10.1016/j.bone.2008.09.001]
Van Caenegem, E., Wierckx, K., Taes, Y., Schreiner, T., Vandewalle, S., Toye, K., Kaufman, J., & T’Sjoen, G. (2014). Preservation of volumetric bone density and geometry in trans women during cross-sex hormonal therapy: a prospective observational study. Osteoporosis International, 26(1), 35–47. [DOI:10.1007/s00198-014-2805-3]
\ No newline at end of file
diff --git a/transfemscience.org/articles/breast-cancer-suppl/index.html b/transfemscience.org/articles/breast-cancer-suppl/index.html
index 59ef4dc2..196ae12b 100644
--- a/transfemscience.org/articles/breast-cancer-suppl/index.html
+++ b/transfemscience.org/articles/breast-cancer-suppl/index.html
@@ -1 +1 @@
-The Possible Role of a Second X Chromosome in Breast Cancer Risk - Transfeminine Science
The Possible Role of a Second X Chromosome in Breast Cancer Risk
By Aly | First published April 25, 2020 | Last modified March 1, 2023
Preface
This is a short supplement article to the section in the main article on breast cancer risk with hormone therapy in transfeminine people that can be found here.
Introduction
The sex chromosomes include the X chromosome and the Y chromosome. Under normal biological circumstances, cisgender women have two X chromosomes (46,XX karyotype) while cisgender men (and transfeminine people) have one X chromosome and one Y chromosome (46,XY karyotype). The sex chromosomes determine whether the gonads will differentiate into ovaries or testes, and hence mediate a large portion of physical sexual dimorphism. However, the sex chromosomes also have effects in terms of sexual dimorphism that are independent of gonadal differentiation. Although hormone therapy appears to increase the risk of breast cancer in transfeminine people, so far breast cancer risk in transfeminine people seems to be much lower than that in cisgender women. There are a variety of possible reasons for this, as touched on elsewhere. One reason may be our lack of a second X chromosome—there is indication that our absence of a second X chromosome may be partially protective against breast cancer.
X Chromosomes and Breast Cancer Risk in Chromosomal Disorders and Intersexuality
A variety of genetic disorders involving abnormal sex-chromosome configurations and/or intersexuality also provide insight on the possible involvement of X chromosomes in breast cancer risk. These conditions include Klinefelter’s syndrome (47,XXY male), complete androgen insensitivity syndrome (46,XY female), Turner syndrome (45,X or mixed 45,X/46,XX female), XX male syndrome (46,XX male), and X trisomy and tetrasomy (47,XXX and 48,XXXX female).
Klinefelter’s Syndrome (47,XXY)
Men with Klinefelter’s syndrome have a 47,XXY karyotype and hence an extra X chromosome. Because of their Y chromosome, men with Klinfelter’s syndrome have testes and develop normally as males. However, they have relatively low testosterone levels (about 260 ng/dL lower than usual), slightly increased estradiol levels (about 6 pg/mL more than normal), an increased ratio of estradiol to testosterone, and negligible progesterone (Wiki). In addition, men with Klinefelter’s syndrome show undermasculinization and sometimes have mild gynecomastia. The risk of breast cancer in men with Klinefelter’s syndrome is strongly increased relative to 46,XY men (Brinton, 2011; Sokol, 2012). The risk is estimated to be 20- to 60-fold higher in men with Klinefelter’s syndrome compared to 46,XY men and only 70% lower than the risk in 46,XX women (Brinton, 2011; Sokol, 2012; Swerdlow et al., 2005). The lifetime risk of breast cancer in men with Klinefelter’s syndrome is 4 to 8% (Sokol, 2012). Breast cancer risk in men with Klinefelter’s syndrome is much higher than that in almost any other known clinical situation in men. The typical age of diagnosis of breast cancer in men with Klinefelter’s syndrome is 58 years (Ferzoco & Ruddy, 2016). It is estimated that 7% of all men with breast cancer have Klinefelter’s syndrome (Ferzoco & Ruddy, 2016). Klinefelter’s syndrome suggests that breast cancer may be dependent on an extra X chromosome (Spatz, Borg, & Feunteun, 2004; Sacchi, 1993; Dawson, 1998), although disordered hormone levels may also be involved.
Complete Androgen Insensitivity Syndrome (46,XY)
Women with complete androgen insensitivity syndrome (CAIS) are individuals with a 46,XY karyotype and intra-abdominal testes who have a defective androgen receptor and hence are insensitive to the effects of androgens like testosterone. Due to their androgen insensitivity, CAIS women develop physically and behaviorally as females instead of as males. CAIS women naturally have male-range levels of sex hormones, including of testosterone, estradiol, and progesterone (Wiki). Relative to 46,XX women, testosterone levels are very high, estradiol levels relatively low (~35 pg/mL), and progesterone levels negligible. During adolescence, CAIS women undergo female puberty due to aromatization of testosterone into estradiol, and this results in feminization and excellent breast development, with breasts that are said to be somewhat above-average compared to those of 46,XX women.
After puberty, all clinically diagnosed CAIS women undergo gonadectomy due to a high risk of testicular cancer that’s associated with their intra-abdominal testes. Following gonadectomy, CAIS women receive estrogen replacement therapy, oftentimes if not usually only at menopausal doses. A progestogen is not usually included since CAIS women don’t have uteruses and hence don’t require the endometrial protection afforded by progestogens. Despite a 1 in 20,000 incidence of CAIS in 46,XY individuals and hence the estimated existence of hundreds of thousands of CAIS women throughout the world, breast cancer has never been reported in a CAIS woman (Hughes et al., 2012; Tiefenbacher & Daxenbichler, 2008; Hughes, 2009). It’s unlikely that breast cancer doesn’t occur in women with CAIS, but rather it’s probable that it’s rare. It’s unknown why this is the case, but major possible reasons include the relatively low estrogen exposure, lack of progesterone, and the lack of a second X chromosome. Their androgen insensitivity is unlikely to be involved as androgens, via activation of the androgen receptor, are thought to be protective in terms of breast cancer risk (Dimitrakakis, 2011). CAIS women are fascinating because they suggest that considerable breast cancer incidence isn’t an inevitable companion of normal breast development.
Turner Syndrome (45,X or Mixed 45,X/46,XX)
Women with Turner syndrome have either a 45,X karyotype or a mosaic of 45,X and 46,XX karyotypes. They often though not always have gonadal failure and hence many of them fail to undergo puberty. Gonadal failure is much more prevalent in those with a 45,X karyotype than in mosaics. When gonadal failure occurs, hormone therapy is required. Women with Turner’s syndrome have uteruses, so those who have pubertal failure receive hormone replacement with both an estrogen and a progestogen. As with CAIS women, this is often with only menopausal replacement doses. The risk of breast cancer appears to be lower in women with Turner syndrome than in regular 46,XX women (Schoemaker et al., 2008; Bösze, Tóth, & Török, 2006; Viuff et al., 2020). For example, in one large cohort study of 3,425 women with Turner syndrome and an average of 17 years of follow up, the risk of breast cancer was significantly lower than in regular 46,XX women. Interestingly, the standardized incidence ratios were 0.2 (0.0–0.9) in those with a 45,X karyotype and 0.9 (0.3–2.0) in those with 45,X and 46,XX mosaicism. In other words, those with mosaicism had a risk of breast cancer similar to that of regular 46,XX women, whereas those with a 45,X chromosome had a significantly lower risk (Gravholt, 2008).
XX Male Syndrome (46,XX)
XX males have a 46,XX karyotype. In about 90% of cases, the condition is caused by translocation of the SRY gene, which encodes the protein testis-determining factor (TDF), onto an X chromosome. As a result of this, XX males develop testes instead of ovaries, in turn developing as males instead of as females. Three case reports of breast cancer in 46,XX males exist (Berglund et al., 2017). On the basis of these cases and the small total number of cases of XX male syndrome that have been reported, it has been said that XX males are at increased risk for breast cancer analogously to men with Klinefelter’s syndrome (Spatz, Borg, & Feunteun, 2004; Giammarini, 1980).
X Trisomy (47,XXX) and Tetrasomy (48,XXXX)
So far there is no evidence of increased breast cancer risk in 47,XXX (X trisomy) or 48,XXXX (X tetrasomy) females (Spatz, Borg, & Feunteun, 2004), although most cases of these syndromes go undetected and the conditions are understudied (Goldschmidt et al., 2010; Wiki). Additional X chromosomes are inactivated in these individuals, limiting—although not fully eliminating—phenotypical abnormalities, possibly including greater breast cancer risk.
Conclusions
Taken together, these findings from chromosomal disorders and intersex conditions suggest that having at least two X chromosomes may greatly increase the risk of breast cancer. In addition, CAIS women suggest that normal breast development with very low breast cancer risk is possible, provided of course that the person has only one X chromosome.
References
Barrios, L., Caballín, M., Miró, R., Fuster, C., Guedea, F., Subias, A., & Egozcue, J. (1991). Chromosomal instability in breast cancer patients. Human Genetics, 88(1), 39–41. [DOI:10.1007/bf00204926]
Berglund, A., Johannsen, T., Stochholm, K., Aksglaede, L., Fedder, J., Viuff, M., Main, K., & Gravholt, C. (2017). Incidence, prevalence, diagnostic delay, morbidity, mortality and socioeconomic status in males with 46,XX disorders of sex development: a nationwide study. Human Reproduction, 32(8), 1751–1760. [DOI:10.1093/humrep/dex210]
Bösze, P., Tóth, A., & Török, M. (2006). Hormone Replacement and the Risk of Breast Cancer in Turner’s Syndrome. New England Journal of Medicine, 355(24), 2599–2600. [DOI:10.1056/nejmc062795]
Brinton, L. A. (2011). Breast cancer risk among patients with Klinefelter syndrome. Acta Paediatrica, 100(6), 814–818. [DOI:10.1111/j.1651-2227.2010.02131.x]
Chaligné, R., & Heard, E. (2014). X-chromosome inactivation in development and cancer. FEBS Letters, 588(15), 2514–2522. [DOI:10.1016/j.febslet.2014.06.023]
Chaligné, R., Popova, T., Mendoza-Parra, M., Saleem, M. M., Gentien, D., Ban, K., Piolot, T., Leroy, O., Mariani, O., Gronemeyer, H., Vincent-Salomon, A., Stern, M., & Heard, E. (2015). The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Research, 25(4), 488–503. [DOI:10.1101/gr.185926.114]
Dawson, P. J. (1998). A history of cancer of the male breast. In Peters, W. P., & Visscher, D. W. (Eds.). Breast Cancer (Advances in Oncobiology, Volume 2) (pp. 229–243). Stamford/London: Jai Press. [DOI:10.1016/s1569-254x(98)80011-3] [Google Books]
Di Oto, E., Monti, V., Cucchi, M. C., Masetti, R., Varga, Z., & Foschini, M. P. (2015). X chromosome gain in male breast cancer. Human Pathology, 46(12), 1908–1912. [DOI:10.1016/j.humpath.2015.08.008]
Di Oto, E., Biserni, G. B., Varga, Z., Morandi, L., Cucchi, M. C., Masetti, R., & Foschini, M. P. (2018). X chromosome gain is related to increased androgen receptor expression in male breast cancer. Virchows Archiv, 473(2), 155–163. [DOI:10.1007/s00428-018-2377-2]
Dimitrakakis, C. (2011). Androgens and Breast Cancer in Men and Women. Endocrinology and Metabolism Clinics of North America, 40(3), 533–547. [DOI:10.1016/j.ecl.2011.05.007]
Ferzoco, R. M., & Ruddy, K. J. (2015). The Epidemiology of Male Breast Cancer. Current Oncology Reports, 18(1), 1. [DOI:10.1007/s11912-015-0487-4]
Giammarini, A., Rocchi, M., Zennaro, W., & Filippi, G. (2008). XX Male with breast cancer. Clinical Genetics, 18(2), 103–108. [DOI:10.1111/j.1399-0004.1980.tb01019.x]
Goldschmidt, E., Márquez, M., Solari, A., Ziembar, M. I., & Laudicina, A. (2010). Variabilidad fenotípica en pacientes 47, XXX: Presentación de cuatro casos nuevos. [Phenotypic variability in 47, XXX patients. Clinical report of four new cases.] Archivos Argentinos de Pediatría, 108(4), e88–e91. [URL]
Gravholt, C. H. (2008). Epidemiology of Turner syndrome. The Lancet Oncology, 9(3), 193–195. [DOI:10.1016/s1470-2045(08)70045-7]
Hughes, I. A. (2010). Evaluation and Management of Disorders of Sex Development. In Brook, C. G. D., Clayton, P. E., & Brown, R. S. (Eds.). Brook’s Clinical Pediatric Endocrinology, 6th Edition (pp. 192–212). Oxford: Wiley-Blackwell. [DOI:10.1002/9781444316728.ch9]
Hughes, I. A., Davies, J. D., Bunch, T. I., Pasterski, V., Mastroyannopoulou, K., & MacDougall, J. (2012). Androgen insensitivity syndrome. The Lancet, 380(9851), 1419–1428. [DOI:10.1016/s0140-6736(12)60071-3]
Jacobs, P. A., Maloney, V., Cooke, R., Crolla, J. A., Ashworth, A., & Swerdlow, A. J. (2013). Male breast cancer, age and sex chromosome aneuploidy. British Journal of Cancer, 108(4), 959–963. [DOI:10.1038/bjc.2012.577]
Lin, I., Chen, D., Chang, Y., Lee, Y., Su, C., Cheng, C., Tsai, Y., Ng, S., Chen, H., Lee, M., Chen, H., Suen, S., Chen, Y., Liu, T., Chang, C., & Hsu, M. (2015). Hierarchical Clustering of Breast Cancer Methylomes Revealed Differentially Methylated and Expressed Breast Cancer Genes. PLOS ONE, 10(2), e0118453. [DOI:10.1371/journal.pone.0118453]
Nakopoulou, L., Panayotopoulou, E. G., Giannopoulou, I., Tsirmpa, I., Katsarou, S., Mylona, E., Alexandrou, P., & Keramopoulos, A. (2006). Extra copies of chromosomes 16 and X in invasive breast carcinomas are related to aggressive phenotype and poor prognosis. Journal of Clinical Pathology, 60(7), 808–815. [DOI:10.1136/jcp.2006.037838]
Richardson, A. L., Wang, Z. C., De Nicolo, A., Lu, X., Brown, M., Miron, A., Liao, X., Iglehart, J. D., Livingston, D. M., & Ganesan, S. (2006). X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell, 9(2), 121–132. [DOI:10.1016/j.ccr.2006.01.013]
Sacchi, N. (1993). Constitutional Chromosome Aneuploidy and Cancer. In Kirsch, I. R. (Ed.). The Causes and Consequences of Chromosomal Aberrations (pp. 191–222). Boca Raton: CRC Press. [Google Books]
Schoemaker, M. J., Swerdlow, A. J., Higgins, C. D., Wright, A. F., & Jacobs, P. A. (2008). Cancer incidence in women with Turner syndrome in Great Britain: a national cohort study. The Lancet Oncology, 9(3), 239–246. [DOI:10.1016/s1470-2045(08)70033-0]
Sirchia, S. M., Tabano, S. M., & Miozzo, M. (2007). BRCA1 and X Chromosome Inactivation: Which is the Link? In Polinsky, K. R. (Ed.). Tumor Suppressor Genes (pp. 5–8). New York: Nova Biomedical Books. [Google Books]
Sokol, R. Z. (2012). It’s not all about the testes: medical issues in Klinefelter patients. Fertility and Sterility, 98(2), 261–265. [DOI:10.1016/j.fertnstert.2012.05.026]
Spatz, A., Borg, C., & Feunteun, J. (2004). X-Chromosome Genetics and Human Cancer. Nature Reviews Cancer, 4(8), 617–629. [DOI:10.1038/nrc1413]
Swerdlow, A. J., Schoemaker, M. J., Higgins, C. D., Wright, A. F., & Jacobs, P. A. (2005). Cancer Incidence and Mortality in Men with Klinefelter Syndrome: A Cohort Study. JNCI: Journal of the National Cancer Institute, 97(16), 1204–1210. [DOI:10.1093/jnci/dji240]
Tiefenbacher, K., & Daxenbichler, G. (2008). The Role of Androgens in Normal and Malignant Breast Tissue. Breast Care, 3(5), 325–331. [DOI:10.1159/000158055]
Viuff, M. H., Stochholm, K., Lin, A., Berglund, A., Juul, S., & Gravholt, C. H. (2021). Cancer occurrence in Turner syndrome and the effect of sex hormone substitution therapy. European Journal of Endocrinology, 184(1), 79–88. [DOI:10.1530/eje-20-0702]
\ No newline at end of file
+The Possible Role of a Second X Chromosome in Breast Cancer Risk - Transfeminine Science
The Possible Role of a Second X Chromosome in Breast Cancer Risk
By Aly | First published April 25, 2020 | Last modified March 1, 2023
Preface
This is a short supplement article to the section in the main article on breast cancer risk with hormone therapy in transfeminine people that can be found here.
Introduction
The sex chromosomes include the X chromosome and the Y chromosome. Under normal biological circumstances, cisgender women have two X chromosomes (46,XX karyotype) while cisgender men (and transfeminine people) have one X chromosome and one Y chromosome (46,XY karyotype). The sex chromosomes determine whether the gonads will differentiate into ovaries or testes, and hence mediate a large portion of physical sexual dimorphism. However, the sex chromosomes also have effects in terms of sexual dimorphism that are independent of gonadal differentiation. Although hormone therapy appears to increase the risk of breast cancer in transfeminine people, so far breast cancer risk in transfeminine people seems to be much lower than that in cisgender women. There are a variety of possible reasons for this, as touched on elsewhere. One reason may be our lack of a second X chromosome—there is indication that our absence of a second X chromosome may be partially protective against breast cancer.
X Chromosomes and Breast Cancer Risk in Chromosomal Disorders and Intersexuality
A variety of genetic disorders involving abnormal sex-chromosome configurations and/or intersexuality also provide insight on the possible involvement of X chromosomes in breast cancer risk. These conditions include Klinefelter’s syndrome (47,XXY male), complete androgen insensitivity syndrome (46,XY female), Turner syndrome (45,X or mixed 45,X/46,XX female), XX male syndrome (46,XX male), and X trisomy and tetrasomy (47,XXX and 48,XXXX female).
Klinefelter’s Syndrome (47,XXY)
Men with Klinefelter’s syndrome have a 47,XXY karyotype and hence an extra X chromosome. Because of their Y chromosome, men with Klinfelter’s syndrome have testes and develop normally as males. However, they have relatively low testosterone levels (about 260 ng/dL lower than usual), slightly increased estradiol levels (about 6 pg/mL more than normal), an increased ratio of estradiol to testosterone, and negligible progesterone (Wiki). In addition, men with Klinefelter’s syndrome show undermasculinization and sometimes have mild gynecomastia. The risk of breast cancer in men with Klinefelter’s syndrome is strongly increased relative to 46,XY men (Brinton, 2011; Sokol, 2012). The risk is estimated to be 20- to 60-fold higher in men with Klinefelter’s syndrome compared to 46,XY men and only 70% lower than the risk in 46,XX women (Brinton, 2011; Sokol, 2012; Swerdlow et al., 2005). The lifetime risk of breast cancer in men with Klinefelter’s syndrome is 4 to 8% (Sokol, 2012). Breast cancer risk in men with Klinefelter’s syndrome is much higher than that in almost any other known clinical situation in men. The typical age of diagnosis of breast cancer in men with Klinefelter’s syndrome is 58 years (Ferzoco & Ruddy, 2016). It is estimated that 7% of all men with breast cancer have Klinefelter’s syndrome (Ferzoco & Ruddy, 2016). Klinefelter’s syndrome suggests that breast cancer may be dependent on an extra X chromosome (Spatz, Borg, & Feunteun, 2004; Sacchi, 1993; Dawson, 1998), although disordered hormone levels may also be involved.
Complete Androgen Insensitivity Syndrome (46,XY)
Women with complete androgen insensitivity syndrome (CAIS) are individuals with a 46,XY karyotype and intra-abdominal testes who have a defective androgen receptor and hence are insensitive to the effects of androgens like testosterone. Due to their androgen insensitivity, CAIS women develop physically and behaviorally as females instead of as males. CAIS women naturally have male-range levels of sex hormones, including of testosterone, estradiol, and progesterone (Wiki). Relative to 46,XX women, testosterone levels are very high, estradiol levels relatively low (~35 pg/mL), and progesterone levels negligible. During adolescence, CAIS women undergo female puberty due to aromatization of testosterone into estradiol, and this results in feminization and excellent breast development, with breasts that are said to be somewhat above-average compared to those of 46,XX women.
After puberty, all clinically diagnosed CAIS women undergo gonadectomy due to a high risk of testicular cancer that’s associated with their intra-abdominal testes. Following gonadectomy, CAIS women receive estrogen replacement therapy, oftentimes if not usually only at menopausal doses. A progestogen is not usually included since CAIS women don’t have uteruses and hence don’t require the endometrial protection afforded by progestogens. Despite a 1 in 20,000 incidence of CAIS in 46,XY individuals and hence the estimated existence of hundreds of thousands of CAIS women throughout the world, breast cancer has never been reported in a CAIS woman (Hughes et al., 2012; Tiefenbacher & Daxenbichler, 2008; Hughes, 2009). It’s unlikely that breast cancer doesn’t occur in women with CAIS, but rather it’s probable that it’s rare. It’s unknown why this is the case, but major possible reasons include the relatively low estrogen exposure, lack of progesterone, and the lack of a second X chromosome. Their androgen insensitivity is unlikely to be involved as androgens, via activation of the androgen receptor, are thought to be protective in terms of breast cancer risk (Dimitrakakis, 2011). CAIS women are fascinating because they suggest that considerable breast cancer incidence isn’t an inevitable companion of normal breast development.
Turner Syndrome (45,X or Mixed 45,X/46,XX)
Women with Turner syndrome have either a 45,X karyotype or a mosaic of 45,X and 46,XX karyotypes. They often though not always have gonadal failure and hence many of them fail to undergo puberty. Gonadal failure is much more prevalent in those with a 45,X karyotype than in mosaics. When gonadal failure occurs, hormone therapy is required. Women with Turner’s syndrome have uteruses, so those who have pubertal failure receive hormone replacement with both an estrogen and a progestogen. As with CAIS women, this is often with only menopausal replacement doses. The risk of breast cancer appears to be lower in women with Turner syndrome than in regular 46,XX women (Schoemaker et al., 2008; Bösze, Tóth, & Török, 2006; Viuff et al., 2020). For example, in one large cohort study of 3,425 women with Turner syndrome and an average of 17 years of follow up, the risk of breast cancer was significantly lower than in regular 46,XX women. Interestingly, the standardized incidence ratios were 0.2 (0.0–0.9) in those with a 45,X karyotype and 0.9 (0.3–2.0) in those with 45,X and 46,XX mosaicism. In other words, those with mosaicism had a risk of breast cancer similar to that of regular 46,XX women, whereas those with a 45,X chromosome had a significantly lower risk (Gravholt, 2008).
XX Male Syndrome (46,XX)
XX males have a 46,XX karyotype. In about 90% of cases, the condition is caused by translocation of the SRY gene, which encodes the protein testis-determining factor (TDF), onto an X chromosome. As a result of this, XX males develop testes instead of ovaries, in turn developing as males instead of as females. Three case reports of breast cancer in 46,XX males exist (Berglund et al., 2017). On the basis of these cases and the small total number of cases of XX male syndrome that have been reported, it has been said that XX males are at increased risk for breast cancer analogously to men with Klinefelter’s syndrome (Spatz, Borg, & Feunteun, 2004; Giammarini, 1980).
X Trisomy (47,XXX) and Tetrasomy (48,XXXX)
So far there is no evidence of increased breast cancer risk in 47,XXX (X trisomy) or 48,XXXX (X tetrasomy) females (Spatz, Borg, & Feunteun, 2004), although most cases of these syndromes go undetected and the conditions are understudied (Goldschmidt et al., 2010; Wiki). Additional X chromosomes are inactivated in these individuals, limiting—although not fully eliminating—phenotypical abnormalities, possibly including greater breast cancer risk.
Conclusions
Taken together, these findings from chromosomal disorders and intersex conditions suggest that having at least two X chromosomes may greatly increase the risk of breast cancer. In addition, CAIS women suggest that normal breast development with very low breast cancer risk is possible, provided of course that the person has only one X chromosome.
References
Aly. (2020). Breast Cancer Risk with Hormone Therapy in Transfeminine People. Transfeminine Science. [URL]
Barrios, L., Caballín, M., Miró, R., Fuster, C., Guedea, F., Subias, A., & Egozcue, J. (1991). Chromosomal instability in breast cancer patients. Human Genetics, 88(1), 39–41. [DOI:10.1007/bf00204926]
Berglund, A., Johannsen, T., Stochholm, K., Aksglaede, L., Fedder, J., Viuff, M., Main, K., & Gravholt, C. (2017). Incidence, prevalence, diagnostic delay, morbidity, mortality and socioeconomic status in males with 46,XX disorders of sex development: a nationwide study. Human Reproduction, 32(8), 1751–1760. [DOI:10.1093/humrep/dex210]
Bösze, P., Tóth, A., & Török, M. (2006). Hormone Replacement and the Risk of Breast Cancer in Turner’s Syndrome. New England Journal of Medicine, 355(24), 2599–2600. [DOI:10.1056/nejmc062795]
Brinton, L. A. (2011). Breast cancer risk among patients with Klinefelter syndrome. Acta Paediatrica, 100(6), 814–818. [DOI:10.1111/j.1651-2227.2010.02131.x]
Chaligné, R., & Heard, E. (2014). X-chromosome inactivation in development and cancer. FEBS Letters, 588(15), 2514–2522. [DOI:10.1016/j.febslet.2014.06.023]
Chaligné, R., Popova, T., Mendoza-Parra, M., Saleem, M. M., Gentien, D., Ban, K., Piolot, T., Leroy, O., Mariani, O., Gronemeyer, H., Vincent-Salomon, A., Stern, M., & Heard, E. (2015). The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Research, 25(4), 488–503. [DOI:10.1101/gr.185926.114]
Dawson, P. J. (1998). A history of cancer of the male breast. In Peters, W. P., & Visscher, D. W. (Eds.). Breast Cancer (Advances in Oncobiology, Volume 2) (pp. 229–243). Stamford/London: Jai Press. [DOI:10.1016/s1569-254x(98)80011-3] [Google Books]
Di Oto, E., Monti, V., Cucchi, M. C., Masetti, R., Varga, Z., & Foschini, M. P. (2015). X chromosome gain in male breast cancer. Human Pathology, 46(12), 1908–1912. [DOI:10.1016/j.humpath.2015.08.008]
Di Oto, E., Biserni, G. B., Varga, Z., Morandi, L., Cucchi, M. C., Masetti, R., & Foschini, M. P. (2018). X chromosome gain is related to increased androgen receptor expression in male breast cancer. Virchows Archiv, 473(2), 155–163. [DOI:10.1007/s00428-018-2377-2]
Dimitrakakis, C. (2011). Androgens and Breast Cancer in Men and Women. Endocrinology and Metabolism Clinics of North America, 40(3), 533–547. [DOI:10.1016/j.ecl.2011.05.007]
Ferzoco, R. M., & Ruddy, K. J. (2015). The Epidemiology of Male Breast Cancer. Current Oncology Reports, 18(1), 1. [DOI:10.1007/s11912-015-0487-4]
Giammarini, A., Rocchi, M., Zennaro, W., & Filippi, G. (2008). XX Male with breast cancer. Clinical Genetics, 18(2), 103–108. [DOI:10.1111/j.1399-0004.1980.tb01019.x]
Goldschmidt, E., Márquez, M., Solari, A., Ziembar, M. I., & Laudicina, A. (2010). Variabilidad fenotípica en pacientes 47, XXX: Presentación de cuatro casos nuevos. [Phenotypic variability in 47, XXX patients. Clinical report of four new cases.] Archivos Argentinos de Pediatría, 108(4), e88–e91. [URL]
Gravholt, C. H. (2008). Epidemiology of Turner syndrome. The Lancet Oncology, 9(3), 193–195. [DOI:10.1016/s1470-2045(08)70045-7]
Hughes, I. A. (2010). Evaluation and Management of Disorders of Sex Development. In Brook, C. G. D., Clayton, P. E., & Brown, R. S. (Eds.). Brook’s Clinical Pediatric Endocrinology, 6th Edition (pp. 192–212). Oxford: Wiley-Blackwell. [DOI:10.1002/9781444316728.ch9]
Hughes, I. A., Davies, J. D., Bunch, T. I., Pasterski, V., Mastroyannopoulou, K., & MacDougall, J. (2012). Androgen insensitivity syndrome. The Lancet, 380(9851), 1419–1428. [DOI:10.1016/s0140-6736(12)60071-3]
Jacobs, P. A., Maloney, V., Cooke, R., Crolla, J. A., Ashworth, A., & Swerdlow, A. J. (2013). Male breast cancer, age and sex chromosome aneuploidy. British Journal of Cancer, 108(4), 959–963. [DOI:10.1038/bjc.2012.577]
Lin, I., Chen, D., Chang, Y., Lee, Y., Su, C., Cheng, C., Tsai, Y., Ng, S., Chen, H., Lee, M., Chen, H., Suen, S., Chen, Y., Liu, T., Chang, C., & Hsu, M. (2015). Hierarchical Clustering of Breast Cancer Methylomes Revealed Differentially Methylated and Expressed Breast Cancer Genes. PLOS ONE, 10(2), e0118453. [DOI:10.1371/journal.pone.0118453]
Nakopoulou, L., Panayotopoulou, E. G., Giannopoulou, I., Tsirmpa, I., Katsarou, S., Mylona, E., Alexandrou, P., & Keramopoulos, A. (2006). Extra copies of chromosomes 16 and X in invasive breast carcinomas are related to aggressive phenotype and poor prognosis. Journal of Clinical Pathology, 60(7), 808–815. [DOI:10.1136/jcp.2006.037838]
Richardson, A. L., Wang, Z. C., De Nicolo, A., Lu, X., Brown, M., Miron, A., Liao, X., Iglehart, J. D., Livingston, D. M., & Ganesan, S. (2006). X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell, 9(2), 121–132. [DOI:10.1016/j.ccr.2006.01.013]
Sacchi, N. (1993). Constitutional Chromosome Aneuploidy and Cancer. In Kirsch, I. R. (Ed.). The Causes and Consequences of Chromosomal Aberrations (pp. 191–222). Boca Raton: CRC Press. [Google Books]
Schoemaker, M. J., Swerdlow, A. J., Higgins, C. D., Wright, A. F., & Jacobs, P. A. (2008). Cancer incidence in women with Turner syndrome in Great Britain: a national cohort study. The Lancet Oncology, 9(3), 239–246. [DOI:10.1016/s1470-2045(08)70033-0]
Sirchia, S. M., Tabano, S. M., & Miozzo, M. (2007). BRCA1 and X Chromosome Inactivation: Which is the Link? In Polinsky, K. R. (Ed.). Tumor Suppressor Genes (pp. 5–8). New York: Nova Biomedical Books. [Google Books]
Sokol, R. Z. (2012). It’s not all about the testes: medical issues in Klinefelter patients. Fertility and Sterility, 98(2), 261–265. [DOI:10.1016/j.fertnstert.2012.05.026]
Spatz, A., Borg, C., & Feunteun, J. (2004). X-Chromosome Genetics and Human Cancer. Nature Reviews Cancer, 4(8), 617–629. [DOI:10.1038/nrc1413]
Swerdlow, A. J., Schoemaker, M. J., Higgins, C. D., Wright, A. F., & Jacobs, P. A. (2005). Cancer Incidence and Mortality in Men with Klinefelter Syndrome: A Cohort Study. JNCI: Journal of the National Cancer Institute, 97(16), 1204–1210. [DOI:10.1093/jnci/dji240]
Tiefenbacher, K., & Daxenbichler, G. (2008). The Role of Androgens in Normal and Malignant Breast Tissue. Breast Care, 3(5), 325–331. [DOI:10.1159/000158055]
Viuff, M. H., Stochholm, K., Lin, A., Berglund, A., Juul, S., & Gravholt, C. H. (2021). Cancer occurrence in Turner syndrome and the effect of sex hormone substitution therapy. European Journal of Endocrinology, 184(1), 79–88. [DOI:10.1530/eje-20-0702]
\ No newline at end of file
diff --git a/transfemscience.org/articles/breast-cancer/index.html b/transfemscience.org/articles/breast-cancer/index.html
index 3c955cec..7292a20b 100644
--- a/transfemscience.org/articles/breast-cancer/index.html
+++ b/transfemscience.org/articles/breast-cancer/index.html
@@ -1 +1 @@
-Breast Cancer Risk with Hormone Therapy in Transfeminine People - Transfeminine Science
Breast Cancer Risk with Hormone Therapy in Transfeminine People
By Aly | First published April 25, 2020 | Last modified May 8, 2023
Abstract / TL;DR
Estrogens and progestogens induce breast development and this may also increase the risk of breast cancer. Breast cancer risk increases exponentially with age in both women and men and is far greater in women than in men. Estrogens and progestogens have been linked to risk of breast cancer in numerous contexts, for instance ovarian activity, menopausal hormone therapy, antiestrogen therapy, and high-dose estrogen therapy, among others. Only a handful of studies have assessed the influence of estrogens and/or progestogens on breast cancer risk in transfeminine people. The available data are limited and have methodological limitations, but the best available evidence suggests that estrogen and progestogen therapy strongly increases the risk of breast cancer in transfeminine people, with the risk lying somewhere intermediate between that of cisgender women and cisgender men. More research is necessary to better-characterize the risk of breast cancer with hormone therapy in transfeminine people, particularly with longer follow-ups and different hormonal regimens (e.g., estrogen alone versus estrogen plus progestogen). Factors likely to modify breast cancer risk in transfeminine people include duration of hormone therapy, age at start of hormone therapy, concomitant long-term progestogen use, and dosages of estrogens and/or progestogens. Having a single X chromosome (as in transfeminine people and cisgender men) instead of having two (as in cisgender women) may be partially protective against breast cancer. Although the risk of breast cancer is likely increased in transfeminine people, the lifetime incidence is likely low, many years of hormonal exposure are likely required for the risk to accumulate, occurrence is usually in older age, and breast cancer is highly treatable. As such, concerns about breast cancer risk do not preclude hormone therapy for transfeminine people. In any case, breast cancer screening may be advisable in transfeminine people as with cisgender women.
Introduction
Estrogens and progestogens induce breast development in humans. This includes major roles for estrogens in pubertal breast development and progestogens in the breast changes that occur during pregnancy in preparation for lactation and breastfeeding. Breast cancer is far more common in cisgender women than in cisgender men, and hormones are well-known to be involved in the development and progression of breast cancer in cisgender women. For these reasons, it’s logical to assume that hormone therapy in transfeminine people might increase the risk of breast cancer. This is a review of this notion and of the available studies on the subject.
Sex and Age as Breast Cancer Risk Factors
The normal lifetime risk of breast cancer in women is about 1 in 8 or 12.5% (Ban & Godellas, 2014), whereas the normal lifetime risk of breast cancer in men is about 1 in 1,000 or 0.1% (Abdelwahab Yousef, 2017). The overall age-standardized risk of breast cancer in men is about 1 in 100,000 person–years (PY) (Abdelwahab Yousef, 2017; Ottini & Capalbo, 2017). In other words, 1 in 100,000 men are diagnosed with breast cancer every year on average. Fewer than 1% of all cases of breast cancer are in men (Abdelwahab Yousef, 2017; Sun et al., 2017). Thus, breast cancer is frequent in women but is extremely rare in men.
After sex, age is the strongest known risk factor for breast cancer (Momenimovahed & Salehiniya, 2018). Breast cancer risk increases exponentially with age in premenopausal women (Dix & Cohen, 1982). The risk of breast cancer in women is about 1 in 600,000 at age 15; 1 in 75,000 at age 20; 1 in 1,500 at age 30; and 1 in 175 at age 40 (Anders et al., 2009). After menopause, which typically occurs at 50 years of age, breast cancer risk continues to increase but more slowly (Dix & Cohen, 1982; Benz, 2008; Colditz & Rosner, 2000). This is assumed to be due to the much lower levels of estradiol and progesterone in postmenopause (Dix & Cohen, 1982). It has been estimated that if it were not for menopause, there would be 6 times as many cases of breast cancer in women, and hence presumably that a majority of women would develop breast cancer in their lifetimes (Brisken, Hess, & Jeitziner, 2015). Similarly to the case in women, breast cancer risk in men increases exponentially up to about age 65, and thereafter increases somewhat more slowly (Ottini & Capalbo, 2017; Cancer Research UK). This may be related to andropause in older age and consequent lower levels of testosterone and estradiol (Satram, 2006). The typical age of diagnosis of breast cancer is around 60 years in women and 65–70 years in men (Ferzoco & Ruddy, 2016; Giordano, 2018). Figure 1 below shows the risk of breast cancer with age in women and men (Ottini & Capalbo, 2017).
Figure 1: Age-specific incidence rates per 100,000 for male and female invasive breast cancer in White Americans from the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute (NCI) (2003–2012) (Ottini & Capalbo, 2017). Note that the male breast cancer and female breast cancer lines use different y-axes and corresponding scales (50× larger y-axis interval in the case of female breast cancer).
Hormone Exposure in Women and Breast Cancer Risk
Along with sex and age, research strongly implicates ovarian activity and ovarian hormones—like estradiol and progesterone—as major risk factors for breast cancer (Ban & Godellas, 2014). Later menarche, earlier menopause, and breastfeeding—all of which result in reduced lifetime ovarian hormone exposure—are associated with decreased risk of breast cancer (Ban & Godellas, 2014; Momenimovahed & Salehiniya, 2018). Surgical removal of the ovaries before 40 years of age is associated with a 50% decrease in lifetime risk of breast cancer (Ban & Godellas, 2014). Antiestrogens including selective estrogen receptor modulators (SERMs) like tamoxifen and aromatase inhibitors (AIs) like anastrozole can be used prophylactically long-term as chemoprevention against breast cancer in high-risk postmenopausal women, providing 50 to 70% reduction in breast cancer risk (Mocellin, Goodwin, & Pasquali, 2019; Nelson et al., 2019). Long-term estrogen and progestogen treatment has been found to produce breast cancer in non-human primates, and high doses may result in breast cancer developing at an earlier age than usual (Cline, 2007).
In accordance with the well-established relationships of ovarian activity and ovarian hormones with breast cancer risk, menopausal hormone therapy is associated with a higher risk of breast cancer in peri- and postmenopausal women (CGHFBC, 2019; Table). This is true both for therapy with an estrogen alone and to a greater extent with the combination of an estrogen and a progestogen (CGHFBC, 2019). In terms of absolute risk of breast cancer with menopausal hormone therapy, the incidence after 20 years of use is about 1.5-fold higher with an estrogen alone and about 2.5-fold higher with an estrogen plus a progestogen per a large recent meta-analysis of all of the available data (CGHFBC, 2019). Hence, the risk of breast cancer is higher with combined estrogen and progestogen therapy than with an estrogen alone. In the past it was thought that progestogens might be protective against breast cancer due to antiestrogenic effects in the breasts (Mauvais-Jarvis, Kuttenn, & Gompel, 1986; Wren & Eden, 1996; Gompel et al., 2002), but this proved not to be the case. The higher risk of breast cancer with menopausal hormone therapy was found to be causal in the Women’s Health Initiative (WHI), at least with the used regimen (specifically conjugated equine estrogens (CEEs) 0.625 mg/day plus medroxyprogesterone acetate (MPA) 2.5 mg/day) (about 1.25-fold higher risk after 5 years of therapy) (WGWHII, 2002; Chlebowski, Aragaki, & Anderson, 2015). The WHI was the largest randomized controlled trial (RCT) of hormone therapy ever conducted and allowed a rare chance to evaluate causation in terms of long-term health effects of hormonal agents.
For a visualization of breast cancer risk with menopausal hormone therapy, Figure 2 below shows breast cancer risk over time based on modeling and data from the observational Nurses’ Health Study (NHS) (Colditz & Rozner, 2000). As can be seen, menopausal hormone therapy is associated with maintenance of the age-related exponential rate of increase in breast-cancer risk that is normally blunted by the onset of menopause.
Bioidentical hormones like estradiol and progesterone are associated with increased risk of breast cancer similarly to non-bioidentical hormonal agents like CEEs and MPA (CGHFBC, 2019; Table). The risk of breast cancer with estradiol does not appear to be different from that with CEEs (CGHFBC, 2019). Oral progesterone has not been associated with an increase in risk of breast cancer with less than 5 years of therapy on the basis of the available data, which is in contrast to progestins (Aly, 2018; CGHFBC, 2019; Stute, Wildt, & Neulen, 2018; Mirkin, 2018; Fournier et al., 2007). However, oral progesterone is associated with increased risk of breast cancer after more than 5 years of therapy, by which point the risk appears to be similar in magnitude to that with MPA (Aly, 2018; CGHFBC, 2019; Stute, Wildt, & Neulen, 2018; Mirkin, 2018; Fournier et al., 2007). The initial lower relative risk with oral progesterone might be related to the low levels of progesterone that oral progesterone achieves and hence its comparatively weak progestogenic strength (Aly, 2018; Kuhl & Schneider, 2013; Davey, 2018; Wiki; Graphs). As a result, a longer duration of therapy (and/or larger sample sizes) may be necessary before an increase in breast cancer risk with oral progesterone can be properly quantified (Aly, 2018; Kuhl & Schneider, 2013; Davey, 2018). Virtually no data are available on breast cancer risk with non-oral routes of progesterone at present (Aly, 2018; Stute, Wildt, & Neulen, 2018). However, non-oral and fully potent progesterone is anticipated by some researchers to have a similar risk of breast cancer as progestins not only with long-term use but also with short-term exposure (Aly, 2018; Kuhl & Schneider, 2013; Davey, 2018).
Although the overall evidence including both prospective observational studies and RCTs shows an increased risk of breast cancer with estrogen alone therapy in menopausal women (CGHFBC, 2019), the available RCTs, namely the WHI, have so far been unable to show increased breast cancer risk with estrogen alone and have instead found a significant decrease in risk (RR 0.77, 95% CI 0.64–0.93) (CGHFBC, 2019; Table). Various hypotheses have been advanced to explain the conflicting findings. For instance, the women in the WHI, who constitute the vast majority of the RCT data, were atypical in that they were of relatively old age at start of hormone therapy (average 64 years) and were relatively overweight and obese (CGHFBC, 2019; Kuhl, 2005). Women who are overweight or obese are already at an increased risk of breast cancer and may experience no further increase with estrogen therapy (Kuhl, 2005). And women who have been deprived of estrogens for many years may experience paradoxical inhibitory effects on breast cancer risk, which is in contrast to women who start at menopause (also seen in the observational data) (Prentice, 2008; CGHFBC, 2019). This has been termed the “gap hypothesis” (Palmieri et al., 2014; Mueck & Ruan, 2011). More RCTs are needed to test these theories and determine the influence of estrogens on menopausal breast cancer risk.
Paradoxically, estrogen and/or progestogen exposure can also decrease breast cancer risk and inhibit breast cancer progression under certain circumstances. For instance, pregnancy lasting more than 34 weeks in young women (<35 years of age) increases breast cancer risk in the short-term (<10 years), but is actually protective against breast cancer in the long-term (>20–25 years), with an up to 50% reduction in lifetime risk (Nichols et al., 2019; Husby et al., 2018). Extremely high levels of estrogens and progesterone occur during pregnancy (e.g., >12,500 pg/mL for estradiol at >34 weeks) (Graph), and animal studies suggest that these hormones are responsible for the protection (Rajkumar et al., 2003; Hilakivi-Clarke et al., 2012). High-dose estrogen therapy is an effective treatment against breast cancer in women who are at least 5 years postmenopausal, with similar effectiveness to antiestrogen therapy (Coelingh Bennink et al., 2017; Wiki). Conversely, high-dose estrogen therapy is not effective for breast cancer in premenopausal women, although it does work if massive doses are given (Coelingh Bennink et al., 2017; Wiki). However, these beneficial effects of estrogens and progestogens against breast cancer risk and progression occur under unusual circumstances (e.g., very high levels/doses and/or prolonged prior sex-hormone deficiency) and are not likely to be of relevance to transfeminine people.
Estrogen Exposure in Men and Breast Cancer Risk
In cisgender men, conditions related to lower androgen exposure and/or higher estrogen exposure are associated with increased risk of breast cancer (Ferzoco & Ruddy, 2016). These conditions include testicular problems and removal as well as liver disease (Abdelwahab Yousef, 2017). Testicular dysfunction and absence result in decreased androgen levels, while liver disease results in higher estrogen levels due to impaired metabolism of estrogens. Both types of conditions result in an increased ratio of estrogens to androgens, in turn causing greater estrogenic stimulation of the breasts. This is presumably responsible for the increased breast cancer risk. Breast conditions in men, like benign breast disease and gynecomastia, have also been associated with breast cancer risk (Sasco, Lowenfels, & Pasker-De Jong, 1993), although findings are mixed for gynecomastia (Giordano, 2005). In addition to health conditions, high-dose estrogen therapy for prostate cancer has been associated with increased breast cancer risk in cisgender men (Thellenberg et al., 2003; Karlsson et al., 2006).
Studies of Breast Cancer Risk in Transfeminine People
A companion doc for this article with a table of available studies on the risk of breast cancer with hormone therapy in transfeminine people can be found here. To date, three large cohorts and several smaller cohorts have evaluated breast cancer risk with transfeminine hormone therapy.
Vrije Universiteit University Medical Center (VUMC) Studies
Most of the studies in the table are by the Vrije Universiteit University Medical Center (VUMC) in Amsterdam, the Netherlands (Asscheman, Gooren, & Eklund, 1989; van Kesteren et al., 1997; Mueller & Gooren, 2008; Asscheman et al., 2011; Gooren et al., 2013; de Blok et al., 2018; de Blok et al., 2019). This clinic treats 95% of transgender people in the Netherlands (de Blok et al., 2019). The studies by the VUMC are all on largely the same evolving cohort of transgender women. For a couple of decades, the VUMC reported a low incidence of breast cancer that was not much higher than the rate expected in cisgender men (only 2 reported cases in 2,300 transgender women over an average duration of hormone therapy of about 20 years) (Gooren et al., 2013). The typical regimen used by the clinic was estrogen and high-dose cyproterone acetate. The studies did not did not use systematic screening for breast cancer and instead presumably relied on patient report for breast cancer diagnosis, raising the possibility of under-detection of cases (Feldman et al., 2016).
In 2019, the VUMC did another follow-up study but this time switched to a new method of obtaining breast cancer diagnoses (de Blok et al., 2019). Instead of simply asking patients whether they’d been diagnosed with breast cancer, the researchers retrieved breast cancer diagnoses using an electronic system of patient records for all of the Netherlands called the Nationwide Network and Registry of Histopathology and Cytopathology in the Netherlands (PALGA). When they did this, their number of breast cancer cases shot up from 2 to 15. This resulted in a relative risk of breast cancer of almost 50-fold the expected risk and an absolute incidence of roughly 0.6% over a mean duration of hormone therapy of about 15 years. These findings supersede all of the previous research by the clinic and indicate that the true number of breast cancer cases in the VUMC cohort had been greatly underestimated in their previous studies. Very large increases in risk, such as the risk of lung cancer with smoking and the nearly 50-fold increase in breast cancer risk in this study, can be considered as likely causal. Accordingly, the increase in risk has been described by researchers using causal language (de Blok et al., 2019; Reactions Weekly, 2019). Prior to this study, it was thought that the increase in risk of breast cancer with hormone therapy in transfeminine people was low, but due to the new data, it’s now known that this isn’t necessarily the case (de Blok, Dreijerink, & Heijer, 2019).
Veterans Health Administration (VHA) Study
Another large cohort is the Veterans Health Administration (VHA) cohort in the United States (Brown & Jones, 2015; Brown, 2015). In 2015, a study was published using data from this cohort. The researchers reported 3 cases of breast cancer in about 3,500 people assigned male at birth with mean follow-up time of approximately 10 years. Hence, there was a rate of about 0.09% over an average duration of 10 years. Although the mean duration of follow up was shorter than in the VUMC cohort, the total follow-up time was similar. The VHA researchers reported the incidence of breast cancer as increased by 33-fold relative to the expected rate. This is somewhat strange in that it’s discrepant with the VUMC’s 2018 numbers (which reported a 46-fold increased risk based on diagnosis in 15 of 2,300 people and mean follow-up time of 15 years). In any case, the VHA concluded that the risk of breast cancer in transfeminine people was higher than that in cisgender men and higher than that reported by the earlier VUMC studies, but still lower than the risk in cisgender women.
The VHA paper provided no specifics on what hormone therapy medications and doses were used, although since it included all patients treated by providers in the VHA system, the regimens are likely to have been quite variable. Because this was the United States, the typical regimen was probably estrogen plus spironolactone and no progestogen.
There are a variety of problems with the VHA study that require discussion:
As with the earlier VUMC studies, the VHA study did not use systematic screening for breast cancer and presumably relied on patient report for breast cancer diagnoses, raising the possibility of under-detection of cases (Feldman et al., 2016).
The study was irrespective of hormone therapy (Feldman et al., 2016). Only some people in the cohort were on hormone therapy while others were not, and the researchers didn’t have full numbers on who was and who wasn’t. Moreover, an unknown percentage of people included in the cohort weren’t actually transgender—the researchers also included all cisgender men in the VHA system with a “transgender-related” diagnosis, which included transvestic fetishism (a.k.a. sexual crossdressing). Most of these individuals presumably were not on hormone therapy.
The follow-up times for those who were definitely on hormone therapy were very short. Of about 1,400 transfeminine people in the VHA cohort who were certainly on hormone therapy, the mean duration of use was only 5.5 years, and was less than 3 years for nearly half of the sample.
The VHA paper was very confusing and unclear. As an example, they misgendered transgender people, referring to them as “male” and “female” per birth sex, and wrote in such a way that made it difficult to understand who was who as well as who was and was not actually on hormone therapy. This was such that even some citing publications reporting on the study mistakenly got the risks for “male” and “female” switched (e.g., Dente et al., 2019).
To add further confusion, the VHA researchers reported 33 total cases of breast cancer in the exact same cohort of 5,100 total people assigned male or female at birth in two other papers published around the same time (Brown & Jones, 2014; Brown & Jones, 2016). For comparison, they only reported 10 total cases in the 2015 paper (3 in those assigned male at birth and 7 in people assigned female at birth). The reason for this discrepancy is unknown (Braun et al., 2017).
Due to the various problems with the VHA study, its findings are of limited usefulness and should likely be considered with caution. The 2019 VUMC findings are of far better-quality data.
Kaiser Permanente in California and Georgia Study
The third and final large cohort study was by Kaiser Permanente in Northern California, Southern California, and Georgia in the United States (Silverberg et al., 2017). They reported the incidences of a variety of cancers in a combined cohort from three Kaiser sites consisting of about 2,800 transfeminine people. The mean duration of follow up in the study was only 4 years. The researchers did not report the incidence of any cancer that had an absolute occurrence of less than 5 cases, and breast cancer in transfeminine people was not included in the relevant table of results nor in the discussion in the paper. Hence, there were presumably fewer than 5 cases of breast cancer in the cohort of transfeminine people. However, T’Sjoen and colleagues, a very reputable group of researchers in the field of transgender medical research, reported in a citing publication that the Kaiser team found a higher risk of breast cancer in transfeminine people compared to cisgender men but a lower risk compared to cisgender women (T’Sjoen et al., 2019). Assuming this is accurate, which it likely is, the information was presumably obtained via personal communication with the Kaiser researchers. Limitations of the Kaiser study include lack of exact figures on breast cancer risk and a very short follow-up duration. In addition, cancer diagnoses were retrieved only from Kaiser’s own systems, and the extent to which screening was systematic, if at all, is unclear.
The Kaiser researchers plan to do further studies in much larger cohorts via expansion to additional sites in the future (Silverberg et al., 2017). This is an exciting development, and should hopefully give us additional data on health risks in transgender people, including of breast cancer risk.
Small Cohort Studies
The remaining investigations of breast cancer risk in transfeminine people were small. One was 50 post-SRS transgender women by the Ghent University Hospital in Belgium who were followed up for an average of 11.4 years each, in whom no cases of breast cancer were seen (Wierckx et al., 2012). Another was 60 transgender women by the University Hospital Erlangen in Germany (Dittrich et al., 2005). No cases were seen in this cohort either, although the treatment period was only 2 years. The last was Harry Benjamin’s patient cohort; he communicated in his 1960s publications that he had treated about 150 transgender women with “medium to fairly large doses of estrogen” for varying periods of time (e.g., 3 months to 12 years) and hadn’t encountered any cases of breast cancer (Benjamin, 1964; Benjamin, 1966; Gooren et al., 2013). These cohorts are all too small and limited to provide meaningful quantification of breast cancer risk in transfeminine people.
Discussion of the Available Studies
Inconclusive Quantification of Breast Cancer Risk Due to Short Follow-Up Times
The available studies on breast cancer risk in transfeminine people are inconclusive due to inadequate follow-up times (de Blok, Dreijerink, & Heijer, 2019; Mueller & Gooren, 2008; Gooren, 2011; Gooren et al., 2013; Brown & Jones, 2015). For instance the average durations of hormone therapy in the VUMC and VHA studies were only 10 to 20 years. Breast cancer incidence in cisgender women increases exponentially over decades during premenopause and is very rare until older age. The typical age of breast cancer diagnosis in cisgender women is 60 years, which includes about 50 years of premenopausal hormone exposure and about 10 years of postmenopausal hormone exposure. (The latter of which is not unimportant, as demonstrated by the greatly decreased risk of breast cancer with prophylactic antiestrogen therapy.) As such, due to the short follow-up times of the available studies of transfeminine hormone therapy and breast cancer risk, the true or lifetime risk of breast cancer in transfeminine people is not yet known (Mueller & Gooren, 2008).
We do know however that hormone therapy in transfeminine people, at least with an estrogen plus a progestogen in the form of cyproterone acetate, appears to strongly increase the risk of breast cancer within about 15 years of use (de Blok, Dreijerink, & Heijer, 2019). Fortunately, the risk is intermediate between that in cisgender women and that in cisgender men (and not, e.g., greater than that in cisgender women). At the same time however, the risk is non-negligible, and the absolute incidence will only increase with longer follow-up times. In terms of lifetime risk, the incidence of breast cancer in transfeminine people is well on-track to be as high as single-digit percentages, at least based on the findings of de Blok et al. (2019) and their particular hormone therapy regimen.
Breast Cancer Risk in Relation to Lifetime Hormone Exposure
Transfeminine people may have a lower risk of breast cancer than cisgender women. This may due at least in part to a more limited amount of lifetime hormone exposure (Mueller & Gooren, 2008). This is based on the fact that, at least historically, transfeminine people have started hormone therapy on average at 30 or 40 years of age, which is decades after the age at which cisgender girls normally undergo puberty (Mueller & Gooren, 2008). Additionally, youth may represent a critical window of susceptibility for breast cancer risk (Biro & Wolff, 2011; Biro & Deardorff, 2013; Biro et al., 2020). However, the age at start of hormone therapy in transfeminine people has been decreasing in recent times (Mueller & Gooren, 2008), and nowadays many start hormones in their teens or early twenties. These individuals will have greater lifetime hormone exposure, and presumably greater breast cancer risk, than many of the transfeminine people of the past (Sutherland et al., 2020). Moreover, transfeminine people may be much less inclined to stop hormone therapy at the normal age of menopause in cisgender women (Mueller & Gooren, 2008). Many transfeminine people will likely stay on hormone therapy their entire lives. This additional exposure may further increase breast cancer risk (Mueller & Gooren, 2008).
Breast Cancer Risk in Relation to Progestogens and Dosage
Based on findings that breast cancer risk is higher with an estrogen plus progestogen relative to an estrogen alone in menopausal women, the risk of breast cancer with hormone therapy in transfeminine people may likewise be higher with combined estrogen and progestogen therapy (de Blok, Dreijerink, & Heijer, 2019). Hence, it’s possible that estrogen therapy without a progestogen could have a lower risk of breast cancer than that observed by the VUMC with an estrogen plus cyproterone acetate (de Blok, Dreijerink, & Heijer, 2019). It’s notable also that the doses of cyproterone acetate used by the VUMC result in rather extreme progestogenic exposure (Aly, 2019). The extent to which breast cancer risk with progestogens is dose-dependent is unknown. Whether or not estrogen dosage influences breast cancer risk, or if higher levels have greater risk, is unclear similarly. In any case, the fact that the use of estrogen–progestogen birth control in premenopausal women is associated with increased risk of breast cancer is suggestive that higher levels of estrogen and/or progestogen exposure may result in some degree of greater risk (Kahlenborn et al., 2006; Zhu et al., 2012; Ji et al., 2019). In addition, an older observational study reported a 4-fold greater risk of breast cancer in menopausal cisgender women treated with injectable estrogens than with oral estrogens or no hormone therapy (Hulka et al., 1982; Coe & Parks, 1989). This is notable as typical doses of injectable estrogens provide much greater estrogenic exposure than oral and other routes of estrogen (Göretzlehner et al., 2002; Aly, 2021). However, the study has important limitations, including only being a single study and being quite old.
X Chromosomes and Breast Cancer Risk
The sex chromosomes include the X chromosome and the Y chromosome. Cisgender women typically have two X chromosomes (46,XX karyotype), while cisgender men (and transfeminine people) typically have one X chromosome and one Y chromosome (46,XY karyotype). It’s possible that a 46,XX karyotype—specifically the presence of a second X chromosome—may be a major risk factor for breast cancer. X-chromosome gain and aberrant X-chromosome inactivation have been associated with breast cancer incidence and aggressiveness (Nakopoulou et al., 2007; Di Oto et al., 2015; Lin et al., 2015; Chaligné et al., 2015; Di Oto et al., 2018). In addition, breast cancer risk is markedly higher in men with Klinefelter’s syndrome (KS), who have a 46,XXY karyotype, than in regular men with a 46,XY karyotype. Conversely, breast cancer has never been reported in women with complete androgen insensitivity syndrome (CAIS), who have a 46,XY karyotype similarly to transfeminine people (Hughes et al., 2012; Tiefenbacher & Daxenbichler, 2008; Hughes, 2009). This is in spite of the fact that CAIS women have excellent breast development (Aly, 2020). However, hormonal abnormalities in people with KS and CAIS may alternatively contribute to differences in breast cancer risk and are thus a confounding variable. It’s notable in particular that CAIS women have relatively low estrogen exposure and little to no progesterone. In any case, the differences in breast cancer risk in conditions like KS and CAIS do not seem to be fully explainable by hormonal abnormalities. Thus, it’s possible that the lack of two X chromosomes in transfeminine people may indeed prove to be partially protective against breast cancer risk. For more in-depth information on this topic, see the supplemental article here.
Summary and Conclusions
Breast cancer risk is far higher in women than in men. The incidence of breast cancer increases exponentially with age and is very rare in younger people. There is strong basis to assume that estrogens and progestogens increase the risk of breast cancer, which can be inferred as a major mechanism for the sex differences in breast cancer risk. The available data do not indicate meaningful differences between bioidentical and non-bioidentical hormones in terms of breast cancer risk.
Older studies of transfeminine hormone therapy and breast cancer risk found low incidences of breast cancer in transfeminine people. However, a recent study with better methodology than previous studies showed a nearly 50-fold increase in breast cancer risk. The hormone therapy regimen used by their cohort was specifically estrogen plus cyproterone acetate. It’s unknown whether other hormonal regimens, for instance estrogen alone or with a non-progestogenic antiandrogen, have similar risks. The available data on breast cancer risk in transfeminine people are limited by inadequate follow-up times. As a result, lifetime breast cancer risk is unknown. Based on available data however, we can project into the future and assume that the lifetime incidence of breast cancer in transfeminine people may be as high as single-digit percentages. This puts the risk of breast cancer in transfeminine people somewhere between that of cisgender men and cisgender women.
Various factors may modify breast cancer risk with transfeminine hormone therapy. Examples include cumulative duration of use, age at start of therapy, long-term progestogen use, and possibly dosages of estrogens and/or progestogens. The lack of a second X chromosome in transfeminine people may be partially protective against breast cancer. Although breast cancer risk with transfeminine hormone therapy is something for transfeminine people to be aware of, particularly the probable contribution of progestogens to the risk, the lifetime incidence is likely to be low and the risk appears to be less than that of cisgender women. In addition, it takes many years of hormone exposure for breast cancer to occur and is generally restricted to old age. Also, breast cancer is a highly treatable disease, with excellent 5- and 10-year survival rates (Cancer.Net). For these reasons, concerns about breast cancer should most certainly not preclude hormone therapy for transfeminine people.
The possibility of breast cancer with hormone therapy in transfeminine people does, however, highlight the importance of routine breast cancer screening in transfeminine people of appropriate age and duration of hormone exposure (Chowdhry & O’Connell, 2020). It’s likely advisable that transfeminine people on long-term hormone therapy follow the same breast cancer screening procedures as those of cisgender women (Chowdhry & O’Connell, 2020).
References
Abdelwahab Yousef, A. J. (2017). Male Breast Cancer: Epidemiology and Risk Factors. Seminars in Oncology, 44(4), 267–272. [DOI:10.1053/j.seminoncol.2017.11.002]
Anders, C. K., Johnson, R., Litton, J., Phillips, M., & Bleyer, A. (2009). Breast Cancer Before Age 40 Years. Seminars in Oncology, 36(3), 237–249. [DOI:10.1053/j.seminoncol.2009.03.001]
Asscheman, H., Gooren, L., & Eklund, P. (1989). Mortality and morbidity in transsexual patients with cross-gender hormone treatment. Metabolism, 38(9), 869–873. [DOI:10.1016/0026-0495(89)90233-3]
Asscheman, H., Giltay, E. J., Megens, J. A., de Ronde, W., van Trotsenburg, M. A., & Gooren, L. J. (2011). A long-term follow-up study of mortality in transsexuals receiving treatment with cross-sex hormones. European Journal of Endocrinology, 164(4), 635–642. [DOI:10.1530/eje-10-1038]
Ban, K. A., & Godellas, C. V. (2014). Epidemiology of Breast Cancer. Surgical Oncology Clinics of North America, 23(3), 409–422. [DOI:10.1016/j.soc.2014.03.011]
Benjamin, H. (1964). Clinical Aspects of Transsexualism in the Male and Female. American Journal of Psychotherapy, 18(3), 458–469. [DOI:10.1176/appi.psychotherapy.1964.18.3.458]
Benz, C. C. (2008). Impact of aging on the biology of breast cancer. Critical Reviews in Oncology/Hematology, 66(1), 65–74. [DOI:10.1016/j.critrevonc.2007.09.001]
Biro, F. M., & Wolff, M. S. (2011). Puberty as a Window of Susceptibility. In Russo, J. (Ed.). Environment and Breast Cancer (pp. 29–41). New York: Springer New York. [DOI:10.1007/978-1-4419-9896-5_2]
Biro, F. M., & Deardorff, J. (2013). Identifying Opportunities for Cancer Prevention During Preadolescence and Adolescence: Puberty as a Window of Susceptibility. Journal of Adolescent Health, 52(5), S15–S20. [DOI:10.1016/j.jadohealth.2012.09.019]
Biro, F. M., Huang, B., Wasserman, H., Gordon, C. M., & Pinney, S. M. (2021). Pubertal Growth, IGF-1, and Windows of Susceptibility: Puberty and Future Breast Cancer Risk. Journal of Adolescent Health, 68(3), 517–522. [DOI:10.1016/j.jadohealth.2020.07.016]
Braun, H., Nash, R., Tangpricha, V., Brockman, J., Ward, K., & Goodman, M. (2017). Cancer in Transgender People: Evidence and Methodological Considerations. Epidemiologic Reviews, 39(1), 93–107. [DOI:10.1093/epirev/mxw003]
Brisken, C., Hess, K., & Jeitziner, R. (2015). Progesterone and Overlooked Endocrine Pathways in Breast Cancer Pathogenesis. Endocrinology, 156(10), 3442–3450. [DOI:10.1210/en.2015-1392]
Brown, G. R., & Jones, K. T. (2014). Incidence of breast cancer in a cohort of 5,135 transgender veterans. Breast Cancer Research and Treatment, 149(1), 191–198. [DOI:10.1007/s10549-014-3213-2]
Brown, G. R., & Jones, K. T. (2014). Racial Health Disparities in a Cohort of 5,135 Transgender Veterans. Journal of Racial and Ethnic Health Disparities, 1(4), 257–266. [DOI:10.1007/s40615-014-0032-4]
Brown, G. R. (2015). Breast Cancer in Transgender Veterans: A Ten-Case Series. LGBT Health, 2(1), 77–80. [DOI:10.1089/lgbt.2014.0123]
Brown, G. R., & Jones, K. T. (2016). Mental Health and Medical Health Disparities in 5135 Transgender Veterans Receiving Healthcare in the Veterans Health Administration: A Case–Control Study. LGBT Health, 3(2), 122–131. [DOI:10.1089/lgbt.2015.0058]
Cancer Research UK. (2020). Breast cancer incidence (invasive) statistics. Cancer Research UK. [URL]
Cancer.Net. (2020). Breast Cancer: Statistics. American Society of Clinical Oncology. [URL]
Chaligné, R., Popova, T., Mendoza-Parra, M., Saleem, M. M., Gentien, D., Ban, K., Piolot, T., Leroy, O., Mariani, O., Gronemeyer, H., Vincent-Salomon, A., Stern, M., & Heard, E. (2015). The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Research, 25(4), 488–503. [DOI:10.1101/gr.185926.114]
Chlebowski, R. T., Aragaki, A. K., & Anderson, G. L. (2015). Menopausal Hormone Therapy Influence on Breast Cancer Outcomes in the Women’s Health Initiative. Journal of the National Comprehensive Cancer Network, 13(7), 917–924. [DOI:10.6004/jnccn.2015.0106]
Chowdhry, D. N., & O’Connell, A. M. (2020). Breast Imaging in Transgender Patients. Journal of Breast Imaging, 2(2), 161–167. [DOI:10.1093/jbi/wbz092]
Cline, J. M. (2007). Assessing the mammary gland of nonhuman primates: effects of endogenous hormones and exogenous hormonal agents and growth factors. Birth Defects Research Part B: Developmental and Reproductive Toxicology, 80(2), 126–146. [DOI:10.1002/bdrb.20112]
Coe, F. L., & Parks, J. H. (1989). The Risks of Oral Contraceptives and Estrogen Replacement Therapy. Perspectives in Biology and Medicine, 33(1), 86–106. [DOI:10.1353/pbm.1990.0026]
Coelingh Bennink, H. J., Verhoeven, C., Dutman, A. E., & Thijssen, J. (2017). The use of high-dose estrogens for the treatment of breast cancer. Maturitas, 95, 11–23. [DOI:10.1016/j.maturitas.2016.10.010]
Colditz, G. A. (2000). Cumulative Risk of Breast Cancer to Age 70 Years According to Risk Factor Status: Data from the Nurses’ Health Study. American Journal of Epidemiology, 152(10), 950–964. [DOI:10.1093/aje/152.10.950]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/s0140-6736(19)31709-x]
Davey, D. A. (2018). Menopausal hormone therapy: a better and safer future. Climacteric, 21(5), 454–461. [DOI:10.1080/13697137.2018.1439915]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, R. H., & den Heijer, M. (2018). Breast cancer in transgender persons receiving gender affirming hormone treatment: results of a nationwide cohort study. Endocrine Abstracts, 56 [20th European Congress of Endocrinology 2018, 19–22 May 2018, Barcelona, Spain], 490–490 (abstract no. P955). [DOI:10.1530/endoabs.56.p955] [PDF]
de Blok, C. J., Dreijerink, K. M., & den Heijer, M. (2019). Cancer Risk in Transgender People. Endocrinology and Metabolism Clinics of North America, 48(2), 441–452. [DOI:10.1016/j.ecl.2019.02.005]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, I. R., & den Heijer, M. (2019). Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in the Netherlands. BMJ, 365, l1652. [DOI:10.1136/bmj.l1652]
Dente, E., Farneth, R., Purks, J., & Torelli, S. (2019). Evaluating risks, reported cases and screening recommendations for breast cancer in transgender patients. Georgetown Medical Review, 3(1). [DOI:10.52504/001c.7774]
Di Oto, E., Monti, V., Cucchi, M. C., Masetti, R., Varga, Z., & Foschini, M. P. (2015). X chromosome gain in male breast cancer. Human Pathology, 46(12), 1908–1912. [DOI:10.1016/j.humpath.2015.08.008]
Di Oto, E., Biserni, G. B., Varga, Z., Morandi, L., Cucchi, M. C., Masetti, R., & Foschini, M. P. (2018). X chromosome gain is related to increased androgen receptor expression in male breast cancer. Virchows Archiv, 473(2), 155–163. [DOI:10.1007/s00428-018-2377-2]
Dix, D., & Cohen, P. (1982). The incidence of breast cancer: analysis of the age dependence. Anticancer Research, 2(1-2), 23–7. [Google Scholar] [PubMed] [PDF]
Dworin, M. (1961). The Rationale of Endocrine Therapy in Breast Cancer. CA: A Cancer Journal for Clinicians, 11(2), 48–54. [DOI:10.3322/canjclin.11.2.48]
Feldman, J., Brown, G. R., Deutsch, M. B., Hembree, W., Meyer, W., Meyer-Bahlburg, H. F., Tangpricha, V., T’Sjoen, G., & Safer, J. D. (2016). Priorities for transgender medical and healthcare research. Current Opinion in Endocrinology, Diabetes & Obesity, 23(2), 180–187. [DOI:10.1097/med.0000000000000231]
Ferzoco, R. M., & Ruddy, K. J. (2016). The Epidemiology of Male Breast Cancer. Current Oncology Reports, 18, 1. [DOI:10.1007/s11912-015-0487-4]
Forrest, A. P. (1989). Endocrine management of breast cancer. Proceedings of the Royal Society of Edinburgh. Section B. Biological Sciences, 95 [Oestrogen and the Human Breast], 1–10. [DOI:10.1017/s0269727000010502]
Fournier, A., Berrino, F., & Clavel-Chapelon, F. (2007). Unequal risks for breast cancer associated with different hormone replacement therapies: results from the E3N cohort study. Breast Cancer Research and Treatment, 107(1), 103–111. [DOI:10.1007/s10549-007-9523-x]
Gellhorn, A., & Jones, L. O. (1949). Chemotherapy of malignant disease. The American Journal of Medicine, 6(2), 188–231. [DOI:10.1016/0002-9343(49)90017-0]
Giordano, S. H. (2005). A Review of the Diagnosis and Management of Male Breast Cancer. The Oncologist, 10(7), 471–479. [DOI:10.1634/theoncologist.10-7-471]
Giordano, S. H. (2018). Breast Cancer in Men. New England Journal of Medicine, 378(24), 2311–2320. [DOI:10.1056/nejmra1707939]
Gompel, A., Levy, D., Chaouat, M., Kazem, A., Hugol, D., Daraı̈, E., Decroix, Y., & Rostène, W. (2002). Is there a place for progestin agents in breast cancer prevention? International Congress Series, 1229, 143–150. [DOI:10.1016/s0531-5131(01)00467-8]
Gooren, L. J. (2011). Care of Transsexual Persons. New England Journal of Medicine, 364(13), 1251–1257. [DOI:10.1056/nejmcp1008161]
Gooren, L. J., van Trotsenburg, M. A., Giltay, E. J., & van Diest, P. J. (2013). Breast Cancer Development in Transsexual Subjects Receiving Cross-Sex Hormone Treatment. The Journal of Sexual Medicine, 10(12), 3129–3134. [DOI:10.1111/jsm.12319]
Göretzlehner, G., Ackermann, W., Angelow, K., Bergmann, G., Bieck, E., Golbs, S., & Kliem, O. (2002). Pharmakokinetik von Estron, Estradiol, FSH, LH und Prolaktin nach intramuskulärer Applikation von 5 mg Estradiolvalerat. [Pharmacokinetics of estradiol valerate in postmenopausal women after intramuscular administration.] Journal für Menopause, 9(2), 46–49. [Google Scholar] [URL] [PDF] [Translation]
Green, R. B., Sethi, R. S., & Lindner, H. H. (1964). Treatment of advanced carcinoma of the breast. The American Journal of Surgery, 108(1), 107–121. [DOI:10.1016/0002-9610(64)90094-7]
Herrmann, J. B. (1947). Effect of estrogenic hormone on advanced carcinoma of the female breast. Archives of Surgery, 54(1), 1–9. [DOI:10.1001/archsurg.1947.01230070004001]
Hilakivi-Clarke, L., de Assis, S., Warri, A., & Luoto, R. (2012). Pregnancy hormonal environment and mother’s breast cancer risk. Hormone Molecular Biology and Clinical Investigation, 9(1), 11–23. [DOI:10.1515/hmbci-2012-0019]
Hughes, I. A. (2010). Evaluation and Management of Disorders of Sex Development. In Brook, C. G. D., Clayton, P. E., & Brown, R. S. (Eds.). Brook’s Clinical Pediatric Endocrinology, 6th Edition (pp. 192–212). Oxford: Wiley-Blackwell. [DOI:10.1002/9781444316728.ch9]
Hughes, I. A., Davies, J. D., Bunch, T. I., Pasterski, V., Mastroyannopoulou, K., & MacDougall, J. (2012). Androgen insensitivity syndrome. The Lancet, 380(9851), 1419–1428. [DOI:10.1016/s0140-6736(12)60071-3]
Hulka, B. S., Chambless, L. E., Deubner, D. C., & Wilkinson, W. E. (1982). Breast cancer and estrogen replacement therapy. American Journal of Obstetrics and Gynecology, 143(6), 638–644. [DOI:10.1016/0002-9378(82)90108-9]
Husby, A., Wohlfahrt, J., Øyen, N., & Melbye, M. (2018). Pregnancy duration and breast cancer risk. Nature Communications, 9(1), 4255. [DOI:10.1038/s41467-018-06748-3]
Ji, L., Jing, C., Zhuang, S., Pan, W., & Hu, X. (2019). Effect of age at first use of oral contraceptives on breast cancer risk. Medicine, 98(36), e15719–e15719. [DOI:10.1097/md.0000000000015719]
Kahlenborn, C., Modugno, F., Potter, D. M., & Severs, W. B. (2006). Oral Contraceptive Use as a Risk Factor for Premenopausal Breast Cancer: A Meta-analysis. Mayo Clinic Proceedings, 81(10), 1290–1302. [DOI:10.4065/81.10.1290]
Karlsson, C. T., Malmer, B., Wiklund, F., & Grönberg, H. (2006). Breast Cancer as a Second Primary in Patients With Prostate Cancer—Estrogen Treatment or Association With Family History of Cancer? Journal of Urology, 176(2), 538–543. [DOI:10.1016/j.juro.2006.03.036]
Kennedy, B.J. (1956). Effects of steroids in women with breast cancer. In Engle, E. T., & Pincus, G. (Eds.). Hormones and the Aging Process: Proceedings of a Conference Held at Arden House, Harriman, New York, 1955 (pp. 253–272). New York: Academic Press. [Google Scholar] [Google Books]
Kennedy, B. J. (1957). The Present Status of Hormone Therapy in Advanced Breast Cancer. Radiology, 69(3), 330–340. [DOI:10.1148/69.3.330]
Kennedy, B. J. (1964). Estrogenic Hormone Therapy in Advanced Breast Cancer. Annals of Internal Medicine, 60(4), 718–718. [DOI:10.7326/0003-4819-60-4-718_1]
Kuhl, H. (2005). Breast cancer risk in the WHI study: The problem of obesity. Maturitas, 51(1), 83–97. [DOI:10.1016/j.maturitas.2005.02.018]
Kuhl, H., & Schneider, H. P. (2013). Progesterone – promoter or inhibitor of breast cancer. Climacteric, 16(Suppl 1), 54–68. [DOI:10.3109/13697137.2013.768806]
Lin, I., Chen, D., Chang, Y., Lee, Y., Su, C., Cheng, C., Tsai, Y., Ng, S., Chen, H., Lee, M., Chen, H., Suen, S., Chen, Y., Liu, T., Chang, C., & Hsu, M. (2015). Hierarchical Clustering of Breast Cancer Methylomes Revealed Differentially Methylated and Expressed Breast Cancer Genes. PLOS ONE, 10(2), e0118453. [DOI:10.1371/journal.pone.0118453]
Lumachi, F. (2015). Current medical treatment of estrogen receptor-positive breast cancer. World Journal of Biological Chemistry, 6(3), 231–239. [DOI:10.4331/wjbc.v6.i3.231]
Mauvais-Jarvis, P., Kuttenn, F., & Gompel, A. (1986). Antiestrogen action of progesterone in breast tissue. Breast Cancer Research and Treatment, 8(3), 179–188. [DOI:10.1007/bf01807330]
Mirkin, S. (2018). Evidence on the use of progesterone in menopausal hormone therapy. Climacteric, 21(4), 346–354. [DOI:10.1080/13697137.2018.1455657]
Mocellin, S., Goodwin, A., & Pasquali, S. (2019). Risk-reducing medications for primary breast cancer: a network meta-analysis. Cochrane Database of Systematic Reviews, 2019(4), CD012191. [DOI:10.1002/14651858.cd012191.pub2]
Momenimovahed, Z., & Salehiniya, H. (2019). Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer: Targets and Therapy, 11, 151–164. [DOI:10.2147/bctt.s176070]
Mueck, A. O., & Ruan, X. (2011). Benefits and risks during HRT: main safety issue breast cancer. Hormone Molecular Biology and Clinical Investigation, 5(2), 105–116. [DOI:10.1515/hmbci.2011.014]
Mueller, A., & Gooren, L. (2008). Hormone-related tumors in transsexuals receiving treatment with cross-sex hormones. European Journal of Endocrinology, 159(3), 197–202. [DOI:10.1530/eje-08-0289]
Nakopoulou, L., Panayotopoulou, E. G., Giannopoulou, I., Tsirmpa, I., Katsarou, S., Mylona, E., Alexandrou, P., & Keramopoulos, A. (2006). Extra copies of chromosomes 16 and X in invasive breast carcinomas are related to aggressive phenotype and poor prognosis. Journal of Clinical Pathology, 60(7), 808–815. [DOI:10.1136/jcp.2006.037838]
Nathanson, I. T. (1947). Hormonal Alteration of Advanced Cancer of the Breast. Surgical Clinics of North America, 27(5), 1144–1150. [DOI:10.1016/s0039-6109(16)32241-1]
Nathanson, I. T. (1951). Sex Hormones and Castration in Advanced Breast Cancer. Radiology, 56(4), 535–552. [DOI:10.1148/56.4.535]
Nelson, H. D., Fu, R., Zakher, B., Pappas, M., & McDonagh, M. (2019). Medication Use for the Risk Reduction of Primary Breast Cancer in Women. JAMA, 322(9), 868–886. [DOI:10.1001/jama.2019.5780]
Nichols, H. B., Schoemaker, M. J., Cai, J., Xu, J., Wright, L. B., Brook, M. N., Jones, M. E., Adami, H., Baglietto, L., Bertrand, K. A., Blot, W. J., Boutron-Ruault, M., Dorronsoro, M., Dossus, L., Eliassen, A. H., Giles, G. G., Gram, I. T., Hankinson, S. E., Hoffman-Bolton, J., Kaaks, R., Key, T. J., Kitahara, C. M., Larsson, S. C., Linet, M., Merritt, M. A., Milne, R. L., Pala, V., Palmer, J. R., Peeters, P. H., Riboli, E., Sund, M., Tamimi, R. M., Tjønneland, A., Trichopoulou, A., Ursin, G., Vatten, L., Visvanathan, K., Weiderpass, E., Wolk, A., Zheng, W., Weinberg, C. R., Swerdlow, A. J., & Sandler, D. P. (2018). Breast Cancer Risk After Recent Childbirth. Annals of Internal Medicine, 170(1), 22–31. [DOI:10.7326/m18-1323]
Ottini, L., & Capalbo, C. (2017). Male Breast Cancer. In Veronesi, U., Goldhirsch, A., Veronesi, P., Gentilini, O. D., & Leonardi, M. C. (Eds.). Breast Cancer: Innovations in Research and Management (pp. 753–762). Cham: Springer. [DOI:10.1007/978-3-319-48848-6_63]
Palmieri, C., Patten, D. K., Januszewski, A., Zucchini, G., & Howell, S. J. (2014). Breast cancer: Current and future endocrine therapies. Molecular and Cellular Endocrinology, 382(1), 695–723. [DOI:10.1016/j.mce.2013.08.001]
Pearson, O. H. (1954). Evaluation of endocrine therapy for advanced breast cancer. JAMA, 154(3), 234–239. [DOI:10.1001/jama.1954.02940370046013]
Pearson, O. H., & Lipsett, M. B. (1956). Endocrine Management of Metastatic Breast Cancer. Medical Clinics of North America, 40(3), 761–772. [DOI:10.1016/s0025-7125(16)34564-3]
Prentice, R. L. (2008). Women’s Health Initiative Studies of Postmenopausal Breast Cancer. In Li, J. J., Li, S. A., Mohla, S., Rochefort, H., & Maudelonde, T. (Eds.). Hormonal Carcinogenesis V (Advances in Experimental Medicine and Biology, Volume 617) (pp. 151–160). New York: Springer New York. [DOI:10.1007/978-0-387-69080-3_14]
Rajkumar, L., Guzman, R. C., Yang, J., Thordarson, G., Talamantes, F., & Nandi, S. (2003). Prevention of mammary carcinogenesis by short-term estrogen and progestin treatments. Breast Cancer Research, 6(1), R31. [DOI:10.1186/bcr734]
Reactions Weekly. (2019). Hormones increase breast cancer risk in Dutch transgender women. Reactions Weekly, 1754, 9–9. [DOI:10.1007/s40278-019-62242-2]
Sasco, A. J., Lowenfels, A. B., & Jong, P. P. (1993). Review article: Epidemiology of male breast cancer. A meta-analysis of published case-control studies and discussion of selected aetiological factors. International Journal of Cancer, 53(4), 538–549. [DOI:10.1002/ijc.2910530403]
Satram, S. (2006). Genetic and environmental factors and male breast cancer risk. (Doctoral dissertation, University of California, Irvine.) [Google Scholar] [URL]
Silverberg, M. J., Nash, R., Becerra-Culqui, T. A., Cromwell, L., Getahun, D., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Slovis, J., Tangpricha, V., & Goodman, M. (2017). Cohort study of cancer risk among insured transgender people. Annals of Epidemiology, 27(8), 499–501. [DOI:10.1016/j.annepidem.2017.07.007]
Stoll, B. A. (1973). Hypothesis: Breast Cancer Regression under Oestrogen Therapy. BMJ, 3(5877), 446–450. [DOI:10.1136/bmj.3.5877.446]
Stute, P., Wildt, L., & Neulen, J. (2018). The impact of micronized progesterone on breast cancer risk: a systematic review. Climacteric, 21(2), 111–122. [DOI:10.1080/13697137.2017.1421925]
Sun, Y., Zhao, Z., Yang, Z., Xu, F., Lu, H., Zhu, Z., Shi, W., Jiang, J., Yao, P., & Zhu, H. (2017). Risk Factors and Preventions of Breast Cancer. International Journal of Biological Sciences, 13(11), 1387–1397. [DOI:10.7150/ijbs.21635]
Sutherland, N., Espinel, W., Grotzke, M., & Colonna, S. (2020). Unanswered Questions: Hereditary breast and gynecological cancer risk assessment in transgender adolescents and young adults. Journal of Genetic Counseling, 29(4), 625–633. [DOI:10.1002/jgc4.1278]
T’Sjoen, G., Arcelus, J., Gooren, L., Klink, D. T., & Tangpricha, V. (2018). Endocrinology of Transgender Medicine. Endocrine Reviews, 40(1), 97–117. [DOI:10.1210/er.2018-00011]
Thellenberg, C., Malmer, B., Tavelin, B., & Grönberg, H. (2003). Second Primary Cancers in Men With Prostate Cancer: An Increased Risk of Male Breast Cancer. Journal of Urology, 169(4), 1345–1348. [DOI:10.1097/01.ju.0000056706.88960.7c]
Tiefenbacher, K., & Daxenbichler, G. (2008). The Role of Androgens in Normal and Malignant Breast Tissue. Breast Care, 3(5), 325–331. [DOI:10.1159/000158055]
Van Kesteren, P. J., Asscheman, H., Megens, J. A., & Gooren, L. J. (1997). Mortality and morbidity in transsexual subjects treated with cross-sex hormones. Clinical Endocrinology, 47(3), 337–343. [DOI:10.1046/j.1365-2265.1997.2601068.x]
Wierckx, K., Mueller, S., Weyers, S., Van Caenegem, E., Roef, G., Heylens, G., & T’Sjoen, G. (2012). Long‐Term Evaluation of Cross‐Sex Hormone Treatment in Transsexual Persons. The Journal of Sexual Medicine, 9(10), 2641–2651. [DOI:10.1111/j.1743-6109.2012.02876.x]
Wren, B. G., & Eden, J. A. (1996). Do Progestogens Reduce The Risk of Breast Cancer? A Review of the Evidence. Menopause, 3(1), 4–12. [DOI:10.1097/00042192-199603010-00003]
Writing Group for the Women’s Health Initiative Investigators. (2002). Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women: Principal Results From the Women’s Health Initiative Randomized Controlled Trial. JAMA, 288(3), 321–333. [DOI:10.1001/jama.288.3.321]
Zhu, H., Lei, X., Feng, J., & Wang, Y. (2012). Oral contraceptive use and risk of breast cancer: A meta-analysis of prospective cohort studies. The European Journal of Contraception & Reproductive Health Care, 17(6), 402–414. [DOI:10.3109/13625187.2012.715357]
\ No newline at end of file
+Breast Cancer Risk with Hormone Therapy in Transfeminine People - Transfeminine Science
Breast Cancer Risk with Hormone Therapy in Transfeminine People
By Aly | First published April 25, 2020 | Last modified May 8, 2023
Abstract / TL;DR
Estrogens and progestogens induce breast development and this may also increase the risk of breast cancer. Breast cancer risk increases exponentially with age in both women and men and is far greater in women than in men. Estrogens and progestogens have been linked to risk of breast cancer in numerous contexts, for instance ovarian activity, menopausal hormone therapy, antiestrogen therapy, and high-dose estrogen therapy, among others. Only a handful of studies have assessed the influence of estrogens and/or progestogens on breast cancer risk in transfeminine people. The available data are limited and have methodological limitations, but the best available evidence suggests that estrogen and progestogen therapy strongly increases the risk of breast cancer in transfeminine people, with the risk lying somewhere intermediate between that of cisgender women and cisgender men. More research is necessary to better-characterize the risk of breast cancer with hormone therapy in transfeminine people, particularly with longer follow-ups and different hormonal regimens (e.g., estrogen alone versus estrogen plus progestogen). Factors likely to modify breast cancer risk in transfeminine people include duration of hormone therapy, age at start of hormone therapy, concomitant long-term progestogen use, and dosages of estrogens and/or progestogens. Having a single X chromosome (as in transfeminine people and cisgender men) instead of having two (as in cisgender women) may be partially protective against breast cancer. Although the risk of breast cancer is likely increased in transfeminine people, the lifetime incidence is likely low, many years of hormonal exposure are likely required for the risk to accumulate, occurrence is usually in older age, and breast cancer is highly treatable. As such, concerns about breast cancer risk do not preclude hormone therapy for transfeminine people. In any case, breast cancer screening may be advisable in transfeminine people as with cisgender women.
Introduction
Estrogens and progestogens induce breast development in humans. This includes major roles for estrogens in pubertal breast development and progestogens in the breast changes that occur during pregnancy in preparation for lactation and breastfeeding. Breast cancer is far more common in cisgender women than in cisgender men, and hormones are well-known to be involved in the development and progression of breast cancer in cisgender women. For these reasons, it’s logical to assume that hormone therapy in transfeminine people might increase the risk of breast cancer. This is a review of this notion and of the available studies on the subject.
Sex and Age as Breast Cancer Risk Factors
The normal lifetime risk of breast cancer in women is about 1 in 8 or 12.5% (Ban & Godellas, 2014), whereas the normal lifetime risk of breast cancer in men is about 1 in 1,000 or 0.1% (Abdelwahab Yousef, 2017). The overall age-standardized risk of breast cancer in men is about 1 in 100,000 person–years (PY) (Abdelwahab Yousef, 2017; Ottini & Capalbo, 2017). In other words, 1 in 100,000 men are diagnosed with breast cancer every year on average. Fewer than 1% of all cases of breast cancer are in men (Abdelwahab Yousef, 2017; Sun et al., 2017). Thus, breast cancer is frequent in women but is extremely rare in men.
After sex, age is the strongest known risk factor for breast cancer (Momenimovahed & Salehiniya, 2018). Breast cancer risk increases exponentially with age in premenopausal women (Dix & Cohen, 1982). The risk of breast cancer in women is about 1 in 600,000 at age 15; 1 in 75,000 at age 20; 1 in 1,500 at age 30; and 1 in 175 at age 40 (Anders et al., 2009). After menopause, which typically occurs at 50 years of age, breast cancer risk continues to increase but more slowly (Dix & Cohen, 1982; Benz, 2008; Colditz & Rosner, 2000). This is assumed to be due to the much lower levels of estradiol and progesterone in postmenopause (Dix & Cohen, 1982). It has been estimated that if it were not for menopause, there would be 6 times as many cases of breast cancer in women, and hence presumably that a majority of women would develop breast cancer in their lifetimes (Brisken, Hess, & Jeitziner, 2015). Similarly to the case in women, breast cancer risk in men increases exponentially up to about age 65, and thereafter increases somewhat more slowly (Ottini & Capalbo, 2017; Cancer Research UK). This may be related to andropause in older age and consequent lower levels of testosterone and estradiol (Satram, 2006). The typical age of diagnosis of breast cancer is around 60 years in women and 65–70 years in men (Ferzoco & Ruddy, 2016; Giordano, 2018). Figure 1 below shows the risk of breast cancer with age in women and men (Ottini & Capalbo, 2017).
Figure 1: Age-specific incidence rates per 100,000 for male and female invasive breast cancer in White Americans from the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute (NCI) (2003–2012) (Ottini & Capalbo, 2017). Note that the male breast cancer and female breast cancer lines use different y-axes and corresponding scales (50× larger y-axis interval in the case of female breast cancer).
Hormone Exposure in Women and Breast Cancer Risk
Along with sex and age, research strongly implicates ovarian activity and ovarian hormones—like estradiol and progesterone—as major risk factors for breast cancer (Ban & Godellas, 2014). Later menarche, earlier menopause, and breastfeeding—all of which result in reduced lifetime ovarian hormone exposure—are associated with decreased risk of breast cancer (Ban & Godellas, 2014; Momenimovahed & Salehiniya, 2018). Surgical removal of the ovaries before 40 years of age is associated with a 50% decrease in lifetime risk of breast cancer (Ban & Godellas, 2014). Antiestrogens including selective estrogen receptor modulators (SERMs) like tamoxifen and aromatase inhibitors (AIs) like anastrozole can be used prophylactically long-term as chemoprevention against breast cancer in high-risk postmenopausal women, providing 50 to 70% reduction in breast cancer risk (Mocellin, Goodwin, & Pasquali, 2019; Nelson et al., 2019). Long-term estrogen and progestogen treatment has been found to produce breast cancer in non-human primates, and high doses may result in breast cancer developing at an earlier age than usual (Cline, 2007).
In accordance with the well-established relationships of ovarian activity and ovarian hormones with breast cancer risk, menopausal hormone therapy is associated with a higher risk of breast cancer in peri- and postmenopausal women (CGHFBC, 2019; Table). This is true both for therapy with an estrogen alone and to a greater extent with the combination of an estrogen and a progestogen (CGHFBC, 2019). In terms of absolute risk of breast cancer with menopausal hormone therapy, the incidence after 20 years of use is about 1.5-fold higher with an estrogen alone and about 2.5-fold higher with an estrogen plus a progestogen per a large recent meta-analysis of all of the available data (CGHFBC, 2019). Hence, the risk of breast cancer is higher with combined estrogen and progestogen therapy than with an estrogen alone. In the past it was thought that progestogens might be protective against breast cancer due to antiestrogenic effects in the breasts (Mauvais-Jarvis, Kuttenn, & Gompel, 1986; Wren & Eden, 1996; Gompel et al., 2002), but this proved not to be the case. The higher risk of breast cancer with menopausal hormone therapy was found to be causal in the Women’s Health Initiative (WHI), at least with the used regimen (specifically conjugated equine estrogens (CEEs) 0.625 mg/day plus medroxyprogesterone acetate (MPA) 2.5 mg/day) (about 1.25-fold higher risk after 5 years of therapy) (WGWHII, 2002; Chlebowski, Aragaki, & Anderson, 2015). The WHI was the largest randomized controlled trial (RCT) of hormone therapy ever conducted and allowed a rare chance to evaluate causation in terms of long-term health effects of hormonal agents.
For a visualization of breast cancer risk with menopausal hormone therapy, Figure 2 below shows breast cancer risk over time based on modeling and data from the observational Nurses’ Health Study (NHS) (Colditz & Rozner, 2000). As can be seen, menopausal hormone therapy is associated with maintenance of the age-related exponential rate of increase in breast-cancer risk that is normally blunted by the onset of menopause.
Bioidentical hormones like estradiol and progesterone are associated with increased risk of breast cancer similarly to non-bioidentical hormonal agents like CEEs and MPA (CGHFBC, 2019; Table). The risk of breast cancer with estradiol does not appear to be different from that with CEEs (CGHFBC, 2019). Oral progesterone has not been associated with an increase in risk of breast cancer with less than 5 years of therapy on the basis of the available data, which is in contrast to progestins (Aly, 2018; CGHFBC, 2019; Stute, Wildt, & Neulen, 2018; Mirkin, 2018; Fournier et al., 2007). However, oral progesterone is associated with increased risk of breast cancer after more than 5 years of therapy, by which point the risk appears to be similar in magnitude to that with MPA (Aly, 2018; CGHFBC, 2019; Stute, Wildt, & Neulen, 2018; Mirkin, 2018; Fournier et al., 2007). The initial lower relative risk with oral progesterone might be related to the low levels of progesterone that oral progesterone achieves and hence its comparatively weak progestogenic strength (Aly, 2018; Kuhl & Schneider, 2013; Davey, 2018; Wiki; Graphs). As a result, a longer duration of therapy (and/or larger sample sizes) may be necessary before an increase in breast cancer risk with oral progesterone can be properly quantified (Aly, 2018; Kuhl & Schneider, 2013; Davey, 2018). Virtually no data are available on breast cancer risk with non-oral routes of progesterone at present (Aly, 2018; Stute, Wildt, & Neulen, 2018). However, non-oral and fully potent progesterone is anticipated by some researchers to have a similar risk of breast cancer as progestins not only with long-term use but also with short-term exposure (Aly, 2018; Kuhl & Schneider, 2013; Davey, 2018).
Although the overall evidence including both prospective observational studies and RCTs shows an increased risk of breast cancer with estrogen alone therapy in menopausal women (CGHFBC, 2019), the available RCTs, namely the WHI, have so far been unable to show increased breast cancer risk with estrogen alone and have instead found a significant decrease in risk (RR 0.77, 95% CI 0.64–0.93) (CGHFBC, 2019; Table). Various hypotheses have been advanced to explain the conflicting findings. For instance, the women in the WHI, who constitute the vast majority of the RCT data, were atypical in that they were of relatively old age at start of hormone therapy (average 64 years) and were relatively overweight and obese (CGHFBC, 2019; Kuhl, 2005). Women who are overweight or obese are already at an increased risk of breast cancer and may experience no further increase with estrogen therapy (Kuhl, 2005). And women who have been deprived of estrogens for many years may experience paradoxical inhibitory effects on breast cancer risk, which is in contrast to women who start at menopause (also seen in the observational data) (Prentice, 2008; CGHFBC, 2019). This has been termed the “gap hypothesis” (Palmieri et al., 2014; Mueck & Ruan, 2011). More RCTs are needed to test these theories and determine the influence of estrogens on menopausal breast cancer risk.
Paradoxically, estrogen and/or progestogen exposure can also decrease breast cancer risk and inhibit breast cancer progression under certain circumstances. For instance, pregnancy lasting more than 34 weeks in young women (<35 years of age) increases breast cancer risk in the short-term (<10 years), but is actually protective against breast cancer in the long-term (>20–25 years), with an up to 50% reduction in lifetime risk (Nichols et al., 2019; Husby et al., 2018). Extremely high levels of estrogens and progesterone occur during pregnancy (e.g., >12,500 pg/mL for estradiol at >34 weeks) (Graph), and animal studies suggest that these hormones are responsible for the protection (Rajkumar et al., 2003; Hilakivi-Clarke et al., 2012). High-dose estrogen therapy is an effective treatment against breast cancer in women who are at least 5 years postmenopausal, with similar effectiveness to antiestrogen therapy (Coelingh Bennink et al., 2017; Wiki). Conversely, high-dose estrogen therapy is not effective for breast cancer in premenopausal women, although it does work if massive doses are given (Coelingh Bennink et al., 2017; Wiki). However, these beneficial effects of estrogens and progestogens against breast cancer risk and progression occur under unusual circumstances (e.g., very high levels/doses and/or prolonged prior sex-hormone deficiency) and are not likely to be of relevance to transfeminine people.
Estrogen Exposure in Men and Breast Cancer Risk
In cisgender men, conditions related to lower androgen exposure and/or higher estrogen exposure are associated with increased risk of breast cancer (Ferzoco & Ruddy, 2016). These conditions include testicular problems and removal as well as liver disease (Abdelwahab Yousef, 2017). Testicular dysfunction and absence result in decreased androgen levels, while liver disease results in higher estrogen levels due to impaired metabolism of estrogens. Both types of conditions result in an increased ratio of estrogens to androgens, in turn causing greater estrogenic stimulation of the breasts. This is presumably responsible for the increased breast cancer risk. Breast conditions in men, like benign breast disease and gynecomastia, have also been associated with breast cancer risk (Sasco, Lowenfels, & Pasker-De Jong, 1993), although findings are mixed for gynecomastia (Giordano, 2005). In addition to health conditions, high-dose estrogen therapy for prostate cancer has been associated with increased breast cancer risk in cisgender men (Thellenberg et al., 2003; Karlsson et al., 2006).
Studies of Breast Cancer Risk in Transfeminine People
A companion doc for this article with a table of available studies on the risk of breast cancer with hormone therapy in transfeminine people can be found here. To date, three large cohorts and several smaller cohorts have evaluated breast cancer risk with transfeminine hormone therapy.
Vrije Universiteit University Medical Center (VUMC) Studies
Most of the studies in the table are by the Vrije Universiteit University Medical Center (VUMC) in Amsterdam, the Netherlands (Asscheman, Gooren, & Eklund, 1989; van Kesteren et al., 1997; Mueller & Gooren, 2008; Asscheman et al., 2011; Gooren et al., 2013; de Blok et al., 2018; de Blok et al., 2019). This clinic treats 95% of transgender people in the Netherlands (de Blok et al., 2019). The studies by the VUMC are all on largely the same evolving cohort of transgender women. For a couple of decades, the VUMC reported a low incidence of breast cancer that was not much higher than the rate expected in cisgender men (only 2 reported cases in 2,300 transgender women over an average duration of hormone therapy of about 20 years) (Gooren et al., 2013). The typical regimen used by the clinic was estrogen and high-dose cyproterone acetate. The studies did not did not use systematic screening for breast cancer and instead presumably relied on patient report for breast cancer diagnosis, raising the possibility of under-detection of cases (Feldman et al., 2016).
In 2019, the VUMC did another follow-up study but this time switched to a new method of obtaining breast cancer diagnoses (de Blok et al., 2019). Instead of simply asking patients whether they’d been diagnosed with breast cancer, the researchers retrieved breast cancer diagnoses using an electronic system of patient records for all of the Netherlands called the Nationwide Network and Registry of Histopathology and Cytopathology in the Netherlands (PALGA). When they did this, their number of breast cancer cases shot up from 2 to 15. This resulted in a relative risk of breast cancer of almost 50-fold the expected risk and an absolute incidence of roughly 0.6% over a mean duration of hormone therapy of about 15 years. These findings supersede all of the previous research by the clinic and indicate that the true number of breast cancer cases in the VUMC cohort had been greatly underestimated in their previous studies. Very large increases in risk, such as the risk of lung cancer with smoking and the nearly 50-fold increase in breast cancer risk in this study, can be considered as likely causal. Accordingly, the increase in risk has been described by researchers using causal language (de Blok et al., 2019; Reactions Weekly, 2019). Prior to this study, it was thought that the increase in risk of breast cancer with hormone therapy in transfeminine people was low, but due to the new data, it’s now known that this isn’t necessarily the case (de Blok, Dreijerink, & Heijer, 2019).
Veterans Health Administration (VHA) Study
Another large cohort is the Veterans Health Administration (VHA) cohort in the United States (Brown & Jones, 2015; Brown, 2015). In 2015, a study was published using data from this cohort. The researchers reported 3 cases of breast cancer in about 3,500 people assigned male at birth with mean follow-up time of approximately 10 years. Hence, there was a rate of about 0.09% over an average duration of 10 years. Although the mean duration of follow up was shorter than in the VUMC cohort, the total follow-up time was similar. The VHA researchers reported the incidence of breast cancer as increased by 33-fold relative to the expected rate. This is somewhat strange in that it’s discrepant with the VUMC’s 2018 numbers (which reported a 46-fold increased risk based on diagnosis in 15 of 2,300 people and mean follow-up time of 15 years). In any case, the VHA concluded that the risk of breast cancer in transfeminine people was higher than that in cisgender men and higher than that reported by the earlier VUMC studies, but still lower than the risk in cisgender women.
The VHA paper provided no specifics on what hormone therapy medications and doses were used, although since it included all patients treated by providers in the VHA system, the regimens are likely to have been quite variable. Because this was the United States, the typical regimen was probably estrogen plus spironolactone and no progestogen.
There are a variety of problems with the VHA study that require discussion:
As with the earlier VUMC studies, the VHA study did not use systematic screening for breast cancer and presumably relied on patient report for breast cancer diagnoses, raising the possibility of under-detection of cases (Feldman et al., 2016).
The study was irrespective of hormone therapy (Feldman et al., 2016). Only some people in the cohort were on hormone therapy while others were not, and the researchers didn’t have full numbers on who was and who wasn’t. Moreover, an unknown percentage of people included in the cohort weren’t actually transgender—the researchers also included all cisgender men in the VHA system with a “transgender-related” diagnosis, which included transvestic fetishism (a.k.a. sexual crossdressing). Most of these individuals presumably were not on hormone therapy.
The follow-up times for those who were definitely on hormone therapy were very short. Of about 1,400 transfeminine people in the VHA cohort who were certainly on hormone therapy, the mean duration of use was only 5.5 years, and was less than 3 years for nearly half of the sample.
The VHA paper was very confusing and unclear. As an example, they misgendered transgender people, referring to them as “male” and “female” per birth sex, and wrote in such a way that made it difficult to understand who was who as well as who was and was not actually on hormone therapy. This was such that even some citing publications reporting on the study mistakenly got the risks for “male” and “female” switched (e.g., Dente et al., 2019).
To add further confusion, the VHA researchers reported 33 total cases of breast cancer in the exact same cohort of 5,100 total people assigned male or female at birth in two other papers published around the same time (Brown & Jones, 2014; Brown & Jones, 2016). For comparison, they only reported 10 total cases in the 2015 paper (3 in those assigned male at birth and 7 in people assigned female at birth). The reason for this discrepancy is unknown (Braun et al., 2017).
Due to the various problems with the VHA study, its findings are of limited usefulness and should likely be considered with caution. The 2019 VUMC findings are of far better-quality data.
Kaiser Permanente in California and Georgia Study
The third and final large cohort study was by Kaiser Permanente in Northern California, Southern California, and Georgia in the United States (Silverberg et al., 2017). They reported the incidences of a variety of cancers in a combined cohort from three Kaiser sites consisting of about 2,800 transfeminine people. The mean duration of follow up in the study was only 4 years. The researchers did not report the incidence of any cancer that had an absolute occurrence of less than 5 cases, and breast cancer in transfeminine people was not included in the relevant table of results nor in the discussion in the paper. Hence, there were presumably fewer than 5 cases of breast cancer in the cohort of transfeminine people. However, T’Sjoen and colleagues, a very reputable group of researchers in the field of transgender medical research, reported in a citing publication that the Kaiser team found a higher risk of breast cancer in transfeminine people compared to cisgender men but a lower risk compared to cisgender women (T’Sjoen et al., 2019). Assuming this is accurate, which it likely is, the information was presumably obtained via personal communication with the Kaiser researchers. Limitations of the Kaiser study include lack of exact figures on breast cancer risk and a very short follow-up duration. In addition, cancer diagnoses were retrieved only from Kaiser’s own systems, and the extent to which screening was systematic, if at all, is unclear.
The Kaiser researchers plan to do further studies in much larger cohorts via expansion to additional sites in the future (Silverberg et al., 2017). This is an exciting development, and should hopefully give us additional data on health risks in transgender people, including of breast cancer risk.
Small Cohort Studies
The remaining investigations of breast cancer risk in transfeminine people were small. One was 50 post-SRS transgender women by the Ghent University Hospital in Belgium who were followed up for an average of 11.4 years each, in whom no cases of breast cancer were seen (Wierckx et al., 2012). Another was 60 transgender women by the University Hospital Erlangen in Germany (Dittrich et al., 2005). No cases were seen in this cohort either, although the treatment period was only 2 years. The last was Harry Benjamin’s patient cohort; he communicated in his 1960s publications that he had treated about 150 transgender women with “medium to fairly large doses of estrogen” for varying periods of time (e.g., 3 months to 12 years) and hadn’t encountered any cases of breast cancer (Benjamin, 1964; Benjamin, 1966; Gooren et al., 2013). These cohorts are all too small and limited to provide meaningful quantification of breast cancer risk in transfeminine people.
Discussion of the Available Studies
Inconclusive Quantification of Breast Cancer Risk Due to Short Follow-Up Times
The available studies on breast cancer risk in transfeminine people are inconclusive due to inadequate follow-up times (de Blok, Dreijerink, & Heijer, 2019; Mueller & Gooren, 2008; Gooren, 2011; Gooren et al., 2013; Brown & Jones, 2015). For instance the average durations of hormone therapy in the VUMC and VHA studies were only 10 to 20 years. Breast cancer incidence in cisgender women increases exponentially over decades during premenopause and is very rare until older age. The typical age of breast cancer diagnosis in cisgender women is 60 years, which includes about 50 years of premenopausal hormone exposure and about 10 years of postmenopausal hormone exposure. (The latter of which is not unimportant, as demonstrated by the greatly decreased risk of breast cancer with prophylactic antiestrogen therapy.) As such, due to the short follow-up times of the available studies of transfeminine hormone therapy and breast cancer risk, the true or lifetime risk of breast cancer in transfeminine people is not yet known (Mueller & Gooren, 2008).
We do know however that hormone therapy in transfeminine people, at least with an estrogen plus a progestogen in the form of cyproterone acetate, appears to strongly increase the risk of breast cancer within about 15 years of use (de Blok, Dreijerink, & Heijer, 2019). Fortunately, the risk is intermediate between that in cisgender women and that in cisgender men (and not, e.g., greater than that in cisgender women). At the same time however, the risk is non-negligible, and the absolute incidence will only increase with longer follow-up times. In terms of lifetime risk, the incidence of breast cancer in transfeminine people is well on-track to be as high as single-digit percentages, at least based on the findings of de Blok et al. (2019) and their particular hormone therapy regimen.
Breast Cancer Risk in Relation to Lifetime Hormone Exposure
Transfeminine people may have a lower risk of breast cancer than cisgender women. This may due at least in part to a more limited amount of lifetime hormone exposure (Mueller & Gooren, 2008). This is based on the fact that, at least historically, transfeminine people have started hormone therapy on average at 30 or 40 years of age, which is decades after the age at which cisgender girls normally undergo puberty (Mueller & Gooren, 2008). Additionally, youth may represent a critical window of susceptibility for breast cancer risk (Biro & Wolff, 2011; Biro & Deardorff, 2013; Biro et al., 2020). However, the age at start of hormone therapy in transfeminine people has been decreasing in recent times (Mueller & Gooren, 2008), and nowadays many start hormones in their teens or early twenties. These individuals will have greater lifetime hormone exposure, and presumably greater breast cancer risk, than many of the transfeminine people of the past (Sutherland et al., 2020). Moreover, transfeminine people may be much less inclined to stop hormone therapy at the normal age of menopause in cisgender women (Mueller & Gooren, 2008). Many transfeminine people will likely stay on hormone therapy their entire lives. This additional exposure may further increase breast cancer risk (Mueller & Gooren, 2008).
Breast Cancer Risk in Relation to Progestogens and Dosage
Based on findings that breast cancer risk is higher with an estrogen plus progestogen relative to an estrogen alone in menopausal women, the risk of breast cancer with hormone therapy in transfeminine people may likewise be higher with combined estrogen and progestogen therapy (de Blok, Dreijerink, & Heijer, 2019). Hence, it’s possible that estrogen therapy without a progestogen could have a lower risk of breast cancer than that observed by the VUMC with an estrogen plus cyproterone acetate (de Blok, Dreijerink, & Heijer, 2019). It’s notable also that the doses of cyproterone acetate used by the VUMC result in rather extreme progestogenic exposure (Aly, 2019). The extent to which breast cancer risk with progestogens is dose-dependent is unknown. Whether or not estrogen dosage influences breast cancer risk, or if higher levels have greater risk, is unclear similarly. In any case, the fact that the use of estrogen–progestogen birth control in premenopausal women is associated with increased risk of breast cancer is suggestive that higher levels of estrogen and/or progestogen exposure may result in some degree of greater risk (Kahlenborn et al., 2006; Zhu et al., 2012; Ji et al., 2019). In addition, an older observational study reported a 4-fold greater risk of breast cancer in menopausal cisgender women treated with injectable estrogens than with oral estrogens or no hormone therapy (Hulka et al., 1982; Coe & Parks, 1989). This is notable as typical doses of injectable estrogens provide much greater estrogenic exposure than oral and other routes of estrogen (Göretzlehner et al., 2002; Aly, 2021). However, the study has important limitations, including only being a single study and being quite old.
X Chromosomes and Breast Cancer Risk
The sex chromosomes include the X chromosome and the Y chromosome. Cisgender women typically have two X chromosomes (46,XX karyotype), while cisgender men (and transfeminine people) typically have one X chromosome and one Y chromosome (46,XY karyotype). It’s possible that a 46,XX karyotype—specifically the presence of a second X chromosome—may be a major risk factor for breast cancer. X-chromosome gain and aberrant X-chromosome inactivation have been associated with breast cancer incidence and aggressiveness (Nakopoulou et al., 2007; Di Oto et al., 2015; Lin et al., 2015; Chaligné et al., 2015; Di Oto et al., 2018). In addition, breast cancer risk is markedly higher in men with Klinefelter’s syndrome (KS), who have a 46,XXY karyotype, than in regular men with a 46,XY karyotype. Conversely, breast cancer has never been reported in women with complete androgen insensitivity syndrome (CAIS), who have a 46,XY karyotype similarly to transfeminine people (Hughes et al., 2012; Tiefenbacher & Daxenbichler, 2008; Hughes, 2009). This is in spite of the fact that CAIS women have excellent breast development (Aly, 2020). However, hormonal abnormalities in people with KS and CAIS may alternatively contribute to differences in breast cancer risk and are thus a confounding variable. It’s notable in particular that CAIS women have relatively low estrogen exposure and little to no progesterone. In any case, the differences in breast cancer risk in conditions like KS and CAIS do not seem to be fully explainable by hormonal abnormalities. Thus, it’s possible that the lack of two X chromosomes in transfeminine people may indeed prove to be partially protective against breast cancer risk. For more in-depth information on this topic, see the supplemental article here.
Summary and Conclusions
Breast cancer risk is far higher in women than in men. The incidence of breast cancer increases exponentially with age and is very rare in younger people. There is strong basis to assume that estrogens and progestogens increase the risk of breast cancer, which can be inferred as a major mechanism for the sex differences in breast cancer risk. The available data do not indicate meaningful differences between bioidentical and non-bioidentical hormones in terms of breast cancer risk.
Older studies of transfeminine hormone therapy and breast cancer risk found low incidences of breast cancer in transfeminine people. However, a recent study with better methodology than previous studies showed a nearly 50-fold increase in breast cancer risk. The hormone therapy regimen used by their cohort was specifically estrogen plus cyproterone acetate. It’s unknown whether other hormonal regimens, for instance estrogen alone or with a non-progestogenic antiandrogen, have similar risks. The available data on breast cancer risk in transfeminine people are limited by inadequate follow-up times. As a result, lifetime breast cancer risk is unknown. Based on available data however, we can project into the future and assume that the lifetime incidence of breast cancer in transfeminine people may be as high as single-digit percentages. This puts the risk of breast cancer in transfeminine people somewhere between that of cisgender men and cisgender women.
Various factors may modify breast cancer risk with transfeminine hormone therapy. Examples include cumulative duration of use, age at start of therapy, long-term progestogen use, and possibly dosages of estrogens and/or progestogens. The lack of a second X chromosome in transfeminine people may be partially protective against breast cancer. Although breast cancer risk with transfeminine hormone therapy is something for transfeminine people to be aware of, particularly the probable contribution of progestogens to the risk, the lifetime incidence is likely to be low and the risk appears to be less than that of cisgender women. In addition, it takes many years of hormone exposure for breast cancer to occur and is generally restricted to old age. Also, breast cancer is a highly treatable disease, with excellent 5- and 10-year survival rates (Cancer.Net). For these reasons, concerns about breast cancer should most certainly not preclude hormone therapy for transfeminine people.
The possibility of breast cancer with hormone therapy in transfeminine people does, however, highlight the importance of routine breast cancer screening in transfeminine people of appropriate age and duration of hormone exposure (Chowdhry & O’Connell, 2020). It’s likely advisable that transfeminine people on long-term hormone therapy follow the same breast cancer screening procedures as those of cisgender women (Chowdhry & O’Connell, 2020).
References
Abdelwahab Yousef, A. J. (2017). Male Breast Cancer: Epidemiology and Risk Factors. Seminars in Oncology, 44(4), 267–272. [DOI:10.1053/j.seminoncol.2017.11.002]
Aly. (2018). Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects. Transfeminine Science. [URL]
Aly. (2019). Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). The Possible Role of a Second X Chromosome in Breast Cancer Risk. Transfeminine Science. [URL]
Aly. (2021). An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations. Transfeminine Science. [URL]
Anders, C. K., Johnson, R., Litton, J., Phillips, M., & Bleyer, A. (2009). Breast Cancer Before Age 40 Years. Seminars in Oncology, 36(3), 237–249. [DOI:10.1053/j.seminoncol.2009.03.001]
Asscheman, H., Gooren, L., & Eklund, P. (1989). Mortality and morbidity in transsexual patients with cross-gender hormone treatment. Metabolism, 38(9), 869–873. [DOI:10.1016/0026-0495(89)90233-3]
Asscheman, H., Giltay, E. J., Megens, J. A., de Ronde, W., van Trotsenburg, M. A., & Gooren, L. J. (2011). A long-term follow-up study of mortality in transsexuals receiving treatment with cross-sex hormones. European Journal of Endocrinology, 164(4), 635–642. [DOI:10.1530/eje-10-1038]
Ban, K. A., & Godellas, C. V. (2014). Epidemiology of Breast Cancer. Surgical Oncology Clinics of North America, 23(3), 409–422. [DOI:10.1016/j.soc.2014.03.011]
Benjamin, H. (1964). Clinical Aspects of Transsexualism in the Male and Female. American Journal of Psychotherapy, 18(3), 458–469. [DOI:10.1176/appi.psychotherapy.1964.18.3.458]
Benz, C. C. (2008). Impact of aging on the biology of breast cancer. Critical Reviews in Oncology/Hematology, 66(1), 65–74. [DOI:10.1016/j.critrevonc.2007.09.001]
Biro, F. M., & Wolff, M. S. (2011). Puberty as a Window of Susceptibility. In Russo, J. (Ed.). Environment and Breast Cancer (pp. 29–41). New York: Springer New York. [DOI:10.1007/978-1-4419-9896-5_2]
Biro, F. M., & Deardorff, J. (2013). Identifying Opportunities for Cancer Prevention During Preadolescence and Adolescence: Puberty as a Window of Susceptibility. Journal of Adolescent Health, 52(5), S15–S20. [DOI:10.1016/j.jadohealth.2012.09.019]
Biro, F. M., Huang, B., Wasserman, H., Gordon, C. M., & Pinney, S. M. (2021). Pubertal Growth, IGF-1, and Windows of Susceptibility: Puberty and Future Breast Cancer Risk. Journal of Adolescent Health, 68(3), 517–522. [DOI:10.1016/j.jadohealth.2020.07.016]
Braun, H., Nash, R., Tangpricha, V., Brockman, J., Ward, K., & Goodman, M. (2017). Cancer in Transgender People: Evidence and Methodological Considerations. Epidemiologic Reviews, 39(1), 93–107. [DOI:10.1093/epirev/mxw003]
Brisken, C., Hess, K., & Jeitziner, R. (2015). Progesterone and Overlooked Endocrine Pathways in Breast Cancer Pathogenesis. Endocrinology, 156(10), 3442–3450. [DOI:10.1210/en.2015-1392]
Brown, G. R., & Jones, K. T. (2014). Incidence of breast cancer in a cohort of 5,135 transgender veterans. Breast Cancer Research and Treatment, 149(1), 191–198. [DOI:10.1007/s10549-014-3213-2]
Brown, G. R., & Jones, K. T. (2014). Racial Health Disparities in a Cohort of 5,135 Transgender Veterans. Journal of Racial and Ethnic Health Disparities, 1(4), 257–266. [DOI:10.1007/s40615-014-0032-4]
Brown, G. R. (2015). Breast Cancer in Transgender Veterans: A Ten-Case Series. LGBT Health, 2(1), 77–80. [DOI:10.1089/lgbt.2014.0123]
Brown, G. R., & Jones, K. T. (2016). Mental Health and Medical Health Disparities in 5135 Transgender Veterans Receiving Healthcare in the Veterans Health Administration: A Case–Control Study. LGBT Health, 3(2), 122–131. [DOI:10.1089/lgbt.2015.0058]
Cancer Research UK. (2020). Breast cancer incidence (invasive) statistics. Cancer Research UK. [URL]
Cancer.Net. (2020). Breast Cancer: Statistics. American Society of Clinical Oncology. [URL]
Chaligné, R., Popova, T., Mendoza-Parra, M., Saleem, M. M., Gentien, D., Ban, K., Piolot, T., Leroy, O., Mariani, O., Gronemeyer, H., Vincent-Salomon, A., Stern, M., & Heard, E. (2015). The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Research, 25(4), 488–503. [DOI:10.1101/gr.185926.114]
Chlebowski, R. T., Aragaki, A. K., & Anderson, G. L. (2015). Menopausal Hormone Therapy Influence on Breast Cancer Outcomes in the Women’s Health Initiative. Journal of the National Comprehensive Cancer Network, 13(7), 917–924. [DOI:10.6004/jnccn.2015.0106]
Chowdhry, D. N., & O’Connell, A. M. (2020). Breast Imaging in Transgender Patients. Journal of Breast Imaging, 2(2), 161–167. [DOI:10.1093/jbi/wbz092]
Cline, J. M. (2007). Assessing the mammary gland of nonhuman primates: effects of endogenous hormones and exogenous hormonal agents and growth factors. Birth Defects Research Part B: Developmental and Reproductive Toxicology, 80(2), 126–146. [DOI:10.1002/bdrb.20112]
Coe, F. L., & Parks, J. H. (1989). The Risks of Oral Contraceptives and Estrogen Replacement Therapy. Perspectives in Biology and Medicine, 33(1), 86–106. [DOI:10.1353/pbm.1990.0026]
Coelingh Bennink, H. J., Verhoeven, C., Dutman, A. E., & Thijssen, J. (2017). The use of high-dose estrogens for the treatment of breast cancer. Maturitas, 95, 11–23. [DOI:10.1016/j.maturitas.2016.10.010]
Colditz, G. A. (2000). Cumulative Risk of Breast Cancer to Age 70 Years According to Risk Factor Status: Data from the Nurses’ Health Study. American Journal of Epidemiology, 152(10), 950–964. [DOI:10.1093/aje/152.10.950]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/s0140-6736(19)31709-x]
Davey, D. A. (2018). Menopausal hormone therapy: a better and safer future. Climacteric, 21(5), 454–461. [DOI:10.1080/13697137.2018.1439915]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, R. H., & den Heijer, M. (2018). Breast cancer in transgender persons receiving gender affirming hormone treatment: results of a nationwide cohort study. Endocrine Abstracts, 56 [20th European Congress of Endocrinology 2018, 19–22 May 2018, Barcelona, Spain], 490–490 (abstract no. P955). [DOI:10.1530/endoabs.56.p955] [PDF]
de Blok, C. J., Dreijerink, K. M., & den Heijer, M. (2019). Cancer Risk in Transgender People. Endocrinology and Metabolism Clinics of North America, 48(2), 441–452. [DOI:10.1016/j.ecl.2019.02.005]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, I. R., & den Heijer, M. (2019). Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in the Netherlands. BMJ, 365, l1652. [DOI:10.1136/bmj.l1652]
Dente, E., Farneth, R., Purks, J., & Torelli, S. (2019). Evaluating risks, reported cases and screening recommendations for breast cancer in transgender patients. Georgetown Medical Review, 3(1). [DOI:10.52504/001c.7774]
Di Oto, E., Monti, V., Cucchi, M. C., Masetti, R., Varga, Z., & Foschini, M. P. (2015). X chromosome gain in male breast cancer. Human Pathology, 46(12), 1908–1912. [DOI:10.1016/j.humpath.2015.08.008]
Di Oto, E., Biserni, G. B., Varga, Z., Morandi, L., Cucchi, M. C., Masetti, R., & Foschini, M. P. (2018). X chromosome gain is related to increased androgen receptor expression in male breast cancer. Virchows Archiv, 473(2), 155–163. [DOI:10.1007/s00428-018-2377-2]
Dix, D., & Cohen, P. (1982). The incidence of breast cancer: analysis of the age dependence. Anticancer Research, 2(1-2), 23–7. [Google Scholar] [PubMed] [PDF]
Dworin, M. (1961). The Rationale of Endocrine Therapy in Breast Cancer. CA: A Cancer Journal for Clinicians, 11(2), 48–54. [DOI:10.3322/canjclin.11.2.48]
Feldman, J., Brown, G. R., Deutsch, M. B., Hembree, W., Meyer, W., Meyer-Bahlburg, H. F., Tangpricha, V., T’Sjoen, G., & Safer, J. D. (2016). Priorities for transgender medical and healthcare research. Current Opinion in Endocrinology, Diabetes & Obesity, 23(2), 180–187. [DOI:10.1097/med.0000000000000231]
Ferzoco, R. M., & Ruddy, K. J. (2016). The Epidemiology of Male Breast Cancer. Current Oncology Reports, 18, 1. [DOI:10.1007/s11912-015-0487-4]
Forrest, A. P. (1989). Endocrine management of breast cancer. Proceedings of the Royal Society of Edinburgh. Section B. Biological Sciences, 95 [Oestrogen and the Human Breast], 1–10. [DOI:10.1017/s0269727000010502]
Fournier, A., Berrino, F., & Clavel-Chapelon, F. (2007). Unequal risks for breast cancer associated with different hormone replacement therapies: results from the E3N cohort study. Breast Cancer Research and Treatment, 107(1), 103–111. [DOI:10.1007/s10549-007-9523-x]
Gellhorn, A., & Jones, L. O. (1949). Chemotherapy of malignant disease. The American Journal of Medicine, 6(2), 188–231. [DOI:10.1016/0002-9343(49)90017-0]
Giordano, S. H. (2005). A Review of the Diagnosis and Management of Male Breast Cancer. The Oncologist, 10(7), 471–479. [DOI:10.1634/theoncologist.10-7-471]
Giordano, S. H. (2018). Breast Cancer in Men. New England Journal of Medicine, 378(24), 2311–2320. [DOI:10.1056/nejmra1707939]
Gompel, A., Levy, D., Chaouat, M., Kazem, A., Hugol, D., Daraı̈, E., Decroix, Y., & Rostène, W. (2002). Is there a place for progestin agents in breast cancer prevention? International Congress Series, 1229, 143–150. [DOI:10.1016/s0531-5131(01)00467-8]
Gooren, L. J. (2011). Care of Transsexual Persons. New England Journal of Medicine, 364(13), 1251–1257. [DOI:10.1056/nejmcp1008161]
Gooren, L. J., van Trotsenburg, M. A., Giltay, E. J., & van Diest, P. J. (2013). Breast Cancer Development in Transsexual Subjects Receiving Cross-Sex Hormone Treatment. The Journal of Sexual Medicine, 10(12), 3129–3134. [DOI:10.1111/jsm.12319]
Göretzlehner, G., Ackermann, W., Angelow, K., Bergmann, G., Bieck, E., Golbs, S., & Kliem, O. (2002). Pharmakokinetik von Estron, Estradiol, FSH, LH und Prolaktin nach intramuskulärer Applikation von 5 mg Estradiolvalerat. [Pharmacokinetics of estradiol valerate in postmenopausal women after intramuscular administration.] Journal für Menopause, 9(2), 46–49. [Google Scholar] [URL] [PDF] [Translation]
Green, R. B., Sethi, R. S., & Lindner, H. H. (1964). Treatment of advanced carcinoma of the breast. The American Journal of Surgery, 108(1), 107–121. [DOI:10.1016/0002-9610(64)90094-7]
Herrmann, J. B. (1947). Effect of estrogenic hormone on advanced carcinoma of the female breast. Archives of Surgery, 54(1), 1–9. [DOI:10.1001/archsurg.1947.01230070004001]
Hilakivi-Clarke, L., de Assis, S., Warri, A., & Luoto, R. (2012). Pregnancy hormonal environment and mother’s breast cancer risk. Hormone Molecular Biology and Clinical Investigation, 9(1), 11–23. [DOI:10.1515/hmbci-2012-0019]
Hughes, I. A. (2010). Evaluation and Management of Disorders of Sex Development. In Brook, C. G. D., Clayton, P. E., & Brown, R. S. (Eds.). Brook’s Clinical Pediatric Endocrinology, 6th Edition (pp. 192–212). Oxford: Wiley-Blackwell. [DOI:10.1002/9781444316728.ch9]
Hughes, I. A., Davies, J. D., Bunch, T. I., Pasterski, V., Mastroyannopoulou, K., & MacDougall, J. (2012). Androgen insensitivity syndrome. The Lancet, 380(9851), 1419–1428. [DOI:10.1016/s0140-6736(12)60071-3]
Hulka, B. S., Chambless, L. E., Deubner, D. C., & Wilkinson, W. E. (1982). Breast cancer and estrogen replacement therapy. American Journal of Obstetrics and Gynecology, 143(6), 638–644. [DOI:10.1016/0002-9378(82)90108-9]
Husby, A., Wohlfahrt, J., Øyen, N., & Melbye, M. (2018). Pregnancy duration and breast cancer risk. Nature Communications, 9(1), 4255. [DOI:10.1038/s41467-018-06748-3]
Ji, L., Jing, C., Zhuang, S., Pan, W., & Hu, X. (2019). Effect of age at first use of oral contraceptives on breast cancer risk. Medicine, 98(36), e15719–e15719. [DOI:10.1097/md.0000000000015719]
Kahlenborn, C., Modugno, F., Potter, D. M., & Severs, W. B. (2006). Oral Contraceptive Use as a Risk Factor for Premenopausal Breast Cancer: A Meta-analysis. Mayo Clinic Proceedings, 81(10), 1290–1302. [DOI:10.4065/81.10.1290]
Karlsson, C. T., Malmer, B., Wiklund, F., & Grönberg, H. (2006). Breast Cancer as a Second Primary in Patients With Prostate Cancer—Estrogen Treatment or Association With Family History of Cancer? Journal of Urology, 176(2), 538–543. [DOI:10.1016/j.juro.2006.03.036]
Kennedy, B.J. (1956). Effects of steroids in women with breast cancer. In Engle, E. T., & Pincus, G. (Eds.). Hormones and the Aging Process: Proceedings of a Conference Held at Arden House, Harriman, New York, 1955 (pp. 253–272). New York: Academic Press. [Google Scholar] [Google Books]
Kennedy, B. J. (1957). The Present Status of Hormone Therapy in Advanced Breast Cancer. Radiology, 69(3), 330–340. [DOI:10.1148/69.3.330]
Kennedy, B. J. (1964). Estrogenic Hormone Therapy in Advanced Breast Cancer. Annals of Internal Medicine, 60(4), 718–718. [DOI:10.7326/0003-4819-60-4-718_1]
Kuhl, H. (2005). Breast cancer risk in the WHI study: The problem of obesity. Maturitas, 51(1), 83–97. [DOI:10.1016/j.maturitas.2005.02.018]
Kuhl, H., & Schneider, H. P. (2013). Progesterone – promoter or inhibitor of breast cancer. Climacteric, 16(Suppl 1), 54–68. [DOI:10.3109/13697137.2013.768806]
Lin, I., Chen, D., Chang, Y., Lee, Y., Su, C., Cheng, C., Tsai, Y., Ng, S., Chen, H., Lee, M., Chen, H., Suen, S., Chen, Y., Liu, T., Chang, C., & Hsu, M. (2015). Hierarchical Clustering of Breast Cancer Methylomes Revealed Differentially Methylated and Expressed Breast Cancer Genes. PLOS ONE, 10(2), e0118453. [DOI:10.1371/journal.pone.0118453]
Lumachi, F. (2015). Current medical treatment of estrogen receptor-positive breast cancer. World Journal of Biological Chemistry, 6(3), 231–239. [DOI:10.4331/wjbc.v6.i3.231]
Mauvais-Jarvis, P., Kuttenn, F., & Gompel, A. (1986). Antiestrogen action of progesterone in breast tissue. Breast Cancer Research and Treatment, 8(3), 179–188. [DOI:10.1007/bf01807330]
Mirkin, S. (2018). Evidence on the use of progesterone in menopausal hormone therapy. Climacteric, 21(4), 346–354. [DOI:10.1080/13697137.2018.1455657]
Mocellin, S., Goodwin, A., & Pasquali, S. (2019). Risk-reducing medications for primary breast cancer: a network meta-analysis. Cochrane Database of Systematic Reviews, 2019(4), CD012191. [DOI:10.1002/14651858.cd012191.pub2]
Momenimovahed, Z., & Salehiniya, H. (2019). Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer: Targets and Therapy, 11, 151–164. [DOI:10.2147/bctt.s176070]
Mueck, A. O., & Ruan, X. (2011). Benefits and risks during HRT: main safety issue breast cancer. Hormone Molecular Biology and Clinical Investigation, 5(2), 105–116. [DOI:10.1515/hmbci.2011.014]
Mueller, A., & Gooren, L. (2008). Hormone-related tumors in transsexuals receiving treatment with cross-sex hormones. European Journal of Endocrinology, 159(3), 197–202. [DOI:10.1530/eje-08-0289]
Nakopoulou, L., Panayotopoulou, E. G., Giannopoulou, I., Tsirmpa, I., Katsarou, S., Mylona, E., Alexandrou, P., & Keramopoulos, A. (2006). Extra copies of chromosomes 16 and X in invasive breast carcinomas are related to aggressive phenotype and poor prognosis. Journal of Clinical Pathology, 60(7), 808–815. [DOI:10.1136/jcp.2006.037838]
Nathanson, I. T. (1947). Hormonal Alteration of Advanced Cancer of the Breast. Surgical Clinics of North America, 27(5), 1144–1150. [DOI:10.1016/s0039-6109(16)32241-1]
Nathanson, I. T. (1951). Sex Hormones and Castration in Advanced Breast Cancer. Radiology, 56(4), 535–552. [DOI:10.1148/56.4.535]
Nelson, H. D., Fu, R., Zakher, B., Pappas, M., & McDonagh, M. (2019). Medication Use for the Risk Reduction of Primary Breast Cancer in Women. JAMA, 322(9), 868–886. [DOI:10.1001/jama.2019.5780]
Nichols, H. B., Schoemaker, M. J., Cai, J., Xu, J., Wright, L. B., Brook, M. N., Jones, M. E., Adami, H., Baglietto, L., Bertrand, K. A., Blot, W. J., Boutron-Ruault, M., Dorronsoro, M., Dossus, L., Eliassen, A. H., Giles, G. G., Gram, I. T., Hankinson, S. E., Hoffman-Bolton, J., Kaaks, R., Key, T. J., Kitahara, C. M., Larsson, S. C., Linet, M., Merritt, M. A., Milne, R. L., Pala, V., Palmer, J. R., Peeters, P. H., Riboli, E., Sund, M., Tamimi, R. M., Tjønneland, A., Trichopoulou, A., Ursin, G., Vatten, L., Visvanathan, K., Weiderpass, E., Wolk, A., Zheng, W., Weinberg, C. R., Swerdlow, A. J., & Sandler, D. P. (2018). Breast Cancer Risk After Recent Childbirth. Annals of Internal Medicine, 170(1), 22–31. [DOI:10.7326/m18-1323]
Ottini, L., & Capalbo, C. (2017). Male Breast Cancer. In Veronesi, U., Goldhirsch, A., Veronesi, P., Gentilini, O. D., & Leonardi, M. C. (Eds.). Breast Cancer: Innovations in Research and Management (pp. 753–762). Cham: Springer. [DOI:10.1007/978-3-319-48848-6_63]
Palmieri, C., Patten, D. K., Januszewski, A., Zucchini, G., & Howell, S. J. (2014). Breast cancer: Current and future endocrine therapies. Molecular and Cellular Endocrinology, 382(1), 695–723. [DOI:10.1016/j.mce.2013.08.001]
Pearson, O. H. (1954). Evaluation of endocrine therapy for advanced breast cancer. JAMA, 154(3), 234–239. [DOI:10.1001/jama.1954.02940370046013]
Pearson, O. H., & Lipsett, M. B. (1956). Endocrine Management of Metastatic Breast Cancer. Medical Clinics of North America, 40(3), 761–772. [DOI:10.1016/s0025-7125(16)34564-3]
Prentice, R. L. (2008). Women’s Health Initiative Studies of Postmenopausal Breast Cancer. In Li, J. J., Li, S. A., Mohla, S., Rochefort, H., & Maudelonde, T. (Eds.). Hormonal Carcinogenesis V (Advances in Experimental Medicine and Biology, Volume 617) (pp. 151–160). New York: Springer New York. [DOI:10.1007/978-0-387-69080-3_14]
Rajkumar, L., Guzman, R. C., Yang, J., Thordarson, G., Talamantes, F., & Nandi, S. (2003). Prevention of mammary carcinogenesis by short-term estrogen and progestin treatments. Breast Cancer Research, 6(1), R31. [DOI:10.1186/bcr734]
Reactions Weekly. (2019). Hormones increase breast cancer risk in Dutch transgender women. Reactions Weekly, 1754, 9–9. [DOI:10.1007/s40278-019-62242-2]
Sasco, A. J., Lowenfels, A. B., & Jong, P. P. (1993). Review article: Epidemiology of male breast cancer. A meta-analysis of published case-control studies and discussion of selected aetiological factors. International Journal of Cancer, 53(4), 538–549. [DOI:10.1002/ijc.2910530403]
Satram, S. (2006). Genetic and environmental factors and male breast cancer risk. (Doctoral dissertation, University of California, Irvine.) [Google Scholar] [URL]
Silverberg, M. J., Nash, R., Becerra-Culqui, T. A., Cromwell, L., Getahun, D., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Slovis, J., Tangpricha, V., & Goodman, M. (2017). Cohort study of cancer risk among insured transgender people. Annals of Epidemiology, 27(8), 499–501. [DOI:10.1016/j.annepidem.2017.07.007]
Stoll, B. A. (1973). Hypothesis: Breast Cancer Regression under Oestrogen Therapy. BMJ, 3(5877), 446–450. [DOI:10.1136/bmj.3.5877.446]
Stute, P., Wildt, L., & Neulen, J. (2018). The impact of micronized progesterone on breast cancer risk: a systematic review. Climacteric, 21(2), 111–122. [DOI:10.1080/13697137.2017.1421925]
Sun, Y., Zhao, Z., Yang, Z., Xu, F., Lu, H., Zhu, Z., Shi, W., Jiang, J., Yao, P., & Zhu, H. (2017). Risk Factors and Preventions of Breast Cancer. International Journal of Biological Sciences, 13(11), 1387–1397. [DOI:10.7150/ijbs.21635]
Sutherland, N., Espinel, W., Grotzke, M., & Colonna, S. (2020). Unanswered Questions: Hereditary breast and gynecological cancer risk assessment in transgender adolescents and young adults. Journal of Genetic Counseling, 29(4), 625–633. [DOI:10.1002/jgc4.1278]
T’Sjoen, G., Arcelus, J., Gooren, L., Klink, D. T., & Tangpricha, V. (2018). Endocrinology of Transgender Medicine. Endocrine Reviews, 40(1), 97–117. [DOI:10.1210/er.2018-00011]
Thellenberg, C., Malmer, B., Tavelin, B., & Grönberg, H. (2003). Second Primary Cancers in Men With Prostate Cancer: An Increased Risk of Male Breast Cancer. Journal of Urology, 169(4), 1345–1348. [DOI:10.1097/01.ju.0000056706.88960.7c]
Tiefenbacher, K., & Daxenbichler, G. (2008). The Role of Androgens in Normal and Malignant Breast Tissue. Breast Care, 3(5), 325–331. [DOI:10.1159/000158055]
Van Kesteren, P. J., Asscheman, H., Megens, J. A., & Gooren, L. J. (1997). Mortality and morbidity in transsexual subjects treated with cross-sex hormones. Clinical Endocrinology, 47(3), 337–343. [DOI:10.1046/j.1365-2265.1997.2601068.x]
Wierckx, K., Mueller, S., Weyers, S., Van Caenegem, E., Roef, G., Heylens, G., & T’Sjoen, G. (2012). Long‐Term Evaluation of Cross‐Sex Hormone Treatment in Transsexual Persons. The Journal of Sexual Medicine, 9(10), 2641–2651. [DOI:10.1111/j.1743-6109.2012.02876.x]
Wren, B. G., & Eden, J. A. (1996). Do Progestogens Reduce The Risk of Breast Cancer? A Review of the Evidence. Menopause, 3(1), 4–12. [DOI:10.1097/00042192-199603010-00003]
Writing Group for the Women’s Health Initiative Investigators. (2002). Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women: Principal Results From the Women’s Health Initiative Randomized Controlled Trial. JAMA, 288(3), 321–333. [DOI:10.1001/jama.288.3.321]
Zhu, H., Lei, X., Feng, J., & Wang, Y. (2012). Oral contraceptive use and risk of breast cancer: A meta-analysis of prospective cohort studies. The European Journal of Contraception & Reproductive Health Care, 17(6), 402–414. [DOI:10.3109/13625187.2012.715357]
\ No newline at end of file
diff --git a/transfemscience.org/articles/buserelin-inexpensive/index.html b/transfemscience.org/articles/buserelin-inexpensive/index.html
index 0b07df8a..c3d70ef3 100644
--- a/transfemscience.org/articles/buserelin-inexpensive/index.html
+++ b/transfemscience.org/articles/buserelin-inexpensive/index.html
@@ -1 +1 @@
-Buserelin, a Gonadotropin-Releasing Hormone Agonist, is Inexpensively Available as a Nasal Spray From Online Pharmacies - Transfeminine Science
Buserelin, a Gonadotropin-Releasing Hormone Agonist, is Inexpensively Available as a Nasal Spray From Online Pharmacies
By Aly | First published August 26, 2018 | Last modified March 1, 2023
Abstract / TL;DR
Buserelin is a gonadotropin-releasing hormone (GnRH) agonist which is formulated as a nasal spray for use two or three times per day. GnRH agonists are the ideal antiandrogens for use in transfeminine hormone therapy. They can completely suppress gonadal testosterone production and have essentially no side effects or risks (when used in combination with estrogen therapy to avoid sex-hormone deficiency). This is in contrast to more commonly used antiandrogenic approaches in transfeminine hormone therapy like spironolactone, cyproterone acetate, and bicalutamide, as well as high-dose estradiol monotherapy, which all have efficacy, tolerability, and/or safety issues depending on the option in question. However, GnRH agonists have historically been extremely costly and consequently inaccessible for most transfeminine people. Recently, certain Eastern European online pharmacies have started selling buserelin nasal spray very inexpensively. Because of this, GnRH agonists, for the first time, appear to be an affordable antiandrogen option for any transfeminine person who desires them.
Introduction
Buserelin, also known by its major brand name Suprefact, is a gonadotropin-releasing hormone (GnRH) agonist. GnRH agonists are powerful functional antiandrogens which are able to reduce testosterone levels in people with testes to the female or castrate range (<50 ng/dL). On account of their effectiveness and selectivity, GnRH agonists—as well as GnRH antagonists—may be considered the ideal antiandrogens for use in transfeminine hormone therapy.
Buserelin is one of two GnRH agonists—the other being nafarelin (Synarel)—that are available in the form of nasal sprays. Both are used three times per day at roughly 8-hour intervals. Nasal spray is an alternative means of administration for GnRH agonists relative to the more common (and invasive) routes of injection and implant. The use of GnRH agonists and antagonists in transfeminine people has traditionally been limited by their high cost, which is typically on the order of US$10,000 per year (Wiki).
Recently, buserelin has become available very inexpensively from a number of Eastern European online pharmacies. The first such pharmacy was known as Cherry Pharmacy (Reddit; Reddit; Reddit; Reddit; Reddit). They sold buserelin nasal spray here (180 × 150-μg doses for US$29.60) and here (140 × 150-μg doses for US$28.50). Buserelin stopped being purchasable from Cherry Pharmacy by February 2019 and the site is now defunct (Reddit; Reddit; Reddit; Reddit; Reddit; Reddit). But other online pharmacies have since begun selling inexpensive buserelin nasal spray as well and as of February 2020 the product is once again available via these sources (Reddit). A site called diyhrt.cafe maintains a list of online pharmacies that sell buserelin and other GnRH agonists.
Dosage and Testosterone Suppression
Dosage information for buserelin nasal spray is as follows (Wiki; see page for sources):
For prostate cancer, the dosage of buserelin by subcutaneous injection is 500 µg three times per day (once every 8 hours, 1,500 µg/day total) for one week and then 200 µg once daily thereafter. If buserelin is used as a nasal spray, the dosage for prostate cancer is 800 µg sprayed into the nostrils three times per day (once every 8 hours, 2,400 µg/day total) for one week followed by 400 µg sprayed into the nostrils three times per day (once every 8 hours, 1,200 µg/day total) thereafter. For endometriosis, buserelin is used specifically as a nasal spray and the dosage is the same as that used for prostate cancer. These dosages of buserelin for both subcutaneous injection and nasal spray have been found to decrease testosterone levels to castrate or near-castrate levels in men with prostate cancer, although suppression was more complete with subcutaneous injection presumably due to suboptimal absorption with intranasal administration.
And suppression of testosterone levels with different buserelin nasal spray dosing regimens is as follows (Wiki; see page for sources):
Buserelin has been found to suppress testosterone levels in men with prostate cancer from 426 ng/dL to 28 ng/dL (by 93.4%) with 200 μg by subcutaneous injection once per day and from 521 ng/dL to 53 ng/dL (by 89.8%) with 400 μg by nasal spray once every 8 hours (1,200 μg/day total). The difference in suppression may have been due to poor compliance [with the nasal spray formulation]. A few small studies have also assessed the suppression of testosterone levels with buserelin nasal spray twice a day instead of three times a day. One such study found that testosterone levels in men with prostate cancer were suppressed during treatment with buserelin from 332 ng/dL to 215 ng/dL (28.9% lower than controls) with 200 μg by nasal spray twice a day (400 μg/day total), from 840 ng/dL to 182 ng/dL (71.4% lower than controls) with 500 μg by nasal spray twice a day (1,000 μg/day total), and from 598 ng/dL to 126 ng/dL (80.4% lower than controls) with 50 μg by subcutaneous injection once a day.
Although the absorption of buserelin as a nasal spray may be suboptimal compared to injectable forms, and although buserelin nasal spray by itself may only be able to achieve near-castrate levels of testosterone (rather than the low castrate levels more typical of castration/GnRH agonists and antagonists), estrogens are also usually taken by transfeminine people and are likewise potent antigonadotropins. It may be the case that if even relatively low doses of estradiol are combined with buserelin, testosterone will be more fully suppressed (into the castrate/female range (<50 ng/dL) or by around 95%) than suggested by studies of buserelin alone. This is notably seen with progestogens, in which high doses are able to maximally suppress testosterone levels by 50 to 70% (to around 200–300 ng/dL on average) but combination with low doses of estrogens are able to suppress testosterone levels by around 95% or into the low castrate range (to around 20–30 ng/dL on average) (Aly, 2019). Similar influences on testosterone levels might be expected with the combination of buserelin and an estrogen.
If for whatever reason an estrogen is not desired (e.g., if the purpose is simple puberty blockade or non-conventional hormone therapy for non-binary transfeminine people; Aly, 2019), buserelin could alternatively be combined with low doses of a progestogen to more fully suppress testosterone levels. Or, as another option, buserelin could be combined with spironolactone or low-dose bicalutamide to block the actions of testosterone that remains unsuppressed.
It’s possible to use buserelin twice a day instead of three times a day for greater convenience. However, this does appear to result in somewhat diminished effectiveness in terms of testosterone suppression (Faure et al., 1982; Tolis et al., 1983). In any case, if buserelin is combined with an estrogen or progestogen, due to the supplementary testosterone suppression that they are expected to provide, more complete suppression of testosterone levels may still be achieved, and hence the reduced effectiveness of twice-daily buserelin might not actually be that consequential.
It should be noted that the Cherry Pharmacy buserelin nasal sprays contain 150 μg buserelin per spray, whereas the dosage information above assumes that the nasal spray can deliver an even 400 μg in sprays total per dose. Obviously, this isn’t possible with a nasal spray that delivers 150 μg per spray. Instead, such a product could be used to provide 300 μg per dose (two sprays) or 450 μg per dose (three sprays). If buserelin is combined with an estrogen or a progestogen, which will help suppress testosterone levels, it might be optimal to opt for the former lower buserelin amount of 300 μg per dose (two sprays). Conversely, if buserelin is not combined with a supplemental antigonadotropin, the latter higher buserelin amount of 450 μg per dose (three sprays) might be advisable. Alternatively, a person could administer a mix of 450 μg for the first dose, 300 μg for the second dose, and 450 μg for the third dose each day to achieve an even dosage of 1,200 μg per day (which would be equal in total dose to 400 μg three times per day and in line with the buserelin dosage information provided above).
Costs of Different Dosing Regimens
The costs of the buserelin nasal sprays from Cherry Pharmacy, dosed the recommended three times per day and not including tax or shipping and handling, break down as follows:
a One dose administered every 8 or so hours, 3 times per day in total.
The most inexpensive dosing regimen would be the US$29.60 option, whereas a good balance between cost and effectiveness might be the US$39.47 option. If a person were to do twice daily administration of buserelin, a regimen of 600 μg in the morning and 600 μg in the evening (12-hour separation), for a total dosage per day of 1,200 μg, might be optimal. This is the same total daily dosage as the US$39.47 option, and hence would be the same cost.
The preceding cost information is obsolete as Cherry Pharmacy is now defunct. However, other sources of buserelin nasal spray may have similar pricing. Hence, the breakdown may still have some relevance.
Initiation of Therapy
A higher dosage of buserelin is recommended during the first week of therapy in buserelin dosing materials. In addition, GnRH agonists cause a flare in testosterone levels during the first week of treatment, with testosterone levels returning to around baseline by the end of that week (Wiki). It takes about 2 to 4 weeks for GnRH agonists to reduce testosterone levels to the female or castrate range (<50 ng/dL) (Wiki). These considerations should be kept in mind when buserelin nasal spray as well as other GnRH agonists are used. The following passage provides more detail on the early dynamics of testosterone suppression with GnRH agonists and antagonists (Wiki; see page for sources):
There are two types of GnRH modulators: GnRH agonists and GnRH antagonists. These medications have the opposite action on the GnRH receptor but paradoxically have the same therapeutic effects. GnRH agonists, such as leuprorelin (Lupron), goserelin (Zoladex), and buserelin (Suprefact), are GnRH receptor superagonists, and work by producing profound desensitization of the GnRH receptor such that the receptor becomes non-functional. This occurs because GnRH is normally released in pulses, but GnRH agonists are continuously present. At the initiation of treatment, GnRH agonists are associated with a “flare” effect on hormone levels due to acute overstimulation of the GnRH receptor. In men, [luteinzing hormone (LH)] levels increase by up to 800% while testosterone levels increase to about 140 to 200% of baseline. Gradually however, the GnRH receptor desensitizes; testosterone levels peak after about 2 to 4 days, return to baseline after about 7 to 8 days, and are reduced to castrate levels within 2 to 4 weeks. Antigonadotropins such as estrogens and cyproterone acetate can be used to diminish or prevent the testosterone flare caused by GnRH agonists. In contrast to GnRH agonists, GnRH antagonists, such as degarelix (Firmagon) and elagolix (Orilissa), work by binding to the GnRH receptor without activating it, thereby displacing GnRH from the receptor and preventing its activation. Unlike with GnRH agonists, there is no initial surge effect with GnRH antagonists, and the therapeutic effects are immediate; sex hormone levels are reduced to castrate levels within a few days.
Discussion and Conclusions
GnRH agonists and antagonists are the ideal antiandrogens for use in transfeminine hormone therapy. They are the most effective, best-tolerated, and safest antiandrogens that are currently available. One could conceptualize them essentially as a readily reversible orchiectomy and little else. When used in combination with an estrogen, GnRH agonists and antagonists have none of the side effects and risks of more mainstream antiandrogens like spironolactone, cyproterone acetate, and bicalutamide. They also have none of the potential efficacy concerns associated with antiandrogens that act mainly as androgen receptor antagonists, such as spironolactone and bicalutamide. GnRH agonists and antagonists allow for the use of essentially any dosage of estradiol without having to worry about testosterone suppression.
Access to GnRH agonists and antagonists has been limited for transfeminine people due to the high costs of these medications and the fact that medical insurance usually denies them. As a result of the availability of inexpensive buserelin nasal spray from online pharmacies however, it would appear that GnRH agonists are now a practical and affordable option for essentially any transfeminine person who desires them. Provided one doesn’t mind the inconvenience of having to use a nasal spray multiple times per day, buserelin may be among the best available options for an antiandrogen in transfeminine people.
References
Faure, N., Labrie, F., Lemay, A., Bélanger, A., Gourdeau, Y., Laroche, B., & Robert, G. (1982). Inhibition of serum androgen levels by chronic intranasal and subcutaneous administration of a potent luteinizing hormone-releasing hormone (LH-RH) agonist in adult men. Fertility and Sterility, 37(3), 416–424. [DOI:10.1016/S0015-0282(16)46107-8]
Tolis, G., Faure, N., Koutsilieris, M., Lemay, A., Klioze, S., Yakabow, A., & Fazekas, A. T. A. (1983). Suppression of testicular steroidogenesis by the GnRH agonistic analogue Buserelin (HOE-766) in patients with prostatic cancer: studies in relation to dose and route of administration. Journal of Steroid Biochemistry, 19(1), 995–998. [DOI:10.1016/0022-4731(83)90045-6]
\ No newline at end of file
+Buserelin, a Gonadotropin-Releasing Hormone Agonist, is Inexpensively Available as a Nasal Spray From Online Pharmacies - Transfeminine Science
Buserelin, a Gonadotropin-Releasing Hormone Agonist, is Inexpensively Available as a Nasal Spray From Online Pharmacies
By Aly | First published August 26, 2018 | Last modified March 1, 2023
Abstract / TL;DR
Buserelin is a gonadotropin-releasing hormone (GnRH) agonist which is formulated as a nasal spray for use two or three times per day. GnRH agonists are the ideal antiandrogens for use in transfeminine hormone therapy. They can completely suppress gonadal testosterone production and have essentially no side effects or risks (when used in combination with estrogen therapy to avoid sex-hormone deficiency). This is in contrast to more commonly used antiandrogenic approaches in transfeminine hormone therapy like spironolactone, cyproterone acetate, and bicalutamide, as well as high-dose estradiol monotherapy, which all have efficacy, tolerability, and/or safety issues depending on the option in question. However, GnRH agonists have historically been extremely costly and consequently inaccessible for most transfeminine people. Recently, certain Eastern European online pharmacies have started selling buserelin nasal spray very inexpensively. Because of this, GnRH agonists, for the first time, appear to be an affordable antiandrogen option for any transfeminine person who desires them.
Introduction
Buserelin, also known by its major brand name Suprefact, is a gonadotropin-releasing hormone (GnRH) agonist. GnRH agonists are powerful functional antiandrogens which are able to reduce testosterone levels in people with testes to the female or castrate range (<50 ng/dL). On account of their effectiveness and selectivity, GnRH agonists—as well as GnRH antagonists—may be considered the ideal antiandrogens for use in transfeminine hormone therapy.
Buserelin is one of two GnRH agonists—the other being nafarelin (Synarel)—that are available in the form of nasal sprays. Both are used three times per day at roughly 8-hour intervals. Nasal spray is an alternative means of administration for GnRH agonists relative to the more common (and invasive) routes of injection and implant. The use of GnRH agonists and antagonists in transfeminine people has traditionally been limited by their high cost, which is typically on the order of US$10,000 per year (Wiki).
Recently, buserelin has become available very inexpensively from a number of Eastern European online pharmacies. The first such pharmacy was known as Cherry Pharmacy (Reddit; Reddit; Reddit; Reddit; Reddit). They sold buserelin nasal spray here (180 × 150-μg doses for US$29.60) and here (140 × 150-μg doses for US$28.50). Buserelin stopped being purchasable from Cherry Pharmacy by February 2019 and the site is now defunct (Reddit; Reddit; Reddit; Reddit; Reddit; Reddit). But other online pharmacies have since begun selling inexpensive buserelin nasal spray as well and as of February 2020 the product is once again available via these sources (Reddit). A site called diyhrt.cafe maintains a list of online pharmacies that sell buserelin and other GnRH agonists.
Dosage and Testosterone Suppression
Dosage information for buserelin nasal spray is as follows (Wiki; see page for sources):
For prostate cancer, the dosage of buserelin by subcutaneous injection is 500 µg three times per day (once every 8 hours, 1,500 µg/day total) for one week and then 200 µg once daily thereafter. If buserelin is used as a nasal spray, the dosage for prostate cancer is 800 µg sprayed into the nostrils three times per day (once every 8 hours, 2,400 µg/day total) for one week followed by 400 µg sprayed into the nostrils three times per day (once every 8 hours, 1,200 µg/day total) thereafter. For endometriosis, buserelin is used specifically as a nasal spray and the dosage is the same as that used for prostate cancer. These dosages of buserelin for both subcutaneous injection and nasal spray have been found to decrease testosterone levels to castrate or near-castrate levels in men with prostate cancer, although suppression was more complete with subcutaneous injection presumably due to suboptimal absorption with intranasal administration.
And suppression of testosterone levels with different buserelin nasal spray dosing regimens is as follows (Wiki; see page for sources):
Buserelin has been found to suppress testosterone levels in men with prostate cancer from 426 ng/dL to 28 ng/dL (by 93.4%) with 200 μg by subcutaneous injection once per day and from 521 ng/dL to 53 ng/dL (by 89.8%) with 400 μg by nasal spray once every 8 hours (1,200 μg/day total). The difference in suppression may have been due to poor compliance [with the nasal spray formulation]. A few small studies have also assessed the suppression of testosterone levels with buserelin nasal spray twice a day instead of three times a day. One such study found that testosterone levels in men with prostate cancer were suppressed during treatment with buserelin from 332 ng/dL to 215 ng/dL (28.9% lower than controls) with 200 μg by nasal spray twice a day (400 μg/day total), from 840 ng/dL to 182 ng/dL (71.4% lower than controls) with 500 μg by nasal spray twice a day (1,000 μg/day total), and from 598 ng/dL to 126 ng/dL (80.4% lower than controls) with 50 μg by subcutaneous injection once a day.
Although the absorption of buserelin as a nasal spray may be suboptimal compared to injectable forms, and although buserelin nasal spray by itself may only be able to achieve near-castrate levels of testosterone (rather than the low castrate levels more typical of castration/GnRH agonists and antagonists), estrogens are also usually taken by transfeminine people and are likewise potent antigonadotropins. It may be the case that if even relatively low doses of estradiol are combined with buserelin, testosterone will be more fully suppressed (into the castrate/female range (<50 ng/dL) or by around 95%) than suggested by studies of buserelin alone. This is notably seen with progestogens, in which high doses are able to maximally suppress testosterone levels by 50 to 70% (to around 200–300 ng/dL on average) but combination with low doses of estrogens are able to suppress testosterone levels by around 95% or into the low castrate range (to around 20–30 ng/dL on average) (Aly, 2019). Similar influences on testosterone levels might be expected with the combination of buserelin and an estrogen.
If for whatever reason an estrogen is not desired (e.g., if the purpose is simple puberty blockade or non-conventional hormone therapy for non-binary transfeminine people; Aly, 2019), buserelin could alternatively be combined with low doses of a progestogen to more fully suppress testosterone levels. Or, as another option, buserelin could be combined with spironolactone or low-dose bicalutamide to block the actions of testosterone that remains unsuppressed.
It’s possible to use buserelin twice a day instead of three times a day for greater convenience. However, this does appear to result in somewhat diminished effectiveness in terms of testosterone suppression (Faure et al., 1982; Tolis et al., 1983). In any case, if buserelin is combined with an estrogen or progestogen, due to the supplementary testosterone suppression that they are expected to provide, more complete suppression of testosterone levels may still be achieved, and hence the reduced effectiveness of twice-daily buserelin might not actually be that consequential.
It should be noted that the Cherry Pharmacy buserelin nasal sprays contain 150 μg buserelin per spray, whereas the dosage information above assumes that the nasal spray can deliver an even 400 μg in sprays total per dose. Obviously, this isn’t possible with a nasal spray that delivers 150 μg per spray. Instead, such a product could be used to provide 300 μg per dose (two sprays) or 450 μg per dose (three sprays). If buserelin is combined with an estrogen or a progestogen, which will help suppress testosterone levels, it might be optimal to opt for the former lower buserelin amount of 300 μg per dose (two sprays). Conversely, if buserelin is not combined with a supplemental antigonadotropin, the latter higher buserelin amount of 450 μg per dose (three sprays) might be advisable. Alternatively, a person could administer a mix of 450 μg for the first dose, 300 μg for the second dose, and 450 μg for the third dose each day to achieve an even dosage of 1,200 μg per day (which would be equal in total dose to 400 μg three times per day and in line with the buserelin dosage information provided above).
Costs of Different Dosing Regimens
The costs of the buserelin nasal sprays from Cherry Pharmacy, dosed the recommended three times per day and not including tax or shipping and handling, break down as follows:
a One dose administered every 8 or so hours, 3 times per day in total.
The most inexpensive dosing regimen would be the US$29.60 option, whereas a good balance between cost and effectiveness might be the US$39.47 option. If a person were to do twice daily administration of buserelin, a regimen of 600 μg in the morning and 600 μg in the evening (12-hour separation), for a total dosage per day of 1,200 μg, might be optimal. This is the same total daily dosage as the US$39.47 option, and hence would be the same cost.
The preceding cost information is obsolete as Cherry Pharmacy is now defunct. However, other sources of buserelin nasal spray may have similar pricing. Hence, the breakdown may still have some relevance.
Initiation of Therapy
A higher dosage of buserelin is recommended during the first week of therapy in buserelin dosing materials. In addition, GnRH agonists cause a flare in testosterone levels during the first week of treatment, with testosterone levels returning to around baseline by the end of that week (Wiki). It takes about 2 to 4 weeks for GnRH agonists to reduce testosterone levels to the female or castrate range (<50 ng/dL) (Wiki). These considerations should be kept in mind when buserelin nasal spray as well as other GnRH agonists are used. The following passage provides more detail on the early dynamics of testosterone suppression with GnRH agonists and antagonists (Wiki; see page for sources):
There are two types of GnRH modulators: GnRH agonists and GnRH antagonists. These medications have the opposite action on the GnRH receptor but paradoxically have the same therapeutic effects. GnRH agonists, such as leuprorelin (Lupron), goserelin (Zoladex), and buserelin (Suprefact), are GnRH receptor superagonists, and work by producing profound desensitization of the GnRH receptor such that the receptor becomes non-functional. This occurs because GnRH is normally released in pulses, but GnRH agonists are continuously present. At the initiation of treatment, GnRH agonists are associated with a “flare” effect on hormone levels due to acute overstimulation of the GnRH receptor. In men, [luteinzing hormone (LH)] levels increase by up to 800% while testosterone levels increase to about 140 to 200% of baseline. Gradually however, the GnRH receptor desensitizes; testosterone levels peak after about 2 to 4 days, return to baseline after about 7 to 8 days, and are reduced to castrate levels within 2 to 4 weeks. Antigonadotropins such as estrogens and cyproterone acetate can be used to diminish or prevent the testosterone flare caused by GnRH agonists. In contrast to GnRH agonists, GnRH antagonists, such as degarelix (Firmagon) and elagolix (Orilissa), work by binding to the GnRH receptor without activating it, thereby displacing GnRH from the receptor and preventing its activation. Unlike with GnRH agonists, there is no initial surge effect with GnRH antagonists, and the therapeutic effects are immediate; sex hormone levels are reduced to castrate levels within a few days.
Discussion and Conclusions
GnRH agonists and antagonists are the ideal antiandrogens for use in transfeminine hormone therapy. They are the most effective, best-tolerated, and safest antiandrogens that are currently available. One could conceptualize them essentially as a readily reversible orchiectomy and little else. When used in combination with an estrogen, GnRH agonists and antagonists have none of the side effects and risks of more mainstream antiandrogens like spironolactone, cyproterone acetate, and bicalutamide. They also have none of the potential efficacy concerns associated with antiandrogens that act mainly as androgen receptor antagonists, such as spironolactone and bicalutamide. GnRH agonists and antagonists allow for the use of essentially any dosage of estradiol without having to worry about testosterone suppression.
Access to GnRH agonists and antagonists has been limited for transfeminine people due to the high costs of these medications and the fact that medical insurance usually denies them. As a result of the availability of inexpensive buserelin nasal spray from online pharmacies however, it would appear that GnRH agonists are now a practical and affordable option for essentially any transfeminine person who desires them. Provided one doesn’t mind the inconvenience of having to use a nasal spray multiple times per day, buserelin may be among the best available options for an antiandrogen in transfeminine people.
References
Aly. (2019). An Exploration of Possibilities for Hormone Therapy in Non-Binary Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People. Transfeminine Science. [URL]
Faure, N., Labrie, F., Lemay, A., Bélanger, A., Gourdeau, Y., Laroche, B., & Robert, G. (1982). Inhibition of serum androgen levels by chronic intranasal and subcutaneous administration of a potent luteinizing hormone-releasing hormone (LH-RH) agonist in adult men. Fertility and Sterility, 37(3), 416–424. [DOI:10.1016/S0015-0282(16)46107-8]
Tolis, G., Faure, N., Koutsilieris, M., Lemay, A., Klioze, S., Yakabow, A., & Fazekas, A. T. A. (1983). Suppression of testicular steroidogenesis by the GnRH agonistic analogue Buserelin (HOE-766) in patients with prostatic cancer: studies in relation to dose and route of administration. Journal of Steroid Biochemistry, 19(1), 995–998. [DOI:10.1016/0022-4731(83)90045-6]
\ No newline at end of file
diff --git a/transfemscience.org/articles/cpa-dosage/index.html b/transfemscience.org/articles/cpa-dosage/index.html
index 0c00b738..9f69bf0c 100644
--- a/transfemscience.org/articles/cpa-dosage/index.html
+++ b/transfemscience.org/articles/cpa-dosage/index.html
@@ -1 +1 @@
-Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People - Transfeminine Science
Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People
By Aly | First published July 1, 2019 | Last modified August 23, 2025
Abstract / TL;DR
Cyproterone acetate (CPA) is a progestogen and antiandrogen which is widely used in transfeminine hormone therapy. It is far more potent as a progestogen than as an androgen receptor antagonist. CPA has typically been used at doses of 1 to 2 mg/day as a progestogen in cisgender women and at doses of 50 to 300 mg/day as an antiandrogen. At typical antiandrogen doses of CPA, there is profound progestogenic overdosage as well as associated side effects and risks. CPA has antigonadotropic effects due to its progestogenic activity and thereby suppresses testosterone levels. By itself, CPA can maximally suppress testosterone levels by 50 to 70%, and in combination with even small amounts of estrogen, it can fully suppress gonadal testosterone production and thereby reduce testosterone levels by about 95%—or well into the female range. Although doses of CPA of 50 to 100 mg/day have been used in transfeminine people historically, it is now clear that 5 to 10 mg/day CPA has maximal or near-maximal effectiveness in terms of suppression of testosterone levels. CPA alone is most commonly available as 50-mg tablets. These tablets can be split with a pill cutter and taken once every day to once every other day to achieve an overall CPA dosage of 6.25 to 12.5 mg/day. These lower doses of CPA are not only much more cost-effective than traditional doses but are also likely to have better tolerability and safety. Due to the retained effectiveness of lower CPA doses and the known dose-dependent risks of CPA, doses of CPA used clinically in transfeminine people have been in a rapid decline.
Introduction
This article is about the dosage of cyproterone acetate (CPA), a progestin and antiandrogen, for use in hormone therapy for transfeminine people. It argues for the use of lower doses of CPA and goes fairly in-depth to justify these doses. If you are only interested in recommended doses of CPA for transfeminine people, they can be found in the Recommended Dosages section below.
Potency, Conventional Dosages, and Health Risks
CPA is a potent progestogen, with an ovulation-inhibiting dosage of about 1 mg/day and endometrial transformation dosage of about 1 to 3 mg/day in cisgender women (Wiki; Table; Endrikat et al., 2011). These dosages of CPA are similar in strength of progestogenic effect to those of normal progesterone production and levels during the luteal phase of the menstrual cycle in premenopausal women (which are about 25 mg/day and 15 ng/mL, respectively). In relation to the preceding, when CPA is used as a progestogen in cisgender women, for instance in birth control pills and menopausal hormone therapy preparations, it is formulated at a dose of 1 or 2 mg per tablet (Wiki).
In contrast to its progestogenic activity, CPA is far less potent as an androgen receptor antagonist (Wiki). When used as an antiandrogen, it is generally given at a dosage of 50 to 300 mg/day, both in cisgender women and men. A dosage of 50 to 100 mg/day is typical for androgen-dependent skin and hair conditions like acne and hirsutism in women and a dosage of 100 to 300 mg/day is typically used for prostate cancer in men (specifically 100–200 mg/day for CPA combined with castration and 200–300 mg/day for CPA monotherapy) (Wiki). As such, CPA is generally formulated at a dose of 50 or 100 mg per tablet for use in androgen-dependent conditions (Wiki). As an antiandrogen, CPA has a dual mechanism of action of both suppressing testosterone levels via its progestogenic activity at low doses and additionally blocking the actions of testosterone directly at the androgen receptor at higher doses.
Because CPA is so much more potent as a progestogen than as an androgen receptor antagonist, there is profound overdosage of progestogenic effect when CPA is used as an antiandrogen at typical clinical dosages. This is described in the following three literature excerpts by Jürgen Hammerstein, one of the scientists who developed CPA (Hammerstein et al., 1975; Hammerstein, 1990; Hammerstein, 1979):
Like chlormadinone acetate, its parent compound, CPA is also a strong progestogen with the endometrial transformation dose of both drugs being between 20 and 30 mg. […] To take full therapeutic advantage of its antiandrogenicity, CPA must be administered in doses per month that are 30 times the physiological equivalent of progesterone production in the cycle. CPA, although the most useful compound available in this field at the moment, cannot be considered therefore an ideal antiandrogen, all the more as some of the side effects may be related to the progestational overdosage rather than to the administered antiandrogenic activity. […] Adverse reactions like tiredness, lassitude, and increase in body weight are possibly due to the enormous overdose of progestational activity in the formula which is necessary to take full advantage of the antiandrogenicity of CPA.
Fixson (1963) tested CPA in ovariectomized women after pre-treatment with oestrogens; with a transformation dose of 20–30 mg this proved a powerful progestogen. The potency of CPA in the menses delay test is not exactly known, but has been estimated to be below 1 mg/day (Miller and Jacobs 1986). In relation to this progestational potency, its antiandrogenicity must be considered rather weak. Thus, in order to take full advantage of the latter, 100 mg CPA must be given daily, i.e. three times the cyclic transformation dose per day (Hammerstein and Cupceancu 1969); notably, this parameter is equivalent to the total progesterone production of a corpus luteum throughout its entire cyclic life span.
CPA may be characterized endocrinologically as possessing strong progestational [and] moderate anti-androgenic […] potencies. […] Its progestational activity, in terms of the transformation dose in the oestrogen-primed human endometrium, is 20–30 mg [per month/cycle] which is comparable to that of chlormadinone acetate and other strong progestogens. To take full clinical advantage of its anti-androgenicity not less than 50–100 mg CPA must be taken orally per day, which totals 2 to 3 times the progestational activity the female organism is exposed to throughout a complete ovulatory menstrual cycle. Thus unless much lower and less efficacious doses of CPA are used, a tremendous progestational overdosage must be accepted. […] As already pointed out CPA is endocrinologically not a well-balanced compound because of the strong preponderance of the progestational over the anti-androgenic potency. A way to avoid the heavy progestogen overdosage inherent with the high-dose reverse sequential therapy would be to combine the low-dose contraceptive formulation just mentioned with a pure anti-androgen such as free cyproterone. […] It must be emphasized that CPA is far from being an ideal drug for the anti-androgenic treatment of hirsutism because its progestational potency is much too strong and it is not effective when administered topically. Therefore it is worthwhile looking for better-balanced anti-androgenic compounds for the future.
The massive overdosage of progestogenic effect that occurs at such doses of CPA is likely responsible for the known adverse effects and risks of higher doses of CPA (Wiki). Examples of these side effects include fatigue, depression, weight gain, high prolactin levels (Wiki), benign brain tumors (Aly, 2020; Wiki; Table; Table), blood clots (Wiki), and cardiovascular problems (Wiki). Such risks are dose-dependent and have not been associated with 1 or 2 mg/day CPA (with the exception of an expected increase in the risk of blood clots in combination with oral estrogens for birth control or menopausal hormone therapy). The risk of liver toxicity with CPA is also dose-dependent, with elevated liver enzymes occurring mostly only at a dosage of 20 mg/day and above and rare cases of liver failure occurring almost exclusively at dosages of 100 mg/day and above (Wiki; Table). As such, there is good rationale for using the lowest possible effective dosage of CPA, an approach that is likely to minimize risks.
In transfeminine people, CPA has historically been used at a dosage of 50 to 100 mg/day (e.g., Moore, Wisniewski, & Dobs, 2003). Some earlier papers have recommended even higher doses of CPA, for instance 100 to 150 mg/day (Asscheman & Gooren, 1993). In 2017, the Endocrine Society published the latest edition of their clinical practice guidelines on hormone therapy for transgender people and reduced their recommended dosage of CPA from 50–100 mg/day to 25–50 mg/day (Hembree et al., 2017; Hembree et al., 2009). This was motivated in part by increasing knowledge and awareness of the risks of higher doses of CPA and by findings that these lower doses of CPA were still effective. However, it is likely that even these new lower dosages are still far in excess of what is really needed.
Testosterone Suppression with Low and High Doses
Progestogens by themselves, including CPA, are able to considerably suppress testosterone levels in gonadally intact people assigned male at birth. Around a dozen small and low-quality but nonetheless notable studies of low-dose CPA from the 1970s and early 1980s found that 5 to 10 mg/day CPA suppressed testosterone levels by about 40 to 70% in healthy young men (Table 1). A couple of individual studies notably reported virtually identical suppression of testosterone levels with 5 mg/day versus 10 mg/day CPA (both ~50% suppression) (Wang & Yeung, 1980; Graph) and with 10 mg/day versus 20 mg/day CPA (both ~60–70% suppression) (Koch et al., 1976; Koch et al., 1975; Graph). This lack of additional testosterone suppression with a doubling of dosage within studies suggests that testosterone suppression with CPA might have actually been maximal at a dosage of only 5 or 10 mg/day. A more modern study, which used a newer and more reliable analytic method for quantification of blood testosterone, found that 10 mg/day CPA suppressed testosterone levels by 66%, from about 600 ± 150 ng/dL to about 185 ng/dL (Meriggiola et al., 2002a; Graph). Similarly, another more modern study found that 10 to 20 mg/day CPA suppressed testosterone levels by 65%, from about 431 ng/dL to about 149 ng/dL, with no reported differences between doses (Zitzmann et al., 2017; Graph).
Table 1: Levels of testosterone and other sex hormones with CPA at low doses (5–30 mg/day):
Treatment and subjects
Findings
Source(s)
30 mg/day CPA in 5 normal males
T decreased “remarkably”. Exact values not given, but has graphs of T levels in a few individuals. After 30 mg/day, 5 mg/day was tried in one case and was not as effective in suppressing sperm production or T. Also reported decreases in gonadotropin excretion.
10 or 20 mg/day CPA in 15 normal healthy fertile males (age 25–35 years) (7 in 10 mg/day group and 8 in 20 mg/day group)
“Androgens (mainly T)” decreased by 60% for both 10 and 20 mg/day. Inconsistent changes in LH and slight decrease in FSH. Exact values not given, except in graphs.
10 mg/day CPA in 10 young healthy fertile men (age mean 27.2 ± 3.2 (range 21–35) years)
T decreased by 70%, DHT by 50%, LH by 30%, and FSH by 40%, while PRL increased by 75%. T was 495 ± 66 ng/dL before, 154 ± 23 ng/dL after 4 weeks, and 187 ± 38 ng/dL after 12 weeks. Also has values and graphs for other hormones.
20 mg/day CPA in 10 healthy males (age 26–55 years)
T decreased by 73% (range 71–75%), from 482 ng/dL (range 410–560 ng/dL) to 130 ng/dL (110–162 ng/dL). DHT decreased by 51% (range 47–55%), LH by 39% (range 34–45%), FSH by 66% (range 47–78%), 17-OH-P4 by 59%, A4 by 30%, TS by 34%, and DHTS by 35%. Also has exact values and graphs for other hormones.
5 or 10 mg/day CPA in 7 males (4 in each group; 1 received both 5 and 10 mg/day CPA at different times)
T change was “−40%” or “–50%”. At 5 mg/day, T was 745 ng/dL before, 460 ng/dL with treatment (–38%), and 668 ng/dL after discontinuation. At 10 mg/day, T was 708 ng/dL before, 398 ng/dL with t (reatment–44%), and 670 ng/dL after discontinuation. Also reported LH and FSH levels.
0, 5, or 10 mg/day CPA in 25 normal healthy males (age 20–51 years); 7 in 5 mg group (mean 37 ± 10 years), 8 in 10 mg group (mean 32 ± 8 years), 10 in 0 mg group (mean 32 ± 10 years)
At 5 mg/day, T decreased from 663 ± 120 ng/dL to 320 ± 160 ng/dL (−52%), and at 10 mg/day, T decreased from 692 ± 180 ng/dL to 340 ± 160 ng/dL (−51%). E2 decreased in parallel to T. At 5 mg/day, LH decreased from 2.1 ± 0.7 IU/L to 1.4 ± 0.5 IU/L (−33%), and at 10 mg/day, LH decreased from 2.3 ± 1.0 IU/L to 1.2 ± 0.5 IU/L (−48%). At 5 mg/day, FSH decreased from 3.1 ± 1.9 IU/L to 1.8 ± 0.9 IU/L (−42%), and at 10 mg/day, FSH decreased from 2.7 ± 1.0 IU/L to 1.5 ± 0.7 IU/L (−44%).
10 or 25 mg/day CPA in 4 healthy men (age 29–37 years); 3 in 10 mg group, 1 in 25 mg group
T “slightly reduced”. E “more significantly lowered”. LH not significantly changed. FSH “reduced” in “more or less all cases”. Exact hormone levels not given, but graphs provided with the values.
10 mg/day CPA (also placebo and 2, 5, and 10 mg/day dienogest) in 5 healthy men in each group
With CPA, T decreased from ~600 ± 150 ng/dL to ~185 ng/dL (–66 ± 4%). Also reported LH, FSH, and SHBG, as well as hormonal changes with placebo and dienogest (2, 5, and 10 mg/day).
10 or 20 mg/day CPA in 14 healthy young men (7 in each group)
T decreased from ~431 ng/dL at baseline to ~149 ng/dL with CPA (–65%) for the 10 and 20 mg/day doses combined. Values for dose subgroups not given. No significant differences between LH/FSH suppression between groups (which is indirectly suggestive of no differences in T suppression as well). Also reported hormone levels with other progestins.
Studies with other progestogens, such as desogestrel, dienogest, and medroxyprogesterone acetate, have consistently found that maximal suppression of testosterone levels in men occurs at a dosage that is between 5 and 10 times that of the ovulation-inhibiting dosage in cisgender women (Wiki; Wiki; Wiki). Another study is likewise suggestive of this for norethisterone acetate and levonorgestrel (Zitzmann et al., 2017; Graph). Along similar lines, doses of progestogens investigated for use in male hormonal contraception, in which the goal is antigonadotropic effects and the lowest fully effective dose is targeted, have been noted as being between 5 and 12 times the doses used in cisgender women (Foegh, 1983). Based on an ovulation-inhibiting dosage of CPA of 1 mg/day, these findings would imply that suppression of testosterone levels with CPA would likely be maximal at a dose of between 5 and 10 mg/day. In accordance, this dose range matches up with the findings of the studies above.
Although progestogens can considerably suppress testosterone levels at maximally effective dosages, it has been found that a “recovery” or “escape phenomenon”, in which testosterone levels eventually increase back to higher levels, occurs when progestogen monotherapy is used on a long-term basis. This has most notably been observed with the related progestogen megestrol acetate (Wiki), but has also been seen with CPA (Goldenberg & Bruchovsky, 1991; Saborowski, 1987; Jacobi, Tunn, & Senge, 1982). In one of these studies, testosterone levels were initially suppressed by CPA by about 70%, but increased back to about 50% of baseline between 6 and 12 months of therapy, remaining stable thereafter up to 24 months. The testosterone escape phenomenon should be kept in mind in the context of progestogen monotherapy for testosterone suppression. In contrast to progestogen monotherapy, this phenomenon has not been associated with combined estrogen and progestogen therapy.
Testosterone Suppression in Combination with Estrogen
CPA is generally used in combination with an estrogen in transfeminine people. Estrogens suppress testosterone levels similarly to progestogens. The combination of an estrogen and a progestogen is synergistic in terms of testosterone suppression and results in suppression of testosterone levels with lower doses than with either an estrogen or progestogen alone (Fink, 1979; Geller & Albert, 1983; Bastianelli et al., 2018). Although estrogens can suppress testosterone levels to an equivalent extent as surgical or medical castration (i.e., orchiectomy or GnRH agonists/antagonists), this usually requires relatively high estrogen levels, for instance in the range of 200 to 500 pg/mL (Wiki; Graphs). Because of the high and supraphysiological estradiol levels required for maximal or near-maximal suppression of testosterone levels, lower doses of estradiol are frequently combined with antiandrogens and/or progestogens to block or suppress remaining testosterone levels instead.
CPA, as mentioned earlier, leads to an incomplete suppression of plasma testosterone levels, which decrease by about 70% and remain at about three times castration values. In a very systematic approach to the problem, Rennie et al. (59) investigated and compared 12 different procedures of androgen deprivation. These authors found that the combination of CPA with an extremely low dose (0.1 mg/d) of [diethylstilbestrol (DES)] led to a very effective withdrawal of androgens in terms of plasma testosterone and tissue dihydrotestosterone. The same group later showed that 200 mg of CPA, and even 100 mg/day, was sufficient to achieve a similar endocrine response, which was correlated to very favorable clinical responses in a Phase II situation (60,61). The approach has many potential advantages, and, from an endocrinological point of view, is very logical: this regimen combines the testosterone-reducing effects of two compounds, therefore, only small amounts of estrogen are required to bring down plasma testosterone to approximately castrate levels. Once castrate levels have been achieved, only low doses of CPA are necessary to counteract remaining androgens, mainly of adrenal origin. The regimen was shown to be associated with few side effects and a very low cost. The combination of low-dose CPA with low-dose DES was never studied in a Phase III situation in comparison to standard management. Considering the endocrine results and the observations in patients treated with this regimen (60), this combination treatment is very likely to be competitive with other standard forms of therapy.
A 2016 study of 50 mg/day CPA and 1 to 2 mg/day transdermal estradiol gel in transfeminine people showed that estradiol levels of about 45 pg/mL with CPA were insufficient to achieve female/castrate levels of testosterone, instead resulting in testosterone levels of about 120 to 190 ng/dL (Gava et al., 2016; Graph). Conversely, estradiol levels of about 85 pg/mL with CPA achieved complete suppression of gonadal testosterone production, with resulting testosterone levels of about 20 ng/dL. As such, a certain minimum level of estradiol with CPA appears to be required for complete testosterone suppression. A 2019 study of CPA and oral estradiol valerate in transfeminine people indicated that testosterone levels were still fully suppressed with median estradiol levels of 76 pg/mL and 25th percentile estradiol levels of 63 pg/mL (Angus et al., 2019; Graph).
Figures 5–7: Testosterone levels with CPA plus low doses/levels of estrogens in men and transfeminine people. Sources: Top-left: Goldenberg et al. (1988). Top-right: Gava et al. (2016). Bottom: Angus et al. (2019). See also on Wikipedia: Gallery. Note for the graph on the top right that the mean transdermal estradiol dosage increased between 6 and 12 months and this was likely responsible for the improvement in testosterone suppression.
Fung and colleagues showed that the combination of either 25 or 50 mg/day CPA with a moderate dosage of oral estradiol (~3.5 mg/day) or transdermal estradiol (~3.5 mg/day gel or ~100 μg/day patch) resulted in equivalent and complete suppression of gonadal testosterone production (~95% suppression of testosterone levels) in transfeminine people (Fung, Hellstern-Layefsky, & Lega, 2017). These dosages of estradiol would be expected to achieve estradiol levels of around 100 pg/mL on average (Aly, 2020; Wiki). This study was notably published 6 months before the 2017 second edition of the Endocrine Society guidelines were released (Hembree et al., 2017), and was probably responsible for the decrease in their recommended dosage of CPA from 50–100 mg/day to 25–50 mg/day.
Few studies to date have assessed testosterone suppression with low-dose CPA in combination with a low or moderate dosage of an estrogen. However, based on the fact that 5 to 10 mg/day CPA alone is probably maximal in terms of suppression of testosterone levels, it is likely that such dosages of CPA will be similarly effective as higher dosages. In accordance, studies of 5 to 12.5 mg/day CPA plus upper physiological replacement dosages of testosterone have demonstrated undetectable gonadotropin levels (<0.5 IU/L) and hence complete suppression of testicular function in healthy young men (Meriggiola et al., 1998; Meriggiola et al., 2002b). Estradiol is a more powerful antigonadotropin than testosterone (Wiki), so these findings probably apply to CPA in combination with physiological replacement levels of estradiol as well (e.g., mean estradiol levels of 100–200 pg/mL).
Accordingly, Meyer et al. (2020) assessed a dosage of CPA in combination with estradiol in 155 transfeminine people and found no difference in testosterone levels with 10, 25, or 50 mg/day CPA; testosterone levels were strongly suppressed with all three doses (to about 15–20 ng/dL on average, or into the lower end of the normal female range). The estradiol forms and doses used in this study were oral estradiol valerate (median 6 mg/day, range 3–10 mg/day), transdermal estradiol gel (median 2.25 mg/day, range 1.5–6 mg/day), and transdermal estradiol patches (100 μg/day in all cases). Estradiol levels were about 100 pg/mL on average, with an interquartile range (i.e., difference between 75th and 25th percentiles) of about 100 pg/mL. This study demonstrates that, provided estradiol levels are adequate, no more than 10 mg/day CPA is needed to fully suppress testosterone levels in transfeminine people. Another study likewise found no difference between <20 mg/day and >50 mg/day CPA in terms of testosterone suppression in transfeminine people (Even-Zohar et al., 2020).
Even doses of CPA lower than 5 mg/day (e.g., 2 mg/day) may be usefully effective for testosterone suppression if combined with sufficient levels of estradiol, although this has not been studied and remains to be validated. But there is certainly precedent for the notion when looking at studies with other progestogens. As an example, one study using 10 mg/day oral medroxyprogesterone acetate (which is roughly equivalent to 1 mg/day CPA in terms of ovulation inhibition in premenopausal women; Table) observed 63% lower testosterone levels (215 ng/dL vs. 79 ng/dL) when added to estradiol and spironolactone therapy in transfeminine people (Jain, Kwan, & Forcier, 2019). Analogous effects on testosterone levels would be anticipated for very-low-dose CPA. Moreover, such dosages of CPA would have the advantage of actually being physiological in terms of progestogenic exposure.
The androgen receptor antagonism of CPA is relatively weak in terms of potency; dosages of CPA of 50 to 300 mg/day seem to be necessary for meaningful or considerable androgen receptor antagonism. Unfortunately, such doses also result in extreme progestogenic overdosage and are associated with considerably greater risks and adverse effects. As a result, the use of such doses of CPA should no longer be considered advisable. Instead, CPA should be used at lower doses simply as a progestogen to suppress testosterone levels. As such, the highest effective dosage of CPA for testosterone suppression, which is probably about 10 mg/day or less (12.5 mg/day also being acceptable), should be around the maximal dosage of CPA that is used in transfeminine people.
It should be emphasized that since the combination of an estrogen and CPA can easily suppress testosterone levels well into the female/castrate range (typically to below average female levels), there isn’t necessarily a requirement for concomitant androgen receptor blockade. In any case, if androgen receptor antagonism to neutralize the remaining female/castrate levels of testosterone is still necessary or desired (e.g., to treat persisting acne or for some other purpose), a low dosage of a non-progestogenic androgen-receptor antagonist like spironolactone (e.g., 100–200 mg/day) or bicalutamide (e.g., 12.5–25 mg/day) can be added to CPA to more safely achieve this than use of higher CPA doses.
Recommended Dosages
Dosage for Testosterone Suppression
Estrogen Plus Cyproterone Acetate
The following recommended dosages of CPA in transfeminine people are for the combination of CPA with an estrogen and are specifically for achieving maximal suppression of testosterone levels:
Table 2: Recommended doses of CPA in combination with estrogen for maximal testosterone suppression in transfeminine people:
Form
Min. dosage
Max. dosage
Amount
10 mg tablets
5 mg/day
10 mg/day
1/2 of a tablet to 1 whole tablet per day
50 mg tablets
6.25 mg/day
12.5 mg/day
1/8th of a tablet to 1/4th of a tablet per day
Start with the minimum dosage of CPA for one month. After one month, have testosterone levels tested and confirm that they are in the normal female/castrate range (<50 ng/dL). Regardless of dosage, a concomitant minimum estradiol level of around 65 pg/mL needs to be attained in order to allow for complete suppression of testosterone levels with CPA. If testosterone levels aren’t sufficiently suppressed after a month and estradiol levels are adequate, increase to the maximum CPA dosage and re-check testosterone levels after another month. Alternatively, the dosage of estradiol can be increased instead; higher estradiol levels result in greater testosterone suppression as well.
Cyproterone Acetate Alone
The use of CPA alone (i.e., as a monotherapy for testosterone suppression) is not recommended due to the risk of decreased bone mineral density and other symptoms of sex-hormone deficiency (Wiki; Aly, 2019). In any case, the recommended dosages for CPA without an estrogen are essentially the same as those listed above of the combination of an estrogen with CPA for testosterone suppression. However, the higher CPA dose (10–12.5 mg/day) may be preferable for good measure in this scenario.
Dosage for Progestogenic Effects
The following recommended dosages of CPA in transfeminine people are for progestogenic effects similar to normal physiological exposure (equivalent of luteal-phase progesterone levels):
Table 3: Recommended doses of CPA for physiological progestogenic effects in transfeminine people:
Form
Dosage
Amount
10 mg tablets
2.5 mg/day
1/4th of a tablet per day
50 mg tablets
3.125 mg/day
1/16th of a tablet per day
Achieving Desired Dosages
CPA is available pharmaceutically most widely as 50-mg tablets. This can make achieving desired low doses of CPA more difficult. For splitting CPA tablets into small fractions, a pill cutter can be used. Additionally, CPA can be taken once every 2 or 3 days instead of once every day to help further divide doses. It is notable that CPA has a relatively long half-life in the body of about 1.5 to 2 days (but possibly up to 4 days) (Wiki; Graph). Hence, taking it once every other day instead of once per day, or even less frequently like once every 3 days, has sound basis and is likely to be entirely viable.
Updates
Update 1: GoLoCypro Study (In-Progress)
The GoLoCypro study (2019–2022) (more info) is being conducted by Dr. Judith Dean at the University of Queensland in Australia. It’s assessing the influence of estradiol plus CPA on testosterone levels at five different CPA dose levels (12.5 mg 2x/week, 12.5 mg/2 days, 12.5 mg/day, 25 mg/day, and 50 mg/day) in a total of 120 to 350 transfeminine people. CPA doses are being titrated to the minimum that maintain testosterone levels within the therapeutic goal range of 0.5 to 1.5 nmol/L (14–43 ng/dL). The study is among the first dose-ranging studies of CPA in transfeminine people to be conducted and is eagerly anticipated due to the valuable information that it should provide in terms of the minimum effective dosage of CPA for adequate testosterone suppression in transfeminine hormone therapy.
Update 2: Kuijpers et al. (2021) and Even Zohar et al. (2021)
Kuijpers, S. M., Wiepjes, C. M., Conemans, E. B., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2021). Toward a lowest effective dose of cyproterone acetate in trans women: Results from the ENIGI study. The Journal of Clinical Endocrinology & Metabolism, 106(10), e3936–e3945. [DOI:10.1210/clinem/dgab427]
The study employed estradiol (2–6 mg/day oral (as estradiol valerate), 50–150 μg/day patch, or gel) plus five different dose levels of CPA—0 mg/day (no CPA), 10 mg/day, 25 mg/day, 50 mg/day, and 100 mg/day. It found incompletely suppressed testosterone in the no CPA group but full and equivalent testosterone suppression with all doses of CPA. The results were as follows:
CPA dosage
0 mg/day
10 mg/day
25 mg/day
50 mg/day
100 mg/day
Initial subjects (n)
34
4
234
599
11
Dose increased (n)
16
1
11
2
0
Dose decreased (n)
0
0
4
40
7
T levels (nmol/L)
5.5
0.9
0.9
1.1
0.9
T levels (ng/dL)
~160
~26
~26
~32
~26
T <2 nmol/L [<~58 ng/dL] (%)
46.3
92.3
96.2
93.4
100.0
Abbreviations: T = testosterone.
The total numbers of subjects and blood tests after CPA dose increases/decreases were not provided. Hence, the exact total number of people and tests for the 10 mg/day group can’t be stated with certainty. The total number of tests for this group was at least 13 based on the testosterone suppression percentage provided however (92.3% or 12/13 but could potentially be 24/26, etc.). Regarding the small number of subjects/tests for the 10 mg/day group, the authors stated the following:
This study is part of the ENIGI initiative, a multicenter prospective cohort study. The main treatment protocol for trans women in this study was 50 mg of CPA daily combined with estrogens. In the first year of study inclusion, a few participants received a dose of 100 mg of CPA. Shortly thereafter, inhospital protocol changed to 50 mg of CPA. As more health concerns related to CPA use were raised over the years, the dose was further lowered from 50 mg to 25 mg and, finally, to 10 mg. However, due to the coronavirus (COVID-19) pandemic, limited results of participants with 10 mg of CPA were available for analysis.
Besides testosterone suppression, the study found that 10 mg/day CPA had less influence on prolactin and high-density lipoprotein (HDL) cholesterol levels than the higher doses of CPA. The study also assessed liver enzyme levels but found no differences between CPA doses.
The authors concluded with the following:
In conclusion, in this cohort of trans women, 10 mg of CPA was found to be effective in lowering testosterone concentrations to the range observed in cis women. A dose of 10 mg was equally effective as higher doses, was found to have less influence on prolactin concentrations and allows higher HDL-C concentrations to be maintained. While GnRH agonists are preferred over CPA due to the fewer associated long-term side effects, this study shows that CPA at a low dose is a viable option when GnRH agonists are contra-indicated, not available, or not reimbursed. Future research should focus on assessing the effectiveness of an even lower dose of CPA (e.g., 5 mg) and the potential long-term side effects.
Around the same that this study was published, Guy T’Sjoen (one of the authors of the study) and other colleagues in a review of optimal hormone therapy for transfeminine people recommended a dosage of no more than 10 or 12.5 mg/day CPA for no longer than 2 years (Glintborg et al., 2021). T’Sjoen is notable in being regarded as one of the foremost experts in transgender medicine and is a coauthor of the Endocrine Society transgender care guidelines (Hembree et al., 2017).
Shortly after the study of Kuijpers and colleagues and also in June 2021, Even Zohar and colleagues in Israel published the following study on low doses of CPA in transfeminine people:
Even Zohar, N., Sofer, Y., Yaish, I., Serebro, M., Tordjman, K., & Greenman, Y. (2021). Low-Dose Cyproterone Acetate Treatment for Transgender Women. The Journal of Sexual Medicine, 18(7), 1292–1298. [10.1016/j.jsxm.2021.04.008]
This study was initially reported as a conference abstract in May 2020 (Even-Zohar et al., 2020).
In the introduction section of the paper, the authors stated the following:
Treatment guidelines published by several organizations are available and assist clinicians in treating transgender women.4,7−9 A wide range of regimens for CPA administration have been proposed. By and large, the recommended doses have decreased over the years: doses of 50–100 mg/day were suggested in the 2009 Endocrine Society Guidelines,10 and amended to 25–50 mg/day in 2017.7 The proposed CPA doses were 12.5–25 mg/day in the 2019 guidelines of the Australian Professional Association for Transgender Health,4 and they were amended to 10–50 mg/day in the 2020 guidelines of the European Society for Sexual Medicine.8 There are no publications on data that compare different doses of CPA for efficacy or safety.
The researchers found that estradiol plus low-dose CPA (10–20 mg/day) suppressed testosterone levels to an equivalent extent as estradiol plus high-dose CPA (50–100 mg/day). Testosterone levels were suppressed into the female/castrate range or near so in both groups (generally ≤2 nmol/L or ≤58 pg/mL). Of the 38 transfeminine people on low-dose CPA, 32 (84%) were on 10 mg/day CPA and 6 (16%) were on 20 mg/day CPA (mean dose 11.6 ± 3.7 mg/day). Estradiol was given as transdermal estradiol patch (mean dose 83.7 ± 36.5 μg/day), transdermal estradiol gel (mean dose 3.8 ± 1.2 g/day), or oral estradiol (mean dose 4.1 ± 1.7 mg/day). Mean estradiol levels ranged from ~110 to 350 pmol/L (~30–95 pg/mL) in the low- and high-dose CPA groups over the follow-up period. Besides showing equivalent testosterone suppression, prolactin levels were significantly lower with low-dose CPA than with high-dose CPA (398 ± 69 mIU/mL vs. 804 ± 121 mIU/mL at 12 months of hormone therapy, respectively).
Based on their findings, the authors stated the following:
We suggest an adjustment of current clinical practice guidelines to recommend lower doses of CPA for the treatment of transgender women.
Both Kuijpers et al. (2021) and Even Zohar et al. (2021) claimed to be the first to demonstrate the efficacy of low-dose CPA in transfeminine people. However, that achievement actually appears to belong to Meyer et al. (2020), who in February 2020 found that estradiol plus 10, 25, or 50 mg/day CPA gave equivalent testosterone suppression across CPA doses in transfeminine people.
Although their study was not about CPA and testosterone suppression, Lim et al. (2020) reported in May/July 2020 that testosterone levels in transfeminine people were median (IQR) 0.6 (0.4–1.0) nmol/L for oral estradiol and 0.9 (0.7–1.6) nmol/L for transdermal estradiol in a mixed group of transfeminine people (n=26 total) on estradiol plus low-dose CPA (12.5 (12.5–18.8) mg/day) (n=14), estradiol alone post-gonadectomy (n=9), and estradiol plus spironolactone (n=3).
In December 2021, the following case report of fatal liver failure with low-dose CPA was published:
Kumar, P., Reddy, S., Kulkarni, A., Sharma, M., & Rao, P. N. (2021). Cyproterone acetate induced Acute liver failure: Case report and review of the literature. Journal of Clinical and Experimental Hepatology, 11(6), 739–741. [DOI:10.1016/j.jceh.2021.01.003]
The case report describes a 30-year-old cisgender woman who was on 25 mg/day CPA for treatment of hirsutism (excessive facial/body hair growth) for 6 months and developed acute liver failure. Four days following hospitalization, she died. This is the second published case report of liver toxicity with CPA at a dosage below 100 mg/day (the first and only other case was at 50 mg/day) (Wiki; Table). It is also the first report of liver failure in a cisgender woman taking CPA. The case indicates that CPA even at a relatively low dose of 25 mg/day is not fully safe in terms of liver toxicity. It further emphasizes the importance of using the lowest effective doses of CPA in transfeminine people (no more than 10–12.5 mg/day).
Update 4: Coleman et al. (2022) [WPATH SOC8 Guidelines]
In September 2022, the World Professional Association for Transgender Health (WPATH) Standards of Care for the Health of Transgender and Gender Diverse People Version 8 (SOC8) were published and made recommendations for transgender hormone therapy for the first time (Coleman et al., 2022). These guidelines recommended a dose of CPA of 10 mg/day in transfeminine people (Coleman et al., 2022). This dose is substantially lower than previous doses recommended by transgender care guidelines and is the first time that major guidelines have recommended a CPA dosage this low. The WPATH SOC8 cited Kuijpers et al. (2021) in support of this recommendation (though notably not Even Zohar et al. (2021) or Meyer et al. (2020)) and also discussed the dose-dependent risks of CPA such as meningiomas and high prolactin levels (Coleman et al., 2022). Considering the key position and importance of the WPATH SOC in transgender health, it is likely that lower CPA doses in transfeminine hormone therapy will now be widely adopted throughout the world. Continued use of higher CPA doses should be considered out of step with current accepted evidence-based practice.
Update 5: Collet et al. (2023)
In October 2022, a study more carefully assessing androgen suppression with estradiol plus CPA in transfeminine people was published:
Collet, S., Gieles, N., Wiepjes, C. M., Heijboer, A. C., Reyns, T., Fiers, T., Lapauw, B., den Heijer, M., & T’Sjoen, G. (2023). Changes in serum testosterone and adrenal androgen levels in transgender women with and without gonadectomy. The Journal of Clinical Endocrinology & Metabolism, 108(2), 331–338. [DOI:10.1210/clinem/dgac576]
In the study, 275 transfeminine people were treated with estradiol plus CPA, and levels of total testosterone, free testosterone, and the adrenal androgensdehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEA-S), and androstenedione (A4) were measured using liquid chromatography–mass spectrometry (LC–MS) at baseline and during follow-ups at 3 months, 12 months, 2 to 4 years, and after surgical gonadal removal (at which time CPA was discontinued). Estradiol was measured both with LC–MS (Amsterdam clinic) and with immunoassays (Ghent clinic). The forms and doses of estradiol used were most commonly oral estradiol valerate 4 mg/day or a transdermal estradiol patch 100 μg/day, while the dosage of CPA was usually 25 or 50 mg/day. About half of the transfeminine people eventually underwent surgical gonadal removal, usually after 2 years of hormone therapy.
Median estradiol levels ranged from 49 to 75 pg/mL (180–275 pmol/L) with LC–MS and from 63 to 69 pg/mL (232–255 pmol/L) with immunoassays at different follow-ups. After 3 months of hormone therapy, total testosterone decreased by 97.1%, from 536 ng/dL (18.6 nmol/L) to 12 ng/dL (0.40 nmol/L), and free testosterone decreased by 98.3%, from 109 pg/mL (378 pmol/L) to 2.0 pg/mL (7.1 pmol/L). Thereafter, total and free testosterone levels remained stable. Levels of DHEA, DHEA-S, and A4 decreased by 24.9 to 28.0%, 20.1 to 23.5%, and 36.5%, respectively, and likewise did not further change after the first 3 to 12 months of hormone therapy. No changes in androgen levels occurred upon surgical gonadal removal with discontinuation of CPA. The authors noted that testosterone levels in the transfeminine people on hormone therapy in the study were similar to or lower than those in cisgender women.
Update 6: Warzywoda et al. (2024) [GoLoCypro Study]
The GoLoCypro study, by Judith Dean and colleagues, was published online in February 2024:
Warzywoda, S., Fowler, J. A., Wood, P., Bisshop, F., Russell, D., Luu, H., Kelly, M., Featherstone, V., & Dean, J. A. (2024). How low can you go? Titrating the lowest effective dose of cyproterone acetate for transgender and gender diverse people who request feminizing hormones. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2317395]
The following are some noteworthy excerpts from the paper:
Of participants who completed the protocol, 74.0% (34/46) were able to achieve the target T-range (0.5–1.5 nmol/L) and 41.3% (19/46) were titrated to the lowest CPA level (12.5 mg cyproterone twice weekly).
Almost all participants who completed the protocol (91.3.0%, 42/46) recorded their CPA levels as level 3 (12.5 mg daily/25 [mg] alternate days) or lower, with 69.0% (29/42) of these being able to achieve the target T-range. Of those that completed, 41.3% (19/46) were able to achieve the lowest CPA level (12.5 mg cyproterone twice week) with 57.9% (11/19) being able to achieve the target T-range.
The study findings showed that for some patients, CPA doses as low as 12.5 mg on alternate days or less can successfully reduce testosterone to pre-menopausal ranges whilst ensuring testosterone was not over-suppressed.
Our study found that doses of CPA lower than the standard dose (12.5 mg CPA daily and/or 25 mg alternate days) were achievable for suppression of testosterone. Several studies have supported this finding that a lower dosage (10 mg CPA daily) is effective in testosterone reduction in individuals undergoing hormone feminization (Even Zohar et al., 2021; Kuijpers et al., 2021). While not all individuals within our study were able to titrate down CPA dosages, almost a quarter of participants who completed the protocol were achieving target T-ranges on 12.5 mg CPA twice weekly (equivalent to 3.5 mg/daily). To our knowledge ours is the first study to demonstrate that doses lower than 10 mg/daily as well as alternate days or twice weekly CPA are clinically effective in maintaining testosterone reduction within target ranges.
Update 7: More New Low-Dose CPA Studies (2023–2025)
Other new studies of low-dose CPA in transfeminine people have also been published in 2023 and 2024:
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (November 2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (November 2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Flamant, T., Vervalcke, J., & T’Sjoen, G. (November 2023). Dose Reduction of Cyproterone Acetate in Trans Women and the Effect on Patient-reported Outcomes: Results from the ENIGI Study. Endocrine Abstracts, 97 [Belgian Endocrine Society 2023], 5–5 (abstract no. 007). [URL] [PDF]
Korpaisarn, S., Arunakul, J., Chaisuksombat, K., & Rattananukrom, T. (2023). A Low Dose Cyproterone Acetate In Feminizing Hormone Treatment. Journal of the Endocrine Society, 7(Suppl 1), A1098–A1099 (abstract no. SAT397/bvad114.2068). [DOI:10.1210/jendso/bvad114.2068]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Bonadonna, S., Amer, M., Foletti, F., Federici, S., Persani, L., Bonomi, M. (2025). Evaluation of Antiandrogen Therapy Effectiveness in Transgender individuals Assigned Male At Birth (AMAB). EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
de Leon-Durango, R., Hernandez-Lazaro, A., Rios-Gomez, C., Santana-Ojeda, B., Molinero-Marcos, I., Arnas-Leon, C., Hernandez-Hernandez, I., Acosta-Calero, C., Kuzior, A., Perez-Rivero, J., Perez-Garcia, M., & Martinez-Martin, F. (2024). P194 Very Low-dose Cyproterone Acetate (12.5 Mg/day) is Effective as Androgen Blocker; Well Tolerated And Not Associated With Hypertension Development in Young Female Transgender People. Journal of Hypertension, 42(Suppl 3), e133–e133. [DOI:10.1097/01.hjh.0001063648.69793.7c]
Korpaisarn, S., Arunakul, J., Chaisuksombat, K., & Rattananukrom, T. (2024). Effectiveness of low dose cyproterone acetate compared to standard dose in feminizing hormone treatment: a single institutional retrospective pilot study. Sexual Medicine, 12(4), qfae063. [DOI:10.1093/sexmed/qfae063]
References
Angus, L., Leemaqz, S., Ooi, O., Cundill, P., Silberstein, N., Locke, P., Zajac, J. D., & Cheung, A. S. (2019). Cyproterone acetate or spironolactone in lowering testosterone concentrations for transgender individuals receiving oestradiol therapy. Endocrine Connections, 8(7), 935–940. [DOI:10.1530/ec-19-0272]
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Asscheman, H., & Gooren, L. J. (1992). Hormone Treatment in Transsexuals. In Bocking, W. O., Coleman, E. (Eds). Gender Dysphoria: Interdisciplinary Approaches in Clinical Management (pp. 39–54). Binghamton: Haworth Press. / Journal of Psychology & Human Sexuality, 5(4), 39–54. [Google Scholar] [Google Books] [DOI:10.1300/J056v05n04_03]
Athanasoulia-Kaspar, A. P., & Stalla, G. K. (2019). Endokrinologische Betreuung von Patienten mit Transsexualität. Geburtshilfe und Frauenheilkunde, 79(7), 672–675. [DOI:10.1055/a-0801-3319]
Bastianelli, C., Farris, M., Rosato, E., Brosens, I., & Benagiano, G. (2018). Pharmacodynamics of combined estrogen-progestin oral contraceptives 3. Inhibition of ovulation. Expert Review of Clinical Pharmacology, 11(11), 1085–1098. [DOI:10.1080/17512433.2018.1536544]
Bonadonna, S., Amer, M., Foletti, F., Federici, S., Persani, L., Bonomi, M. (2025). Evaluation of Antiandrogen Therapy Effectiveness in Transgender individuals Assigned Male At Birth (AMAB). EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
Bourns, A. (2019). Guidelines for Gender-Affirming Primary Care with Trans and Non-Binary Patients, 4th Edition. Toronto: Rainbow Health Ontario/Sherbourne Health. [URL] [PDF]
Bruchovsky, N., Larry Goldenberg, S., Akakura, K., & Rennie, P. S. (1993). Luteinizing hormone-releasing hormone agonists in prostate cancer. Elimination of flare reaction by pretreatment with cyproterone acetate and low-dose diethylstilbestrol. Cancer, 72(5), 1685–1691. [DOI:10.1002/1097-0142(19930901)72:5<1685::aid-cncr2820720532>3.0.co;2-3]
Bultynck, C., Pas, C., Defreyne, J., Cosyns, M., den Heijer, M., & T’Sjoen, G. (2017). Self-perception of voice in transgender persons during cross-sex hormone therapy. The Laryngoscope, 127(12), 2796–2804. [DOI:10.1002/lary.26716]
Chen, H., Wiepjes, C. M., van Schoor, N. M., Heijboer, A. C., de Jongh, R. T., den Heijer, M., & Lips, P. (2019). Changes of Vitamin D-Binding Protein, and Total, Bioavailable, and Free 25-Hydroxyvitamin D in Transgender People. The Journal of Clinical Endocrinology & Metabolism, 104(7), 2728–2734. [DOI:10.1210/jc.2018-02602]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Collet, S., Gieles, N., Wiepjes, C. M., Heijboer, A. C., Reyns, T., Fiers, T., Lapauw, B., den Heijer, M., & T’Sjoen, G. (2023). Changes in serum testosterone and adrenal androgen levels in transgender women with and without gonadectomy. The Journal of Clinical Endocrinology & Metabolism, 108(2), 331–338. [DOI:10.1210/clinem/dgac576]
Damgaard-Pedersen, F., & Føgh, M. (1980). The effect of cyproterone acetate on serum lipids in normal men. Acta Endocrinologica, 94(2), 280–283. [DOI:10.1530/acta.0.0940280]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de Leon-Durango, R., Hernandez-Lazaro, A., Rios-Gomez, C., Santana-Ojeda, B., Molinero-Marcos, I., Arnas-Leon, C., Hernandez-Hernandez, I., Acosta-Calero, C., Kuzior, A., Perez-Rivero, J., Perez-Garcia, M., & Martinez-Martin, F. (2024). P194 Very Low-dose Cyproterone Acetate (12.5 Mg/day) is Effective as Androgen Blocker; Well Tolerated And Not Associated With Hypertension Development in Young Female Transgender People. Journal of Hypertension, 42(Suppl 3), e133–e133. [DOI:10.1097/01.hjh.0001063648.69793.7c]
Defreyne, J., Vantomme, B., Van Caenegem, E., Wierckx, K., De Blok, C., Klaver, M., Nota, N. M., Van Dijk, D., Wiepjes, C. M., Den Heijer, M., & T’Sjoen, G. (2018). Prospective evaluation of hematocrit in gender-affirming hormone treatment: results from European Network for the Investigation of Gender Incongruence. Andrology, 6(3), 446–454. [DOI:10.1111/andr.12485]
Endrikat, J., Gerlinger, C., Richard, S., Rosenbaum, P., & Düsterberg, B. (2011). Ovulation inhibition doses of progestins: a systematic review of the available literature and of marketed preparations worldwide. Contraception, 84(6), 549–557. [DOI:10.1016/j.contraception.2011.04.009]
Even-Zohar, N., Sofer, Y., Yaish, I., Serebro, M., Tordjman, K., & Greenman, Y. (2020). SUN-042 Low Dose Cyproterone Acetate for the Treatment of Transgender Women - a Retrospective Study. Journal of the Endocrine Society, 4(Suppl 1), A715–A715. [DOI:10.1210/jendso/bvaa046.1412]
Even Zohar, N., Sofer, Y., Yaish, I., Serebro, M., Tordjman, K., & Greenman, Y. (2021). Low-Dose Cyproterone Acetate Treatment for Transgender Women. The Journal of Sexual Medicine, 18(7), 1292–1298. [DOI:10.1016/j.jsxm.2021.04.008]
Fink, G. (1979). Feedback Actions of Target Hormones on Hypothalamus and Pituitary With Special Reference to Gonadal Steroids. Annual Review of Physiology, 41(1), 571–585. [DOI:10.1146/annurev.ph.41.030179.003035]
Flamant, T., Vervalcke, J., & T’Sjoen, G. (2023). Dose Reduction of Cyproterone Acetate in Trans Women and the Effect on Patient-reported Outcomes: Results from the ENIGI Study. Endocrine Abstracts, 97 [Belgian Endocrine Society 2023], 5–5 (abstract no. 007). [URL] [PDF]
Føgh, M., Corker, C. S., Hunter, W. M., McLean, H., Philip, J., Schou, G., & Shakkebæk, N. E. (1979). The effects of low doses of cyproterone acetate on some functions of the reproductive system in normal men. Acta Endocrinologica, 91(3), 545–552. [DOI:10.1530/acta.0.0910545]
Føgh, M., Knudsen, J. B., & Gormsen, J. (1980). Effect of cyproterone acetate on platelet aggregability, fibrinolytic activity and fibrinolytic capacity in normal men. Acta Endocrinologica, 94(3), 430–432. [DOI:10.1530/acta.0.0940430]
Foegh, M. (1983). Evaluation of Steroids as COntraceptives in Men. Acta Endocrinologica, 104(3 Suppl b), S9–S48. [DOI:10.1530/acta.0.104s009]
Fredricsson, B., & Carlström, K. (2009). Effects of Low Doses of Cyproterone Acetate on Sperm Morphology and some other Parameters of Reproduction in Normal Men. Andrologia, 13(4), 369–375. [DOI:10.1111/j.1439-0272.1981.tb00067.x]
Fung, R., Hellstern-Layefsky, M., & Lega, I. (2017). Is a lower dose of cyproterone acetate as effective at testosterone suppression in transgender women as higher doses? International Journal of Transgenderism, 18(2), 123–128. [DOI:10.1080/15532739.2017.1290566]
Fuss, J., Hellweg, R., Van Caenegem, E., Briken, P., Stalla, G. K., T’Sjoen, G., & Auer, M. K. (2015). Cross-sex hormone treatment in male-to-female transsexual persons reduces serum brain-derived neurotrophic factor (BDNF). European Neuropsychopharmacology, 25(1), 95–99. [DOI:10.1016/j.euroneuro.2014.11.019]
Fuss, J., Claro, L., Ising, M., Biedermann, S. V., Wiedemann, K., Stalla, G. K., Briken, P., & Auer, M. K. (2019). Does sex hormone treatment reverse the sex-dependent stress regulation? A longitudinal study on hypothalamus-pituitary-adrenal (HPA) axis activity in transgender individuals. Psychoneuroendocrinology, 104, 228–237. [DOI:10.1016/j.psyneuen.2019.02.023]
Gava, G., Cerpolini, S., Martelli, V., Battista, G., Seracchioli, R., & Meriggiola, M. C. (2016). Cyproterone acetatevsleuprolide acetate in combination with transdermal oestradiol in transwomen: a comparison of safety and effectiveness. Clinical Endocrinology, 85(2), 239–246. [DOI:10.1111/cen.13050]
Gava, G., Mancini, I., Alvisi, S., Seracchioli, R., & Meriggiola, M. C. (2020). A comparison of 5-year administration of cyproterone acetate or leuprolide acetate in combination with estradiol in transwomen. European Journal of Endocrinology, 183(6), 561–569. [DOI:10.1530/eje-20-0370]
Geller, J., Albert, J., Yen, S. S., Geller, S., & Loza, D. (1981). Medical Castration of Males with Megestrol Acetate and Small Doses of Diethylstilbestrol*. The Journal of Clinical Endocrinology & Metabolism, 52(3), 576–580. [DOI:10.1210/jcem-52-3-576]
Geller, J., Albert, J., Yen, S. S., Geller, S., & Loza, D. (1981). Medical castration with megestrol acetate and minidose of diethylstilbestrol. Urology, 17(4 Suppl), 27–33. [Google Scholar] [PubMed]
Geller, J., & Albert, J. D. (1983). Comparison of various hormonal therapies for prostatic carcinoma. Seminars in Oncology, 10(4 Suppl 4), 34–41. [Google Scholar] [PubMed] [PDF]
Geller, J. (1988). Megestrol acetate and minidose estrogen in prostatic carcinoma. Urology, 32(3), 281–282. [DOI:10.1016/0090-4295(88)90402-5]
Geller J. (1991). Megestrol Acetate Plus Low-Dose Estrogen in the Management of Advanced Prostatic Carcinoma. The Urologic Clinics of North America, 18(1), 83–91. [DOI:10.1016/S0094-0143(21)01395-1] [Archive.org]
Giltay, E. J., & Gooren, L. J. (2000). Effects of Sex Steroid Deprivation/Administration on Hair Growth and Skin Sebum Production in Transsexual Males and Females. The Journal of Clinical Endocrinology & Metabolism, 85(8), 2913–2921. [DOI:10.1210/jcem.85.8.6710]
Giltay, E. J., Gooren, L. J., Emeis, J. J., Kooistra, T., & Stehouwer, C. D. (2000). Oral, but Not Transdermal, Administration of Estrogens Lowers Tissue-Type Plasminogen Activator Levels in Humans Without Affecting Endothelial Synthesis. Arteriosclerosis, Thrombosis, and Vascular Biology, 20(5), 1396–1403. [DOI:10.1161/01.atv.20.5.1396]
Giltay, E. J., Verhoef, P., Gooren, L. J., Geleijnse, J. M., Schouten, E. G., & Stehouwer, C. D. (2003). Oral and transdermal estrogens both lower plasma total homocysteine in male-to-female transsexuals. Atherosclerosis, 168(1), 139–146. [DOI:10.1016/s0021-9150(03)00090-x]
Giltay, E. J., Gooren, L. J., Toorians, A. W., Katan, M. B., & Zock, P. L. (2004). Docosahexaenoic acid concentrations are higher in women than in men because of estrogenic effects. The American Journal of Clinical Nutrition, 80(5), 1167–1174. [DOI:10.1093/ajcn/80.5.1167]
Glintborg, D., T’Sjoen, G., Ravn, P., & Andersen, M. S. (2021). MANAGEMENT OF ENDOCRINE DISEASE: Optimal feminizing hormone treatment in transgender people. European Journal of Endocrinology, 185(2), R49–R63. [DOI:10.1530/eje-21-0059]
Goldenberg, S. L., Bruchovsky, N., Rennie, P. S., & Coppin, C. M. (1988). The Combination of Cyproterone Acetate and Low Dose Diethylstilbestrol in the Treatment of Advanced Prostatic Carcinoma. Journal of Urology, 140(6), 1460–1465. [DOI:10.1016/s0022-5347(17)42073-8]
Goldenberg, S. L., & Bruchovsky, N. (1991). Use of Cyproterone Acetate in Prostate Cancer. The Urologic Clinics of North America, 18(1), 111–122. [DOI:10.1016/S0094-0143(21)01398-7] [Archive.org]
Goldenberg, S., Bruchovsky, N., Gleave, M., & Sullivan, L. (1996). Low-dose cyproterone acetate plus mini-dose diethylstilbestrol—A protocol for reversible medical castration. Urology, 47(6), 882–884. [DOI:10.1016/s0090-4295(96)00048-9]
Gooren, L. J., Giltay, E. J., & Bunck, M. C. (2008). Long-Term Treatment of Transsexuals with Cross-Sex Hormones: Extensive Personal Experience. The Journal of Clinical Endocrinology & Metabolism, 93(1), 19–25. [DOI:10.1210/jc.2007-1809]
Gräf, K., Brotherton, J., & Neumann, F. (1974). Clinical Uses of Antiandrogens. In Hughes, A., Hasan, S. H., Oertel, G. W., Voss, H. E., Bahner, F., Neumann, F., Steinbeck, H., Gräf, K.-J., Brotherton, J., Horn, H. J., & Wagner, R. K. (Eds.). Androgens II and Antiandrogens / Androgene II und Antiandrogene (Handbuch der experimentellen Pharmakologie/Handbook of Experimental Pharmacology, Volume 35, Part 2) (pp. 485–542). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-80859-3_7]
Hammerstein, J., Meckies, J., Leo-Rossberg, I., Moltz, L., & Zielske, F. (1975). Use of cyproterone acetate (CPA) in the treatment of acne, hirsutism and virilism. Journal of Steroid Biochemistry, 6(6), 827–836. [DOI:10.1016/0022-4731(75)90311-8]
Hammerstein, J. (1979). Cyproterone Acetate. In Jacobs, H. S. (Ed.). Advances in Gynaecological Endocrinology: Proceedings of the Sixth Study Group of the Royal College of Obstetricians and Gynaecologists, 18th and 19th October, 1978 (pp. 367–382). London: The College. [Google Scholar] [Google Books] [PDF]
Hammerstein, J. (1990). Antiandrogens: Clinical Aspects. In Orfanos, C. E., & Happle, R. (Eds.). Hair and Hair Diseases (pp. 827–886). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-74612-3_35]
Heath, R. A., & Wynne, K. (2019). A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (p. 122). Santa Barbara: Praeger/ABC-CLIO. [Google Books]
Hembree, W. C., Cohen-Kettenis, P., Delemarre-Van De Waal, H. A., Gooren, L. J., Meyer III, W. J., Spack, N. P., Tangpricha, V., & Montori, V. M. (2009). Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism, 94(9), 3132–3154. [DOI:10.1210/jc.2009-0345]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society* Clinical Practice Guideline [2nd Version]. The Journal of Clinical Endocrinology & Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Jacobeit, J. W. (2019). Die hormonelle Behandlung von adulten Trans*Personen (in Deutschland). [Hormonal treatment of adult trans* persons (in Germany).] Journal für Klinische Endokrinologie und Stoffwechsel, 12(3), 102–110. [DOI:10.1007/s41969-019-00080-x]
Jacobi, G. H., Altwein, J. E., Kurth, K. H., Basting, R., & Hohenfellner, R. (1980). Treatment of Advanced Prostatic Cancer with Parenteral Cyproterone Acetate: A Phase III Randomised Trial*. British Journal of Urology, 52(3), 208–215. [DOI:10.1111/j.1464-410x.1980.tb02961.x]
Jacobi, G. H., Tunn, U., & Senge, T. (1982). Clinical experience with cyproterone acetate for palliation of inoperable prostate cancer. In Jacobi, G. H., & Hohenfellner, R. (Eds.). Prostate Cancer (International Perspectives in Urology, Volume 3) (pp. 305–319). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Jequier, A. M., Bullimore, N. J., & Bishop, M. J. (2009). Cyproterone Acetate and a Small Dose of Oestrogen in the Pre-operative Management of Male Transsexuals. A Report of Three Cases. [Cyproteronacetat und kleine Östrogendosis in dem präoperativen Management männlicher Transsexueller. Bericht über drei Fälle.] Andrologia, 21(5), 456–461. [DOI:10.1111/j.1439-0272.1989.tb02447.x]
Johnson, D. E., Babaian, R. J., Swanson, D. A., Von Eschenbach, A. C., Wishnow, K. I., & Tenney, D. (1988). Medical castration using megestrol acetate and minidose estrogen. Urology, 31(5), 371–374. [DOI:10.1016/0090-4295(88)90726-1]
Knuth, U. A., Hano, R., & Nieschlag, E. (1984). Effect of Flutamide or Cyproterone Acetate on Pituitary and Testicular Hormones in Normal Men. The Journal of Clinical Endocrinology & Metabolism, 59(5), 963–969. [DOI:10.1210/jcem-59-5-963]
Koch, U. J., Lorenz, F., Danehl, K., & Hammerstein, J. (1975). Über die Verwendbarkeit von Cyproteronacetat zur Fertilitätshemmung beim Mann. Morphologische Veränderungen und Einflüsse auf die Spermienmotilität. Archiv für Gynäkologie, 219(1–4), 581–582. [DOI:10.1007/bf00669258]
Koch, U., Lorenz, F., Danehl, K., Ericsson, R., Hasan, S., Keyserlingk, D., Lübke, K., Mehring, M., Römmler, A., Schwartz, U., & Hammerstein, J. (1976). Continuous oral low-dosage cyproterone acetate for fertility regulation in the male? A trend analysis in 15 volunteers. Contraception, 14(2), 117–135. [DOI:10.1016/0010-7824(76)90081-0]
Korpaisarn, S., Arunakul, J., Chaisuksombat, K., & Rattananukrom, T. (2023). A Low Dose Cyproterone Acetate In Feminizing Hormone Treatment. Journal of the Endocrine Society, 7(Suppl 1), A1098–A1099 (abstract no. SAT397/bvad114.2068). [DOI:10.1210/jendso/bvad114.2068]
Korpaisarn, S., Arunakul, J., Chaisuksombat, K., & Rattananukrom, T. (2024). Effectiveness of low dose cyproterone acetate compared to standard dose in feminizing hormone treatment: a single institutional retrospective pilot study. Sexual Medicine, 12(4), qfae063. [DOI:10.1093/sexmed/qfae063]
Kranz, G. S., Seiger, R., Kaufmann, U., Hummer, A., Hahn, A., Ganger, S., Tik, M., Windischberger, C., Kasper, S., & Lanzenberger, R. (2017). Effects of sex hormone treatment on white matter microstructure in individuals with gender dysphoria. NeuroImage, 150, 60–67. [DOI:10.1016/j.neuroimage.2017.02.027]
Kranz, G. S., Kaufmann, U., & Lanzenberger, R. (2019). Probing the Impact of Gender-Affirming Hormone Treatment on Odor Perception. Chemical Senses, 45(1), 37–44. [DOI:10.1093/chemse/bjz069]
Kuijpers, S. M., Wiepjes, C. M., Conemans, E. B., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2021). Toward a Lowest Effective Dose of Cyproterone Acetate in Trans Women: Results From the ENIGI Study. The Journal of Clinical Endocrinology & Metabolism, 106(10), e3936–e3945. [DOI:10.1210/clinem/dgab427]
Kumar, P., Reddy, S., Kulkarni, A., Sharma, M., & Rao, P. N. (2021). Cyproterone acetate induced acute liver failure: case report and review of the literature. Journal of Clinical and Experimental Hepatology, 11(6), 739–741. [DOI:10.1016/j.jceh.2021.01.003]
Lederbogen, S. (2009). Hormonbehandlung. PiD - Psychotherapie im Dialog, 10(1), 41–43. [DOI:10.1055/s-0028-1090190]
Lim, H. Y., Leemaqz, S. Y., Torkamani, N., Grossmann, M., Zajac, J. D., Nandurkar, H., Ho, P., & Cheung, A. S. (2020). Global Coagulation Assays in Transgender Women on Oral and Transdermal Estradiol Therapy. The Journal of Clinical Endocrinology & Metabolism, 105(7), e2369–e2377. [DOI:10.1210/clinem/dgaa262]
Liu, P. Y., Takahashi, P., & Veldhuis, J. D. (2017). An Ensemble Perspective of Aging-Related Hypoandrogenemia in Men. In Winters, S. J., & Huhtaniemi, I. T. (Eds.). Male Hypogonadism: Basic, Clinical and Therapeutic Principles, 2nd Edition (pp. 325–347). Cham: Springer. [DOI:10.1007/978-3-319-53298-1_16]
Mahfouda, S., Moore, J. K., Siafarikas, A., Hewitt, T., Ganti, U., Lin, A., & Zepf, F. D. (2019). Gender-affirming hormones and surgery in transgender children and adolescents. The Lancet Diabetes & Endocrinology, 7(6), 484–498. [DOI:10.1016/s2213-8587(18)30305-x]
Meriggiola, M. C., Bremner, W. J., Costantino, A., Di Cintio, G., & Flamigni, C. (1998). Low dose of cyproterone acetate and testosterone enanthate for contraception in men. Human Reproduction, 13(5), 1225–1229. [DOI:10.1093/humrep/13.5.1225]
Meriggiola, M. C., Bremner, W. J., Costantino, A., Bertaccini, A., Morselli-Labate, A. M., Huebler, D., Kaufmann, G., Oettel, M., & Flamigni, C. (2002). Twenty-One Day Administration of Dienogest Reversibly Suppresses Gonadotropins and Testosterone in Normal Men. The Journal of Clinical Endocrinology & Metabolism, 87(5), 2107–2113. [DOI:10.1210/jcem.87.5.8514]
Meriggiola, M. C., Costantino, A., Bremner, W. J., & Morselli-Labate, A. M. (2002). Higher Testosterone Dose Impairs Sperm Suppression Induced by a Combined Androgen‐Progestin Regimen. Journal of Andrology, 23(5), 684–690. [DOI:10.1002/j.1939-4640.2002.tb02311.x]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463] [PDF]
Moltz, L., Römmler, A., Schwartz, U., & Hammerstein, J. (1978). Effects of Cyproterone Acetate (CPA) on Pituitary Gonadotrophin Release and on Androgen Secretion Before and After LH-RH Double Stimulation Tests in Men. International Journal of Andrology, 1(Suppl 2b) [5th Annual Workshop on the Testis, Geilo, Norway, April 1978, Endocrine Approach to Male Contraception], 713–719. [DOI:10.1111/j.1365-2605.1978.tb00518.x]
Moltz, L., Römmler, A., Schwartz, U., Post, K., & Hammerstein, J. (1978). Cyproterone acetate (CPA)—a potential male contraceptive: further studies on the interactions with endocrine parameters. Journal of Steroid Biochemistry, 9(9), 865–865 (abstract no. 252). [DOI:10.1016/0022-4731(78)90952-4]
Moltz, L., Römmler, A., Post, K., Schwartz, U., & Hammerstein, J. (1980). Medium dose cyproterone acetate (CPA): Effects on hormone secretion and on spermatogenesis in men. Contraception, 21(4), 393–413. [DOI:10.1016/s0010-7824(80)80017-5]
Moltz, L., Koch, U., Schwartz, U., Rommler, A., & Hammerstein, J. (1982). Male fertility regulation with cyproterone acetate (CPA). Contraceptive Delivery Systems, 3(3/4) [Retroproductive Health Care International Symposium, October 10-15 1982 Maui, Hawaii, USA, Expanded Abstracts], 298–298 (abstract no. 293). [Google Scholar] [PDF]
Moore, E., Wisniewski, A., & Dobs, A. (2003). Endocrine Treatment of Transsexual People: A Review of Treatment Regimens, Outcomes, and Adverse Effects. The Journal of Clinical Endocrinology & Metabolism, 88(8), 3467–3473. [DOI:10.1210/jc.2002-021967]
Nelson, J. B. (2012). Hormone Therapy for Prostate Cancer. In Wein, A. J., Kavoussi, L. R., Novick, A. C., Partin, A. W., & Peters, C. A. (Eds.). Campbell-Walsh Urology, 10th Edition, Volume 2 (pp. 2920–2953). Philadelphia: Elsevier/Saunders. [Google Scholar] [Google Books]
Nota, N. M., den Heijer, M., Gooren, L. J. (2019). Evaluation and Treatment of Gender-Dysphoric/Gender Incongruent Adults. [Updated 2019 Jul 21]. In Feingold, K. R., Anawalt, B., Blackman, M. R., et al. (Eds.). Endotext [Internet]. South Dartmouth, Massachusetts: MDText.com. [PubMed]
Oliphant, J., Veale, J., Macdonald, J., Carroll, R., Johnson, R., Harte, M., Stephenson, C. & Bullock, J. (2018). Guidelines for Gender Affirming Healthcare for Gender Diverse and Transgender Children, Young People and Adults in Aotearoa New Zealand. Waikato: Transgender Health Research Lab/University of Waikato. [URL] [PDF]
Ott, J., Aust, S., Promberger, R., Huber, J. C., & Kaufmann, U. (2011). Cross‐Sex Hormone Therapy Alters the Serum Lipid Profile: A Retrospective Cohort Study in 169 Transsexuals. The Journal of Sexual Medicine, 8(8), 2361–2369. [DOI:10.1111/j.1743-6109.2011.02311.x]
Petry, R., Mauss, J., Senge, T., & Rausch-Stroomann, J. (1970). Über den Einfluß von Cyproteronacetat, Norethisteronönanthat und Gestonoroncapronat auf die Hypophysen-Gonadenachse beim Mann. [Influence of Cyproterone-acetate, Norethisterone-enanthate and Gestonorone-capronate on the Hypophyseal-Gonadal-Axis in the Male.] In Kracht, J. (Ed.). Endokrinologie der Entwicklung und Reifung, 16. Symposion, Ulm, 26.-28. Februar 1970 (Symposion der Deutschen Gesellschaft für Endokrinologie, Volume 16) (pp. 428–430). Berlin: Springer. [Google Books] [DOI:10.1007/978-3-642-80591-2_118] [WorldCat]
Petry, R., Rausch-Stroomann, J.-G., Berthold, K. Mauss, J., Ai, M., Senge, Th., & Vermeulen, A. (1970). Untersuchungen zum Wirkungsmechanismus der Antiandrogene Cyproteron und Cyproteronacetat beim Menschen (Gonadotropin-, Plasma-testosteron- und morphologische Keimdrüsenuntersuchungen). [Investigations on the mechanism of action of the antiandrogens cyproterone and cyproterone acetate in humans (gonadotropin, plasma testosterone, and morphological gonad investigations).] In Schlegel, B. (Ed.). Verhandlungen der Deutschen Gesellschaft für Innere Medizin: Sechsundsiebzigster Kongress Gehalten zu Wiesbaden vom 6. April – 9. April 1970 (Verhandlungen der Deutschen Gesellschaft für Innere Medizin, Volume 76) (pp. 873–876). München: Bergmann. [Google Scholar] [Google Books] [DOI:10.1007/978-3-642-85446-0] [WorldCat]
Petry, R., Rausch-Stroomann, J. G., Mauss, J., Senge, Th., Ai, M., & Berthold, K. (1970). Investigations on the mode of action of the antiandrogens cyproterone and cyproterone acetate in man. / Investigations on the mechanism of action of anti androgenic cyproterone and cyproterone acetate in humans (gonadotropin, plasma testosterone, and morphological generative gland investigations). Medizinische Welt, 29, 1336–. [EurekaMag] [Cited by Koch et al. (1976)]
Petry, R., Mauss, J., Rausch-Stroomann, J. G., & Vermeulen, A. (1972). Reversible inhibition of spermatogenesis in men. Hormone and Metabolic Research, 4(5), 386–388. [DOI:10.1055/s-0028-1094040]
Roy, S., Chatterjee, S., Prasad, M., Poddar, A., Pandey, D., Pandey, H., & Jadhav, Y. (1976). Effects of cyproterone acetate on reproductive functions in normal human males. Contraception, 14(4), 403–423. [DOI:10.1016/s0010-7824(76)80055-8]
Roy, S., & Chatterjee, S. (1979). Studies with cyproterone acetate for male contraception. In James, V. H. T., & Pasqualini, J. R. (Eds.). Hormonal Steroids: Proceedings of the Fifth International Congress on Hormonal Steroids, New Delhi, India, October/November 1978 (pp. 675–680). Oxford: Pergamon Press. [DOI:10.1016/b978-0-08-023796-1.50099-2]
Roy, S., & Chatterjee, S. (1979). The Role of Antiandrogenic Action in Cyproterone Acetate-Induced Morphologic and Biochemical Changes in Human Semen. Fertility and Sterility, 32(1), 93–95. [DOI:10.1016/s0015-0282(16)44122-1]
Saborowski, K.-J. (1987). Konservative Therapie mit Cyproteronacetat und Estradiolundecylat beim Fortgeschrittenen Prostatacarcinom: Eine 5-Jahres-Studie. [Conservative Therapy with Cyproterone Acetate and Estradiol Undecylate in Advanced Prostate Cancer: A 5-Year Study.] (Doctoral dissertation, Ruhr-University Bochum.) [58 pages] [Google Scholar] [Google Books] [WorldCat] [PDF] [Translation]
Scharff, M., Wiepjes, C. M., Klaver, M., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2019). Change in grip strength in trans people and its association with lean body mass and bone density. Endocrine Connections, 8(7), 1020–1028. [DOI:10.1530/ec-19-0196]
Schröder, F. H., & Radlmaier, A. (2002). Steroidal Antiandrogens. In Jordan, C. V., & Furr, B. J. A. (Eds.). Hormone Therapy in Breast and Prostate Cancer (pp. 325–346). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-59259-152-7_15]
Slagter, M. H., Gooren, L. J., de Ronde, W., Soosaipillai, A., Scorilas, A., Giltay, E. J., Paliouras, M., & Diamandis, E. P. (2006). Serum and Urine Tissue Kallikrein Concentrations in Male-to-Female Transsexuals Treated with Antiandrogens and Estrogens. Clinical Chemistry, 52(7), 1356–1365. [DOI:10.1373/clinchem.2006.068932]
Sofer, Y., Yaish, I., Yaron, M., Bach, M. Y., Stern, N., & Greenman, Y. (2020). Differential Endocrine and Metabolic Effects of Testosterone Suppressive Agents in Transgender Women. Endocrine Practice, 26(8), 883–890. [DOI:10.4158/ep-2020-0032]
T’Sjoen, G. G., Beguin, Y., Feyen, E., Rubens, R., Kaufman, J., & Gooren, L. (2005). Influence of exogenous oestrogen or (anti-) androgen administration on soluble transferrin receptor in human plasma. Journal of Endocrinology, 186(1), 61–67. [DOI:10.1677/joe.1.06112]
T’Sjoen, G., Weyers, S., Taes, Y., Lapauw, B., Toye, K., Goemaere, S., & Kaufman, J. (2009). Prevalence of Low Bone Mass in Relation to Estrogen Treatment and Body Composition in Male-to-Female Transsexual Persons. Journal of Clinical Densitometry, 12(3), 306–313. [DOI:10.1016/j.jocd.2008.11.002]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine Position Statement “Assessment and Hormonal Management in Adolescent and Adult Trans People, with Attention for Sexual Function and Satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
Tack, L. J., Heyse, R., Craen, M., Dhondt, K., Bossche, H. V., Laridaen, J., & Cools, M. (2017). Consecutive Cyproterone Acetate and Estradiol Treatment in Late-Pubertal Transgender Female Adolescents. The Journal of Sexual Medicine, 14(5), 747–757. [DOI:10.1016/j.jsxm.2017.03.251]
Toorians, A. W., Thomassen, M. C., Zweegman, S., Magdeleyns, E. J., Tans, G., Gooren, L. J., & Rosing, J. (2003). Venous Thrombosis and Changes of Hemostatic Variables during Cross-Sex Hormone Treatment in Transsexual People. The Journal of Clinical Endocrinology & Metabolism, 88(12), 5723–5729. [DOI:10.1210/jc.2003-030520]
Torre, B. l., Norén, S., Hedman, M., & Diczfalusy, E. (1979). Effect of cyproterone acetate (CPA) on gonadal and adrenal function in men. Contraception, 20(4), 377–396. [DOI:10.1016/s0010-7824(79)80048-7]
Van Caenegem, E., Wierckx, K., Taes, Y., Schreiner, T., Vandewalle, S., Toye, K., Kaufman, J., & T’Sjoen, G. (2014). Preservation of volumetric bone density and geometry in trans women during cross-sex hormonal therapy: a prospective observational study. Osteoporosis International, 26(1), 35–47. [DOI:10.1007/s00198-014-2805-3]
van Dijk, D., Dekker, M. J., Conemans, E. B., Wiepjes, C. M., de Goeij, E. G., Overbeek, K. A., Fisher, A. D., den Heijer, M., & T’Sjoen, G. (2019). Explorative Prospective Evaluation of Short-Term Subjective Effects of Hormonal Treatment in Trans People—Results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 16(8), 1297–1309. [DOI:10.1016/j.jsxm.2019.05.009]
van Velzen, D. M., Paldino, A., Klaver, M., Nota, N. M., Defreyne, J., Hovingh, G. K., Thijs, A., Simsek, S., T’Sjoen, G., & den Heijer, M. (2019). Cardiometabolic Effects of Testosterone in Transmen and Estrogen Plus Cyproterone Acetate in Transwomen. The Journal of Clinical Endocrinology & Metabolism, 104(6), 1937–1947. [DOI:10.1210/jc.2018-02138]
Venner, P. M., Klotz, P. G., Klotz, L. H., Stewart, D. J., Davis, I. R., Orovan, W. L., & Ramsey, E. W. (1988). Megestrol acetate plus minidose diethylstilbestrol in the treatment of carcinoma of the prostate. Seminars in Oncology, 15(2 Suppl 1), 62–67. [Google Scholar] [PubMed]
Vereecke, G. (2019). Characterisation of testicular function and spermatogenesis in transgender women. (Master’s thesis, Ghent University.) [PDF]
Vereecke, G., Defreyne, J., Van Saen, D., Collet, S., Van Dorpe, J., T’Sjoen, G., & Goossens, E. (2020). Characterisation of testicular function and spermatogenesis in transgender women. Human Reproduction, 36(1), 5–15. [DOI:10.1093/humrep/deaa254]
Vita, R., Settineri, S., Liotta, M., Benvenga, S., & Trimarchi, F. (2018). Changes in hormonal and metabolic parameters in transgender subjects on cross-sex hormone therapy: A cohort study. Maturitas, 107, 92–96. [DOI:10.1016/j.maturitas.2017.10.012]
Vlot, M. C., Wiepjes, C. M., Jongh, R. T., T’Sjoen, G., Heijboer, A. C., & den Heijer, M. (2019). Gender‐Affirming Hormone Treatment Decreases Bone Turnover in Transwomen and Older Transmen. Journal of Bone and Mineral Research, 34(10), 1862–1872. [DOI:10.1002/jbmr.3762]
Wang, C., & Yeung, K. (1980). Use of low-dosage oral cyproterone acetate as a male contraceptive. Contraception, 21(3), 245–272. [DOI:10.1016/0010-7824(80)90005-0]
Warzywoda, S., Fowler, J. A., Wood, P., Bisshop, F., Russell, D., Luu, H., Kelly, M., Featherstone, V., & Dean, J. A. (2024). How low can you go? Titrating the lowest effective dose of cyproterone acetate for transgender and gender diverse people who request feminizing hormones. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2317395]
Wiepjes, C. M., Vlot, M. C., Klaver, M., Nota, N. M., de Blok, C. J., de Jongh, R. T., Lips, P., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Bone Mineral Density Increases in Trans Persons After 1 Year of Hormonal Treatment: A Multicenter Prospective Observational Study. Journal of Bone and Mineral Research, 32(6), 1252–1260. [DOI:10.1002/jbmr.3102]
Wiepjes, C. M., Vlot, M. C., de Blok, C. J., Nota, N. M., de Jongh, R. T., & den Heijer, M. (2019). Bone geometry and trabecular bone score in transgender people before and after short- and long-term hormonal treatment. Bone, 127, 280–286. [DOI:10.1016/j.bone.2019.06.029]
Wierckx, K., Mueller, S., Weyers, S., Van Caenegem, E., Roef, G., Heylens, G., & T’Sjoen, G. (2012). Long‐Term Evaluation of Cross‐Sex Hormone Treatment in Transsexual Persons. The Journal of Sexual Medicine, 9(10), 2641–2651. [DOI:10.1111/j.1743-6109.2012.02876.x]
Wierckx, K., Van Caenegem, E., Schreiner, T., Haraldsen, I., Fisher, A., Toye, K., Kaufman, J. M., & T’Sjoen, G. (2014). Cross‐Sex Hormone Therapy in Trans Persons Is Safe and Effective at Short‐Time Follow‐Up: Results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 11(8), 1999–2011. [DOI:10.1111/jsm.12571]
Winkler-Crepaz, K., Müller, A., Böttcher, B., & Wildt, L. (2017). Hormonbehandlung bei Transgenderpatienten. [Hormone treatment of transgender patients.] Gynäkologische Endokrinologie, 15(1), 39–42. [DOI:10.1007/s10304-016-0116-9]
Winters, S. J., Wang, C., & (2009). LH and Non-SHBG Testosterone and Estradiol Levels During Testosterone Replacement of Hypogonadal Men: Further Evidence That Steroid Negative Feedback Increases as Men Grow Older. Journal of Andrology, 31(3), 281–287. [DOI:10.2164/jandrol.109.009035]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Zitzmann, M., Rohayem, J., Raidt, J., Kliesch, S., Kumar, N., Sitruk-Ware, R., & Nieschlag, E. (2017). Impact of various progestins with or without transdermal testosterone on gonadotropin levels for non-invasive hormonal male contraception: a randomized clinical trial. Andrology, 5(3), 516–526. [DOI:10.1111/andr.12328]
Zubiaurre-Elorza, L., Junque, C., Gómez-Gil, E., & Guillamon, A. (2014). Effects of Cross-Sex Hormone Treatment on Cortical Thickness in Transsexual Individuals. The Journal of Sexual Medicine, 11(5), 1248–1261. [DOI:10.1111/jsm.12491]
\ No newline at end of file
+Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People - Transfeminine Science
Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People
By Aly | First published July 1, 2019 | Last modified August 23, 2025
Abstract / TL;DR
Cyproterone acetate (CPA) is a progestogen and antiandrogen which is widely used in transfeminine hormone therapy. It is far more potent as a progestogen than as an androgen receptor antagonist. CPA has typically been used at doses of 1 to 2 mg/day as a progestogen in cisgender women and at doses of 50 to 300 mg/day as an antiandrogen. At typical antiandrogen doses of CPA, there is profound progestogenic overdosage as well as associated side effects and risks. CPA has antigonadotropic effects due to its progestogenic activity and thereby suppresses testosterone levels. By itself, CPA can maximally suppress testosterone levels by 50 to 70%, and in combination with even small amounts of estrogen, it can fully suppress gonadal testosterone production and thereby reduce testosterone levels by about 95%—or well into the female range. Although doses of CPA of 50 to 100 mg/day have been used in transfeminine people historically, it is now clear that 5 to 10 mg/day CPA has maximal or near-maximal effectiveness in terms of suppression of testosterone levels. CPA alone is most commonly available as 50-mg tablets. These tablets can be split with a pill cutter and taken once every day to once every other day to achieve an overall CPA dosage of 6.25 to 12.5 mg/day. These lower doses of CPA are not only much more cost-effective than traditional doses but are also likely to have better tolerability and safety. Due to the retained effectiveness of lower CPA doses and the known dose-dependent risks of CPA, doses of CPA used clinically in transfeminine people have been in a rapid decline.
Introduction
This article is about the dosage of cyproterone acetate (CPA), a progestin and antiandrogen, for use in hormone therapy for transfeminine people. It argues for the use of lower doses of CPA and goes fairly in-depth to justify these doses. If you are only interested in recommended doses of CPA for transfeminine people, they can be found in the Recommended Dosages section below.
Potency, Conventional Dosages, and Health Risks
CPA is a potent progestogen, with an ovulation-inhibiting dosage of about 1 mg/day and endometrial transformation dosage of about 1 to 3 mg/day in cisgender women (Wiki; Table; Endrikat et al., 2011). These dosages of CPA are similar in strength of progestogenic effect to those of normal progesterone production and levels during the luteal phase of the menstrual cycle in premenopausal women (which are about 25 mg/day and 15 ng/mL, respectively). In relation to the preceding, when CPA is used as a progestogen in cisgender women, for instance in birth control pills and menopausal hormone therapy preparations, it is formulated at a dose of 1 or 2 mg per tablet (Wiki).
In contrast to its progestogenic activity, CPA is far less potent as an androgen receptor antagonist (Wiki). When used as an antiandrogen, it is generally given at a dosage of 50 to 300 mg/day, both in cisgender women and men. A dosage of 50 to 100 mg/day is typical for androgen-dependent skin and hair conditions like acne and hirsutism in women and a dosage of 100 to 300 mg/day is typically used for prostate cancer in men (specifically 100–200 mg/day for CPA combined with castration and 200–300 mg/day for CPA monotherapy) (Wiki). As such, CPA is generally formulated at a dose of 50 or 100 mg per tablet for use in androgen-dependent conditions (Wiki). As an antiandrogen, CPA has a dual mechanism of action of both suppressing testosterone levels via its progestogenic activity at low doses and additionally blocking the actions of testosterone directly at the androgen receptor at higher doses.
Because CPA is so much more potent as a progestogen than as an androgen receptor antagonist, there is profound overdosage of progestogenic effect when CPA is used as an antiandrogen at typical clinical dosages. This is described in the following three literature excerpts by Jürgen Hammerstein, one of the scientists who developed CPA (Hammerstein et al., 1975; Hammerstein, 1990; Hammerstein, 1979):
Like chlormadinone acetate, its parent compound, CPA is also a strong progestogen with the endometrial transformation dose of both drugs being between 20 and 30 mg. […] To take full therapeutic advantage of its antiandrogenicity, CPA must be administered in doses per month that are 30 times the physiological equivalent of progesterone production in the cycle. CPA, although the most useful compound available in this field at the moment, cannot be considered therefore an ideal antiandrogen, all the more as some of the side effects may be related to the progestational overdosage rather than to the administered antiandrogenic activity. […] Adverse reactions like tiredness, lassitude, and increase in body weight are possibly due to the enormous overdose of progestational activity in the formula which is necessary to take full advantage of the antiandrogenicity of CPA.
Fixson (1963) tested CPA in ovariectomized women after pre-treatment with oestrogens; with a transformation dose of 20–30 mg this proved a powerful progestogen. The potency of CPA in the menses delay test is not exactly known, but has been estimated to be below 1 mg/day (Miller and Jacobs 1986). In relation to this progestational potency, its antiandrogenicity must be considered rather weak. Thus, in order to take full advantage of the latter, 100 mg CPA must be given daily, i.e. three times the cyclic transformation dose per day (Hammerstein and Cupceancu 1969); notably, this parameter is equivalent to the total progesterone production of a corpus luteum throughout its entire cyclic life span.
CPA may be characterized endocrinologically as possessing strong progestational [and] moderate anti-androgenic […] potencies. […] Its progestational activity, in terms of the transformation dose in the oestrogen-primed human endometrium, is 20–30 mg [per month/cycle] which is comparable to that of chlormadinone acetate and other strong progestogens. To take full clinical advantage of its anti-androgenicity not less than 50–100 mg CPA must be taken orally per day, which totals 2 to 3 times the progestational activity the female organism is exposed to throughout a complete ovulatory menstrual cycle. Thus unless much lower and less efficacious doses of CPA are used, a tremendous progestational overdosage must be accepted. […] As already pointed out CPA is endocrinologically not a well-balanced compound because of the strong preponderance of the progestational over the anti-androgenic potency. A way to avoid the heavy progestogen overdosage inherent with the high-dose reverse sequential therapy would be to combine the low-dose contraceptive formulation just mentioned with a pure anti-androgen such as free cyproterone. […] It must be emphasized that CPA is far from being an ideal drug for the anti-androgenic treatment of hirsutism because its progestational potency is much too strong and it is not effective when administered topically. Therefore it is worthwhile looking for better-balanced anti-androgenic compounds for the future.
The massive overdosage of progestogenic effect that occurs at such doses of CPA is likely responsible for the known adverse effects and risks of higher doses of CPA (Wiki). Examples of these side effects include fatigue, depression, weight gain, high prolactin levels (Wiki), benign brain tumors (Aly, 2020; Wiki; Table; Table), blood clots (Wiki), and cardiovascular problems (Wiki). Such risks are dose-dependent and have not been associated with 1 or 2 mg/day CPA (with the exception of an expected increase in the risk of blood clots in combination with oral estrogens for birth control or menopausal hormone therapy). The risk of liver toxicity with CPA is also dose-dependent, with elevated liver enzymes occurring mostly only at a dosage of 20 mg/day and above and rare cases of liver failure occurring almost exclusively at dosages of 100 mg/day and above (Wiki; Table). As such, there is good rationale for using the lowest possible effective dosage of CPA, an approach that is likely to minimize risks.
In transfeminine people, CPA has historically been used at a dosage of 50 to 100 mg/day (e.g., Moore, Wisniewski, & Dobs, 2003). Some earlier papers have recommended even higher doses of CPA, for instance 100 to 150 mg/day (Asscheman & Gooren, 1993). In 2017, the Endocrine Society published the latest edition of their clinical practice guidelines on hormone therapy for transgender people and reduced their recommended dosage of CPA from 50–100 mg/day to 25–50 mg/day (Hembree et al., 2017; Hembree et al., 2009). This was motivated in part by increasing knowledge and awareness of the risks of higher doses of CPA and by findings that these lower doses of CPA were still effective. However, it is likely that even these new lower dosages are still far in excess of what is really needed.
Testosterone Suppression with Low and High Doses
Progestogens by themselves, including CPA, are able to considerably suppress testosterone levels in gonadally intact people assigned male at birth. Around a dozen small and low-quality but nonetheless notable studies of low-dose CPA from the 1970s and early 1980s found that 5 to 10 mg/day CPA suppressed testosterone levels by about 40 to 70% in healthy young men (Table 1). A couple of individual studies notably reported virtually identical suppression of testosterone levels with 5 mg/day versus 10 mg/day CPA (both ~50% suppression) (Wang & Yeung, 1980; Graph) and with 10 mg/day versus 20 mg/day CPA (both ~60–70% suppression) (Koch et al., 1976; Koch et al., 1975; Graph). This lack of additional testosterone suppression with a doubling of dosage within studies suggests that testosterone suppression with CPA might have actually been maximal at a dosage of only 5 or 10 mg/day. A more modern study, which used a newer and more reliable analytic method for quantification of blood testosterone, found that 10 mg/day CPA suppressed testosterone levels by 66%, from about 600 ± 150 ng/dL to about 185 ng/dL (Meriggiola et al., 2002a; Graph). Similarly, another more modern study found that 10 to 20 mg/day CPA suppressed testosterone levels by 65%, from about 431 ng/dL to about 149 ng/dL, with no reported differences between doses (Zitzmann et al., 2017; Graph).
Table 1: Levels of testosterone and other sex hormones with CPA at low doses (5–30 mg/day):
Treatment and subjects
Findings
Source(s)
30 mg/day CPA in 5 normal males
T decreased “remarkably”. Exact values not given, but has graphs of T levels in a few individuals. After 30 mg/day, 5 mg/day was tried in one case and was not as effective in suppressing sperm production or T. Also reported decreases in gonadotropin excretion.
10 or 20 mg/day CPA in 15 normal healthy fertile males (age 25–35 years) (7 in 10 mg/day group and 8 in 20 mg/day group)
“Androgens (mainly T)” decreased by 60% for both 10 and 20 mg/day. Inconsistent changes in LH and slight decrease in FSH. Exact values not given, except in graphs.
10 mg/day CPA in 10 young healthy fertile men (age mean 27.2 ± 3.2 (range 21–35) years)
T decreased by 70%, DHT by 50%, LH by 30%, and FSH by 40%, while PRL increased by 75%. T was 495 ± 66 ng/dL before, 154 ± 23 ng/dL after 4 weeks, and 187 ± 38 ng/dL after 12 weeks. Also has values and graphs for other hormones.
20 mg/day CPA in 10 healthy males (age 26–55 years)
T decreased by 73% (range 71–75%), from 482 ng/dL (range 410–560 ng/dL) to 130 ng/dL (110–162 ng/dL). DHT decreased by 51% (range 47–55%), LH by 39% (range 34–45%), FSH by 66% (range 47–78%), 17-OH-P4 by 59%, A4 by 30%, TS by 34%, and DHTS by 35%. Also has exact values and graphs for other hormones.
5 or 10 mg/day CPA in 7 males (4 in each group; 1 received both 5 and 10 mg/day CPA at different times)
T change was “−40%” or “–50%”. At 5 mg/day, T was 745 ng/dL before, 460 ng/dL with treatment (–38%), and 668 ng/dL after discontinuation. At 10 mg/day, T was 708 ng/dL before, 398 ng/dL with t (reatment–44%), and 670 ng/dL after discontinuation. Also reported LH and FSH levels.
0, 5, or 10 mg/day CPA in 25 normal healthy males (age 20–51 years); 7 in 5 mg group (mean 37 ± 10 years), 8 in 10 mg group (mean 32 ± 8 years), 10 in 0 mg group (mean 32 ± 10 years)
At 5 mg/day, T decreased from 663 ± 120 ng/dL to 320 ± 160 ng/dL (−52%), and at 10 mg/day, T decreased from 692 ± 180 ng/dL to 340 ± 160 ng/dL (−51%). E2 decreased in parallel to T. At 5 mg/day, LH decreased from 2.1 ± 0.7 IU/L to 1.4 ± 0.5 IU/L (−33%), and at 10 mg/day, LH decreased from 2.3 ± 1.0 IU/L to 1.2 ± 0.5 IU/L (−48%). At 5 mg/day, FSH decreased from 3.1 ± 1.9 IU/L to 1.8 ± 0.9 IU/L (−42%), and at 10 mg/day, FSH decreased from 2.7 ± 1.0 IU/L to 1.5 ± 0.7 IU/L (−44%).
10 or 25 mg/day CPA in 4 healthy men (age 29–37 years); 3 in 10 mg group, 1 in 25 mg group
T “slightly reduced”. E “more significantly lowered”. LH not significantly changed. FSH “reduced” in “more or less all cases”. Exact hormone levels not given, but graphs provided with the values.
10 mg/day CPA (also placebo and 2, 5, and 10 mg/day dienogest) in 5 healthy men in each group
With CPA, T decreased from ~600 ± 150 ng/dL to ~185 ng/dL (–66 ± 4%). Also reported LH, FSH, and SHBG, as well as hormonal changes with placebo and dienogest (2, 5, and 10 mg/day).
10 or 20 mg/day CPA in 14 healthy young men (7 in each group)
T decreased from ~431 ng/dL at baseline to ~149 ng/dL with CPA (–65%) for the 10 and 20 mg/day doses combined. Values for dose subgroups not given. No significant differences between LH/FSH suppression between groups (which is indirectly suggestive of no differences in T suppression as well). Also reported hormone levels with other progestins.
Studies with other progestogens, such as desogestrel, dienogest, and medroxyprogesterone acetate, have consistently found that maximal suppression of testosterone levels in men occurs at a dosage that is between 5 and 10 times that of the ovulation-inhibiting dosage in cisgender women (Wiki; Wiki; Wiki). Another study is likewise suggestive of this for norethisterone acetate and levonorgestrel (Zitzmann et al., 2017; Graph). Along similar lines, doses of progestogens investigated for use in male hormonal contraception, in which the goal is antigonadotropic effects and the lowest fully effective dose is targeted, have been noted as being between 5 and 12 times the doses used in cisgender women (Foegh, 1983). Based on an ovulation-inhibiting dosage of CPA of 1 mg/day, these findings would imply that suppression of testosterone levels with CPA would likely be maximal at a dose of between 5 and 10 mg/day. In accordance, this dose range matches up with the findings of the studies above.
Although progestogens can considerably suppress testosterone levels at maximally effective dosages, it has been found that a “recovery” or “escape phenomenon”, in which testosterone levels eventually increase back to higher levels, occurs when progestogen monotherapy is used on a long-term basis. This has most notably been observed with the related progestogen megestrol acetate (Wiki), but has also been seen with CPA (Goldenberg & Bruchovsky, 1991; Saborowski, 1987; Jacobi, Tunn, & Senge, 1982). In one of these studies, testosterone levels were initially suppressed by CPA by about 70%, but increased back to about 50% of baseline between 6 and 12 months of therapy, remaining stable thereafter up to 24 months. The testosterone escape phenomenon should be kept in mind in the context of progestogen monotherapy for testosterone suppression. In contrast to progestogen monotherapy, this phenomenon has not been associated with combined estrogen and progestogen therapy.
Testosterone Suppression in Combination with Estrogen
CPA is generally used in combination with an estrogen in transfeminine people. Estrogens suppress testosterone levels similarly to progestogens. The combination of an estrogen and a progestogen is synergistic in terms of testosterone suppression and results in suppression of testosterone levels with lower doses than with either an estrogen or progestogen alone (Fink, 1979; Geller & Albert, 1983; Bastianelli et al., 2018). Although estrogens can suppress testosterone levels to an equivalent extent as surgical or medical castration (i.e., orchiectomy or GnRH agonists/antagonists), this usually requires relatively high estrogen levels, for instance in the range of 200 to 500 pg/mL (Wiki; Graphs). Because of the high and supraphysiological estradiol levels required for maximal or near-maximal suppression of testosterone levels, lower doses of estradiol are frequently combined with antiandrogens and/or progestogens to block or suppress remaining testosterone levels instead.
CPA, as mentioned earlier, leads to an incomplete suppression of plasma testosterone levels, which decrease by about 70% and remain at about three times castration values. In a very systematic approach to the problem, Rennie et al. (59) investigated and compared 12 different procedures of androgen deprivation. These authors found that the combination of CPA with an extremely low dose (0.1 mg/d) of [diethylstilbestrol (DES)] led to a very effective withdrawal of androgens in terms of plasma testosterone and tissue dihydrotestosterone. The same group later showed that 200 mg of CPA, and even 100 mg/day, was sufficient to achieve a similar endocrine response, which was correlated to very favorable clinical responses in a Phase II situation (60,61). The approach has many potential advantages, and, from an endocrinological point of view, is very logical: this regimen combines the testosterone-reducing effects of two compounds, therefore, only small amounts of estrogen are required to bring down plasma testosterone to approximately castrate levels. Once castrate levels have been achieved, only low doses of CPA are necessary to counteract remaining androgens, mainly of adrenal origin. The regimen was shown to be associated with few side effects and a very low cost. The combination of low-dose CPA with low-dose DES was never studied in a Phase III situation in comparison to standard management. Considering the endocrine results and the observations in patients treated with this regimen (60), this combination treatment is very likely to be competitive with other standard forms of therapy.
A 2016 study of 50 mg/day CPA and 1 to 2 mg/day transdermal estradiol gel in transfeminine people showed that estradiol levels of about 45 pg/mL with CPA were insufficient to achieve female/castrate levels of testosterone, instead resulting in testosterone levels of about 120 to 190 ng/dL (Gava et al., 2016; Graph). Conversely, estradiol levels of about 85 pg/mL with CPA achieved complete suppression of gonadal testosterone production, with resulting testosterone levels of about 20 ng/dL. As such, a certain minimum level of estradiol with CPA appears to be required for complete testosterone suppression. A 2019 study of CPA and oral estradiol valerate in transfeminine people indicated that testosterone levels were still fully suppressed with median estradiol levels of 76 pg/mL and 25th percentile estradiol levels of 63 pg/mL (Angus et al., 2019; Graph).
Figures 5–7: Testosterone levels with CPA plus low doses/levels of estrogens in men and transfeminine people. Sources: Top-left: Goldenberg et al. (1988). Top-right: Gava et al. (2016). Bottom: Angus et al. (2019). See also on Wikipedia: Gallery. Note for the graph on the top right that the mean transdermal estradiol dosage increased between 6 and 12 months and this was likely responsible for the improvement in testosterone suppression.
Fung and colleagues showed that the combination of either 25 or 50 mg/day CPA with a moderate dosage of oral estradiol (~3.5 mg/day) or transdermal estradiol (~3.5 mg/day gel or ~100 μg/day patch) resulted in equivalent and complete suppression of gonadal testosterone production (~95% suppression of testosterone levels) in transfeminine people (Fung, Hellstern-Layefsky, & Lega, 2017). These dosages of estradiol would be expected to achieve estradiol levels of around 100 pg/mL on average (Aly, 2020; Wiki). This study was notably published 6 months before the 2017 second edition of the Endocrine Society guidelines were released (Hembree et al., 2017), and was probably responsible for the decrease in their recommended dosage of CPA from 50–100 mg/day to 25–50 mg/day.
Few studies to date have assessed testosterone suppression with low-dose CPA in combination with a low or moderate dosage of an estrogen. However, based on the fact that 5 to 10 mg/day CPA alone is probably maximal in terms of suppression of testosterone levels, it is likely that such dosages of CPA will be similarly effective as higher dosages. In accordance, studies of 5 to 12.5 mg/day CPA plus upper physiological replacement dosages of testosterone have demonstrated undetectable gonadotropin levels (<0.5 IU/L) and hence complete suppression of testicular function in healthy young men (Meriggiola et al., 1998; Meriggiola et al., 2002b). Estradiol is a more powerful antigonadotropin than testosterone (Wiki), so these findings probably apply to CPA in combination with physiological replacement levels of estradiol as well (e.g., mean estradiol levels of 100–200 pg/mL).
Accordingly, Meyer et al. (2020) assessed a dosage of CPA in combination with estradiol in 155 transfeminine people and found no difference in testosterone levels with 10, 25, or 50 mg/day CPA; testosterone levels were strongly suppressed with all three doses (to about 15–20 ng/dL on average, or into the lower end of the normal female range). The estradiol forms and doses used in this study were oral estradiol valerate (median 6 mg/day, range 3–10 mg/day), transdermal estradiol gel (median 2.25 mg/day, range 1.5–6 mg/day), and transdermal estradiol patches (100 μg/day in all cases). Estradiol levels were about 100 pg/mL on average, with an interquartile range (i.e., difference between 75th and 25th percentiles) of about 100 pg/mL. This study demonstrates that, provided estradiol levels are adequate, no more than 10 mg/day CPA is needed to fully suppress testosterone levels in transfeminine people. Another study likewise found no difference between <20 mg/day and >50 mg/day CPA in terms of testosterone suppression in transfeminine people (Even-Zohar et al., 2020).
Even doses of CPA lower than 5 mg/day (e.g., 2 mg/day) may be usefully effective for testosterone suppression if combined with sufficient levels of estradiol, although this has not been studied and remains to be validated. But there is certainly precedent for the notion when looking at studies with other progestogens. As an example, one study using 10 mg/day oral medroxyprogesterone acetate (which is roughly equivalent to 1 mg/day CPA in terms of ovulation inhibition in premenopausal women; Table) observed 63% lower testosterone levels (215 ng/dL vs. 79 ng/dL) when added to estradiol and spironolactone therapy in transfeminine people (Jain, Kwan, & Forcier, 2019). Analogous effects on testosterone levels would be anticipated for very-low-dose CPA. Moreover, such dosages of CPA would have the advantage of actually being physiological in terms of progestogenic exposure.
The androgen receptor antagonism of CPA is relatively weak in terms of potency; dosages of CPA of 50 to 300 mg/day seem to be necessary for meaningful or considerable androgen receptor antagonism. Unfortunately, such doses also result in extreme progestogenic overdosage and are associated with considerably greater risks and adverse effects. As a result, the use of such doses of CPA should no longer be considered advisable. Instead, CPA should be used at lower doses simply as a progestogen to suppress testosterone levels. As such, the highest effective dosage of CPA for testosterone suppression, which is probably about 10 mg/day or less (12.5 mg/day also being acceptable), should be around the maximal dosage of CPA that is used in transfeminine people.
It should be emphasized that since the combination of an estrogen and CPA can easily suppress testosterone levels well into the female/castrate range (typically to below average female levels), there isn’t necessarily a requirement for concomitant androgen receptor blockade. In any case, if androgen receptor antagonism to neutralize the remaining female/castrate levels of testosterone is still necessary or desired (e.g., to treat persisting acne or for some other purpose), a low dosage of a non-progestogenic androgen-receptor antagonist like spironolactone (e.g., 100–200 mg/day) or bicalutamide (e.g., 12.5–25 mg/day) can be added to CPA to more safely achieve this than use of higher CPA doses.
Recommended Dosages
Dosage for Testosterone Suppression
Estrogen Plus Cyproterone Acetate
The following recommended dosages of CPA in transfeminine people are for the combination of CPA with an estrogen and are specifically for achieving maximal suppression of testosterone levels:
Table 2: Recommended doses of CPA in combination with estrogen for maximal testosterone suppression in transfeminine people:
Form
Min. dosage
Max. dosage
Amount
10 mg tablets
5 mg/day
10 mg/day
1/2 of a tablet to 1 whole tablet per day
50 mg tablets
6.25 mg/day
12.5 mg/day
1/8th of a tablet to 1/4th of a tablet per day
Start with the minimum dosage of CPA for one month. After one month, have testosterone levels tested and confirm that they are in the normal female/castrate range (<50 ng/dL). Regardless of dosage, a concomitant minimum estradiol level of around 65 pg/mL needs to be attained in order to allow for complete suppression of testosterone levels with CPA. If testosterone levels aren’t sufficiently suppressed after a month and estradiol levels are adequate, increase to the maximum CPA dosage and re-check testosterone levels after another month. Alternatively, the dosage of estradiol can be increased instead; higher estradiol levels result in greater testosterone suppression as well.
Cyproterone Acetate Alone
The use of CPA alone (i.e., as a monotherapy for testosterone suppression) is not recommended due to the risk of decreased bone mineral density and other symptoms of sex-hormone deficiency (Wiki; Aly, 2019). In any case, the recommended dosages for CPA without an estrogen are essentially the same as those listed above of the combination of an estrogen with CPA for testosterone suppression. However, the higher CPA dose (10–12.5 mg/day) may be preferable for good measure in this scenario.
Dosage for Progestogenic Effects
The following recommended dosages of CPA in transfeminine people are for progestogenic effects similar to normal physiological exposure (equivalent of luteal-phase progesterone levels):
Table 3: Recommended doses of CPA for physiological progestogenic effects in transfeminine people:
Form
Dosage
Amount
10 mg tablets
2.5 mg/day
1/4th of a tablet per day
50 mg tablets
3.125 mg/day
1/16th of a tablet per day
Achieving Desired Dosages
CPA is available pharmaceutically most widely as 50-mg tablets. This can make achieving desired low doses of CPA more difficult. For splitting CPA tablets into small fractions, a pill cutter can be used. Additionally, CPA can be taken once every 2 or 3 days instead of once every day to help further divide doses. It is notable that CPA has a relatively long half-life in the body of about 1.5 to 2 days (but possibly up to 4 days) (Wiki; Graph). Hence, taking it once every other day instead of once per day, or even less frequently like once every 3 days, has sound basis and is likely to be entirely viable.
Updates
Update 1: GoLoCypro Study (In-Progress)
The GoLoCypro study (2019–2022) (more info) is being conducted by Dr. Judith Dean at the University of Queensland in Australia. It’s assessing the influence of estradiol plus CPA on testosterone levels at five different CPA dose levels (12.5 mg 2x/week, 12.5 mg/2 days, 12.5 mg/day, 25 mg/day, and 50 mg/day) in a total of 120 to 350 transfeminine people. CPA doses are being titrated to the minimum that maintain testosterone levels within the therapeutic goal range of 0.5 to 1.5 nmol/L (14–43 ng/dL). The study is among the first dose-ranging studies of CPA in transfeminine people to be conducted and is eagerly anticipated due to the valuable information that it should provide in terms of the minimum effective dosage of CPA for adequate testosterone suppression in transfeminine hormone therapy.
Update 2: Kuijpers et al. (2021) and Even Zohar et al. (2021)
Kuijpers, S. M., Wiepjes, C. M., Conemans, E. B., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2021). Toward a lowest effective dose of cyproterone acetate in trans women: Results from the ENIGI study. The Journal of Clinical Endocrinology & Metabolism, 106(10), e3936–e3945. [DOI:10.1210/clinem/dgab427]
The study employed estradiol (2–6 mg/day oral (as estradiol valerate), 50–150 μg/day patch, or gel) plus five different dose levels of CPA—0 mg/day (no CPA), 10 mg/day, 25 mg/day, 50 mg/day, and 100 mg/day. It found incompletely suppressed testosterone in the no CPA group but full and equivalent testosterone suppression with all doses of CPA. The results were as follows:
CPA dosage
0 mg/day
10 mg/day
25 mg/day
50 mg/day
100 mg/day
Initial subjects (n)
34
4
234
599
11
Dose increased (n)
16
1
11
2
0
Dose decreased (n)
0
0
4
40
7
T levels (nmol/L)
5.5
0.9
0.9
1.1
0.9
T levels (ng/dL)
~160
~26
~26
~32
~26
T <2 nmol/L [<~58 ng/dL] (%)
46.3
92.3
96.2
93.4
100.0
Abbreviations: T = testosterone.
The total numbers of subjects and blood tests after CPA dose increases/decreases were not provided. Hence, the exact total number of people and tests for the 10 mg/day group can’t be stated with certainty. The total number of tests for this group was at least 13 based on the testosterone suppression percentage provided however (92.3% or 12/13 but could potentially be 24/26, etc.). Regarding the small number of subjects/tests for the 10 mg/day group, the authors stated the following:
This study is part of the ENIGI initiative, a multicenter prospective cohort study. The main treatment protocol for trans women in this study was 50 mg of CPA daily combined with estrogens. In the first year of study inclusion, a few participants received a dose of 100 mg of CPA. Shortly thereafter, inhospital protocol changed to 50 mg of CPA. As more health concerns related to CPA use were raised over the years, the dose was further lowered from 50 mg to 25 mg and, finally, to 10 mg. However, due to the coronavirus (COVID-19) pandemic, limited results of participants with 10 mg of CPA were available for analysis.
Besides testosterone suppression, the study found that 10 mg/day CPA had less influence on prolactin and high-density lipoprotein (HDL) cholesterol levels than the higher doses of CPA. The study also assessed liver enzyme levels but found no differences between CPA doses.
The authors concluded with the following:
In conclusion, in this cohort of trans women, 10 mg of CPA was found to be effective in lowering testosterone concentrations to the range observed in cis women. A dose of 10 mg was equally effective as higher doses, was found to have less influence on prolactin concentrations and allows higher HDL-C concentrations to be maintained. While GnRH agonists are preferred over CPA due to the fewer associated long-term side effects, this study shows that CPA at a low dose is a viable option when GnRH agonists are contra-indicated, not available, or not reimbursed. Future research should focus on assessing the effectiveness of an even lower dose of CPA (e.g., 5 mg) and the potential long-term side effects.
Around the same that this study was published, Guy T’Sjoen (one of the authors of the study) and other colleagues in a review of optimal hormone therapy for transfeminine people recommended a dosage of no more than 10 or 12.5 mg/day CPA for no longer than 2 years (Glintborg et al., 2021). T’Sjoen is notable in being regarded as one of the foremost experts in transgender medicine and is a coauthor of the Endocrine Society transgender care guidelines (Hembree et al., 2017).
Shortly after the study of Kuijpers and colleagues and also in June 2021, Even Zohar and colleagues in Israel published the following study on low doses of CPA in transfeminine people:
Even Zohar, N., Sofer, Y., Yaish, I., Serebro, M., Tordjman, K., & Greenman, Y. (2021). Low-Dose Cyproterone Acetate Treatment for Transgender Women. The Journal of Sexual Medicine, 18(7), 1292–1298. [10.1016/j.jsxm.2021.04.008]
This study was initially reported as a conference abstract in May 2020 (Even-Zohar et al., 2020).
In the introduction section of the paper, the authors stated the following:
Treatment guidelines published by several organizations are available and assist clinicians in treating transgender women.4,7−9 A wide range of regimens for CPA administration have been proposed. By and large, the recommended doses have decreased over the years: doses of 50–100 mg/day were suggested in the 2009 Endocrine Society Guidelines,10 and amended to 25–50 mg/day in 2017.7 The proposed CPA doses were 12.5–25 mg/day in the 2019 guidelines of the Australian Professional Association for Transgender Health,4 and they were amended to 10–50 mg/day in the 2020 guidelines of the European Society for Sexual Medicine.8 There are no publications on data that compare different doses of CPA for efficacy or safety.
The researchers found that estradiol plus low-dose CPA (10–20 mg/day) suppressed testosterone levels to an equivalent extent as estradiol plus high-dose CPA (50–100 mg/day). Testosterone levels were suppressed into the female/castrate range or near so in both groups (generally ≤2 nmol/L or ≤58 pg/mL). Of the 38 transfeminine people on low-dose CPA, 32 (84%) were on 10 mg/day CPA and 6 (16%) were on 20 mg/day CPA (mean dose 11.6 ± 3.7 mg/day). Estradiol was given as transdermal estradiol patch (mean dose 83.7 ± 36.5 μg/day), transdermal estradiol gel (mean dose 3.8 ± 1.2 g/day), or oral estradiol (mean dose 4.1 ± 1.7 mg/day). Mean estradiol levels ranged from ~110 to 350 pmol/L (~30–95 pg/mL) in the low- and high-dose CPA groups over the follow-up period. Besides showing equivalent testosterone suppression, prolactin levels were significantly lower with low-dose CPA than with high-dose CPA (398 ± 69 mIU/mL vs. 804 ± 121 mIU/mL at 12 months of hormone therapy, respectively).
Based on their findings, the authors stated the following:
We suggest an adjustment of current clinical practice guidelines to recommend lower doses of CPA for the treatment of transgender women.
Both Kuijpers et al. (2021) and Even Zohar et al. (2021) claimed to be the first to demonstrate the efficacy of low-dose CPA in transfeminine people. However, that achievement actually appears to belong to Meyer et al. (2020), who in February 2020 found that estradiol plus 10, 25, or 50 mg/day CPA gave equivalent testosterone suppression across CPA doses in transfeminine people.
Although their study was not about CPA and testosterone suppression, Lim et al. (2020) reported in May/July 2020 that testosterone levels in transfeminine people were median (IQR) 0.6 (0.4–1.0) nmol/L for oral estradiol and 0.9 (0.7–1.6) nmol/L for transdermal estradiol in a mixed group of transfeminine people (n=26 total) on estradiol plus low-dose CPA (12.5 (12.5–18.8) mg/day) (n=14), estradiol alone post-gonadectomy (n=9), and estradiol plus spironolactone (n=3).
In December 2021, the following case report of fatal liver failure with low-dose CPA was published:
Kumar, P., Reddy, S., Kulkarni, A., Sharma, M., & Rao, P. N. (2021). Cyproterone acetate induced Acute liver failure: Case report and review of the literature. Journal of Clinical and Experimental Hepatology, 11(6), 739–741. [DOI:10.1016/j.jceh.2021.01.003]
The case report describes a 30-year-old cisgender woman who was on 25 mg/day CPA for treatment of hirsutism (excessive facial/body hair growth) for 6 months and developed acute liver failure. Four days following hospitalization, she died. This is the second published case report of liver toxicity with CPA at a dosage below 100 mg/day (the first and only other case was at 50 mg/day) (Wiki; Table). It is also the first report of liver failure in a cisgender woman taking CPA. The case indicates that CPA even at a relatively low dose of 25 mg/day is not fully safe in terms of liver toxicity. It further emphasizes the importance of using the lowest effective doses of CPA in transfeminine people (no more than 10–12.5 mg/day).
Update 4: Coleman et al. (2022) [WPATH SOC8 Guidelines]
In September 2022, the World Professional Association for Transgender Health (WPATH) Standards of Care for the Health of Transgender and Gender Diverse People Version 8 (SOC8) were published and made recommendations for transgender hormone therapy for the first time (Coleman et al., 2022). These guidelines recommended a dose of CPA of 10 mg/day in transfeminine people (Coleman et al., 2022). This dose is substantially lower than previous doses recommended by transgender care guidelines and is the first time that major guidelines have recommended a CPA dosage this low. The WPATH SOC8 cited Kuijpers et al. (2021) in support of this recommendation (though notably not Even Zohar et al. (2021) or Meyer et al. (2020)) and also discussed the dose-dependent risks of CPA such as meningiomas and high prolactin levels (Coleman et al., 2022). Considering the key position and importance of the WPATH SOC in transgender health, it is likely that lower CPA doses in transfeminine hormone therapy will now be widely adopted throughout the world. Continued use of higher CPA doses should be considered out of step with current accepted evidence-based practice.
Update 5: Collet et al. (2023)
In October 2022, a study more carefully assessing androgen suppression with estradiol plus CPA in transfeminine people was published:
Collet, S., Gieles, N., Wiepjes, C. M., Heijboer, A. C., Reyns, T., Fiers, T., Lapauw, B., den Heijer, M., & T’Sjoen, G. (2023). Changes in serum testosterone and adrenal androgen levels in transgender women with and without gonadectomy. The Journal of Clinical Endocrinology & Metabolism, 108(2), 331–338. [DOI:10.1210/clinem/dgac576]
In the study, 275 transfeminine people were treated with estradiol plus CPA, and levels of total testosterone, free testosterone, and the adrenal androgensdehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEA-S), and androstenedione (A4) were measured using liquid chromatography–mass spectrometry (LC–MS) at baseline and during follow-ups at 3 months, 12 months, 2 to 4 years, and after surgical gonadal removal (at which time CPA was discontinued). Estradiol was measured both with LC–MS (Amsterdam clinic) and with immunoassays (Ghent clinic). The forms and doses of estradiol used were most commonly oral estradiol valerate 4 mg/day or a transdermal estradiol patch 100 μg/day, while the dosage of CPA was usually 25 or 50 mg/day. About half of the transfeminine people eventually underwent surgical gonadal removal, usually after 2 years of hormone therapy.
Median estradiol levels ranged from 49 to 75 pg/mL (180–275 pmol/L) with LC–MS and from 63 to 69 pg/mL (232–255 pmol/L) with immunoassays at different follow-ups. After 3 months of hormone therapy, total testosterone decreased by 97.1%, from 536 ng/dL (18.6 nmol/L) to 12 ng/dL (0.40 nmol/L), and free testosterone decreased by 98.3%, from 109 pg/mL (378 pmol/L) to 2.0 pg/mL (7.1 pmol/L). Thereafter, total and free testosterone levels remained stable. Levels of DHEA, DHEA-S, and A4 decreased by 24.9 to 28.0%, 20.1 to 23.5%, and 36.5%, respectively, and likewise did not further change after the first 3 to 12 months of hormone therapy. No changes in androgen levels occurred upon surgical gonadal removal with discontinuation of CPA. The authors noted that testosterone levels in the transfeminine people on hormone therapy in the study were similar to or lower than those in cisgender women.
Update 6: Warzywoda et al. (2024) [GoLoCypro Study]
The GoLoCypro study, by Judith Dean and colleagues, was published online in February 2024:
Warzywoda, S., Fowler, J. A., Wood, P., Bisshop, F., Russell, D., Luu, H., Kelly, M., Featherstone, V., & Dean, J. A. (2024). How low can you go? Titrating the lowest effective dose of cyproterone acetate for transgender and gender diverse people who request feminizing hormones. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2317395]
The following are some noteworthy excerpts from the paper:
Of participants who completed the protocol, 74.0% (34/46) were able to achieve the target T-range (0.5–1.5 nmol/L) and 41.3% (19/46) were titrated to the lowest CPA level (12.5 mg cyproterone twice weekly).
Almost all participants who completed the protocol (91.3.0%, 42/46) recorded their CPA levels as level 3 (12.5 mg daily/25 [mg] alternate days) or lower, with 69.0% (29/42) of these being able to achieve the target T-range. Of those that completed, 41.3% (19/46) were able to achieve the lowest CPA level (12.5 mg cyproterone twice week) with 57.9% (11/19) being able to achieve the target T-range.
The study findings showed that for some patients, CPA doses as low as 12.5 mg on alternate days or less can successfully reduce testosterone to pre-menopausal ranges whilst ensuring testosterone was not over-suppressed.
Our study found that doses of CPA lower than the standard dose (12.5 mg CPA daily and/or 25 mg alternate days) were achievable for suppression of testosterone. Several studies have supported this finding that a lower dosage (10 mg CPA daily) is effective in testosterone reduction in individuals undergoing hormone feminization (Even Zohar et al., 2021; Kuijpers et al., 2021). While not all individuals within our study were able to titrate down CPA dosages, almost a quarter of participants who completed the protocol were achieving target T-ranges on 12.5 mg CPA twice weekly (equivalent to 3.5 mg/daily). To our knowledge ours is the first study to demonstrate that doses lower than 10 mg/daily as well as alternate days or twice weekly CPA are clinically effective in maintaining testosterone reduction within target ranges.
Update 7: More New Low-Dose CPA Studies (2023–2025)
Other new studies of low-dose CPA in transfeminine people have also been published in 2023 and 2024:
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (November 2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (November 2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Flamant, T., Vervalcke, J., & T’Sjoen, G. (November 2023). Dose Reduction of Cyproterone Acetate in Trans Women and the Effect on Patient-reported Outcomes: Results from the ENIGI Study. Endocrine Abstracts, 97 [Belgian Endocrine Society 2023], 5–5 (abstract no. 007). [URL] [PDF]
Korpaisarn, S., Arunakul, J., Chaisuksombat, K., & Rattananukrom, T. (2023). A Low Dose Cyproterone Acetate In Feminizing Hormone Treatment. Journal of the Endocrine Society, 7(Suppl 1), A1098–A1099 (abstract no. SAT397/bvad114.2068). [DOI:10.1210/jendso/bvad114.2068]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Bonadonna, S., Amer, M., Foletti, F., Federici, S., Persani, L., Bonomi, M. (2025). Evaluation of Antiandrogen Therapy Effectiveness in Transgender individuals Assigned Male At Birth (AMAB). EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
de Leon-Durango, R., Hernandez-Lazaro, A., Rios-Gomez, C., Santana-Ojeda, B., Molinero-Marcos, I., Arnas-Leon, C., Hernandez-Hernandez, I., Acosta-Calero, C., Kuzior, A., Perez-Rivero, J., Perez-Garcia, M., & Martinez-Martin, F. (2024). P194 Very Low-dose Cyproterone Acetate (12.5 Mg/day) is Effective as Androgen Blocker; Well Tolerated And Not Associated With Hypertension Development in Young Female Transgender People. Journal of Hypertension, 42(Suppl 3), e133–e133. [DOI:10.1097/01.hjh.0001063648.69793.7c]
Korpaisarn, S., Arunakul, J., Chaisuksombat, K., & Rattananukrom, T. (2024). Effectiveness of low dose cyproterone acetate compared to standard dose in feminizing hormone treatment: a single institutional retrospective pilot study. Sexual Medicine, 12(4), qfae063. [DOI:10.1093/sexmed/qfae063]
References
Aly. (2019). An Exploration of Possibilities for Hormone Therapy in Non-Binary Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Aly. (2020). Recent Developments on Cyproterone Acetate and Meningioma Risk Out of France and Implications for Transfeminine People. Transfeminine Science. [URL]
Angus, L., Leemaqz, S., Ooi, O., Cundill, P., Silberstein, N., Locke, P., Zajac, J. D., & Cheung, A. S. (2019). Cyproterone acetate or spironolactone in lowering testosterone concentrations for transgender individuals receiving oestradiol therapy. Endocrine Connections, 8(7), 935–940. [DOI:10.1530/ec-19-0272]
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Asscheman, H., & Gooren, L. J. (1992). Hormone Treatment in Transsexuals. In Bocking, W. O., Coleman, E. (Eds). Gender Dysphoria: Interdisciplinary Approaches in Clinical Management (pp. 39–54). Binghamton: Haworth Press. / Journal of Psychology & Human Sexuality, 5(4), 39–54. [Google Scholar] [Google Books] [DOI:10.1300/J056v05n04_03]
Athanasoulia-Kaspar, A. P., & Stalla, G. K. (2019). Endokrinologische Betreuung von Patienten mit Transsexualität. Geburtshilfe und Frauenheilkunde, 79(7), 672–675. [DOI:10.1055/a-0801-3319]
Bastianelli, C., Farris, M., Rosato, E., Brosens, I., & Benagiano, G. (2018). Pharmacodynamics of combined estrogen-progestin oral contraceptives 3. Inhibition of ovulation. Expert Review of Clinical Pharmacology, 11(11), 1085–1098. [DOI:10.1080/17512433.2018.1536544]
Bonadonna, S., Amer, M., Foletti, F., Federici, S., Persani, L., Bonomi, M. (2025). Evaluation of Antiandrogen Therapy Effectiveness in Transgender individuals Assigned Male At Birth (AMAB). EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
Bourns, A. (2019). Guidelines for Gender-Affirming Primary Care with Trans and Non-Binary Patients, 4th Edition. Toronto: Rainbow Health Ontario/Sherbourne Health. [URL] [PDF]
Bruchovsky, N., Larry Goldenberg, S., Akakura, K., & Rennie, P. S. (1993). Luteinizing hormone-releasing hormone agonists in prostate cancer. Elimination of flare reaction by pretreatment with cyproterone acetate and low-dose diethylstilbestrol. Cancer, 72(5), 1685–1691. [DOI:10.1002/1097-0142(19930901)72:5<1685::aid-cncr2820720532>3.0.co;2-3]
Bultynck, C., Pas, C., Defreyne, J., Cosyns, M., den Heijer, M., & T’Sjoen, G. (2017). Self-perception of voice in transgender persons during cross-sex hormone therapy. The Laryngoscope, 127(12), 2796–2804. [DOI:10.1002/lary.26716]
Chen, H., Wiepjes, C. M., van Schoor, N. M., Heijboer, A. C., de Jongh, R. T., den Heijer, M., & Lips, P. (2019). Changes of Vitamin D-Binding Protein, and Total, Bioavailable, and Free 25-Hydroxyvitamin D in Transgender People. The Journal of Clinical Endocrinology & Metabolism, 104(7), 2728–2734. [DOI:10.1210/jc.2018-02602]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Collet, S., Gieles, N., Wiepjes, C. M., Heijboer, A. C., Reyns, T., Fiers, T., Lapauw, B., den Heijer, M., & T’Sjoen, G. (2023). Changes in serum testosterone and adrenal androgen levels in transgender women with and without gonadectomy. The Journal of Clinical Endocrinology & Metabolism, 108(2), 331–338. [DOI:10.1210/clinem/dgac576]
Damgaard-Pedersen, F., & Føgh, M. (1980). The effect of cyproterone acetate on serum lipids in normal men. Acta Endocrinologica, 94(2), 280–283. [DOI:10.1530/acta.0.0940280]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de Leon-Durango, R., Hernandez-Lazaro, A., Rios-Gomez, C., Santana-Ojeda, B., Molinero-Marcos, I., Arnas-Leon, C., Hernandez-Hernandez, I., Acosta-Calero, C., Kuzior, A., Perez-Rivero, J., Perez-Garcia, M., & Martinez-Martin, F. (2024). P194 Very Low-dose Cyproterone Acetate (12.5 Mg/day) is Effective as Androgen Blocker; Well Tolerated And Not Associated With Hypertension Development in Young Female Transgender People. Journal of Hypertension, 42(Suppl 3), e133–e133. [DOI:10.1097/01.hjh.0001063648.69793.7c]
Defreyne, J., Vantomme, B., Van Caenegem, E., Wierckx, K., De Blok, C., Klaver, M., Nota, N. M., Van Dijk, D., Wiepjes, C. M., Den Heijer, M., & T’Sjoen, G. (2018). Prospective evaluation of hematocrit in gender-affirming hormone treatment: results from European Network for the Investigation of Gender Incongruence. Andrology, 6(3), 446–454. [DOI:10.1111/andr.12485]
Endrikat, J., Gerlinger, C., Richard, S., Rosenbaum, P., & Düsterberg, B. (2011). Ovulation inhibition doses of progestins: a systematic review of the available literature and of marketed preparations worldwide. Contraception, 84(6), 549–557. [DOI:10.1016/j.contraception.2011.04.009]
Even-Zohar, N., Sofer, Y., Yaish, I., Serebro, M., Tordjman, K., & Greenman, Y. (2020). SUN-042 Low Dose Cyproterone Acetate for the Treatment of Transgender Women - a Retrospective Study. Journal of the Endocrine Society, 4(Suppl 1), A715–A715. [DOI:10.1210/jendso/bvaa046.1412]
Even Zohar, N., Sofer, Y., Yaish, I., Serebro, M., Tordjman, K., & Greenman, Y. (2021). Low-Dose Cyproterone Acetate Treatment for Transgender Women. The Journal of Sexual Medicine, 18(7), 1292–1298. [DOI:10.1016/j.jsxm.2021.04.008]
Fink, G. (1979). Feedback Actions of Target Hormones on Hypothalamus and Pituitary With Special Reference to Gonadal Steroids. Annual Review of Physiology, 41(1), 571–585. [DOI:10.1146/annurev.ph.41.030179.003035]
Flamant, T., Vervalcke, J., & T’Sjoen, G. (2023). Dose Reduction of Cyproterone Acetate in Trans Women and the Effect on Patient-reported Outcomes: Results from the ENIGI Study. Endocrine Abstracts, 97 [Belgian Endocrine Society 2023], 5–5 (abstract no. 007). [URL] [PDF]
Føgh, M., Corker, C. S., Hunter, W. M., McLean, H., Philip, J., Schou, G., & Shakkebæk, N. E. (1979). The effects of low doses of cyproterone acetate on some functions of the reproductive system in normal men. Acta Endocrinologica, 91(3), 545–552. [DOI:10.1530/acta.0.0910545]
Føgh, M., Knudsen, J. B., & Gormsen, J. (1980). Effect of cyproterone acetate on platelet aggregability, fibrinolytic activity and fibrinolytic capacity in normal men. Acta Endocrinologica, 94(3), 430–432. [DOI:10.1530/acta.0.0940430]
Foegh, M. (1983). Evaluation of Steroids as COntraceptives in Men. Acta Endocrinologica, 104(3 Suppl b), S9–S48. [DOI:10.1530/acta.0.104s009]
Fredricsson, B., & Carlström, K. (2009). Effects of Low Doses of Cyproterone Acetate on Sperm Morphology and some other Parameters of Reproduction in Normal Men. Andrologia, 13(4), 369–375. [DOI:10.1111/j.1439-0272.1981.tb00067.x]
Fung, R., Hellstern-Layefsky, M., & Lega, I. (2017). Is a lower dose of cyproterone acetate as effective at testosterone suppression in transgender women as higher doses? International Journal of Transgenderism, 18(2), 123–128. [DOI:10.1080/15532739.2017.1290566]
Fuss, J., Hellweg, R., Van Caenegem, E., Briken, P., Stalla, G. K., T’Sjoen, G., & Auer, M. K. (2015). Cross-sex hormone treatment in male-to-female transsexual persons reduces serum brain-derived neurotrophic factor (BDNF). European Neuropsychopharmacology, 25(1), 95–99. [DOI:10.1016/j.euroneuro.2014.11.019]
Fuss, J., Claro, L., Ising, M., Biedermann, S. V., Wiedemann, K., Stalla, G. K., Briken, P., & Auer, M. K. (2019). Does sex hormone treatment reverse the sex-dependent stress regulation? A longitudinal study on hypothalamus-pituitary-adrenal (HPA) axis activity in transgender individuals. Psychoneuroendocrinology, 104, 228–237. [DOI:10.1016/j.psyneuen.2019.02.023]
Gava, G., Cerpolini, S., Martelli, V., Battista, G., Seracchioli, R., & Meriggiola, M. C. (2016). Cyproterone acetatevsleuprolide acetate in combination with transdermal oestradiol in transwomen: a comparison of safety and effectiveness. Clinical Endocrinology, 85(2), 239–246. [DOI:10.1111/cen.13050]
Gava, G., Mancini, I., Alvisi, S., Seracchioli, R., & Meriggiola, M. C. (2020). A comparison of 5-year administration of cyproterone acetate or leuprolide acetate in combination with estradiol in transwomen. European Journal of Endocrinology, 183(6), 561–569. [DOI:10.1530/eje-20-0370]
Geller, J., Albert, J., Yen, S. S., Geller, S., & Loza, D. (1981). Medical Castration of Males with Megestrol Acetate and Small Doses of Diethylstilbestrol*. The Journal of Clinical Endocrinology & Metabolism, 52(3), 576–580. [DOI:10.1210/jcem-52-3-576]
Geller, J., Albert, J., Yen, S. S., Geller, S., & Loza, D. (1981). Medical castration with megestrol acetate and minidose of diethylstilbestrol. Urology, 17(4 Suppl), 27–33. [Google Scholar] [PubMed]
Geller, J., & Albert, J. D. (1983). Comparison of various hormonal therapies for prostatic carcinoma. Seminars in Oncology, 10(4 Suppl 4), 34–41. [Google Scholar] [PubMed] [PDF]
Geller, J. (1988). Megestrol acetate and minidose estrogen in prostatic carcinoma. Urology, 32(3), 281–282. [DOI:10.1016/0090-4295(88)90402-5]
Geller J. (1991). Megestrol Acetate Plus Low-Dose Estrogen in the Management of Advanced Prostatic Carcinoma. The Urologic Clinics of North America, 18(1), 83–91. [DOI:10.1016/S0094-0143(21)01395-1] [Archive.org]
Giltay, E. J., & Gooren, L. J. (2000). Effects of Sex Steroid Deprivation/Administration on Hair Growth and Skin Sebum Production in Transsexual Males and Females. The Journal of Clinical Endocrinology & Metabolism, 85(8), 2913–2921. [DOI:10.1210/jcem.85.8.6710]
Giltay, E. J., Gooren, L. J., Emeis, J. J., Kooistra, T., & Stehouwer, C. D. (2000). Oral, but Not Transdermal, Administration of Estrogens Lowers Tissue-Type Plasminogen Activator Levels in Humans Without Affecting Endothelial Synthesis. Arteriosclerosis, Thrombosis, and Vascular Biology, 20(5), 1396–1403. [DOI:10.1161/01.atv.20.5.1396]
Giltay, E. J., Verhoef, P., Gooren, L. J., Geleijnse, J. M., Schouten, E. G., & Stehouwer, C. D. (2003). Oral and transdermal estrogens both lower plasma total homocysteine in male-to-female transsexuals. Atherosclerosis, 168(1), 139–146. [DOI:10.1016/s0021-9150(03)00090-x]
Giltay, E. J., Gooren, L. J., Toorians, A. W., Katan, M. B., & Zock, P. L. (2004). Docosahexaenoic acid concentrations are higher in women than in men because of estrogenic effects. The American Journal of Clinical Nutrition, 80(5), 1167–1174. [DOI:10.1093/ajcn/80.5.1167]
Glintborg, D., T’Sjoen, G., Ravn, P., & Andersen, M. S. (2021). MANAGEMENT OF ENDOCRINE DISEASE: Optimal feminizing hormone treatment in transgender people. European Journal of Endocrinology, 185(2), R49–R63. [DOI:10.1530/eje-21-0059]
Goldenberg, S. L., Bruchovsky, N., Rennie, P. S., & Coppin, C. M. (1988). The Combination of Cyproterone Acetate and Low Dose Diethylstilbestrol in the Treatment of Advanced Prostatic Carcinoma. Journal of Urology, 140(6), 1460–1465. [DOI:10.1016/s0022-5347(17)42073-8]
Goldenberg, S. L., & Bruchovsky, N. (1991). Use of Cyproterone Acetate in Prostate Cancer. The Urologic Clinics of North America, 18(1), 111–122. [DOI:10.1016/S0094-0143(21)01398-7] [Archive.org]
Goldenberg, S., Bruchovsky, N., Gleave, M., & Sullivan, L. (1996). Low-dose cyproterone acetate plus mini-dose diethylstilbestrol—A protocol for reversible medical castration. Urology, 47(6), 882–884. [DOI:10.1016/s0090-4295(96)00048-9]
Gooren, L. J., Giltay, E. J., & Bunck, M. C. (2008). Long-Term Treatment of Transsexuals with Cross-Sex Hormones: Extensive Personal Experience. The Journal of Clinical Endocrinology & Metabolism, 93(1), 19–25. [DOI:10.1210/jc.2007-1809]
Gräf, K., Brotherton, J., & Neumann, F. (1974). Clinical Uses of Antiandrogens. In Hughes, A., Hasan, S. H., Oertel, G. W., Voss, H. E., Bahner, F., Neumann, F., Steinbeck, H., Gräf, K.-J., Brotherton, J., Horn, H. J., & Wagner, R. K. (Eds.). Androgens II and Antiandrogens / Androgene II und Antiandrogene (Handbuch der experimentellen Pharmakologie/Handbook of Experimental Pharmacology, Volume 35, Part 2) (pp. 485–542). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-80859-3_7]
Hammerstein, J., Meckies, J., Leo-Rossberg, I., Moltz, L., & Zielske, F. (1975). Use of cyproterone acetate (CPA) in the treatment of acne, hirsutism and virilism. Journal of Steroid Biochemistry, 6(6), 827–836. [DOI:10.1016/0022-4731(75)90311-8]
Hammerstein, J. (1979). Cyproterone Acetate. In Jacobs, H. S. (Ed.). Advances in Gynaecological Endocrinology: Proceedings of the Sixth Study Group of the Royal College of Obstetricians and Gynaecologists, 18th and 19th October, 1978 (pp. 367–382). London: The College. [Google Scholar] [Google Books] [PDF]
Hammerstein, J. (1990). Antiandrogens: Clinical Aspects. In Orfanos, C. E., & Happle, R. (Eds.). Hair and Hair Diseases (pp. 827–886). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-74612-3_35]
Heath, R. A., & Wynne, K. (2019). A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (p. 122). Santa Barbara: Praeger/ABC-CLIO. [Google Books]
Hembree, W. C., Cohen-Kettenis, P., Delemarre-Van De Waal, H. A., Gooren, L. J., Meyer III, W. J., Spack, N. P., Tangpricha, V., & Montori, V. M. (2009). Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism, 94(9), 3132–3154. [DOI:10.1210/jc.2009-0345]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society* Clinical Practice Guideline [2nd Version]. The Journal of Clinical Endocrinology & Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Jacobeit, J. W. (2019). Die hormonelle Behandlung von adulten Trans*Personen (in Deutschland). [Hormonal treatment of adult trans* persons (in Germany).] Journal für Klinische Endokrinologie und Stoffwechsel, 12(3), 102–110. [DOI:10.1007/s41969-019-00080-x]
Jacobi, G. H., Altwein, J. E., Kurth, K. H., Basting, R., & Hohenfellner, R. (1980). Treatment of Advanced Prostatic Cancer with Parenteral Cyproterone Acetate: A Phase III Randomised Trial*. British Journal of Urology, 52(3), 208–215. [DOI:10.1111/j.1464-410x.1980.tb02961.x]
Jacobi, G. H., Tunn, U., & Senge, T. (1982). Clinical experience with cyproterone acetate for palliation of inoperable prostate cancer. In Jacobi, G. H., & Hohenfellner, R. (Eds.). Prostate Cancer (International Perspectives in Urology, Volume 3) (pp. 305–319). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Jequier, A. M., Bullimore, N. J., & Bishop, M. J. (2009). Cyproterone Acetate and a Small Dose of Oestrogen in the Pre-operative Management of Male Transsexuals. A Report of Three Cases. [Cyproteronacetat und kleine Östrogendosis in dem präoperativen Management männlicher Transsexueller. Bericht über drei Fälle.] Andrologia, 21(5), 456–461. [DOI:10.1111/j.1439-0272.1989.tb02447.x]
Johnson, D. E., Babaian, R. J., Swanson, D. A., Von Eschenbach, A. C., Wishnow, K. I., & Tenney, D. (1988). Medical castration using megestrol acetate and minidose estrogen. Urology, 31(5), 371–374. [DOI:10.1016/0090-4295(88)90726-1]
Knuth, U. A., Hano, R., & Nieschlag, E. (1984). Effect of Flutamide or Cyproterone Acetate on Pituitary and Testicular Hormones in Normal Men. The Journal of Clinical Endocrinology & Metabolism, 59(5), 963–969. [DOI:10.1210/jcem-59-5-963]
Koch, U. J., Lorenz, F., Danehl, K., & Hammerstein, J. (1975). Über die Verwendbarkeit von Cyproteronacetat zur Fertilitätshemmung beim Mann. Morphologische Veränderungen und Einflüsse auf die Spermienmotilität. Archiv für Gynäkologie, 219(1–4), 581–582. [DOI:10.1007/bf00669258]
Koch, U., Lorenz, F., Danehl, K., Ericsson, R., Hasan, S., Keyserlingk, D., Lübke, K., Mehring, M., Römmler, A., Schwartz, U., & Hammerstein, J. (1976). Continuous oral low-dosage cyproterone acetate for fertility regulation in the male? A trend analysis in 15 volunteers. Contraception, 14(2), 117–135. [DOI:10.1016/0010-7824(76)90081-0]
Korpaisarn, S., Arunakul, J., Chaisuksombat, K., & Rattananukrom, T. (2023). A Low Dose Cyproterone Acetate In Feminizing Hormone Treatment. Journal of the Endocrine Society, 7(Suppl 1), A1098–A1099 (abstract no. SAT397/bvad114.2068). [DOI:10.1210/jendso/bvad114.2068]
Korpaisarn, S., Arunakul, J., Chaisuksombat, K., & Rattananukrom, T. (2024). Effectiveness of low dose cyproterone acetate compared to standard dose in feminizing hormone treatment: a single institutional retrospective pilot study. Sexual Medicine, 12(4), qfae063. [DOI:10.1093/sexmed/qfae063]
Kranz, G. S., Seiger, R., Kaufmann, U., Hummer, A., Hahn, A., Ganger, S., Tik, M., Windischberger, C., Kasper, S., & Lanzenberger, R. (2017). Effects of sex hormone treatment on white matter microstructure in individuals with gender dysphoria. NeuroImage, 150, 60–67. [DOI:10.1016/j.neuroimage.2017.02.027]
Kranz, G. S., Kaufmann, U., & Lanzenberger, R. (2019). Probing the Impact of Gender-Affirming Hormone Treatment on Odor Perception. Chemical Senses, 45(1), 37–44. [DOI:10.1093/chemse/bjz069]
Kuijpers, S. M., Wiepjes, C. M., Conemans, E. B., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2021). Toward a Lowest Effective Dose of Cyproterone Acetate in Trans Women: Results From the ENIGI Study. The Journal of Clinical Endocrinology & Metabolism, 106(10), e3936–e3945. [DOI:10.1210/clinem/dgab427]
Kumar, P., Reddy, S., Kulkarni, A., Sharma, M., & Rao, P. N. (2021). Cyproterone acetate induced acute liver failure: case report and review of the literature. Journal of Clinical and Experimental Hepatology, 11(6), 739–741. [DOI:10.1016/j.jceh.2021.01.003]
Lederbogen, S. (2009). Hormonbehandlung. PiD - Psychotherapie im Dialog, 10(1), 41–43. [DOI:10.1055/s-0028-1090190]
Lim, H. Y., Leemaqz, S. Y., Torkamani, N., Grossmann, M., Zajac, J. D., Nandurkar, H., Ho, P., & Cheung, A. S. (2020). Global Coagulation Assays in Transgender Women on Oral and Transdermal Estradiol Therapy. The Journal of Clinical Endocrinology & Metabolism, 105(7), e2369–e2377. [DOI:10.1210/clinem/dgaa262]
Liu, P. Y., Takahashi, P., & Veldhuis, J. D. (2017). An Ensemble Perspective of Aging-Related Hypoandrogenemia in Men. In Winters, S. J., & Huhtaniemi, I. T. (Eds.). Male Hypogonadism: Basic, Clinical and Therapeutic Principles, 2nd Edition (pp. 325–347). Cham: Springer. [DOI:10.1007/978-3-319-53298-1_16]
Mahfouda, S., Moore, J. K., Siafarikas, A., Hewitt, T., Ganti, U., Lin, A., & Zepf, F. D. (2019). Gender-affirming hormones and surgery in transgender children and adolescents. The Lancet Diabetes & Endocrinology, 7(6), 484–498. [DOI:10.1016/s2213-8587(18)30305-x]
Meriggiola, M. C., Bremner, W. J., Costantino, A., Di Cintio, G., & Flamigni, C. (1998). Low dose of cyproterone acetate and testosterone enanthate for contraception in men. Human Reproduction, 13(5), 1225–1229. [DOI:10.1093/humrep/13.5.1225]
Meriggiola, M. C., Bremner, W. J., Costantino, A., Bertaccini, A., Morselli-Labate, A. M., Huebler, D., Kaufmann, G., Oettel, M., & Flamigni, C. (2002). Twenty-One Day Administration of Dienogest Reversibly Suppresses Gonadotropins and Testosterone in Normal Men. The Journal of Clinical Endocrinology & Metabolism, 87(5), 2107–2113. [DOI:10.1210/jcem.87.5.8514]
Meriggiola, M. C., Costantino, A., Bremner, W. J., & Morselli-Labate, A. M. (2002). Higher Testosterone Dose Impairs Sperm Suppression Induced by a Combined Androgen‐Progestin Regimen. Journal of Andrology, 23(5), 684–690. [DOI:10.1002/j.1939-4640.2002.tb02311.x]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463] [PDF]
Moltz, L., Römmler, A., Schwartz, U., & Hammerstein, J. (1978). Effects of Cyproterone Acetate (CPA) on Pituitary Gonadotrophin Release and on Androgen Secretion Before and After LH-RH Double Stimulation Tests in Men. International Journal of Andrology, 1(Suppl 2b) [5th Annual Workshop on the Testis, Geilo, Norway, April 1978, Endocrine Approach to Male Contraception], 713–719. [DOI:10.1111/j.1365-2605.1978.tb00518.x]
Moltz, L., Römmler, A., Schwartz, U., Post, K., & Hammerstein, J. (1978). Cyproterone acetate (CPA)—a potential male contraceptive: further studies on the interactions with endocrine parameters. Journal of Steroid Biochemistry, 9(9), 865–865 (abstract no. 252). [DOI:10.1016/0022-4731(78)90952-4]
Moltz, L., Römmler, A., Post, K., Schwartz, U., & Hammerstein, J. (1980). Medium dose cyproterone acetate (CPA): Effects on hormone secretion and on spermatogenesis in men. Contraception, 21(4), 393–413. [DOI:10.1016/s0010-7824(80)80017-5]
Moltz, L., Koch, U., Schwartz, U., Rommler, A., & Hammerstein, J. (1982). Male fertility regulation with cyproterone acetate (CPA). Contraceptive Delivery Systems, 3(3/4) [Retroproductive Health Care International Symposium, October 10-15 1982 Maui, Hawaii, USA, Expanded Abstracts], 298–298 (abstract no. 293). [Google Scholar] [PDF]
Moore, E., Wisniewski, A., & Dobs, A. (2003). Endocrine Treatment of Transsexual People: A Review of Treatment Regimens, Outcomes, and Adverse Effects. The Journal of Clinical Endocrinology & Metabolism, 88(8), 3467–3473. [DOI:10.1210/jc.2002-021967]
Nelson, J. B. (2012). Hormone Therapy for Prostate Cancer. In Wein, A. J., Kavoussi, L. R., Novick, A. C., Partin, A. W., & Peters, C. A. (Eds.). Campbell-Walsh Urology, 10th Edition, Volume 2 (pp. 2920–2953). Philadelphia: Elsevier/Saunders. [Google Scholar] [Google Books]
Nota, N. M., den Heijer, M., Gooren, L. J. (2019). Evaluation and Treatment of Gender-Dysphoric/Gender Incongruent Adults. [Updated 2019 Jul 21]. In Feingold, K. R., Anawalt, B., Blackman, M. R., et al. (Eds.). Endotext [Internet]. South Dartmouth, Massachusetts: MDText.com. [PubMed]
Oliphant, J., Veale, J., Macdonald, J., Carroll, R., Johnson, R., Harte, M., Stephenson, C. & Bullock, J. (2018). Guidelines for Gender Affirming Healthcare for Gender Diverse and Transgender Children, Young People and Adults in Aotearoa New Zealand. Waikato: Transgender Health Research Lab/University of Waikato. [URL] [PDF]
Ott, J., Aust, S., Promberger, R., Huber, J. C., & Kaufmann, U. (2011). Cross‐Sex Hormone Therapy Alters the Serum Lipid Profile: A Retrospective Cohort Study in 169 Transsexuals. The Journal of Sexual Medicine, 8(8), 2361–2369. [DOI:10.1111/j.1743-6109.2011.02311.x]
Petry, R., Mauss, J., Senge, T., & Rausch-Stroomann, J. (1970). Über den Einfluß von Cyproteronacetat, Norethisteronönanthat und Gestonoroncapronat auf die Hypophysen-Gonadenachse beim Mann. [Influence of Cyproterone-acetate, Norethisterone-enanthate and Gestonorone-capronate on the Hypophyseal-Gonadal-Axis in the Male.] In Kracht, J. (Ed.). Endokrinologie der Entwicklung und Reifung, 16. Symposion, Ulm, 26.-28. Februar 1970 (Symposion der Deutschen Gesellschaft für Endokrinologie, Volume 16) (pp. 428–430). Berlin: Springer. [Google Books] [DOI:10.1007/978-3-642-80591-2_118] [WorldCat]
Petry, R., Rausch-Stroomann, J.-G., Berthold, K. Mauss, J., Ai, M., Senge, Th., & Vermeulen, A. (1970). Untersuchungen zum Wirkungsmechanismus der Antiandrogene Cyproteron und Cyproteronacetat beim Menschen (Gonadotropin-, Plasma-testosteron- und morphologische Keimdrüsenuntersuchungen). [Investigations on the mechanism of action of the antiandrogens cyproterone and cyproterone acetate in humans (gonadotropin, plasma testosterone, and morphological gonad investigations).] In Schlegel, B. (Ed.). Verhandlungen der Deutschen Gesellschaft für Innere Medizin: Sechsundsiebzigster Kongress Gehalten zu Wiesbaden vom 6. April – 9. April 1970 (Verhandlungen der Deutschen Gesellschaft für Innere Medizin, Volume 76) (pp. 873–876). München: Bergmann. [Google Scholar] [Google Books] [DOI:10.1007/978-3-642-85446-0] [WorldCat]
Petry, R., Rausch-Stroomann, J. G., Mauss, J., Senge, Th., Ai, M., & Berthold, K. (1970). Investigations on the mode of action of the antiandrogens cyproterone and cyproterone acetate in man. / Investigations on the mechanism of action of anti androgenic cyproterone and cyproterone acetate in humans (gonadotropin, plasma testosterone, and morphological generative gland investigations). Medizinische Welt, 29, 1336–. [EurekaMag] [Cited by Koch et al. (1976)]
Petry, R., Mauss, J., Rausch-Stroomann, J. G., & Vermeulen, A. (1972). Reversible inhibition of spermatogenesis in men. Hormone and Metabolic Research, 4(5), 386–388. [DOI:10.1055/s-0028-1094040]
Roy, S., Chatterjee, S., Prasad, M., Poddar, A., Pandey, D., Pandey, H., & Jadhav, Y. (1976). Effects of cyproterone acetate on reproductive functions in normal human males. Contraception, 14(4), 403–423. [DOI:10.1016/s0010-7824(76)80055-8]
Roy, S., & Chatterjee, S. (1979). Studies with cyproterone acetate for male contraception. In James, V. H. T., & Pasqualini, J. R. (Eds.). Hormonal Steroids: Proceedings of the Fifth International Congress on Hormonal Steroids, New Delhi, India, October/November 1978 (pp. 675–680). Oxford: Pergamon Press. [DOI:10.1016/b978-0-08-023796-1.50099-2]
Roy, S., & Chatterjee, S. (1979). The Role of Antiandrogenic Action in Cyproterone Acetate-Induced Morphologic and Biochemical Changes in Human Semen. Fertility and Sterility, 32(1), 93–95. [DOI:10.1016/s0015-0282(16)44122-1]
Saborowski, K.-J. (1987). Konservative Therapie mit Cyproteronacetat und Estradiolundecylat beim Fortgeschrittenen Prostatacarcinom: Eine 5-Jahres-Studie. [Conservative Therapy with Cyproterone Acetate and Estradiol Undecylate in Advanced Prostate Cancer: A 5-Year Study.] (Doctoral dissertation, Ruhr-University Bochum.) [58 pages] [Google Scholar] [Google Books] [WorldCat] [PDF] [Translation]
Scharff, M., Wiepjes, C. M., Klaver, M., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2019). Change in grip strength in trans people and its association with lean body mass and bone density. Endocrine Connections, 8(7), 1020–1028. [DOI:10.1530/ec-19-0196]
Schröder, F. H., & Radlmaier, A. (2002). Steroidal Antiandrogens. In Jordan, C. V., & Furr, B. J. A. (Eds.). Hormone Therapy in Breast and Prostate Cancer (pp. 325–346). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-59259-152-7_15]
Slagter, M. H., Gooren, L. J., de Ronde, W., Soosaipillai, A., Scorilas, A., Giltay, E. J., Paliouras, M., & Diamandis, E. P. (2006). Serum and Urine Tissue Kallikrein Concentrations in Male-to-Female Transsexuals Treated with Antiandrogens and Estrogens. Clinical Chemistry, 52(7), 1356–1365. [DOI:10.1373/clinchem.2006.068932]
Sofer, Y., Yaish, I., Yaron, M., Bach, M. Y., Stern, N., & Greenman, Y. (2020). Differential Endocrine and Metabolic Effects of Testosterone Suppressive Agents in Transgender Women. Endocrine Practice, 26(8), 883–890. [DOI:10.4158/ep-2020-0032]
T’Sjoen, G. G., Beguin, Y., Feyen, E., Rubens, R., Kaufman, J., & Gooren, L. (2005). Influence of exogenous oestrogen or (anti-) androgen administration on soluble transferrin receptor in human plasma. Journal of Endocrinology, 186(1), 61–67. [DOI:10.1677/joe.1.06112]
T’Sjoen, G., Weyers, S., Taes, Y., Lapauw, B., Toye, K., Goemaere, S., & Kaufman, J. (2009). Prevalence of Low Bone Mass in Relation to Estrogen Treatment and Body Composition in Male-to-Female Transsexual Persons. Journal of Clinical Densitometry, 12(3), 306–313. [DOI:10.1016/j.jocd.2008.11.002]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine Position Statement “Assessment and Hormonal Management in Adolescent and Adult Trans People, with Attention for Sexual Function and Satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
Tack, L. J., Heyse, R., Craen, M., Dhondt, K., Bossche, H. V., Laridaen, J., & Cools, M. (2017). Consecutive Cyproterone Acetate and Estradiol Treatment in Late-Pubertal Transgender Female Adolescents. The Journal of Sexual Medicine, 14(5), 747–757. [DOI:10.1016/j.jsxm.2017.03.251]
Toorians, A. W., Thomassen, M. C., Zweegman, S., Magdeleyns, E. J., Tans, G., Gooren, L. J., & Rosing, J. (2003). Venous Thrombosis and Changes of Hemostatic Variables during Cross-Sex Hormone Treatment in Transsexual People. The Journal of Clinical Endocrinology & Metabolism, 88(12), 5723–5729. [DOI:10.1210/jc.2003-030520]
Torre, B. l., Norén, S., Hedman, M., & Diczfalusy, E. (1979). Effect of cyproterone acetate (CPA) on gonadal and adrenal function in men. Contraception, 20(4), 377–396. [DOI:10.1016/s0010-7824(79)80048-7]
Van Caenegem, E., Wierckx, K., Taes, Y., Schreiner, T., Vandewalle, S., Toye, K., Kaufman, J., & T’Sjoen, G. (2014). Preservation of volumetric bone density and geometry in trans women during cross-sex hormonal therapy: a prospective observational study. Osteoporosis International, 26(1), 35–47. [DOI:10.1007/s00198-014-2805-3]
van Dijk, D., Dekker, M. J., Conemans, E. B., Wiepjes, C. M., de Goeij, E. G., Overbeek, K. A., Fisher, A. D., den Heijer, M., & T’Sjoen, G. (2019). Explorative Prospective Evaluation of Short-Term Subjective Effects of Hormonal Treatment in Trans People—Results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 16(8), 1297–1309. [DOI:10.1016/j.jsxm.2019.05.009]
van Velzen, D. M., Paldino, A., Klaver, M., Nota, N. M., Defreyne, J., Hovingh, G. K., Thijs, A., Simsek, S., T’Sjoen, G., & den Heijer, M. (2019). Cardiometabolic Effects of Testosterone in Transmen and Estrogen Plus Cyproterone Acetate in Transwomen. The Journal of Clinical Endocrinology & Metabolism, 104(6), 1937–1947. [DOI:10.1210/jc.2018-02138]
Venner, P. M., Klotz, P. G., Klotz, L. H., Stewart, D. J., Davis, I. R., Orovan, W. L., & Ramsey, E. W. (1988). Megestrol acetate plus minidose diethylstilbestrol in the treatment of carcinoma of the prostate. Seminars in Oncology, 15(2 Suppl 1), 62–67. [Google Scholar] [PubMed]
Vereecke, G. (2019). Characterisation of testicular function and spermatogenesis in transgender women. (Master’s thesis, Ghent University.) [PDF]
Vereecke, G., Defreyne, J., Van Saen, D., Collet, S., Van Dorpe, J., T’Sjoen, G., & Goossens, E. (2020). Characterisation of testicular function and spermatogenesis in transgender women. Human Reproduction, 36(1), 5–15. [DOI:10.1093/humrep/deaa254]
Vita, R., Settineri, S., Liotta, M., Benvenga, S., & Trimarchi, F. (2018). Changes in hormonal and metabolic parameters in transgender subjects on cross-sex hormone therapy: A cohort study. Maturitas, 107, 92–96. [DOI:10.1016/j.maturitas.2017.10.012]
Vlot, M. C., Wiepjes, C. M., Jongh, R. T., T’Sjoen, G., Heijboer, A. C., & den Heijer, M. (2019). Gender‐Affirming Hormone Treatment Decreases Bone Turnover in Transwomen and Older Transmen. Journal of Bone and Mineral Research, 34(10), 1862–1872. [DOI:10.1002/jbmr.3762]
Wang, C., & Yeung, K. (1980). Use of low-dosage oral cyproterone acetate as a male contraceptive. Contraception, 21(3), 245–272. [DOI:10.1016/0010-7824(80)90005-0]
Warzywoda, S., Fowler, J. A., Wood, P., Bisshop, F., Russell, D., Luu, H., Kelly, M., Featherstone, V., & Dean, J. A. (2024). How low can you go? Titrating the lowest effective dose of cyproterone acetate for transgender and gender diverse people who request feminizing hormones. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2317395]
Wiepjes, C. M., Vlot, M. C., Klaver, M., Nota, N. M., de Blok, C. J., de Jongh, R. T., Lips, P., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Bone Mineral Density Increases in Trans Persons After 1 Year of Hormonal Treatment: A Multicenter Prospective Observational Study. Journal of Bone and Mineral Research, 32(6), 1252–1260. [DOI:10.1002/jbmr.3102]
Wiepjes, C. M., Vlot, M. C., de Blok, C. J., Nota, N. M., de Jongh, R. T., & den Heijer, M. (2019). Bone geometry and trabecular bone score in transgender people before and after short- and long-term hormonal treatment. Bone, 127, 280–286. [DOI:10.1016/j.bone.2019.06.029]
Wierckx, K., Mueller, S., Weyers, S., Van Caenegem, E., Roef, G., Heylens, G., & T’Sjoen, G. (2012). Long‐Term Evaluation of Cross‐Sex Hormone Treatment in Transsexual Persons. The Journal of Sexual Medicine, 9(10), 2641–2651. [DOI:10.1111/j.1743-6109.2012.02876.x]
Wierckx, K., Van Caenegem, E., Schreiner, T., Haraldsen, I., Fisher, A., Toye, K., Kaufman, J. M., & T’Sjoen, G. (2014). Cross‐Sex Hormone Therapy in Trans Persons Is Safe and Effective at Short‐Time Follow‐Up: Results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 11(8), 1999–2011. [DOI:10.1111/jsm.12571]
Winkler-Crepaz, K., Müller, A., Böttcher, B., & Wildt, L. (2017). Hormonbehandlung bei Transgenderpatienten. [Hormone treatment of transgender patients.] Gynäkologische Endokrinologie, 15(1), 39–42. [DOI:10.1007/s10304-016-0116-9]
Winters, S. J., Wang, C., & (2009). LH and Non-SHBG Testosterone and Estradiol Levels During Testosterone Replacement of Hypogonadal Men: Further Evidence That Steroid Negative Feedback Increases as Men Grow Older. Journal of Andrology, 31(3), 281–287. [DOI:10.2164/jandrol.109.009035]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Zitzmann, M., Rohayem, J., Raidt, J., Kliesch, S., Kumar, N., Sitruk-Ware, R., & Nieschlag, E. (2017). Impact of various progestins with or without transdermal testosterone on gonadotropin levels for non-invasive hormonal male contraception: a randomized clinical trial. Andrology, 5(3), 516–526. [DOI:10.1111/andr.12328]
Zubiaurre-Elorza, L., Junque, C., Gómez-Gil, E., & Guillamon, A. (2014). Effects of Cross-Sex Hormone Treatment on Cortical Thickness in Transsexual Individuals. The Journal of Sexual Medicine, 11(5), 1248–1261. [DOI:10.1111/jsm.12491]
\ No newline at end of file
diff --git a/transfemscience.org/articles/cpa-meningioma/index.html b/transfemscience.org/articles/cpa-meningioma/index.html
index c98d2460..8957e1e7 100644
--- a/transfemscience.org/articles/cpa-meningioma/index.html
+++ b/transfemscience.org/articles/cpa-meningioma/index.html
@@ -1 +1 @@
-Recent Developments on Cyproterone Acetate and Meningioma Risk Out of France and Implications for Transfeminine People - Transfeminine Science
Recent Developments on Cyproterone Acetate and Meningioma Risk Out of France and Implications for Transfeminine People
By Aly | First published April 24, 2020 | Last modified March 30, 2024
Abstract / TL;DR
Cyproterone acetate is a progestogen and antiandrogen commonly used in transfeminine hormone therapy. At the doses typically used in transfeminine people, it has extremely strong progestogenic effects. Whereas doses of around 2 or 3 mg/day have similar progestogenic effect as normal progesterone exposure during the luteal phase of the menstrual cycle in women, doses of cyproterone acetate of 25 to 100 mg/day have typically been used in transfeminine people. In 2018, the French government published findings from a large epidemiological study showing a strong and dose-dependent increase in the risk of meningiomas, a type of hormone-sensitive brain tumor, with typical high doses of cyproterone acetate. With cyproterone acetate used cyclically (20 days per month) at a dose of 50 mg/day for >5 years or 25 mg/day for >10 years, the risk of surgically operated meningioma was found to be increased by 20-fold. Subsequent studies have since replicated these results. Consequent to these findings, clinical use of cyproterone acetate has been restricted and its doses have been greatly reduced. Fortunately, cyproterone acetate is still a highly effective means of testosterone suppression in transfeminine people at much lower doses (e.g., 5–10 mg/day). Nonetheless, due to the risks of meningioma as well as various other complications, it is recommended that cyproterone acetate now be used cautiously and in a limited fashion in transfeminine people. For instance, it should only be used at low doses for a limited duration (e.g., 2 years) in people without known risk factors for meningiomas.
Introduction
Cyproterone acetate (CPA) is a progestogen and antiandrogen commonly used in transfeminine hormone therapy. Typical doses of CPA used in transfeminine hormone therapy have very strong progestogenic strength. Consequent to the strong progestogenic exposure, CPA is known to have a risk of meningioma, a rare and benign (non-cancerous) form of brain tumor. Meningiomas are tumors of the meninges, the membranes enveloping the brain and spinal cord. While not typically malignant (cancerous), meningiomas can nonetheless result in complications like visual disturbances, headaches, and seizures. They may necessitate brain surgery. Death has also resulted from CPA-induced meningioma. There is some more information on the topic of CPA and meningioma here on Wikipedia. A table of published case reports of meningioma with CPA can be found here on Wikipedia.
Recent findings out of France indicate that the risk of meningioma with CPA is much higher than previously thought (ASNM/CNAM). This article discusses these findings and the implications for use of CPA in transfeminine people, including recommendations for use of CPA more safely.
Recent French Findings
Case reports of meningioma with CPA started in 2007. These reports caused the French government to add a warning to the CPA production information in 2011 and to undertake a large epidemiological investigation into the risk of meningioma with CPA. They announced their findings in 2018 and reported the following (ASNM/CNAM):
The study was very large with 290,000 person–years of CPA-treated follow up.
The risk of meningioma with CPA was increased 7-fold with >6 months of treatment at an average dosage ≥25 mg/day. It was increased 20-fold with 50 mg/day for 20 days per month for >5 years. It was likewise increased 20-fold with 25 mg/day for 20 days per month for >10 years. As such, the risk is strongly exposure-dependent in terms of dosage and duration of therapy.
The risk of surgically operated meningioma with CPA was around 4 in 1,000 patients per year with 50 mg/day for 20 days per month for >5 years. This underestimates the true incidence of meningioma with CPA since it only includes operated cases.
There were more than 500 operated cases of meningioma in association with CPA in France between 2007 and 2015. The unoperated incidence is unknown but likely to be much higher.
The population incidence of meningioma in general is strongly age-dependent. It’s 0.14 in 10,000 individuals per year at under 20 years of age and 4.9 in 10,000 individuals per year at above 85 years of age (a 35-fold difference). The average risk is 0.8 in 10,000 individuals per year. Accordingly, the risk of meningioma with CPA was found to be highly age-dependent as well.
The risk of meningioma decreases strongly after discontinuation of CPA.
As a result of their findings, the following occurred (ASNM/CNAM):
A toll-free phone number was set up in France for inquiries about CPA and meningioma.
Recommendations were made such that all patients starting CPA should be given an MRI within 6 months of initiation of therapy. Subsequently, all patients taking CPA should get another MRI after 5 years of therapy and then an additional MRI every 2 years of therapy thereafter.
It was recommended that CPA not be used in postmenopausal women due to their age.
Whether CPA should be withdrawn from the market was discussed but was decided against. Part of this was related to the reliance of transfeminine people on CPA for hormone therapy.
CPA sales in France plummeted by 73% from September 2018 to September 2019. Sales of spironolactone increased by a lesser magnitude over the same time period.
France is also looking into meningioma risk with other high-dose progestogen formulations, including nomegestrol acetate (Lutenyl) and chlormadinone acetate (Luteran).
France requested that the European Medicines Agency (EMA) conduct a review of CPA and meningioma. This review began in mid-2019 (Chustecka, 2019). In February 2020, the EMA announced restrictions on the clinical CPA use in the European Union (EMA, 2020). These included use of CPA only after failure of other treatments and use of the lowest effective maintenance dose of CPA (EMA, 2020; Chustecka, 2020). The restrictions applied to formulations of CPA containing 10 mg/day CPA or more (EMA, 2020; Chustecka, 2019). The Medicines and Healthcare products Regulatory Agency (MHRA) in the United Kingdom published a drug safety update for cyproterone acetate in June 2020 (MHRA, 2020).
This 2018 French news article (translated from French to English via Google Translate) succinctly discusses some of the developments. Several recent literature publications have also discussed the topic (Schmutz, 2018; Gerson, 2018; Coubret et al., 2019; Plu-Bureau, 2019; Senofonte, Pallotti, & Lombardo, 2020). And long-form reports of the epidemiological study by the French government here (PDF; 128 pages) and here (PDF; 106 pages) provide more information. A more succinct report by the French government can also be found here (PDF; 9 pages; English translation here). Unfortunately however, many of these materials are in French or other non-English languages and translations are unavailable for English speakers (Google Translate is useful however).
Implications for Transfeminine People
CPA tends to be used at very high doses in transfeminine people. A dose of CPA of 2 or 3 mg/day is similar in progestogenic exposure to that occurring during the luteal phase of the menstrual cycle in women. Conversely, doses of CPA recommended by guidelines for use in transfeminine people typically range from 25 to 100 mg/day. This progestogenic exposure is similar to or greater than that during pregnancy in women, when progesterone levels increase by up to 20-fold relative to luteal levels. As such, it’s highly supraphysiological relative to typical circumstances in women. In addition to meningioma, high doses of CPA have been associated with blood clots, prolactinomas, and liver toxicity (Wiki; Aly, 2019). Long-term therapy with progestogens like CPA may increase the risk of breast cancer (Aly, 2020; Table; CGHFBC, 2019; de Blok et al., 2019) and cardiovascular problems as well (Table; Stefanick, 2010). There is also a great deal still unknown about high doses of CPA and their safety, as evidenced by the fact that we’re only uncovering the risk of meningioma with CPA now. It’s likely that using the lowest possible doses of CPA will minimize its various health risks.
The doses of CPA used in transfeminine people tend to be far more than are necessary. CPA appears to have maximal effectiveness in terms of testosterone suppression at a dose of only 5 to 10 mg/day. See my article here for a review of this topic. The effectiveness of lower doses of CPA was recently validated by Meyer et al. (2020), who showed that in combination with estrogen, 10 mg/day CPA was no different from 50 mg/day CPA in terms of testosterone suppression in transfeminine people. It seems clear that transfeminine people should not use a dose of CPA higher than 10 or 12.5 mg/day at most. With 50-mg CPA tablets and a pill cutter, a total dose of CPA of 6.25 to 12.5 mg/day can be achieved by taking one-fourth of a tablet once every other day to once every day. As a result of new findings, published CPA dose recommendations for transfeminine people have gone down in the last few years (Aly, 2019). It is likely that this will continue to a greater extent in the future.
Meningiomas express progesterone receptors and activation of these receptors is thought to stimulate growth of meningiomas (Marosi et al., 2008; Li et al., 2013). Other progestogens besides CPA as well as pregnancy have also been associated with meningioma, and spontaneous regression of meningiomas has been reported after pregnancy (Li et al., 2013; Wiki). The incidence of meningiomas is 2- to 3-fold higher in women than in men, and the female-to-male incidence ratio is greatest during the peak female reproductive years (Marosi et al., 2008; Wiemels, Wrensch, & Claus, 2010; Li et al., 2013; Wiki). The luteal phase of the menstrual cycle, when progesterone levels are increased, has been associated with exacerbation of meningiomas in women (Marosi et al., 2008; Li et al., 2013; Wiki). Meningioma has also been associated with birth control pills and menopausal hormone therapy (estrogen–progestogen preparations) (Li et al., 2013). As such, assuming these associations are indeed due to progesterone receptor activation, there may be no fully risk-free dosage of progestogens or CPA in terms of meningioma. Indeed, meningiomas have been reported even with 5 to 10 mg/day CPA, despite the relatively uncommon use of such doses historically (Table). In any case, the data indicate that the risk is dose-dependent, and hence that lower doses should have the least risk. A dose of 2 to 3 mg/day CPA continuously would presumably have minimal excess risk of meningioma relative to normal female physiological circumstances.
Recommendations for Transfeminine People
In light of the risks of CPA and its high testosterone-suppressing effectiveness even at low doses, the following recommendations for use of CPA in transfeminine people seem appropriate:
CPA should be used at the lowest effective dosage (generally 5–12.5 mg/day and no greater).
CPA should be used for a limited duration (e.g., a few years) and not long-term.
Other safer but similarly effective antiandrogenic approaches should be used instead of CPA where possible (e.g., GnRH modulators, gonadectomy, estradiol monotherapy, etc.).
CPA should be avoided in older individuals (e.g., >50 years of age).
CPA should not be used in people with a personal or family history of meningioma.
If possible, CPA should be used under supervision of a physician and people taking CPA should follow monitoring guidelines (e.g., regular MRIs as in France).
At the same time it should be noted that the absolute incidence of meningioma with CPA is low. The risk is likely to be especially small in young people and with low CPA doses (e.g., 5–12.5 mg/day). Use of CPA in a more cautious and restricted fashion may allow for more acceptable safety. In any case, meningiomas and brain surgery are serious, so CPA should not be taken carelessly.
Updates
Update 1: Weill et al. (2021) [French Government Study]
The French government study was finally published in the formal scientific literature in February 2021:
Weill, A., Nguyen, P., Labidi, M., Cadier, B., Passeri, T., Duranteau, L., Bernat, A., Yoldjian, I., Fontanel, S., Froelich, S., & Coste, J. (2021). Use of High Dose Cyproterone Acetate and Risk of Intracranial Meningioma in Women: Cohort Study. BMJ, 372, n37. [DOI:10.1136/bmj.n37]
A Medscape article here covers the study and discusses consequent restrictions on CPA use.
Update 2: Mikkelsen et al. (2021) [Danish National Study]
A national study in Denmark was published in June 2021 and has replicated the findings:
Mikkelsen, A. P., Greiber, I. K., Scheller, N. M., Hilden, M., & Lidegaard, Ø. (2021). Cyproterone acetate and risk of meningioma: a nationwide cohort study. Journal of Neurology, Neurosurgery & Psychiatry, 93(2), 222–223. [DOI:10.1136/jnnp-2021-326138]
Update 3: VUMC Switching From CPA to GnRH Agonists
As of August 2020, the Vrije Universiteit Medical Center (VUMC) in Amsterdam is switching antiandrogen therapy of all transgender patients from cyproterone acetate to gonadotropin-releasing hormone (GnRH) agonists (VUMC, 2020). This was due to a Dutch Medicines Authority warning about meningiomas with CPA (VUMC, 2020). The VUMC is the home of the Center of Expertise on Gender Dysphoria, one of the largest care and research institutes for transgender hormone therapy in the world, and is highly influential in this area (Wiepjes et al., 2018; Bakker, 2021). The VUMC is said to be the provider of hormone therapy for about 95% of hormone-seeking transgender people in the Netherlands, and has treated about 10,000 transgender people since its gender clinic opened in 1972 (Wiepjes et al., 2018; Bakker, 2021).
Update 4: Millward et al. (2021)
In October 2021, a systematic review of meningiomas with CPA in transfeminine people was published:
Millward, C. P., Keshwara, S. M., Islim, A. I., Jenkinson, M. D., Alalade, A. F., & Gilkes, C. E. (2022). Development and Growth of Intracranial Meningiomas in Transgender Women Taking Cyproterone Acetate as Gender-Affirming Progestogen Therapy: A Systematic Review. Transgender Health, 7(6), 473–483. [DOI:10.1089/trgh.2021.0025]
Update 5: Lee et al. (2022)
In February 2022, a systematic review and meta-analysis of meningiomas with CPA was published:
Lee, K. S., Zhang, J., Kirollos, R., Santarius, T., Nga, V., & Yeo, T. T. (2022). A systematic review and meta-analysis of the association between cyproterone acetate and intracranial meningiomas. Scientific Reports, 12(1), 1942. [DOI:10.1038/s41598-022-05773-z]
In September 2022, the following French study of meningioma incidence in people treated with high-dose progestin therapy following institution of systematic screening was published:
Samoyeau, T., Provost, C., Roux, A., Legrand, L., Dezamis, E., Plu-Bureau, G., Pallud, J., Oppenheim, C., & Benzakoun, J. (2022). Meningioma in patients exposed to progestin drugs: results from a real-life screening program. Journal of Neuro-Oncology, 160(1), 127–136. [DOI:10.1007/s11060-022-04124-2]
The absolute incidence of meningiomas with CPA was 13 of 103 or a striking 13% of individuals. This was much higher than in the general population, in which rates of incidental meningiomas of about 1 to 2% have been observed. Moreover, the authors noted that the 13% figure observed in this study was likely an underestimate, as most cases of meningiomas with CPA were likely already diagnosed prior to the launch of systematic screening. Relative to the high absolute incidence, the year-adjusted incidence rate of meningiomas with CPA was 13.2 per 100,000 person–years. As with other studies, meningioma incidence was associated with longer CPA treatment duration, greater cumulative CPA dose, and older age. The incidence of meningiomas was much lower with two other progestins, nomegestrol acetate and chlormadinone acetate. These progestins tend to be used at much lower doses than CPA.
Update 7: Roland et al. (2024)
In March 2024, the following French study assessed meningioma risk with various progestogens:
Roland, N., Neumann, A., Hoisnard, L., Duranteau, L., Froelich, S., Zureik, M., & Weill, A. (2024). Use of progestogens and the risk of intracranial meningioma: national case-control study. BMJ, 384, e078078. [DOI:10.1136/bmj-2023-078078]
The study found increased risk of surgically operated meningiomas not only with CPA but also with a variety of other progestogens, including nomegestrol acetate, chlormadinone acetate, medrogestone, injectable medroxyprogesterone acetate, and promegestone. Conversely, progesterone, dydrogesterone, and levonorgestrelintrauterine devices showed no increased risk of surgically operated meningiomas. No conclusions could be drawn for dienogest or hydroxyprogesterone caproate due to the small numbers of individuals who received these progestogens. Many other progestogens, such as most 19-nortestosterone derivatives, were not included or assessed in the study. CPA, related to the high doses used in its case, had by far the greatest risk of surgically operated meningiomas. These findings indicate that meningiomas are a dose-dependent risk of many different progestogens.
References
Bakker, A. (2021). Een halve eeuw transgenderzorg aan de VU [Half a Century of Transgender Care at the VU]. Amsterdam: Boom. [Google Books] [WorldCat] [URL]
Bême. (2018 September 21). Androcur et risque de méningiomes: un numéro vert pour répondre aux patients. [Androcur and risk of meningiomas: a toll-free number to answer patients.] Doctissimo. [URL] [Translation]
Chustecka, Z. (2019 July 12). Risk of Meningioma With Cyproterone: EMA Review Begins. Medscape. [URL]
Chustecka, Z. (2020 February 14). EU Restricts High-Dose Cyproterone Because of Meningioma Risk. Medscape. [URL]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/s0140-6736(19)31709-x]
Coubret, A., Geniaux, H., & Buxeraud, J. (2019). Méningiomes et acétate de cyprotérone. Actualités Pharmaceutiques, 58(590), 55–56. [DOI:10.1016/j.actpha.2019.09.013]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, I. R., & den Heijer, M. (2019). Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in the Netherlands. BMJ, , l1652–l1652. [DOI:10.1136/bmj.l1652]
European Medicines Agency (EMA). (2020 February 14). Restrictions in use of cyproterone due to meningioma risk. European Medicines Agency. [URL] [PDF]
Lee, K. S., Zhang, J., Kirollos, R., Santarius, T., Nga, V., & Yeo, T. T. (2022). A systematic review and meta-analysis of the association between cyproterone acetate and intracranial meningiomas. Scientific Reports, 12(1), 1942. [DOI:10.1038/s41598-022-05773-z]
Li, Q., Coulson, H., Klaassen, Z., Sharma, S., Ramalingam, P., Moses, K. A., & Terris, M. K. (2013). Emerging association between androgen deprivation therapy and male meningioma: significant expression of luteinizing hormone-releasing hormone receptor in male meningioma. Prostate Cancer and Prostatic Diseases, 16(4), 387–390. [DOI:10.1038/pcan.2013.45]
Marosi, C., Hassler, M., Roessler, K., Reni, M., Sant, M., Mazza, E., & Vecht, C. (2008). Meningioma. Critical Reviews in Oncology/Hematology, 67(2), 153–171. [DOI:10.1016/j.critrevonc.2008.01.010]
Medicines and Healthcare products Regulatory Agency (MHRA). (2020 June 29). Cyproterone acetate: new advice to minimise risk of meningioma. GOV.UK. [URL]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463] [PDF]
Mikkelsen, A. P., Greiber, I. K., Scheller, N. M., Hilden, M., & Lidegaard, Ø. (2021). Cyproterone acetate and risk of meningioma: a nationwide cohort study. Journal of Neurology, Neurosurgery & Psychiatry, 93(2), 222–223. [DOI:10.1136/jnnp-2021-326138]
Millward, C. P., Keshwara, S. M., Islim, A. I., Jenkinson, M. D., Alalade, A. F., & Gilkes, C. E. (2022). Development and Growth of Intracranial Meningiomas in Transgender Women Taking Cyproterone Acetate as Gender-Affirming Progestogen Therapy: A Systematic Review. Transgender Health, 7(6), 473–483. [DOI:10.1089/trgh.2021.0025]
Nelson, R. (2021 February 3). Increased Risk of Meningioma With Cyproterone Acetate Use. Medscape. [URL]
Plu-Bureau, G. (2019). Faut-il rayer l’acétate de cyprotérone de nos prescriptions ? Gynécologie Obstétrique Fertilité & Sénologie , 47(12), 823–824. [DOI:10.1016/j.gofs.2019.10.006]
Roland, N., Neumann, A., Hoisnard, L., Duranteau, L., Froelich, S., Zureik, M., & Weill, A. (2024). Use of progestogens and the risk of intracranial meningioma: national case-control study. BMJ, 384, e078078. [DOI:10.1136/bmj-2023-078078]
Samoyeau, T., Provost, C., Roux, A., Legrand, L., Dezamis, E., Plu-Bureau, G., Pallud, J., Oppenheim, C., & Benzakoun, J. (2022). Meningioma in patients exposed to progestin drugs: results from a real-life screening program. Journal of Neuro-Oncology, 160(1), 127–136. [DOI:10.1007/s11060-022-04124-2]
Schmutz, J. (2018). Méningiomes et acétate de cyprotérone : mise au point. Annales de Dermatologie et de Vénéréologie, 145(5), 390–391. [DOI:10.1016/j.annder.2018.04.001]
Senofonte, G., Pallotti, F., & Lombardo, F. (2020). Ciproterone acetato e meningiomi: lo stato dell’arte. L’Endocrinologo, 21(3), 171–175. [DOI:10.1007/s40619-020-00746-8]
Stefanick, M. (2010). Postmenopausal hormone therapy and cardiovascular disease in women. Nutrition, Metabolism and Cardiovascular Diseases, 20(6), 451–458. [DOI:10.1016/j.numecd.2010.02.015]
University of Bristol. (2022 February 4). Widely-used hormone drug associated with increased risk of benign brain tumor at high doses. University of Bristol. [ScienceDaily] [Medical Xpress]
Vrije Universiteit Medical Center (VUMC). (2020). Wijzigingen medisch beleid hormoonbehandeling. [Changes in medical policy on hormone treatment.] Vrije Universiteit Medical Center. [URL]
Weill, A., Nguyen, P., Labidi, M., Cadier, B., Passeri, T., Duranteau, L., Bernat, A., Yoldjian, I., Fontanel, S., Froelich, S., & Coste, J. (2021). Use of High Dose Cyproterone Acetate and Risk of Intracranial Meningioma in Women: Cohort Study. BMJ, 372, n37. [DOI:10.1136/bmj.n37]
Wiemels, J., Wrensch, M., & Claus, E. B. (2010). Epidemiology and etiology of meningioma. Journal of Neuro-Oncology, 99(3), 307–314. [DOI:10.1007/s11060-010-0386-3]
Wiepjes, C. M., Nota, N. M., de Blok, C. J., Klaver, M., de Vries, A. L., Wensing-Kruger, S. A., de Jongh, R. T., Bouman, M., Steensma, T. D., Cohen-Kettenis, P., Gooren, L. J., Kreukels, B. P., & den Heijer, M. (2018). The Amsterdam Cohort of Gender Dysphoria Study (1972–2015): Trends in Prevalence, Treatment, and Regrets. The Journal of Sexual Medicine, 15(4), 582–590. [DOI:10.1016/j.jsxm.2018.01.016]
\ No newline at end of file
+Recent Developments on Cyproterone Acetate and Meningioma Risk Out of France and Implications for Transfeminine People - Transfeminine Science
Recent Developments on Cyproterone Acetate and Meningioma Risk Out of France and Implications for Transfeminine People
By Aly | First published April 24, 2020 | Last modified March 30, 2024
Abstract / TL;DR
Cyproterone acetate is a progestogen and antiandrogen commonly used in transfeminine hormone therapy. At the doses typically used in transfeminine people, it has extremely strong progestogenic effects. Whereas doses of around 2 or 3 mg/day have similar progestogenic effect as normal progesterone exposure during the luteal phase of the menstrual cycle in women, doses of cyproterone acetate of 25 to 100 mg/day have typically been used in transfeminine people. In 2018, the French government published findings from a large epidemiological study showing a strong and dose-dependent increase in the risk of meningiomas, a type of hormone-sensitive brain tumor, with typical high doses of cyproterone acetate. With cyproterone acetate used cyclically (20 days per month) at a dose of 50 mg/day for >5 years or 25 mg/day for >10 years, the risk of surgically operated meningioma was found to be increased by 20-fold. Subsequent studies have since replicated these results. Consequent to these findings, clinical use of cyproterone acetate has been restricted and its doses have been greatly reduced. Fortunately, cyproterone acetate is still a highly effective means of testosterone suppression in transfeminine people at much lower doses (e.g., 5–10 mg/day). Nonetheless, due to the risks of meningioma as well as various other complications, it is recommended that cyproterone acetate now be used cautiously and in a limited fashion in transfeminine people. For instance, it should only be used at low doses for a limited duration (e.g., 2 years) in people without known risk factors for meningiomas.
Introduction
Cyproterone acetate (CPA) is a progestogen and antiandrogen commonly used in transfeminine hormone therapy. Typical doses of CPA used in transfeminine hormone therapy have very strong progestogenic strength. Consequent to the strong progestogenic exposure, CPA is known to have a risk of meningioma, a rare and benign (non-cancerous) form of brain tumor. Meningiomas are tumors of the meninges, the membranes enveloping the brain and spinal cord. While not typically malignant (cancerous), meningiomas can nonetheless result in complications like visual disturbances, headaches, and seizures. They may necessitate brain surgery. Death has also resulted from CPA-induced meningioma. There is some more information on the topic of CPA and meningioma here on Wikipedia. A table of published case reports of meningioma with CPA can be found here on Wikipedia.
Recent findings out of France indicate that the risk of meningioma with CPA is much higher than previously thought (ASNM/CNAM). This article discusses these findings and the implications for use of CPA in transfeminine people, including recommendations for use of CPA more safely.
Recent French Findings
Case reports of meningioma with CPA started in 2007. These reports caused the French government to add a warning to the CPA production information in 2011 and to undertake a large epidemiological investigation into the risk of meningioma with CPA. They announced their findings in 2018 and reported the following (ASNM/CNAM):
The study was very large with 290,000 person–years of CPA-treated follow up.
The risk of meningioma with CPA was increased 7-fold with >6 months of treatment at an average dosage ≥25 mg/day. It was increased 20-fold with 50 mg/day for 20 days per month for >5 years. It was likewise increased 20-fold with 25 mg/day for 20 days per month for >10 years. As such, the risk is strongly exposure-dependent in terms of dosage and duration of therapy.
The risk of surgically operated meningioma with CPA was around 4 in 1,000 patients per year with 50 mg/day for 20 days per month for >5 years. This underestimates the true incidence of meningioma with CPA since it only includes operated cases.
There were more than 500 operated cases of meningioma in association with CPA in France between 2007 and 2015. The unoperated incidence is unknown but likely to be much higher.
The population incidence of meningioma in general is strongly age-dependent. It’s 0.14 in 10,000 individuals per year at under 20 years of age and 4.9 in 10,000 individuals per year at above 85 years of age (a 35-fold difference). The average risk is 0.8 in 10,000 individuals per year. Accordingly, the risk of meningioma with CPA was found to be highly age-dependent as well.
The risk of meningioma decreases strongly after discontinuation of CPA.
As a result of their findings, the following occurred (ASNM/CNAM):
A toll-free phone number was set up in France for inquiries about CPA and meningioma.
Recommendations were made such that all patients starting CPA should be given an MRI within 6 months of initiation of therapy. Subsequently, all patients taking CPA should get another MRI after 5 years of therapy and then an additional MRI every 2 years of therapy thereafter.
It was recommended that CPA not be used in postmenopausal women due to their age.
Whether CPA should be withdrawn from the market was discussed but was decided against. Part of this was related to the reliance of transfeminine people on CPA for hormone therapy.
CPA sales in France plummeted by 73% from September 2018 to September 2019. Sales of spironolactone increased by a lesser magnitude over the same time period.
France is also looking into meningioma risk with other high-dose progestogen formulations, including nomegestrol acetate (Lutenyl) and chlormadinone acetate (Luteran).
France requested that the European Medicines Agency (EMA) conduct a review of CPA and meningioma. This review began in mid-2019 (Chustecka, 2019). In February 2020, the EMA announced restrictions on the clinical CPA use in the European Union (EMA, 2020). These included use of CPA only after failure of other treatments and use of the lowest effective maintenance dose of CPA (EMA, 2020; Chustecka, 2020). The restrictions applied to formulations of CPA containing 10 mg/day CPA or more (EMA, 2020; Chustecka, 2019). The Medicines and Healthcare products Regulatory Agency (MHRA) in the United Kingdom published a drug safety update for cyproterone acetate in June 2020 (MHRA, 2020).
This 2018 French news article (translated from French to English via Google Translate) succinctly discusses some of the developments. Several recent literature publications have also discussed the topic (Schmutz, 2018; Gerson, 2018; Coubret et al., 2019; Plu-Bureau, 2019; Senofonte, Pallotti, & Lombardo, 2020). And long-form reports of the epidemiological study by the French government here (PDF; 128 pages) and here (PDF; 106 pages) provide more information. A more succinct report by the French government can also be found here (PDF; 9 pages; English translation here). Unfortunately however, many of these materials are in French or other non-English languages and translations are unavailable for English speakers (Google Translate is useful however).
Implications for Transfeminine People
CPA tends to be used at very high doses in transfeminine people. A dose of CPA of 2 or 3 mg/day is similar in progestogenic exposure to that occurring during the luteal phase of the menstrual cycle in women. Conversely, doses of CPA recommended by guidelines for use in transfeminine people typically range from 25 to 100 mg/day. This progestogenic exposure is similar to or greater than that during pregnancy in women, when progesterone levels increase by up to 20-fold relative to luteal levels. As such, it’s highly supraphysiological relative to typical circumstances in women. In addition to meningioma, high doses of CPA have been associated with blood clots, prolactinomas, and liver toxicity (Wiki; Aly, 2019). Long-term therapy with progestogens like CPA may increase the risk of breast cancer (Aly, 2020; Table; CGHFBC, 2019; de Blok et al., 2019) and cardiovascular problems as well (Table; Stefanick, 2010). There is also a great deal still unknown about high doses of CPA and their safety, as evidenced by the fact that we’re only uncovering the risk of meningioma with CPA now. It’s likely that using the lowest possible doses of CPA will minimize its various health risks.
The doses of CPA used in transfeminine people tend to be far more than are necessary. CPA appears to have maximal effectiveness in terms of testosterone suppression at a dose of only 5 to 10 mg/day. See my article here for a review of this topic. The effectiveness of lower doses of CPA was recently validated by Meyer et al. (2020), who showed that in combination with estrogen, 10 mg/day CPA was no different from 50 mg/day CPA in terms of testosterone suppression in transfeminine people. It seems clear that transfeminine people should not use a dose of CPA higher than 10 or 12.5 mg/day at most. With 50-mg CPA tablets and a pill cutter, a total dose of CPA of 6.25 to 12.5 mg/day can be achieved by taking one-fourth of a tablet once every other day to once every day. As a result of new findings, published CPA dose recommendations for transfeminine people have gone down in the last few years (Aly, 2019). It is likely that this will continue to a greater extent in the future.
Meningiomas express progesterone receptors and activation of these receptors is thought to stimulate growth of meningiomas (Marosi et al., 2008; Li et al., 2013). Other progestogens besides CPA as well as pregnancy have also been associated with meningioma, and spontaneous regression of meningiomas has been reported after pregnancy (Li et al., 2013; Wiki). The incidence of meningiomas is 2- to 3-fold higher in women than in men, and the female-to-male incidence ratio is greatest during the peak female reproductive years (Marosi et al., 2008; Wiemels, Wrensch, & Claus, 2010; Li et al., 2013; Wiki). The luteal phase of the menstrual cycle, when progesterone levels are increased, has been associated with exacerbation of meningiomas in women (Marosi et al., 2008; Li et al., 2013; Wiki). Meningioma has also been associated with birth control pills and menopausal hormone therapy (estrogen–progestogen preparations) (Li et al., 2013). As such, assuming these associations are indeed due to progesterone receptor activation, there may be no fully risk-free dosage of progestogens or CPA in terms of meningioma. Indeed, meningiomas have been reported even with 5 to 10 mg/day CPA, despite the relatively uncommon use of such doses historically (Table). In any case, the data indicate that the risk is dose-dependent, and hence that lower doses should have the least risk. A dose of 2 to 3 mg/day CPA continuously would presumably have minimal excess risk of meningioma relative to normal female physiological circumstances.
Recommendations for Transfeminine People
In light of the risks of CPA and its high testosterone-suppressing effectiveness even at low doses, the following recommendations for use of CPA in transfeminine people seem appropriate:
CPA should be used at the lowest effective dosage (generally 5–12.5 mg/day and no greater).
CPA should be used for a limited duration (e.g., a few years) and not long-term.
Other safer but similarly effective antiandrogenic approaches should be used instead of CPA where possible (e.g., GnRH modulators, gonadectomy, estradiol monotherapy, etc.).
CPA should be avoided in older individuals (e.g., >50 years of age).
CPA should not be used in people with a personal or family history of meningioma.
If possible, CPA should be used under supervision of a physician and people taking CPA should follow monitoring guidelines (e.g., regular MRIs as in France).
At the same time it should be noted that the absolute incidence of meningioma with CPA is low. The risk is likely to be especially small in young people and with low CPA doses (e.g., 5–12.5 mg/day). Use of CPA in a more cautious and restricted fashion may allow for more acceptable safety. In any case, meningiomas and brain surgery are serious, so CPA should not be taken carelessly.
Updates
Update 1: Weill et al. (2021) [French Government Study]
The French government study was finally published in the formal scientific literature in February 2021:
Weill, A., Nguyen, P., Labidi, M., Cadier, B., Passeri, T., Duranteau, L., Bernat, A., Yoldjian, I., Fontanel, S., Froelich, S., & Coste, J. (2021). Use of High Dose Cyproterone Acetate and Risk of Intracranial Meningioma in Women: Cohort Study. BMJ, 372, n37. [DOI:10.1136/bmj.n37]
A Medscape article here covers the study and discusses consequent restrictions on CPA use.
Update 2: Mikkelsen et al. (2021) [Danish National Study]
A national study in Denmark was published in June 2021 and has replicated the findings:
Mikkelsen, A. P., Greiber, I. K., Scheller, N. M., Hilden, M., & Lidegaard, Ø. (2021). Cyproterone acetate and risk of meningioma: a nationwide cohort study. Journal of Neurology, Neurosurgery & Psychiatry, 93(2), 222–223. [DOI:10.1136/jnnp-2021-326138]
Update 3: VUMC Switching From CPA to GnRH Agonists
As of August 2020, the Vrije Universiteit Medical Center (VUMC) in Amsterdam is switching antiandrogen therapy of all transgender patients from cyproterone acetate to gonadotropin-releasing hormone (GnRH) agonists (VUMC, 2020). This was due to a Dutch Medicines Authority warning about meningiomas with CPA (VUMC, 2020). The VUMC is the home of the Center of Expertise on Gender Dysphoria, one of the largest care and research institutes for transgender hormone therapy in the world, and is highly influential in this area (Wiepjes et al., 2018; Bakker, 2021). The VUMC is said to be the provider of hormone therapy for about 95% of hormone-seeking transgender people in the Netherlands, and has treated about 10,000 transgender people since its gender clinic opened in 1972 (Wiepjes et al., 2018; Bakker, 2021).
Update 4: Millward et al. (2021)
In October 2021, a systematic review of meningiomas with CPA in transfeminine people was published:
Millward, C. P., Keshwara, S. M., Islim, A. I., Jenkinson, M. D., Alalade, A. F., & Gilkes, C. E. (2022). Development and Growth of Intracranial Meningiomas in Transgender Women Taking Cyproterone Acetate as Gender-Affirming Progestogen Therapy: A Systematic Review. Transgender Health, 7(6), 473–483. [DOI:10.1089/trgh.2021.0025]
Update 5: Lee et al. (2022)
In February 2022, a systematic review and meta-analysis of meningiomas with CPA was published:
Lee, K. S., Zhang, J., Kirollos, R., Santarius, T., Nga, V., & Yeo, T. T. (2022). A systematic review and meta-analysis of the association between cyproterone acetate and intracranial meningiomas. Scientific Reports, 12(1), 1942. [DOI:10.1038/s41598-022-05773-z]
In September 2022, the following French study of meningioma incidence in people treated with high-dose progestin therapy following institution of systematic screening was published:
Samoyeau, T., Provost, C., Roux, A., Legrand, L., Dezamis, E., Plu-Bureau, G., Pallud, J., Oppenheim, C., & Benzakoun, J. (2022). Meningioma in patients exposed to progestin drugs: results from a real-life screening program. Journal of Neuro-Oncology, 160(1), 127–136. [DOI:10.1007/s11060-022-04124-2]
The absolute incidence of meningiomas with CPA was 13 of 103 or a striking 13% of individuals. This was much higher than in the general population, in which rates of incidental meningiomas of about 1 to 2% have been observed. Moreover, the authors noted that the 13% figure observed in this study was likely an underestimate, as most cases of meningiomas with CPA were likely already diagnosed prior to the launch of systematic screening. Relative to the high absolute incidence, the year-adjusted incidence rate of meningiomas with CPA was 13.2 per 100,000 person–years. As with other studies, meningioma incidence was associated with longer CPA treatment duration, greater cumulative CPA dose, and older age. The incidence of meningiomas was much lower with two other progestins, nomegestrol acetate and chlormadinone acetate. These progestins tend to be used at much lower doses than CPA.
Update 7: Roland et al. (2024)
In March 2024, the following French study assessed meningioma risk with various progestogens:
Roland, N., Neumann, A., Hoisnard, L., Duranteau, L., Froelich, S., Zureik, M., & Weill, A. (2024). Use of progestogens and the risk of intracranial meningioma: national case-control study. BMJ, 384, e078078. [DOI:10.1136/bmj-2023-078078]
The study found increased risk of surgically operated meningiomas not only with CPA but also with a variety of other progestogens, including nomegestrol acetate, chlormadinone acetate, medrogestone, injectable medroxyprogesterone acetate, and promegestone. Conversely, progesterone, dydrogesterone, and levonorgestrelintrauterine devices showed no increased risk of surgically operated meningiomas. No conclusions could be drawn for dienogest or hydroxyprogesterone caproate due to the small numbers of individuals who received these progestogens. Many other progestogens, such as most 19-nortestosterone derivatives, were not included or assessed in the study. CPA, related to the high doses used in its case, had by far the greatest risk of surgically operated meningiomas. These findings indicate that meningiomas are a dose-dependent risk of many different progestogens.
References
Aly. (2019). Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Breast Cancer Risk with Hormone Therapy in Transfeminine People. Transfeminine Science. [URL]
Bakker, A. (2021). Een halve eeuw transgenderzorg aan de VU [Half a Century of Transgender Care at the VU]. Amsterdam: Boom. [Google Books] [WorldCat] [URL]
Bême. (2018 September 21). Androcur et risque de méningiomes: un numéro vert pour répondre aux patients. [Androcur and risk of meningiomas: a toll-free number to answer patients.] Doctissimo. [URL] [Translation]
Chustecka, Z. (2019 July 12). Risk of Meningioma With Cyproterone: EMA Review Begins. Medscape. [URL]
Chustecka, Z. (2020 February 14). EU Restricts High-Dose Cyproterone Because of Meningioma Risk. Medscape. [URL]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/s0140-6736(19)31709-x]
Coubret, A., Geniaux, H., & Buxeraud, J. (2019). Méningiomes et acétate de cyprotérone. Actualités Pharmaceutiques, 58(590), 55–56. [DOI:10.1016/j.actpha.2019.09.013]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, I. R., & den Heijer, M. (2019). Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in the Netherlands. BMJ, , l1652–l1652. [DOI:10.1136/bmj.l1652]
European Medicines Agency (EMA). (2020 February 14). Restrictions in use of cyproterone due to meningioma risk. European Medicines Agency. [URL] [PDF]
Lee, K. S., Zhang, J., Kirollos, R., Santarius, T., Nga, V., & Yeo, T. T. (2022). A systematic review and meta-analysis of the association between cyproterone acetate and intracranial meningiomas. Scientific Reports, 12(1), 1942. [DOI:10.1038/s41598-022-05773-z]
Li, Q., Coulson, H., Klaassen, Z., Sharma, S., Ramalingam, P., Moses, K. A., & Terris, M. K. (2013). Emerging association between androgen deprivation therapy and male meningioma: significant expression of luteinizing hormone-releasing hormone receptor in male meningioma. Prostate Cancer and Prostatic Diseases, 16(4), 387–390. [DOI:10.1038/pcan.2013.45]
Marosi, C., Hassler, M., Roessler, K., Reni, M., Sant, M., Mazza, E., & Vecht, C. (2008). Meningioma. Critical Reviews in Oncology/Hematology, 67(2), 153–171. [DOI:10.1016/j.critrevonc.2008.01.010]
Medicines and Healthcare products Regulatory Agency (MHRA). (2020 June 29). Cyproterone acetate: new advice to minimise risk of meningioma. GOV.UK. [URL]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463] [PDF]
Mikkelsen, A. P., Greiber, I. K., Scheller, N. M., Hilden, M., & Lidegaard, Ø. (2021). Cyproterone acetate and risk of meningioma: a nationwide cohort study. Journal of Neurology, Neurosurgery & Psychiatry, 93(2), 222–223. [DOI:10.1136/jnnp-2021-326138]
Millward, C. P., Keshwara, S. M., Islim, A. I., Jenkinson, M. D., Alalade, A. F., & Gilkes, C. E. (2022). Development and Growth of Intracranial Meningiomas in Transgender Women Taking Cyproterone Acetate as Gender-Affirming Progestogen Therapy: A Systematic Review. Transgender Health, 7(6), 473–483. [DOI:10.1089/trgh.2021.0025]
Nelson, R. (2021 February 3). Increased Risk of Meningioma With Cyproterone Acetate Use. Medscape. [URL]
Plu-Bureau, G. (2019). Faut-il rayer l’acétate de cyprotérone de nos prescriptions ? Gynécologie Obstétrique Fertilité & Sénologie , 47(12), 823–824. [DOI:10.1016/j.gofs.2019.10.006]
Roland, N., Neumann, A., Hoisnard, L., Duranteau, L., Froelich, S., Zureik, M., & Weill, A. (2024). Use of progestogens and the risk of intracranial meningioma: national case-control study. BMJ, 384, e078078. [DOI:10.1136/bmj-2023-078078]
Samoyeau, T., Provost, C., Roux, A., Legrand, L., Dezamis, E., Plu-Bureau, G., Pallud, J., Oppenheim, C., & Benzakoun, J. (2022). Meningioma in patients exposed to progestin drugs: results from a real-life screening program. Journal of Neuro-Oncology, 160(1), 127–136. [DOI:10.1007/s11060-022-04124-2]
Schmutz, J. (2018). Méningiomes et acétate de cyprotérone : mise au point. Annales de Dermatologie et de Vénéréologie, 145(5), 390–391. [DOI:10.1016/j.annder.2018.04.001]
Senofonte, G., Pallotti, F., & Lombardo, F. (2020). Ciproterone acetato e meningiomi: lo stato dell’arte. L’Endocrinologo, 21(3), 171–175. [DOI:10.1007/s40619-020-00746-8]
Stefanick, M. (2010). Postmenopausal hormone therapy and cardiovascular disease in women. Nutrition, Metabolism and Cardiovascular Diseases, 20(6), 451–458. [DOI:10.1016/j.numecd.2010.02.015]
University of Bristol. (2022 February 4). Widely-used hormone drug associated with increased risk of benign brain tumor at high doses. University of Bristol. [ScienceDaily] [Medical Xpress]
Vrije Universiteit Medical Center (VUMC). (2020). Wijzigingen medisch beleid hormoonbehandeling. [Changes in medical policy on hormone treatment.] Vrije Universiteit Medical Center. [URL]
Weill, A., Nguyen, P., Labidi, M., Cadier, B., Passeri, T., Duranteau, L., Bernat, A., Yoldjian, I., Fontanel, S., Froelich, S., & Coste, J. (2021). Use of High Dose Cyproterone Acetate and Risk of Intracranial Meningioma in Women: Cohort Study. BMJ, 372, n37. [DOI:10.1136/bmj.n37]
Wiemels, J., Wrensch, M., & Claus, E. B. (2010). Epidemiology and etiology of meningioma. Journal of Neuro-Oncology, 99(3), 307–314. [DOI:10.1007/s11060-010-0386-3]
Wiepjes, C. M., Nota, N. M., de Blok, C. J., Klaver, M., de Vries, A. L., Wensing-Kruger, S. A., de Jongh, R. T., Bouman, M., Steensma, T. D., Cohen-Kettenis, P., Gooren, L. J., Kreukels, B. P., & den Heijer, M. (2018). The Amsterdam Cohort of Gender Dysphoria Study (1972–2015): Trends in Prevalence, Treatment, and Regrets. The Journal of Sexual Medicine, 15(4), 582–590. [DOI:10.1016/j.jsxm.2018.01.016]
\ No newline at end of file
diff --git a/transfemscience.org/articles/e2-equivalent-doses/index.html b/transfemscience.org/articles/e2-equivalent-doses/index.html
index 1c46aa1d..cbe95923 100644
--- a/transfemscience.org/articles/e2-equivalent-doses/index.html
+++ b/transfemscience.org/articles/e2-equivalent-doses/index.html
@@ -1 +1 @@
-Approximate Comparable Dosages of Estradiol by Different Routes - Transfeminine Science
Approximate Comparable Dosages of Estradiol by Different Routes
By Aly | First published April 19, 2020 | Last modified April 13, 2023
Preface
Transfeminine people often ask about equivalent dosages of estradiol by different routes of administration. This page was put together to help facilitate answering these questions.
Table of Comparable Estradiol Dosages
The following is a table of estimated equivalent dosages of estradiol by different routes:
a For oral estradiol. Oral estradiol 1.5 mg is equivalent to about 2 mg oral estradiol valerate (Wiki). b Based on sublingual estradiol having ~2- to 5-fold greater bioavailability than oral estradiol per studies (Wiki; Sam, 2021). c Much lower doses of transdermal estradiol can be used in the case of genital application relative to conventional skin sites (potentially e.g. 5-fold lower doses for similar estradiol levels) (Aly., 2019). d Different patch brands may result in differing estradiol levels (Rohr, Nauert, & Stehle, 1999; Langley et al., 2008; Farahmand & Maibach, 2009; Langley et al., 2015). e For i.m. or s.c. injection, total dose per week of an estradiol ester like estradiol valerate, estradiol cypionate, estradiol enanthate, or estradiol benzoate. Differences in molecular weight between these esters are minor (Table) and can be ignored for simplicity. Optimal injection intervals vary depending on the ester and doses should be scaled by injection interval to match the listed total dose per week (Aly, 2021).
Notes
These doses are not absolute and should be considered only a rough guideline. They represent a generalized model based on many different studies with often very different individual findings. There is also an assumption that estradiol levels scale linearly or proportionally with dose, which may or may not actually be the case. One study found lower bioavailability of oral estradiol at high doses for instance (Kuhnz, Gansau, & Mahler, 1993).
These doses are approximate equivalent or comparable doses and don’t necessarily correspond to typical or recommended clinical doses. Injectable estradiol formulations are generally used at higher doses than other routes and forms of estradiol for instance (Aly, 2021).
The comparable doses are based on total estradiol exposure rather than therapeutic estrogenic potency. Time-related variations in sex hormone levels have been reported to modify potency of sex hormone preparations (Aly, 2021). However, this has not been factored in to the table here due to a lack of available data and analysis on the influence. In any case, it may be relevant to routes with large fluctuations in estradiol levels like sublingual administration and shorter-acting injections.
A transdermal estradiol spray sold under the brand name Lenzetto is available. In a study in postmenopausal women, mean baseline-adjusted estradiol levels with Lenzetto over the course of a week following achievement of steady state were about 6 pg/mL pre-treatment, 13 pg/mL with 1 spray/day (1.53 mg/day), 19 pg/mL with 2 sprays/day (3.06 mg/day), and 26 pg/mL with 3 sprays/day (4.59 mg/day) (Morton et al., 2009; Graph). Hence, this form of estradiol appears to achieve relatively low estradiol levels that likely aren’t well-suited for transfeminine people. No data are available on higher doses (i.e., more sprays per day) and so this formulation has not been included in the table.
Discussion
For reference, mean integrated levels of estradiol during the normal menstrual cycle in premenopausal cisgender women are about 100 pg/mL and during the luteal phase (the latter half of the menstrual cycle) are around 150 pg/mL (Graph; Graph; Aly, 2018). The total production of estradiol over a single menstrual cycle (i.e., one month) is about 6 mg on average (Rosenfield et al., 2008; Aly, 2018). Slightly higher doses are required for injectable estradiol esters relative to endogenous production amounts since they contain less estradiol by weight due to the ester components (Table).
Note that there is high interindividual variability (i.e., variability between individuals) in terms of estradiol levels achieved with different forms and routes of estradiol. As an example, some people may get relatively low estradiol levels with the oral route and others may get relatively low levels with the transdermal route. Conversely, some people may get relatively high levels with the transdermal route or with injections. For data showing the substantial variability even with injections, see Aly (2021). Due to the variability in estradiol levels between individuals, the appropriate doses will often not be the same for different people. Doses should be adjusted as necessary based on blood work. It should also be noted that there are large time-dependent changes in estradiol levels with certain routes, namely sublingual or buccal administration (Graphs; Graph; Sam, 2021) and intramuscular or subcutaneous injection (Graphs; Aly, 2021). Due to these fluctuations, estradiol levels will be vastly different when measured at different time points with these routes (e.g., around peak versus around trough).
For transfeminine people who have not yet undergone or do not plan to undergo gonadectomy, a high to very high dose of estradiol can be used to achieve strong suppression of testosterone levels. On average, the high dose will suppress testosterone levels by about 90%, to around 50 ng/dL (Wiki; Aly, 2018). Hence, the high to very high doses are indicated for estradiol monotherapy (i.e., estradiol alone without an antiandrogen). After gonadectomy, testosterone suppression is no longer needed and lower doses of estradiol, such as the moderate doses, can be used instead. High doses of estradiol are not necessarily required if estradiol is used in combination with an adequately effective antiandrogen, for instance cyproterone acetate, bicalutamide, or a gonadotropin-releasing hormone agonist or antagonist. Spironolactone, on the other hand, may often not be fully effective for opposing androgens (Aly, 2018).
References
General Sources
Aly. (2019). Analysis of Estradiol and Testosterone Levels with Oral Estradiol in Transfeminine People Based on Leinung et al. (2018). Transfeminine Science, oral-e2-leinung-2018. [Google Scholar] [URL]
Aly. (2021). An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations. Transfeminine Science, injectable-e2-meta-analysis. [Google Scholar] [URL]
Armston, A., & Wood, P. (2002). Hormone replacement therapy (oestradiol-only preparations): can the laboratory recommend a concentration of plasma oestradiol to protect against osteoporosis? Annals of Clinical Biochemistry, 39(3), 184–193. [DOI:10.1258/0004563021902107]
Baker, V. L. (1994). Alternatives to Oral Estrogen Replacement: Transdermal Patches, Percutaneous Gels, Vaginal Creams and Rings, Implants, and Other Methods of Delivery. Obstetrics and Gynecology Clinics of North America, 21(2) [Primary Care of the Mature Woman], 271–297. [DOI:10.1016/S0889-8545(21)00629-X]
Bruni, V., & Pampaloni, F. (2019). Hormone Replacement Therapy in Premature Ovarian Insufficiency. In Berga, S. L., Genazzani, A. R., Naftolin, F., & Petraglia, F. (Eds.). Menstrual Cycle Related Disorders (pp. 111–142). Cham: Springer. [DOI:10.1007/978-3-030-14358-9_10]
Farahmand, S., & Maibach, H. I. (2009). Transdermal drug pharmacokinetics in man: Interindividual variability and partial prediction. International Journal of Pharmaceutics, 367(1–2), 1–15. [DOI:10.1016/j.ijpharm.2008.11.020]
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of Subcutaneous and Intramuscular Estradiol Regimens as part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Lobo, R. A., & Cassidenti, D. L. (1992). Pharmacokinetics of Oral 17 β-Estradiol. The Journal of Reproductive Medicine, 37(1), 77–84. [Google Scholar] [PubMed] [PDF]
Sam. (2021). An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People. Transfeminine Science, sublingual-e2-transfem. [Google Scholar] [URL]
Simon, J. A., & Snabes, M. C. (2007). Menopausal hormone therapy for vasomotor symptoms: balancing the risks and benefits with ultra-low doses of estrogen. Expert Opinion on Investigational Drugs, 16(12), 2005–2020. [DOI:10.1517/13543784.16.12.2005]
Taboada, M., Santen, R., Lima, J., Hossain, J., Singh, R., Klein, K. O., & Mauras, N. (2011). Pharmacokinetics and pharmacodynamics of oral and transdermal 17β estradiol in girls with Turner syndrome. The Journal of Clinical Endocrinology & Metabolism, 96(11), 3502–3510. [DOI:10.1210/jc.2011-1449]
Wikipedia. (2020, August 29). Pharmacokinetics of estradiol. Wikimedia Foundation. [URL]
Wikipedia. (2020, October 2). Template:Estrogen dosages for menopausal hormone therapy. Wikimedia Foundation. [URL]
Inline Citations
Farahmand, S., & Maibach, H. I. (2009). Transdermal drug pharmacokinetics in man: Interindividual variability and partial prediction. International Journal of Pharmaceutics, 367(1–2), 1–15. [DOI:10.1016/j.ijpharm.2008.11.020]
Kuhnz, W., Gansau, C., & Mahler, M. (1993). Pharmacokinetics of estradiol, free and total estrone, in young women following single intravenous and oral administration of 17β-estradiol. Arzneimittelforschung, 43(9), 966–973. [Google Scholar] [PubMed] [PDF]
Langley, R. E., Godsland, I. F., Kynaston, H., Clarke, N. W., Rosen, S. D., Morgan, R. C., Pollock, P., Kockelbergh, R., Lalani, E., Dearnaley, D., Parmar, M., & Abel, P. D. (2008). Early hormonal data from a multicentre phase II trial using transdermal oestrogen patches as first-line hormonal therapy in patients with locally advanced or metastatic prostate cancer. BJU International, 102(4), 442–445. [DOI:10.1111/j.1464-410x.2008.07583.x]
Langley, R. E., Duong, T., Godsland, I. F., Kynaston, H., Kockelbergh, R., Rosen, S. D., Alhasso, A. A., Dearnaley, D. P., Clarke, N. W., Jovic, G., Carpenter, R., Bara, A., Welland, A., Parmar, M. K., & Abel, P. D. (2015). Oestrogen patches (OP) to treat prostate cancer (PC) – Are different commercial brands interchangeable? Maturitas, 81(1), 210–211. [DOI:10.1016/j.maturitas.2015.02.320]
Morton, T. L., Gattermeir, D. J., Petersen, C. A., Day, W. W., & Schumacher, R. J. (2009). Steady-State Pharmacokinetics Following Application of a Novel Transdermal Estradiol Spray in Healthy Postmenopausal Women. The Journal of Clinical Pharmacology, 49(9), 1037–1046. [DOI:10.1177/0091270009339187]
Rohr, U. D., Nauert, C., & Stehle, B. (1999). 17β-Estradiol delivered by three different matrix patches 50 μg/day. Maturitas, 33(1), 45–58. [DOI:10.1016/s0378-5122(99)00039-0]
Rosenfield, R. L., Cooke, D. W., & Radovick, S. (2008). Puberty and Its Disorders in the Female. In Sperling, M. A. (Ed.). Pediatric Endocrinology, 3rd Edition (pp. 530–609). Philadelphia: Saunders/Elsevier. [Google Scholar] [Google Books] [DOI:10.1016/b978-141604090-3.50019-3] [Archive.org]
\ No newline at end of file
+Approximate Comparable Dosages of Estradiol by Different Routes - Transfeminine Science
Approximate Comparable Dosages of Estradiol by Different Routes
By Aly | First published April 19, 2020 | Last modified April 13, 2023
Preface
Transfeminine people often ask about equivalent dosages of estradiol by different routes of administration. This page was put together to help facilitate answering these questions.
Table of Comparable Estradiol Dosages
The following is a table of estimated equivalent dosages of estradiol by different routes:
a For oral estradiol. Oral estradiol 1.5 mg is equivalent to about 2 mg oral estradiol valerate (Wiki). b Based on sublingual estradiol having ~2- to 5-fold greater bioavailability than oral estradiol per studies (Wiki; Sam, 2021). c Much lower doses of transdermal estradiol can be used in the case of genital application relative to conventional skin sites (potentially e.g. 5-fold lower doses for similar estradiol levels) (Aly., 2019). d Different patch brands may result in differing estradiol levels (Rohr, Nauert, & Stehle, 1999; Langley et al., 2008; Farahmand & Maibach, 2009; Langley et al., 2015). e For i.m. or s.c. injection, total dose per week of an estradiol ester like estradiol valerate, estradiol cypionate, estradiol enanthate, or estradiol benzoate. Differences in molecular weight between these esters are minor (Table) and can be ignored for simplicity. Optimal injection intervals vary depending on the ester and doses should be scaled by injection interval to match the listed total dose per week (Aly, 2021).
Notes
These doses are not absolute and should be considered only a rough guideline. They represent a generalized model based on many different studies with often very different individual findings. There is also an assumption that estradiol levels scale linearly or proportionally with dose, which may or may not actually be the case. One study found lower bioavailability of oral estradiol at high doses for instance (Kuhnz, Gansau, & Mahler, 1993).
These doses are approximate equivalent or comparable doses and don’t necessarily correspond to typical or recommended clinical doses. Injectable estradiol formulations are generally used at higher doses than other routes and forms of estradiol for instance (Aly, 2021).
The comparable doses are based on total estradiol exposure rather than therapeutic estrogenic potency. Time-related variations in sex hormone levels have been reported to modify potency of sex hormone preparations (Aly, 2021). However, this has not been factored in to the table here due to a lack of available data and analysis on the influence. In any case, it may be relevant to routes with large fluctuations in estradiol levels like sublingual administration and shorter-acting injections.
A transdermal estradiol spray sold under the brand name Lenzetto is available. In a study in postmenopausal women, mean baseline-adjusted estradiol levels with Lenzetto over the course of a week following achievement of steady state were about 6 pg/mL pre-treatment, 13 pg/mL with 1 spray/day (1.53 mg/day), 19 pg/mL with 2 sprays/day (3.06 mg/day), and 26 pg/mL with 3 sprays/day (4.59 mg/day) (Morton et al., 2009; Graph). Hence, this form of estradiol appears to achieve relatively low estradiol levels that likely aren’t well-suited for transfeminine people. No data are available on higher doses (i.e., more sprays per day) and so this formulation has not been included in the table.
Discussion
For reference, mean integrated levels of estradiol during the normal menstrual cycle in premenopausal cisgender women are about 100 pg/mL and during the luteal phase (the latter half of the menstrual cycle) are around 150 pg/mL (Graph; Graph; Aly, 2018). The total production of estradiol over a single menstrual cycle (i.e., one month) is about 6 mg on average (Rosenfield et al., 2008; Aly, 2018). Slightly higher doses are required for injectable estradiol esters relative to endogenous production amounts since they contain less estradiol by weight due to the ester components (Table).
Note that there is high interindividual variability (i.e., variability between individuals) in terms of estradiol levels achieved with different forms and routes of estradiol. As an example, some people may get relatively low estradiol levels with the oral route and others may get relatively low levels with the transdermal route. Conversely, some people may get relatively high levels with the transdermal route or with injections. For data showing the substantial variability even with injections, see Aly (2021). Due to the variability in estradiol levels between individuals, the appropriate doses will often not be the same for different people. Doses should be adjusted as necessary based on blood work. It should also be noted that there are large time-dependent changes in estradiol levels with certain routes, namely sublingual or buccal administration (Graphs; Graph; Sam, 2021) and intramuscular or subcutaneous injection (Graphs; Aly, 2021). Due to these fluctuations, estradiol levels will be vastly different when measured at different time points with these routes (e.g., around peak versus around trough).
For transfeminine people who have not yet undergone or do not plan to undergo gonadectomy, a high to very high dose of estradiol can be used to achieve strong suppression of testosterone levels. On average, the high dose will suppress testosterone levels by about 90%, to around 50 ng/dL (Wiki; Aly, 2018). Hence, the high to very high doses are indicated for estradiol monotherapy (i.e., estradiol alone without an antiandrogen). After gonadectomy, testosterone suppression is no longer needed and lower doses of estradiol, such as the moderate doses, can be used instead. High doses of estradiol are not necessarily required if estradiol is used in combination with an adequately effective antiandrogen, for instance cyproterone acetate, bicalutamide, or a gonadotropin-releasing hormone agonist or antagonist. Spironolactone, on the other hand, may often not be fully effective for opposing androgens (Aly, 2018).
References
General Sources
Aly. (2019). Analysis of Estradiol and Testosterone Levels with Oral Estradiol in Transfeminine People Based on Leinung et al. (2018). Transfeminine Science, oral-e2-leinung-2018. [Google Scholar] [URL]
Aly. (2021). An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations. Transfeminine Science, injectable-e2-meta-analysis. [Google Scholar] [URL]
Armston, A., & Wood, P. (2002). Hormone replacement therapy (oestradiol-only preparations): can the laboratory recommend a concentration of plasma oestradiol to protect against osteoporosis? Annals of Clinical Biochemistry, 39(3), 184–193. [DOI:10.1258/0004563021902107]
Baker, V. L. (1994). Alternatives to Oral Estrogen Replacement: Transdermal Patches, Percutaneous Gels, Vaginal Creams and Rings, Implants, and Other Methods of Delivery. Obstetrics and Gynecology Clinics of North America, 21(2) [Primary Care of the Mature Woman], 271–297. [DOI:10.1016/S0889-8545(21)00629-X]
Bruni, V., & Pampaloni, F. (2019). Hormone Replacement Therapy in Premature Ovarian Insufficiency. In Berga, S. L., Genazzani, A. R., Naftolin, F., & Petraglia, F. (Eds.). Menstrual Cycle Related Disorders (pp. 111–142). Cham: Springer. [DOI:10.1007/978-3-030-14358-9_10]
Farahmand, S., & Maibach, H. I. (2009). Transdermal drug pharmacokinetics in man: Interindividual variability and partial prediction. International Journal of Pharmaceutics, 367(1–2), 1–15. [DOI:10.1016/j.ijpharm.2008.11.020]
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of Subcutaneous and Intramuscular Estradiol Regimens as part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Lobo, R. A., & Cassidenti, D. L. (1992). Pharmacokinetics of Oral 17 β-Estradiol. The Journal of Reproductive Medicine, 37(1), 77–84. [Google Scholar] [PubMed] [PDF]
Sam. (2021). An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People. Transfeminine Science, sublingual-e2-transfem. [Google Scholar] [URL]
Simon, J. A., & Snabes, M. C. (2007). Menopausal hormone therapy for vasomotor symptoms: balancing the risks and benefits with ultra-low doses of estrogen. Expert Opinion on Investigational Drugs, 16(12), 2005–2020. [DOI:10.1517/13543784.16.12.2005]
Taboada, M., Santen, R., Lima, J., Hossain, J., Singh, R., Klein, K. O., & Mauras, N. (2011). Pharmacokinetics and pharmacodynamics of oral and transdermal 17β estradiol in girls with Turner syndrome. The Journal of Clinical Endocrinology & Metabolism, 96(11), 3502–3510. [DOI:10.1210/jc.2011-1449]
Wikipedia. (2020, August 29). Pharmacokinetics of estradiol. Wikimedia Foundation. [URL]
Wikipedia. (2020, October 2). Template:Estrogen dosages for menopausal hormone therapy. Wikimedia Foundation. [URL]
Inline Citations
Aly. (2018). A Review of Studies on Spironolactone and Testosterone Suppression in Cisgender Men, Cisgender Women, and Transfeminine People. Transfeminine Science. [URL]
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). Genital Application via the Scrotum and Neolabia for Greatly Enhanced Absorption of Transdermal Estradiol in Transfeminine People. Transfeminine Science. [URL]
Farahmand, S., & Maibach, H. I. (2009). Transdermal drug pharmacokinetics in man: Interindividual variability and partial prediction. International Journal of Pharmaceutics, 367(1–2), 1–15. [DOI:10.1016/j.ijpharm.2008.11.020]
Kuhnz, W., Gansau, C., & Mahler, M. (1993). Pharmacokinetics of estradiol, free and total estrone, in young women following single intravenous and oral administration of 17β-estradiol. Arzneimittelforschung, 43(9), 966–973. [Google Scholar] [PubMed] [PDF]
Langley, R. E., Godsland, I. F., Kynaston, H., Clarke, N. W., Rosen, S. D., Morgan, R. C., Pollock, P., Kockelbergh, R., Lalani, E., Dearnaley, D., Parmar, M., & Abel, P. D. (2008). Early hormonal data from a multicentre phase II trial using transdermal oestrogen patches as first-line hormonal therapy in patients with locally advanced or metastatic prostate cancer. BJU International, 102(4), 442–445. [DOI:10.1111/j.1464-410x.2008.07583.x]
Langley, R. E., Duong, T., Godsland, I. F., Kynaston, H., Kockelbergh, R., Rosen, S. D., Alhasso, A. A., Dearnaley, D. P., Clarke, N. W., Jovic, G., Carpenter, R., Bara, A., Welland, A., Parmar, M. K., & Abel, P. D. (2015). Oestrogen patches (OP) to treat prostate cancer (PC) – Are different commercial brands interchangeable? Maturitas, 81(1), 210–211. [DOI:10.1016/j.maturitas.2015.02.320]
Morton, T. L., Gattermeir, D. J., Petersen, C. A., Day, W. W., & Schumacher, R. J. (2009). Steady-State Pharmacokinetics Following Application of a Novel Transdermal Estradiol Spray in Healthy Postmenopausal Women. The Journal of Clinical Pharmacology, 49(9), 1037–1046. [DOI:10.1177/0091270009339187]
Rohr, U. D., Nauert, C., & Stehle, B. (1999). 17β-Estradiol delivered by three different matrix patches 50 μg/day. Maturitas, 33(1), 45–58. [DOI:10.1016/s0378-5122(99)00039-0]
Rosenfield, R. L., Cooke, D. W., & Radovick, S. (2008). Puberty and Its Disorders in the Female. In Sperling, M. A. (Ed.). Pediatric Endocrinology, 3rd Edition (pp. 530–609). Philadelphia: Saunders/Elsevier. [Google Scholar] [Google Books] [DOI:10.1016/b978-141604090-3.50019-3] [Archive.org]
\ No newline at end of file
diff --git a/transfemscience.org/articles/ec508/index.html b/transfemscience.org/articles/ec508/index.html
index 428da4f7..9f6eb453 100644
--- a/transfemscience.org/articles/ec508/index.html
+++ b/transfemscience.org/articles/ec508/index.html
@@ -1 +1 @@
-EC508 (Estradiol Aminosulfonylbenzoylproline), a Unique and Highly Promising Estradiol Ester and Possible Oral Estradiol Form of the Future - Transfeminine Science
EC508 (Estradiol Aminosulfonylbenzoylproline), a Unique and Highly Promising Estradiol Ester and Possible Oral Estradiol Form of the Future
By Aly | First published August 2, 2018 | Last modified September 29, 2021
EC508, also known as estradiol 17β-(1-(4-(aminosulfonyl)benzoyl)-L-proline), or somewhat more simply as estradiol aminosulfonylbenzoylproline, is an estradiol ester and prodrug of estradiol which is under development by a small pharmaceutical company called Evestra for potential medical use in menopausal hormone therapy and as a form of hormonal birth control in cisgender women. It is intended for oral administration similarly to certain other orally used estradiol esters and prodrugs like estradiol valerate and estradiol acetate. However, EC508 is very different from conventional estradiol esters, and is far more promising in comparison. This is because from a theoretical standpoint, despite the fact that it is intended for use as an oral form of estradiol, EC508 has a pharmacological profile by this route that is much more similar to that of non-oral estradiol forms like transdermal estradiol and injectable estradiol than to oral administration of estradiol or conventional estradiol esters.
A “first pass” through the intestines and liver normally occurs with oral administration of drugs before they reach the bloodstream. Estradiol is highly susceptible to metabolism in the liver and intestines. For this reason, when estradiol is taken orally, it is overwhelmingly converted into metabolites such as estrone, estrone sulfate, various other estrogen conjugates, and hydroxylated metabolites. These metabolites of estradiol are mostly inactive as estrogens, and with oral administration, the major metabolites like estrone and estrone sulfate circulate in the blood at much higher levels than does estradiol. Although not currently known to be therapeutically consequential, the circulating metabolite profile of oral estradiol is very unphysiological relative to normal hormonal circumstances. The metabolism of estradiol with the oral first pass is so substantial that the oral bioavailability of estradiol, or the amount of an oral dose that reaches the blood as estradiol when compared to the same dose of estradiol infused directly into the blood, is only about 5%. Moreover, there is substantial variability between people in the bioavailability of oral estradiol, with one pharmacokinetic study reporting a range of 0.1 to 12% in different individuals (Wiki). The estradiol levels that are achieved with the same dose of oral estradiol vary considerably in different people, and varying degrees of first-pass metabolism certainly contribute to this. Conventional estradiol esters like estradiol valerate are little different than estradiol with oral administration, due to rapid and probably complete conversion into estradiol during the first pass.
Another consequence of the first pass with oral estradiol is that much higher levels of estradiol are present in the liver than normal or with non-oral estradiol (Wiki). It is estimated that the exposure of the liver to estradiol is around 5 times greater with oral estradiol than with non-oral estradiol. This is of importance because the liver is an estrogen-sensitive organ, with significant expression of estrogen receptors. The liver synthesizes a large number of important proteins and other products that are then released into the bloodstream and regulate biological functions throughout the body. Through activation of estrogen receptors, estrogens can modulate the production of many such liver-derived products (Table). Some of the more well-known examples include sex hormone-binding globulin (SHBG) and blood lipids like cholesterol and triglycerides. Of particular therapeutic concern however is the influence of estradiol and other estrogens on the synthesis of coagulation factors. By modifying the liver synthesis of these proteins, estrogens can have procoagulatory effects and can increase the risk of blood clots and associated cardiovascular problems like stroke and heart attack (Aly, 2020). Physiological levels of estradiol normally only have a small influence on liver synthesis, and effects like strong procoagulation are specific to states of extremely high estradiol levels like pregnancy. However, oral estradiol, due to the liver first pass and particularly at high doses, can result in pregnancy-like changes in liver synthesis and hence on coagulation and associated health risks. This is not something that normally occurs with physiological doses of non-oral estradiol forms, which have a minimal influence even at higher levels (although very high doses of non-oral estradiol can certainly still have such effects).
Because of the first pass and its low bioavailability, substantial variability, unphysiological metabolite pattern, greater impact on liver synthesis, and greater health risks, oral estradiol is a less favorable form of estradiol than are non-oral estradiol forms like transdermal and injectable estradiol. Consequently, it is generally recommended that non-oral estradiol be used instead of oral estradiol where possible. However, oral estradiol is less expensive than certain non-oral forms (e.g., transdermal), is very easy and convenient to use, and has ubiquitous availability (unlike e.g. injectables). As such, it continues to see widespread use, and remains probably the most commonly used form of estradiol in medicine.
EC508 is special because although it is intended as an oral form of estradiol, it is not like oral estradiol or conventional orally used estradiol esters like estradiol valerate. EC508 is different because it appears to bypass the first pass that normally occurs with oral administration. Due to its unique ester, it seems to be resistant to metabolism in the intestines and liver, which limits its conversion into estradiol and estradiol metabolites at this time. This is also notably seen with certain other estrogen esters like estrone sulfate and estramustine phosphate (estradiol normustine phosphate). In addition, and more importantly, because of its unusual ester moiety, EC508 binds with high affinity to a protein called carbonic anhydrase II (CAII) in red blood cells. This binding causes EC508 to be rapidly accumulated and sequestered into blood cells. Estradiol normally goes through the hepatic portal vein, a blood vessel that carries blood from the gastrointestinal tract to the liver, to reach the liver. With EC508, it is taken up into blood cells so quickly that little ultimately seems to reach the liver. Instead, the red blood cells carry EC508 through the portal vein to the bloodstream, which it is then slowly released into. As with other estradiol esters, EC508 has little or no estrogenic activity itself and is only active after being converted into estradiol.
In accordance with the fact that it bypasses the oral first pass, EC508 shows 100% bioavailability in animals. This can be compared in humans to about 5% for oral estradiol and about 43% for ethinylestradiol (EE)—a syntheticrelative of estradiol with strongly improved metabolic stability (but also far greater influence on liver synthesis and related health risks than estradiol). As a result of its complete bioavailability, EC508 has 100 times the oral estrogenic potency of estradiol and 10 times that of ethinylestradiol in animals. As such, EC508 would be expected to be active in humans at microgram (μg) doses, similarly to the estimated delivery rates of transdermal estradiol patches (e.g., 25–100 μg/day) as well as to typical doses of ethinylestradiol (e.g., 5–50 μg/day) but in contrast to the 100-fold higher usual doses of oral estradiol (e.g., 0.5–10 mg/day). Further in accordance with it bypassing the oral first pass, EC508 and non-oral estradiol showed little or no effect on liver synthesis at assessed doses in animals, whereas oral estradiol and ethinylestradiol showed marked effects. Lastly, EC508 had a biological half-life of 5 hours in rats, which is relatively long for this species. For this reason, a long duration may be anticipated with EC508 in humans, in contrast to estradiol injected directly into the blood (which has a very short half-life) but similarly to oral, transdermal, and injectable estradiol.
An earlier drug candidate called estradiol sulfamate (E2MATE; J995), which is likewise a special estradiol ester that binds to CAII and bypasses the oral first pass, was discovered by the same research group in the 1990s, and was likewise developed as a new form of oral estradiol. However, it was subsequently found that E2MATE is additionally a highly potent inhibitor of steroid sulfatase (STS) and prevents its own activation by this enzyme into estradiol in humans. This served to abolish its estrogenic effects in the case of humans and resulted in it being not useful for clinical estrogen therapy. E2MATE was also undesirably found to be substantially converted into estrone sulfamate (E1MATE; J994) during the first pass with oral administration. Consequent to these findings, E2MATE was eventually repurposed as an estrogen synthesis inhibitor for treatment of endometriosis, and as of 2017, remains under development for this indication. In contrast to E2MATE, EC508 is not converted into the corresponding estrone variant, and is not thought to be an STS inhibitor, so the problems that E2MATE had are not expected with it.
Taken together, EC508 shows a preclinical and theoretical profile of being an oral form of estradiol that bypasses the first pass with oral administration. As a result, EC508 with oral administration undergoes little or no first-pass metabolism, has high bioavailability, and does not result in excessive estrogenic exposure in the liver. Consequently, EC508 is expected to be orally active at much lower doses than estradiol, to have reduced variability compared to estradiol, to lack an unphysiological metabolite pattern, and to not have disproportionate influence on estrogen-sensitive liver synthesis and associated health risks. In these regards, EC508 shows a favorable and advantageous profile relative to oral estradiol that is analogous to that with non-oral forms of estradiol like transdermal and injectable estradiol but with the convenience of oral administration. For these reasons, EC508 is a pharmaceutical candidate with the potential to supersede not only oral estradiol in clinical practice but also non-oral estradiol and even other clinically used estrogens like ethinylestradiol (e.g., in birth control pills). If it’s successfully developed and approved, EC508 would naturally one day be used in transfeminine hormone therapy as well.
In addition to EC508, the researchers at Evestra synthesized a testosterone equivalent known as EC586 (testosterone 17β-(1-((5-(aminosulfonyl)-2-pyridinyl)carbonyl)-L-proline or slightly more simply as testosterone aminosulfonylpyridinylcarbonylproline). This testosterone ester shows analogous properties to those of EC508, and hence has the potential to be a non-oral-like form of oral testosterone with high bioavailability and potency, a long duration, and lack of disproportionate liver effects—and hence with major advantages over oral testosterone forms available today. In parallel to EC508, EC586 (or similar drugs) could someday be used in transmasculine hormone therapy. The prodrug approach used to create EC508 and EC586 also notably has the potential to be employed with many other drugs.
Update: No Further Development
Patents for EC508 were filed in 2014 and 2017 and publications describing it were published in the scientific literature in 2017. An Investigational New Drug (IND) application for EC508 was tentatively supposed to be filed with the Food and Drug Administration (FDA) in the United States in the second quarter of 2018 (Evestra). However, as of September 2021, there have been no further updates on the development of EC508. In addition, Evestra’s website has since been redesigned and its research pipeline page listing the status of EC508 has been removed. It’s unknown at this time if EC508 and EC586 are still under development or whether further updates on these compounds can be expected.
References
Ahmed, G., Elger, W., Meece, F., Nair, H. B., Schneider, B., Wyrwa, R., & Nickisch, K. (2017). A prodrug design for improved oral absorption and reduced hepatic interaction. Bioorganic & Medicinal Chemistry, 25(20), 5569–5575. [DOI:10.1016/j.bmc.2017.08.027]
Elger, W., Schwarz, S., Hedden, A., Reddersen, G., & Schneider, B. (1995). Sulfamates of various estrogens are prodrugs with increased systemic and reduced hepatic estrogenicity at oral application. The Journal of Steroid Biochemistry and Molecular Biology, 55(3-4), 395–403. [DOI:10.1016/0960-0760(95)00214-6]
Elger, W., Palme, H. J., & Schwarz, S. (1998). Novel oestrogen sulfamates: a new approach to oral hormone therapy. Expert Opinion on Investigational Drugs, 7(4), 575–589. [DOI:10.1517/13543784.7.4.575]
Elger, W., Barth, A., Hedden, A., Reddersen, G., Ritter, P., Schneider, B., Züchner, J., Krahl, E., Müller, K., Oettel, M., & Schwarz, S. (2001). Estrogen Sulfamates: A New Approach to Oral Estrogen Therapy. Reproduction, Fertility, and Development, 13(4), 297–305. [DOI:10.1071/rd01029]
Elger, W., Wyrwa, R., Ahmed, G., Meece, F., Nair, H. B., Santhamma, B., Killeen, Z., Schneider, B., Meister, R., Schubert, H., & Nickisch, K. (2017). Estradiol prodrugs (EP) for efficient oral estrogen treatment and abolished effects on estrogen modulated liver functions. The Journal of Steroid Biochemistry and Molecular Biology, 165, 305–311. [DOI:10.1016/j.jsbmb.2016.07.008]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Nickisch, K., Santhamma, B., Ahmed, G., Meece, F., Elger, W., Wyrwa, R., & Nair, H. (2017). U.S. Patent No. 9,745,338. Washington, DC: U.S. Patent and Trademark Office. [US9745338B2]
Nickisch, K., Santhamma, B., Ahmed, G., Meece, F., Elger, W., Wyrwa, R., & Nair, H. (2019). U.S. Patent No. 10,273,263. Washington, DC: U.S. Patent and Trademark Office. [US10273263B2]
Potter, B. V. (2018). SULFATION PATHWAYS: Steroid sulphatase inhibition via aryl sulphamates: clinical progress, mechanism and future prospects. Journal of Molecular Endocrinology, 61(2), T233–T252. [DOI:10.1530/JME-18-0045]
Stanczyk, F. Z., Archer, D. F., & Bhavnani, B. R. (2013). Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception, 87(6), 706–727. [DOI:10.1016/j.contraception.2012.12.011]
Thomas, M. P., & Potter, B. V. (2015). Discovery and development of the aryl O-sulfamate pharmacophore for oncology and women’s health. Journal of Medicinal Chemistry, 58(19), 7634–7658. [DOI:10.1021/acs.jmedchem.5b00386]
Thomas, M. P., & Potter, B. V. (2015). Estrogen O-sulfamates and their analogues: clinical steroid sulfatase inhibitors with broad potential. The Journal of Steroid Biochemistry and Molecular Biology, 153, 160–169. [DOI:10.1016/j.jsbmb.2015.03.012]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
\ No newline at end of file
+EC508 (Estradiol Aminosulfonylbenzoylproline), a Unique and Highly Promising Estradiol Ester and Possible Oral Estradiol Form of the Future - Transfeminine Science
EC508 (Estradiol Aminosulfonylbenzoylproline), a Unique and Highly Promising Estradiol Ester and Possible Oral Estradiol Form of the Future
By Aly | First published August 2, 2018 | Last modified September 29, 2021
EC508, also known as estradiol 17β-(1-(4-(aminosulfonyl)benzoyl)-L-proline), or somewhat more simply as estradiol aminosulfonylbenzoylproline, is an estradiol ester and prodrug of estradiol which is under development by a small pharmaceutical company called Evestra for potential medical use in menopausal hormone therapy and as a form of hormonal birth control in cisgender women. It is intended for oral administration similarly to certain other orally used estradiol esters and prodrugs like estradiol valerate and estradiol acetate. However, EC508 is very different from conventional estradiol esters, and is far more promising in comparison. This is because from a theoretical standpoint, despite the fact that it is intended for use as an oral form of estradiol, EC508 has a pharmacological profile by this route that is much more similar to that of non-oral estradiol forms like transdermal estradiol and injectable estradiol than to oral administration of estradiol or conventional estradiol esters.
A “first pass” through the intestines and liver normally occurs with oral administration of drugs before they reach the bloodstream. Estradiol is highly susceptible to metabolism in the liver and intestines. For this reason, when estradiol is taken orally, it is overwhelmingly converted into metabolites such as estrone, estrone sulfate, various other estrogen conjugates, and hydroxylated metabolites. These metabolites of estradiol are mostly inactive as estrogens, and with oral administration, the major metabolites like estrone and estrone sulfate circulate in the blood at much higher levels than does estradiol. Although not currently known to be therapeutically consequential, the circulating metabolite profile of oral estradiol is very unphysiological relative to normal hormonal circumstances. The metabolism of estradiol with the oral first pass is so substantial that the oral bioavailability of estradiol, or the amount of an oral dose that reaches the blood as estradiol when compared to the same dose of estradiol infused directly into the blood, is only about 5%. Moreover, there is substantial variability between people in the bioavailability of oral estradiol, with one pharmacokinetic study reporting a range of 0.1 to 12% in different individuals (Wiki). The estradiol levels that are achieved with the same dose of oral estradiol vary considerably in different people, and varying degrees of first-pass metabolism certainly contribute to this. Conventional estradiol esters like estradiol valerate are little different than estradiol with oral administration, due to rapid and probably complete conversion into estradiol during the first pass.
Another consequence of the first pass with oral estradiol is that much higher levels of estradiol are present in the liver than normal or with non-oral estradiol (Wiki). It is estimated that the exposure of the liver to estradiol is around 5 times greater with oral estradiol than with non-oral estradiol. This is of importance because the liver is an estrogen-sensitive organ, with significant expression of estrogen receptors. The liver synthesizes a large number of important proteins and other products that are then released into the bloodstream and regulate biological functions throughout the body. Through activation of estrogen receptors, estrogens can modulate the production of many such liver-derived products (Table). Some of the more well-known examples include sex hormone-binding globulin (SHBG) and blood lipids like cholesterol and triglycerides. Of particular therapeutic concern however is the influence of estradiol and other estrogens on the synthesis of coagulation factors. By modifying the liver synthesis of these proteins, estrogens can have procoagulatory effects and can increase the risk of blood clots and associated cardiovascular problems like stroke and heart attack (Aly, 2020). Physiological levels of estradiol normally only have a small influence on liver synthesis, and effects like strong procoagulation are specific to states of extremely high estradiol levels like pregnancy. However, oral estradiol, due to the liver first pass and particularly at high doses, can result in pregnancy-like changes in liver synthesis and hence on coagulation and associated health risks. This is not something that normally occurs with physiological doses of non-oral estradiol forms, which have a minimal influence even at higher levels (although very high doses of non-oral estradiol can certainly still have such effects).
Because of the first pass and its low bioavailability, substantial variability, unphysiological metabolite pattern, greater impact on liver synthesis, and greater health risks, oral estradiol is a less favorable form of estradiol than are non-oral estradiol forms like transdermal and injectable estradiol. Consequently, it is generally recommended that non-oral estradiol be used instead of oral estradiol where possible. However, oral estradiol is less expensive than certain non-oral forms (e.g., transdermal), is very easy and convenient to use, and has ubiquitous availability (unlike e.g. injectables). As such, it continues to see widespread use, and remains probably the most commonly used form of estradiol in medicine.
EC508 is special because although it is intended as an oral form of estradiol, it is not like oral estradiol or conventional orally used estradiol esters like estradiol valerate. EC508 is different because it appears to bypass the first pass that normally occurs with oral administration. Due to its unique ester, it seems to be resistant to metabolism in the intestines and liver, which limits its conversion into estradiol and estradiol metabolites at this time. This is also notably seen with certain other estrogen esters like estrone sulfate and estramustine phosphate (estradiol normustine phosphate). In addition, and more importantly, because of its unusual ester moiety, EC508 binds with high affinity to a protein called carbonic anhydrase II (CAII) in red blood cells. This binding causes EC508 to be rapidly accumulated and sequestered into blood cells. Estradiol normally goes through the hepatic portal vein, a blood vessel that carries blood from the gastrointestinal tract to the liver, to reach the liver. With EC508, it is taken up into blood cells so quickly that little ultimately seems to reach the liver. Instead, the red blood cells carry EC508 through the portal vein to the bloodstream, which it is then slowly released into. As with other estradiol esters, EC508 has little or no estrogenic activity itself and is only active after being converted into estradiol.
In accordance with the fact that it bypasses the oral first pass, EC508 shows 100% bioavailability in animals. This can be compared in humans to about 5% for oral estradiol and about 43% for ethinylestradiol (EE)—a syntheticrelative of estradiol with strongly improved metabolic stability (but also far greater influence on liver synthesis and related health risks than estradiol). As a result of its complete bioavailability, EC508 has 100 times the oral estrogenic potency of estradiol and 10 times that of ethinylestradiol in animals. As such, EC508 would be expected to be active in humans at microgram (μg) doses, similarly to the estimated delivery rates of transdermal estradiol patches (e.g., 25–100 μg/day) as well as to typical doses of ethinylestradiol (e.g., 5–50 μg/day) but in contrast to the 100-fold higher usual doses of oral estradiol (e.g., 0.5–10 mg/day). Further in accordance with it bypassing the oral first pass, EC508 and non-oral estradiol showed little or no effect on liver synthesis at assessed doses in animals, whereas oral estradiol and ethinylestradiol showed marked effects. Lastly, EC508 had a biological half-life of 5 hours in rats, which is relatively long for this species. For this reason, a long duration may be anticipated with EC508 in humans, in contrast to estradiol injected directly into the blood (which has a very short half-life) but similarly to oral, transdermal, and injectable estradiol.
An earlier drug candidate called estradiol sulfamate (E2MATE; J995), which is likewise a special estradiol ester that binds to CAII and bypasses the oral first pass, was discovered by the same research group in the 1990s, and was likewise developed as a new form of oral estradiol. However, it was subsequently found that E2MATE is additionally a highly potent inhibitor of steroid sulfatase (STS) and prevents its own activation by this enzyme into estradiol in humans. This served to abolish its estrogenic effects in the case of humans and resulted in it being not useful for clinical estrogen therapy. E2MATE was also undesirably found to be substantially converted into estrone sulfamate (E1MATE; J994) during the first pass with oral administration. Consequent to these findings, E2MATE was eventually repurposed as an estrogen synthesis inhibitor for treatment of endometriosis, and as of 2017, remains under development for this indication. In contrast to E2MATE, EC508 is not converted into the corresponding estrone variant, and is not thought to be an STS inhibitor, so the problems that E2MATE had are not expected with it.
Taken together, EC508 shows a preclinical and theoretical profile of being an oral form of estradiol that bypasses the first pass with oral administration. As a result, EC508 with oral administration undergoes little or no first-pass metabolism, has high bioavailability, and does not result in excessive estrogenic exposure in the liver. Consequently, EC508 is expected to be orally active at much lower doses than estradiol, to have reduced variability compared to estradiol, to lack an unphysiological metabolite pattern, and to not have disproportionate influence on estrogen-sensitive liver synthesis and associated health risks. In these regards, EC508 shows a favorable and advantageous profile relative to oral estradiol that is analogous to that with non-oral forms of estradiol like transdermal and injectable estradiol but with the convenience of oral administration. For these reasons, EC508 is a pharmaceutical candidate with the potential to supersede not only oral estradiol in clinical practice but also non-oral estradiol and even other clinically used estrogens like ethinylestradiol (e.g., in birth control pills). If it’s successfully developed and approved, EC508 would naturally one day be used in transfeminine hormone therapy as well.
In addition to EC508, the researchers at Evestra synthesized a testosterone equivalent known as EC586 (testosterone 17β-(1-((5-(aminosulfonyl)-2-pyridinyl)carbonyl)-L-proline or slightly more simply as testosterone aminosulfonylpyridinylcarbonylproline). This testosterone ester shows analogous properties to those of EC508, and hence has the potential to be a non-oral-like form of oral testosterone with high bioavailability and potency, a long duration, and lack of disproportionate liver effects—and hence with major advantages over oral testosterone forms available today. In parallel to EC508, EC586 (or similar drugs) could someday be used in transmasculine hormone therapy. The prodrug approach used to create EC508 and EC586 also notably has the potential to be employed with many other drugs.
Update: No Further Development
Patents for EC508 were filed in 2014 and 2017 and publications describing it were published in the scientific literature in 2017. An Investigational New Drug (IND) application for EC508 was tentatively supposed to be filed with the Food and Drug Administration (FDA) in the United States in the second quarter of 2018 (Evestra). However, as of September 2021, there have been no further updates on the development of EC508. In addition, Evestra’s website has since been redesigned and its research pipeline page listing the status of EC508 has been removed. It’s unknown at this time if EC508 and EC586 are still under development or whether further updates on these compounds can be expected.
References
Ahmed, G., Elger, W., Meece, F., Nair, H. B., Schneider, B., Wyrwa, R., & Nickisch, K. (2017). A prodrug design for improved oral absorption and reduced hepatic interaction. Bioorganic & Medicinal Chemistry, 25(20), 5569–5575. [DOI:10.1016/j.bmc.2017.08.027]
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
Elger, W., Schwarz, S., Hedden, A., Reddersen, G., & Schneider, B. (1995). Sulfamates of various estrogens are prodrugs with increased systemic and reduced hepatic estrogenicity at oral application. The Journal of Steroid Biochemistry and Molecular Biology, 55(3-4), 395–403. [DOI:10.1016/0960-0760(95)00214-6]
Elger, W., Palme, H. J., & Schwarz, S. (1998). Novel oestrogen sulfamates: a new approach to oral hormone therapy. Expert Opinion on Investigational Drugs, 7(4), 575–589. [DOI:10.1517/13543784.7.4.575]
Elger, W., Barth, A., Hedden, A., Reddersen, G., Ritter, P., Schneider, B., Züchner, J., Krahl, E., Müller, K., Oettel, M., & Schwarz, S. (2001). Estrogen Sulfamates: A New Approach to Oral Estrogen Therapy. Reproduction, Fertility, and Development, 13(4), 297–305. [DOI:10.1071/rd01029]
Elger, W., Wyrwa, R., Ahmed, G., Meece, F., Nair, H. B., Santhamma, B., Killeen, Z., Schneider, B., Meister, R., Schubert, H., & Nickisch, K. (2017). Estradiol prodrugs (EP) for efficient oral estrogen treatment and abolished effects on estrogen modulated liver functions. The Journal of Steroid Biochemistry and Molecular Biology, 165, 305–311. [DOI:10.1016/j.jsbmb.2016.07.008]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Nickisch, K., Santhamma, B., Ahmed, G., Meece, F., Elger, W., Wyrwa, R., & Nair, H. (2017). U.S. Patent No. 9,745,338. Washington, DC: U.S. Patent and Trademark Office. [US9745338B2]
Nickisch, K., Santhamma, B., Ahmed, G., Meece, F., Elger, W., Wyrwa, R., & Nair, H. (2019). U.S. Patent No. 10,273,263. Washington, DC: U.S. Patent and Trademark Office. [US10273263B2]
Potter, B. V. (2018). SULFATION PATHWAYS: Steroid sulphatase inhibition via aryl sulphamates: clinical progress, mechanism and future prospects. Journal of Molecular Endocrinology, 61(2), T233–T252. [DOI:10.1530/JME-18-0045]
Stanczyk, F. Z., Archer, D. F., & Bhavnani, B. R. (2013). Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception, 87(6), 706–727. [DOI:10.1016/j.contraception.2012.12.011]
Thomas, M. P., & Potter, B. V. (2015). Discovery and development of the aryl O-sulfamate pharmacophore for oncology and women’s health. Journal of Medicinal Chemistry, 58(19), 7634–7658. [DOI:10.1021/acs.jmedchem.5b00386]
Thomas, M. P., & Potter, B. V. (2015). Estrogen O-sulfamates and their analogues: clinical steroid sulfatase inhibitors with broad potential. The Journal of Steroid Biochemistry and Molecular Biology, 153, 160–169. [DOI:10.1016/j.jsbmb.2015.03.012]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
\ No newline at end of file
diff --git a/transfemscience.org/articles/endo-system-muscle/index.html b/transfemscience.org/articles/endo-system-muscle/index.html
index 32f1caaa..d8d53c89 100644
--- a/transfemscience.org/articles/endo-system-muscle/index.html
+++ b/transfemscience.org/articles/endo-system-muscle/index.html
@@ -1 +1 @@
-The Relationship Between the Endocrine System and Muscle Mass - Transfeminine Science
As someone who has recently undergone dramatic physiological changes prior to gender-affirming hormone therapy (GAHT) and who recently began taking antiandrogens and estradiol I was extremely interested in the effects of GAHT on muscle mass. Looking through traditional trans resources and talking to various general providers and transcare specialists I largely received anecdote related to “you’ll lose muscle mass” when suppressing androgens and/or taking estradiol. I thought and hope others will find the digging I’ve done to try and collate research on this useful or interesting as well.
Background
A bit of basic background on muscles:
There are generally speaking two types of skeletal muscle fiber: type 1 (slow twitch) and type 2 (fast twitch).
Muscle cells called myocytes are filled with myofibrils – protein chains that contract, have large stores of glycogen as well as oxygen.
Muscular hypertrophy is the growth of muscle mass.
Most muscle mass building is via anaerobic (non-oxygenated) exercise.
While I initially sought out to find the effects of estrogens on muscle mass I was largely at a loss. I could find little information until I had a moment of clarity and realized that while estrogens do have interactions with the formation of muscle mass and muscle cells – the seemingly more important hormones are androgens and that is the presence or absence of testosterone that seems to have a far greater impact on muscle mass than the presence of estrogen. Something I would of course love to see would be studies similar to ones I look at below but with estrogens added in addition to androgens suppressed.
Summary
Testosterone from heavily androgen suppressed levels to above normal doses in assigned male at birth (AMAB) individuals seems to have a linear relationship to muscle mass. This seems to occur while maintaining the pre-existing distribution of both major skeletal muscle types (type I and type II). Further this seems to occur by an increase in the number of muscle cells (myonuclear number) as well as significant changes to their cellular structure – they add more myofibrils. This seems to be due to testosterone induced protein synthesis and prevention of protein degradation.
Below I have included a number of papers I found interesting and drew these conclusions from, with takeaways, quotes I found interesting as well as graphs.
The main takeaway from this paper is to illustrate that even in AMAB individuals without suppressed androgens – increasing testosterone is useful in increasing muscle mass – as measured by looking at fat free mass (FFM) and also a concordant decrease in body fat (BF) percentage.
Our results show that supraphysiologic doses of testosterone, especially when combined with strength training, increase fat-free mass, muscle size, and strength in normal men when potentially confounding variables, such as nutritional intake and exercise stimulus, are standardized. The combination of strength training and testosterone produced greater increases in muscle size and strength than were achieved with either intervention alone. The combined regimen of testosterone and exercise led to an increase of 6.1 kg in fat-free mass over the course of 10 weeks; this increase entirely accounted for the changes in body weight.
This paper was the first that I found that really illustrated that suppression of testosterone down to levels that people on an antiandrogen often seek. You can see that fat free mass (bone and muscle mass) decreases while body fat percentage increased.
Young men treated with a GnRHa were less efficient in their oxidation of fat, with a consequent decrease in the resting energy expenditure, which probably explains the increase in adiposity and the decrease in lean body mass observed during these experiments. The mechanisms for these findings are not completely understood; however, several considerations apply.
In summary, severe androgen deficiency in young men was associated with decreased lean body mass and increased adiposity, decreased lipid oxidation and energy expenditure rates, decreased rates of whole body protein synthesis, and decreased leg muscle strength. These findings were not associated with changes in circulating amino acid concentrations. These changes were associated with decreased gene expression for IGF-I in muscle, but no peripheral decreases in GH and IGF-I production. We conclude that androgens can directly affect systemic protein synthesis, independent of the effect of peripheral GH and IGF-I. The latter may be important when an anabolic effect is the desired effect in the treatment of both elderly and young men.
Possibly one of the most interesting papers I found. Not only did they suppress AMAB individuals testosterone with a gonadotropin-releasing hormone agonist they then gave different groups varying levels of it so that you could actually plot a dose response curve. They measured a number of things outside of fat free mass such as leg press strength, fat mass, thigh muscle volume, etc..
[…] 61 eugonadal men, 18–35 yr, were randomized to one of five groups to receive monthly injections of a long-acting gonadotropin-releasing hormone (GnRH) agonist, to suppress endogenous testosterone secretion, and weekly injections of 25, 50, 125, 300, or 600 mg of testosterone enanthate for 20 wk. Energy and protein intakes were standardized. The administration of the GnRH agonist plus graded doses of testosterone resulted in mean nadir testosterone concentrations of 253, 306, 542, 1,345, and 2,370 ng/dl at the 25-, 50-, 125-, 300-, and 600-mg doses, respectively. Fat-free mass increased dose dependently in men receiving 125, 300, or 600 mg of testosterone weekly (change 13.4, 5.2, and 7.9 kg, respectively). The changes in fat-free mass were highly dependent on testosterone dose (P = 0.0001) and correlated with log testosterone concentrations (r = 0.73, P = 0.0001). Changes in leg press strength, leg power, thigh and quadriceps muscle volumes, hemoglobin, and IGF-I were positively correlated with testosterone concentrations, whereas changes in fat mass and plasma high-density lipoprotein (HDL) cholesterol were negatively correlated.
While the previous papers looked at the relationship between muscle mass and other factors and testosterone – this paper looks at how that occurs. They again supress testosterone and then give different groups varying levels of it – only this time they take muscle biopsies and look at how the actual muscle groups differ.
In healthy, young men, in whom testicular testosterone production had been suppressed by a GnRH agonist, administration of graded doses of testosterone was associated with dose dependent changes in circulating concentrations of total and free testosterone (7).
Data presented in this manuscript demonstrate that testosterone-induced gains in muscle size were associated with a significant increase in muscle fiber cross-sectional area. The cross-sectional areas of both type I and type II fibers increased in proportion to testosterone concentrations. The relative proportion of type I and II fibers did not change significantly. We therefore conclude that testosterone increases skeletal muscle size primarily by inducing muscle fiber hypertrophy.
In our study, the myonuclear number increased in direct relation to the increase in muscle fiber diameter. Therefore, it is possible that muscle fiber hypertrophy and increase in myonuclear number were preceded by testosterone induced increase in satellite cell number and their fusion with muscle fibers.
A paper by the same research group as previous, this paper does a great job summarizing the effects of testosterone on muscle mass and its role in muscular hypertrophy. I’ve included here their table of its effects relevant to muscle mass.
The change in percent satellite cell number correlated with changes in total and free testosterone concentrations (10). Satellite cell and mitochondrial areas were significantly higher and the nuclear to cytoplasmic ratio lower after treatment with 300 mg and 600 mg doses (10). These data demonstrate that testosterone-induced muscle fiber hypertrophy is associated with an increase in satellite cell number, a proportionate increase in myonuclear number, and changes in satellite cell ultrastructure. These alterations in satellite cell number and ultrastructure and muscle morphology cannot be explained by the muscle protein synthesis hypothesis.
Testosterone has been reported to inhibit lipid uptake and lipoprotein lipase activity in adipocytes, and stimulate lipolysis (21), in part by increasing the number of lipolytic beta-adrenergic receptors.
Table 1. Summary of the Observed Effects of Testosterone on Body Composition
Effects on Fat-Free Compartment
Increase in fat-free and lean body mass (2–14)
Increase in bone mass (52)
Increase in nitrogen retention in castrated male mammals, eunuchoidal men, women, and prepubertal boys (25–28)
Changes in Muscle Histomorphology
Increase in cross-sectional areas of both types I and II skeletal muscle fibers (9)
Increase in the number of myonuclei (10)
Increase in the number of satellite cells (10)
Changes in Protein Dynamics
Increase in nonoxidative leucine disappearance rate (14)
Increase in fractional synthesis rates of mixed skeletal muscle protein (11–14)
No net increase in protein balance, although net protein balance becomes less negative in fasting state (15,16)
Decrease in protein degradation by the arteriovenous difference method (15)
Decrease in proteasome-mediated protein degradation (15)
Effects of Fat Compartment
Decrease in whole-body fat mass in hypogonadal men treated with replacement doses of testosterone (3–6)
Decrease in whole-body fat mass in eugonadal men with supraphysiological doses of testosterone and other androgens (7)
Decrease in intra-abdominal fat mass in middle-aged men with low normal testosterone levels (25,26)
Increased lipolysis (26)
Regulation of lipoprotein lipase activity (52,53)
Decreased triglyceride assimilation in abdominal fat compartment (26)
Inhibits preadipocyte to adipocyte differentiation (52)
Note: Numbers in parentheses are reference numbers.
References
Bhasin, S., Storer, T. W., Berman, N., Callegari, C., Clevenger, B., Phillips, J., Bunnell, T. J., Tricker, R., Shirazi, A., & Casaburi, R. (1996). The Effects of Supraphysiologic Doses of Testosterone on Muscle Size and Strength in Normal Men. New England Journal of Medicine, 335(1), 1–7. [DOI:10.1056/nejm199607043350101]
Bhasin, S., Woodhouse, L., Casaburi, R., Singh, A. B., Bhasin, D., Berman, N., Chen, X., Yarasheski, K. E., Magliano, L., Dzekov, C., Dzekov, J., Bross, R., Phillips, J., Sinha-Hikim, I., Shen, R., & Storer, T. W. (2001). Testosterone dose-response relationships in healthy young men. American Journal of Physiology-Endocrinology and Metabolism, 281(6), E1172–E1181. [DOI:10.1152/ajpendo.2001.281.6.e1172]
Bhasin, S., Taylor, W. E., Singh, R., Artaza, J., Sinha-Hikim, I., Jasuja, R., Choi, H., & Gonzalez-Cadavid, N. F. (2003). The Mechanisms of Androgen Effects on Body Composition: Mesenchymal Pluripotent Cell as the Target of Androgen Action. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 58(12), M1103–M1110. [DOI:10.1093/gerona/58.12.m1103]
Mauras, N., Hayes, V., Welch, S., Rini, A., Helgeson, K., Dokler, M., Veldhuis, J. D., & Urban, R. J. (1998). Testosterone Deficiency in Young Men: Marked Alterations in Whole Body Protein Kinetics, Strength, and Adiposity. The Journal of Clinical Endocrinology & Metabolism, 83(6), 1886–1892. [DOI:10.1210/jcem.83.6.4892]
Sinha-Hikim, I., Artaza, J., Woodhouse, L., Gonzalez-Cadavid, N., Singh, A. B., Lee, M. I., Storer, T. W., Casaburi, R., Shen, R., & Bhasin, S. (2002). Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. American Journal of Physiology-Endocrinology and Metabolism, 283(1), E154–E164. [DOI:10.1152/ajpendo.00502.2001]
\ No newline at end of file
+The Relationship Between the Endocrine System and Muscle Mass - Transfeminine Science
As someone who has recently undergone dramatic physiological changes prior to gender-affirming hormone therapy (GAHT) and who recently began taking antiandrogens and estradiol I was extremely interested in the effects of GAHT on muscle mass. Looking through traditional trans resources and talking to various general providers and transcare specialists I largely received anecdote related to “you’ll lose muscle mass” when suppressing androgens and/or taking estradiol. I thought and hope others will find the digging I’ve done to try and collate research on this useful or interesting as well.
Background
A bit of basic background on muscles:
There are generally speaking two types of skeletal muscle fiber: type 1 (slow twitch) and type 2 (fast twitch).
Muscle cells called myocytes are filled with myofibrils – protein chains that contract, have large stores of glycogen as well as oxygen.
Muscular hypertrophy is the growth of muscle mass.
Most muscle mass building is via anaerobic (non-oxygenated) exercise.
While I initially sought out to find the effects of estrogens on muscle mass I was largely at a loss. I could find little information until I had a moment of clarity and realized that while estrogens do have interactions with the formation of muscle mass and muscle cells – the seemingly more important hormones are androgens and that is the presence or absence of testosterone that seems to have a far greater impact on muscle mass than the presence of estrogen. Something I would of course love to see would be studies similar to ones I look at below but with estrogens added in addition to androgens suppressed.
Summary
Testosterone from heavily androgen suppressed levels to above normal doses in assigned male at birth (AMAB) individuals seems to have a linear relationship to muscle mass. This seems to occur while maintaining the pre-existing distribution of both major skeletal muscle types (type I and type II). Further this seems to occur by an increase in the number of muscle cells (myonuclear number) as well as significant changes to their cellular structure – they add more myofibrils. This seems to be due to testosterone induced protein synthesis and prevention of protein degradation.
Below I have included a number of papers I found interesting and drew these conclusions from, with takeaways, quotes I found interesting as well as graphs.
The main takeaway from this paper is to illustrate that even in AMAB individuals without suppressed androgens – increasing testosterone is useful in increasing muscle mass – as measured by looking at fat free mass (FFM) and also a concordant decrease in body fat (BF) percentage.
Our results show that supraphysiologic doses of testosterone, especially when combined with strength training, increase fat-free mass, muscle size, and strength in normal men when potentially confounding variables, such as nutritional intake and exercise stimulus, are standardized. The combination of strength training and testosterone produced greater increases in muscle size and strength than were achieved with either intervention alone. The combined regimen of testosterone and exercise led to an increase of 6.1 kg in fat-free mass over the course of 10 weeks; this increase entirely accounted for the changes in body weight.
This paper was the first that I found that really illustrated that suppression of testosterone down to levels that people on an antiandrogen often seek. You can see that fat free mass (bone and muscle mass) decreases while body fat percentage increased.
Young men treated with a GnRHa were less efficient in their oxidation of fat, with a consequent decrease in the resting energy expenditure, which probably explains the increase in adiposity and the decrease in lean body mass observed during these experiments. The mechanisms for these findings are not completely understood; however, several considerations apply.
In summary, severe androgen deficiency in young men was associated with decreased lean body mass and increased adiposity, decreased lipid oxidation and energy expenditure rates, decreased rates of whole body protein synthesis, and decreased leg muscle strength. These findings were not associated with changes in circulating amino acid concentrations. These changes were associated with decreased gene expression for IGF-I in muscle, but no peripheral decreases in GH and IGF-I production. We conclude that androgens can directly affect systemic protein synthesis, independent of the effect of peripheral GH and IGF-I. The latter may be important when an anabolic effect is the desired effect in the treatment of both elderly and young men.
Possibly one of the most interesting papers I found. Not only did they suppress AMAB individuals testosterone with a gonadotropin-releasing hormone agonist they then gave different groups varying levels of it so that you could actually plot a dose response curve. They measured a number of things outside of fat free mass such as leg press strength, fat mass, thigh muscle volume, etc..
[…] 61 eugonadal men, 18–35 yr, were randomized to one of five groups to receive monthly injections of a long-acting gonadotropin-releasing hormone (GnRH) agonist, to suppress endogenous testosterone secretion, and weekly injections of 25, 50, 125, 300, or 600 mg of testosterone enanthate for 20 wk. Energy and protein intakes were standardized. The administration of the GnRH agonist plus graded doses of testosterone resulted in mean nadir testosterone concentrations of 253, 306, 542, 1,345, and 2,370 ng/dl at the 25-, 50-, 125-, 300-, and 600-mg doses, respectively. Fat-free mass increased dose dependently in men receiving 125, 300, or 600 mg of testosterone weekly (change 13.4, 5.2, and 7.9 kg, respectively). The changes in fat-free mass were highly dependent on testosterone dose (P = 0.0001) and correlated with log testosterone concentrations (r = 0.73, P = 0.0001). Changes in leg press strength, leg power, thigh and quadriceps muscle volumes, hemoglobin, and IGF-I were positively correlated with testosterone concentrations, whereas changes in fat mass and plasma high-density lipoprotein (HDL) cholesterol were negatively correlated.
While the previous papers looked at the relationship between muscle mass and other factors and testosterone – this paper looks at how that occurs. They again supress testosterone and then give different groups varying levels of it – only this time they take muscle biopsies and look at how the actual muscle groups differ.
In healthy, young men, in whom testicular testosterone production had been suppressed by a GnRH agonist, administration of graded doses of testosterone was associated with dose dependent changes in circulating concentrations of total and free testosterone (7).
Data presented in this manuscript demonstrate that testosterone-induced gains in muscle size were associated with a significant increase in muscle fiber cross-sectional area. The cross-sectional areas of both type I and type II fibers increased in proportion to testosterone concentrations. The relative proportion of type I and II fibers did not change significantly. We therefore conclude that testosterone increases skeletal muscle size primarily by inducing muscle fiber hypertrophy.
In our study, the myonuclear number increased in direct relation to the increase in muscle fiber diameter. Therefore, it is possible that muscle fiber hypertrophy and increase in myonuclear number were preceded by testosterone induced increase in satellite cell number and their fusion with muscle fibers.
A paper by the same research group as previous, this paper does a great job summarizing the effects of testosterone on muscle mass and its role in muscular hypertrophy. I’ve included here their table of its effects relevant to muscle mass.
The change in percent satellite cell number correlated with changes in total and free testosterone concentrations (10). Satellite cell and mitochondrial areas were significantly higher and the nuclear to cytoplasmic ratio lower after treatment with 300 mg and 600 mg doses (10). These data demonstrate that testosterone-induced muscle fiber hypertrophy is associated with an increase in satellite cell number, a proportionate increase in myonuclear number, and changes in satellite cell ultrastructure. These alterations in satellite cell number and ultrastructure and muscle morphology cannot be explained by the muscle protein synthesis hypothesis.
Testosterone has been reported to inhibit lipid uptake and lipoprotein lipase activity in adipocytes, and stimulate lipolysis (21), in part by increasing the number of lipolytic beta-adrenergic receptors.
Table 1. Summary of the Observed Effects of Testosterone on Body Composition
Effects on Fat-Free Compartment
Increase in fat-free and lean body mass (2–14)
Increase in bone mass (52)
Increase in nitrogen retention in castrated male mammals, eunuchoidal men, women, and prepubertal boys (25–28)
Changes in Muscle Histomorphology
Increase in cross-sectional areas of both types I and II skeletal muscle fibers (9)
Increase in the number of myonuclei (10)
Increase in the number of satellite cells (10)
Changes in Protein Dynamics
Increase in nonoxidative leucine disappearance rate (14)
Increase in fractional synthesis rates of mixed skeletal muscle protein (11–14)
No net increase in protein balance, although net protein balance becomes less negative in fasting state (15,16)
Decrease in protein degradation by the arteriovenous difference method (15)
Decrease in proteasome-mediated protein degradation (15)
Effects of Fat Compartment
Decrease in whole-body fat mass in hypogonadal men treated with replacement doses of testosterone (3–6)
Decrease in whole-body fat mass in eugonadal men with supraphysiological doses of testosterone and other androgens (7)
Decrease in intra-abdominal fat mass in middle-aged men with low normal testosterone levels (25,26)
Increased lipolysis (26)
Regulation of lipoprotein lipase activity (52,53)
Decreased triglyceride assimilation in abdominal fat compartment (26)
Inhibits preadipocyte to adipocyte differentiation (52)
Note: Numbers in parentheses are reference numbers.
References
Bhasin, S., Storer, T. W., Berman, N., Callegari, C., Clevenger, B., Phillips, J., Bunnell, T. J., Tricker, R., Shirazi, A., & Casaburi, R. (1996). The Effects of Supraphysiologic Doses of Testosterone on Muscle Size and Strength in Normal Men. New England Journal of Medicine, 335(1), 1–7. [DOI:10.1056/nejm199607043350101]
Bhasin, S., Woodhouse, L., Casaburi, R., Singh, A. B., Bhasin, D., Berman, N., Chen, X., Yarasheski, K. E., Magliano, L., Dzekov, C., Dzekov, J., Bross, R., Phillips, J., Sinha-Hikim, I., Shen, R., & Storer, T. W. (2001). Testosterone dose-response relationships in healthy young men. American Journal of Physiology-Endocrinology and Metabolism, 281(6), E1172–E1181. [DOI:10.1152/ajpendo.2001.281.6.e1172]
Bhasin, S., Taylor, W. E., Singh, R., Artaza, J., Sinha-Hikim, I., Jasuja, R., Choi, H., & Gonzalez-Cadavid, N. F. (2003). The Mechanisms of Androgen Effects on Body Composition: Mesenchymal Pluripotent Cell as the Target of Androgen Action. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 58(12), M1103–M1110. [DOI:10.1093/gerona/58.12.m1103]
Mauras, N., Hayes, V., Welch, S., Rini, A., Helgeson, K., Dokler, M., Veldhuis, J. D., & Urban, R. J. (1998). Testosterone Deficiency in Young Men: Marked Alterations in Whole Body Protein Kinetics, Strength, and Adiposity. The Journal of Clinical Endocrinology & Metabolism, 83(6), 1886–1892. [DOI:10.1210/jcem.83.6.4892]
Sinha-Hikim, I., Artaza, J., Woodhouse, L., Gonzalez-Cadavid, N., Singh, A. B., Lee, M. I., Storer, T. W., Casaburi, R., Shen, R., & Bhasin, S. (2002). Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. American Journal of Physiology-Endocrinology and Metabolism, 283(1), E154–E164. [DOI:10.1152/ajpendo.00502.2001]
\ No newline at end of file
diff --git a/transfemscience.org/articles/estrogens-blood-clots/index.html b/transfemscience.org/articles/estrogens-blood-clots/index.html
index fe457c68..6d77deb5 100644
--- a/transfemscience.org/articles/estrogens-blood-clots/index.html
+++ b/transfemscience.org/articles/estrogens-blood-clots/index.html
@@ -1 +1 @@
-Estrogens and Their Influences on Coagulation and Risk of Blood Clots - Transfeminine Science
Estrogens and Their Influences on Coagulation and Risk of Blood Clots
By Aly | First published October 20, 2020 | Last modified March 28, 2023
Abstract / TL;DR
Estrogens increase coagulation by activating estrogen receptors in the liver and thereby modulating the production of a variety of circulating coagulation factors. With sufficiently high exposure, this can result in an increase in the risk of blood clots as well as coagulation-associated cardiovascular complications like heart attack and stroke. However, the degrees of risk vary depending on the estrogen type, route, and dose. Non-bioidentical estrogens like ethinylestradiol have greater strength in the liver due to their relative resistance to metabolism and increase blood clot risk more readily than bioidentical estradiol, while oral administration of estradiol results in a first pass through the liver and has greater impact on blood clot risk than non-oral estradiol. Physiological estradiol levels with non-oral estradiol appear to have minimal to no risk of blood clots, whereas oral estradiol has significant risk and at high doses may have risk similar to that of the doses of ethinylestradiol in modern birth control pills. Higher estradiol levels with non-oral estradiol seem to have significant risk of blood clots and cardiovascular problems as well, although the risks appear to be lower than with ethinylestradiol-containing birth control pills. Absolute risks of blood clots are low but accumulate with time and add up on a population scale. In addition, a variety of risk factors, such as age, physical inactivity, concomitant progestogen use, and often-unknown thrombophilic abnormalities, can substantially augment risk. Due to their higher risks of blood clots, oral estradiol as well as excessive doses of non-oral estradiol should ideally be avoided in transfeminine people. This is particularly applicable in those with risk factors for blood clots. In any case, therapeutic considerations for transfeminine people include not only safety but also effectiveness, other factors like cost and convenience, and the natures of the alternative therapeutic options.
Introduction
Estrogens increase coagulation (blood clotting) and the risk of thrombosis, a cardiovascular event otherwise known as a blood clot. There are two major types of blood clots, which are categorized depending on whether they happen in a vein or in an artery: (1) venous thrombosis or venous thromboembolism (VTE); and (2) arterial thrombosis. VTE is a blood clot in a vein, a blood vessel that carries blood towards the heart. It comprises two different subtypes: (1) deep vein thrombosis (DVT), a clot in a vein of the leg or pelvic region; and (2) pulmonary embolism (PE), a clot that has broken free and blocked an artery in the lungs. Arterial thrombosis is a blood clot in an artery, a blood vessel that carries blood away from the heart. Arterial thrombosis can lead to myocardial infarction (MI; also known as heart attack) or cerebrovascular accident (CVA; also known as stroke). Blood clots are major health problems that can cause serious complications and even death. Estrogens, via increased coagulation with sufficiently high exposure, have the potential to heighten the risk of both venous and arterial thrombosis and hence to increase all of the aforementioned risks. The risk of blood clots with estrogens serves as a limiting factor in their use due to the potential health consequences.
Estrogens are selectiveagonists of the estrogen receptors (ERs). They are thought to increase coagulation and hence blood clot risk by activating ERs. However, the impact on coagulation and risk of blood clots with estrogens varies due to factors like estrogen type, route, and dose. In addition, other factors, like concomitant progestogen use and a variety of non-hormonal factors, are known to modify the risk. The purpose of this article is to review the risks of blood clots with estrogens, the mechanisms underlying increased coagulation and blood clot risk with estrogens, and the reasons for differences among estrogens in terms of risk. Exploring these topics can inform estrogen dosing considerations in transfeminine people and help to minimize risks and optimize safety. Moreover, higher levels of estrogens are therapeutically useful for suppressing testosterone production in transfeminine people but may increase blood clot risk, and risk–benefit analysis is warranted in this context.
Blood Clot Risks with Estrogens and Progestogens
A variety of estrogens have been used in medicine. These include bioidentical estrogens like estradiol as well as non-bioidentical estrogens like conjugated estrogens (CEEs; Premarin), ethinylestradiol (EE), and diethylstilbestrol (DES). Estradiol is the major natural estrogen in the human body. CEEs deliver primarily estradiol as the active estrogen, but also contain significant quantities of naturally occurring equine (horse) estrogens such as equilin (7-dehydroestrone) and 17β-dihydroequilin (7-dehydroestradiol). EE and DES are synthetic estrogens that were created by humans and do not occur naturally. DES was discontinued decades ago and is relatively little-known today, but has significant historical importance. Estradiol is used in both oral and non-oral forms (e.g., transdermal patches), while the non-bioidentical estrogens have typically been used orally. For context, the table below shows some approximate comparable doses of these estrogens in terms of general estrogenicity.
Table 1: Approximate or estimated comparable doses of estrogens in terms of general/systemic estrogenicity (Aly, 2020; Kuhl, 2005; Table; Table; Table):
Estrogen type/route
Very low dose a
Low dose a
Moderate dose b
High dose
Oral estradiol
1 mg/day
2 mg/day
4 mg/day
8 mg/day
Transdermal estradiolc
25 μg/day
50 μg/day
100 μg/day
200 μg/day
Oral conjugated estrogens
0.625 mg/day
1.25 mg/day
2.5 mg/day
5 mg/day
Oral ethinylestradiol
7.5 μg/day
15 μg/day
30 μg/day
60 μg/day
Oral diethylstilbestrol
0.375 mg/day
0.75 mg/day
1.5 mg/day
3 mg/day
Comparable estradiol level
~25 pg/mL
~50 pg/mL
~100 pg/mL
~200 pg/mL
a Menopausal replacement dosages. b Similar to normal mean/integrated estrogenic exposure during the menstrual cycle in premenopausal women (Aly, 2018). c Specifically transdermal patches.
Estrogens were first associated with blood clots and associated cardiovascular complications in the 1960s and 1970s. Significant to substantial increases in these risks were found in clinical trials of high-dose DES (5 mg/day) for prostate cancer in men (VACURG, 1967; Byar, 1973; Turo et al., 2014), trials of moderate-dose CEEs (2.5–5 mg/day) for prevention of heart disease in men (Coronary Drug Project Research Group, 1970; Coronary Drug Project Research Group, 1973; Luria, 1989; Sudhir & Komesaroff, 1999; Dutra et al., 2019), and studies of early high-dose EE-containing birth control pills (50–150 μg/day) in premenopausal women (Gerstman et al., 1991; PCASRM, 2017; Table). The increase in cardiovascular events with DES in men with prostate cancer was sufficiently great that it actually cancelled out the benefits of its effects against prostate cancer in terms of overall mortality. The large increases in blood clots and cardiovascular problems seen in these studies resulted in alarm and concern about the safety of estrogens. Consequent to these events, estrogen doses were lowered. DES for prostate cancer was decreased to 1 to 3 mg/day and EE in birth control pills was decreased to 20 to 35 μg/day. Estrogens were also reduced to lower doses for other indications, such as menopausal hormone therapy. The dose reductions helped to lower the risks, although it did not eliminate them.
Whereas the WHI demonstrated causation for oral CEEs alone in terms of blood clot risk, no adequately powered RCTs have been conducted with oral or transdermal estradiol alone to establish causation in terms of blood clot risk at this time. Only very large and expensive trials would be able to show this due to the rarity of blood clots, and these studies have not been conducted to date. For similar reasons, RCTs demonstrating increased risk of blood clots with EE-containing birth control pills have also not been conducted at this time (Moores, Bilello, & Murin, 2004). In any case, causation has clearly been demonstrated with estrogens in other contexts, and this can be assumed as likely in the case of oral estradiol similarly. In addition, the Estrogen in Venous Thromboembolism Trial (EVTET), an RCT of low-dose (2 mg/day) oral estradiol plus the progestogen norethisterone acetate (NETA) versus placebo in postmenopausal women with history of previous blood clots, found that this hormone therapy regimen significantly increased coagulation and the incidence of blood clots (10.7% incidence with hormone therapy and 2.3% with placebo; P = 0.04) (Høibraaten et al., 2000; Høibraaten et al., 2001).
Estradiol levels appear to not be associated with blood clot risk in premenopausal women (Holmegard et al., 2014). The fact that transdermal estradiol patches at 100 μg/day in menopausal women haven’t been associated with a greater risk of blood clots is notable as this dose achieves estradiol levels of around 100 pg/mL on average, which are similar to the mean integrated levels of estradiol during the normal menstrual cycle in premenopausal women (Aly, 2018; Wiki). Rates of blood clots are also similar between men—who have relatively low estradiol levels—and women after controlling for atypical hormonal states like pregnancy and use of birth control pills in women (Moores, Bilello, & Murin, 2004; Rosendaal, 2005; Montagnana et al., 2010; Roach et al., 2013). Interestingly however, men have a consistently higher incidence of recurrent blood clots than women (Roach et al., 2013). These findings suggest that physiological levels of estradiol and progesterone in premenopausal women may not meaningfully increase coagulation or blood clot risk. However, the available data are mixed, with some studies suggesting that estradiol and/or progesterone levels within physiological ranges may indeed influence coagulation (Chaireti et al., 2013) and blood clot risk in premenopausal and/or perimenopausal women (Simon et al., 2006; Canonico et al., 2014; Scheres et al., 2019).
Modern combined birth control pills contain EE at moderately estrogenic doses (20–35 μg/day) and a physiological dose of a progestogen. They increase the risk of blood clots by several-fold (Konkle & Sood, 2019; Vinogradova, Coupland, & Hippisley-Cox, 2015; Table). In addition, they are associated with about a 1.5- to 2-fold increase in risk of heart attack and stroke (Lidegaard, 2014; Konkle & Sood, 2019). However, overall mortality is not increased with birth control pills—at least in the relatively young women in whom they are used (Hannaford et al., 2010). Per studies of menopausal hormone therapy, it is likely that the progestogen in EE-containing birth control pills augments the risk of blood clots with EE. Early high-dose birth control pills (50–100 μg/day) had as much as twice the risk of blood clots of modern birth control pills (Gerstman et al., 1991; PCASRM, 2017; Table). In contrast to the different blood clot risks between oral and transdermal estradiol, non-oral birth control forms containing EE, for instance transdermal birth control patches and vaginal birth control rings, are associated with similar increases in blood clot risk as EE-containing birth control pills (Plu-Bureau et al., 2013; PCASRM, 2017; Konkle & Sood, 2019; Abou-Ismail, Sridhar, & Nayak, 2020). Hence, unlike with estradiol, route of administration does not appear to modify blood clot risk with EE based on available data.
High-dose estrogen therapy using oral synthetic estrogens like DES and EE in people with breast or prostate cancer has been found to strongly increase the risk of blood clots and associated cardiovascular complications (Phillips et al., 2014; Turo et al., 2014; Coelingh Bennink et al., 2017). This has also been the case with estramustine phosphate (EMP; estradiol normustine phosphate), an estradiol ester that is used at massive doses in prostate cancer (e.g., 140–1,400 mg/day orally) and that results in pregnancy levels of estradiol (Kitamura, 2001 [Graph]; Ravery et al., 2011). In the 1980s however, it was found that high-dose non-oral estradiol did not have the same cardiovascular risks as high-dose estrogen therapy with oral synthetic estrogens or EMP (von Schoultz et al., 1989; Ockrim & Abel, 2009). This included studies with polyestradiol phosphate (PEP), a long-lasting injectable prodrug of estradiol, and with high-dose transdermal estradiol gel (von Schoultz et al., 1989; Aly, 2019). However, subsequent larger and higher-quality studies found that although the cardiovascular risks with PEP were much lower than with high-dose oral synthetic estrogen therapy, they were nonetheless still increased (Hedlung et al., 2008; Ockrim & Abel, 2009; Hedlund et al., 2011; Sam, 2020). This includes an approximate 2-fold increase in the risk of blood clots with estradiol levels in the range of roughly 300 to 500 pg/mL (Sam, 2020). Studies using high-dose transdermal estradiol patches have not found significantly increased cardiovascular complications as of present (Langley et al., 2013; Sam, 2020). However, these studies have been relatively underpowered, which limits their interpretation. In any case, increased coagulation has been observed with high-dose transdermal estradiol patches (achieving estradiol levels of 350 to 500 pg/mL) (Bland et al., 2005) similarly to PEP (Mikkola et al., 1999). More data on the risk of blood clots and cardiovascular issues with high-dose transdermal estradiol patches should come in the future with PATCH and STAMPEDE—two large-scale clinical studies in the United Kingdom that are evaluating this form of estradiol for prostate cancer (Gilbert et al., 2018; Singla, Ghandour, & Raj, 2019).
Injections of short-acting estradiol esters like estradiol valerate and estradiol cypionate are notable in that they are often used by transfeminine people and are generally used at doses that achieve high estradiol levels. As with high-dose transdermal estradiol patches, little to no quality data on the risk of blood clots exists for these preparations at present. Pyra and colleagues found that the risk of blood clots with injectable estradiol valerate in transfeminine people was increased by around 2-fold, but the confidence intervals were very wide and statistical significance was not reached (Pyra et al., 2020). The doses used in the whole population for the study were not provided, but in the actual VTE cases, the doses of injectable estradiol valerate were described and ranged from 4 to 20 mg once per week and 10 to 40 mg once every 2 weeks (Pyra et al., 2020). Studies have also assessed and found increased coagulation with high doses of estradiol valerate by injection in the range of 10 to 40 mg once every 2 weeks in men with prostate cancer (Kohli & McClellan, 2001; Kohli et al., 2004; Kohli, 2005). Increased coagulation has additionally been observed with the combination of 5 mg estradiol valerate and a progestogen once per month as a combined injectable contraceptive in premenopausal women (Meng et al., 1990; UN/WHO et al., 2003). It is unclear whether the high peaks in estradiol levels associated with short-acting injectable forms of estradiol are harmful in terms of coagulation and blood clot risk (Hembree et al., 2017). However, the increased risk of polycythemia with short-acting injectable testosterone esters relative to other non-oral forms of testosterone (Ohlander, Varghese, & Pastuszak, 2018) is indirectly suggestive that this could be the case. Accordingly, a study found increased coagulation in premenopausal women with a combined injectable contraceptive containing estradiol valerate but not with one employing the more prolonged and stable estradiol cypionate at the same dose (UN/WHO et al., 2003).
Pregnancy is a time when estradiol and progesterone levels increase to extremely high concentrations (Graphs). Estradiol levels increase progressively throughout pregnancy to around 2,000 pg/mL on average at the end of the first trimester, to about 10,000 pg/mL on average at the end of the second trimester, and to around 20,000 pg/mL on average at the end of the third trimester (Kerlan et al., 1994 [Graph]; Schock et al., 2016). Coagulation is greatly increased during pregnancy, and the risk of blood clots is likewise strongly increased (Heit et al., 2000; Abdul Sultan et al., 2015; Heit, Spencer, & White, 2016; Table). Estradiol and progesterone levels are strongly correlated with the increases in coagulation during pregnancy (Bagot et al., 2019). The risk of blood clots with modern birth control pills is similar to that with pregnancy as a whole (Heit, Spencer, & White, 2016), while the increases in risk of blood clots with early high-dose EE-containing birth control pills and with high-dose oral synthetic estrogen therapy for breast and prostate cancer are comparable to the risk increase during late pregnancy. Estradiol levels also increase to very high concentrations during ovarian stimulation for in-vitro fertilization in premenopausal women, and this has been associated with increased coagulation and risk of blood clots as well (Westerlund et al., 2012; Rova, Passmark, & Lindqvist, 2012; Kasum et al., 2014).
Due to their greater risks of cardiovascular problems as well as other concerns, DES has been virtually abandoned while EE has been discontinued for almost all indications except birth control. EE continues to be used in birth control because it is resistant to metabolism in the uterus and controls menstrual bleeding better than oral estradiol does (Stanczyk, Archer, & Bhavnani, 2013). CEEs are also being increasingly superseded by estradiol in medicine, although significant use of CEEs for hormone therapy in cisgender women continues. Transdermal estradiol is gaining momentum over oral estradiol in menopausal hormone therapy as well. Major transgender hormone therapy guidelines (see also Aly, 2020) recommend against the use of EE and CEEs in transfeminine people due to their greater risks and the inability to accurately monitor blood estrogen levels with these preparations (Coleman et al., 2012; Deutsch, 2016; Hembree et al., 2017). Estradiol is the estrogen that is almost exclusively used in transfeminine people today. Besides estrogen type, it has been recommended that transdermal estradiol be used instead of oral estradiol in transfeminine people who are over 40 or 45 years of age or are otherwise at risk for blood clots (Deutsch, 2016; Iwamoto et al., 2019; Glintborg et al., 2021). Menopausal hormone therapy guidelines similarly recommend the use of transdermal estradiol over oral estrogens in cisgender women who are at higher risk for blood clots (e.g., Stuenkel et al., 2015).
In a historically notable study conducted by the Center of Expertise on Gender Dysphoria (CEGD) at the Vrije Universiteit Medical Center (VUMC) in Amsterdam, the Netherlands in the 1980s, it was reported that the risk of blood clots with high-dose EE and CPA in transfeminine people was increased by 45-fold relative to the expected incidence in the general population (Asscheman, Gooren, & Eklund, 1989; Asscheman et al., 2014). Mortality also appeared to be elevated and other health risks were increased as well (Asscheman, Gooren, & Eklund, 1989; Gooren & T’Sjoen, 2018). A subsequent study in transfeminine people by the CEGD confirmed strongly increased coagulation with EE but much lower increases with oral or transdermal estradiol (Toorians et al., 2003). Upon the CEGD switching transfeminine people from high-dose EE to physiological doses of oral or transdermal estradiol (also usually in combination with CPA), the risks decreased considerably (van Kesteren et al., 1997; Asscheman et al., 2011; Asscheman et al., 2014). These findings were of major importance in the replacement of EE with estradiol in transfeminine hormone therapy, and have surely contributed significantly to apprehension about the use of high doses of estrogens in transfeminine people.
Taken together, estrogens of all kinds have been shown to dose-dependently increase or be associated with increased risk of blood clots. These findings suggest that, provided of course sufficient exposure occurs, increased coagulation and blood clot risk are common properties of estrogens. However, synthetic and non-bioidentical estrogens have greater risk of blood clots than estradiol, and oral estradiol shows greater risk than non-oral estradiol. In fact, physiological estradiol levels in women and low to moderate doses of transdermal estradiol may have no significant risk of blood clots at all. Nonetheless, non-oral estradiol with sufficiently high exposure can increase blood clot risk just the same as other forms of estrogen. Concomitant therapy with progestogens appears to augment the risk of blood clots with estrogens and high doses may particularly amplify the risk.
Risks with Different Hormonal Exposures
The table below provides relative risk increases for blood clots with different types, routes, and doses of estrogens, as well as with SERMs, pregnancy, and high-dose CPA. It shows the greater risks of blood clots with (1) oral estradiol relative to non-oral estradiol; (2) estradiol compared to non-bioidentical estrogens; and (3) lower estrogen levels/doses relative to higher estrogen levels/doses.
Table 2: Relative risks of blood clots with different hormonal exposures (see also Machin & Ragni, 2020):
Footnotes:a At typical menopausal replacement doses (i.e., not very high—probably no more than double the given dose). b MPA, norethisterone, norgestrel, or drospirenone. c Modern EE + P birth control contains 20–35 μg/day EE, while high-dose EE + P birth control used in the 1960s and 1970s contained 50–150 μg/day EE. d Risk around twice as high as modern birth control pills. e Unpublished original research/analysis with borderline statistical significance (95% CI 0.99–4.22). f Excluding the postpartum period. With the postpartum period included, the risk of blood clots with pregnancy is 5–10× (McLintock, 2014). Abbreviations: E2 = Estradiol; CEEs = Conjugated estrogens; EE = Ethinylestradiol; DES = Diethylstilbestrol; EMP = Estramustine phosphate; PEP = Polyestradiol phosphate; SERMs = Selective estrogen receptor modulators; CPA = Cyproterone acetate; P = Progestogen.
Note that the values in the table are associations mostly from observational studies rather than from RCTs. Hence, in many cases, causation has not been definitively established. In addition, the values represent rough average values with often wide 95% confidence intervals. As a result, precision and accuracy of the estimates may in some cases be low. Also note that quantified blood clot risk will vary depending on the study and its definitions and methodology (including factors like sampling error, approach to control of confounding variables, and residual confounding influences).
Mechanisms of Increased Coagulation with Estrogens
Aside from coagulation factors, estrogens also modulate the synthesis of numerous other liver products (Kuhl, 1999; Kuhl, 2005; Table). Examples include sex hormone-binding globulin (SHBG), corticosteroid-binding globulin (CBG), various other circulating binding proteins, angiotensinogen, lipoproteins, and triglycerides, among others. In accordance with the mechanisms underlying increased coagulation and blood clot risk with estrogens, the differences in risk of blood clots with different types and routes of estrogens are mirrored in their influences on estrogen-sensitive liver products. Put another way, different estrogens have different relative potency in the liver when compared to their estrogenic potency elsewhere in the body. Synthetic and non-bioidentical estrogens have greater impact on liver synthesis than estradiol, while oral administration of estradiol has greater influence on liver synthesis than non-oral routes like transdermal administration or intramuscular injection, and this is likely to explain the observed differences in coagulation and blood clot risk with these different estrogens. The table below shows the liver potency of different estrogenic exposures as measured by influence specifically on SHBG levels, one of the most sensitive and well-characterized estrogen-modulated liver products.
Table 3: Relative increases in SHBG levels with different estrogenic exposures (see also Aly, 2020):
Footnotes:a Due to differences in molecular weight, estradiol valerate has about 75% of the amount of estradiol as regular estradiol. Hence, 6 mg/day estradiol valerate is approximately equivalent to 4.5 mg/day estradiol. b Modern EE + P birth control contains 20–35 μg/day EE, while high-dose EE + P birth control used in the 1960s and 1970s contained 50–150 μg/day EE. c In the form of 320 mg/month PEP (~700 pg/mL estradiol), 100 mg/month estradiol undecylate (~500–600 pg/mL estradiol), or 10 mg/10 days estradiol valerate (~500–1,200 pg/mL peak estradiol; Graphs). Abbreviations: E2 = Estradiol; EV = Estradiol valerate; CEEs = Conjugated estrogens; EE = Ethinylestradiol; DES = Diethylstilbestrol; EMP = Estramustine phosphate; PEP = Polyestradiol phosphate; P = Progestogen.
The increase in SHBG levels with estrogen therapy correlates with increases in coagulation and blood clot risk and can serve as a reliable surrogate indicator of these effects (Odlind et al., 2002; van Rooijen et al., 2004; van Vliet et al., 2005; Tchaikovski & Rosing, 2010; Raps et al., 2012; Stegeman et al., 2013; Hugon-Rodin et al., 2017; Eilertsen et al., 2019). The increases in SHBG levels and blood clot risk even appear quite similar to each other with modern birth control pills (both ~4-fold), high-dose oral synthetic estrogen therapy (both ~5–10-fold), and late pregnancy (both ~5–10-fold). When data on blood clot risk with a given estrogen route or dose are limited or unavailable—for instance with high-dose oral estradiol or high-dose estradiol ester injections—changes in SHBG levels can be used as a rough proxy or surrogate instead to estimate overall liver impact, magnitude of change in coagulation systems, and blood clot risk. It should be noted however that progestogens may augment the blood clot risk with estrogens without necessarily affecting SHBG levels or even while decreasing SHBG levels via concomitant androgenic activity (Kuhl, 2005; Vinogradova, Coupland, & Hippisley-Cox, 2019).
Physiological levels of estradiol appear to have relatively minimal influence on liver synthesis (Eisenfeld & Aten, 1979; Lax, 1987; Kuhl, 2005). This is in accordance with the limited influence or non-influence of physiological estradiol levels in women on blood clot risk. It is thought that under normal physiological circumstances, estradiol is only supposed to considerably affect liver synthesis at very high levels—namely during pregnancy. The changes in synthesis of liver products during pregnancy presumably have important biological roles at this time (Eisenfeld & Aten, 1979). One of these is considered to be increased coagulation, as coagulation limits blood loss with childbirth and hence has survival benefits. Conversely, there is no obvious benefit to increased coagulation outside of pregnancy.
Estradiol and the Liver First Pass with Oral Administration
The oralroute of administration is subject to a first pass through the liver via the hepatic portal vein which is not present with non-oral routes of administration (Pond & Tozer, 1984; Back & Rogers, 1987). As such, oral estradiol is subject to a hepatic first pass while this does not occur with non-oral forms of estradiol such as transdermal estradiol and injectable estradiol (Kuhl, 1998; Kuhl, 2005). This first pass results in disproportionate exposure of the liver to estradiol as well as disproportionate estrogenic impact on liver protein synthesis (Kuhl, 2005). Oral estradiol likewise has disproportionate estrogenic impact on the hepatic synthesis of coagulation factors (Kuhl, 1998; Kuhl, 2005). Due to the first pass, it is estimated that there is a 4- or 5-fold greater estrogenic impact of oral estradiol in the liver relative to non-oral estradiol (Kuhl, 2005). Due to the absence of the hepatic first pass with most non-oral routes, there is strong biological plausibility for the lower risk of blood clots that has been found with transdermal estradiol in comparison to oral estradiol in observational studies (Baber et al., 2016).
Although oral estradiol has a much higher relative potential for blood clots due to the liver first pass, sufficiently high levels of estradiol will diffuse into the liver from the blood to act on this tissue regardless of route of administration. Hence, high levels of estradiol via non-oral routes (or produced by the body itself) can increase coagulation and blood clot risk similarly to the oral route. This is clearly evidenced by hyperestrogenic situations like pregnancy and ovarian stimulation for in-vitro fertilization, when estradiol levels increase to very high concentrations and substantially influence liver protein synthesis.
Non-Bioidentical Estrogens and Resistance to Liver Metabolism
Non-bioidentical estrogens such as EE, DES, and CEEs have greater impact on liver protein synthesis and risk of blood clots than either oral estradiol or non-oral estradiol (Kuhl, 1998; Kuhl, 2005; Phillips et al., 2014; Turo et al., 2014; Table). This is because the liver strongly metabolizes and inactivates estradiol, whereas non-bioidentical estrogens have differences in their chemical structures relative to estradiol that result in them being much more resistant to liver metabolism (Kuhl, 1998; Kuhl, 2005; Connors & Middeldorp, 2019; Swee, Javaid, & Quinton, 2019).
EE can be considered as a case example. The oral bioavailability of EE is around 45%, while that of estradiol is only about 5% (Kuhl, 2005; Stanczyk, Archer, & Bhavnani, 2013). In addition, the blood half-life of EE is in the range of 5 to 30 hours, compared to less than 1 hour in the case of estradiol (White et al., 1998; Kuhl, 2005; Stanczyk, Archer, & Bhavnani, 2013). As a result of these and other differences, EE is approximately 120 times as potent as estradiol by weight in terms of general estrogenic effect (Kuhl, 2005; Table). Hence, EE is used clinically in μg doses whereas oral estradiol is used at over 100-fold higher mg doses. The pharmacokinetic differences between EE and estradiol reflect the strong resistance of EE to liver metabolism (Kuhl, 2005). EE, or 17α-ethynylestradiol, shows resistance to liver metabolism because of an ethynyl group at the C17α position which has been added to what is the otherwise unchanged structure of estradiol (Kuhl, 2005). This modification results in steric hindrance which blocks 17β-hydroxysteroid dehydrogenases (17β-HSDs) as well as conjugating enzymes like sulfotransferases and glucuronosyltransferases from metabolizing EE at the C17β hydroxyl group. 17β-HSDs normally convert estradiol into the weakly active estrone while the conjugating enzymes convert estradiol into inactive C17β estrogen sulfate and glucuronide conjugates like estrone sulfate (Kuhl, 2005). An “ethinylestrone” metabolite is in fact a structural impossibility due to the requirement of a double bond for a C17 ketone group—the needed C17α position is already occupied in EE by its ethynyl group. As such, the metabolism of estradiol into weakly active or inactive metabolites like estrone and estrone sulfate in the liver is protective against activation of hepatic ERs and procoagulation, and the lack of this with EE is responsible for its greater blood clot risk (Kuhl, 2005; Russell et al., 2017).
Figure 2: Chemical structures of selected estrogens. The C17 position in the case of the steroidal estrogens (E2, E1, and EE) is at the top right of the steroid nucleus.
Due to the marked resistance of EE to hepatic metabolism and inactivation, it persists for a long time in the liver—often cycling through it many times before finally being broken down. Moreover, EE shows several-fold disproportionate impact on liver protein synthesis at otherwise equivalent doses relative to oral estradiol (Kuhl, 2005; Table). Consequently, whereas EE has around 120-fold the general potency of oral estradiol, the liver potency of EE is around 350 to 1,500 times greater than that of oral estradiol (von Schoultz et al., 1989; Kuhl, 2005). A dose of EE of as little as 1 μg/day has been shown to impact liver metabolism (Speroff et al., 1996; Trémollieres, 2012). In addition, the fact that EE shows similar hepatic impact and risk of blood clots regardless of whether it is administered orally, transdermally, or vaginally indicates that unlike oral estradiol, the first pass through the liver with oral administration is not necessary for blood clot risk with EE (Plu-Bureau et al., 2013; PCASRM, 2017; Konkle & Sood, 2019). EE is so resistant to metabolism that it does not seem to matter how it is administered—the liver impact is substantial regardless of route. The greatly increased liver potency of EE results in its influence on coagulation and blood clot risk being much higher than that of estradiol at equivalent doses.
CEEs show a few-fold disproportionate estrogenic impact on liver protein synthesis relative to oral estradiol but less than that of EE (Kuhl, 2005; Table). This can be attributed to the equine (horse) estrogens in CEEs, which humans are presumably not adapted to and which show resistance to liver metabolism in humans. DES, on the other hand, shows even greater estrogenic influence on the liver than EE (Kuhl, 2005; Table). The more disproportionate impact on liver synthesis of DES relative to EE or CEEs may be attributable to the fact that it is a nonsteroidal estrogen and is far removed in structure from steroidal estrogens. This is relevant as steroidal estrogens are susceptible to varying extents to robust steroid-metabolizing enzymes in the liver (Kuhl, 2005). As with EE, 17β-HSDs have no affinity for DES and the hydroxyl groups of DES are not oxidized to form estrone-like ketone metabolites (Jensen et al., 2010). Consequent to their resistance to liver metabolism relative to estradiol, CEEs and nonsteroidal estrogens like DES have greater impacts on coagulation and blood clot risk than equivalent doses of estradiol similarly to EE although to varying extents.
When compared to transdermal estradiol rather than oral estradiol, the disproportionate influence of oral non-bioidentical estrogens on estrogen-modulated liver protein synthesis becomes extreme. With a little math, it quickly becomes apparent why high doses of these estrogens have influences on liver proteins and blood clot risks that are comparable to those during pregnancy. The table below shows some roughly calculated estimates for comparative liver strength of the different estrogens.
Table 4: Roughly calculated ratios of liver estrogenic potency to general/systemic estrogenic potency with estrogens based on a selection of liver products (e.g., SHBG, others) (Kuhl, 2005; Table):
Estrogen
Comparative liver potency
Relative to oral E2
Relative to transdermal E2
Transdermal E2
~0.25×a
1.0×a
Oral E2
1.0×
~4.0×
Oral CEEs
1.3–4.5×
~5.2–18×
Oral EE
2.9–5.0×
~12–20×
Oral DES
5.7–7.5×
~23–30×
a Based on a study that found oral estradiol to have 4-fold greater effect on SHBG levels than transdermal estradiol when used at doses that produced similar estradiol levels (Nachtigall et al., 2000).
Changes in liver protein synthesis induced by estrogens don’t scale linearly with dose or relative liver potency. There is progressive saturation in terms of changes in levels of SHBG and other liver products with estrogen dose—that is, higher doses have relatively diminished effect compared to lower doses (Kuhl, 1990; Kuhl, 1999). As an example, oral EE shows the following dose-dependent increases in SHBG levels: 2.0-fold at 5 μg/day, 3.0-fold at 10 μg/day, 3.4-fold at 20 μg/day, and 4.0-fold at 50 μg/day (Kuhl, 1998; Kuhl, 1999). These findings can be attributed to saturation of the competitive binding and/or activation of liver ERs by high estrogen concentrations (Kuhl, 1990). An implication of this dose-dependent saturation is that although for instance oral EE has much stronger potency in the liver than oral estradiol, oral estradiol can more quickly “catch up” to oral EE and other non-bioidentical estrogens in terms of liver impact than might be initially anticipated. Accordingly, oral estradiol has shown the following dose-dependent increases in SHBG levels: 1.6-fold at 1 mg/day, 2.2-fold at 2 mg/day, and 1.9- to 3.2-fold at 4 mg/day (Fåhraeus & Larsson-Cohn, 1982; Kuhl, 1998; Gibney et al., 2005; Ropponen et al., 2005). Hence, although oral EE may have roughly 3- to 5-fold higher liver potency than oral estradiol, a dose of oral estradiol near-equivalent to that of oral EE in terms of general estrogenic effect can increase SHBG levels to an extent that is only somewhat lower in comparison.
Selective Estrogen Receptor Modulators and Metabolism Resistance
SERMs like tamoxifen and raloxifene are essentially partial agonists of the ER. This is in contrast to estrogens—like estradiol, CEEs, EE, and DES—which act as full agonists of the ER. Similarly to nonsteroidal estrogens like DES, the clinically used SERMs are all nonsteroidal in structure and are strongly resistant to hepatic metabolism. In fact, certain SERMs, like tamoxifen and clomifene, are structurally related to and were derived from DES. SERMs show tissue differences in their ER-mediated effects, with estrogenic effects in some tissues (e.g., bone) and antiestrogenic effects in other tissues (e.g., breasts) (Lain, 2019; Table). Although there is variation between SERMs in terms of their effects in certain tissues (e.g., uterus), they are uniformly estrogenic in the liver. Consequently, SERMs show similar increases in blood clot risk as estrogens (Park & Jordan, 2002; Fabian & Kimler, 2005). As with non-bioidentical estrogens, the greater risk of blood clots with SERMs compared to oral estradiol can be attributed to their resistance to liver metabolism and hence to greater hepatic estrogenic potency. The SERMs that are used medically belong to diverse structural families (e.g., triphenylethylenes like tamoxifen and benzothiophenes like raloxifene). The only way in which SERMs of different structural classes are known to be related is in their shared interactions with the ERs.
Figure 3: Chemical structures of selected SERMs. They are nonsteroidal in structure and include tamoxifen (a triphenylethylene) and raloxifene (a benzothiophene).
Activation of the Estrogen Receptor is Specifically Responsible for Increased Coagulation with Estrogens and SERMs
Findings from preclinical and genetic research provide direct evidence for ER activation being responsible for the increased blood clot risk with estrogens. In an important animal study, EE was administered to mice and changes in procoagulant and anticoagulant biomarkers were measured (Cleuren et al., 2010). EE caused changes in levels of a variety of coagulation factors (Cleuren et al., 2010). The researchers also assessed estradiol and observed comparable changes (Cleuren et al., 2010). Co-administration of the selective ER full antagonist fulvestrant with EE neutralized all of the EE-induced coagulatory changes (Cleuren et al., 2010). Additionally, EE showed no effect on coagulation factors in ERα knockout mice (Cleuren et al., 2010). These findings are consistent with human and mouse genome-wide association studies which have found estrogen response elements (EREs)—DNA sequences that act as binding sites for genes regulated by the ER—embedded in a large number of genes involved in coagulatory pathways (Cleuren et al., 2010; Stanczyk, Mathews, & Cortessis, 2017).
The preceding findings are consistent with ER activation being responsible for increased coagulation and blood clot risk with estrogens and SERMs. This is in accordance with the fact that blood clot risk is a shared effect of selective ER agonists with highly diverse chemical structures, providing strong circumstantial support against a non-ER-mediated action of some sort being responsible (e.g., the weakly estrogenic metabolite estrone somehow mediating the blood clot risk with estradiol—Bagot et al., 2010). Increased coagulation and blood clot risk can thus be regarded as class effects of estrogens and SERMs—provided sufficiently high liver exposure. Due to differences in susceptibility to liver metabolism however, different ER agonists show differences in their relative impact on coagulation. Owing to estradiol’s lack of resistance to metabolism and its robust inactivation in the liver, the dosage requirements for increased coagulation and blood clot risk with estradiol—particularly in the case of non-oral estradiol—are greater than with non-bioidentical estrogens. Hence, estradiol, especially when administered via non-oral routes, is a safer form of estrogen therapy than other estrogens.
Absolute Incidences and Risk Factors
States of estrogen and/or progestogen exposure, such as exogenous hormone administration and pregnancy, are of course established risk factors for blood clots in women. In healthy young individuals without relevant risk factors for blood clots however, the incidence of blood clots is rare even in situations of considerably increased risk due to hormones (Rosendaal, 2005). The absolute incidence of VTE in non-pregnant women is only 1 to 5 of every 10,000 women each year (i.e., 0.01–0.05% per year) (PCASRM, 2017; Konkle & Sood, 2019). EE-containing birth control pills, which on average increase VTE risk by about 4-fold, are associated with an incidence of VTE of only 3 to 9 of every 10,000 women each year (i.e., 0.03–0.09% per year) (Konkle & Sood, 2019). Likewise, the absolute risk of blood clots during pregnancy, when estradiol and progesterone levels increase to extremely high concentrations and VTE risk is increased up to 7-fold (Abdul Sultan et al., 2015), is about 5 to 20 of every 10,000 women each year (i.e., 0.05–0.2% per year) (PCASRM, 2017; Konkle & Sood, 2019).
a 1–2/10,000 per year at <19 years of age, 2–3/10,000 per year at 20–29 years of age, 3–4/10,000 per year at 30–39 years of age, 5–7/10,000 per year at 40–49 years of age; roughly 3–4/10,000 per year for age 15–49 years overall (Rabe et al., 2011).
In addition to time and population considerations, there are, besides estrogen and progestogen exposure, a variety of other known risk factors for blood clots, and these risk factors can substantially augment blood clot risk (Heit et al., 2000; Rosendaal, 2005). Age is among the strongest of the known risk factors (Rosendaal, 2005; Montagnana et al., 2010). Moreover, age is uniquely notable as a risk factor in that it is one that eventually becomes relevant to everyone. The risk of blood clots increases on the order of 100-fold going from ≤15 years of age (incidence <0.005–0.01% per year) to ≥80 years of age (incidence ~0.5–1.0% per year) (Rosendaal, 2005; Montagnana et al., 2010; Rabe et al., 2011). The figure below provides a graphical representation of the influence of age on risk of blood clots.
Figure 4: Risk of first-incidence VTE (per 100,000 per year) by age group (in years) in men (black bars) and women (gray bars) (Oger, 2000; Rosendaal, 2005; Rosendaal, 2016).
Other established risk factors for blood clots and associated cardiovascular problems include physical inactivity (due to, e.g., bed rest, long-distance travel, etc.), obesity, smoking, thrombophilic abnormalities, cancer, surgery, and HIV, among many others (Baron et al., 1998; Heit et al., 2000; Rosendaal, 2005; Lijfering, Rosendaal, & Cannegieter, 2010; Timp et al., 2013). In addition to age, physical inactivity is one of the most important risk factors for blood clots and mediates the risk increases for many of the others (Rosendaal, 2005). Smoking on its own is not consistently associated with increased risk of VTE (Lijfering, Rosendaal, & Cannegieter, 2010), but in combination with EE-containing birth control pills has been associated with a synergistic increase in VTE risk (Pomp, Rosendaal, & Doggen, 2008) as well as large increases in risk of heart attack—for instance 20-fold higher risk in heavy smokers (Kuhl, 1999). The table below shows the influence of a selection of known risk factors for VTE:
Thrombophilias, heritable and acquired, exist in significant percentages of the population and can lead to large increases in blood clot risk (Lijfering, Rosendaal, & Cannegieter, 2010). Moreover, they are often if not usually unknown (Morimont, Dogné, & Douxfils, 2020). This is due to the fact that screening for heritable thrombophilias is mainly based on family history, which has low sensitivity and poor predictive value for identifying people with these abnormalities (Morimont, Dogné, & Douxfils, 2020). Hence, many people are at increased risk of blood clots without realizing it. The table below shows the prevalences of a variety of thrombophilic abnormalities and their impacts on blood clot risk.
Blood clots are considered to be a multicausal disease (Rosendaal, 2005). The risk of blood clots and associated cardiovascular complications with hormonal exposure is highest when multiple risk factors combine in a given individual. Under what are among the most extreme of circumstances in terms of risk—elderly people with cancer who are on high-dose oral synthetic estrogen therapy (e.g., DES)—blood clot incidence can be as high as 15 to 28% and overall incidence of cardiovascular complications as great as 35% (Phillips et al., 2014; Sciarria et al., 2014; Turo et al., 2014). These adverse effects contribute to substantial morbidity and incidence of death in these populations. Most people are however at nowhere near as great of risk. Risk factors like age are why pregnant women can have massive levels of estradiol and progesterone with relatively little issue whereas elderly cancer patients on high-dose oral synthetic estrogen therapy have a considerable risk of death.
In the VUMC studies that found 20- to 45-fold increased incidence of blood clots with high-dose EE and CPA over 5 to 10 years in transfeminine people, the absolute incidence of blood clots was approximately 6.3% (142/10,000 people per year) in the 1989 report and 5.5% (58/10,000 people per year) in the 1997 follow up (Asscheman, Gooren, & Eklund, 1989; van Kesteren et al., 1997; Asscheman et al., 2014; Goldstein et al., 2019; Min & Hopkins, 2021). In keeping with the known influence of age on blood clot risk, the absolute incidence was 2.1% in those under 40 years of age and 12% in those over 40 years of age in the 1989 study (Asscheman, Gooren, & Eklund, 1989; Asscheman et al., 2014). In about 70% of cases, there were—aside from age—no known risk factors for blood clots (Asscheman, Gooren, & Eklund, 1989; Asscheman et al., 2014). Following subsequent replacement of EE with low-to-moderate-dose transdermal estradiol in those over 40 years of age, the incidence of blood clots decreased substantially (with only one event occurring in the transdermal estradiol group) (van Kesteren et al., 1997; Asscheman et al., 2014; Min & Hopkins, 2021). A later study in 2013 by the Ghent University Hospital in Belgium observed a blood clot incidence of 5.1% in transfeminine people using mostly oral or transdermal estradiol with or without CPA over an average treatment period of 7.7 years (range 3 months to 35 years) (Wierckx et al., 2013; Min & Hopkins, 2021). Those who had blood clots often had other risk factors such as older age, smoking, immoblization due to surgery, or hypercoagulability (Wierckx et al., 2013; Min & Hopkins, 2021). In addition to cumulative exposure time, these studies further highlight the converging impact of multiple risk factors—with estrogen type, route, and dose, progestogen exposure, and age included among them—on the risk of blood clots.
Therapeutic Implications for Transfeminine People
Due to their greater risk of blood clots and cardiovascular problems, non-bioidentical estrogens like EE and CEEs are mostly no longer used in transfeminine people. Instead, estradiol, both in oral and non-oral forms, is used. Transgender clinical guidelines generally recommend keeping estradiol levels within normal physiological ranges for non-pregnant females of around 100 to 200 pg/mL regardless of whether the route of administration of estradiol is oral or non-oral (Aly, 2018). Higher estradiol levels are not currently known to have greater therapeutic benefit in terms of feminization or breast development (Nolan & Cheung, 2020). However, higher levels, in the range of 200 to 500 pg/mL, can provide additional therapeutic effect in the area of testosterone suppression—which can be indirectly beneficial to feminization if otherwise inadequate (Aly, 2018). Despite their recommendations for keeping estradiol levels in physiological ranges, transgender clinical guidelines notably recommend doses of estradiol ester injections that reach and even greatly exceed estradiol levels of 200 pg/mL (Aly, 2021).
Based on the available research (e.g., the risk of blood clots with lower doses, comparative SHBG increases), it would not be surprising if high-dose oral estradiol (e.g., 8 mg/day) had similar risk of blood clots as the relatively lower amounts of EE in birth control pills. The risk is likely to be particularly great in combination with progestogens (e.g., CPA). Due to its greater and unnecessary risk of blood clots relative to non-oral estradiol, oral estradiol should ideally be avoided in transfeminine people—particularly in those with risk factors for blood clots such as older age (e.g., >40 years) or concomitant progestogen use. However, the convenience of oral estradiol and its relative inexpensiveness (compared to e.g. transdermal forms) are significant advantages that will also be considered by transfeminine people and their clinicians. In contrast to oral estradiol, non-oral estradiol—with estradiol levels kept in physiological ranges of for instance 100 to 200 pg/mL—appears to have minimal to no risk of blood clots. Hence, non-oral estradiol at these levels can be used in transfeminine people with little concern.
In terms of higher estradiol levels delivered non-orally, the estimated 2-fold increase in risk of blood clots with estradiol levels of approximately 300 to 500 pg/mL (Sam, 2020) is notably lower than the average 4-fold increase in risk with widely used EE-containing birth control pills. Based on the usefulness of these levels for suppressing testosterone production and the widespread usage of EE-based birth control in cisgender women throughout the world, the degree of blood clot risk with high-dose non-oral estradiol, in reasonable amounts, could be considered therapeutically acceptable in transfeminine people (Haupt et al., 2020). This may be particularly true when high-dose non-oral estradiol monotherapy is compared to combination of estradiol with antiandrogens like spironolactone, CPA, or bicalutamide, which all have their own unique risks and drawbacks. In any case, as with oral estradiol, high estradiol levels with non-oral estradiol should ideally be avoided due to the additional risk they pose, and this is especially true in those with relevant risk factors for blood clots (e.g., older age). In addition, very high doses of non-oral estradiol resulting in estradiol levels above those required for testosterone suppression are difficult to justify as they pose further unnecessary risk and offer no clear additional therapeutic benefit.
Prevention of Blood Clots
The best way to prevent blood clots from happening is to avoid risk altogether. Avoiding use of oral estradiol, excessively high doses of non-oral estradiol, and progestogens when feasible and opting for safer therapeutic choices is recommended in this regard. In addition, avoiding use of such therapies in those with risk factors like older age (>40 years), known thrombophilic abnormalities, and sedentary lifestyle is advocated. Proactive behaviors like physical activity (e.g., walking, exercise), quitting smoking, and weight loss may help to reduce the risk of blood clots (Hibbs, 2008).
Certain anticoagulant and antiplatelet medications are used to help prevent blood clots in high-risk individuals. Examples include low-dose aspirin (Mekaj, Daci, & Mekaj, 2015; Matharu et al., 2020), direct factor Xa inhibitors like rivaroxaban (Xarelto) (Blondon, 2020), and direct thrombin inhibitors like dabigatran (Pradaxa), among others. Aspirin has been found to be effective in the prevention of blood clots (Mekaj, Daci, & Mekaj, 2015; Matharu et al., 2020) and has been recommended for use specifically in transfeminine people on hormone therapy (Feldman & Goldberg, 2006; Deutsch, 2016). However, evidence is limited and conflicting for prevention of blood clots related to hormone therapy (Grady et al., 2000; Cushman et al., 2004) and use of aspirin in transfeminine people for such purposes has been recommended against by others (Shatzel, Connelly, & DeLoughery, 2017). Rivaroxaban has been associated with more than completely offset risk of blood clots with oral menopausal hormonal therapy (Blondon, 2020). In any case, no anticoagulants are currently approved or well-supported for preventing risk of blood clots with hormone therapy. Accordingly, clinical guidelines state that there is insufficient evidence to guide decision-making in this area at this time (e.g., McLintock, 2014). It should also be cautioned that anticoagulants have side effects and risks of their own and should be used carefully.
Temporary discontinuation of estrogen therapy before surgery has traditionally been thought to help reduce the risk of blood clots during recovery based on theory and has been advised as well as mandated for transfeminine people undergoing surgical procedures (e.g., Asscheman et al., 2014). However, evidence is limited and inconclusive on this strategy at present and more research is needed to determine whether it is actually beneficial or not (Boskey, Taghinia, & Ganor, 2019; Nolan & Cheung, 2020; Haveles et al., 2021; Hontscharuk et al., 2021; Kozato et al., 2021; Nolan et al., 2021; Zucker, Reisman, & Safer, 2021). Recent studies have not found reduction in risk of blood clots with discontinuation of hormone therapy before surgery in transfeminine people but these studies have been underpowered and larger studies are needed (Blasdel et al., 2021). Temporarily stopping hormone therapy can be distressing for many transfeminine people and this should be weighed accordingly. A potential alternative to discontinuation of hormone therapy is temporary use of transdermal estradiol at physiological doses which has no known blood clot risk and is more likely to be safe.
Updates
Update 1: Langley et al. (2021) [PATCH Study Results]
In February 2021, a report on long-term cardiovascular outcomes for the Prostate Adenocarcinoma: TransCutaneous Hormones (PATCH) trial was published (Langley et al., 2021). The PATCH trial is a large ongoing phase 2/3 randomized controlled trial of high-dose transdermal estradiol patches versus GnRH agonists for the treatment of prostate cancer in men (Langley et al., 2021). The estradiol patch dosage employed is specifically three to four 100 μg/day FemSeven or Progynova TS patches (Langley et al., 2021). In the February 2021 report of the study, 1,694 men were enrolled and randomized, with 790 included in the analysis for the GnRH agonist group and 904 included in the analysis for the estradiol patch group (Langley et al., 2021).
In those given estradiol, the median estradiol level was around 215 pg/mL (5%–95% range ~100–550 pg/mL) (Langley et al., 2021). About 93% of the men in this group achieved suppression of testosterone levels into the castrate range (<50 ng/dL), which was notably equal to the rate of suppression in the GnRH agonist group (~93%) (Langley et al., 2021). However, actual testosterone levels—as opposed to rates of testosterone suppression—were not provided in this report and hence comparison between groups is unavailable for this metric (Langley et al., 2021). After about 4 years median follow up, there were no significant differences on a variety of cardiovascular outcomes between the estradiol group and the GnRH agonist group (Langley et al., 2021). Among these outcomes included VTE, thromboembolic stroke, and other arterial embolic events (Langley et al., 2021). These results are in contrast to previous large clinical trials of PEP in prostate cancer, which found increased cardiovascular morbidity and risk of VTE but notably involved higher estradiol levels than employed in the PATCH trial (Ockrim & Abel, 2009; Sam, 2020). Based on their promising safety findings, the PATCH researchers stated that transdermal estrogen should be reconsidered for the treatment of prostate cancer (Langley et al., 2021).
These findings are reassuring and suggest that limitedly high levels of estradiol (e.g., 200–300 pg/mL perhaps) may likewise be acceptably safe in terms of blood clot and cardiovascular risk in transfeminine people. It should be noted however that the sample size of the trial, while large relative to previous clinical studies in this area, was underpowered for assessing risk of blood clots—which are relatively rare events that require very large samples to thoroughly quantify. Studies precisely assessing blood clot risk in peri- and postmenopausal women have included tens of thousands of individuals for instance. As such, while substantial increases in risk are not likely based on this trial, smaller increases in risk still cannot be ruled out at this time. It should additionally be noted that the robust testosterone suppression at the used doses in this study might not generalize to transfeminine people as a whole, as the men were mostly elderly and testosterone levels are known to decrease with age.
Update 2: Totaro et al. (2021) and Kotamarti et al. (2021)
Totaro, M., Palazzi, S., Castellini, C., Parisi, A., D’Amato, F., Tienforti, D., Baroni, M. G., Francavilla, S., & Barbonetti, A. (2021). Risk of Venous Thromboembolism in Transgender People Undergoing Hormone Feminizing Therapy: A Prevalence Meta-Analysis and Meta-Regression Study. Frontiers in Endocrinology, 12, 741866. [DOI:10.3389/fendo.2021.741866]
This study is the largest of its kind that has been conducted to date. The meta-analysis included 18 studies totaling 11,542 transfeminine people on hormone therapy. The pooled prevalence of VTE was 2% with a 95% confidence interval of 1 to 3%. However, there was large variability between studies. In the meta-regression analysis, older age and longer length of estrogen therapy were significantly positively associated with VTE prevalence. When analysis was restricted to those greater than or equal to 37.5 years of age, the prevalence of VTE was 3% (95% CI: 0–5%). Conversely, in those less than 37.5 years of age, the prevalence of VTE was 0% (95% CI: 0–2%). VTE prevalence was 1% (95% CI: 0–3%) with greater than or equal to 4.4 years of estrogen therapy and was 0% (95% CI: 0–3%) with less than 4.4 years of estrogen therapy. With regard to the 0% estimates, it is not the case that there is truly no risk of VTE in these instances but rather it can be assumed that the risks are sufficiently low that the meta-analysis was not powered well enough to detect and quantify them.
A limitation of the meta-analysis was that subgroup analyses based on estrogen type (i.e., estradiol vs. CEEs vs. EE) and route (e.g., oral estrogens or oral estradiol vs. transdermal estradiol) were said to not be possible due to insufficient data and hence were not performed. However, another recent meta-analysis published in July 2021, which analyzed much of the same literature as Totaro et al. (2021), did perform subgroup analyses by estrogen type and route. This publication is as follows:
Kotamarti, V. S., Greige, N., Heiman, A. J., Patel, A., & Ricci, J. A. (2021). Risk for Venous Thromboembolism in Transgender Patients Undergoing Cross-Sex Hormone Treatment: A Systematic Review. The Journal of Sexual Medicine, 18(7), 1280–1291. [DOI:10.1016/j.jsxm.2021.04.006]
And this is what they reported in terms of subgroup analyses for estrogen type and route:
Because varying VTE rates have been reported with different estrogen regimens, analyses of VTE incidence were performed comparing oral or transdermal delivery, or the specific estrogen formulation. As many studies reported populations using mixed estrogen formulations or did not report the type of estrogen regimen, further statistical analysis could not be performed.
Route of estrogen administration appeared to play a role in the AMAB population. [Oral] estrogens (7 studies; 34.0 VTE per 10,000 person-years) vs transdermal estrogens (3 studies, 11.2 VTE per 10,000 person-years). Additionally, estrogen formulation also appeared to have a difference VTE incidence. Ethinyl estradiol was also associated with increased VTE incidence (3 studies, 293.1 VTE per 10,000 person-years) followed by conjugated equine estrogens (1 study, 49.0 VTE per 10,000 person-years) and estradiol valerate (4 studies, 31.5 VTE per 10,000 person-years).
It is unclear how accurate these precise numbers are due to the quality limitations of the underlying data. Moreover, antiandrogens (e.g., CPA) were not controlled for and as discussed by this article are likely to additionally influence VTE risk. In any case, the reported numbers are interesting and are in accordance with different estrogen types and routes varying in terms of VTE risk.
References
Abdul Sultan, A., West, J., Stephansson, O., Grainge, M. J., Tata, L. J., Fleming, K. M., Humes, D., & Ludvigsson, J. F. (2015). Defining venous thromboembolism and measuring its incidence using Swedish health registries: a nationwide pregnancy cohort study. BMJ Open, 5(11), e008864. [DOI:10.1136/bmjopen-2015-008864]
Abou-Ismail, M. Y., Citla Sridhar, D., & Nayak, L. (2020). Estrogen and thrombosis: A bench to bedside review. Thrombosis Research, 192, 40–51. [DOI:10.1016/j.thromres.2020.05.008]
Amitrano, L., Guardascione, M. A., Brancaccio, V., & Balzano, A. (2002). Coagulation Disorders in Liver Disease. Seminars in Liver Disease, 22(1), 83–96. [DOI:10.1055/s-2002-23205]
Anderson, G. L., Limacher, M., Assaf, A. R., Bassford, T., Beresford, S. A., Black, H., Bonds, D., Brunner, R., Brzyski, R., Caan, B., Chlebowski, R., Curb, D., Gass, M., Hays, J., Heiss, G., Hendrix, S., Howard, B. V., Hsia, J., Hubbell, A., Jackson, R., … & Women’s Health Initiative Steering Committee. (2004). Effects of Conjugated Equine Estrogen in Postmenopausal Women With Hysterectomy: The Women’s Health Initiative Randomized Controlled Trial. JAMA, 291(14), 1701–1712. [DOI:10.1001/jama.291.14.1701]
Arnold, J. D., Sarkodie, E. P., Coleman, M. E., & Goldstein, D. A. (2016). Incidence of Venous Thromboembolism in Transgender Women Receiving Oral Estradiol. The Journal of Sexual Medicine, 13(11), 1773–1777. [DOI:10.1016/j.jsxm.2016.09.001]
Asscheman, H., Gooren, L., & Eklund, P. (1989). Mortality and morbidity in transsexual patients with cross-gender hormone treatment. Metabolism, 38(9), 869–873. [DOI:10.1016/0026-0495(89)90233-3]
Asscheman, H., Giltay, E. J., Megens, J. A., de Ronde, W. (Pim), van Trotsenburg, M. A., & Gooren, L. J. (2011). A long-term follow-up study of mortality in transsexuals receiving treatment with cross-sex hormones. European Journal of Endocrinology, 164(4), 635–642. [DOI:10.1530/eje-10-1038]
Asscheman, H., T’Sjoen, G., Lemaire, A., Mas, M., Meriggiola, M. C., Mueller, A., Kuhn, A., Dhejne, C., Morel-Journel, N., & Gooren, L. J. (2013). Venous thrombo-embolism as a complication of cross-sex hormone treatment of male-to-female transsexual subjects: a review. Andrologia, 46(7), 791–795. [DOI:10.1111/and.12150]
Baber, R. J., Panay, N., & Fenton, A. (2016). 2016 IMS Recommendations on women’s midlife health and menopause hormone therapy. Climacteric, 19(2), 109–150. [DOI:10.3109/13697137.2015.1129166]
Back, D. J., & Rogers, S. M. (2007). Review: first-pass metabolism by the gastrointestinal mucosa. Alimentary Pharmacology & Therapeutics, 1(5), 339–357. [DOI:10.1111/j.1365-2036.1987.tb00634.x]
Bagot, C., Marsh, M., Whitehead, M., Sherwood, R., Roberts, L., Patel, R., & Arya, R. (2010). The effect of estrone on thrombin generation may explain the different thrombotic risk between oral and transdermal hormone replacement therapy. Journal of Thrombosis and Haemostasis, 8(8), 1736–1744. [DOI:10.1111/j.1538-7836.2010.03953.x]
Bagot, C., Leishman, E., Onyiaodike, C., Jordan, F., Gibson, V., & Freeman, D. (2019). Changes in laboratory markers of thrombotic risk early in the first trimester of pregnancy may be linked to an increase in estradiol and progesterone. Thrombosis Research, 178, 47–53. [DOI:10.1016/j.thromres.2019.03.015]
Baron, J. A., Gridley, G., Weiderpass, E., Nyren, O., & Linet, M. (1998). Venous thromboembolism and cancer. The Lancet, 351(9109), 1077–1080. [DOI:10.1016/s0140-6736(97)10018-6]
Bezwada, P., Shaikh, A., & Misra, D. (2017). The Effect of Transdermal Estrogen Patch Use on Cardiovascular Outcomes: A Systematic Review. Journal of Women’s Health, 26(12), 1319–1325. [DOI:10.1089/jwh.2016.6151]
Blanco-Molina, M., Lozano, M., Cano, A., Cristobal, I., Pallardo, L., & Lete, I. (2012). Progestin-only contraception and venous thromboembolism. Thrombosis Research, 129(5), e257–e262. [DOI:10.1016/j.thromres.2012.02.042]
Bland, L. B., Garzotto, M., DeLoughery, T. G., Ryan, C. W., Schuff, K. G., Wersinger, E. M., Lemmon, D., & Beer, T. M. (2005). Phase II study of transdermal estradiol in androgen-independent prostate carcinoma. Cancer, 103(4), 717–723. [DOI:10.1002/cncr.20857]
Blasdel, G., Shakir, N., Parker, A., Bluebond-Langner, R., & Zhao, L. (2021). Letter to the Editor from Blasdel et al: “No Venous Thromboembolism Increase Among Transgender Female Patients Remaining on Estrogen for Gender-affirming Surgery”. The Journal of Clinical Endocrinology & Metabolism, 106(9), e3783–e3784. [DOI:10.1210/clinem/dgab243]
Blondon, M. (2020). Update On Oral Contraception And Venous Thromboembolism. HemaSphere Educational Updates in Hematology Book: 25th Congress of the European Hematology Association, Virtual Edition 2020, 4(S2). European Hematology Association. [Google Scholar] [DOI:10.1097/HS9.0000000000000444] [PDF]
Boskey, E. R., Taghinia, A. H., & Ganor, O. (2019). Association of Surgical Risk With Exogenous Hormone Use in Transgender Patients. JAMA Surgery, 154(2), 159–169. [DOI:10.1001/jamasurg.2018.4598]
Canonico, M., Plu-Bureau, G., Lowe, G. D., & Scarabin, P. (2008). Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systematic review and meta-analysis. BMJ, 336(7655), 1227–1231. [DOI:10.1136/bmj.39555.441944.be]
Canonico, M., Plu-Bureau, G., O’Sullivan, M. J., Stefanick, M. L., Cochrane, B., Scarabin, P., & Manson, J. E. (2014). Age at menopause, reproductive history, and venous thromboembolism risk among postmenopausal women. Menopause, 21(3), 214–220. [DOI:10.1097/gme.0b013e31829752e0]
Cassidy, A., & Minihane, A. (2017). The role of metabolism (and the microbiome) in defining the clinical efficacy of dietary flavonoids. The American Journal of Clinical Nutrition, 105(1), 10–22. [DOI:10.3945/ajcn.116.136051]
Chaireti, R., Gustafsson, K. M., Bystrom, B., Bremme, K., & Lindahl, T. L. (2013). Endogenous thrombin potential is higher during the luteal phase than during the follicular phase of a normal menstrual cycle. Human Reproduction, 28(7), 1846–1852. [DOI:10.1093/humrep/det092]
Choi, J., Kim, D., Park, S., Lee, H., Kim, K., Kim, K., Kim, M., Kim, S., & Kim, S. (2015). Anti-thrombotic effect of rutin isolated from Dendropanax morbifera Leveille. Journal of Bioscience and Bioengineering, 120(2), 181–186. [DOI:10.1016/j.jbiosc.2014.12.012]
Cleuren, A., Van der Linden, I., de Visser, Y., Wagenaar, G., Reitsma, P., & van Vlijmen, B. (2010). 17α‐Ethinylestradiol rapidly alters transcript levels of murine coagulation genes via estrogen receptor α. Journal of Thrombosis and Haemostasis, 8(8), 1838–1846. [DOI:10.1111/j.1538-7836.2010.03930.x]
Coelingh Bennink, H. J., Verhoeven, C., Dutman, A. E., & Thijssen, J. (2017). The use of high-dose estrogens for the treatment of breast cancer. Maturitas, 95, 11–23. [DOI:10.1016/j.maturitas.2016.10.010]
Coleman, E., Bockting, W., Botzer, M., Cohen-Kettenis, P., DeCuypere, G., Feldman, J., Fraser, L., Green, J., Knudson, G., Meyer, W. J., Monstrey, S., Adler, R. K., Brown, G. R., Devor, A. H., Ehrbar, R., Ettner, R., Eyler, E., Garofalo, R., Karasic, D. H., … & Zucker, K. (2012). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, Version 7. International Journal of Transgenderism, 13(4), 165–232. [DOI:10.1080/15532739.2011.700873] [URL] [PDF]
Conard, J., Plu-Bureau, G., Bahi, N., Horellou, M., Pelissier, C., & Thalabard, J. (2004). Progestogen-only contraception in women at high risk of venous thromboembolism. Contraception, 70(6), 437–441. [DOI:10.1016/j.contraception.2004.07.009]
Connelly, P. J., Marie Freel, E., Perry, C., Ewan, J., Touyz, R. M., Currie, G., & Delles, C. (2019). Gender-Affirming Hormone Therapy, Vascular Health and Cardiovascular Disease in Transgender Adults. Hypertension, 74(6), 1266–1274. [DOI:10.1161/hypertensionaha.119.13080]
Connors, J. M., & Middeldorp, S. (2019). Transgender patients and the role of the coagulation clinician. Journal of Thrombosis and Haemostasis, 17(11), 1790–1797. [DOI:10.1111/jth.14626]
Curb, J. D., Prentice, R. L., Bray, P. F., Langer, R. D., Van Horn, L., Barnabei, V. M., Bloch, M. J., Cyr, M. G., Gass, M., Lepine, L., Rodabough, R. J., Sidney, S., Uwaifo, G. I., & Rosendaal, F. R. (2006). Venous Thrombosis and Conjugated Equine Estrogen in Women Without a Uterus. Archives of Internal Medicine, 166(7), 772–772. [DOI:10.1001/archinte.166.7.772]
Cushman, M. (2004). Estrogen Plus Progestin and Risk of Venous Thrombosis. JAMA, 292(13), 1573–1580. [DOI:10.1001/jama.292.13.1573]
Deitcher, S. R., & Gomes, M. P. (2004). The risk of venous thromboembolic disease associated with adjuvant hormone therapy for breast carcinoma. Cancer, 101(3), 439–449. [DOI:10.1002/cncr.20347]
DeLoughery, T. G. (2011). Estrogen and thrombosis: Controversies and common sense. Reviews in Endocrine and Metabolic Disorders, 12(2), 77–84. [DOI:10.1007/s11154-011-9178-0]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Dinger, J., Do Minh, T., & Heinemann, K. (2016). Impact of estrogen type on cardiovascular safety of combined oral contraceptives. Contraception, 94(4), 328–339. [DOI:10.1016/j.contraception.2016.06.010]
Dittrich, R., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Mueller, A. (2005). Endocrine Treatment of Male-to-Female Transsexuals Using Gonadotropin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 113(10), 586–592. [DOI:10.1055/s-2005-865900]
Douxfils, J., Morimont, L., & Bouvy, C. (2020). Oral Contraceptives and Venous Thromboembolism: Focus on Testing that May Enable Prediction and Assessment of the Risk. Seminars in Thrombosis and Hemostasis, 46(8), 872–886. [DOI:10.1055/s-0040-1714140]
Douxfils, J., Klipping, C., Duijkers, I., Kinet, V., Mawet, M., Maillard, C., Jost, M., Rosing, J., & Foidart, J. (2020). Evaluation of the effect of a new oral contraceptive containing estetrol and drospirenone on hemostasis parameters. Contraception, 102(6), 396–402. [DOI:10.1016/j.contraception.2020.08.015]
Dutra, E., Lee, J., Torbati, T., Garcia, M., Merz, C. N., & Shufelt, C. (2019). Cardiovascular implications of gender-affirming hormone treatment in the transgender population. Maturitas, 129, 45–49. [DOI:10.1016/j.maturitas.2019.08.010]
Eilertsen, A. L., Dahm, A. E., Høibraaten, E., Lofthus, C. M., Mowinckel, M., & Sandset, P. M. (2019). Relationship between sex hormone binding globulin and blood coagulation in women on postmenopausal hormone treatment. Blood Coagulation & Fibrinolysis, 30(1), 17–23. [DOI:10.1097/mbc.0000000000000784]
Eisenfeld, A. J., & Aten, R. F. (1979). Estrogen receptor in the mammalian liver. In Briggs, M. H., & Corbin, A. (Eds.). Advances in Steroid Biochemistry and Pharmacology, 7, 91–117. London: Academic Press. [Google Scholar] [PubMed]
Eisenfeld, A. J., & Aten, R. F. (1987). Estrogen receptors and androgen receptors in the mammalian liver. Journal of Steroid Biochemistry, 27(4–6), 1109–1118. [DOI:10.1016/0022-4731(87)90197-x]
Fabian, C. J., & Kimler, B. F. (2005). Selective Estrogen-Receptor Modulators for Primary Prevention of Breast Cancer. Journal of Clinical Oncology, 23(8), 1644–1655. [DOI:10.1200/jco.2005.11.005]
Fåhraeus, L., & Larsson-Cohn, U. (1982). Oestrogens, gonadotrophins and SHBG during oral and cutaneous administration of oestradiol-17β to menopausal women. Acta Endocrinologica, 101(4), 592–596. [DOI:10.1530/acta.0.1010592]
Feldman, J. L., & Goldberg, J. (2006). Transgender Primary Medical Care: Suggested Guidelines for Clinicians in British Columbia. crhc/csac/Transcend Transgender Support & Education Society/Vancouver Coastal Health. [Google Scholar] [PDF]
Franchini, M., & Mannucci, P. M. (2015). Classic thrombophilic gene variants. Thrombosis and Haemostasis, 114(11), 885–889. [DOI:10.1160/th15-02-0141]
Fruzzetti, F., & Cagnacci, A. (2018). Venous thrombosis and hormonal contraception: what’s new with estradiol-based hormonal contraceptives? Open Access Journal of Contraception, 9, 75–79. [DOI:10.2147/oajc.s179673]
Gerstman, B. B., Piper, J. M., Tomita, D. K., Ferguson, W. J., Stadel, B. V., & Lundin, F. E. (1991). Oral Contraceptive Estrogen Dose and the Risk of Deep Venous Thromboembolic Disease. American Journal of Epidemiology, 133(1), 32–37. [DOI:10.1093/oxfordjournals.aje.a115799]
Getahun, D., Nash, R., Flanders, W. D., Baird, T. C., Becerra-Culqui, T. A., Cromwell, L., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Silverberg, M. J., Safer, J., Slovis, J., Tangpricha, V., & Goodman, M. (2018). Cross-sex Hormones and Acute Cardiovascular Events in Transgender Persons. Annals of Internal Medicine, 169(4), 205. [DOI:10.7326/m17-2785]
Gibney, J., Johannsson, G., Leung, K., & Ho, K. K. (2005). Comparison of the Metabolic Effects of Raloxifene and Oral Estrogen in Postmenopausal and Growth Hormone-Deficient Women. The Journal of Clinical Endocrinology & Metabolism, 90(7), 3897–3903. [DOI:10.1210/jc.2005-0173]
Gilbert, D. C., Duong, T., Sydes, M., Bara, A., Clarke, N., Abel, P., James, N., Langley, R., Parmar, M., & (2018). Transdermal oestradiol as a method of androgen suppression for prostate cancer within the STAMPEDE trial platform. BJU International, 121(5), 680–683. [DOI:10.1111/bju.14153]
Glintborg, D., T’Sjoen, G., Ravn, P., & Andersen, M. S. (2021). MANAGEMENT OF ENDOCRINE DISEASE: Optimal feminizing hormone treatment in transgender people. European Journal of Endocrinology, 185(2), R49–R63. [DOI:10.1530/eje-21-0059]
Goldstein, Z., Khan, M., Reisman, T., & Safer, J. D. (2019). Managing the risk of venous thromboembolism in transgender adults undergoing hormone therapy. Journal of Blood Medicine, 10, 209–216. [DOI:10.2147/jbm.s166780]
Gooren, L. J., & T’Sjoen, G. (2018). Endocrine treatment of aging transgender people. Reviews in Endocrine and Metabolic Disorders, 19(3), 253–262. [DOI:10.1007/s11154-018-9449-0]
Gourdy, P., Bachelot, A., Catteau-Jonard, S., Chabbert-Buffet, N., Christin-Maître, S., Conard, J., Fredenrich, A., Gompel, A., Lamiche-Lorenzini, F., Moreau, C., Plu-Bureau, G., Vambergue, A., Vergès, B., & Kerlan, V. (2012). Hormonal contraception in women at risk of vascular and metabolic disorders: Guidelines of the French Society of Endocrinology. Annales d’Endocrinologie, 73(5), 469–487. [DOI:10.1016/j.ando.2012.09.001]
Grady, D., Wenger, N. K., Herrington, D., Khan, S., Furberg, C., Hunninghake, D., Vittinghoff, E., Hulley, S., & (2000). Postmenopausal Hormone Therapy Increases Risk for Venous Thromboembolic Disease: The Heart and Estrogen/progestin Replacement Study. Annals of Internal Medicine, 132(9), 689–696. [DOI:10.7326/0003-4819-132-9-200005020-00002]
Grandi, G., Facchinetti, F., & Bitzer, J. (2017). Estradiol in hormonal contraception: real evolution or just same old wine in a new bottle? The European Journal of Contraception & Reproductive Health Care, 22(4), 245–246. [DOI:10.1080/13625187.2017.1372571]
Grandi, G., Barra, F., Ferrero, S., & Facchinetti, F. (2019). Estradiol in non-oral hormonal contraception: a “long and winding road”. Expert Review of Endocrinology & Metabolism, 14(3), 153–155. [DOI:10.1080/17446651.2019.1604217]
Grandi, G., Del Savio, M. C., Lopes da Silva-Filho, A., & Facchinetti, F. (2020). Estetrol (E4): the new estrogenic component of combined oral contraceptives. Expert Review of Clinical Pharmacology, 13(4), 327–330. [DOI:10.1080/17512433.2020.1750365]
Grandi, G., Facchinetti, F., & Bitzer, J. (2022). Confirmation of the safety of combined oral contraceptives containing oestradiol on the risk of venous thromboembolism. The European Journal of Contraception & Reproductive Health Care, 27(2), 83–84. [DOI:10.1080/13625187.2022.2029397]
Grossmann, M., Wierman, M. E., Angus, P., & Handelsman, D. J. (2018). Reproductive Endocrinology of Nonalcoholic Fatty Liver Disease. Endocrine Reviews, 40(2), 417–446. [DOI:10.1210/er.2018-00158]
Hammond, G. L. (2017). Sex Hormone-Binding Globulin and the Metabolic Syndrome. In Winters, S. J., & Huhtaniemi, I. T. (Eds.). Male Hypogonadism: Basic, Clinical and Therapeutic Principles, 2nd Edition (pp. 305–324). Cham: Springer. [DOI:10.1007/978-3-319-53298-1_15]
Hannaford, P. C., Iversen, L., Macfarlane, T. V., Elliott, A. M., Angus, V., & Lee, A. J. (2010). Mortality among contraceptive pill users: cohort evidence from Royal College of General Practitioners’ Oral Contraception Study. BMJ, 340, c927. [DOI:10.1136/bmj.c927]
Haupt, C., Henke, M., Kutschmar, A., Hauser, B., Baldinger, S., Saenz, S. R., & Schreiber, G. (2020). Antiandrogen or estradiol treatment or both during hormone therapy in transitioning transgender women. Cochrane Database of Systematic Reviews, 2020(11), CD013138. [DOI:10.1002/14651858.cd013138.pub2]
Haveles, C. S., Wang, M. M., Arjun, A., Zaila, K. E., & Lee, J. C. (2020). Effect of Cross-Sex Hormone Therapy on Venous Thromboembolism Risk in Male-to-Female Gender-Affirming Surgery. Annals of Plastic Surgery, 86(1), 109–114. [DOI:10.1097/sap.0000000000002300]
Hedlund, P. O., Johansson, R., Damber, J. E., Hagerman, I., Henriksson, P., Iversen, P., Klarskov, P., Mogensen, P., Rasmussen, F., & Varenhorst, E. (2011). Significance of pretreatment cardiovascular morbidity as a risk factor during treatment with parenteral oestrogen or combined androgen deprivation of 915 patients with metastasized prostate cancer: Evaluation of cardiovascular events in a randomized trial. Scandinavian Journal of Urology and Nephrology, 45(5), 346–353. [DOI:10.3109/00365599.2011.585820]
Heit, J. A., Silverstein, M. D., Mohr, D. N., Petterson, T. M., O’Fallon, W. M., & Melton, L. J. (2000). Risk Factors for Deep Vein Thrombosis and Pulmonary Embolism. Archives of Internal Medicine, 160(6), 809–815. [DOI:10.1001/archinte.160.6.809]
Heit, J. A., Spencer, F. A., & White, R. H. (2016). The epidemiology of venous thromboembolism. Journal of Thrombosis and Thrombolysis, 41(1), 3–14. [DOI:10.1007/s11239-015-1311-6]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society* Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658]
Hemelaar, M., van der Mooren, M. J., Rad, M., Kluft, C., & Kenemans, P. (2008). Effects of non-oral postmenopausal hormone therapy on markers of cardiovascular risk: a systematic review. Fertility and Sterility, 90(3), 642–672. [DOI:10.1016/j.fertnstert.2007.07.1298]
Hibbs, D. (2008). Hormone replacement and the treatment of the transgender patient: A critical literature review. de Chesnay, M., & Anderson, B. (Eds.). Caring for the Vulnerable: Perspectives in Nursing Theory, Practice, and Research, 2nd Edition (pp. 351–362). Sudbury, Massachusetts: Jones & Barlett. [Google Scholar] [Google Books]
Higdon, J., Drake, V. J., Delage, B., & Crozier, A. (2016). Flavonoids. Corvallis, Oregon: Micronutrient Information Center, Linus Pauling Institute, Oregon State University. [URL]
Høibraaten, E., Qvigstad, E., Arnesen, H., Larsen, S., Wickstrøm, E., & Sandset, P. M. (2000). Increased Risk of Recurrent Venous Thromboembolism during Hormone Replacement Therapy. Thrombosis and Haemostasis, 84(12), 961–967. [DOI:10.1055/s-0037-1614156]
Høibraaten, E., Qvigstad, E., Andersen, T. O., Mowinckel, M., & Sandset, P. M. (2001). The Effects of Hormone Replacement Therapy (HRT) on Hemostatic Variables in Women with Previous Venous Thromboembolism – Results from a Randomized, Double-Blind, Clinical Trial. Thrombosis and Haemostasis, 85(5), 775–781. [DOI:10.1055/s-0037-1615717]
Holmegard, H., Nordestgaard, B., Schnohr, P., Tybjærg‐Hansen, A., & Benn, M. (2014). Endogenous sex hormones and risk of venous thromboembolism in women and men. Journal of Thrombosis and Haemostasis, 12(3), 297–305. [DOI:10.1111/jth.12484]
Hontscharuk, R., Alba, B., Manno, C., Pine, E., Deutsch, M. B., Coon, D., & Schechter, L. (2021). Perioperative Transgender Hormone Management: Avoiding Venous Thromboembolism and Other Complications. Plastic & Reconstructive Surgery, 147(4), 1008–1017. [DOI:10.1097/prs.0000000000007786]
Hugon-Rodin, J., Alhenc-Gelas, M., Hemker, H. C., Brailly-Tabard, S., Guiochon-Mantel, A., Plu-Bureau, G., & Scarabin, P. (2016). Sex hormone-binding globulin and thrombin generation in women using hormonal contraception. Biomarkers, 22(1), 81–85. [DOI:10.1080/1354750x.2016.1204010]
Hulley, S. (1998). Randomized Trial of Estrogen Plus Progestin for Secondary Prevention of Coronary Heart Disease in Postmenopausal Women. JAMA, 280(7), 605–613. [DOI:10.1001/jama.280.7.605]
Iqbal, J., Ginsburg, O. M., Wijeratne, T. D., Howell, A., Evans, G., Sestak, I., & Narod, S. A. (2012). Endometrial cancer and venous thromboembolism in women under age 50 who take tamoxifen for prevention of breast cancer: A systematic review. Cancer Treatment Reviews, 38(4), 318–328. [DOI:10.1016/j.ctrv.2011.06.009]
Irwig, M. S. (2018). Cardiovascular health in transgender people. Reviews in Endocrine and Metabolic Disorders, 19(3), 243–251. [DOI:10.1007/s11154-018-9454-3]
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 204201881987116. [DOI:10.1177/2042018819871166]
Jasuja, R., Passam, F. H., Kennedy, D. R., Kim, S. H., van Hessem, L., Lin, L., Bowley, S. R., Joshi, S. S., Dilks, J. R., Furie, B., Furie, B. C., & Flaumenhaft, R. (2012). Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. Journal of Clinical Investigation, 122(6), 2104–2113. [DOI:10.1172/jci61228]
Jensen, E. V., Jacobson, H. I., Walf, A. A., & Frye, C. A. (2010). Estrogen action: A historic perspective on the implications of considering alternative approaches. Physiology & Behavior, 99(2), 151–162. [DOI:10.1016/j.physbeh.2009.08.013]
Kaemmle, L. M., Stadler, A., Janka, H., von Wolff, M., & Stute, P. (2022). The impact of micronized progesterone on cardiovascular events – a systematic review. Climacteric, 25(4), 327–336. [DOI:10.1080/13697137.2021.2022644]
Kasum, M., Danolić, D., Orešković, S., Ježek, D., Beketić-Orešković, L., & Pekez, M. (2014). Thrombosis following ovarian hyperstimulation syndrome. Gynecological Endocrinology, 30(11), 764–768. [DOI:10.3109/09513590.2014.927858]
Keenan, L., Kerr, T., Duane, M., & Van Gundy, K. (2018). Systematic Review of Hormonal Contraception and Risk of Venous Thrombosis. The Linacre Quarterly, 85(4), 470–477. [DOI:10.1177/0024363918816683]
Kerlan, V., Nahoul, K., Martelot, M., & Bercovici, J. (1994). Longitudinal study of maternal plasma bioavailable testosterone and androstanediol glucuronide levels during pregnancy. Clinical Endocrinology, 40(2), 263–267. [DOI:10.1111/j.1365-2265.1994.tb02478.x]
Khan, J., Schmidt, R. L., Spittal, M. J., Goldstein, Z., Smock, K. J., & Greene, D. N. (2019). Venous Thrombotic Risk in Transgender Women Undergoing Estrogen Therapy: A Systematic Review and Metaanalysis. Clinical Chemistry, 65(1), 57–66. [DOI:10.1373/clinchem.2018.288316]
Kitamura, T. (2001). Necessity of re‐evaluation of estramustine phosphate sodium (EMP) as a treatment option for first‐line monotherapy in advanced prostate cancer. International Journal of Urology, 8(2), 33–36. [DOI:10.1046/j.1442-2042.2001.00254.x]
Klil-Drori, A. J., Yin, H., Tagalakis, V., Aprikian, A., & Azoulay, L. (2016). Androgen Deprivation Therapy for Prostate Cancer and the Risk of Venous Thromboembolism. European Urology, 70(1), 56–61. [DOI:10.1016/j.eururo.2015.06.022]
Kohli, M., & McClellan, J. (2001). Parenteral Estrogen Therapy in Advanced Prostate Cancer: Retrospective Analysis of Intra-Muscular Estradiol Valerate in “Hormone Refractory” Prostate Disease. In Grunberg, S. M. (Ed.). Proceedings of the American Society of Clinical Oncology [Proc Am Soc Clin Oncol / Proceedings of ASCO], 20 [Thirty-Seventh Annual Meeting of the American Society of Clinical Oncology, May 12–15, 2001, San Francisco, California], 164b–164b (abstract no. 2407). Baltimore: Lippincott Williams & Wilkins. [ISSN:1081-0641] [ISBN-10:0-9664495-3-3] [Google Scholar] [WorldCat 1] [WorldCat 2] [WorldCat 3] [PDF]
Kohli, M., Alikhan, M. A., Spencer, H. J., & Carter, G. (2004). Phase I trial of intramuscular estradiol valerate (I/M-E) in hormone refractory prostate cancer. Journal of Clinical Oncology, 22(14 Suppl) [40th Annual Meeting of the American Society of Clinical Oncology: June 5–8, 2004, Ernest N. Morial Convention Center, New Orleans, Louisiana, Annual Meeting Proceedings], 436–436 (abstract no. 4726). [DOI:10.1200/jco.2004.22.90140.4726] [Google Books] [PDF]
Kohli, M. (2006). Phase II study of transdermal estradiol in androgen-independent prostate carcinoma. Cancer, 106(1), 234–235. [DOI:10.1002/cncr.21528]
Konkle, B. A., & Sood, S. L. (2019). Thrombotic Risk of Contraceptives and Other Hormonal Therapies. In Kitchens, C. S., Kessler, C. M., Konkle, B. A., Streiff, M. B., & Garcia, D. A. (Eds.). Consultative Hemostasis and Thrombosis, 4th Edition (pp. 637–650). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-46202-0.00031-5]
Kotamarti, V. S., Greige, N., Heiman, A. J., Patel, A., & Ricci, J. A. (2021). Risk for Venous Thromboembolism in Transgender Patients Undergoing Cross-Sex Hormone Treatment: A Systematic Review. The Journal of Sexual Medicine, 18(7), 1280–1291. [DOI:10.1016/j.jsxm.2021.04.006]
Kozato, A., Fox, G. W., Yong, P. C., Shin, S. J., Avanessian, B. K., Ting, J., Ling, Y., Karim, S., Safer, J. D., & Pang, J. H. (2021). No Venous Thromboembolism Increase Among Transgender Female Patients Remaining on Estrogen for Gender-Affirming Surgery. The Journal of Clinical Endocrinology & Metabolism, 106(4), 1586–1590. [DOI:10.1210/clinem/dgaa966]
Kronawitter, D., Gooren, L. J., Zollver, H., Oppelt, P. G., Beckmann, M. W., Dittrich, R., & Mueller, A. (2009). Effects of transdermal testosterone or oral dydrogesterone on hypoactive sexual desire disorder in transsexual women: results of a pilot study. European Journal of Endocrinology, 161(2), 363–368. [DOI:10.1530/eje-09-0265] [Table]
Kuhl, H. (1990). Ovulationshemmer: Die Bedeutung der Östrogendosis. [Ovulation Preventives: The Significance of the Estrogen Dose. / Ovulation Inhibitors: The Significance of Estrogen Dose.] Geburtshilfe und Frauenheilkunde, 50(12), 910–922. [Google Scholar 1] [Google Scholar 2] [PubMed] [DOI:10.1055/s-2008-1026392] [Translation]
Kuhl, H. (1996). Effects of progestogens on haemostasis. Maturitas, 24(1–2), 1–19. [DOI:10.1016/0378-5122(96)00994-2]
Kuhl, H. (1997). Metabolische Effekte der Östrogene und Gestagene. [Metabolic Effects of Estrogens and Progestogens.] Der Gynäkologe, 30(4), 357–369. [DOI:10.1007/pl00003042]
Kuhl, H. (1998). Adverse effects of estrogen treatment: natural vs. synthetic estrogens. In Lippert, T. H., Mueck, A. O., & Ginsburg, J. (Eds.). Sex Steroids and the Cardiovascular System: The Proceedings of the 1st Interdisciplinary Workshop, Tuebingen, Germany, October 1996. Parthenon Publishing Group, New York, London (pp. 201–210). London/New York: Parthenon. [Google Scholar] [Google Books] [PDF]
Kuhl, H. (1999). Hormonal contraception. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 363–407). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_18]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H., & Stevenson, J. (2006). The effect of medroxyprogesterone acetate on estrogen-dependent risks and benefits – an attempt to interpret the Women’s Health Initiative results. Gynecological Endocrinology, 22(6), 303–317. [DOI:10.1080/09513590600717368]
Langley, R. E., Cafferty, F. H., Alhasso, A. A., Rosen, S. D., Sundaram, S. K., Freeman, S. C., Pollock, P., Jinks, R. C., Godsland, I. F., Kockelbergh, R., Clarke, N. W., Kynaston, H. G., Parmar, M. K., & Abel, P. D. (2013). Cardiovascular outcomes in patients with locally advanced and metastatic prostate cancer treated with luteinising-hormone-releasing-hormone agonists or transdermal oestrogen: the randomised, phase 2 MRC PATCH trial (PR09). The Lancet Oncology, 14(4), 306–316. [DOI:10.1016/s1470-2045(13)70025-1]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., Paez, E., Robinson, A., McFarlane, J., Deighan, J. V., Marshall, J., Forcat, S., Weiss, M., Kockelbergh, R., Alhasso, A., Kynaston, H., & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/s0140-6736(21)00100-8]
Lax, E. (1987). Mechanisms of physiological and pharmacological sex hormone action on the mammalian liver. Journal of Steroid Biochemistry, 27(4–6), 1119–1128. [DOI:10.1016/0022-4731(87)90198-1]
Lidegaard, Ø. (2014). Hormonal contraception, thrombosis and age. Expert Opinion on Drug Safety, 13(10), 1353–1360. [DOI:10.1517/14740338.2014.950654]
Lijfering, W. M., Rosendaal, F. R., & Cannegieter, S. C. (2010). Risk factors for venous thrombosis - current understanding from an epidemiological point of view. British Journal of Haematology, 149(6), 824–833. [DOI:10.1111/j.1365-2141.2010.08206.x]
Lim, H. Y., Leemaqz, S. Y., Torkamani, N., Grossmann, M., Zajac, J. D., Nandurkar, H., Ho, P., & Cheung, A. S. (2020). Global Coagulation Assays in Transgender Women on Oral and Transdermal Estradiol Therapy. The Journal of Clinical Endocrinology & Metabolism, 105(7), e2369–e2377. [DOI:10.1210/clinem/dgaa262]
Luria, M. H. (1989). Estrogen and coronary arterial disease in men. International Journal of Cardiology, 25(2), 159–166. [DOI:10.1016/0167-5273(89)90102-2]
Ma, Y., Zeng, M., Sun, R., & Hu, M. (2015). Disposition of Flavonoids Impacts their Efficacy and Safety. Current Drug Metabolism, 15(9), 841–864. [DOI:10.2174/1389200216666150206123719]
Machin, N., & Ragni, M. V. (2020). Hormones and thrombosis: risk across the reproductive years and beyond. Translational Research, 225, 9–19. [DOI:10.1016/j.trsl.2020.06.011]
Mammen, E. F. (1992). Coagulation Abnormalities in Liver Disease. Hematology/Oncology Clinics of North America, 6(6), 1247–1257. [DOI:10.1016/s0889-8588(18)30273-9]
Mantha, S., Karp, R., Raghavan, V., Terrin, N., Bauer, K. A., & Zwicker, J. I. (2012). Assessing the risk of venous thromboembolic events in women taking progestin-only contraception: a meta-analysis. BMJ, 345, e4944. [DOI:10.1136/bmj.e4944]
Martinelli, I., Passamonti, S. M., & Bucciarelli, P. (2014). Thrombophilic states. In Biller, J., & Ferro, J. M. (Eds.). Neurologic Aspects of Systemic Disease Part II (Handbook of Clinical Neurology, Volume 120) (pp. 1061–1071). Philadelphia: Elsevier. [DOI:10.1016/b978-0-7020-4087-0.00071-1]
Martinez-Zapata, M. J., Vernooij, R. W., Uriona Tuma, S. M., Stein, A. T., Moreno, R. M., Vargas, E., Capellà, D., & Bonfill Cosp, X. (2016). Phlebotonics for venous insufficiency. Cochrane Database of Systematic Reviews, 2016(4), CD003229. [DOI:10.1002/14651858.cd003229.pub3]
Matharu, G. S., Kunutsor, S. K., Judge, A., Blom, A. W., & Whitehouse, M. R. (2020). Clinical Effectiveness and Safety of Aspirin for Venous Thromboembolism Prophylaxis After Total Hip and Knee Replacement. JAMA Internal Medicine, 180(3), 376–384. [DOI:10.1001/jamainternmed.2019.6108]
McLintock, C. (2014). Thromboembolism in pregnancy: Challenges and controversies in the prevention of pregnancy-associated venous thromboembolism and management of anticoagulation in women with mechanical prosthetic heart valves. Best Practice & Research Clinical Obstetrics & Gynaecology, 28(4), 519–536. [DOI:10.1016/j.bpobgyn.2014.03.001]
Mekaj, A., Mekaj, Y., & Daci, F. (2015). New insights into the mechanisms of action of aspirin and its use in the prevention and treatment of arterial and venous thromboembolism. Therapeutics and Clinical Risk Management, 11, 1449–1456. [DOI:10.2147/tcrm.s92222]
Mellinger, G. T., Bailar, J. C., Arduino, L. J., & the Veterans Administration Cooperative Urological Research Group. (1967). Treatment and survival of patients with cancer of the prostate. The Veterans Administration Co-operative Urological Research Group. Surgery Gynecology & Obstetrics, 124(5), 1011–1017. [Google Scholar] [PubMed]
Meng, Y., Jiang, H., Chen, A., Lu, F., Yang, H., Shen, M. Z., Sun, D., Shao, Q., & Fotherby, K. (1990). Hemostatic changes in women using a monthly injectable contraceptive for one year. Contraception, 42(4), 455–466. [DOI:10.1016/0010-7824(90)90052-w]
Mikkola, A., Ruutu, M., Aro, J., Rannikko, S., & Salo, J. (1999). The role of parenteral polyestradiol phosphate in the treatment of advanced prostatic cancer on the threshold of the new millennium. Annales Chirurgiae et Gynaecologiae, 88(1), 18–21. [Google Scholar] [PubMed] [PDF]
Min, L. L., & Hopkins, R. (2021). Endocrinological Care for Patients Undergoing Gender Affirmation (Including Risk of Thromboembolic Events). In Nikolavsky, D., & Blakely, S. A. (Eds.). Urological Care for the Transgender Patient: A Comprehensive Guide (pp. 23–36). Cham: Springer. [DOI:10.1007/978-3-030-18533-6_3]
Mohammed, K., Abu Dabrh, A. M., Benkhadra, K., Al Nofal, A., Carranza Leon, B. G., Prokop, L. J., Montori, V. M., Faubion, S. S., & Murad, M. H. (2015). Oral vs Transdermal Estrogen Therapy and Vascular Events: A Systematic Review and Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism, 100(11), 4012–4020. [DOI:10.1210/jc.2015-2237]
Montagnana, M., Favaloro, E. J., Franchini, M., Guidi, G. C., & Lippi, G. (2009). The role of ethnicity, age and gender in venous thromboembolism. Journal of Thrombosis and Thrombolysis, 29(4), 489–496. [DOI:10.1007/s11239-009-0365-8]
Moores, L., Bilello, K. L., & Murin, S. (2004). Sex and gender issues and venous thromboembolism. Clinics in Chest Medicine, 25(2), 281–297. [DOI:10.1016/j.ccm.2004.01.013]
Morimont, L., Dogné, J., & Douxfils, J. (2020). Letter to the Editors-in-Chief in response to the article of Abou-Ismail, et al. entitled “Estrogen and thrombosis: A bench to bedside review” (Thrombosis Research 192 (2020) 40–51). Thrombosis Research, 193, 221–223. [DOI:10.1016/j.thromres.2020.08.006]
Morimont, L., Haguet, H., Dogné, J., Gaspard, U., & Douxfils, J. (2021). Combined Oral Contraceptives and Venous Thromboembolism: Review and Perspective to Mitigate the Risk. Frontiers in Endocrinology, 12, 769187. [DOI:10.3389/fendo.2021.769187]
Morling, J. R., Broderick, C., Yeoh, S. E., & Kolbach, D. N. (2018). Rutosides for treatment of post-thrombotic syndrome. Cochrane Database of Systematic Reviews, 2018(11), CD005625. [DOI:10.1002/14651858.cd005625.pub4]
Mueller, A., Dittrich, R., Binder, H., Kuehnel, W., Maltaris, T., Hoffmann, I., & Beckmann, M. W. (2005). High dose estrogen treatment increases bone mineral density in male-to-female transsexuals receiving gonadotropin-releasing hormone agonist in the absence of testosterone. European Journal of Endocrinology, 153(1), 107–113. [DOI:10.1530/eje.1.01943]
Mueller, A., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Dittrich, R. (2006). Effects on the Male Endocrine System of Long-term Treatment with Gonadotropin-releasing Hormone Agonists and Estrogens in Male-to-Female Transsexuals. Hormone and Metabolic Research, 38(3), 183–187. [DOI:10.1055/s-2006-925198]
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body composition and bone mineral density in male-to-female transsexuals during cross-sex hormone therapy using gonadotrophin-releasing hormone agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074] [Table]
Nachtigall, L. E., Raju, U., Banerjee, S., Wan, L., & Levitz, M. (2000). Serum Estradiol-Binding Profiles in Postmenopausal Women Undergoing Three Common Estrogen Replacement Therapies. Menopause, 7(4), 243–250. [DOI:10.1097/00042192-200007040-00006]
Nelson, M. D., Szczepaniak, L. S., Wei, J., Szczepaniak, E., Sánchez, F. J., Vilain, E., Stern, J. H., Bergman, R. N., Bairey Merz, C. N., & Clegg, D. J. (2016). Transwomen and the Metabolic Syndrome: Is Orchiectomy Protective? Transgender Health, 1(1), 165–171. [DOI:10.1089/trgh.2016.0016]
Nolan, B. J., & Cheung, A. S. (2020). Estradiol Therapy in the Perioperative Period: Implications for Transgender People Undergoing Feminizing Hormone Therapy. The Yale Journal of Biology and Medicine, 93(4), 539–548. [PubMed] [PubMed Central]
Nolan, B. J., & Cheung, A. S. (2021). Relationship Between Serum Estradiol Concentrations and Clinical Outcomes in Transgender Individuals Undergoing Feminizing Hormone Therapy: A Narrative Review. Transgender Health, 6(3), 125–131. [DOI:10.1089/trgh.2020.0077]
Nolan, I. T., Haley, C., Morrison, S. D., Pannucci, C. J., & Satterwhite, T. (2021). Estrogen Continuation and Venous Thromboembolism in Penile Inversion Vaginoplasty. The Journal of Sexual Medicine, 18(1), 193–200. [DOI:10.1016/j.jsxm.2020.10.018]
Ockrim, J., & Abel, P. D. (2009). Androgen deprivation therapy for prostate cancer – the potential of parenteral estrogen. Central European Journal of Urology, 62, 132–140. [DOI:10.5173/ceju.2009.03.art1]
Odlind, V., Milsom, I., Persson, I., & Victor, A. (2002). Can changes in sex hormone binding globulin predict the risk of venous thromboembolism with combined oral contraceptive pills?: A discussion based on recent recommendations from the European agency for evaluation of medicinal products regarding third generation oral contraceptive pills. Acta Obstetricia et Gynecologica Scandinavica, 81(6), 482–490. [DOI:10.1080/j.1600-0412.2002.810603.x] [URL]
Oger, E., & (2000). Incidence of Venous Thromboembolism: A Community-based Study in Western France. Thrombosis and Haemostasis, 83(5), 657–660. [DOI:10.1055/s-0037-1613887]
Ohlander, S. J., Varghese, B., & Pastuszak, A. W. (2018). Erythrocytosis Following Testosterone Therapy. Sexual Medicine Reviews, 6(1), 77–85. [DOI:10.1016/j.sxmr.2017.04.001]
Olié, V., Canonico, M., & Scarabin, P. (2010). Risk of venous thrombosis with oral versus transdermal estrogen therapy among postmenopausal women. Current Opinion in Hematology, 17(5), 457–463. [DOI:10.1097/moh.0b013e32833c07bc]
Olié, V., Plu-Bureau, G., Conard, J., Horellou, M., Canonico, M., & Scarabin, P. (2011). Hormone therapy and recurrence of venous thromboembolism among postmenopausal women. Menopause, 18(5), 488–493. [DOI:10.1097/gme.0b013e3181f9f7c3]
Oliver-Williams, C., Glisic, M., Shahzad, S., Brown, E., Pellegrino Baena, C., Chadni, M., Chowdhury, R., Franco, O. H., & Muka, T. (2018). The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Human Reproduction Update, 25(2), 257–271. [DOI:10.1093/humupd/dmy039]
Olov Hedlund, P., Damber, J., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Johansson, R., Klarskov, P., Lundbeck, F., Rasmussen, F., Varenhorst, E., Viitanen, J., Olov Hedlund, P., Damber, J., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Johansson, R., Klarskov, P., Lundbeck, F., Rasmussen, F., Varenhorst, E., Viitanen, J., & The SPCG-5 Study Group. (2008). Parenteral estrogen versus combined androgen deprivation in the treatment of metastatic prostatic cancer: Part 2. Final evaluation of the Scandinavian Prostatic Cancer Group (SPCG) Study No. 5. Scandinavian Journal of Urology and Nephrology, 42(3), 220–229. [DOI:10.1080/00365590801943274]
Park, W. (2002). Selective estrogen receptor modulators (SERMS) and their roles in breast cancer prevention. Trends in Molecular Medicine, 8(2), 82–88. [DOI:10.1016/s1471-4914(02)02282-7]
Patel, K. T., Adeel, S., Rodrigues Miragaya, J., & Tangpricha, V. (2022). Progestogen Use in Gender-Affirming Hormone Therapy: A Systematic Review. Endocrine Practice, 28(12), 1244–1252. [DOI:10.1016/j.eprac.2022.08.012]
Peck-Radosavljevic, M. (2007). Review article: coagulation disorders in chronic liver disease. Alimentary Pharmacology & Therapeutics, 26, 21–28. [DOI:10.1111/j.1365-2036.2007.03509.x]
Pfeifer, S., Butts, S., Dumesic, D., Fossum, G., Gracia, C., La Barbera, A., Mersereau, J., Odem, R., Penzias, A., Pisarska, M., Rebar, R., Reindollar, R., Rosen, M., Sandlow, J., Sokol, R., Vernon, M., & Widra, E. (2017). Combined hormonal contraception and the risk of venous thromboembolism: a guideline. Fertility and Sterility, 107(1), 43–51. [DOI:10.1016/j.fertnstert.2016.09.027]
Phillips, I., Shah, S. I., Duong, T., Abel, P., & Langley, R. E. (2014). Androgen Deprivation Therapy and the Re-emergence of Parenteral Estrogen in Prostate Cancer. Oncology & Hematology Review, 10(1), 42–47. [PubMed Central] [DOI:10.17925/ohr.2014.10.1.42]
Plu-Bureau, G., Maitrot-Mantelet, L., Hugon-Rodin, J., & Canonico, M. (2013). Hormonal contraceptives and venous thromboembolism: An epidemiological update. Best Practice & Research Clinical Endocrinology & Metabolism, 27(1), 25–34. [DOI:10.1016/j.beem.2012.11.002]
Pomp, E. R., Rosendaal, F. R., & Doggen, C. J. (2008). Smoking increases the risk of venous thrombosis and acts synergistically with oral contraceptive use. American Journal of Hematology, 83(2), 97–102. [DOI:10.1002/ajh.21059]
Pond, S. M., & Tozer, T. N. (1984). First-Pass Elimination. Clinical Pharmacokinetics, 9(1), 1–25. [DOI:10.2165/00003088-198409010-00001]
Prentice, R. L., & Anderson, G. L. (2008). The Women’s Health Initiative: Lessons Learned. Annual Review of Public Health, 29(1), 131–150. [DOI:10.1146/annurev.publhealth.29.020907.090947]
Prentice, R. (2014). Postmenopausal Hormone Therapy and the Risks of Coronary Heart Disease, Breast Cancer, and Stroke. Seminars in Reproductive Medicine, 32(6), 419–425. [DOI:10.1055/s-0034-1384624]
Pyra, M., Casimiro, I., Rusie, L., Ross, N., Blum, C., Keglovitz Baker, K., Baker, A., & Schneider, J. (2020). An Observational Study of Hypertension and Thromboembolism Among Transgender Patients Using Gender-Affirming Hormone Therapy. Transgender Health, 5(1), 1–9. [DOI:10.1089/trgh.2019.0061]
Quinton, R., & Swee, D. S. (2019). Hormone replacement therapy: transgender studies show safety of estradiol. BMJ, 364, l600. [DOI:10.1136/bmj.l600]
Rabe, T., Luxembourg, B., Ludwig, M., Dinger, J. C., Bauersachs, R., Rott, H., Mueck, A. O., & Albring, C. (2011). Contraception and Thrombophilia - A statement from the German Society of Gynecological Endocrinology and Reproductive Medicine (DGGEF e. V.) and the Professional Association of the German Gynaecologists (BVF e. V.). Journal für Reproduktionsmedizin und Endokrinologie-Journal of Reproductive Medicine and Endocrinology, 8(Special Issue 1), 178–218. [URL]
Raps, M., Helmerhorst, F., Fleischer, K., Thomassen, S., Rosendaal, F., Rosing, J., Ballieux, B., & Van Vliet, H. (2012). Sex hormone‐binding globulin as a marker for the thrombotic risk of hormonal contraceptives. Journal of Thrombosis and Haemostasis, 10(6), 992–997. [DOI:10.1111/j.1538-7836.2012.04720.x]
Ravery, V., Fizazi, K., Oudard, S., Drouet, L., Eymard, J., Culine, S., Gravis, G., Hennequin, C., & Zerbib, M. (2011). The use of estramustine phosphate in the modern management of advanced prostate cancer. BJU International, 108(11), 1782–1786. [DOI:10.1111/j.1464-410x.2011.10201.x]
Reda, S., Morimont, L., Douxfils, J., & Rühl, H. (2020). Can We Measure the Individual Prothrombotic or Prohemorrhagic Tendency by Global Coagulation Tests? Hämostaseologie, 40(3), 364–378. [DOI:10.1055/a-1153-5824]
Renoux, C., Dell’Aniello, S., & Suissa, S. (2010). Hormone replacement therapy and the risk of venous thromboembolism: a population-based study. Journal of Thrombosis and Haemostasis, 8(5), 979–986. [DOI:10.1111/j.1538-7836.2010.03839.x]
Renoux, C., Dell’Aniello, S., Garbe, E., & Suissa, S. (2010). Transdermal and oral hormone replacement therapy and the risk of stroke: a nested case-control study. BMJ, 340, c2519. [DOI:10.1136/bmj.c2519]
Roach, R., Lijfering, W., Helmerhorst, F., Cannegieter, S., Rosendaal, F., & van Hylckama Vlieg, A. (2013). The risk of venous thrombosis in women over 50 years old using oral contraception or postmenopausal hormone therapy. Journal of Thrombosis and Haemostasis, 11(1), 124–131. [DOI:10.1111/jth.12060]
Roach, R. E., Lijfering, W. M., Rosendaal, F. R., Cannegieter, S. C., & le Cessie, S. (2014). Sex Difference in Risk of Second but Not of First Venous Thrombosis. Circulation, 129(1), 51–56. [DOI:10.1161/circulationaha.113.004768]
Rooijen, M., Silveira, A., Hamsten, A., & Bremme, K. (2004). Sex hormone–binding globulin—A surrogate marker for the prothrombotic effects of combined oral contraceptives. American Journal of Obstetrics and Gynecology, 190(2), 332–337. [DOI:10.1016/s0002-9378(03)00950-5]
Ropponen, A., Aittomäki, K., Vihma, V., Tikkanen, M. J., & Ylikorkala, O. (2005). Effects of Oral and Transdermal Estradiol Administration on Levels of Sex Hormone-Binding Globulin in Postmenopausal Women with and without a History of Intrahepatic Cholestasis of Pregnancy. The Journal of Clinical Endocrinology & Metabolism, 90(6), 3431–3434. [DOI:10.1210/jc.2005-0352]
Rosendaal, F. R. (2005). Venous thrombosis: the role of genes, environment, and behavior. Hematology American Society of Hematology Education Program, 2005(1), 1–12. [Google Scholar] [DOI:10.1182/asheducation-2005.1.1] [URL]
Rosendaal, F. R. (2016). Causes of venous thrombosis. Thrombosis Journal, 14(Suppl 1) [State of the Art 2016: Research and Review from the 9th Congress of the Asian-Pacific Society on Thrombosis and Hemostasis], 24. [DOI:10.1186/s12959-016-0108-y]
Rott, H. (2019). Birth Control Pills and Thrombotic Risks: Differences of Contraception Methods with and without Estrogen. Hämostaseologie, 39(1), 42–48. [DOI:10.1055/s-0039-1677806]
Rova, K., Passmark, H., & Lindqvist, P. G. (2012). Venous thromboembolism in relation to in vitro fertilization: an approach to determining the incidence and increase in risk in successful cycles. Fertility and Sterility, 97(1), 95–100. [DOI:10.1016/j.fertnstert.2011.10.038]
Rovinski, D., Ramos, R. B., Fighera, T. M., Casanova, G. K., & Spritzer, P. M. (2018). Risk of venous thromboembolism events in postmenopausal women using oral versus non-oral hormone therapy: A systematic review and meta-analysis. Thrombosis Research, 168, 83–95. [DOI:10.1016/j.thromres.2018.06.014]
Ruiz Garcia, V., López-Briz, E., Carbonell Sanchis, R., Gonzalvez Perales, J. L., & Bort-Martí, S. (2013). Megestrol acetate for treatment of anorexia-cachexia syndrome. Cochrane Database of Systematic Reviews, 2019(3), CD004310. [DOI:10.1002/14651858.cd004310.pub3]
Russell, N., Cheung, A., & Grossmann, M. (2017). Estradiol for the mitigation of adverse effects of androgen deprivation therapy. Endocrine-Related Cancer, 24(8), R297–R313. [DOI:10.1530/erc-17-0153]
Sahlin, L., & Schoultz, B. V. (1999). Liver Inclusive Protein, Lipid and Carbohydrate Metabolism. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 163–178). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_8]
Scarabin, P. (2018). Progestogens and venous thromboembolism in menopausal women: an updated oral versus transdermal estrogen meta-analysis. Climacteric, 21(4), 341–345. [DOI:10.1080/13697137.2018.1446931]
Scarabin, P., Canonico, M., Plu-Bureau, G., & Oger, E. (2020). Menopause and hormone therapy in the 21st century: why promote transdermal estradiol and progesterone? Heart, 106(16), 1278–1278. [DOI:10.1136/heartjnl-2020-316907]
Scheres, L. J., van Hylckama Vlieg, A., Ballieux, B. E., Fauser, B. C., Rosendaal, F. R., Middeldorp, S., & Cannegieter, S. C. (2019). Endogenous sex hormones and risk of venous thromboembolism in young women. Journal of Thrombosis and Haemostasis, 17(8), 1297–1304. [DOI:10.1111/jth.14474]
Scheres, L. J., Selier, N. L., Nota, N. M., van Diemen, J. J., Cannegieter, S. C., & den Heijer, M. (2021). Effect of gender‐affirming hormone use on coagulation profiles in transmen and transwomen. Journal of Thrombosis and Haemostasis, 19(4), 1029–1037. [DOI:10.1111/jth.15256]
Schindler, A. E. (2003). Differential effects of progestins on hemostasis. Maturitas, 46, 31–37. [DOI:10.1016/j.maturitas.2003.09.016]
Schock, H., Zeleniuch-Jacquotte, A., Lundin, E., Grankvist, K., Lakso, H., Idahl, A., Lehtinen, M., Surcel, H., & Fortner, R. T. (2016). Hormone concentrations throughout uncomplicated pregnancies: a longitudinal study. BMC Pregnancy and Childbirth, 16(1), 146. [DOI:10.1186/s12884-016-0937-5]
Schröder, F. H., & Radlmaier, A. (2002). Steroidal Antiandrogens. In Jordan, C. V., & Furr, B. J. A. (Eds.). Hormone Therapy in Breast and Prostate Cancer (pp. 325–346). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-59259-152-7_15]
Sciarra, A., Gentile, V., Cattarino, S., Gentilucci, A., Alfarone, A., D’Eramo, G., & Salciccia, S. (2014). Oral ethinylestradiol in castration-resistant prostate cancer: A 10-year experience. International Journal of Urology, 22(1), 98–103. [DOI:10.1111/iju.12613]
Seaman, H. E., Langley, S. E., Farmer, R. D., & de Vries, C. S. (2007). Venous thromboembolism and cyproterone acetate in men with prostate cancer: a study using the General Practice Research Database. BJU International, 99(6), 1398–1403. [DOI:10.1111/j.1464-410x.2007.06859.x]
Shatzel, J. J., Connelly, K. J., & DeLoughery, T. G. (2017). Thrombotic issues in transgender medicine: A review. American Journal of Hematology, 92(2), 204–208. [DOI:10.1002/ajh.24593]
Shifren, J. L., Rifai, N., Desindes, S., McIlwain, M., Doros, G., & Mazer, N. A. (2008). A Comparison of the Short-Term Effects of Oral Conjugated Equine Estrogens Versus Transdermal Estradiol on C-Reactive Protein, Other Serum Markers of Inflammation, and Other Hepatic Proteins in Naturally Menopausal Women. The Journal of Clinical Endocrinology & Metabolism, 93(5), 1702–1710. [DOI:10.1210/jc.2007-2193]
Simon, T., De Jonage-Canonico, M. B., Oger, E., Wahl, D., Conard, J., Meyer, G., Emmerich, J., Barrellier, M., Guiraud, A., & Scarabin, P. (2006). Indicators of lifetime endogenous estrogen exposure and risk of venous thromboembolism. Journal of Thrombosis and Haemostasis, 4(1), 71–76. [DOI:10.1111/j.1538-7836.2005.01693.x]
Singla, N., Ghandour, R. A., & Raj, G. V. (2019). Investigational luteinizing hormone releasing hormone (LHRH) agonists and other hormonal agents in early stage clinical trials for prostate cancer. Expert Opinion on Investigational Drugs, 28(3), 249–259. [DOI:10.1080/13543784.2019.1570130]
Sitruk-Ware, R., & Nath, A. (2011). Metabolic effects of contraceptive steroids. Reviews in Endocrine and Metabolic Disorders, 12(2), 63–75. [DOI:10.1007/s11154-011-9182-4]
Sitruk-Ware, R., & Nath, A. (2013). Characteristics and metabolic effects of estrogen and progestins contained in oral contraceptive pills. Best Practice & Research Clinical Endocrinology & Metabolism, 27(1), 13–24. [DOI:10.1016/j.beem.2012.09.004]
Skouby, S. O., & Sidelmann, J. J. (2018). Impact of progestogens on hemostasis. Hormone Molecular Biology and Clinical Investigation, 37(2), 20180041. [DOI:10.1515/hmbci-2018-0041]
Smith, K., Galazi, M., Openshaw, M. R., Wilson, P., Sarker, S. J., O’Brien, N., Alifrangis, C., Stebbing, J., & Shamash, J. (2020). The Use of Transdermal Estrogen in Castrate-resistant, Steroid-refractory Prostate Cancer. Clinical Genitourinary Cancer, 18(3), e217–e223. [DOI:10.1016/j.clgc.2019.09.019]
Smith, N. L. (2004). Esterified Estrogens and Conjugated Equine Estrogens and the Risk of Venous Thrombosis. JAMA, 292(13), 1581–1587. [DOI:10.1001/jama.292.13.1581]
Smith, N. L., Blondon, M., Wiggins, K. L., Harrington, L. B., van Hylckama Vlieg, A., Floyd, J. S., Hwang, M., Bis, J. C., McKnight, B., Rice, K. M., Lumley, T., Rosendaal, F. R., Heckbert, S. R., & Psaty, B. M. (2014). Lower Risk of Cardiovascular Events in Postmenopausal Women Taking Oral Estradiol Compared With Oral Conjugated Equine Estrogens. JAMA Internal Medicine, 174(1), 25–34. [DOI:10.1001/jamainternmed.2013.11074]
Speroff, L. (1996). The Comparative Effect on Bone Density, Endometrium, and Lipids of Continuous Hormones as Replacement Therapy (CHART Study). JAMA, 276(17), 1397–1403. [DOI:10.1001/jama.1996.03540170041030]
Stanczyk, F. Z., Archer, D. F., & Bhavnani, B. R. (2013). Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception, 87(6), 706–727. [DOI:10.1016/j.contraception.2012.12.011]
Stanczyk, F. Z., Mathews, B. W., & Cortessis, V. K. (2017). Does the type of progestin influence the production of clotting factors? Contraception, 95(2), 113–116. [DOI:10.1016/j.contraception.2016.07.007]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., & Pousette, A. (1988). Single drug polyestradiol phosphate therapy in prostatic cancer. American Journal of Clinical Oncology, 11(Suppl 2), S101–S103. [DOI:10.1097/00000421-198801102-00024] [PDF]
Stegeman, B., Raps, M., Helmerhorst, F., Vos, H., van Vliet, H., Rosendaal, F., & van Hylckama Vlieg, A. (2013). Effect of ethinylestradiol dose and progestagen in combined oral contraceptives on plasma sex hormone‐binding globulin levels in premenopausal women. Journal of Thrombosis and Haemostasis, 11(1), 203–205. [DOI:10.1111/jth.12054]
Stuenkel, C. A., Davis, S. R., Gompel, A., Lumsden, M. A., Murad, M. H., Pinkerton, J. V., & Santen, R. J. (2015). Treatment of Symptoms of the Menopause: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism, 100(11), 3975–4011. [DOI:10.1210/jc.2015-2236]
Sudhir, K., & Komesaroff, P. A. (1999). Cardiovascular Actions of Estrogens in Men. The Journal of Clinical Endocrinology & Metabolism, 84(10), 3411–3415. [DOI:10.1210/jcem.84.10.5954]
Swee, D. S., Javaid, U., & Quinton, R. (2019). Estrogen Replacement in Young Hypogonadal Women—Transferrable Lessons From the Literature Related to the Care of Young Women With Premature Ovarian Failure and Transgender Women. Frontiers in Endocrinology, 10, 685. [DOI:10.3389/fendo.2019.00685]
Tadd, W., & Bayer, A. (2006). Dignity in health and social care for older Europeans: implications of a European project. Aging Health, 2(5), 771–779. [DOI:10.2217/1745509x.2.5.771]
Taylor, J. K., & Pendleton, N. (2016). Progesterone therapy for the treatment of non-cancer cachexia: a systematic review. BMJ Supportive & Palliative Care, 6(3), 276–286. [DOI:10.1136/bmjspcare-2015-001041]
Tchaikovski, S. N., & Rosing, J. (2010). Mechanisms of Estrogen-Induced Venous Thromboembolism. Thrombosis Research, 126(1), 5–11. [DOI:10.1016/j.thromres.2010.01.045]
Tepper, N. K., Whiteman, M. K., Marchbanks, P. A., James, A. H., & Curtis, K. M. (2016). Progestin-only contraception and thromboembolism: A systematic review. Contraception, 94(6), 678–700. [DOI:10.1016/j.contraception.2016.04.014]
Tepper, N. K., Jeng, G., Curtis, K. M., Boutot, M. E., Boulet, S. L., & Whiteman, M. K. (2019). Venous Thromboembolism Among Women Initiating Depot Medroxyprogesterone Acetate Immediately Postpartum. Obstetrics & Gynecology, 133(3), 533–540. [DOI:10.1097/aog.0000000000003135]
The Coronary Drug Project Research Group. (1970). The Coronary Drug Project. JAMA, 214(7), 1303–1313. [DOI:10.1001/jama.1970.03180070069012]
The Coronary Drug Project Research Group. (1973). The Coronary Drug Project. JAMA, 226(6), 652–657. [DOI:10.1001/jama.1973.03230060030009]
The Oral Contraceptive and Hemostasis Study Group. (1999). An open label, randomized study to evaluate the effects of seven monophasic oral contraceptive regimens on hemostatic variables. Contraception, 59(6), 345–355. [DOI:10.1016/s0010-7824(99)00044-x]
Timp, J. F., Braekkan, S. K., Versteeg, H. H., & Cannegieter, S. C. (2013). Epidemiology of cancer-associated venous thrombosis. Blood, 122(10), 1712–1723. [DOI:10.1182/blood-2013-04-460121]
Toorians, A. W., Thomassen, M. C., Zweegman, S., Magdeleyns, E. J., Tans, G., Gooren, L. J., & Rosing, J. (2003). Venous Thrombosis and Changes of Hemostatic Variables during Cross-Sex Hormone Treatment in Transsexual People. The Journal of Clinical Endocrinology & Metabolism, 88(12), 5723–5729. [DOI:10.1210/jc.2003-030520]
Totaro, M., Palazzi, S., Castellini, C., Parisi, A., D’Amato, F., Tienforti, D., Baroni, M. G., Francavilla, S., & Barbonetti, A. (2021). Risk of Venous Thromboembolism in Transgender People Undergoing Hormone Feminizing Therapy: A Prevalence Meta-Analysis and Meta-Regression Study. Frontiers in Endocrinology, 12, 741866. [DOI:10.3389/fendo.2021.741866]
Trémollieres, F. (2012). Contraception orale estro-progestative: quelle différence entre éthinylestradiol et estradiol? [Oral combined contraception: Is there any difference between ethinyl-estradiol and estradiol?] Gynécologie Obstétrique & Fertilité, 40(2), 109–115. [DOI:10.1016/j.gyobfe.2011.10.009]
Turo, R., Smolski, M., Esler, R., Kujawa, M. L., Bromage, S. J., Oakley, N., Adeyoju, A., Brown, S. C., Brough, R., Sinclair, A., & Collins, G. N. (2013). Diethylstilboestrol for the treatment of prostate cancer: past, present and future. Scandinavian Journal of Urology, 48(1), 4–14. [DOI:10.3109/21681805.2013.861508]
United Nations Development Programme/United Nations Population Fund/World Health Organization/World Bank Special Programme of Research, Development and Research Training in Human Reproduction, & Task Force on Long-acting Systemic Agents for Fertility Regulation. (2003). Comparative study of the effects of two once-a-month injectable contraceptives (Cyclofem® and Mesigyna®) and one oral contraceptive (Ortho-Novum 1/35®) on coagulation and fibrinolysis. Contraception, 68(3), 159–176. [DOI:10.1016/s0010-7824(03)00164-1]
van Hylckama Vlieg, A., Helmerhorst, F. M., & Rosendaal, F. R. (2010). The Risk of Deep Venous Thrombosis Associated With Injectable Depot–Medroxyprogesterone Acetate Contraceptives or a Levonorgestrel Intrauterine Device. Arteriosclerosis, Thrombosis, and Vascular Biology, 30(11), 2297–2300. [DOI:10.1161/atvbaha.110.211482]
Van Kesteren, P. J., Asscheman, H., Megens, J. A., & Gooren, L. J. (1997). Mortality and morbidity in transsexual subjects treated with cross-sex hormones. Clinical Endocrinology, 47(3), 337–343. [DOI:10.1046/j.1365-2265.1997.2601068.x]
van Vliet, H. A., Frolich, M., Christella, M., Thomassen, L., Doggen, C. J., Rosendaal, F. R., Rosing, J., & Helmerhorst, F. M. (2005). Association between sex hormone-binding globulin levels and activated protein C resistance in explaining the risk of thrombosis in users of oral contraceptives containing different progestogens. Human Reproduction, 20(2), 563–568. [DOI:10.1093/humrep/deh612]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2015). Use of combined oral contraceptives and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 350, h2135. [DOI:10.1136/bmj.h2135]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2019). Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 364, k4810. [DOI:10.1136/bmj.k4810]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
Walker, I. D. (2010). Obstetrics, Contraception, and Estrogen Replacement. In Key, N., Makris, M., O’Shaughnessy, D., & Lillicrap, D. (Eds.). Practical Hemostasis and Thrombosis, 2nd Edition (pp. 247–257). Oxford: Wiley-Blackwell. [DOI:10.1002/9781444306286.ch24]
Weinand, J. D., & Safer, J. D. (2015). Hormone therapy in transgender adults is safe with provider supervision; A review of hormone therapy sequelae for transgender individuals. Journal of Clinical & Translational Endocrinology, 2(2), 55–60. [DOI:10.1016/j.jcte.2015.02.003]
Westerlund, E., Henriksson, P., Wallén, H., Hovatta, O., Wallberg, K. R., & Antovic, A. (2012). Detection of a procoagulable state during controlled ovarian hyperstimulation for in vitro fertilization with global assays of haemostasis. Thrombosis Research, 130(4), 649–653. [DOI:10.1016/j.thromres.2011.11.024]
White, C. M., Ferraro‐Borgida, M. J., Fossati, A. T., McGill, C. C., Ahlberg, A. W., Feng, Y. J., Heller, G. V., & Chow, M. S. (1998). The Pharmacokinetics of Intravenous Estradiol—A Preliminary Study. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 18(6), 1343–1346. [DOI:10.1002/j.1875-9114.1998.tb03157.x]
Wiegratz, I., & Kuhl, H. (2006). Metabolic and clinical effects of progestogens. The European Journal of Contraception & Reproductive Health Care, 11(3), 153–161. [DOI:10.1080/13625180600772741]
Wierckx, K., Elaut, E., Declercq, E., Heylens, G., De Cuypere, G., Taes, Y., Kaufman, J. M., & T’Sjoen, G. (2013). Prevalence of cardiovascular disease and cancer during cross-sex hormone therapy in a large cohort of trans persons: a case–control study. European Journal of Endocrinology, 169(4), 471–478. [DOI:10.1530/eje-13-0493]
Writing Group for the Women’s Health Initiative Investigators. (2002). Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women: Principal Results From the Women’s Health Initiative Randomized Controlled Trial. JAMA, 288(3), 321–333. [DOI:10.1001/jama.288.3.321]
Zhang, M., Zhu, S., Yang, W., Huang, Q., & Ho, C. (2021). The biological fate and bioefficacy of citrus flavonoids: bioavailability, biotransformation, and delivery systems. Food & Function, 12(8), 3307–3323. [DOI:10.1039/d0fo03403g]
Zhao, J., Yang, J., & Xie, Y. (2019). Improvement strategies for the oral bioavailability of poorly water-soluble flavonoids: An overview. International Journal of Pharmaceutics, 570, 118642. [DOI:10.1016/j.ijpharm.2019.118642]
Zucker, R., Reisman, T., & Safer, J. D. (2021). Minimizing Venous Thromboembolism in Feminizing Hormone Therapy: Applying Lessons From Cisgender Women and Previous Data. Endocrine Practice, 27(6), 621–625. [DOI:10.1016/j.eprac.2021.03.010]
\ No newline at end of file
+Estrogens and Their Influences on Coagulation and Risk of Blood Clots - Transfeminine Science
Estrogens and Their Influences on Coagulation and Risk of Blood Clots
By Aly | First published October 20, 2020 | Last modified March 28, 2023
Abstract / TL;DR
Estrogens increase coagulation by activating estrogen receptors in the liver and thereby modulating the production of a variety of circulating coagulation factors. With sufficiently high exposure, this can result in an increase in the risk of blood clots as well as coagulation-associated cardiovascular complications like heart attack and stroke. However, the degrees of risk vary depending on the estrogen type, route, and dose. Non-bioidentical estrogens like ethinylestradiol have greater strength in the liver due to their relative resistance to metabolism and increase blood clot risk more readily than bioidentical estradiol, while oral administration of estradiol results in a first pass through the liver and has greater impact on blood clot risk than non-oral estradiol. Physiological estradiol levels with non-oral estradiol appear to have minimal to no risk of blood clots, whereas oral estradiol has significant risk and at high doses may have risk similar to that of the doses of ethinylestradiol in modern birth control pills. Higher estradiol levels with non-oral estradiol seem to have significant risk of blood clots and cardiovascular problems as well, although the risks appear to be lower than with ethinylestradiol-containing birth control pills. Absolute risks of blood clots are low but accumulate with time and add up on a population scale. In addition, a variety of risk factors, such as age, physical inactivity, concomitant progestogen use, and often-unknown thrombophilic abnormalities, can substantially augment risk. Due to their higher risks of blood clots, oral estradiol as well as excessive doses of non-oral estradiol should ideally be avoided in transfeminine people. This is particularly applicable in those with risk factors for blood clots. In any case, therapeutic considerations for transfeminine people include not only safety but also effectiveness, other factors like cost and convenience, and the natures of the alternative therapeutic options.
Introduction
Estrogens increase coagulation (blood clotting) and the risk of thrombosis, a cardiovascular event otherwise known as a blood clot. There are two major types of blood clots, which are categorized depending on whether they happen in a vein or in an artery: (1) venous thrombosis or venous thromboembolism (VTE); and (2) arterial thrombosis. VTE is a blood clot in a vein, a blood vessel that carries blood towards the heart. It comprises two different subtypes: (1) deep vein thrombosis (DVT), a clot in a vein of the leg or pelvic region; and (2) pulmonary embolism (PE), a clot that has broken free and blocked an artery in the lungs. Arterial thrombosis is a blood clot in an artery, a blood vessel that carries blood away from the heart. Arterial thrombosis can lead to myocardial infarction (MI; also known as heart attack) or cerebrovascular accident (CVA; also known as stroke). Blood clots are major health problems that can cause serious complications and even death. Estrogens, via increased coagulation with sufficiently high exposure, have the potential to heighten the risk of both venous and arterial thrombosis and hence to increase all of the aforementioned risks. The risk of blood clots with estrogens serves as a limiting factor in their use due to the potential health consequences.
Estrogens are selectiveagonists of the estrogen receptors (ERs). They are thought to increase coagulation and hence blood clot risk by activating ERs. However, the impact on coagulation and risk of blood clots with estrogens varies due to factors like estrogen type, route, and dose. In addition, other factors, like concomitant progestogen use and a variety of non-hormonal factors, are known to modify the risk. The purpose of this article is to review the risks of blood clots with estrogens, the mechanisms underlying increased coagulation and blood clot risk with estrogens, and the reasons for differences among estrogens in terms of risk. Exploring these topics can inform estrogen dosing considerations in transfeminine people and help to minimize risks and optimize safety. Moreover, higher levels of estrogens are therapeutically useful for suppressing testosterone production in transfeminine people but may increase blood clot risk, and risk–benefit analysis is warranted in this context.
Blood Clot Risks with Estrogens and Progestogens
A variety of estrogens have been used in medicine. These include bioidentical estrogens like estradiol as well as non-bioidentical estrogens like conjugated estrogens (CEEs; Premarin), ethinylestradiol (EE), and diethylstilbestrol (DES). Estradiol is the major natural estrogen in the human body. CEEs deliver primarily estradiol as the active estrogen, but also contain significant quantities of naturally occurring equine (horse) estrogens such as equilin (7-dehydroestrone) and 17β-dihydroequilin (7-dehydroestradiol). EE and DES are synthetic estrogens that were created by humans and do not occur naturally. DES was discontinued decades ago and is relatively little-known today, but has significant historical importance. Estradiol is used in both oral and non-oral forms (e.g., transdermal patches), while the non-bioidentical estrogens have typically been used orally. For context, the table below shows some approximate comparable doses of these estrogens in terms of general estrogenicity.
Table 1: Approximate or estimated comparable doses of estrogens in terms of general/systemic estrogenicity (Aly, 2020; Kuhl, 2005; Table; Table; Table):
Estrogen type/route
Very low dose a
Low dose a
Moderate dose b
High dose
Oral estradiol
1 mg/day
2 mg/day
4 mg/day
8 mg/day
Transdermal estradiolc
25 μg/day
50 μg/day
100 μg/day
200 μg/day
Oral conjugated estrogens
0.625 mg/day
1.25 mg/day
2.5 mg/day
5 mg/day
Oral ethinylestradiol
7.5 μg/day
15 μg/day
30 μg/day
60 μg/day
Oral diethylstilbestrol
0.375 mg/day
0.75 mg/day
1.5 mg/day
3 mg/day
Comparable estradiol level
~25 pg/mL
~50 pg/mL
~100 pg/mL
~200 pg/mL
a Menopausal replacement dosages. b Similar to normal mean/integrated estrogenic exposure during the menstrual cycle in premenopausal women (Aly, 2018). c Specifically transdermal patches.
Estrogens were first associated with blood clots and associated cardiovascular complications in the 1960s and 1970s. Significant to substantial increases in these risks were found in clinical trials of high-dose DES (5 mg/day) for prostate cancer in men (VACURG, 1967; Byar, 1973; Turo et al., 2014), trials of moderate-dose CEEs (2.5–5 mg/day) for prevention of heart disease in men (Coronary Drug Project Research Group, 1970; Coronary Drug Project Research Group, 1973; Luria, 1989; Sudhir & Komesaroff, 1999; Dutra et al., 2019), and studies of early high-dose EE-containing birth control pills (50–150 μg/day) in premenopausal women (Gerstman et al., 1991; PCASRM, 2017; Table). The increase in cardiovascular events with DES in men with prostate cancer was sufficiently great that it actually cancelled out the benefits of its effects against prostate cancer in terms of overall mortality. The large increases in blood clots and cardiovascular problems seen in these studies resulted in alarm and concern about the safety of estrogens. Consequent to these events, estrogen doses were lowered. DES for prostate cancer was decreased to 1 to 3 mg/day and EE in birth control pills was decreased to 20 to 35 μg/day. Estrogens were also reduced to lower doses for other indications, such as menopausal hormone therapy. The dose reductions helped to lower the risks, although it did not eliminate them.
Whereas the WHI demonstrated causation for oral CEEs alone in terms of blood clot risk, no adequately powered RCTs have been conducted with oral or transdermal estradiol alone to establish causation in terms of blood clot risk at this time. Only very large and expensive trials would be able to show this due to the rarity of blood clots, and these studies have not been conducted to date. For similar reasons, RCTs demonstrating increased risk of blood clots with EE-containing birth control pills have also not been conducted at this time (Moores, Bilello, & Murin, 2004). In any case, causation has clearly been demonstrated with estrogens in other contexts, and this can be assumed as likely in the case of oral estradiol similarly. In addition, the Estrogen in Venous Thromboembolism Trial (EVTET), an RCT of low-dose (2 mg/day) oral estradiol plus the progestogen norethisterone acetate (NETA) versus placebo in postmenopausal women with history of previous blood clots, found that this hormone therapy regimen significantly increased coagulation and the incidence of blood clots (10.7% incidence with hormone therapy and 2.3% with placebo; P = 0.04) (Høibraaten et al., 2000; Høibraaten et al., 2001).
Estradiol levels appear to not be associated with blood clot risk in premenopausal women (Holmegard et al., 2014). The fact that transdermal estradiol patches at 100 μg/day in menopausal women haven’t been associated with a greater risk of blood clots is notable as this dose achieves estradiol levels of around 100 pg/mL on average, which are similar to the mean integrated levels of estradiol during the normal menstrual cycle in premenopausal women (Aly, 2018; Wiki). Rates of blood clots are also similar between men—who have relatively low estradiol levels—and women after controlling for atypical hormonal states like pregnancy and use of birth control pills in women (Moores, Bilello, & Murin, 2004; Rosendaal, 2005; Montagnana et al., 2010; Roach et al., 2013). Interestingly however, men have a consistently higher incidence of recurrent blood clots than women (Roach et al., 2013). These findings suggest that physiological levels of estradiol and progesterone in premenopausal women may not meaningfully increase coagulation or blood clot risk. However, the available data are mixed, with some studies suggesting that estradiol and/or progesterone levels within physiological ranges may indeed influence coagulation (Chaireti et al., 2013) and blood clot risk in premenopausal and/or perimenopausal women (Simon et al., 2006; Canonico et al., 2014; Scheres et al., 2019).
Modern combined birth control pills contain EE at moderately estrogenic doses (20–35 μg/day) and a physiological dose of a progestogen. They increase the risk of blood clots by several-fold (Konkle & Sood, 2019; Vinogradova, Coupland, & Hippisley-Cox, 2015; Table). In addition, they are associated with about a 1.5- to 2-fold increase in risk of heart attack and stroke (Lidegaard, 2014; Konkle & Sood, 2019). However, overall mortality is not increased with birth control pills—at least in the relatively young women in whom they are used (Hannaford et al., 2010). Per studies of menopausal hormone therapy, it is likely that the progestogen in EE-containing birth control pills augments the risk of blood clots with EE. Early high-dose birth control pills (50–100 μg/day) had as much as twice the risk of blood clots of modern birth control pills (Gerstman et al., 1991; PCASRM, 2017; Table). In contrast to the different blood clot risks between oral and transdermal estradiol, non-oral birth control forms containing EE, for instance transdermal birth control patches and vaginal birth control rings, are associated with similar increases in blood clot risk as EE-containing birth control pills (Plu-Bureau et al., 2013; PCASRM, 2017; Konkle & Sood, 2019; Abou-Ismail, Sridhar, & Nayak, 2020). Hence, unlike with estradiol, route of administration does not appear to modify blood clot risk with EE based on available data.
High-dose estrogen therapy using oral synthetic estrogens like DES and EE in people with breast or prostate cancer has been found to strongly increase the risk of blood clots and associated cardiovascular complications (Phillips et al., 2014; Turo et al., 2014; Coelingh Bennink et al., 2017). This has also been the case with estramustine phosphate (EMP; estradiol normustine phosphate), an estradiol ester that is used at massive doses in prostate cancer (e.g., 140–1,400 mg/day orally) and that results in pregnancy levels of estradiol (Kitamura, 2001 [Graph]; Ravery et al., 2011). In the 1980s however, it was found that high-dose non-oral estradiol did not have the same cardiovascular risks as high-dose estrogen therapy with oral synthetic estrogens or EMP (von Schoultz et al., 1989; Ockrim & Abel, 2009). This included studies with polyestradiol phosphate (PEP), a long-lasting injectable prodrug of estradiol, and with high-dose transdermal estradiol gel (von Schoultz et al., 1989; Aly, 2019). However, subsequent larger and higher-quality studies found that although the cardiovascular risks with PEP were much lower than with high-dose oral synthetic estrogen therapy, they were nonetheless still increased (Hedlung et al., 2008; Ockrim & Abel, 2009; Hedlund et al., 2011; Sam, 2020). This includes an approximate 2-fold increase in the risk of blood clots with estradiol levels in the range of roughly 300 to 500 pg/mL (Sam, 2020). Studies using high-dose transdermal estradiol patches have not found significantly increased cardiovascular complications as of present (Langley et al., 2013; Sam, 2020). However, these studies have been relatively underpowered, which limits their interpretation. In any case, increased coagulation has been observed with high-dose transdermal estradiol patches (achieving estradiol levels of 350 to 500 pg/mL) (Bland et al., 2005) similarly to PEP (Mikkola et al., 1999). More data on the risk of blood clots and cardiovascular issues with high-dose transdermal estradiol patches should come in the future with PATCH and STAMPEDE—two large-scale clinical studies in the United Kingdom that are evaluating this form of estradiol for prostate cancer (Gilbert et al., 2018; Singla, Ghandour, & Raj, 2019).
Injections of short-acting estradiol esters like estradiol valerate and estradiol cypionate are notable in that they are often used by transfeminine people and are generally used at doses that achieve high estradiol levels. As with high-dose transdermal estradiol patches, little to no quality data on the risk of blood clots exists for these preparations at present. Pyra and colleagues found that the risk of blood clots with injectable estradiol valerate in transfeminine people was increased by around 2-fold, but the confidence intervals were very wide and statistical significance was not reached (Pyra et al., 2020). The doses used in the whole population for the study were not provided, but in the actual VTE cases, the doses of injectable estradiol valerate were described and ranged from 4 to 20 mg once per week and 10 to 40 mg once every 2 weeks (Pyra et al., 2020). Studies have also assessed and found increased coagulation with high doses of estradiol valerate by injection in the range of 10 to 40 mg once every 2 weeks in men with prostate cancer (Kohli & McClellan, 2001; Kohli et al., 2004; Kohli, 2005). Increased coagulation has additionally been observed with the combination of 5 mg estradiol valerate and a progestogen once per month as a combined injectable contraceptive in premenopausal women (Meng et al., 1990; UN/WHO et al., 2003). It is unclear whether the high peaks in estradiol levels associated with short-acting injectable forms of estradiol are harmful in terms of coagulation and blood clot risk (Hembree et al., 2017). However, the increased risk of polycythemia with short-acting injectable testosterone esters relative to other non-oral forms of testosterone (Ohlander, Varghese, & Pastuszak, 2018) is indirectly suggestive that this could be the case. Accordingly, a study found increased coagulation in premenopausal women with a combined injectable contraceptive containing estradiol valerate but not with one employing the more prolonged and stable estradiol cypionate at the same dose (UN/WHO et al., 2003).
Pregnancy is a time when estradiol and progesterone levels increase to extremely high concentrations (Graphs). Estradiol levels increase progressively throughout pregnancy to around 2,000 pg/mL on average at the end of the first trimester, to about 10,000 pg/mL on average at the end of the second trimester, and to around 20,000 pg/mL on average at the end of the third trimester (Kerlan et al., 1994 [Graph]; Schock et al., 2016). Coagulation is greatly increased during pregnancy, and the risk of blood clots is likewise strongly increased (Heit et al., 2000; Abdul Sultan et al., 2015; Heit, Spencer, & White, 2016; Table). Estradiol and progesterone levels are strongly correlated with the increases in coagulation during pregnancy (Bagot et al., 2019). The risk of blood clots with modern birth control pills is similar to that with pregnancy as a whole (Heit, Spencer, & White, 2016), while the increases in risk of blood clots with early high-dose EE-containing birth control pills and with high-dose oral synthetic estrogen therapy for breast and prostate cancer are comparable to the risk increase during late pregnancy. Estradiol levels also increase to very high concentrations during ovarian stimulation for in-vitro fertilization in premenopausal women, and this has been associated with increased coagulation and risk of blood clots as well (Westerlund et al., 2012; Rova, Passmark, & Lindqvist, 2012; Kasum et al., 2014).
Due to their greater risks of cardiovascular problems as well as other concerns, DES has been virtually abandoned while EE has been discontinued for almost all indications except birth control. EE continues to be used in birth control because it is resistant to metabolism in the uterus and controls menstrual bleeding better than oral estradiol does (Stanczyk, Archer, & Bhavnani, 2013). CEEs are also being increasingly superseded by estradiol in medicine, although significant use of CEEs for hormone therapy in cisgender women continues. Transdermal estradiol is gaining momentum over oral estradiol in menopausal hormone therapy as well. Major transgender hormone therapy guidelines (see also Aly, 2020) recommend against the use of EE and CEEs in transfeminine people due to their greater risks and the inability to accurately monitor blood estrogen levels with these preparations (Coleman et al., 2012; Deutsch, 2016; Hembree et al., 2017). Estradiol is the estrogen that is almost exclusively used in transfeminine people today. Besides estrogen type, it has been recommended that transdermal estradiol be used instead of oral estradiol in transfeminine people who are over 40 or 45 years of age or are otherwise at risk for blood clots (Deutsch, 2016; Iwamoto et al., 2019; Glintborg et al., 2021). Menopausal hormone therapy guidelines similarly recommend the use of transdermal estradiol over oral estrogens in cisgender women who are at higher risk for blood clots (e.g., Stuenkel et al., 2015).
In a historically notable study conducted by the Center of Expertise on Gender Dysphoria (CEGD) at the Vrije Universiteit Medical Center (VUMC) in Amsterdam, the Netherlands in the 1980s, it was reported that the risk of blood clots with high-dose EE and CPA in transfeminine people was increased by 45-fold relative to the expected incidence in the general population (Asscheman, Gooren, & Eklund, 1989; Asscheman et al., 2014). Mortality also appeared to be elevated and other health risks were increased as well (Asscheman, Gooren, & Eklund, 1989; Gooren & T’Sjoen, 2018). A subsequent study in transfeminine people by the CEGD confirmed strongly increased coagulation with EE but much lower increases with oral or transdermal estradiol (Toorians et al., 2003). Upon the CEGD switching transfeminine people from high-dose EE to physiological doses of oral or transdermal estradiol (also usually in combination with CPA), the risks decreased considerably (van Kesteren et al., 1997; Asscheman et al., 2011; Asscheman et al., 2014). These findings were of major importance in the replacement of EE with estradiol in transfeminine hormone therapy, and have surely contributed significantly to apprehension about the use of high doses of estrogens in transfeminine people.
Taken together, estrogens of all kinds have been shown to dose-dependently increase or be associated with increased risk of blood clots. These findings suggest that, provided of course sufficient exposure occurs, increased coagulation and blood clot risk are common properties of estrogens. However, synthetic and non-bioidentical estrogens have greater risk of blood clots than estradiol, and oral estradiol shows greater risk than non-oral estradiol. In fact, physiological estradiol levels in women and low to moderate doses of transdermal estradiol may have no significant risk of blood clots at all. Nonetheless, non-oral estradiol with sufficiently high exposure can increase blood clot risk just the same as other forms of estrogen. Concomitant therapy with progestogens appears to augment the risk of blood clots with estrogens and high doses may particularly amplify the risk.
Risks with Different Hormonal Exposures
The table below provides relative risk increases for blood clots with different types, routes, and doses of estrogens, as well as with SERMs, pregnancy, and high-dose CPA. It shows the greater risks of blood clots with (1) oral estradiol relative to non-oral estradiol; (2) estradiol compared to non-bioidentical estrogens; and (3) lower estrogen levels/doses relative to higher estrogen levels/doses.
Table 2: Relative risks of blood clots with different hormonal exposures (see also Machin & Ragni, 2020):
Footnotes:a At typical menopausal replacement doses (i.e., not very high—probably no more than double the given dose). b MPA, norethisterone, norgestrel, or drospirenone. c Modern EE + P birth control contains 20–35 μg/day EE, while high-dose EE + P birth control used in the 1960s and 1970s contained 50–150 μg/day EE. d Risk around twice as high as modern birth control pills. e Unpublished original research/analysis with borderline statistical significance (95% CI 0.99–4.22). f Excluding the postpartum period. With the postpartum period included, the risk of blood clots with pregnancy is 5–10× (McLintock, 2014). Abbreviations: E2 = Estradiol; CEEs = Conjugated estrogens; EE = Ethinylestradiol; DES = Diethylstilbestrol; EMP = Estramustine phosphate; PEP = Polyestradiol phosphate; SERMs = Selective estrogen receptor modulators; CPA = Cyproterone acetate; P = Progestogen.
Note that the values in the table are associations mostly from observational studies rather than from RCTs. Hence, in many cases, causation has not been definitively established. In addition, the values represent rough average values with often wide 95% confidence intervals. As a result, precision and accuracy of the estimates may in some cases be low. Also note that quantified blood clot risk will vary depending on the study and its definitions and methodology (including factors like sampling error, approach to control of confounding variables, and residual confounding influences).
Mechanisms of Increased Coagulation with Estrogens
Aside from coagulation factors, estrogens also modulate the synthesis of numerous other liver products (Kuhl, 1999; Kuhl, 2005; Table). Examples include sex hormone-binding globulin (SHBG), corticosteroid-binding globulin (CBG), various other circulating binding proteins, angiotensinogen, lipoproteins, and triglycerides, among others. In accordance with the mechanisms underlying increased coagulation and blood clot risk with estrogens, the differences in risk of blood clots with different types and routes of estrogens are mirrored in their influences on estrogen-sensitive liver products. Put another way, different estrogens have different relative potency in the liver when compared to their estrogenic potency elsewhere in the body. Synthetic and non-bioidentical estrogens have greater impact on liver synthesis than estradiol, while oral administration of estradiol has greater influence on liver synthesis than non-oral routes like transdermal administration or intramuscular injection, and this is likely to explain the observed differences in coagulation and blood clot risk with these different estrogens. The table below shows the liver potency of different estrogenic exposures as measured by influence specifically on SHBG levels, one of the most sensitive and well-characterized estrogen-modulated liver products.
Table 3: Relative increases in SHBG levels with different estrogenic exposures (see also Aly, 2020):
Footnotes:a Due to differences in molecular weight, estradiol valerate has about 75% of the amount of estradiol as regular estradiol. Hence, 6 mg/day estradiol valerate is approximately equivalent to 4.5 mg/day estradiol. b Modern EE + P birth control contains 20–35 μg/day EE, while high-dose EE + P birth control used in the 1960s and 1970s contained 50–150 μg/day EE. c In the form of 320 mg/month PEP (~700 pg/mL estradiol), 100 mg/month estradiol undecylate (~500–600 pg/mL estradiol), or 10 mg/10 days estradiol valerate (~500–1,200 pg/mL peak estradiol; Graphs). Abbreviations: E2 = Estradiol; EV = Estradiol valerate; CEEs = Conjugated estrogens; EE = Ethinylestradiol; DES = Diethylstilbestrol; EMP = Estramustine phosphate; PEP = Polyestradiol phosphate; P = Progestogen.
The increase in SHBG levels with estrogen therapy correlates with increases in coagulation and blood clot risk and can serve as a reliable surrogate indicator of these effects (Odlind et al., 2002; van Rooijen et al., 2004; van Vliet et al., 2005; Tchaikovski & Rosing, 2010; Raps et al., 2012; Stegeman et al., 2013; Hugon-Rodin et al., 2017; Eilertsen et al., 2019). The increases in SHBG levels and blood clot risk even appear quite similar to each other with modern birth control pills (both ~4-fold), high-dose oral synthetic estrogen therapy (both ~5–10-fold), and late pregnancy (both ~5–10-fold). When data on blood clot risk with a given estrogen route or dose are limited or unavailable—for instance with high-dose oral estradiol or high-dose estradiol ester injections—changes in SHBG levels can be used as a rough proxy or surrogate instead to estimate overall liver impact, magnitude of change in coagulation systems, and blood clot risk. It should be noted however that progestogens may augment the blood clot risk with estrogens without necessarily affecting SHBG levels or even while decreasing SHBG levels via concomitant androgenic activity (Kuhl, 2005; Vinogradova, Coupland, & Hippisley-Cox, 2019).
Physiological levels of estradiol appear to have relatively minimal influence on liver synthesis (Eisenfeld & Aten, 1979; Lax, 1987; Kuhl, 2005). This is in accordance with the limited influence or non-influence of physiological estradiol levels in women on blood clot risk. It is thought that under normal physiological circumstances, estradiol is only supposed to considerably affect liver synthesis at very high levels—namely during pregnancy. The changes in synthesis of liver products during pregnancy presumably have important biological roles at this time (Eisenfeld & Aten, 1979). One of these is considered to be increased coagulation, as coagulation limits blood loss with childbirth and hence has survival benefits. Conversely, there is no obvious benefit to increased coagulation outside of pregnancy.
Estradiol and the Liver First Pass with Oral Administration
The oralroute of administration is subject to a first pass through the liver via the hepatic portal vein which is not present with non-oral routes of administration (Pond & Tozer, 1984; Back & Rogers, 1987). As such, oral estradiol is subject to a hepatic first pass while this does not occur with non-oral forms of estradiol such as transdermal estradiol and injectable estradiol (Kuhl, 1998; Kuhl, 2005). This first pass results in disproportionate exposure of the liver to estradiol as well as disproportionate estrogenic impact on liver protein synthesis (Kuhl, 2005). Oral estradiol likewise has disproportionate estrogenic impact on the hepatic synthesis of coagulation factors (Kuhl, 1998; Kuhl, 2005). Due to the first pass, it is estimated that there is a 4- or 5-fold greater estrogenic impact of oral estradiol in the liver relative to non-oral estradiol (Kuhl, 2005). Due to the absence of the hepatic first pass with most non-oral routes, there is strong biological plausibility for the lower risk of blood clots that has been found with transdermal estradiol in comparison to oral estradiol in observational studies (Baber et al., 2016).
Although oral estradiol has a much higher relative potential for blood clots due to the liver first pass, sufficiently high levels of estradiol will diffuse into the liver from the blood to act on this tissue regardless of route of administration. Hence, high levels of estradiol via non-oral routes (or produced by the body itself) can increase coagulation and blood clot risk similarly to the oral route. This is clearly evidenced by hyperestrogenic situations like pregnancy and ovarian stimulation for in-vitro fertilization, when estradiol levels increase to very high concentrations and substantially influence liver protein synthesis.
Non-Bioidentical Estrogens and Resistance to Liver Metabolism
Non-bioidentical estrogens such as EE, DES, and CEEs have greater impact on liver protein synthesis and risk of blood clots than either oral estradiol or non-oral estradiol (Kuhl, 1998; Kuhl, 2005; Phillips et al., 2014; Turo et al., 2014; Table). This is because the liver strongly metabolizes and inactivates estradiol, whereas non-bioidentical estrogens have differences in their chemical structures relative to estradiol that result in them being much more resistant to liver metabolism (Kuhl, 1998; Kuhl, 2005; Connors & Middeldorp, 2019; Swee, Javaid, & Quinton, 2019).
EE can be considered as a case example. The oral bioavailability of EE is around 45%, while that of estradiol is only about 5% (Kuhl, 2005; Stanczyk, Archer, & Bhavnani, 2013). In addition, the blood half-life of EE is in the range of 5 to 30 hours, compared to less than 1 hour in the case of estradiol (White et al., 1998; Kuhl, 2005; Stanczyk, Archer, & Bhavnani, 2013). As a result of these and other differences, EE is approximately 120 times as potent as estradiol by weight in terms of general estrogenic effect (Kuhl, 2005; Table). Hence, EE is used clinically in μg doses whereas oral estradiol is used at over 100-fold higher mg doses. The pharmacokinetic differences between EE and estradiol reflect the strong resistance of EE to liver metabolism (Kuhl, 2005). EE, or 17α-ethynylestradiol, shows resistance to liver metabolism because of an ethynyl group at the C17α position which has been added to what is the otherwise unchanged structure of estradiol (Kuhl, 2005). This modification results in steric hindrance which blocks 17β-hydroxysteroid dehydrogenases (17β-HSDs) as well as conjugating enzymes like sulfotransferases and glucuronosyltransferases from metabolizing EE at the C17β hydroxyl group. 17β-HSDs normally convert estradiol into the weakly active estrone while the conjugating enzymes convert estradiol into inactive C17β estrogen sulfate and glucuronide conjugates like estrone sulfate (Kuhl, 2005). An “ethinylestrone” metabolite is in fact a structural impossibility due to the requirement of a double bond for a C17 ketone group—the needed C17α position is already occupied in EE by its ethynyl group. As such, the metabolism of estradiol into weakly active or inactive metabolites like estrone and estrone sulfate in the liver is protective against activation of hepatic ERs and procoagulation, and the lack of this with EE is responsible for its greater blood clot risk (Kuhl, 2005; Russell et al., 2017).
Figure 2: Chemical structures of selected estrogens. The C17 position in the case of the steroidal estrogens (E2, E1, and EE) is at the top right of the steroid nucleus.
Due to the marked resistance of EE to hepatic metabolism and inactivation, it persists for a long time in the liver—often cycling through it many times before finally being broken down. Moreover, EE shows several-fold disproportionate impact on liver protein synthesis at otherwise equivalent doses relative to oral estradiol (Kuhl, 2005; Table). Consequently, whereas EE has around 120-fold the general potency of oral estradiol, the liver potency of EE is around 350 to 1,500 times greater than that of oral estradiol (von Schoultz et al., 1989; Kuhl, 2005). A dose of EE of as little as 1 μg/day has been shown to impact liver metabolism (Speroff et al., 1996; Trémollieres, 2012). In addition, the fact that EE shows similar hepatic impact and risk of blood clots regardless of whether it is administered orally, transdermally, or vaginally indicates that unlike oral estradiol, the first pass through the liver with oral administration is not necessary for blood clot risk with EE (Plu-Bureau et al., 2013; PCASRM, 2017; Konkle & Sood, 2019). EE is so resistant to metabolism that it does not seem to matter how it is administered—the liver impact is substantial regardless of route. The greatly increased liver potency of EE results in its influence on coagulation and blood clot risk being much higher than that of estradiol at equivalent doses.
CEEs show a few-fold disproportionate estrogenic impact on liver protein synthesis relative to oral estradiol but less than that of EE (Kuhl, 2005; Table). This can be attributed to the equine (horse) estrogens in CEEs, which humans are presumably not adapted to and which show resistance to liver metabolism in humans. DES, on the other hand, shows even greater estrogenic influence on the liver than EE (Kuhl, 2005; Table). The more disproportionate impact on liver synthesis of DES relative to EE or CEEs may be attributable to the fact that it is a nonsteroidal estrogen and is far removed in structure from steroidal estrogens. This is relevant as steroidal estrogens are susceptible to varying extents to robust steroid-metabolizing enzymes in the liver (Kuhl, 2005). As with EE, 17β-HSDs have no affinity for DES and the hydroxyl groups of DES are not oxidized to form estrone-like ketone metabolites (Jensen et al., 2010). Consequent to their resistance to liver metabolism relative to estradiol, CEEs and nonsteroidal estrogens like DES have greater impacts on coagulation and blood clot risk than equivalent doses of estradiol similarly to EE although to varying extents.
When compared to transdermal estradiol rather than oral estradiol, the disproportionate influence of oral non-bioidentical estrogens on estrogen-modulated liver protein synthesis becomes extreme. With a little math, it quickly becomes apparent why high doses of these estrogens have influences on liver proteins and blood clot risks that are comparable to those during pregnancy. The table below shows some roughly calculated estimates for comparative liver strength of the different estrogens.
Table 4: Roughly calculated ratios of liver estrogenic potency to general/systemic estrogenic potency with estrogens based on a selection of liver products (e.g., SHBG, others) (Kuhl, 2005; Table):
Estrogen
Comparative liver potency
Relative to oral E2
Relative to transdermal E2
Transdermal E2
~0.25×a
1.0×a
Oral E2
1.0×
~4.0×
Oral CEEs
1.3–4.5×
~5.2–18×
Oral EE
2.9–5.0×
~12–20×
Oral DES
5.7–7.5×
~23–30×
a Based on a study that found oral estradiol to have 4-fold greater effect on SHBG levels than transdermal estradiol when used at doses that produced similar estradiol levels (Nachtigall et al., 2000).
Changes in liver protein synthesis induced by estrogens don’t scale linearly with dose or relative liver potency. There is progressive saturation in terms of changes in levels of SHBG and other liver products with estrogen dose—that is, higher doses have relatively diminished effect compared to lower doses (Kuhl, 1990; Kuhl, 1999). As an example, oral EE shows the following dose-dependent increases in SHBG levels: 2.0-fold at 5 μg/day, 3.0-fold at 10 μg/day, 3.4-fold at 20 μg/day, and 4.0-fold at 50 μg/day (Kuhl, 1998; Kuhl, 1999). These findings can be attributed to saturation of the competitive binding and/or activation of liver ERs by high estrogen concentrations (Kuhl, 1990). An implication of this dose-dependent saturation is that although for instance oral EE has much stronger potency in the liver than oral estradiol, oral estradiol can more quickly “catch up” to oral EE and other non-bioidentical estrogens in terms of liver impact than might be initially anticipated. Accordingly, oral estradiol has shown the following dose-dependent increases in SHBG levels: 1.6-fold at 1 mg/day, 2.2-fold at 2 mg/day, and 1.9- to 3.2-fold at 4 mg/day (Fåhraeus & Larsson-Cohn, 1982; Kuhl, 1998; Gibney et al., 2005; Ropponen et al., 2005). Hence, although oral EE may have roughly 3- to 5-fold higher liver potency than oral estradiol, a dose of oral estradiol near-equivalent to that of oral EE in terms of general estrogenic effect can increase SHBG levels to an extent that is only somewhat lower in comparison.
Selective Estrogen Receptor Modulators and Metabolism Resistance
SERMs like tamoxifen and raloxifene are essentially partial agonists of the ER. This is in contrast to estrogens—like estradiol, CEEs, EE, and DES—which act as full agonists of the ER. Similarly to nonsteroidal estrogens like DES, the clinically used SERMs are all nonsteroidal in structure and are strongly resistant to hepatic metabolism. In fact, certain SERMs, like tamoxifen and clomifene, are structurally related to and were derived from DES. SERMs show tissue differences in their ER-mediated effects, with estrogenic effects in some tissues (e.g., bone) and antiestrogenic effects in other tissues (e.g., breasts) (Lain, 2019; Table). Although there is variation between SERMs in terms of their effects in certain tissues (e.g., uterus), they are uniformly estrogenic in the liver. Consequently, SERMs show similar increases in blood clot risk as estrogens (Park & Jordan, 2002; Fabian & Kimler, 2005). As with non-bioidentical estrogens, the greater risk of blood clots with SERMs compared to oral estradiol can be attributed to their resistance to liver metabolism and hence to greater hepatic estrogenic potency. The SERMs that are used medically belong to diverse structural families (e.g., triphenylethylenes like tamoxifen and benzothiophenes like raloxifene). The only way in which SERMs of different structural classes are known to be related is in their shared interactions with the ERs.
Figure 3: Chemical structures of selected SERMs. They are nonsteroidal in structure and include tamoxifen (a triphenylethylene) and raloxifene (a benzothiophene).
Activation of the Estrogen Receptor is Specifically Responsible for Increased Coagulation with Estrogens and SERMs
Findings from preclinical and genetic research provide direct evidence for ER activation being responsible for the increased blood clot risk with estrogens. In an important animal study, EE was administered to mice and changes in procoagulant and anticoagulant biomarkers were measured (Cleuren et al., 2010). EE caused changes in levels of a variety of coagulation factors (Cleuren et al., 2010). The researchers also assessed estradiol and observed comparable changes (Cleuren et al., 2010). Co-administration of the selective ER full antagonist fulvestrant with EE neutralized all of the EE-induced coagulatory changes (Cleuren et al., 2010). Additionally, EE showed no effect on coagulation factors in ERα knockout mice (Cleuren et al., 2010). These findings are consistent with human and mouse genome-wide association studies which have found estrogen response elements (EREs)—DNA sequences that act as binding sites for genes regulated by the ER—embedded in a large number of genes involved in coagulatory pathways (Cleuren et al., 2010; Stanczyk, Mathews, & Cortessis, 2017).
The preceding findings are consistent with ER activation being responsible for increased coagulation and blood clot risk with estrogens and SERMs. This is in accordance with the fact that blood clot risk is a shared effect of selective ER agonists with highly diverse chemical structures, providing strong circumstantial support against a non-ER-mediated action of some sort being responsible (e.g., the weakly estrogenic metabolite estrone somehow mediating the blood clot risk with estradiol—Bagot et al., 2010). Increased coagulation and blood clot risk can thus be regarded as class effects of estrogens and SERMs—provided sufficiently high liver exposure. Due to differences in susceptibility to liver metabolism however, different ER agonists show differences in their relative impact on coagulation. Owing to estradiol’s lack of resistance to metabolism and its robust inactivation in the liver, the dosage requirements for increased coagulation and blood clot risk with estradiol—particularly in the case of non-oral estradiol—are greater than with non-bioidentical estrogens. Hence, estradiol, especially when administered via non-oral routes, is a safer form of estrogen therapy than other estrogens.
Absolute Incidences and Risk Factors
States of estrogen and/or progestogen exposure, such as exogenous hormone administration and pregnancy, are of course established risk factors for blood clots in women. In healthy young individuals without relevant risk factors for blood clots however, the incidence of blood clots is rare even in situations of considerably increased risk due to hormones (Rosendaal, 2005). The absolute incidence of VTE in non-pregnant women is only 1 to 5 of every 10,000 women each year (i.e., 0.01–0.05% per year) (PCASRM, 2017; Konkle & Sood, 2019). EE-containing birth control pills, which on average increase VTE risk by about 4-fold, are associated with an incidence of VTE of only 3 to 9 of every 10,000 women each year (i.e., 0.03–0.09% per year) (Konkle & Sood, 2019). Likewise, the absolute risk of blood clots during pregnancy, when estradiol and progesterone levels increase to extremely high concentrations and VTE risk is increased up to 7-fold (Abdul Sultan et al., 2015), is about 5 to 20 of every 10,000 women each year (i.e., 0.05–0.2% per year) (PCASRM, 2017; Konkle & Sood, 2019).
a 1–2/10,000 per year at <19 years of age, 2–3/10,000 per year at 20–29 years of age, 3–4/10,000 per year at 30–39 years of age, 5–7/10,000 per year at 40–49 years of age; roughly 3–4/10,000 per year for age 15–49 years overall (Rabe et al., 2011).
In addition to time and population considerations, there are, besides estrogen and progestogen exposure, a variety of other known risk factors for blood clots, and these risk factors can substantially augment blood clot risk (Heit et al., 2000; Rosendaal, 2005). Age is among the strongest of the known risk factors (Rosendaal, 2005; Montagnana et al., 2010). Moreover, age is uniquely notable as a risk factor in that it is one that eventually becomes relevant to everyone. The risk of blood clots increases on the order of 100-fold going from ≤15 years of age (incidence <0.005–0.01% per year) to ≥80 years of age (incidence ~0.5–1.0% per year) (Rosendaal, 2005; Montagnana et al., 2010; Rabe et al., 2011). The figure below provides a graphical representation of the influence of age on risk of blood clots.
Figure 4: Risk of first-incidence VTE (per 100,000 per year) by age group (in years) in men (black bars) and women (gray bars) (Oger, 2000; Rosendaal, 2005; Rosendaal, 2016).
Other established risk factors for blood clots and associated cardiovascular problems include physical inactivity (due to, e.g., bed rest, long-distance travel, etc.), obesity, smoking, thrombophilic abnormalities, cancer, surgery, and HIV, among many others (Baron et al., 1998; Heit et al., 2000; Rosendaal, 2005; Lijfering, Rosendaal, & Cannegieter, 2010; Timp et al., 2013). In addition to age, physical inactivity is one of the most important risk factors for blood clots and mediates the risk increases for many of the others (Rosendaal, 2005). Smoking on its own is not consistently associated with increased risk of VTE (Lijfering, Rosendaal, & Cannegieter, 2010), but in combination with EE-containing birth control pills has been associated with a synergistic increase in VTE risk (Pomp, Rosendaal, & Doggen, 2008) as well as large increases in risk of heart attack—for instance 20-fold higher risk in heavy smokers (Kuhl, 1999). The table below shows the influence of a selection of known risk factors for VTE:
Thrombophilias, heritable and acquired, exist in significant percentages of the population and can lead to large increases in blood clot risk (Lijfering, Rosendaal, & Cannegieter, 2010). Moreover, they are often if not usually unknown (Morimont, Dogné, & Douxfils, 2020). This is due to the fact that screening for heritable thrombophilias is mainly based on family history, which has low sensitivity and poor predictive value for identifying people with these abnormalities (Morimont, Dogné, & Douxfils, 2020). Hence, many people are at increased risk of blood clots without realizing it. The table below shows the prevalences of a variety of thrombophilic abnormalities and their impacts on blood clot risk.
Blood clots are considered to be a multicausal disease (Rosendaal, 2005). The risk of blood clots and associated cardiovascular complications with hormonal exposure is highest when multiple risk factors combine in a given individual. Under what are among the most extreme of circumstances in terms of risk—elderly people with cancer who are on high-dose oral synthetic estrogen therapy (e.g., DES)—blood clot incidence can be as high as 15 to 28% and overall incidence of cardiovascular complications as great as 35% (Phillips et al., 2014; Sciarria et al., 2014; Turo et al., 2014). These adverse effects contribute to substantial morbidity and incidence of death in these populations. Most people are however at nowhere near as great of risk. Risk factors like age are why pregnant women can have massive levels of estradiol and progesterone with relatively little issue whereas elderly cancer patients on high-dose oral synthetic estrogen therapy have a considerable risk of death.
In the VUMC studies that found 20- to 45-fold increased incidence of blood clots with high-dose EE and CPA over 5 to 10 years in transfeminine people, the absolute incidence of blood clots was approximately 6.3% (142/10,000 people per year) in the 1989 report and 5.5% (58/10,000 people per year) in the 1997 follow up (Asscheman, Gooren, & Eklund, 1989; van Kesteren et al., 1997; Asscheman et al., 2014; Goldstein et al., 2019; Min & Hopkins, 2021). In keeping with the known influence of age on blood clot risk, the absolute incidence was 2.1% in those under 40 years of age and 12% in those over 40 years of age in the 1989 study (Asscheman, Gooren, & Eklund, 1989; Asscheman et al., 2014). In about 70% of cases, there were—aside from age—no known risk factors for blood clots (Asscheman, Gooren, & Eklund, 1989; Asscheman et al., 2014). Following subsequent replacement of EE with low-to-moderate-dose transdermal estradiol in those over 40 years of age, the incidence of blood clots decreased substantially (with only one event occurring in the transdermal estradiol group) (van Kesteren et al., 1997; Asscheman et al., 2014; Min & Hopkins, 2021). A later study in 2013 by the Ghent University Hospital in Belgium observed a blood clot incidence of 5.1% in transfeminine people using mostly oral or transdermal estradiol with or without CPA over an average treatment period of 7.7 years (range 3 months to 35 years) (Wierckx et al., 2013; Min & Hopkins, 2021). Those who had blood clots often had other risk factors such as older age, smoking, immoblization due to surgery, or hypercoagulability (Wierckx et al., 2013; Min & Hopkins, 2021). In addition to cumulative exposure time, these studies further highlight the converging impact of multiple risk factors—with estrogen type, route, and dose, progestogen exposure, and age included among them—on the risk of blood clots.
Therapeutic Implications for Transfeminine People
Due to their greater risk of blood clots and cardiovascular problems, non-bioidentical estrogens like EE and CEEs are mostly no longer used in transfeminine people. Instead, estradiol, both in oral and non-oral forms, is used. Transgender clinical guidelines generally recommend keeping estradiol levels within normal physiological ranges for non-pregnant females of around 100 to 200 pg/mL regardless of whether the route of administration of estradiol is oral or non-oral (Aly, 2018). Higher estradiol levels are not currently known to have greater therapeutic benefit in terms of feminization or breast development (Nolan & Cheung, 2020). However, higher levels, in the range of 200 to 500 pg/mL, can provide additional therapeutic effect in the area of testosterone suppression—which can be indirectly beneficial to feminization if otherwise inadequate (Aly, 2018). Despite their recommendations for keeping estradiol levels in physiological ranges, transgender clinical guidelines notably recommend doses of estradiol ester injections that reach and even greatly exceed estradiol levels of 200 pg/mL (Aly, 2021).
Based on the available research (e.g., the risk of blood clots with lower doses, comparative SHBG increases), it would not be surprising if high-dose oral estradiol (e.g., 8 mg/day) had similar risk of blood clots as the relatively lower amounts of EE in birth control pills. The risk is likely to be particularly great in combination with progestogens (e.g., CPA). Due to its greater and unnecessary risk of blood clots relative to non-oral estradiol, oral estradiol should ideally be avoided in transfeminine people—particularly in those with risk factors for blood clots such as older age (e.g., >40 years) or concomitant progestogen use. However, the convenience of oral estradiol and its relative inexpensiveness (compared to e.g. transdermal forms) are significant advantages that will also be considered by transfeminine people and their clinicians. In contrast to oral estradiol, non-oral estradiol—with estradiol levels kept in physiological ranges of for instance 100 to 200 pg/mL—appears to have minimal to no risk of blood clots. Hence, non-oral estradiol at these levels can be used in transfeminine people with little concern.
In terms of higher estradiol levels delivered non-orally, the estimated 2-fold increase in risk of blood clots with estradiol levels of approximately 300 to 500 pg/mL (Sam, 2020) is notably lower than the average 4-fold increase in risk with widely used EE-containing birth control pills. Based on the usefulness of these levels for suppressing testosterone production and the widespread usage of EE-based birth control in cisgender women throughout the world, the degree of blood clot risk with high-dose non-oral estradiol, in reasonable amounts, could be considered therapeutically acceptable in transfeminine people (Haupt et al., 2020). This may be particularly true when high-dose non-oral estradiol monotherapy is compared to combination of estradiol with antiandrogens like spironolactone, CPA, or bicalutamide, which all have their own unique risks and drawbacks. In any case, as with oral estradiol, high estradiol levels with non-oral estradiol should ideally be avoided due to the additional risk they pose, and this is especially true in those with relevant risk factors for blood clots (e.g., older age). In addition, very high doses of non-oral estradiol resulting in estradiol levels above those required for testosterone suppression are difficult to justify as they pose further unnecessary risk and offer no clear additional therapeutic benefit.
Prevention of Blood Clots
The best way to prevent blood clots from happening is to avoid risk altogether. Avoiding use of oral estradiol, excessively high doses of non-oral estradiol, and progestogens when feasible and opting for safer therapeutic choices is recommended in this regard. In addition, avoiding use of such therapies in those with risk factors like older age (>40 years), known thrombophilic abnormalities, and sedentary lifestyle is advocated. Proactive behaviors like physical activity (e.g., walking, exercise), quitting smoking, and weight loss may help to reduce the risk of blood clots (Hibbs, 2008).
Certain anticoagulant and antiplatelet medications are used to help prevent blood clots in high-risk individuals. Examples include low-dose aspirin (Mekaj, Daci, & Mekaj, 2015; Matharu et al., 2020), direct factor Xa inhibitors like rivaroxaban (Xarelto) (Blondon, 2020), and direct thrombin inhibitors like dabigatran (Pradaxa), among others. Aspirin has been found to be effective in the prevention of blood clots (Mekaj, Daci, & Mekaj, 2015; Matharu et al., 2020) and has been recommended for use specifically in transfeminine people on hormone therapy (Feldman & Goldberg, 2006; Deutsch, 2016). However, evidence is limited and conflicting for prevention of blood clots related to hormone therapy (Grady et al., 2000; Cushman et al., 2004) and use of aspirin in transfeminine people for such purposes has been recommended against by others (Shatzel, Connelly, & DeLoughery, 2017). Rivaroxaban has been associated with more than completely offset risk of blood clots with oral menopausal hormonal therapy (Blondon, 2020). In any case, no anticoagulants are currently approved or well-supported for preventing risk of blood clots with hormone therapy. Accordingly, clinical guidelines state that there is insufficient evidence to guide decision-making in this area at this time (e.g., McLintock, 2014). It should also be cautioned that anticoagulants have side effects and risks of their own and should be used carefully.
Temporary discontinuation of estrogen therapy before surgery has traditionally been thought to help reduce the risk of blood clots during recovery based on theory and has been advised as well as mandated for transfeminine people undergoing surgical procedures (e.g., Asscheman et al., 2014). However, evidence is limited and inconclusive on this strategy at present and more research is needed to determine whether it is actually beneficial or not (Boskey, Taghinia, & Ganor, 2019; Nolan & Cheung, 2020; Haveles et al., 2021; Hontscharuk et al., 2021; Kozato et al., 2021; Nolan et al., 2021; Zucker, Reisman, & Safer, 2021). Recent studies have not found reduction in risk of blood clots with discontinuation of hormone therapy before surgery in transfeminine people but these studies have been underpowered and larger studies are needed (Blasdel et al., 2021). Temporarily stopping hormone therapy can be distressing for many transfeminine people and this should be weighed accordingly. A potential alternative to discontinuation of hormone therapy is temporary use of transdermal estradiol at physiological doses which has no known blood clot risk and is more likely to be safe.
Updates
Update 1: Langley et al. (2021) [PATCH Study Results]
In February 2021, a report on long-term cardiovascular outcomes for the Prostate Adenocarcinoma: TransCutaneous Hormones (PATCH) trial was published (Langley et al., 2021). The PATCH trial is a large ongoing phase 2/3 randomized controlled trial of high-dose transdermal estradiol patches versus GnRH agonists for the treatment of prostate cancer in men (Langley et al., 2021). The estradiol patch dosage employed is specifically three to four 100 μg/day FemSeven or Progynova TS patches (Langley et al., 2021). In the February 2021 report of the study, 1,694 men were enrolled and randomized, with 790 included in the analysis for the GnRH agonist group and 904 included in the analysis for the estradiol patch group (Langley et al., 2021).
In those given estradiol, the median estradiol level was around 215 pg/mL (5%–95% range ~100–550 pg/mL) (Langley et al., 2021). About 93% of the men in this group achieved suppression of testosterone levels into the castrate range (<50 ng/dL), which was notably equal to the rate of suppression in the GnRH agonist group (~93%) (Langley et al., 2021). However, actual testosterone levels—as opposed to rates of testosterone suppression—were not provided in this report and hence comparison between groups is unavailable for this metric (Langley et al., 2021). After about 4 years median follow up, there were no significant differences on a variety of cardiovascular outcomes between the estradiol group and the GnRH agonist group (Langley et al., 2021). Among these outcomes included VTE, thromboembolic stroke, and other arterial embolic events (Langley et al., 2021). These results are in contrast to previous large clinical trials of PEP in prostate cancer, which found increased cardiovascular morbidity and risk of VTE but notably involved higher estradiol levels than employed in the PATCH trial (Ockrim & Abel, 2009; Sam, 2020). Based on their promising safety findings, the PATCH researchers stated that transdermal estrogen should be reconsidered for the treatment of prostate cancer (Langley et al., 2021).
These findings are reassuring and suggest that limitedly high levels of estradiol (e.g., 200–300 pg/mL perhaps) may likewise be acceptably safe in terms of blood clot and cardiovascular risk in transfeminine people. It should be noted however that the sample size of the trial, while large relative to previous clinical studies in this area, was underpowered for assessing risk of blood clots—which are relatively rare events that require very large samples to thoroughly quantify. Studies precisely assessing blood clot risk in peri- and postmenopausal women have included tens of thousands of individuals for instance. As such, while substantial increases in risk are not likely based on this trial, smaller increases in risk still cannot be ruled out at this time. It should additionally be noted that the robust testosterone suppression at the used doses in this study might not generalize to transfeminine people as a whole, as the men were mostly elderly and testosterone levels are known to decrease with age.
Update 2: Totaro et al. (2021) and Kotamarti et al. (2021)
Totaro, M., Palazzi, S., Castellini, C., Parisi, A., D’Amato, F., Tienforti, D., Baroni, M. G., Francavilla, S., & Barbonetti, A. (2021). Risk of Venous Thromboembolism in Transgender People Undergoing Hormone Feminizing Therapy: A Prevalence Meta-Analysis and Meta-Regression Study. Frontiers in Endocrinology, 12, 741866. [DOI:10.3389/fendo.2021.741866]
This study is the largest of its kind that has been conducted to date. The meta-analysis included 18 studies totaling 11,542 transfeminine people on hormone therapy. The pooled prevalence of VTE was 2% with a 95% confidence interval of 1 to 3%. However, there was large variability between studies. In the meta-regression analysis, older age and longer length of estrogen therapy were significantly positively associated with VTE prevalence. When analysis was restricted to those greater than or equal to 37.5 years of age, the prevalence of VTE was 3% (95% CI: 0–5%). Conversely, in those less than 37.5 years of age, the prevalence of VTE was 0% (95% CI: 0–2%). VTE prevalence was 1% (95% CI: 0–3%) with greater than or equal to 4.4 years of estrogen therapy and was 0% (95% CI: 0–3%) with less than 4.4 years of estrogen therapy. With regard to the 0% estimates, it is not the case that there is truly no risk of VTE in these instances but rather it can be assumed that the risks are sufficiently low that the meta-analysis was not powered well enough to detect and quantify them.
A limitation of the meta-analysis was that subgroup analyses based on estrogen type (i.e., estradiol vs. CEEs vs. EE) and route (e.g., oral estrogens or oral estradiol vs. transdermal estradiol) were said to not be possible due to insufficient data and hence were not performed. However, another recent meta-analysis published in July 2021, which analyzed much of the same literature as Totaro et al. (2021), did perform subgroup analyses by estrogen type and route. This publication is as follows:
Kotamarti, V. S., Greige, N., Heiman, A. J., Patel, A., & Ricci, J. A. (2021). Risk for Venous Thromboembolism in Transgender Patients Undergoing Cross-Sex Hormone Treatment: A Systematic Review. The Journal of Sexual Medicine, 18(7), 1280–1291. [DOI:10.1016/j.jsxm.2021.04.006]
And this is what they reported in terms of subgroup analyses for estrogen type and route:
Because varying VTE rates have been reported with different estrogen regimens, analyses of VTE incidence were performed comparing oral or transdermal delivery, or the specific estrogen formulation. As many studies reported populations using mixed estrogen formulations or did not report the type of estrogen regimen, further statistical analysis could not be performed.
Route of estrogen administration appeared to play a role in the AMAB population. [Oral] estrogens (7 studies; 34.0 VTE per 10,000 person-years) vs transdermal estrogens (3 studies, 11.2 VTE per 10,000 person-years). Additionally, estrogen formulation also appeared to have a difference VTE incidence. Ethinyl estradiol was also associated with increased VTE incidence (3 studies, 293.1 VTE per 10,000 person-years) followed by conjugated equine estrogens (1 study, 49.0 VTE per 10,000 person-years) and estradiol valerate (4 studies, 31.5 VTE per 10,000 person-years).
It is unclear how accurate these precise numbers are due to the quality limitations of the underlying data. Moreover, antiandrogens (e.g., CPA) were not controlled for and as discussed by this article are likely to additionally influence VTE risk. In any case, the reported numbers are interesting and are in accordance with different estrogen types and routes varying in terms of VTE risk.
References
Abdul Sultan, A., West, J., Stephansson, O., Grainge, M. J., Tata, L. J., Fleming, K. M., Humes, D., & Ludvigsson, J. F. (2015). Defining venous thromboembolism and measuring its incidence using Swedish health registries: a nationwide pregnancy cohort study. BMJ Open, 5(11), e008864. [DOI:10.1136/bmjopen-2015-008864]
Abou-Ismail, M. Y., Citla Sridhar, D., & Nayak, L. (2020). Estrogen and thrombosis: A bench to bedside review. Thrombosis Research, 192, 40–51. [DOI:10.1016/j.thromres.2020.05.008]
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2018). Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects. Transfeminine Science. [URL]
Aly. (2019). A Review of Studies on Estradiol Levels and Testosterone Suppression with High-Dose Transdermal Estradiol Gel and Ointment in Cisgender Men with Prostate Cancer. Transfeminine Science. [URL]
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Aly. (2020). Clinical Guidelines with Information on Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Aly. (2020). The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Aly. (2021). An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations. Transfeminine Science. [URL]
Amitrano, L., Guardascione, M. A., Brancaccio, V., & Balzano, A. (2002). Coagulation Disorders in Liver Disease. Seminars in Liver Disease, 22(1), 83–96. [DOI:10.1055/s-2002-23205]
Anderson, G. L., Limacher, M., Assaf, A. R., Bassford, T., Beresford, S. A., Black, H., Bonds, D., Brunner, R., Brzyski, R., Caan, B., Chlebowski, R., Curb, D., Gass, M., Hays, J., Heiss, G., Hendrix, S., Howard, B. V., Hsia, J., Hubbell, A., Jackson, R., … & Women’s Health Initiative Steering Committee. (2004). Effects of Conjugated Equine Estrogen in Postmenopausal Women With Hysterectomy: The Women’s Health Initiative Randomized Controlled Trial. JAMA, 291(14), 1701–1712. [DOI:10.1001/jama.291.14.1701]
Arnold, J. D., Sarkodie, E. P., Coleman, M. E., & Goldstein, D. A. (2016). Incidence of Venous Thromboembolism in Transgender Women Receiving Oral Estradiol. The Journal of Sexual Medicine, 13(11), 1773–1777. [DOI:10.1016/j.jsxm.2016.09.001]
Asscheman, H., Gooren, L., & Eklund, P. (1989). Mortality and morbidity in transsexual patients with cross-gender hormone treatment. Metabolism, 38(9), 869–873. [DOI:10.1016/0026-0495(89)90233-3]
Asscheman, H., Giltay, E. J., Megens, J. A., de Ronde, W. (Pim), van Trotsenburg, M. A., & Gooren, L. J. (2011). A long-term follow-up study of mortality in transsexuals receiving treatment with cross-sex hormones. European Journal of Endocrinology, 164(4), 635–642. [DOI:10.1530/eje-10-1038]
Asscheman, H., T’Sjoen, G., Lemaire, A., Mas, M., Meriggiola, M. C., Mueller, A., Kuhn, A., Dhejne, C., Morel-Journel, N., & Gooren, L. J. (2013). Venous thrombo-embolism as a complication of cross-sex hormone treatment of male-to-female transsexual subjects: a review. Andrologia, 46(7), 791–795. [DOI:10.1111/and.12150]
Baber, R. J., Panay, N., & Fenton, A. (2016). 2016 IMS Recommendations on women’s midlife health and menopause hormone therapy. Climacteric, 19(2), 109–150. [DOI:10.3109/13697137.2015.1129166]
Back, D. J., & Rogers, S. M. (2007). Review: first-pass metabolism by the gastrointestinal mucosa. Alimentary Pharmacology & Therapeutics, 1(5), 339–357. [DOI:10.1111/j.1365-2036.1987.tb00634.x]
Bagot, C., Marsh, M., Whitehead, M., Sherwood, R., Roberts, L., Patel, R., & Arya, R. (2010). The effect of estrone on thrombin generation may explain the different thrombotic risk between oral and transdermal hormone replacement therapy. Journal of Thrombosis and Haemostasis, 8(8), 1736–1744. [DOI:10.1111/j.1538-7836.2010.03953.x]
Bagot, C., Leishman, E., Onyiaodike, C., Jordan, F., Gibson, V., & Freeman, D. (2019). Changes in laboratory markers of thrombotic risk early in the first trimester of pregnancy may be linked to an increase in estradiol and progesterone. Thrombosis Research, 178, 47–53. [DOI:10.1016/j.thromres.2019.03.015]
Baron, J. A., Gridley, G., Weiderpass, E., Nyren, O., & Linet, M. (1998). Venous thromboembolism and cancer. The Lancet, 351(9109), 1077–1080. [DOI:10.1016/s0140-6736(97)10018-6]
Bezwada, P., Shaikh, A., & Misra, D. (2017). The Effect of Transdermal Estrogen Patch Use on Cardiovascular Outcomes: A Systematic Review. Journal of Women’s Health, 26(12), 1319–1325. [DOI:10.1089/jwh.2016.6151]
Blanco-Molina, M., Lozano, M., Cano, A., Cristobal, I., Pallardo, L., & Lete, I. (2012). Progestin-only contraception and venous thromboembolism. Thrombosis Research, 129(5), e257–e262. [DOI:10.1016/j.thromres.2012.02.042]
Bland, L. B., Garzotto, M., DeLoughery, T. G., Ryan, C. W., Schuff, K. G., Wersinger, E. M., Lemmon, D., & Beer, T. M. (2005). Phase II study of transdermal estradiol in androgen-independent prostate carcinoma. Cancer, 103(4), 717–723. [DOI:10.1002/cncr.20857]
Blasdel, G., Shakir, N., Parker, A., Bluebond-Langner, R., & Zhao, L. (2021). Letter to the Editor from Blasdel et al: “No Venous Thromboembolism Increase Among Transgender Female Patients Remaining on Estrogen for Gender-affirming Surgery”. The Journal of Clinical Endocrinology & Metabolism, 106(9), e3783–e3784. [DOI:10.1210/clinem/dgab243]
Blondon, M. (2020). Update On Oral Contraception And Venous Thromboembolism. HemaSphere Educational Updates in Hematology Book: 25th Congress of the European Hematology Association, Virtual Edition 2020, 4(S2). European Hematology Association. [Google Scholar] [DOI:10.1097/HS9.0000000000000444] [PDF]
Boskey, E. R., Taghinia, A. H., & Ganor, O. (2019). Association of Surgical Risk With Exogenous Hormone Use in Transgender Patients. JAMA Surgery, 154(2), 159–169. [DOI:10.1001/jamasurg.2018.4598]
Canonico, M., Plu-Bureau, G., Lowe, G. D., & Scarabin, P. (2008). Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systematic review and meta-analysis. BMJ, 336(7655), 1227–1231. [DOI:10.1136/bmj.39555.441944.be]
Canonico, M., Plu-Bureau, G., O’Sullivan, M. J., Stefanick, M. L., Cochrane, B., Scarabin, P., & Manson, J. E. (2014). Age at menopause, reproductive history, and venous thromboembolism risk among postmenopausal women. Menopause, 21(3), 214–220. [DOI:10.1097/gme.0b013e31829752e0]
Cassidy, A., & Minihane, A. (2017). The role of metabolism (and the microbiome) in defining the clinical efficacy of dietary flavonoids. The American Journal of Clinical Nutrition, 105(1), 10–22. [DOI:10.3945/ajcn.116.136051]
Chaireti, R., Gustafsson, K. M., Bystrom, B., Bremme, K., & Lindahl, T. L. (2013). Endogenous thrombin potential is higher during the luteal phase than during the follicular phase of a normal menstrual cycle. Human Reproduction, 28(7), 1846–1852. [DOI:10.1093/humrep/det092]
Choi, J., Kim, D., Park, S., Lee, H., Kim, K., Kim, K., Kim, M., Kim, S., & Kim, S. (2015). Anti-thrombotic effect of rutin isolated from Dendropanax morbifera Leveille. Journal of Bioscience and Bioengineering, 120(2), 181–186. [DOI:10.1016/j.jbiosc.2014.12.012]
Cleuren, A., Van der Linden, I., de Visser, Y., Wagenaar, G., Reitsma, P., & van Vlijmen, B. (2010). 17α‐Ethinylestradiol rapidly alters transcript levels of murine coagulation genes via estrogen receptor α. Journal of Thrombosis and Haemostasis, 8(8), 1838–1846. [DOI:10.1111/j.1538-7836.2010.03930.x]
Coelingh Bennink, H. J., Verhoeven, C., Dutman, A. E., & Thijssen, J. (2017). The use of high-dose estrogens for the treatment of breast cancer. Maturitas, 95, 11–23. [DOI:10.1016/j.maturitas.2016.10.010]
Coleman, E., Bockting, W., Botzer, M., Cohen-Kettenis, P., DeCuypere, G., Feldman, J., Fraser, L., Green, J., Knudson, G., Meyer, W. J., Monstrey, S., Adler, R. K., Brown, G. R., Devor, A. H., Ehrbar, R., Ettner, R., Eyler, E., Garofalo, R., Karasic, D. H., … & Zucker, K. (2012). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, Version 7. International Journal of Transgenderism, 13(4), 165–232. [DOI:10.1080/15532739.2011.700873] [URL] [PDF]
Conard, J., Plu-Bureau, G., Bahi, N., Horellou, M., Pelissier, C., & Thalabard, J. (2004). Progestogen-only contraception in women at high risk of venous thromboembolism. Contraception, 70(6), 437–441. [DOI:10.1016/j.contraception.2004.07.009]
Connelly, P. J., Marie Freel, E., Perry, C., Ewan, J., Touyz, R. M., Currie, G., & Delles, C. (2019). Gender-Affirming Hormone Therapy, Vascular Health and Cardiovascular Disease in Transgender Adults. Hypertension, 74(6), 1266–1274. [DOI:10.1161/hypertensionaha.119.13080]
Connors, J. M., & Middeldorp, S. (2019). Transgender patients and the role of the coagulation clinician. Journal of Thrombosis and Haemostasis, 17(11), 1790–1797. [DOI:10.1111/jth.14626]
Curb, J. D., Prentice, R. L., Bray, P. F., Langer, R. D., Van Horn, L., Barnabei, V. M., Bloch, M. J., Cyr, M. G., Gass, M., Lepine, L., Rodabough, R. J., Sidney, S., Uwaifo, G. I., & Rosendaal, F. R. (2006). Venous Thrombosis and Conjugated Equine Estrogen in Women Without a Uterus. Archives of Internal Medicine, 166(7), 772–772. [DOI:10.1001/archinte.166.7.772]
Cushman, M. (2004). Estrogen Plus Progestin and Risk of Venous Thrombosis. JAMA, 292(13), 1573–1580. [DOI:10.1001/jama.292.13.1573]
Deitcher, S. R., & Gomes, M. P. (2004). The risk of venous thromboembolic disease associated with adjuvant hormone therapy for breast carcinoma. Cancer, 101(3), 439–449. [DOI:10.1002/cncr.20347]
DeLoughery, T. G. (2011). Estrogen and thrombosis: Controversies and common sense. Reviews in Endocrine and Metabolic Disorders, 12(2), 77–84. [DOI:10.1007/s11154-011-9178-0]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Dinger, J., Do Minh, T., & Heinemann, K. (2016). Impact of estrogen type on cardiovascular safety of combined oral contraceptives. Contraception, 94(4), 328–339. [DOI:10.1016/j.contraception.2016.06.010]
Dittrich, R., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Mueller, A. (2005). Endocrine Treatment of Male-to-Female Transsexuals Using Gonadotropin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 113(10), 586–592. [DOI:10.1055/s-2005-865900]
Douxfils, J., Morimont, L., & Bouvy, C. (2020). Oral Contraceptives and Venous Thromboembolism: Focus on Testing that May Enable Prediction and Assessment of the Risk. Seminars in Thrombosis and Hemostasis, 46(8), 872–886. [DOI:10.1055/s-0040-1714140]
Douxfils, J., Klipping, C., Duijkers, I., Kinet, V., Mawet, M., Maillard, C., Jost, M., Rosing, J., & Foidart, J. (2020). Evaluation of the effect of a new oral contraceptive containing estetrol and drospirenone on hemostasis parameters. Contraception, 102(6), 396–402. [DOI:10.1016/j.contraception.2020.08.015]
Dutra, E., Lee, J., Torbati, T., Garcia, M., Merz, C. N., & Shufelt, C. (2019). Cardiovascular implications of gender-affirming hormone treatment in the transgender population. Maturitas, 129, 45–49. [DOI:10.1016/j.maturitas.2019.08.010]
Eilertsen, A. L., Dahm, A. E., Høibraaten, E., Lofthus, C. M., Mowinckel, M., & Sandset, P. M. (2019). Relationship between sex hormone binding globulin and blood coagulation in women on postmenopausal hormone treatment. Blood Coagulation & Fibrinolysis, 30(1), 17–23. [DOI:10.1097/mbc.0000000000000784]
Eisenfeld, A. J., & Aten, R. F. (1979). Estrogen receptor in the mammalian liver. In Briggs, M. H., & Corbin, A. (Eds.). Advances in Steroid Biochemistry and Pharmacology, 7, 91–117. London: Academic Press. [Google Scholar] [PubMed]
Eisenfeld, A. J., & Aten, R. F. (1987). Estrogen receptors and androgen receptors in the mammalian liver. Journal of Steroid Biochemistry, 27(4–6), 1109–1118. [DOI:10.1016/0022-4731(87)90197-x]
Fabian, C. J., & Kimler, B. F. (2005). Selective Estrogen-Receptor Modulators for Primary Prevention of Breast Cancer. Journal of Clinical Oncology, 23(8), 1644–1655. [DOI:10.1200/jco.2005.11.005]
Fåhraeus, L., & Larsson-Cohn, U. (1982). Oestrogens, gonadotrophins and SHBG during oral and cutaneous administration of oestradiol-17β to menopausal women. Acta Endocrinologica, 101(4), 592–596. [DOI:10.1530/acta.0.1010592]
Feldman, J. L., & Goldberg, J. (2006). Transgender Primary Medical Care: Suggested Guidelines for Clinicians in British Columbia. crhc/csac/Transcend Transgender Support & Education Society/Vancouver Coastal Health. [Google Scholar] [PDF]
Franchini, M., & Mannucci, P. M. (2015). Classic thrombophilic gene variants. Thrombosis and Haemostasis, 114(11), 885–889. [DOI:10.1160/th15-02-0141]
Fruzzetti, F., & Cagnacci, A. (2018). Venous thrombosis and hormonal contraception: what’s new with estradiol-based hormonal contraceptives? Open Access Journal of Contraception, 9, 75–79. [DOI:10.2147/oajc.s179673]
Gerstman, B. B., Piper, J. M., Tomita, D. K., Ferguson, W. J., Stadel, B. V., & Lundin, F. E. (1991). Oral Contraceptive Estrogen Dose and the Risk of Deep Venous Thromboembolic Disease. American Journal of Epidemiology, 133(1), 32–37. [DOI:10.1093/oxfordjournals.aje.a115799]
Getahun, D., Nash, R., Flanders, W. D., Baird, T. C., Becerra-Culqui, T. A., Cromwell, L., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Silverberg, M. J., Safer, J., Slovis, J., Tangpricha, V., & Goodman, M. (2018). Cross-sex Hormones and Acute Cardiovascular Events in Transgender Persons. Annals of Internal Medicine, 169(4), 205. [DOI:10.7326/m17-2785]
Gibney, J., Johannsson, G., Leung, K., & Ho, K. K. (2005). Comparison of the Metabolic Effects of Raloxifene and Oral Estrogen in Postmenopausal and Growth Hormone-Deficient Women. The Journal of Clinical Endocrinology & Metabolism, 90(7), 3897–3903. [DOI:10.1210/jc.2005-0173]
Gilbert, D. C., Duong, T., Sydes, M., Bara, A., Clarke, N., Abel, P., James, N., Langley, R., Parmar, M., & (2018). Transdermal oestradiol as a method of androgen suppression for prostate cancer within the STAMPEDE trial platform. BJU International, 121(5), 680–683. [DOI:10.1111/bju.14153]
Glintborg, D., T’Sjoen, G., Ravn, P., & Andersen, M. S. (2021). MANAGEMENT OF ENDOCRINE DISEASE: Optimal feminizing hormone treatment in transgender people. European Journal of Endocrinology, 185(2), R49–R63. [DOI:10.1530/eje-21-0059]
Goldstein, Z., Khan, M., Reisman, T., & Safer, J. D. (2019). Managing the risk of venous thromboembolism in transgender adults undergoing hormone therapy. Journal of Blood Medicine, 10, 209–216. [DOI:10.2147/jbm.s166780]
Gooren, L. J., & T’Sjoen, G. (2018). Endocrine treatment of aging transgender people. Reviews in Endocrine and Metabolic Disorders, 19(3), 253–262. [DOI:10.1007/s11154-018-9449-0]
Gourdy, P., Bachelot, A., Catteau-Jonard, S., Chabbert-Buffet, N., Christin-Maître, S., Conard, J., Fredenrich, A., Gompel, A., Lamiche-Lorenzini, F., Moreau, C., Plu-Bureau, G., Vambergue, A., Vergès, B., & Kerlan, V. (2012). Hormonal contraception in women at risk of vascular and metabolic disorders: Guidelines of the French Society of Endocrinology. Annales d’Endocrinologie, 73(5), 469–487. [DOI:10.1016/j.ando.2012.09.001]
Grady, D., Wenger, N. K., Herrington, D., Khan, S., Furberg, C., Hunninghake, D., Vittinghoff, E., Hulley, S., & (2000). Postmenopausal Hormone Therapy Increases Risk for Venous Thromboembolic Disease: The Heart and Estrogen/progestin Replacement Study. Annals of Internal Medicine, 132(9), 689–696. [DOI:10.7326/0003-4819-132-9-200005020-00002]
Grandi, G., Facchinetti, F., & Bitzer, J. (2017). Estradiol in hormonal contraception: real evolution or just same old wine in a new bottle? The European Journal of Contraception & Reproductive Health Care, 22(4), 245–246. [DOI:10.1080/13625187.2017.1372571]
Grandi, G., Barra, F., Ferrero, S., & Facchinetti, F. (2019). Estradiol in non-oral hormonal contraception: a “long and winding road”. Expert Review of Endocrinology & Metabolism, 14(3), 153–155. [DOI:10.1080/17446651.2019.1604217]
Grandi, G., Del Savio, M. C., Lopes da Silva-Filho, A., & Facchinetti, F. (2020). Estetrol (E4): the new estrogenic component of combined oral contraceptives. Expert Review of Clinical Pharmacology, 13(4), 327–330. [DOI:10.1080/17512433.2020.1750365]
Grandi, G., Facchinetti, F., & Bitzer, J. (2022). Confirmation of the safety of combined oral contraceptives containing oestradiol on the risk of venous thromboembolism. The European Journal of Contraception & Reproductive Health Care, 27(2), 83–84. [DOI:10.1080/13625187.2022.2029397]
Grossmann, M., Wierman, M. E., Angus, P., & Handelsman, D. J. (2018). Reproductive Endocrinology of Nonalcoholic Fatty Liver Disease. Endocrine Reviews, 40(2), 417–446. [DOI:10.1210/er.2018-00158]
Hammond, G. L. (2017). Sex Hormone-Binding Globulin and the Metabolic Syndrome. In Winters, S. J., & Huhtaniemi, I. T. (Eds.). Male Hypogonadism: Basic, Clinical and Therapeutic Principles, 2nd Edition (pp. 305–324). Cham: Springer. [DOI:10.1007/978-3-319-53298-1_15]
Hannaford, P. C., Iversen, L., Macfarlane, T. V., Elliott, A. M., Angus, V., & Lee, A. J. (2010). Mortality among contraceptive pill users: cohort evidence from Royal College of General Practitioners’ Oral Contraception Study. BMJ, 340, c927. [DOI:10.1136/bmj.c927]
Haupt, C., Henke, M., Kutschmar, A., Hauser, B., Baldinger, S., Saenz, S. R., & Schreiber, G. (2020). Antiandrogen or estradiol treatment or both during hormone therapy in transitioning transgender women. Cochrane Database of Systematic Reviews, 2020(11), CD013138. [DOI:10.1002/14651858.cd013138.pub2]
Haveles, C. S., Wang, M. M., Arjun, A., Zaila, K. E., & Lee, J. C. (2020). Effect of Cross-Sex Hormone Therapy on Venous Thromboembolism Risk in Male-to-Female Gender-Affirming Surgery. Annals of Plastic Surgery, 86(1), 109–114. [DOI:10.1097/sap.0000000000002300]
Hedlund, P. O., Johansson, R., Damber, J. E., Hagerman, I., Henriksson, P., Iversen, P., Klarskov, P., Mogensen, P., Rasmussen, F., & Varenhorst, E. (2011). Significance of pretreatment cardiovascular morbidity as a risk factor during treatment with parenteral oestrogen or combined androgen deprivation of 915 patients with metastasized prostate cancer: Evaluation of cardiovascular events in a randomized trial. Scandinavian Journal of Urology and Nephrology, 45(5), 346–353. [DOI:10.3109/00365599.2011.585820]
Heit, J. A., Silverstein, M. D., Mohr, D. N., Petterson, T. M., O’Fallon, W. M., & Melton, L. J. (2000). Risk Factors for Deep Vein Thrombosis and Pulmonary Embolism. Archives of Internal Medicine, 160(6), 809–815. [DOI:10.1001/archinte.160.6.809]
Heit, J. A., Spencer, F. A., & White, R. H. (2016). The epidemiology of venous thromboembolism. Journal of Thrombosis and Thrombolysis, 41(1), 3–14. [DOI:10.1007/s11239-015-1311-6]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society* Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658]
Hemelaar, M., van der Mooren, M. J., Rad, M., Kluft, C., & Kenemans, P. (2008). Effects of non-oral postmenopausal hormone therapy on markers of cardiovascular risk: a systematic review. Fertility and Sterility, 90(3), 642–672. [DOI:10.1016/j.fertnstert.2007.07.1298]
Hibbs, D. (2008). Hormone replacement and the treatment of the transgender patient: A critical literature review. de Chesnay, M., & Anderson, B. (Eds.). Caring for the Vulnerable: Perspectives in Nursing Theory, Practice, and Research, 2nd Edition (pp. 351–362). Sudbury, Massachusetts: Jones & Barlett. [Google Scholar] [Google Books]
Higdon, J., Drake, V. J., Delage, B., & Crozier, A. (2016). Flavonoids. Corvallis, Oregon: Micronutrient Information Center, Linus Pauling Institute, Oregon State University. [URL]
Høibraaten, E., Qvigstad, E., Arnesen, H., Larsen, S., Wickstrøm, E., & Sandset, P. M. (2000). Increased Risk of Recurrent Venous Thromboembolism during Hormone Replacement Therapy. Thrombosis and Haemostasis, 84(12), 961–967. [DOI:10.1055/s-0037-1614156]
Høibraaten, E., Qvigstad, E., Andersen, T. O., Mowinckel, M., & Sandset, P. M. (2001). The Effects of Hormone Replacement Therapy (HRT) on Hemostatic Variables in Women with Previous Venous Thromboembolism – Results from a Randomized, Double-Blind, Clinical Trial. Thrombosis and Haemostasis, 85(5), 775–781. [DOI:10.1055/s-0037-1615717]
Holmegard, H., Nordestgaard, B., Schnohr, P., Tybjærg‐Hansen, A., & Benn, M. (2014). Endogenous sex hormones and risk of venous thromboembolism in women and men. Journal of Thrombosis and Haemostasis, 12(3), 297–305. [DOI:10.1111/jth.12484]
Hontscharuk, R., Alba, B., Manno, C., Pine, E., Deutsch, M. B., Coon, D., & Schechter, L. (2021). Perioperative Transgender Hormone Management: Avoiding Venous Thromboembolism and Other Complications. Plastic & Reconstructive Surgery, 147(4), 1008–1017. [DOI:10.1097/prs.0000000000007786]
Hugon-Rodin, J., Alhenc-Gelas, M., Hemker, H. C., Brailly-Tabard, S., Guiochon-Mantel, A., Plu-Bureau, G., & Scarabin, P. (2016). Sex hormone-binding globulin and thrombin generation in women using hormonal contraception. Biomarkers, 22(1), 81–85. [DOI:10.1080/1354750x.2016.1204010]
Hulley, S. (1998). Randomized Trial of Estrogen Plus Progestin for Secondary Prevention of Coronary Heart Disease in Postmenopausal Women. JAMA, 280(7), 605–613. [DOI:10.1001/jama.280.7.605]
Iqbal, J., Ginsburg, O. M., Wijeratne, T. D., Howell, A., Evans, G., Sestak, I., & Narod, S. A. (2012). Endometrial cancer and venous thromboembolism in women under age 50 who take tamoxifen for prevention of breast cancer: A systematic review. Cancer Treatment Reviews, 38(4), 318–328. [DOI:10.1016/j.ctrv.2011.06.009]
Irwig, M. S. (2018). Cardiovascular health in transgender people. Reviews in Endocrine and Metabolic Disorders, 19(3), 243–251. [DOI:10.1007/s11154-018-9454-3]
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 204201881987116. [DOI:10.1177/2042018819871166]
Jasuja, R., Passam, F. H., Kennedy, D. R., Kim, S. H., van Hessem, L., Lin, L., Bowley, S. R., Joshi, S. S., Dilks, J. R., Furie, B., Furie, B. C., & Flaumenhaft, R. (2012). Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. Journal of Clinical Investigation, 122(6), 2104–2113. [DOI:10.1172/jci61228]
Jensen, E. V., Jacobson, H. I., Walf, A. A., & Frye, C. A. (2010). Estrogen action: A historic perspective on the implications of considering alternative approaches. Physiology & Behavior, 99(2), 151–162. [DOI:10.1016/j.physbeh.2009.08.013]
Kaemmle, L. M., Stadler, A., Janka, H., von Wolff, M., & Stute, P. (2022). The impact of micronized progesterone on cardiovascular events – a systematic review. Climacteric, 25(4), 327–336. [DOI:10.1080/13697137.2021.2022644]
Kasum, M., Danolić, D., Orešković, S., Ježek, D., Beketić-Orešković, L., & Pekez, M. (2014). Thrombosis following ovarian hyperstimulation syndrome. Gynecological Endocrinology, 30(11), 764–768. [DOI:10.3109/09513590.2014.927858]
Keenan, L., Kerr, T., Duane, M., & Van Gundy, K. (2018). Systematic Review of Hormonal Contraception and Risk of Venous Thrombosis. The Linacre Quarterly, 85(4), 470–477. [DOI:10.1177/0024363918816683]
Kerlan, V., Nahoul, K., Martelot, M., & Bercovici, J. (1994). Longitudinal study of maternal plasma bioavailable testosterone and androstanediol glucuronide levels during pregnancy. Clinical Endocrinology, 40(2), 263–267. [DOI:10.1111/j.1365-2265.1994.tb02478.x]
Khan, J., Schmidt, R. L., Spittal, M. J., Goldstein, Z., Smock, K. J., & Greene, D. N. (2019). Venous Thrombotic Risk in Transgender Women Undergoing Estrogen Therapy: A Systematic Review and Metaanalysis. Clinical Chemistry, 65(1), 57–66. [DOI:10.1373/clinchem.2018.288316]
Kitamura, T. (2001). Necessity of re‐evaluation of estramustine phosphate sodium (EMP) as a treatment option for first‐line monotherapy in advanced prostate cancer. International Journal of Urology, 8(2), 33–36. [DOI:10.1046/j.1442-2042.2001.00254.x]
Klil-Drori, A. J., Yin, H., Tagalakis, V., Aprikian, A., & Azoulay, L. (2016). Androgen Deprivation Therapy for Prostate Cancer and the Risk of Venous Thromboembolism. European Urology, 70(1), 56–61. [DOI:10.1016/j.eururo.2015.06.022]
Kohli, M., & McClellan, J. (2001). Parenteral Estrogen Therapy in Advanced Prostate Cancer: Retrospective Analysis of Intra-Muscular Estradiol Valerate in “Hormone Refractory” Prostate Disease. In Grunberg, S. M. (Ed.). Proceedings of the American Society of Clinical Oncology [Proc Am Soc Clin Oncol / Proceedings of ASCO], 20 [Thirty-Seventh Annual Meeting of the American Society of Clinical Oncology, May 12–15, 2001, San Francisco, California], 164b–164b (abstract no. 2407). Baltimore: Lippincott Williams & Wilkins. [ISSN:1081-0641] [ISBN-10:0-9664495-3-3] [Google Scholar] [WorldCat 1] [WorldCat 2] [WorldCat 3] [PDF]
Kohli, M., Alikhan, M. A., Spencer, H. J., & Carter, G. (2004). Phase I trial of intramuscular estradiol valerate (I/M-E) in hormone refractory prostate cancer. Journal of Clinical Oncology, 22(14 Suppl) [40th Annual Meeting of the American Society of Clinical Oncology: June 5–8, 2004, Ernest N. Morial Convention Center, New Orleans, Louisiana, Annual Meeting Proceedings], 436–436 (abstract no. 4726). [DOI:10.1200/jco.2004.22.90140.4726] [Google Books] [PDF]
Kohli, M. (2006). Phase II study of transdermal estradiol in androgen-independent prostate carcinoma. Cancer, 106(1), 234–235. [DOI:10.1002/cncr.21528]
Konkle, B. A., & Sood, S. L. (2019). Thrombotic Risk of Contraceptives and Other Hormonal Therapies. In Kitchens, C. S., Kessler, C. M., Konkle, B. A., Streiff, M. B., & Garcia, D. A. (Eds.). Consultative Hemostasis and Thrombosis, 4th Edition (pp. 637–650). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-46202-0.00031-5]
Kotamarti, V. S., Greige, N., Heiman, A. J., Patel, A., & Ricci, J. A. (2021). Risk for Venous Thromboembolism in Transgender Patients Undergoing Cross-Sex Hormone Treatment: A Systematic Review. The Journal of Sexual Medicine, 18(7), 1280–1291. [DOI:10.1016/j.jsxm.2021.04.006]
Kozato, A., Fox, G. W., Yong, P. C., Shin, S. J., Avanessian, B. K., Ting, J., Ling, Y., Karim, S., Safer, J. D., & Pang, J. H. (2021). No Venous Thromboembolism Increase Among Transgender Female Patients Remaining on Estrogen for Gender-Affirming Surgery. The Journal of Clinical Endocrinology & Metabolism, 106(4), 1586–1590. [DOI:10.1210/clinem/dgaa966]
Kronawitter, D., Gooren, L. J., Zollver, H., Oppelt, P. G., Beckmann, M. W., Dittrich, R., & Mueller, A. (2009). Effects of transdermal testosterone or oral dydrogesterone on hypoactive sexual desire disorder in transsexual women: results of a pilot study. European Journal of Endocrinology, 161(2), 363–368. [DOI:10.1530/eje-09-0265] [Table]
Kuhl, H. (1990). Ovulationshemmer: Die Bedeutung der Östrogendosis. [Ovulation Preventives: The Significance of the Estrogen Dose. / Ovulation Inhibitors: The Significance of Estrogen Dose.] Geburtshilfe und Frauenheilkunde, 50(12), 910–922. [Google Scholar 1] [Google Scholar 2] [PubMed] [DOI:10.1055/s-2008-1026392] [Translation]
Kuhl, H. (1996). Effects of progestogens on haemostasis. Maturitas, 24(1–2), 1–19. [DOI:10.1016/0378-5122(96)00994-2]
Kuhl, H. (1997). Metabolische Effekte der Östrogene und Gestagene. [Metabolic Effects of Estrogens and Progestogens.] Der Gynäkologe, 30(4), 357–369. [DOI:10.1007/pl00003042]
Kuhl, H. (1998). Adverse effects of estrogen treatment: natural vs. synthetic estrogens. In Lippert, T. H., Mueck, A. O., & Ginsburg, J. (Eds.). Sex Steroids and the Cardiovascular System: The Proceedings of the 1st Interdisciplinary Workshop, Tuebingen, Germany, October 1996. Parthenon Publishing Group, New York, London (pp. 201–210). London/New York: Parthenon. [Google Scholar] [Google Books] [PDF]
Kuhl, H. (1999). Hormonal contraception. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 363–407). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_18]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H., & Stevenson, J. (2006). The effect of medroxyprogesterone acetate on estrogen-dependent risks and benefits – an attempt to interpret the Women’s Health Initiative results. Gynecological Endocrinology, 22(6), 303–317. [DOI:10.1080/09513590600717368]
Lain. (2019). A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Langley, R. E., Cafferty, F. H., Alhasso, A. A., Rosen, S. D., Sundaram, S. K., Freeman, S. C., Pollock, P., Jinks, R. C., Godsland, I. F., Kockelbergh, R., Clarke, N. W., Kynaston, H. G., Parmar, M. K., & Abel, P. D. (2013). Cardiovascular outcomes in patients with locally advanced and metastatic prostate cancer treated with luteinising-hormone-releasing-hormone agonists or transdermal oestrogen: the randomised, phase 2 MRC PATCH trial (PR09). The Lancet Oncology, 14(4), 306–316. [DOI:10.1016/s1470-2045(13)70025-1]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., Paez, E., Robinson, A., McFarlane, J., Deighan, J. V., Marshall, J., Forcat, S., Weiss, M., Kockelbergh, R., Alhasso, A., Kynaston, H., & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/s0140-6736(21)00100-8]
Lax, E. (1987). Mechanisms of physiological and pharmacological sex hormone action on the mammalian liver. Journal of Steroid Biochemistry, 27(4–6), 1119–1128. [DOI:10.1016/0022-4731(87)90198-1]
Lidegaard, Ø. (2014). Hormonal contraception, thrombosis and age. Expert Opinion on Drug Safety, 13(10), 1353–1360. [DOI:10.1517/14740338.2014.950654]
Lijfering, W. M., Rosendaal, F. R., & Cannegieter, S. C. (2010). Risk factors for venous thrombosis - current understanding from an epidemiological point of view. British Journal of Haematology, 149(6), 824–833. [DOI:10.1111/j.1365-2141.2010.08206.x]
Lim, H. Y., Leemaqz, S. Y., Torkamani, N., Grossmann, M., Zajac, J. D., Nandurkar, H., Ho, P., & Cheung, A. S. (2020). Global Coagulation Assays in Transgender Women on Oral and Transdermal Estradiol Therapy. The Journal of Clinical Endocrinology & Metabolism, 105(7), e2369–e2377. [DOI:10.1210/clinem/dgaa262]
Luria, M. H. (1989). Estrogen and coronary arterial disease in men. International Journal of Cardiology, 25(2), 159–166. [DOI:10.1016/0167-5273(89)90102-2]
Ma, Y., Zeng, M., Sun, R., & Hu, M. (2015). Disposition of Flavonoids Impacts their Efficacy and Safety. Current Drug Metabolism, 15(9), 841–864. [DOI:10.2174/1389200216666150206123719]
Machin, N., & Ragni, M. V. (2020). Hormones and thrombosis: risk across the reproductive years and beyond. Translational Research, 225, 9–19. [DOI:10.1016/j.trsl.2020.06.011]
Mammen, E. F. (1992). Coagulation Abnormalities in Liver Disease. Hematology/Oncology Clinics of North America, 6(6), 1247–1257. [DOI:10.1016/s0889-8588(18)30273-9]
Mantha, S., Karp, R., Raghavan, V., Terrin, N., Bauer, K. A., & Zwicker, J. I. (2012). Assessing the risk of venous thromboembolic events in women taking progestin-only contraception: a meta-analysis. BMJ, 345, e4944. [DOI:10.1136/bmj.e4944]
Martinelli, I., Passamonti, S. M., & Bucciarelli, P. (2014). Thrombophilic states. In Biller, J., & Ferro, J. M. (Eds.). Neurologic Aspects of Systemic Disease Part II (Handbook of Clinical Neurology, Volume 120) (pp. 1061–1071). Philadelphia: Elsevier. [DOI:10.1016/b978-0-7020-4087-0.00071-1]
Martinez-Zapata, M. J., Vernooij, R. W., Uriona Tuma, S. M., Stein, A. T., Moreno, R. M., Vargas, E., Capellà, D., & Bonfill Cosp, X. (2016). Phlebotonics for venous insufficiency. Cochrane Database of Systematic Reviews, 2016(4), CD003229. [DOI:10.1002/14651858.cd003229.pub3]
Matharu, G. S., Kunutsor, S. K., Judge, A., Blom, A. W., & Whitehouse, M. R. (2020). Clinical Effectiveness and Safety of Aspirin for Venous Thromboembolism Prophylaxis After Total Hip and Knee Replacement. JAMA Internal Medicine, 180(3), 376–384. [DOI:10.1001/jamainternmed.2019.6108]
McLintock, C. (2014). Thromboembolism in pregnancy: Challenges and controversies in the prevention of pregnancy-associated venous thromboembolism and management of anticoagulation in women with mechanical prosthetic heart valves. Best Practice & Research Clinical Obstetrics & Gynaecology, 28(4), 519–536. [DOI:10.1016/j.bpobgyn.2014.03.001]
Mekaj, A., Mekaj, Y., & Daci, F. (2015). New insights into the mechanisms of action of aspirin and its use in the prevention and treatment of arterial and venous thromboembolism. Therapeutics and Clinical Risk Management, 11, 1449–1456. [DOI:10.2147/tcrm.s92222]
Mellinger, G. T., Bailar, J. C., Arduino, L. J., & the Veterans Administration Cooperative Urological Research Group. (1967). Treatment and survival of patients with cancer of the prostate. The Veterans Administration Co-operative Urological Research Group. Surgery Gynecology & Obstetrics, 124(5), 1011–1017. [Google Scholar] [PubMed]
Meng, Y., Jiang, H., Chen, A., Lu, F., Yang, H., Shen, M. Z., Sun, D., Shao, Q., & Fotherby, K. (1990). Hemostatic changes in women using a monthly injectable contraceptive for one year. Contraception, 42(4), 455–466. [DOI:10.1016/0010-7824(90)90052-w]
Mikkola, A., Ruutu, M., Aro, J., Rannikko, S., & Salo, J. (1999). The role of parenteral polyestradiol phosphate in the treatment of advanced prostatic cancer on the threshold of the new millennium. Annales Chirurgiae et Gynaecologiae, 88(1), 18–21. [Google Scholar] [PubMed] [PDF]
Min, L. L., & Hopkins, R. (2021). Endocrinological Care for Patients Undergoing Gender Affirmation (Including Risk of Thromboembolic Events). In Nikolavsky, D., & Blakely, S. A. (Eds.). Urological Care for the Transgender Patient: A Comprehensive Guide (pp. 23–36). Cham: Springer. [DOI:10.1007/978-3-030-18533-6_3]
Mohammed, K., Abu Dabrh, A. M., Benkhadra, K., Al Nofal, A., Carranza Leon, B. G., Prokop, L. J., Montori, V. M., Faubion, S. S., & Murad, M. H. (2015). Oral vs Transdermal Estrogen Therapy and Vascular Events: A Systematic Review and Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism, 100(11), 4012–4020. [DOI:10.1210/jc.2015-2237]
Montagnana, M., Favaloro, E. J., Franchini, M., Guidi, G. C., & Lippi, G. (2009). The role of ethnicity, age and gender in venous thromboembolism. Journal of Thrombosis and Thrombolysis, 29(4), 489–496. [DOI:10.1007/s11239-009-0365-8]
Moores, L., Bilello, K. L., & Murin, S. (2004). Sex and gender issues and venous thromboembolism. Clinics in Chest Medicine, 25(2), 281–297. [DOI:10.1016/j.ccm.2004.01.013]
Morimont, L., Dogné, J., & Douxfils, J. (2020). Letter to the Editors-in-Chief in response to the article of Abou-Ismail, et al. entitled “Estrogen and thrombosis: A bench to bedside review” (Thrombosis Research 192 (2020) 40–51). Thrombosis Research, 193, 221–223. [DOI:10.1016/j.thromres.2020.08.006]
Morimont, L., Haguet, H., Dogné, J., Gaspard, U., & Douxfils, J. (2021). Combined Oral Contraceptives and Venous Thromboembolism: Review and Perspective to Mitigate the Risk. Frontiers in Endocrinology, 12, 769187. [DOI:10.3389/fendo.2021.769187]
Morling, J. R., Broderick, C., Yeoh, S. E., & Kolbach, D. N. (2018). Rutosides for treatment of post-thrombotic syndrome. Cochrane Database of Systematic Reviews, 2018(11), CD005625. [DOI:10.1002/14651858.cd005625.pub4]
Mueller, A., Dittrich, R., Binder, H., Kuehnel, W., Maltaris, T., Hoffmann, I., & Beckmann, M. W. (2005). High dose estrogen treatment increases bone mineral density in male-to-female transsexuals receiving gonadotropin-releasing hormone agonist in the absence of testosterone. European Journal of Endocrinology, 153(1), 107–113. [DOI:10.1530/eje.1.01943]
Mueller, A., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Dittrich, R. (2006). Effects on the Male Endocrine System of Long-term Treatment with Gonadotropin-releasing Hormone Agonists and Estrogens in Male-to-Female Transsexuals. Hormone and Metabolic Research, 38(3), 183–187. [DOI:10.1055/s-2006-925198]
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body composition and bone mineral density in male-to-female transsexuals during cross-sex hormone therapy using gonadotrophin-releasing hormone agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074] [Table]
Nachtigall, L. E., Raju, U., Banerjee, S., Wan, L., & Levitz, M. (2000). Serum Estradiol-Binding Profiles in Postmenopausal Women Undergoing Three Common Estrogen Replacement Therapies. Menopause, 7(4), 243–250. [DOI:10.1097/00042192-200007040-00006]
Nelson, M. D., Szczepaniak, L. S., Wei, J., Szczepaniak, E., Sánchez, F. J., Vilain, E., Stern, J. H., Bergman, R. N., Bairey Merz, C. N., & Clegg, D. J. (2016). Transwomen and the Metabolic Syndrome: Is Orchiectomy Protective? Transgender Health, 1(1), 165–171. [DOI:10.1089/trgh.2016.0016]
Nolan, B. J., & Cheung, A. S. (2020). Estradiol Therapy in the Perioperative Period: Implications for Transgender People Undergoing Feminizing Hormone Therapy. The Yale Journal of Biology and Medicine, 93(4), 539–548. [PubMed] [PubMed Central]
Nolan, B. J., & Cheung, A. S. (2021). Relationship Between Serum Estradiol Concentrations and Clinical Outcomes in Transgender Individuals Undergoing Feminizing Hormone Therapy: A Narrative Review. Transgender Health, 6(3), 125–131. [DOI:10.1089/trgh.2020.0077]
Nolan, I. T., Haley, C., Morrison, S. D., Pannucci, C. J., & Satterwhite, T. (2021). Estrogen Continuation and Venous Thromboembolism in Penile Inversion Vaginoplasty. The Journal of Sexual Medicine, 18(1), 193–200. [DOI:10.1016/j.jsxm.2020.10.018]
Ockrim, J., & Abel, P. D. (2009). Androgen deprivation therapy for prostate cancer – the potential of parenteral estrogen. Central European Journal of Urology, 62, 132–140. [DOI:10.5173/ceju.2009.03.art1]
Odlind, V., Milsom, I., Persson, I., & Victor, A. (2002). Can changes in sex hormone binding globulin predict the risk of venous thromboembolism with combined oral contraceptive pills?: A discussion based on recent recommendations from the European agency for evaluation of medicinal products regarding third generation oral contraceptive pills. Acta Obstetricia et Gynecologica Scandinavica, 81(6), 482–490. [DOI:10.1080/j.1600-0412.2002.810603.x] [URL]
Oger, E., & (2000). Incidence of Venous Thromboembolism: A Community-based Study in Western France. Thrombosis and Haemostasis, 83(5), 657–660. [DOI:10.1055/s-0037-1613887]
Ohlander, S. J., Varghese, B., & Pastuszak, A. W. (2018). Erythrocytosis Following Testosterone Therapy. Sexual Medicine Reviews, 6(1), 77–85. [DOI:10.1016/j.sxmr.2017.04.001]
Olié, V., Canonico, M., & Scarabin, P. (2010). Risk of venous thrombosis with oral versus transdermal estrogen therapy among postmenopausal women. Current Opinion in Hematology, 17(5), 457–463. [DOI:10.1097/moh.0b013e32833c07bc]
Olié, V., Plu-Bureau, G., Conard, J., Horellou, M., Canonico, M., & Scarabin, P. (2011). Hormone therapy and recurrence of venous thromboembolism among postmenopausal women. Menopause, 18(5), 488–493. [DOI:10.1097/gme.0b013e3181f9f7c3]
Oliver-Williams, C., Glisic, M., Shahzad, S., Brown, E., Pellegrino Baena, C., Chadni, M., Chowdhury, R., Franco, O. H., & Muka, T. (2018). The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Human Reproduction Update, 25(2), 257–271. [DOI:10.1093/humupd/dmy039]
Olov Hedlund, P., Damber, J., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Johansson, R., Klarskov, P., Lundbeck, F., Rasmussen, F., Varenhorst, E., Viitanen, J., Olov Hedlund, P., Damber, J., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Johansson, R., Klarskov, P., Lundbeck, F., Rasmussen, F., Varenhorst, E., Viitanen, J., & The SPCG-5 Study Group. (2008). Parenteral estrogen versus combined androgen deprivation in the treatment of metastatic prostatic cancer: Part 2. Final evaluation of the Scandinavian Prostatic Cancer Group (SPCG) Study No. 5. Scandinavian Journal of Urology and Nephrology, 42(3), 220–229. [DOI:10.1080/00365590801943274]
Park, W. (2002). Selective estrogen receptor modulators (SERMS) and their roles in breast cancer prevention. Trends in Molecular Medicine, 8(2), 82–88. [DOI:10.1016/s1471-4914(02)02282-7]
Patel, K. T., Adeel, S., Rodrigues Miragaya, J., & Tangpricha, V. (2022). Progestogen Use in Gender-Affirming Hormone Therapy: A Systematic Review. Endocrine Practice, 28(12), 1244–1252. [DOI:10.1016/j.eprac.2022.08.012]
Peck-Radosavljevic, M. (2007). Review article: coagulation disorders in chronic liver disease. Alimentary Pharmacology & Therapeutics, 26, 21–28. [DOI:10.1111/j.1365-2036.2007.03509.x]
Pfeifer, S., Butts, S., Dumesic, D., Fossum, G., Gracia, C., La Barbera, A., Mersereau, J., Odem, R., Penzias, A., Pisarska, M., Rebar, R., Reindollar, R., Rosen, M., Sandlow, J., Sokol, R., Vernon, M., & Widra, E. (2017). Combined hormonal contraception and the risk of venous thromboembolism: a guideline. Fertility and Sterility, 107(1), 43–51. [DOI:10.1016/j.fertnstert.2016.09.027]
Phillips, I., Shah, S. I., Duong, T., Abel, P., & Langley, R. E. (2014). Androgen Deprivation Therapy and the Re-emergence of Parenteral Estrogen in Prostate Cancer. Oncology & Hematology Review, 10(1), 42–47. [PubMed Central] [DOI:10.17925/ohr.2014.10.1.42]
Plu-Bureau, G., Maitrot-Mantelet, L., Hugon-Rodin, J., & Canonico, M. (2013). Hormonal contraceptives and venous thromboembolism: An epidemiological update. Best Practice & Research Clinical Endocrinology & Metabolism, 27(1), 25–34. [DOI:10.1016/j.beem.2012.11.002]
Pomp, E. R., Rosendaal, F. R., & Doggen, C. J. (2008). Smoking increases the risk of venous thrombosis and acts synergistically with oral contraceptive use. American Journal of Hematology, 83(2), 97–102. [DOI:10.1002/ajh.21059]
Pond, S. M., & Tozer, T. N. (1984). First-Pass Elimination. Clinical Pharmacokinetics, 9(1), 1–25. [DOI:10.2165/00003088-198409010-00001]
Prentice, R. L., & Anderson, G. L. (2008). The Women’s Health Initiative: Lessons Learned. Annual Review of Public Health, 29(1), 131–150. [DOI:10.1146/annurev.publhealth.29.020907.090947]
Prentice, R. (2014). Postmenopausal Hormone Therapy and the Risks of Coronary Heart Disease, Breast Cancer, and Stroke. Seminars in Reproductive Medicine, 32(6), 419–425. [DOI:10.1055/s-0034-1384624]
Pyra, M., Casimiro, I., Rusie, L., Ross, N., Blum, C., Keglovitz Baker, K., Baker, A., & Schneider, J. (2020). An Observational Study of Hypertension and Thromboembolism Among Transgender Patients Using Gender-Affirming Hormone Therapy. Transgender Health, 5(1), 1–9. [DOI:10.1089/trgh.2019.0061]
Quinton, R., & Swee, D. S. (2019). Hormone replacement therapy: transgender studies show safety of estradiol. BMJ, 364, l600. [DOI:10.1136/bmj.l600]
Rabe, T., Luxembourg, B., Ludwig, M., Dinger, J. C., Bauersachs, R., Rott, H., Mueck, A. O., & Albring, C. (2011). Contraception and Thrombophilia - A statement from the German Society of Gynecological Endocrinology and Reproductive Medicine (DGGEF e. V.) and the Professional Association of the German Gynaecologists (BVF e. V.). Journal für Reproduktionsmedizin und Endokrinologie-Journal of Reproductive Medicine and Endocrinology, 8(Special Issue 1), 178–218. [URL]
Raps, M., Helmerhorst, F., Fleischer, K., Thomassen, S., Rosendaal, F., Rosing, J., Ballieux, B., & Van Vliet, H. (2012). Sex hormone‐binding globulin as a marker for the thrombotic risk of hormonal contraceptives. Journal of Thrombosis and Haemostasis, 10(6), 992–997. [DOI:10.1111/j.1538-7836.2012.04720.x]
Ravery, V., Fizazi, K., Oudard, S., Drouet, L., Eymard, J., Culine, S., Gravis, G., Hennequin, C., & Zerbib, M. (2011). The use of estramustine phosphate in the modern management of advanced prostate cancer. BJU International, 108(11), 1782–1786. [DOI:10.1111/j.1464-410x.2011.10201.x]
Reda, S., Morimont, L., Douxfils, J., & Rühl, H. (2020). Can We Measure the Individual Prothrombotic or Prohemorrhagic Tendency by Global Coagulation Tests? Hämostaseologie, 40(3), 364–378. [DOI:10.1055/a-1153-5824]
Renoux, C., Dell’Aniello, S., & Suissa, S. (2010). Hormone replacement therapy and the risk of venous thromboembolism: a population-based study. Journal of Thrombosis and Haemostasis, 8(5), 979–986. [DOI:10.1111/j.1538-7836.2010.03839.x]
Renoux, C., Dell’Aniello, S., Garbe, E., & Suissa, S. (2010). Transdermal and oral hormone replacement therapy and the risk of stroke: a nested case-control study. BMJ, 340, c2519. [DOI:10.1136/bmj.c2519]
Roach, R., Lijfering, W., Helmerhorst, F., Cannegieter, S., Rosendaal, F., & van Hylckama Vlieg, A. (2013). The risk of venous thrombosis in women over 50 years old using oral contraception or postmenopausal hormone therapy. Journal of Thrombosis and Haemostasis, 11(1), 124–131. [DOI:10.1111/jth.12060]
Roach, R. E., Lijfering, W. M., Rosendaal, F. R., Cannegieter, S. C., & le Cessie, S. (2014). Sex Difference in Risk of Second but Not of First Venous Thrombosis. Circulation, 129(1), 51–56. [DOI:10.1161/circulationaha.113.004768]
Rooijen, M., Silveira, A., Hamsten, A., & Bremme, K. (2004). Sex hormone–binding globulin—A surrogate marker for the prothrombotic effects of combined oral contraceptives. American Journal of Obstetrics and Gynecology, 190(2), 332–337. [DOI:10.1016/s0002-9378(03)00950-5]
Ropponen, A., Aittomäki, K., Vihma, V., Tikkanen, M. J., & Ylikorkala, O. (2005). Effects of Oral and Transdermal Estradiol Administration on Levels of Sex Hormone-Binding Globulin in Postmenopausal Women with and without a History of Intrahepatic Cholestasis of Pregnancy. The Journal of Clinical Endocrinology & Metabolism, 90(6), 3431–3434. [DOI:10.1210/jc.2005-0352]
Rosendaal, F. R. (2005). Venous thrombosis: the role of genes, environment, and behavior. Hematology American Society of Hematology Education Program, 2005(1), 1–12. [Google Scholar] [DOI:10.1182/asheducation-2005.1.1] [URL]
Rosendaal, F. R. (2016). Causes of venous thrombosis. Thrombosis Journal, 14(Suppl 1) [State of the Art 2016: Research and Review from the 9th Congress of the Asian-Pacific Society on Thrombosis and Hemostasis], 24. [DOI:10.1186/s12959-016-0108-y]
Rott, H. (2019). Birth Control Pills and Thrombotic Risks: Differences of Contraception Methods with and without Estrogen. Hämostaseologie, 39(1), 42–48. [DOI:10.1055/s-0039-1677806]
Rova, K., Passmark, H., & Lindqvist, P. G. (2012). Venous thromboembolism in relation to in vitro fertilization: an approach to determining the incidence and increase in risk in successful cycles. Fertility and Sterility, 97(1), 95–100. [DOI:10.1016/j.fertnstert.2011.10.038]
Rovinski, D., Ramos, R. B., Fighera, T. M., Casanova, G. K., & Spritzer, P. M. (2018). Risk of venous thromboembolism events in postmenopausal women using oral versus non-oral hormone therapy: A systematic review and meta-analysis. Thrombosis Research, 168, 83–95. [DOI:10.1016/j.thromres.2018.06.014]
Ruiz Garcia, V., López-Briz, E., Carbonell Sanchis, R., Gonzalvez Perales, J. L., & Bort-Martí, S. (2013). Megestrol acetate for treatment of anorexia-cachexia syndrome. Cochrane Database of Systematic Reviews, 2019(3), CD004310. [DOI:10.1002/14651858.cd004310.pub3]
Russell, N., Cheung, A., & Grossmann, M. (2017). Estradiol for the mitigation of adverse effects of androgen deprivation therapy. Endocrine-Related Cancer, 24(8), R297–R313. [DOI:10.1530/erc-17-0153]
Sahlin, L., & Schoultz, B. V. (1999). Liver Inclusive Protein, Lipid and Carbohydrate Metabolism. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 163–178). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_8]
Sam. (2020). Analysis of Cardiovascular and Thromboembolic Toxicity with High Dose Parenteral Polyestradiol Phosphate in the Treatment of Prostate Cancer. Transfeminine Science. [URL]
Scarabin, P. (2018). Progestogens and venous thromboembolism in menopausal women: an updated oral versus transdermal estrogen meta-analysis. Climacteric, 21(4), 341–345. [DOI:10.1080/13697137.2018.1446931]
Scarabin, P., Canonico, M., Plu-Bureau, G., & Oger, E. (2020). Menopause and hormone therapy in the 21st century: why promote transdermal estradiol and progesterone? Heart, 106(16), 1278–1278. [DOI:10.1136/heartjnl-2020-316907]
Scheres, L. J., van Hylckama Vlieg, A., Ballieux, B. E., Fauser, B. C., Rosendaal, F. R., Middeldorp, S., & Cannegieter, S. C. (2019). Endogenous sex hormones and risk of venous thromboembolism in young women. Journal of Thrombosis and Haemostasis, 17(8), 1297–1304. [DOI:10.1111/jth.14474]
Scheres, L. J., Selier, N. L., Nota, N. M., van Diemen, J. J., Cannegieter, S. C., & den Heijer, M. (2021). Effect of gender‐affirming hormone use on coagulation profiles in transmen and transwomen. Journal of Thrombosis and Haemostasis, 19(4), 1029–1037. [DOI:10.1111/jth.15256]
Schindler, A. E. (2003). Differential effects of progestins on hemostasis. Maturitas, 46, 31–37. [DOI:10.1016/j.maturitas.2003.09.016]
Schock, H., Zeleniuch-Jacquotte, A., Lundin, E., Grankvist, K., Lakso, H., Idahl, A., Lehtinen, M., Surcel, H., & Fortner, R. T. (2016). Hormone concentrations throughout uncomplicated pregnancies: a longitudinal study. BMC Pregnancy and Childbirth, 16(1), 146. [DOI:10.1186/s12884-016-0937-5]
Schröder, F. H., & Radlmaier, A. (2002). Steroidal Antiandrogens. In Jordan, C. V., & Furr, B. J. A. (Eds.). Hormone Therapy in Breast and Prostate Cancer (pp. 325–346). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-59259-152-7_15]
Sciarra, A., Gentile, V., Cattarino, S., Gentilucci, A., Alfarone, A., D’Eramo, G., & Salciccia, S. (2014). Oral ethinylestradiol in castration-resistant prostate cancer: A 10-year experience. International Journal of Urology, 22(1), 98–103. [DOI:10.1111/iju.12613]
Seaman, H. E., Langley, S. E., Farmer, R. D., & de Vries, C. S. (2007). Venous thromboembolism and cyproterone acetate in men with prostate cancer: a study using the General Practice Research Database. BJU International, 99(6), 1398–1403. [DOI:10.1111/j.1464-410x.2007.06859.x]
Shatzel, J. J., Connelly, K. J., & DeLoughery, T. G. (2017). Thrombotic issues in transgender medicine: A review. American Journal of Hematology, 92(2), 204–208. [DOI:10.1002/ajh.24593]
Shifren, J. L., Rifai, N., Desindes, S., McIlwain, M., Doros, G., & Mazer, N. A. (2008). A Comparison of the Short-Term Effects of Oral Conjugated Equine Estrogens Versus Transdermal Estradiol on C-Reactive Protein, Other Serum Markers of Inflammation, and Other Hepatic Proteins in Naturally Menopausal Women. The Journal of Clinical Endocrinology & Metabolism, 93(5), 1702–1710. [DOI:10.1210/jc.2007-2193]
Simon, T., De Jonage-Canonico, M. B., Oger, E., Wahl, D., Conard, J., Meyer, G., Emmerich, J., Barrellier, M., Guiraud, A., & Scarabin, P. (2006). Indicators of lifetime endogenous estrogen exposure and risk of venous thromboembolism. Journal of Thrombosis and Haemostasis, 4(1), 71–76. [DOI:10.1111/j.1538-7836.2005.01693.x]
Singla, N., Ghandour, R. A., & Raj, G. V. (2019). Investigational luteinizing hormone releasing hormone (LHRH) agonists and other hormonal agents in early stage clinical trials for prostate cancer. Expert Opinion on Investigational Drugs, 28(3), 249–259. [DOI:10.1080/13543784.2019.1570130]
Sitruk-Ware, R., & Nath, A. (2011). Metabolic effects of contraceptive steroids. Reviews in Endocrine and Metabolic Disorders, 12(2), 63–75. [DOI:10.1007/s11154-011-9182-4]
Sitruk-Ware, R., & Nath, A. (2013). Characteristics and metabolic effects of estrogen and progestins contained in oral contraceptive pills. Best Practice & Research Clinical Endocrinology & Metabolism, 27(1), 13–24. [DOI:10.1016/j.beem.2012.09.004]
Skouby, S. O., & Sidelmann, J. J. (2018). Impact of progestogens on hemostasis. Hormone Molecular Biology and Clinical Investigation, 37(2), 20180041. [DOI:10.1515/hmbci-2018-0041]
Smith, K., Galazi, M., Openshaw, M. R., Wilson, P., Sarker, S. J., O’Brien, N., Alifrangis, C., Stebbing, J., & Shamash, J. (2020). The Use of Transdermal Estrogen in Castrate-resistant, Steroid-refractory Prostate Cancer. Clinical Genitourinary Cancer, 18(3), e217–e223. [DOI:10.1016/j.clgc.2019.09.019]
Smith, N. L. (2004). Esterified Estrogens and Conjugated Equine Estrogens and the Risk of Venous Thrombosis. JAMA, 292(13), 1581–1587. [DOI:10.1001/jama.292.13.1581]
Smith, N. L., Blondon, M., Wiggins, K. L., Harrington, L. B., van Hylckama Vlieg, A., Floyd, J. S., Hwang, M., Bis, J. C., McKnight, B., Rice, K. M., Lumley, T., Rosendaal, F. R., Heckbert, S. R., & Psaty, B. M. (2014). Lower Risk of Cardiovascular Events in Postmenopausal Women Taking Oral Estradiol Compared With Oral Conjugated Equine Estrogens. JAMA Internal Medicine, 174(1), 25–34. [DOI:10.1001/jamainternmed.2013.11074]
Speroff, L. (1996). The Comparative Effect on Bone Density, Endometrium, and Lipids of Continuous Hormones as Replacement Therapy (CHART Study). JAMA, 276(17), 1397–1403. [DOI:10.1001/jama.1996.03540170041030]
Stanczyk, F. Z., Archer, D. F., & Bhavnani, B. R. (2013). Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception, 87(6), 706–727. [DOI:10.1016/j.contraception.2012.12.011]
Stanczyk, F. Z., Mathews, B. W., & Cortessis, V. K. (2017). Does the type of progestin influence the production of clotting factors? Contraception, 95(2), 113–116. [DOI:10.1016/j.contraception.2016.07.007]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., & Pousette, A. (1988). Single drug polyestradiol phosphate therapy in prostatic cancer. American Journal of Clinical Oncology, 11(Suppl 2), S101–S103. [DOI:10.1097/00000421-198801102-00024] [PDF]
Stegeman, B., Raps, M., Helmerhorst, F., Vos, H., van Vliet, H., Rosendaal, F., & van Hylckama Vlieg, A. (2013). Effect of ethinylestradiol dose and progestagen in combined oral contraceptives on plasma sex hormone‐binding globulin levels in premenopausal women. Journal of Thrombosis and Haemostasis, 11(1), 203–205. [DOI:10.1111/jth.12054]
Stuenkel, C. A., Davis, S. R., Gompel, A., Lumsden, M. A., Murad, M. H., Pinkerton, J. V., & Santen, R. J. (2015). Treatment of Symptoms of the Menopause: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism, 100(11), 3975–4011. [DOI:10.1210/jc.2015-2236]
Sudhir, K., & Komesaroff, P. A. (1999). Cardiovascular Actions of Estrogens in Men. The Journal of Clinical Endocrinology & Metabolism, 84(10), 3411–3415. [DOI:10.1210/jcem.84.10.5954]
Swee, D. S., Javaid, U., & Quinton, R. (2019). Estrogen Replacement in Young Hypogonadal Women—Transferrable Lessons From the Literature Related to the Care of Young Women With Premature Ovarian Failure and Transgender Women. Frontiers in Endocrinology, 10, 685. [DOI:10.3389/fendo.2019.00685]
Tadd, W., & Bayer, A. (2006). Dignity in health and social care for older Europeans: implications of a European project. Aging Health, 2(5), 771–779. [DOI:10.2217/1745509x.2.5.771]
Taylor, J. K., & Pendleton, N. (2016). Progesterone therapy for the treatment of non-cancer cachexia: a systematic review. BMJ Supportive & Palliative Care, 6(3), 276–286. [DOI:10.1136/bmjspcare-2015-001041]
Tchaikovski, S. N., & Rosing, J. (2010). Mechanisms of Estrogen-Induced Venous Thromboembolism. Thrombosis Research, 126(1), 5–11. [DOI:10.1016/j.thromres.2010.01.045]
Tepper, N. K., Whiteman, M. K., Marchbanks, P. A., James, A. H., & Curtis, K. M. (2016). Progestin-only contraception and thromboembolism: A systematic review. Contraception, 94(6), 678–700. [DOI:10.1016/j.contraception.2016.04.014]
Tepper, N. K., Jeng, G., Curtis, K. M., Boutot, M. E., Boulet, S. L., & Whiteman, M. K. (2019). Venous Thromboembolism Among Women Initiating Depot Medroxyprogesterone Acetate Immediately Postpartum. Obstetrics & Gynecology, 133(3), 533–540. [DOI:10.1097/aog.0000000000003135]
The Coronary Drug Project Research Group. (1970). The Coronary Drug Project. JAMA, 214(7), 1303–1313. [DOI:10.1001/jama.1970.03180070069012]
The Coronary Drug Project Research Group. (1973). The Coronary Drug Project. JAMA, 226(6), 652–657. [DOI:10.1001/jama.1973.03230060030009]
The Oral Contraceptive and Hemostasis Study Group. (1999). An open label, randomized study to evaluate the effects of seven monophasic oral contraceptive regimens on hemostatic variables. Contraception, 59(6), 345–355. [DOI:10.1016/s0010-7824(99)00044-x]
Timp, J. F., Braekkan, S. K., Versteeg, H. H., & Cannegieter, S. C. (2013). Epidemiology of cancer-associated venous thrombosis. Blood, 122(10), 1712–1723. [DOI:10.1182/blood-2013-04-460121]
Toorians, A. W., Thomassen, M. C., Zweegman, S., Magdeleyns, E. J., Tans, G., Gooren, L. J., & Rosing, J. (2003). Venous Thrombosis and Changes of Hemostatic Variables during Cross-Sex Hormone Treatment in Transsexual People. The Journal of Clinical Endocrinology & Metabolism, 88(12), 5723–5729. [DOI:10.1210/jc.2003-030520]
Totaro, M., Palazzi, S., Castellini, C., Parisi, A., D’Amato, F., Tienforti, D., Baroni, M. G., Francavilla, S., & Barbonetti, A. (2021). Risk of Venous Thromboembolism in Transgender People Undergoing Hormone Feminizing Therapy: A Prevalence Meta-Analysis and Meta-Regression Study. Frontiers in Endocrinology, 12, 741866. [DOI:10.3389/fendo.2021.741866]
Trémollieres, F. (2012). Contraception orale estro-progestative: quelle différence entre éthinylestradiol et estradiol? [Oral combined contraception: Is there any difference between ethinyl-estradiol and estradiol?] Gynécologie Obstétrique & Fertilité, 40(2), 109–115. [DOI:10.1016/j.gyobfe.2011.10.009]
Turo, R., Smolski, M., Esler, R., Kujawa, M. L., Bromage, S. J., Oakley, N., Adeyoju, A., Brown, S. C., Brough, R., Sinclair, A., & Collins, G. N. (2013). Diethylstilboestrol for the treatment of prostate cancer: past, present and future. Scandinavian Journal of Urology, 48(1), 4–14. [DOI:10.3109/21681805.2013.861508]
United Nations Development Programme/United Nations Population Fund/World Health Organization/World Bank Special Programme of Research, Development and Research Training in Human Reproduction, & Task Force on Long-acting Systemic Agents for Fertility Regulation. (2003). Comparative study of the effects of two once-a-month injectable contraceptives (Cyclofem® and Mesigyna®) and one oral contraceptive (Ortho-Novum 1/35®) on coagulation and fibrinolysis. Contraception, 68(3), 159–176. [DOI:10.1016/s0010-7824(03)00164-1]
van Hylckama Vlieg, A., Helmerhorst, F. M., & Rosendaal, F. R. (2010). The Risk of Deep Venous Thrombosis Associated With Injectable Depot–Medroxyprogesterone Acetate Contraceptives or a Levonorgestrel Intrauterine Device. Arteriosclerosis, Thrombosis, and Vascular Biology, 30(11), 2297–2300. [DOI:10.1161/atvbaha.110.211482]
Van Kesteren, P. J., Asscheman, H., Megens, J. A., & Gooren, L. J. (1997). Mortality and morbidity in transsexual subjects treated with cross-sex hormones. Clinical Endocrinology, 47(3), 337–343. [DOI:10.1046/j.1365-2265.1997.2601068.x]
van Vliet, H. A., Frolich, M., Christella, M., Thomassen, L., Doggen, C. J., Rosendaal, F. R., Rosing, J., & Helmerhorst, F. M. (2005). Association between sex hormone-binding globulin levels and activated protein C resistance in explaining the risk of thrombosis in users of oral contraceptives containing different progestogens. Human Reproduction, 20(2), 563–568. [DOI:10.1093/humrep/deh612]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2015). Use of combined oral contraceptives and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 350, h2135. [DOI:10.1136/bmj.h2135]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2019). Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 364, k4810. [DOI:10.1136/bmj.k4810]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
Walker, I. D. (2010). Obstetrics, Contraception, and Estrogen Replacement. In Key, N., Makris, M., O’Shaughnessy, D., & Lillicrap, D. (Eds.). Practical Hemostasis and Thrombosis, 2nd Edition (pp. 247–257). Oxford: Wiley-Blackwell. [DOI:10.1002/9781444306286.ch24]
Weinand, J. D., & Safer, J. D. (2015). Hormone therapy in transgender adults is safe with provider supervision; A review of hormone therapy sequelae for transgender individuals. Journal of Clinical & Translational Endocrinology, 2(2), 55–60. [DOI:10.1016/j.jcte.2015.02.003]
Westerlund, E., Henriksson, P., Wallén, H., Hovatta, O., Wallberg, K. R., & Antovic, A. (2012). Detection of a procoagulable state during controlled ovarian hyperstimulation for in vitro fertilization with global assays of haemostasis. Thrombosis Research, 130(4), 649–653. [DOI:10.1016/j.thromres.2011.11.024]
White, C. M., Ferraro‐Borgida, M. J., Fossati, A. T., McGill, C. C., Ahlberg, A. W., Feng, Y. J., Heller, G. V., & Chow, M. S. (1998). The Pharmacokinetics of Intravenous Estradiol—A Preliminary Study. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 18(6), 1343–1346. [DOI:10.1002/j.1875-9114.1998.tb03157.x]
Wiegratz, I., & Kuhl, H. (2006). Metabolic and clinical effects of progestogens. The European Journal of Contraception & Reproductive Health Care, 11(3), 153–161. [DOI:10.1080/13625180600772741]
Wierckx, K., Elaut, E., Declercq, E., Heylens, G., De Cuypere, G., Taes, Y., Kaufman, J. M., & T’Sjoen, G. (2013). Prevalence of cardiovascular disease and cancer during cross-sex hormone therapy in a large cohort of trans persons: a case–control study. European Journal of Endocrinology, 169(4), 471–478. [DOI:10.1530/eje-13-0493]
Writing Group for the Women’s Health Initiative Investigators. (2002). Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women: Principal Results From the Women’s Health Initiative Randomized Controlled Trial. JAMA, 288(3), 321–333. [DOI:10.1001/jama.288.3.321]
Zhang, M., Zhu, S., Yang, W., Huang, Q., & Ho, C. (2021). The biological fate and bioefficacy of citrus flavonoids: bioavailability, biotransformation, and delivery systems. Food & Function, 12(8), 3307–3323. [DOI:10.1039/d0fo03403g]
Zhao, J., Yang, J., & Xie, Y. (2019). Improvement strategies for the oral bioavailability of poorly water-soluble flavonoids: An overview. International Journal of Pharmaceutics, 570, 118642. [DOI:10.1016/j.ijpharm.2019.118642]
Zucker, R., Reisman, T., & Safer, J. D. (2021). Minimizing Venous Thromboembolism in Feminizing Hormone Therapy: Applying Lessons From Cisgender Women and Previous Data. Endocrine Practice, 27(6), 621–625. [DOI:10.1016/j.eprac.2021.03.010]
\ No newline at end of file
diff --git a/transfemscience.org/articles/genital-e2-application/index.html b/transfemscience.org/articles/genital-e2-application/index.html
index 75f37ed0..a499bcc0 100644
--- a/transfemscience.org/articles/genital-e2-application/index.html
+++ b/transfemscience.org/articles/genital-e2-application/index.html
@@ -1 +1 @@
-Genital Application via the Scrotum and Neolabia for Greatly Enhanced Absorption of Transdermal Estradiol in Transfeminine People - Transfeminine Science
Genital Application via the Scrotum and Neolabia for Greatly Enhanced Absorption of Transdermal Estradiol in Transfeminine People
By Aly | First published March 29, 2019 | Last modified April 27, 2022
Abstract / TL;DR
Genital skin, such as scrotal, penile, and labial skin, appears to have enhanced capacity for absorption relative to other skin areas. Hormonal medications like estradiol and testosterone used transdermally are normally applied to skin sites like the arms, buttocks, and abdomen. These medications have been limitedly studied by genital application. A single 100 μg/day transdermal estradiol patch normally achieves estradiol levels of around 100 pg/mL on average. An Argentinian clinical study of scrotally applied transdermal estradiol patches in men with prostate cancer found that scrotal administration resulted in much higher estradiol levels than forearm application and that a single 100 μg/day transdermal estradiol patch applied to the scrotum produced mean estradiol levels of 500 pg/mL. This is in line with roughly 5-fold higher estradiol levels with scrotal application relative to conventional skin sites. Genital application of transdermal estradiol gel has not been evaluated, but studies of testosterone gel and cream applied scrotally have found 5- to 8-fold higher testosterone levels than with application to conventional skin sites. Based on the chemical similarity of estradiol to testosterone and the comparably much higher estradiol levels with scrotal estradiol patches, this is likely to generalize to estradiol gel. The enhanced absorption of estradiol afforded by genital administration may be useful in transfeminine hormone therapy. Genital application may be used for achieving better estradiol levels in those who have less satisfactory estradiol levels with the transdermal route and for achieving higher estradiol levels for purposes of testosterone suppression, for instance with high-dose estradiol monotherapy.
Introduction
Estradiol is available for use in a variety of forms and by different routes (Wiki). One of these routes is transdermal administration, which includes estradiol patches, estradiol gels, and estradiol emulsions and sprays (Wiki). Estradiol patches and gels are by far the most commonly used forms by this route however. Conventional skin sites by which transdermal estradiol is administered include the arms and the abdomen. Typically, estradiol patches achieve estradiol levels of around 50 to 100 pg/mL per 100 μg/day patch, while estradiol gel achieves estradiol levels of around 100 pg/mL per 3 mg/day estradiol (Aly, 2020; Wiki). However, there is substantial variability between people in estradiol levels achieved with both estradiol patches and gels (Wiki). Some individuals obtain estradiol levels higher or lower than others with the same form and dose of transdermal estradiol. In a subset of people, estradiol levels may be too low for optimal therapeutic efficacy and testosterone suppression may consequently be inadequate. It may also be difficult to obtain the high estradiol levels needed for high-dose estradiol monotherapy with transdermal estradiol in people who opt for this particular therapeutic approach.
In the 1960s, a clinical study of the transdermal absorption of hydrocortisone applied to different skin sites in humans was published (Feldmann & Maibach, 1967 [Materials]). Hydrocortisone is also known as cortisol and is notable in being a steroid hormone closely related structurally to sex hormones like estradiol and testosterone. In the study, a solution of radiolabeled hydrocortisone was applied to a variety of different skin sites in men and then subsequent urinary excretion of the radioactive material (hydrocortisone and its metabolites) was quantified (Feldmann & Maibach, 1967). The researchers found that the radioactivity excreted varied considerably by application site and was dramatically higher than with the forearm and other sites in the case of application to the scrotum (Feldmann & Maibach, 1967). Relative to the forearm (1.0), the recovery of radioactivity from greatest to least was scrotum (42.0), jaw angle (13.0), forehead (6.0), underarm (3.6), scalp (3.5), back (1.7), palm of hand (0.8), ankle (0.4), and sole of foot (0.1) (Feldmann & Maibach, 1967). This study was the first to indicate that transdermal absorption via genital skin such as scrotum can be markedly higher compared to other skin areas.
Findings that hydrocortisone and other medications are absorbed much better by scrotal application raised the question of whether estradiol and testosterone would similarly have greater bioavailability by genital administration. This was eventually confirmed in the case of testosterone and led to the development of scrotal testosterone patches (brand name Testoderm) in the 1980s and their marketing approval in the United States in 1993 (Place et al., 1990; Mazer et al., 1992; Atkinson, Chang, & Snyder, 1998). Conversely, scrotal application of transdermal estradiol, perhaps owing to the much more limited use of estrogens in men, was seemingly never investigated. Recently however, the present author came upon the following obscure study on scrotum application of transdermal estradiol patches as a form of high-dose estrogen therapy for men with prostate cancer:
Premoli, F., Re, I., Asenjo, G., Maximino, G., & Micheletti, L. (2005). Tratamiento del Cáncer de Próstata Avanzado con Estrógenos Transdérmicos Escrotales (ETE). [Transdermal Scrotal Estrogen Patches (TSEP) in the Treatment of Advanced Prostate Cancer.] Revista Argentina de Urología, 70(4), 231–241. [Google Scholar] [URL] [PDF] [Translation]
This study would appear to be the first and currently only study to investigate scrotal application of transdermal estradiol. Previous searches had missed it due to the paper being in a non-English language. Before the findings of the study are discussed, some further background and reasoning for the concept of scrotal administration of transdermal estradiol will be provided. Interested readers who would prefer to just go straight to the results of the study can skip to the Study Findings section below.
For prostate cancer, injections of estradiol esters are effective, but the long-acting estradiol esters like polyestradiol phosphate and estradiol undecylate that have traditionally been used to treat prostate cancer have been discontinued. Shorter-acting estradiol esters like estradiol valerate and estradiol cypionate can be used but need to be injected frequently (e.g., weekly) and have limited availability in many parts of the world. Injections are also inconvenient and can be anxiety-provoking and painful. Transdermal estradiol patches can be effective, but generally three or four large 100 μg/day patches are required for adequate testosterone suppression (Ockrim, Lalani, & Abel, 2006; Langley et al., 2008; Langley et al., 2021). Using this many patches at the same time can be uncomfortable and expensive. Transdermal estradiol gel has been used to treat prostate cancer as well, but very high doses of estradiol have been required by this route (≥6 mg/day), and even then, only limitedly high estradiol levels and incomplete suppression of testosterone levels have been achieved (Aly, 2019).
Transdermally administered medications are absorbed through the skin. The capacity for absorption of skin varies for different skin areas. It has been known for decades that scrotal skin has a far higher capacity for absorption of medications than do most other skin sites (Feldmann & Maibach, 1967 [Materials]; Wiki). It is notable in this regard that the first transdermal testosterone patches to be introduced for medical use were scrotal patches. This was due to the large amounts of hormone that needed to be delivered in men in the case of testosterone. Non-scrotal testosterone patches were only introduced later, and had to be larger in size in comparison to scrotal testosterone patches to deliver the same amount of hormone (Behre & Nieschlag, 2012; Khera, 2013). Based on the preceding, and with estradiol closely related to testosterone structurally, scrotal application of transdermal estradiol formulations may likewise have much greater absorption than with conventional skin sites.
Premoli et al. (2005), the study cited in the Introduction section of this article, assessed the use of a single transdermal estradiol patch worn on the scrotum to treat prostate cancer. These researchers are from Argentina, a country in which poverty is high. Many people have considerable difficulty affording medications in this part of the world. In addition to the advantages of estrogen therapy over conventional antiandrogen therapy, the economic aspect was the motivation for their research. Essentially, their goal was to achieve, via a parenteral and hence less toxic route, high levels of estradiol that could fully suppress testosterone levels and treat prostate cancer while remaining affordable and reasonably convenient. The therapeutic goals of many transfeminine people, aside from treating prostate cancer, are quite similar. Hence, this therapy could be of value for transfeminine hormone therapy as well.
Sublingual estradiol via sublingual administration of oral estradiol tablets serves the purpose of achieving higher estradiol levels and testosterone suppression relative to oral estradiol for many transfeminine people. But sublingual administration of estradiol has some drawbacks, for instance a short duration, large fluctuations in estradiol levels, and some exposure of the liver to excessive estradiol levels in turn likely resulting in greater health risks like blood clots and cardiovascular problems (Sam, 2021; Wiki). Hence, an alternative option with more steady and sustained estradiol levels would be favorable.
Study Findings
First, Premoli et al. (2005) conducted a crossoverproof-of-conceptpilot experiment with two men with prostate cancer to assess whether scrotal transdermal estradiol is absorbed better than non-scrotal transdermal estradiol. One of the men used a 50 μg/day transdermal estradiol patch on the forearm first and subsequently on the scrotum, while the other man used a 100 μg/day transdermal estradiol patch on the forearm initially and then on the scrotum. The researchers found that maximal estradiol levels were much higher with scrotal application compared to application to the forearm:
Table 1: Estradiol levels with estradiol patches applied to the forearm versus scrotum in a pilot study:
Patient
Estradiol patch dose
Application site
Maximal estradiol level
Difference
Patient #1
50 μg/day
Forearm
55 pg/mL
–
Scrotum
200 pg/mL
3.6-fold
Patient #2
100 μg/day
Forearm
180 pg/mL
–
Scrotum
500 pg/mL
2.8-fold
Here is a graph of the results of the initial pilot “mini study” with the full levels (n = 2, crossover design):
Figure 1: Estradiol levels with transdermal estradiol patches applied to the scrotum in two men with prostate cancer in a crossover design (Premoli et al., 2005). Following this initial pilot experiment, estradiol levels were measured with continuous scrotally applied 100 μg/day transdermal estradiol patches in a larger sample of 35 men with prostate cancer and mean estradiol levels of around 500 pg/mL were observed (Premoli et al., 2005).
After the pilot “mini study”, Premoli et al. (2005) conducted a full prostate cancer study with 35 patients, each patient wearing one 100 μg/day transdermal estradiol patch on the scrotum. Analogously to the pilot study, estradiol levels of around 500 pg/mL were produced on average in the full sample of men, with a range of estradiol levels across patients of about 125 to 1,200 pg/mL. This wide range is consistent with the high interindividual variability in estradiol levels achieved with estradiol by the transdermal route in general (Wiki). With application to conventional skin sites, a single 100 μg/day estradiol patch will only achieve estradiol levels of around 100 pg/mL on average (Wiki; Graphs). In addition, two to six estradiol patches were reported in one study to achieve mean estradiol levels of only about 200 to 400 pg/mL (Graph; Ockrim, Lalani, & Abel, 2006). Taken together, it seems that scrotal application of transdermal estradiol patches may result in at least about 5-fold or greater bioavailability compared to placement of the patches on non-scrotal skin. This marked increase in bioavailability is analogous to the increase in bioavailability afforded by taking oral estradiol tablets sublingually (about 2- to 5-fold increase) (Wiki).
The mean levels of estradiol observed in the study (i.e., 500 pg/mL) are known to be sufficient for strong suppression of gonadal testosterone production and by extension circulating testosterone levels, with estradiol levels of ≥200 pg/mL known to suppress testosterone levels by ≥90% on average (Aly, 2018; Wiki). Unfortunately, Premoli and colleagues were not able to obtain data on testosterone suppression in most of the men in their study because at the start of the study almost all of the men already had low pre-treatment testosterone levels (mean 28 ng/dL, range 10–90 ng/dL). This was due to concomitant androgen deprivation therapy with conventional approaches like gonadectomy and GnRH modulators. In any case, the researchers reported that in two men who were not on conventional androgen deprivation therapy and who had male-range initial testosterone levels, treatment with intermittent scrotal transdermal estradiol patches resulted in testosterone levels that were 10 to 30 ng/dL during the “on” periods and 200 to 600 ng/dL during the “off” periods. This is consistent with strong suppression of testosterone levels by high estradiol levels as expected based on other clinical studies.
Implications for Transfeminine People
The findings of Premoli et al. (2005) are important as they provide valuable information supporting an additional option for enhancing the effectiveness of estradiol in transfeminine hormone therapy. Transfeminine people who have not undergone vaginoplasty can apply transdermal estradiol patches to the scrotum and those who have undergone vaginoplasty may be able to apply transdermal estradiol to the neolabia. Scrotal or neolabial application of transdermal estradiol may be useful for improving estradiol levels in those with low estradiol levels when applied to conventional skin sites, for allowing for lower doses and hence reduced costs, or for achieving higher estradiol levels for the purpose of testosterone suppression as in for instance high-dose estradiol monotherapy. Moreover, this approach for achieving higher estradiol levels may be advantageous relative to alternatives like sublingual and rectal estradiol in terms of considerations like stability of estradiol levels and potentially convenience.
Aside from transdermal estradiol patches, other transdermal forms of estradiol like transdermal estradiol gel may also achieve much higher estradiol levels by genital application similarly. It is also notable that it may not be necessary to apply estradiol gel to a large area of skin as a study of transdermal estradiol gel found that the smaller the area of application, the greater the estradiol levels achieved (Järvinen et al., 1997; Graph). Higher estradiol levels with estradiol gel by the scrotal or neolabial route still remains to be tested and confirmed however. In any case, if it does work, it has a major advantage relative to patches of only needing to be applied once a day instead of having to be worn constantly. It is notable that 100 μg/day estradiol patches can be quite large (Table). Scrotal testosterone patches were discontinued because they were too large and irritating, which is easy to imagine in practice. However, lower-dose estradiol patches like 50 or 75 μg/day are smaller in size (Table) and may be more practical and tolerable than higher-dose patches for scrotal use while still potentially achieving high levels of estradiol.
Another interesting thought relates to potential therapeutic use of transdermal progesterone. Very low but nonetheless significant circulating levels of progesterone (~0.75 ng/mL) have been observed with transdermal progesterone creams (Wiki). Because of the low progesterone levels achieved with transdermal progesterone and its lack of clear clinical effectiveness, no transdermal forms of progesterone are approved for medical use at this time. Transdermal progesterone could potentially be made more effective by scrotal application similarly to estradiol and testosterone. Whether this would translate into transdermal progesterone having actual therapeutic usefulness is unknown and possibly unlikely however—the circulating levels of progesterone that occur with transdermal progesterone may just be too low even with scrotal administration. Another issue is the genital skin has high expression of 5α-reductase, a major metabolizing enzyme for progesterone. In any case, it would nonetheless be very interesting for scrotal application of transdermal progesterone formulations to be evaluated.
In summary, Premoli et al. (2005) has shown that scrotal application of transdermal estradiol achieves much higher estradiol levels than non-scrotal application. This was already known to be the case for testosterone, but prior to this study there were no published data on this issue for estradiol.
Additional Topics on Genital Application of Estradiol
Patch Sizes and Selection
There are widely different sizes of transdermal estradiol patches in terms of brands (e.g., Climara, Vivelle, Vivelle-Dot, etc.), doses (i.e., 14 to 100 μg/day), and durations (i.e., designed for once weekly or twice weekly use) (Table). Estradiol patches range in size from smaller than a United States dime (1.65 cm2) to almost as large as a typical coffee cup base (44 cm2). This is a more than 25-fold range in size! In the case of 50 μg/day estradiol patches, sizes range from 3.3 to 22 cm2, and in the case of 100 μg/day patches, sizes range from 6.6 to 44 cm2. Wearing very large transdermal estradiol patches on the scrotum is obviously not going to be easy nor comfortable. The preceding linked table may be useful for helping to determine what the best patch brand and dose for a given person and their personal circumstances would be. Patches may also be cut and this may also be helpful.
Matrix patches are self-adhesive and release approximately 25 µg 17β-oestradiol/24 hours. Since the oestradiol is evenly distributed throughout the patch, the patches can be cut to provide the required dose. Practically, patches are cut into half or quarter as more complex divisions would be prone to inaccuracies and impracticable. Unused patch fractions may be stored in their packaging in the fridge for up to 1 week. The patch (or patch fraction) should be applied to clean dry skin over the buttocks or hips using Opsite® (a transparent adhesive film) if necessary to ensure good adhesion. […] Transdermal patches may be more difficult to use particularly when cutting patches to small sizes as they may fall off and require tape support.
Currently, the lowest-dose patch commercially available delivers 14 μg/d E2, and the most widely used low-dose patches deliver 25 μg/d. One method to deliver lower doses is to cut the patch in smaller pieces. Patches with a matrix design can be easily cut, whereas patches with a reservoir technology should not be cut. The disadvantages of cutting patches are that handling the smaller pieces may be difficult and cutting the patches is not recommended by the products’ labels. However, there is clinical experience with this, especially in Scandinavia. There, a group showed that a fractionated patch dose (one-quarter patch of a 25-μg dose approximately equals 6.2 μg or even less) applied overnight mimicked the normal, early-morning serum E2 peak and fell back to baseline within a few hours of patch removal (17).
For transdermal administration, gel and patches are available; dosing by cutting patches is more reliable when titrating the E2 serum level, and the delivery can be stopped in the morning by just taking the patch away when mimicking the circadian rhythm seen in early spontaneous puberty (Fig. 1). […] Our present recommendation is to use E2 matrix patches when available. Matrix patches are stable and have a homogenous E2 layer over the total surface and can therefore be cut into individualized doses. However, matrix patches are available with different patch areas for the same dose from different brands. The larger the patch area, the easier to cut the patch into smaller pieces for the target dose.
This is to be compared to the reservoir patches which have a separate drug layer with a liquid compartment containing a drug solution or suspension separated by the adhesive layer. By cutting such a patch, the liquid compartment will be destroyed and the drug will leak out.
It is unknown how to best deliver estradiol doses below 14 μg daily. While a European matrix patch can apparently be cut into quarters so the estrogen can be delivered overnight, the US manufacturers recommend against this and our limited experience has yielded erratic plasma estradiol levels, which suggests that these patches may not be uniformly impregnated with estradiol.
Almost all estradiol patches available today are matrix patches. Estraderm, a reservoir patch, is one of the only exceptions. It is discontinued in the United States but may still be used in the United Kingdom.
Tegaderm, a transparent film dressing, is another option for helping to hold patches in place (Reddit).
Penile Skin Application
Penile skin seems to have similar absorptive characteristics to scrotal skin and hence may also allow for improved absorption with transdermal application. As such, it could be useful as a supplement or alternative to scrotal application of transdermal estradiol. See the following literature excerpt for some more information on penile application (Hairston, Becher, & McVary, 2006):
[…] topical penile therapy has a unique set of anatomic and physiological issues that must be considered. There are several anatomic/fascial layers between the penile skin and the corpora cavernosa. The tunica albuginea is presumed to be difficult to penetrate because of its thick layers of collagen. Therefore, topical treatment trials have emphasized exposure to the glans penis because it has direct venous communication to the corpora cavernosa (40,41). The skin itself is a relatively impermeable tissue because of the stratum corneum. The horny cells at the stratum corneum are bonded with a very tight intercellular lipid matrix bilayer that makes the passage of drugs challenging (42). To overcome this barrier, investigators have used penetration enhancers that permeate this layer and reach the subdermis. Fortunately, the penis and scrotum are unique in that their stratum corneum is the most permeable of all anatomic locations tested. Depending on the molecular structure of the agent tested, there can be nearly 100% absorption of topical agents applied to these areas. Exposure to the glans affords a more easily “breached” layer. Other skin regions (e.g., back and palms) are particularly impermeable (43). An additional factor confounding efficient delivery of drug is the rich vasculature of the deep dermis that may “steal” the drugs to the systemic circulation.
Hence, if more skin area is desired for application of estradiol gel or patches, use of penile skin could possibly work as well. This may notably also be relevant to vaginal administration for most post-vaginoplasty transfeminine people (i.e., via penile inversion) as the neovaginal lining is penile skin. However, no studies exist on the absorptive effectiveness of neovaginal administration.
Genital Application of Estradiol Gel and Cream
Two clinical studies on scrotal application of transdermal testosterone gel and cream in men have been published:
Iyer, R., Mok, S. F., Savkovic, S., Turner, L., Fraser, G., Desai, R., Jayadev, V., Conway, A. J., & Handelsman, D. J. (2017). Pharmacokinetics of testosterone cream applied to scrotal skin. Andrology, 5(4), 725–731. [DOI:10.1111/andr.12357]
Kuhnert, B., Byrne, M., Simoni, M., Kopcke, W., Gerss, J., Lemmnitz, G., & Nieschlag, E. (2005). Testosterone substitution with a new transdermal, hydroalcoholic gel applied to scrotal or non-scrotal skin: a multicentre trial. European Journal of Endocrinology, 153(2), 317–326. [DOI:10.1530/eje.1.01964]
Here are some relevant excerpts from the first paper (which also briefly describe the findings of the second paper):
Scrotal skin is thin and has high steroid permeability, but the pharmacokinetics of testosterone via the scrotal skin route has not been studied in detail. The aim of this study was to define the pharmacokinetics of testosterone [cream] delivered via the scrotal skin route. The study was a single‐center, three‐phase cross‐over pharmacokinetic study of three single doses (12.5, 25, 50 mg) of testosterone cream administered in random sequence on different days with at least 2 days between doses to healthy eugonadal volunteers with endogenous testosterone suppressed by administration of nandrolone decanoate.
The bioavailability of testosterone via the scrotal skin is striking[ly] higher than for abdominal skin. Using the same testosterone cream and steroid LC-MS assay measurements, in this study a Cmax (4.6 ng/mL, 16.0 nM) was achieved with the lowest dose (12.5 mg) applied to the scrotal skin whereas applying 100 mg testosterone cream to the abdominal skin produced a Cmax of 16.3 nmol/L (4.7 ng/mL). This suggests an about eightfold increase in testosterone bioavailability, using the scrotal compared with abdominal skin routes.
One previous study has reported that the pharmacokinetics of scrotal application of testosterone gel was similar to that of a scrotal testosterone patch or a fivefold larger dose of non-scrotal testosterone gel, consistent with at least a fivefold higher transdermal bioavailability of testosterone (Kuhnert et al., 2005). Other studies assessing pharmacokinetics of testosterone application to non-scrotal skin have yielded variable time of peak concentration (Tmax) ranging from 6–16 h (Marbury et al., 2003; Miller et al., 2011; Olsson et al., 2014) but similar peak concentration (Cmax) as scrotal skin application (Rolf et al., 2002; Bouloux, 2005; Olsson et al., 2014).
We conclude that the scrotal administration of testosterone in a cream formulation provides high bioavailability, dose-dependent peak serum testosterone concentration, and tolerability with a much lower dose relative to the non-scrotal transdermal route.
Based on the findings of these studies, maximal testosterone levels with testosterone gel or cream are about 5- to 8-fold higher with with scrotal application than with application to conventional skin sites (e.g., abdomen). This is analogous to the 5-fold higher testosterone levels that have been achieved with scrotal testosterone patches compared to non-scrotal testosterone patches.
Estradiol is very similar to testosterone in chemical structure and chemical properties (e.g., lipophilicity and likely absorption characteristics). Hence, although there is currently no research assessing scrotal/neolabial application of transdermal estradiol gel or cream, or showing definitively that it achieves greater estradiol levels than conventional transdermal application, we can infer that because this has been demonstrated for testosterone, it is likely to be the case with estradiol as well. This is particularly true considering that far higher estradiol levels have already been shown with scrotal application of transdermal estradiol patches compared to non-scrotal application of such patches.
Consequently, scrotal/neolabial administration of transdermal estradiol gel or cream is likely to be a powerful alternative option for achieving high estradiol levels similarly to scrotal/neolabial application of transdermal estradiol patches. Scrotal/neolabial application of transdermal estradiol preparations may allow for greater efficacy and considerably reduced cost compared to conventional transdermal application of such formulations. In addition, whereas patches must be worn constantly and scrotal/neolabial application of such patches may be uncomfortable and inconvenient compared to conventional transdermal application, gels and creams are applied once per day and dry rapidly. Scrotal/neolabial application of patches also requires hair removal and can leave difficult-to-remove adhesive residues as well as cause adhesion-related local skin reactions. In contrast, this is not the case with gels or creams either. As such, estradiol preparations like gel or cream may be the preferred type of transdermal formulation for scrotal/neolabial use.
One possible caveat to scrotal/neolabial application is that transdermal estradiol gels are hydroalcoholic gels and consequently have the potential to irritate the skin and burn or sting when applied to the genital region. A potential solution to this is the use of transdermal estradiol preparations containing less or no alcohol, such as creams (e.g., compounded and over-the-counter products) and emulsions (e.g., Estrasorb), instead of hydroalcoholic gels. There is also a transdermal estradiol aerosol spray product (brand name Evamist) that may be another alternative option.
Taken together, transdermal testosterone gel or cream applied to scrotal skin is absorbed much better and results in approximately 5- to 8-fold higher testosterone levels than when applied to conventional skin sites. The same is likely also true for estradiol due to the close structural similarity of estradiol to testosterone and due to estradiol patches having been shown to be absorbed substantially better when applied to scrotal skin than to conventional skin sites.
Safety of Genital Application of Estradiol
Some have expressed concern as to the safety of genital application of estradiol. For instance, it has been asked whether estradiol applied to the scrotum might increase risk of testicular cancer or prostate cancer due to the high local estradiol concentrations that might result. Large observational studies of transfeminine people on standard hormone therapy have found no increase in risk of testicular cancer (Bensley et al., 2021; de Nie et al., 2021) or prostate cancer (de Nie et al., 2020). In fact, risk of prostate cancer in transfeminine people appears to be substantially decreased with standard hormone therapy (de Nie et al., 2020). Additionally, androgen deprivation reduces prostate cancer risk and progression in cisgender men, and high-dose estrogen therapy is effective for the treatment of this cancer, with a longstanding history of use (Ockrim, Lalani, & Abel, 2006; Norman et al., 2008; Langley et al., 2021). Hence, while we do not have the data needed to answer the question of whether very high local concentrations of estradiol in the testes or prostate might increase cancer risk in these tissues, the available circumstantial clinical evidence is not suggestive of such and this is reassuring.
References
Atkinson, L. E., Chang, Y. L., & Snyder, P. J. (1998). Long-term experience with testosterone replacement through scrotal skin. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 365–388). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_13]
Behre, H. M., & Nieschlag, E. (2012). Testosterone preparations for clinical use in males. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Testosterone: Action · Deficiency · Substitution, 4th Edition (pp. 309–335). Cambridge/New York: Cambridge University Press. [DOI:10.1017/CBO9781139003353.016]
Bensley, J. G., Cheung, A. S., Grossmann, M., & Papa, N. (2022). Testicular Cancer in Trans People Using Feminising Hormone Therapy–A Brief Review. Urology, 160, 1–4. [DOI:10.1016/j.urology.2021.11.014]
de Nie, I., de Blok, C., van der Sluis, T. M., Barbé, E., Pigot, G., Wiepjes, C. M., Nota, N. M., van Mello, N. M., Valkenburg, N. E., Huirne, J., Gooren, L., van Moorselaar, R., Dreijerink, K., & den Heijer, M. (2020). Prostate Cancer Incidence under Androgen Deprivation: Nationwide Cohort Study in Trans Women Receiving Hormone Treatment. The Journal of Clinical Endocrinology and Metabolism, 105(9), e3293–e3299. [DOI:10.1210/clinem/dgaa412]
de Nie, I., Wiepjes, C. M., Blok, C. J., Moorselaar, R. J., Pigot, G. L., van der Sluis, T. M., Barbé, E., van der Voorn, P., van Mello, N. M., Huirne, J., & den Heijer, M. (2021). Incidence of testicular cancer in trans women using gender‐affirming hormonal treatment: a nationwide cohort study. BJU International, 129(4), 491–497. [DOI:10.1111/bju.15575]
Feldmann, R. J., & Maibach, H. I. (1967). Regional variation in percutaneous penetration of 14C cortisol in man. The Journal of Investigative Dermatology, 48(2), 181–183. [DOI:10.1038/jid.1967.29]
Hairston, J. C., Becher, E., & McVary, K. T. (2006). Topical and Intra-Urethral Therapy. In Mulcahy, J. J. (Ed.). Male Sexual Function: A Guide to Clinical Management, 2nd Edition (Current Clinical Urology) (pp. 303–321). Totowa: Humana Press. [DOI:10.1007/978-1-59745-155-0_14] [Google Books]
Iyer, R., Mok, S. F., Savkovic, S., Turner, L., Fraser, G., Desai, R., Jayadev, V., Conway, A. J., & Handelsman, D. J. (2017). Pharmacokinetics of testosterone cream applied to scrotal skin. Andrology, 5(4), 725–731. [DOI:10.1111/andr.12357]
Järvinen, A., Granander, M., Nykänen, S., Laine, T., Geurts, P., & Viitanen, A. (1997). Steady-state pharmacokinetics of oestradiol gel in post-menopausal women: effects of application area and washing. British Journal of Obstetrics and Gynaecology, 104(Suppl 16), 14–18. [DOI:10.1111/j.1471-0528.1997.tb11562.x]
Khera, M. (2013). Treatment Options for Testosterone Replacement Therapy. In Hellstrom, W. J. G. (Ed.). Androgen Deficiency and Testosterone Replacement: Current Controversies and Strategies (pp. 129–139). Totowa: Humana Press. [DOI:10.1007/978-1-62703-179-0_10]
Klein, K. O., Rosenfield, R. L., Santen, R. J., Gawlik, A. M., Backeljauw, P. F., Gravholt, C. H., Sas, T., & Mauras, N. (2018). Estrogen replacement in Turner syndrome: literature review and practical considerations. The Journal of Clinical Endocrinology & Metabolism, 103(5), 1790–1803. [DOI:10.1210/jc.2017-02183]
Kuhnert, B., Byrne, M., Simoni, M., Kopcke, W., Gerss, J., Lemmnitz, G., & Nieschlag, E. (2005). Testosterone substitution with a new transdermal, hydroalcoholic gel applied to scrotal or non-scrotal skin: a multicentre trial. European Journal of Endocrinology, 153(2), 317–326. [DOI:10.1530/eje.1.01964]
Langley, R. E., Godsland, I. F., Kynaston, H., Clarke, N. W., Rosen, S. D., Morgan, R. C., Pollock, P., Kockelbergh, R., Lalani, e., Dearnaley, D., Parmar, M., & Abel, P. D. (2008). Early hormonal data from a multicentre phase II trial using transdermal oestrogen patches as first‐line hormonal therapy in patients with locally advanced or metastatic prostate cancer. BJU International, 102(4), 442–445. [DOI:10.1111/j.1464-410X.2008.07583.x]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., … & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/S0140-6736(21)00100-8]
Matthews, D., Bath, L., Högler, W., Mason, A., Smyth, A., & Skae, M. (2017). Hormone supplementation for pubertal induction in girls. Archives of Disease in Childhood, 102(10), 975–980. [DOI:10.1136/archdischild-2016-311372]
Mazer, N. A., Heiber, W. E., Moellmer, J. F., Meikle, A. W., Stringham, J. D., Sanders, S. W., Tolman, K. G., & Odell, W. D. (1992). Enhanced transdermal delivery of testosterone: a new physiological approach for androgen replacement in hypogonadal men. Journal of Controlled Release, 19(1–3), 347–361. [DOI:10.1016/0168-3659(92)90089-A]
Norjavaara, E., Ankarberg-Lindgren, C., & Kriström, B. (2016). Sex steroid replacement therapy in female hypogonadism from childhood to young adulthood. In Bourguignon J.-P., Parent A.-S. (Eds.). Puberty from Bench to Clinic. Lessons for Clinical Management of Pubertal Disorders (Endocrine Development, Volume29) (pp. 198–213). Basel: Karger. [DOI:10.1159/000438892]
Norman, G., Dean, M. E., Langley, R. E., Hodges, Z. C., Ritchie, G., Parmar, M. K., Sydes, M. R., Abel, P., & Eastwood, A. J. (2008). Parenteral oestrogen in the treatment of prostate cancer: a systematic review. British Journal of Cancer, 98(4), 697–707. [DOI:10.1038/sj.bjc.6604230]
Ockrim, J., Lalani, E. N., & Abel, P. (2006). Therapy insight: parenteral estrogen treatment for prostate cancer—a new dawn for an old therapy. Nature clinical practice Oncology, 3(10), 552–563. [DOI:10.1038/ncponc0602]
Place, V. A., Atkinson, L., Prather, D. A., Trunnell, N., & Yates, F. E. (1990). Transdermal testosterone replacement through genital skin. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 1st Edition (pp. 165–181). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-662-00814-0_9]
Premoli, F., Re, I., Asenjo, G., Maximino, G., & Micheletti, L. (2005). Tratamiento del Cáncer de Próstata Avanzado con Estrógenos Transdérmicos Escrotales (ETE). [Transdermal Scrotal Estrogen Patches (TSEP) in the Treatment of Advanced Prostate Cancer.] Revista Argentina de Urología, 70(4), 231–241. [Google Scholar] [URL] [PDF] [Translation]
Rosenfield, R. L., Kiess, W., & de Muinck Keizer-Schrama, S. (2006). Physiologic induction of puberty in Turner syndrome with very low-dose estradiol. International Congress Series, 1298, 71–79. [DOI:10.1016/j.ics.2006.07.003]
\ No newline at end of file
+Genital Application via the Scrotum and Neolabia for Greatly Enhanced Absorption of Transdermal Estradiol in Transfeminine People - Transfeminine Science
Genital Application via the Scrotum and Neolabia for Greatly Enhanced Absorption of Transdermal Estradiol in Transfeminine People
By Aly | First published March 29, 2019 | Last modified April 27, 2022
Abstract / TL;DR
Genital skin, such as scrotal, penile, and labial skin, appears to have enhanced capacity for absorption relative to other skin areas. Hormonal medications like estradiol and testosterone used transdermally are normally applied to skin sites like the arms, buttocks, and abdomen. These medications have been limitedly studied by genital application. A single 100 μg/day transdermal estradiol patch normally achieves estradiol levels of around 100 pg/mL on average. An Argentinian clinical study of scrotally applied transdermal estradiol patches in men with prostate cancer found that scrotal administration resulted in much higher estradiol levels than forearm application and that a single 100 μg/day transdermal estradiol patch applied to the scrotum produced mean estradiol levels of 500 pg/mL. This is in line with roughly 5-fold higher estradiol levels with scrotal application relative to conventional skin sites. Genital application of transdermal estradiol gel has not been evaluated, but studies of testosterone gel and cream applied scrotally have found 5- to 8-fold higher testosterone levels than with application to conventional skin sites. Based on the chemical similarity of estradiol to testosterone and the comparably much higher estradiol levels with scrotal estradiol patches, this is likely to generalize to estradiol gel. The enhanced absorption of estradiol afforded by genital administration may be useful in transfeminine hormone therapy. Genital application may be used for achieving better estradiol levels in those who have less satisfactory estradiol levels with the transdermal route and for achieving higher estradiol levels for purposes of testosterone suppression, for instance with high-dose estradiol monotherapy.
Introduction
Estradiol is available for use in a variety of forms and by different routes (Wiki). One of these routes is transdermal administration, which includes estradiol patches, estradiol gels, and estradiol emulsions and sprays (Wiki). Estradiol patches and gels are by far the most commonly used forms by this route however. Conventional skin sites by which transdermal estradiol is administered include the arms and the abdomen. Typically, estradiol patches achieve estradiol levels of around 50 to 100 pg/mL per 100 μg/day patch, while estradiol gel achieves estradiol levels of around 100 pg/mL per 3 mg/day estradiol (Aly, 2020; Wiki). However, there is substantial variability between people in estradiol levels achieved with both estradiol patches and gels (Wiki). Some individuals obtain estradiol levels higher or lower than others with the same form and dose of transdermal estradiol. In a subset of people, estradiol levels may be too low for optimal therapeutic efficacy and testosterone suppression may consequently be inadequate. It may also be difficult to obtain the high estradiol levels needed for high-dose estradiol monotherapy with transdermal estradiol in people who opt for this particular therapeutic approach.
In the 1960s, a clinical study of the transdermal absorption of hydrocortisone applied to different skin sites in humans was published (Feldmann & Maibach, 1967 [Materials]). Hydrocortisone is also known as cortisol and is notable in being a steroid hormone closely related structurally to sex hormones like estradiol and testosterone. In the study, a solution of radiolabeled hydrocortisone was applied to a variety of different skin sites in men and then subsequent urinary excretion of the radioactive material (hydrocortisone and its metabolites) was quantified (Feldmann & Maibach, 1967). The researchers found that the radioactivity excreted varied considerably by application site and was dramatically higher than with the forearm and other sites in the case of application to the scrotum (Feldmann & Maibach, 1967). Relative to the forearm (1.0), the recovery of radioactivity from greatest to least was scrotum (42.0), jaw angle (13.0), forehead (6.0), underarm (3.6), scalp (3.5), back (1.7), palm of hand (0.8), ankle (0.4), and sole of foot (0.1) (Feldmann & Maibach, 1967). This study was the first to indicate that transdermal absorption via genital skin such as scrotum can be markedly higher compared to other skin areas.
Findings that hydrocortisone and other medications are absorbed much better by scrotal application raised the question of whether estradiol and testosterone would similarly have greater bioavailability by genital administration. This was eventually confirmed in the case of testosterone and led to the development of scrotal testosterone patches (brand name Testoderm) in the 1980s and their marketing approval in the United States in 1993 (Place et al., 1990; Mazer et al., 1992; Atkinson, Chang, & Snyder, 1998). Conversely, scrotal application of transdermal estradiol, perhaps owing to the much more limited use of estrogens in men, was seemingly never investigated. Recently however, the present author came upon the following obscure study on scrotum application of transdermal estradiol patches as a form of high-dose estrogen therapy for men with prostate cancer:
Premoli, F., Re, I., Asenjo, G., Maximino, G., & Micheletti, L. (2005). Tratamiento del Cáncer de Próstata Avanzado con Estrógenos Transdérmicos Escrotales (ETE). [Transdermal Scrotal Estrogen Patches (TSEP) in the Treatment of Advanced Prostate Cancer.] Revista Argentina de Urología, 70(4), 231–241. [Google Scholar] [URL] [PDF] [Translation]
This study would appear to be the first and currently only study to investigate scrotal application of transdermal estradiol. Previous searches had missed it due to the paper being in a non-English language. Before the findings of the study are discussed, some further background and reasoning for the concept of scrotal administration of transdermal estradiol will be provided. Interested readers who would prefer to just go straight to the results of the study can skip to the Study Findings section below.
For prostate cancer, injections of estradiol esters are effective, but the long-acting estradiol esters like polyestradiol phosphate and estradiol undecylate that have traditionally been used to treat prostate cancer have been discontinued. Shorter-acting estradiol esters like estradiol valerate and estradiol cypionate can be used but need to be injected frequently (e.g., weekly) and have limited availability in many parts of the world. Injections are also inconvenient and can be anxiety-provoking and painful. Transdermal estradiol patches can be effective, but generally three or four large 100 μg/day patches are required for adequate testosterone suppression (Ockrim, Lalani, & Abel, 2006; Langley et al., 2008; Langley et al., 2021). Using this many patches at the same time can be uncomfortable and expensive. Transdermal estradiol gel has been used to treat prostate cancer as well, but very high doses of estradiol have been required by this route (≥6 mg/day), and even then, only limitedly high estradiol levels and incomplete suppression of testosterone levels have been achieved (Aly, 2019).
Transdermally administered medications are absorbed through the skin. The capacity for absorption of skin varies for different skin areas. It has been known for decades that scrotal skin has a far higher capacity for absorption of medications than do most other skin sites (Feldmann & Maibach, 1967 [Materials]; Wiki). It is notable in this regard that the first transdermal testosterone patches to be introduced for medical use were scrotal patches. This was due to the large amounts of hormone that needed to be delivered in men in the case of testosterone. Non-scrotal testosterone patches were only introduced later, and had to be larger in size in comparison to scrotal testosterone patches to deliver the same amount of hormone (Behre & Nieschlag, 2012; Khera, 2013). Based on the preceding, and with estradiol closely related to testosterone structurally, scrotal application of transdermal estradiol formulations may likewise have much greater absorption than with conventional skin sites.
Premoli et al. (2005), the study cited in the Introduction section of this article, assessed the use of a single transdermal estradiol patch worn on the scrotum to treat prostate cancer. These researchers are from Argentina, a country in which poverty is high. Many people have considerable difficulty affording medications in this part of the world. In addition to the advantages of estrogen therapy over conventional antiandrogen therapy, the economic aspect was the motivation for their research. Essentially, their goal was to achieve, via a parenteral and hence less toxic route, high levels of estradiol that could fully suppress testosterone levels and treat prostate cancer while remaining affordable and reasonably convenient. The therapeutic goals of many transfeminine people, aside from treating prostate cancer, are quite similar. Hence, this therapy could be of value for transfeminine hormone therapy as well.
Sublingual estradiol via sublingual administration of oral estradiol tablets serves the purpose of achieving higher estradiol levels and testosterone suppression relative to oral estradiol for many transfeminine people. But sublingual administration of estradiol has some drawbacks, for instance a short duration, large fluctuations in estradiol levels, and some exposure of the liver to excessive estradiol levels in turn likely resulting in greater health risks like blood clots and cardiovascular problems (Sam, 2021; Wiki). Hence, an alternative option with more steady and sustained estradiol levels would be favorable.
Study Findings
First, Premoli et al. (2005) conducted a crossoverproof-of-conceptpilot experiment with two men with prostate cancer to assess whether scrotal transdermal estradiol is absorbed better than non-scrotal transdermal estradiol. One of the men used a 50 μg/day transdermal estradiol patch on the forearm first and subsequently on the scrotum, while the other man used a 100 μg/day transdermal estradiol patch on the forearm initially and then on the scrotum. The researchers found that maximal estradiol levels were much higher with scrotal application compared to application to the forearm:
Table 1: Estradiol levels with estradiol patches applied to the forearm versus scrotum in a pilot study:
Patient
Estradiol patch dose
Application site
Maximal estradiol level
Difference
Patient #1
50 μg/day
Forearm
55 pg/mL
–
Scrotum
200 pg/mL
3.6-fold
Patient #2
100 μg/day
Forearm
180 pg/mL
–
Scrotum
500 pg/mL
2.8-fold
Here is a graph of the results of the initial pilot “mini study” with the full levels (n = 2, crossover design):
Figure 1: Estradiol levels with transdermal estradiol patches applied to the scrotum in two men with prostate cancer in a crossover design (Premoli et al., 2005). Following this initial pilot experiment, estradiol levels were measured with continuous scrotally applied 100 μg/day transdermal estradiol patches in a larger sample of 35 men with prostate cancer and mean estradiol levels of around 500 pg/mL were observed (Premoli et al., 2005).
After the pilot “mini study”, Premoli et al. (2005) conducted a full prostate cancer study with 35 patients, each patient wearing one 100 μg/day transdermal estradiol patch on the scrotum. Analogously to the pilot study, estradiol levels of around 500 pg/mL were produced on average in the full sample of men, with a range of estradiol levels across patients of about 125 to 1,200 pg/mL. This wide range is consistent with the high interindividual variability in estradiol levels achieved with estradiol by the transdermal route in general (Wiki). With application to conventional skin sites, a single 100 μg/day estradiol patch will only achieve estradiol levels of around 100 pg/mL on average (Wiki; Graphs). In addition, two to six estradiol patches were reported in one study to achieve mean estradiol levels of only about 200 to 400 pg/mL (Graph; Ockrim, Lalani, & Abel, 2006). Taken together, it seems that scrotal application of transdermal estradiol patches may result in at least about 5-fold or greater bioavailability compared to placement of the patches on non-scrotal skin. This marked increase in bioavailability is analogous to the increase in bioavailability afforded by taking oral estradiol tablets sublingually (about 2- to 5-fold increase) (Wiki).
The mean levels of estradiol observed in the study (i.e., 500 pg/mL) are known to be sufficient for strong suppression of gonadal testosterone production and by extension circulating testosterone levels, with estradiol levels of ≥200 pg/mL known to suppress testosterone levels by ≥90% on average (Aly, 2018; Wiki). Unfortunately, Premoli and colleagues were not able to obtain data on testosterone suppression in most of the men in their study because at the start of the study almost all of the men already had low pre-treatment testosterone levels (mean 28 ng/dL, range 10–90 ng/dL). This was due to concomitant androgen deprivation therapy with conventional approaches like gonadectomy and GnRH modulators. In any case, the researchers reported that in two men who were not on conventional androgen deprivation therapy and who had male-range initial testosterone levels, treatment with intermittent scrotal transdermal estradiol patches resulted in testosterone levels that were 10 to 30 ng/dL during the “on” periods and 200 to 600 ng/dL during the “off” periods. This is consistent with strong suppression of testosterone levels by high estradiol levels as expected based on other clinical studies.
Implications for Transfeminine People
The findings of Premoli et al. (2005) are important as they provide valuable information supporting an additional option for enhancing the effectiveness of estradiol in transfeminine hormone therapy. Transfeminine people who have not undergone vaginoplasty can apply transdermal estradiol patches to the scrotum and those who have undergone vaginoplasty may be able to apply transdermal estradiol to the neolabia. Scrotal or neolabial application of transdermal estradiol may be useful for improving estradiol levels in those with low estradiol levels when applied to conventional skin sites, for allowing for lower doses and hence reduced costs, or for achieving higher estradiol levels for the purpose of testosterone suppression as in for instance high-dose estradiol monotherapy. Moreover, this approach for achieving higher estradiol levels may be advantageous relative to alternatives like sublingual and rectal estradiol in terms of considerations like stability of estradiol levels and potentially convenience.
Aside from transdermal estradiol patches, other transdermal forms of estradiol like transdermal estradiol gel may also achieve much higher estradiol levels by genital application similarly. It is also notable that it may not be necessary to apply estradiol gel to a large area of skin as a study of transdermal estradiol gel found that the smaller the area of application, the greater the estradiol levels achieved (Järvinen et al., 1997; Graph). Higher estradiol levels with estradiol gel by the scrotal or neolabial route still remains to be tested and confirmed however. In any case, if it does work, it has a major advantage relative to patches of only needing to be applied once a day instead of having to be worn constantly. It is notable that 100 μg/day estradiol patches can be quite large (Table). Scrotal testosterone patches were discontinued because they were too large and irritating, which is easy to imagine in practice. However, lower-dose estradiol patches like 50 or 75 μg/day are smaller in size (Table) and may be more practical and tolerable than higher-dose patches for scrotal use while still potentially achieving high levels of estradiol.
Another interesting thought relates to potential therapeutic use of transdermal progesterone. Very low but nonetheless significant circulating levels of progesterone (~0.75 ng/mL) have been observed with transdermal progesterone creams (Wiki). Because of the low progesterone levels achieved with transdermal progesterone and its lack of clear clinical effectiveness, no transdermal forms of progesterone are approved for medical use at this time. Transdermal progesterone could potentially be made more effective by scrotal application similarly to estradiol and testosterone. Whether this would translate into transdermal progesterone having actual therapeutic usefulness is unknown and possibly unlikely however—the circulating levels of progesterone that occur with transdermal progesterone may just be too low even with scrotal administration. Another issue is the genital skin has high expression of 5α-reductase, a major metabolizing enzyme for progesterone. In any case, it would nonetheless be very interesting for scrotal application of transdermal progesterone formulations to be evaluated.
In summary, Premoli et al. (2005) has shown that scrotal application of transdermal estradiol achieves much higher estradiol levels than non-scrotal application. This was already known to be the case for testosterone, but prior to this study there were no published data on this issue for estradiol.
Additional Topics on Genital Application of Estradiol
Patch Sizes and Selection
There are widely different sizes of transdermal estradiol patches in terms of brands (e.g., Climara, Vivelle, Vivelle-Dot, etc.), doses (i.e., 14 to 100 μg/day), and durations (i.e., designed for once weekly or twice weekly use) (Table). Estradiol patches range in size from smaller than a United States dime (1.65 cm2) to almost as large as a typical coffee cup base (44 cm2). This is a more than 25-fold range in size! In the case of 50 μg/day estradiol patches, sizes range from 3.3 to 22 cm2, and in the case of 100 μg/day patches, sizes range from 6.6 to 44 cm2. Wearing very large transdermal estradiol patches on the scrotum is obviously not going to be easy nor comfortable. The preceding linked table may be useful for helping to determine what the best patch brand and dose for a given person and their personal circumstances would be. Patches may also be cut and this may also be helpful.
Matrix patches are self-adhesive and release approximately 25 µg 17β-oestradiol/24 hours. Since the oestradiol is evenly distributed throughout the patch, the patches can be cut to provide the required dose. Practically, patches are cut into half or quarter as more complex divisions would be prone to inaccuracies and impracticable. Unused patch fractions may be stored in their packaging in the fridge for up to 1 week. The patch (or patch fraction) should be applied to clean dry skin over the buttocks or hips using Opsite® (a transparent adhesive film) if necessary to ensure good adhesion. […] Transdermal patches may be more difficult to use particularly when cutting patches to small sizes as they may fall off and require tape support.
Currently, the lowest-dose patch commercially available delivers 14 μg/d E2, and the most widely used low-dose patches deliver 25 μg/d. One method to deliver lower doses is to cut the patch in smaller pieces. Patches with a matrix design can be easily cut, whereas patches with a reservoir technology should not be cut. The disadvantages of cutting patches are that handling the smaller pieces may be difficult and cutting the patches is not recommended by the products’ labels. However, there is clinical experience with this, especially in Scandinavia. There, a group showed that a fractionated patch dose (one-quarter patch of a 25-μg dose approximately equals 6.2 μg or even less) applied overnight mimicked the normal, early-morning serum E2 peak and fell back to baseline within a few hours of patch removal (17).
For transdermal administration, gel and patches are available; dosing by cutting patches is more reliable when titrating the E2 serum level, and the delivery can be stopped in the morning by just taking the patch away when mimicking the circadian rhythm seen in early spontaneous puberty (Fig. 1). […] Our present recommendation is to use E2 matrix patches when available. Matrix patches are stable and have a homogenous E2 layer over the total surface and can therefore be cut into individualized doses. However, matrix patches are available with different patch areas for the same dose from different brands. The larger the patch area, the easier to cut the patch into smaller pieces for the target dose.
This is to be compared to the reservoir patches which have a separate drug layer with a liquid compartment containing a drug solution or suspension separated by the adhesive layer. By cutting such a patch, the liquid compartment will be destroyed and the drug will leak out.
It is unknown how to best deliver estradiol doses below 14 μg daily. While a European matrix patch can apparently be cut into quarters so the estrogen can be delivered overnight, the US manufacturers recommend against this and our limited experience has yielded erratic plasma estradiol levels, which suggests that these patches may not be uniformly impregnated with estradiol.
Almost all estradiol patches available today are matrix patches. Estraderm, a reservoir patch, is one of the only exceptions. It is discontinued in the United States but may still be used in the United Kingdom.
Tegaderm, a transparent film dressing, is another option for helping to hold patches in place (Reddit).
Penile Skin Application
Penile skin seems to have similar absorptive characteristics to scrotal skin and hence may also allow for improved absorption with transdermal application. As such, it could be useful as a supplement or alternative to scrotal application of transdermal estradiol. See the following literature excerpt for some more information on penile application (Hairston, Becher, & McVary, 2006):
[…] topical penile therapy has a unique set of anatomic and physiological issues that must be considered. There are several anatomic/fascial layers between the penile skin and the corpora cavernosa. The tunica albuginea is presumed to be difficult to penetrate because of its thick layers of collagen. Therefore, topical treatment trials have emphasized exposure to the glans penis because it has direct venous communication to the corpora cavernosa (40,41). The skin itself is a relatively impermeable tissue because of the stratum corneum. The horny cells at the stratum corneum are bonded with a very tight intercellular lipid matrix bilayer that makes the passage of drugs challenging (42). To overcome this barrier, investigators have used penetration enhancers that permeate this layer and reach the subdermis. Fortunately, the penis and scrotum are unique in that their stratum corneum is the most permeable of all anatomic locations tested. Depending on the molecular structure of the agent tested, there can be nearly 100% absorption of topical agents applied to these areas. Exposure to the glans affords a more easily “breached” layer. Other skin regions (e.g., back and palms) are particularly impermeable (43). An additional factor confounding efficient delivery of drug is the rich vasculature of the deep dermis that may “steal” the drugs to the systemic circulation.
Hence, if more skin area is desired for application of estradiol gel or patches, use of penile skin could possibly work as well. This may notably also be relevant to vaginal administration for most post-vaginoplasty transfeminine people (i.e., via penile inversion) as the neovaginal lining is penile skin. However, no studies exist on the absorptive effectiveness of neovaginal administration.
Genital Application of Estradiol Gel and Cream
Two clinical studies on scrotal application of transdermal testosterone gel and cream in men have been published:
Iyer, R., Mok, S. F., Savkovic, S., Turner, L., Fraser, G., Desai, R., Jayadev, V., Conway, A. J., & Handelsman, D. J. (2017). Pharmacokinetics of testosterone cream applied to scrotal skin. Andrology, 5(4), 725–731. [DOI:10.1111/andr.12357]
Kuhnert, B., Byrne, M., Simoni, M., Kopcke, W., Gerss, J., Lemmnitz, G., & Nieschlag, E. (2005). Testosterone substitution with a new transdermal, hydroalcoholic gel applied to scrotal or non-scrotal skin: a multicentre trial. European Journal of Endocrinology, 153(2), 317–326. [DOI:10.1530/eje.1.01964]
Here are some relevant excerpts from the first paper (which also briefly describe the findings of the second paper):
Scrotal skin is thin and has high steroid permeability, but the pharmacokinetics of testosterone via the scrotal skin route has not been studied in detail. The aim of this study was to define the pharmacokinetics of testosterone [cream] delivered via the scrotal skin route. The study was a single‐center, three‐phase cross‐over pharmacokinetic study of three single doses (12.5, 25, 50 mg) of testosterone cream administered in random sequence on different days with at least 2 days between doses to healthy eugonadal volunteers with endogenous testosterone suppressed by administration of nandrolone decanoate.
The bioavailability of testosterone via the scrotal skin is striking[ly] higher than for abdominal skin. Using the same testosterone cream and steroid LC-MS assay measurements, in this study a Cmax (4.6 ng/mL, 16.0 nM) was achieved with the lowest dose (12.5 mg) applied to the scrotal skin whereas applying 100 mg testosterone cream to the abdominal skin produced a Cmax of 16.3 nmol/L (4.7 ng/mL). This suggests an about eightfold increase in testosterone bioavailability, using the scrotal compared with abdominal skin routes.
One previous study has reported that the pharmacokinetics of scrotal application of testosterone gel was similar to that of a scrotal testosterone patch or a fivefold larger dose of non-scrotal testosterone gel, consistent with at least a fivefold higher transdermal bioavailability of testosterone (Kuhnert et al., 2005). Other studies assessing pharmacokinetics of testosterone application to non-scrotal skin have yielded variable time of peak concentration (Tmax) ranging from 6–16 h (Marbury et al., 2003; Miller et al., 2011; Olsson et al., 2014) but similar peak concentration (Cmax) as scrotal skin application (Rolf et al., 2002; Bouloux, 2005; Olsson et al., 2014).
We conclude that the scrotal administration of testosterone in a cream formulation provides high bioavailability, dose-dependent peak serum testosterone concentration, and tolerability with a much lower dose relative to the non-scrotal transdermal route.
Based on the findings of these studies, maximal testosterone levels with testosterone gel or cream are about 5- to 8-fold higher with with scrotal application than with application to conventional skin sites (e.g., abdomen). This is analogous to the 5-fold higher testosterone levels that have been achieved with scrotal testosterone patches compared to non-scrotal testosterone patches.
Estradiol is very similar to testosterone in chemical structure and chemical properties (e.g., lipophilicity and likely absorption characteristics). Hence, although there is currently no research assessing scrotal/neolabial application of transdermal estradiol gel or cream, or showing definitively that it achieves greater estradiol levels than conventional transdermal application, we can infer that because this has been demonstrated for testosterone, it is likely to be the case with estradiol as well. This is particularly true considering that far higher estradiol levels have already been shown with scrotal application of transdermal estradiol patches compared to non-scrotal application of such patches.
Consequently, scrotal/neolabial administration of transdermal estradiol gel or cream is likely to be a powerful alternative option for achieving high estradiol levels similarly to scrotal/neolabial application of transdermal estradiol patches. Scrotal/neolabial application of transdermal estradiol preparations may allow for greater efficacy and considerably reduced cost compared to conventional transdermal application of such formulations. In addition, whereas patches must be worn constantly and scrotal/neolabial application of such patches may be uncomfortable and inconvenient compared to conventional transdermal application, gels and creams are applied once per day and dry rapidly. Scrotal/neolabial application of patches also requires hair removal and can leave difficult-to-remove adhesive residues as well as cause adhesion-related local skin reactions. In contrast, this is not the case with gels or creams either. As such, estradiol preparations like gel or cream may be the preferred type of transdermal formulation for scrotal/neolabial use.
One possible caveat to scrotal/neolabial application is that transdermal estradiol gels are hydroalcoholic gels and consequently have the potential to irritate the skin and burn or sting when applied to the genital region. A potential solution to this is the use of transdermal estradiol preparations containing less or no alcohol, such as creams (e.g., compounded and over-the-counter products) and emulsions (e.g., Estrasorb), instead of hydroalcoholic gels. There is also a transdermal estradiol aerosol spray product (brand name Evamist) that may be another alternative option.
Taken together, transdermal testosterone gel or cream applied to scrotal skin is absorbed much better and results in approximately 5- to 8-fold higher testosterone levels than when applied to conventional skin sites. The same is likely also true for estradiol due to the close structural similarity of estradiol to testosterone and due to estradiol patches having been shown to be absorbed substantially better when applied to scrotal skin than to conventional skin sites.
Safety of Genital Application of Estradiol
Some have expressed concern as to the safety of genital application of estradiol. For instance, it has been asked whether estradiol applied to the scrotum might increase risk of testicular cancer or prostate cancer due to the high local estradiol concentrations that might result. Large observational studies of transfeminine people on standard hormone therapy have found no increase in risk of testicular cancer (Bensley et al., 2021; de Nie et al., 2021) or prostate cancer (de Nie et al., 2020). In fact, risk of prostate cancer in transfeminine people appears to be substantially decreased with standard hormone therapy (de Nie et al., 2020). Additionally, androgen deprivation reduces prostate cancer risk and progression in cisgender men, and high-dose estrogen therapy is effective for the treatment of this cancer, with a longstanding history of use (Ockrim, Lalani, & Abel, 2006; Norman et al., 2008; Langley et al., 2021). Hence, while we do not have the data needed to answer the question of whether very high local concentrations of estradiol in the testes or prostate might increase cancer risk in these tissues, the available circumstantial clinical evidence is not suggestive of such and this is reassuring.
References
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). A Review of Studies on Estradiol Levels and Testosterone Suppression with High-Dose Transdermal Estradiol Gel and Ointment in Cisgender Men with Prostate Cancer. Transfeminine Science. [URL]
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
Atkinson, L. E., Chang, Y. L., & Snyder, P. J. (1998). Long-term experience with testosterone replacement through scrotal skin. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 365–388). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_13]
Behre, H. M., & Nieschlag, E. (2012). Testosterone preparations for clinical use in males. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Testosterone: Action · Deficiency · Substitution, 4th Edition (pp. 309–335). Cambridge/New York: Cambridge University Press. [DOI:10.1017/CBO9781139003353.016]
Bensley, J. G., Cheung, A. S., Grossmann, M., & Papa, N. (2022). Testicular Cancer in Trans People Using Feminising Hormone Therapy–A Brief Review. Urology, 160, 1–4. [DOI:10.1016/j.urology.2021.11.014]
de Nie, I., de Blok, C., van der Sluis, T. M., Barbé, E., Pigot, G., Wiepjes, C. M., Nota, N. M., van Mello, N. M., Valkenburg, N. E., Huirne, J., Gooren, L., van Moorselaar, R., Dreijerink, K., & den Heijer, M. (2020). Prostate Cancer Incidence under Androgen Deprivation: Nationwide Cohort Study in Trans Women Receiving Hormone Treatment. The Journal of Clinical Endocrinology and Metabolism, 105(9), e3293–e3299. [DOI:10.1210/clinem/dgaa412]
de Nie, I., Wiepjes, C. M., Blok, C. J., Moorselaar, R. J., Pigot, G. L., van der Sluis, T. M., Barbé, E., van der Voorn, P., van Mello, N. M., Huirne, J., & den Heijer, M. (2021). Incidence of testicular cancer in trans women using gender‐affirming hormonal treatment: a nationwide cohort study. BJU International, 129(4), 491–497. [DOI:10.1111/bju.15575]
Feldmann, R. J., & Maibach, H. I. (1967). Regional variation in percutaneous penetration of 14C cortisol in man. The Journal of Investigative Dermatology, 48(2), 181–183. [DOI:10.1038/jid.1967.29]
Hairston, J. C., Becher, E., & McVary, K. T. (2006). Topical and Intra-Urethral Therapy. In Mulcahy, J. J. (Ed.). Male Sexual Function: A Guide to Clinical Management, 2nd Edition (Current Clinical Urology) (pp. 303–321). Totowa: Humana Press. [DOI:10.1007/978-1-59745-155-0_14] [Google Books]
Iyer, R., Mok, S. F., Savkovic, S., Turner, L., Fraser, G., Desai, R., Jayadev, V., Conway, A. J., & Handelsman, D. J. (2017). Pharmacokinetics of testosterone cream applied to scrotal skin. Andrology, 5(4), 725–731. [DOI:10.1111/andr.12357]
Järvinen, A., Granander, M., Nykänen, S., Laine, T., Geurts, P., & Viitanen, A. (1997). Steady-state pharmacokinetics of oestradiol gel in post-menopausal women: effects of application area and washing. British Journal of Obstetrics and Gynaecology, 104(Suppl 16), 14–18. [DOI:10.1111/j.1471-0528.1997.tb11562.x]
Khera, M. (2013). Treatment Options for Testosterone Replacement Therapy. In Hellstrom, W. J. G. (Ed.). Androgen Deficiency and Testosterone Replacement: Current Controversies and Strategies (pp. 129–139). Totowa: Humana Press. [DOI:10.1007/978-1-62703-179-0_10]
Klein, K. O., Rosenfield, R. L., Santen, R. J., Gawlik, A. M., Backeljauw, P. F., Gravholt, C. H., Sas, T., & Mauras, N. (2018). Estrogen replacement in Turner syndrome: literature review and practical considerations. The Journal of Clinical Endocrinology & Metabolism, 103(5), 1790–1803. [DOI:10.1210/jc.2017-02183]
Kuhnert, B., Byrne, M., Simoni, M., Kopcke, W., Gerss, J., Lemmnitz, G., & Nieschlag, E. (2005). Testosterone substitution with a new transdermal, hydroalcoholic gel applied to scrotal or non-scrotal skin: a multicentre trial. European Journal of Endocrinology, 153(2), 317–326. [DOI:10.1530/eje.1.01964]
Langley, R. E., Godsland, I. F., Kynaston, H., Clarke, N. W., Rosen, S. D., Morgan, R. C., Pollock, P., Kockelbergh, R., Lalani, e., Dearnaley, D., Parmar, M., & Abel, P. D. (2008). Early hormonal data from a multicentre phase II trial using transdermal oestrogen patches as first‐line hormonal therapy in patients with locally advanced or metastatic prostate cancer. BJU International, 102(4), 442–445. [DOI:10.1111/j.1464-410X.2008.07583.x]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., … & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/S0140-6736(21)00100-8]
Matthews, D., Bath, L., Högler, W., Mason, A., Smyth, A., & Skae, M. (2017). Hormone supplementation for pubertal induction in girls. Archives of Disease in Childhood, 102(10), 975–980. [DOI:10.1136/archdischild-2016-311372]
Mazer, N. A., Heiber, W. E., Moellmer, J. F., Meikle, A. W., Stringham, J. D., Sanders, S. W., Tolman, K. G., & Odell, W. D. (1992). Enhanced transdermal delivery of testosterone: a new physiological approach for androgen replacement in hypogonadal men. Journal of Controlled Release, 19(1–3), 347–361. [DOI:10.1016/0168-3659(92)90089-A]
Norjavaara, E., Ankarberg-Lindgren, C., & Kriström, B. (2016). Sex steroid replacement therapy in female hypogonadism from childhood to young adulthood. In Bourguignon J.-P., Parent A.-S. (Eds.). Puberty from Bench to Clinic. Lessons for Clinical Management of Pubertal Disorders (Endocrine Development, Volume29) (pp. 198–213). Basel: Karger. [DOI:10.1159/000438892]
Norman, G., Dean, M. E., Langley, R. E., Hodges, Z. C., Ritchie, G., Parmar, M. K., Sydes, M. R., Abel, P., & Eastwood, A. J. (2008). Parenteral oestrogen in the treatment of prostate cancer: a systematic review. British Journal of Cancer, 98(4), 697–707. [DOI:10.1038/sj.bjc.6604230]
Ockrim, J., Lalani, E. N., & Abel, P. (2006). Therapy insight: parenteral estrogen treatment for prostate cancer—a new dawn for an old therapy. Nature clinical practice Oncology, 3(10), 552–563. [DOI:10.1038/ncponc0602]
Place, V. A., Atkinson, L., Prather, D. A., Trunnell, N., & Yates, F. E. (1990). Transdermal testosterone replacement through genital skin. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 1st Edition (pp. 165–181). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-662-00814-0_9]
Premoli, F., Re, I., Asenjo, G., Maximino, G., & Micheletti, L. (2005). Tratamiento del Cáncer de Próstata Avanzado con Estrógenos Transdérmicos Escrotales (ETE). [Transdermal Scrotal Estrogen Patches (TSEP) in the Treatment of Advanced Prostate Cancer.] Revista Argentina de Urología, 70(4), 231–241. [Google Scholar] [URL] [PDF] [Translation]
Rosenfield, R. L., Kiess, W., & de Muinck Keizer-Schrama, S. (2006). Physiologic induction of puberty in Turner syndrome with very low-dose estradiol. International Congress Series, 1298, 71–79. [DOI:10.1016/j.ics.2006.07.003]
Sam. (2021). An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People. Transfeminine Science. [URL]
\ No newline at end of file
diff --git a/transfemscience.org/articles/hair-loss/index.html b/transfemscience.org/articles/hair-loss/index.html
index 1e880687..bab23055 100644
--- a/transfemscience.org/articles/hair-loss/index.html
+++ b/transfemscience.org/articles/hair-loss/index.html
@@ -1 +1 @@
-A Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People - Transfeminine Science
A Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People
By Sam | First published September 8, 2025 | Last modified September 9, 2025
Abstract / TL;DR
Androgenetic alopecia is a common condition affecting a significant proportion of transfeminine people at onset of hormonal transition. Although several studies have assessed the influence of gender-affirming hormone therapy on scalp hair, there are no robust data to guide optimal dosing regimens. In the wider population, 5α-reductase inhibitors and other treatments, such as minoxidil, have been found to act dose-dependently and additively or synergistically with each other to halt and partially reverse hair loss caused by androgens. Some transfeminine people prefer to add these treatments to their regimens to try to regrow more hair or as prophylaxis against further hair loss. Dutasteride is a superior 5α-reductase inhibitor to finasteride in terms of efficacy and has similar adverse effects. For transfeminine people who wish to use minoxidil, the route of administration should be considered and determined on an individual basis. Other agents, such as spironolactone, may also provide benefit. Research into new therapies which could one day result in new pharmaceutical options for transfeminine people is currently ongoing in the general population.
Introduction
Scalp hair loss, particularly androgenetic alopecia (AGA), is a condition that affects millions of people worldwide and that carries significant psychosocial implications. Hair is a major component of human identity and is often integral to gender expression. As such, hair loss can be particularly distressing for transfeminine people (Marks & Senna, 2020; Gao et al., 2023a; Tang et al., 2023). Over the last decade, there has been a surge in interest in pharmaceutical treatments for treating AGA among both the scientific community and the general population. However, considerably less attention has been devoted to treating AGA in transfeminine people, specifically.
A few studies have evaluated the effects of feminising hormone therapy on scalp hair. A multicentre prospective study found a slight but statistically significant reduction in average Norwood–Hamilton score after 12 months in transfeminine people treated with estradiol and cyproterone acetate (Cocchetti et al., 2023). In another study, duration of estradiol use with or without spironolactone was associated with a −0.07 cm (95% CI: [−0.10, −0.04]) reduction in lateral forehead length per year in the first few years of treatment (Nguyen et al., 2025). Finally, a prospective study demonstrated statistically significant increases in follicular density and total hair count at both the midfrontal and vertex (crown) scalp after 24 weeks in transfeminine people on an unspecified regimen (Tang et al., 2025a). This was accompanied by a reduction in average hair shaft diameter, driven by increases in intermediate and vellus hair density, but with no significant change in terminal hair density. Individual case studies and series showing improvement have also been reported in the literature (Dewhurst & Underhill, 1979; Adenuga, Summers, & Bergfel, 2012; Stevenson, Wixon, & Safer, 2016). Overall, it appears that AGA progression is usually halted and partially reversed due to reduced or suppressed testosterone levels. However, there is a lack of data to inform long-term outcomes. Reversal of AGA may be incomplete in many transfeminine people, particularly in advanced stages, due to irreversible follicular miniaturisation.
The most common therapies for AGA in cisgender people include 5α-reductase inhibitors (5-ARIs) and minoxidil (Gao et al., 2023b; Devjani et al., 2023). Minoxidil use in transfeminine people has been examined by a small study (Zaminski et al., 2025). A study also found that concomitant use of finasteride, dutasteride, and/or minoxidil was associated with a lower hairline compared to treatment with estradiol alone or estradiol and spironolactone alone (Nguyen et al., 2025). However, limitations of these studies include risk of bias due to study design, possible confounding due to secondary factors, and in the case of the former study, lack of a control group. As such, there is a paucity of reliable data to show if these treatments, especially 5-ARIs, provide additional benefit over gender-affirming hormone therapy alone (T’Sjoen et al., 2019; Prince & Safer, 2020; Irwig, 2021; Coleman et al., 2022; Gao et al., 2023b). Despite this, a considerable subset of transfeminine people opt to use 5α-reductase inhibitors and minoxidil (Leinung, Feustel, & Joseph, 2018; Nguyen et al., 2025).
The purpose of this literature review is to critically summarise various pharmaceutical interventions that have been shown to have an acceptable safety profile in the general population and that may therefore be used as adjunct therapy to treat and as prophylaxis against hair loss in transfeminine people if desired. Non-pharmaceutical interventions, such as microneedling and hair transplantation, are outside the scope of this article. Specifically, this review focuses predominantly on 5-ARIs and minoxidil but also discusses androgen receptor antagonists, in addition to some further therapies that may be available in the future.
Etiology of Androgenetic Scalp Hair Loss
Androgenetic alopecia, commonly referred to as male-pattern or female-pattern hair loss, is a polygenic condition characterised by progressive miniaturisation of scalp hair follicles in a pattern-specific distribution (Anastassakis, 2022; Ovcharenko, Khobzei, & Lortkipanidze, 2025). The pathophysiology of male-pattern hair loss is fundamentally androgen-dependent. Specifically, dihydrotestosterone (DHT) binds to the androgen receptors in susceptible hair follicles, in turn initiating a cascade of transcriptional changes that alter the follicular growth cycle. This includes shortening of the anagen (growth) phase, prolongation of the telogen (resting) phase, and eventually, follicular miniaturisation (Dhurat & Daruwalla, 2021). Notably, while androgens are necessary for the development of AGA, they are not sufficient on their own; individuals with high circulating androgens may not develop AGA if their follicles lack the required sensitivity level (Khaled et al. 2020).
The genetic architecture of AGA is complex and involves multiple loci, although the AR gene on the X chromosome is generally thought to be a significant contributor (Sadasivam et al., 2024). Polymorphisms in the AR gene, particularly those affecting receptor sensitivity, have been associated with increased risk of AGA. Additionally, epigenetic factors, including methylation patterns and histone modifications, may also play a role in regulating gene expression relevant to hair follicle cycling. Large genome-wide association studies (GWAS) have identified other loci which modulate follicular response to androgens in the scalp (Pirastu et al., 2017; Chen et al., 2022; Janivara et al., 2025).
Not all scalp follicles are equally sensitive to androgens. Susceptibility is region-specific and genetically determined, with the vertex and frontal scalp being most affected (Severi et al., 2003; Fujimaki et al., 2024). The expression of 5α-reductase, the enzyme responsible for converting testosterone to DHT, is elevated in these regions, which serves to further amplify local androgenic signalling (Dhurat & Daruwalla, 2021). While androgens promote hair growth in areas such as the beard and chest, they paradoxically cause scalp hair loss in genetically predisposed individuals (Anastassakis, 2022; Ovcharenko, Khobzei, & Lortkipanidze, 2025). Miniaturised scalp hair follicles in AGA undergo a progressive transformation. There may be a critical window for intervention in which current therapeutic strategies may reverse or significantly slow progression. Ultimately, the follicles eventually enter a state of dormancy or senescence, rendering them unable to produce cosmetically significant hair. However, the follicles themselves do appear to remain in situ.
Although pattern hair loss is often divided into “male-“ or “female-“ pattern hair loss in the literature, the presentation is often similar. Studies of men with pattern hair loss have consistently shown the role of DHT (Vierhapper et al., 2001; Ryu et al., 2006; Olsen et al., 2006). There are three isoforms of 5α-reductase: type I, II, and III, all of which are thought to contribute to AGA. However, the mechanism of type III 5α-reductase is less well understood. It is notable that men with 5α-reductase type II deficiency do not experience AGA (Imperato-McGinley & Zhu, 2002). Consequently, reducing intracellular DHT concentrations can prevent AGA. However, the involvement of DHT in female AGA is less clear. A study of women with pattern hair loss found that mean average concentrations of testosterone and DHT were higher than in controls without AGA, but still within the female range (Vierhapper et al., 2003). Hence, female concentrations of testosterone and DHT appear to be associated with pattern hair loss in a certain subset of women. A case of a woman with complete androgen insensitivity syndrome (CAIS) with female pattern hair loss has been reported in the literature (Cousen & Messenger, 2010).
5α-Reductase Inhibitors
5α-Reductase inhibitors are a class of medications developed to treat conditions caused by the effects of DHT, such as benign prostatic hyperplasia and androgenetic alopecia. As implied by their name, these medications function by inhibiting 5α-reductase enzyme activity. Whilst not curative, 5-ARIs appear to halt or substantially slow hair loss in cisgender men. Globally, the two most widely used 5-ARIs include finasteride and dutasteride.
Efficacy of Finasteride Compared to Dutasteride in AGA
In the United States, finasteride prescriptions have increased exponentially in recent years, largely driven by its use in the treatment of hair loss (AHLA, 2024). Dutasteride prescriptions are far fewer, estimated in the hundreds of thousands, but also growing due to increased use for AGA. This difference is largely due to finasteride being widely licensed for this indication throughout the world, whilst dutasteride remains off-label in most countries (Altendorf et al., 2025). However, dutasteride is licensed for use in the treatment of AGA in South Korea, Japan and Mexico. Increasing interest is driving further adoption and research into its use.
Despite its use for AGA being mostly off-label, dutasteride is now widely regarded as a more efficacious and hence superior 5-ARI than finasteride. Numerous studies have established that dutasteride results in greater suppression of serum DHT concentrations (i.e., about 70% with finasteride vs 90–95% with dutasteride) (Clark et al., 2004; Olsen et al., 2006; Amory et al., 2007; Upreti et al., 2015). Another study directly comparing scalp tissue concentrations found that dutasteride reduced DHT substantially more than finasteride (mean reduction of about 65% with finasteride versus 90% with dutasteride, though with wide interindividual variation). (Hobo et al., 2023). These differences have been primarily attributed to its broader inhibition of the 5α-reductase enzyme. More specifically, finasteride is a selective inhibitor of type II and III 5α-reductase, whereas dutasteride indiscriminately acts on all three isoforms (Gisleskog et al., 1998; Keam & Scott, 2008; Yamana, Labrie, & Luu-The, 2010). Dutasteride has also been theorised to accumulate inside certain tissues, further enhancing its therapeutic effect.
In accordance with the above, two large network meta-analysis studies found that oral dutasteride is superior to oral and topical finasteride in the treatment of male AGA in terms of both total hair density and terminal hair density (Gupta et al., 2024a; Gupta et al., 2025a). These studies also found the effects of finasteride and dutasteride to be dose-dependent. On average, there was no difference between treatment groups using oral and topical finasteride. A systematic review found that dutasteride was superior to finasteride in some studies in terms of hair thickness (Almudimeegh et al., 2024). However, in contrast to the above findings in the case of male AGA, a network meta-analysis of studies investigating different interventions for female AGA found that clinical trials assessing the effectiveness of dutasteride do not yet exist (Gupta et al., 2024b). Notably, oral finasteride given at a dose of 1 mg/day was not found to be effective in treating female AGA, yet oral finasteride used at a dose of 5 mg/day outperformed all other single-agent interventions. Because of the dose-dependent effects of 5-ARIs, it could well be that dutasteride might be more efficacious than finasteride in the treatment of female AGA, as in male AGA. Hopefully, future clinical trials will shed light on this.
Adverse Effects of 5α-Reductase Inhibitors
5-ARIs are associated with certain adverse effects in a subset of individuals. These include, but are not limited to decreased libido, erectile dysfunction, reduced ejaculate volume and, possibly, fatigue and depression (Gupta, Vujcic, & Gupta, 2020; Choi et al., 2022; Zhong et al., 2025; Cilio et al., 2025). In the case of transfeminine hormone therapy, some of these effects may actually be desirable.
Currently, there are no large cohort studies or randomised controlled trials that have investigated the incidence of certain adverse effects of hormone therapy with 5-ARIs in transfeminine people. Studies of the general cisgender population may provide some limited insight. A meta-analysis of clinical trials found that the odds of hypoactive sexual desire and erectile dysfunction were each approximately 1.5-fold greater, in male users of 5-ARIs versus placebo (Corona et al., 2017). No difference was found between finasteride and dutasteride in their effects on sexual desire. This is in accordance with findings from more recent publications (Zhou et al., 2019; Zahkem et al., 2019; Estill et al., 2023; Neubauer, Ong, & Lipner, 2025). It is possible that 5-ARIs could produce further reduction in sexual function combined with other hormone therapy medications in transfeminine people, but there are no data to confirm or refute this.
Some studies have found that 5-ARIs are associated with slightly increased circulating testosterone levels (Amory et al., 2007; Stanczyk, Azen, & Pike, 2013; Maeda et al., 2018). However, a 2019 meta-analysis of studies of cisgender men concluded that finasteride and dutasteride use did not unequivocally result in statistically significant increases in serum testosterone levels (Traish et al., 2019). The relevance of marginally raised testosterone in individuals receiving gender-affirming hormone therapy is unclear because exogenous estrogen and antiandrogen therapy typically suppresses endogenous testosterone production to concentrations far below the male range. Nevertheless, a retrospective analysis of transfeminine people using oral estradiol and spironolactone did find that finasteride use appeared to have a moderately deleterious effect on testosterone suppression (Leinung, Feustel, & Joseph, 2018). Studies have generally found that spironolactone does not, by itself, actually lower testosterone concentrations in transfeminine people (Angus et al., 2021). As such, these findings might not be applicable to other antigonadotrophic antiandrogens such as GnRH agonists and progestogens including cyproterone acetate. The influence of 5-ARIs on testosterone concentrations in transfeminine people would be an interesting point for more studies to explore in the future.
The possible effect of 5-ARIs on cognitive function, mood, depression, and suicide risk is controversial. Androgen deprivation therapy, in general, results in an increased risk of psychiatric complications (Izard & Siemens, 2020; Siebert, Lapping-Carr, & Morgans, 2020). These effects have been attributed to the depletion of circulating testosterone available for conversion into estrogens and hence can be mitigated with the use of concomitant estradiol administration (Coelingh Bennink et al., 2024). However, in some studies, 5-ARIs have been associated with increased rates of depression despite testosterone levels remaining well within the male range. Several large pharmacovigilance studies have found signals for reduced cognitive function, depression, and suicidality in users of finasteride and dutasteride (Nguyen et al., 2021; Cho et al., 2024; Zhong et al., 2025; Gupta et al., 2025b; Lee at al., 2025). In some of these studies, the signals have been quite strong. These data have prompted some countries to mandate warnings about possible long-term side effects on product labels. The psychiatric effects of 5-ARIs have been hypothesised to be a consequence of this class of drugs also inhibiting the synthesis of neuroactive steroids such as allopregnanolone. These neurosteroids may have antidepressant and anxiolytic effects. In contrast to the findings of pharmacovigilance studies, a 2024 meta-analysis incorporating prospective study data from around 2 million users of 5-ARIs did not find associations for depression (aHR: 1.30, 95% CI: 0.85–2.00) or suicide (aHR 1.30, 95% CI: 0.65–2.61) (Uleri et al., 2024). Subgroup analyses for finasteride and dutasteride yielded similar findings. It should be noted that despite the extremely large sample size, the confidence intervals in the pooled risk estimates were still wide and hence could not rule out marginal risk increases. Overall, these findings may be both concerning and reassuring at the same time. Whilst the involvement of 5-ARIs in mood and depression still remains unclear, the preponderance of evidence strongly suggests that any excess risk is likely to be small.
Post-Finasteride Syndrome
“Post-finasteride syndrome” (PFS) is an even more controversial and poorly understood condition characterised by persistent sexual, neurological, and psychological symptoms that arise during or after the use of finasteride or dutasteride (Cilio et al., 2025; Leliefeld, Debruyne, & Reisman, 2025). PFS has gained increasing attention since it was popularised in the literature in 2012 following a subset of 5-ARI users reporting enduring adverse effects after discontinuation (Irwig, 2012).
Various case reports and studies have been published reporting associations between 5-ARIs and PFS symptoms (Traish et al., 2011; Irwig & Kolukula, 2011; Irwig, 2012; Irwig, 2014; Caruso et al, 2015; Ali, Heran, & Etminan, 2015; Guo et al., 2016; Kiguradze et al., 2017; Pereira & Coelho, 2020). Notably, one apparently well-designed study reported altered levels of neuroactive steroids in cerebrospinal fluid and plasma after discontinuation of finasteride in men who reported suffering from PFS symptoms (Caruso et al, 2015). A retrospective analysis also found that rates of erectile dysfunction were higher in men with cumulatively greater exposure to finasteride and dutasteride (Kiguradze et al., 2017). However, overall these data have been of low-quality, at high risk of bias, may have been confounded by secondary variables, and universally suffer from the lack of a placebo control group to establish causation (Hirshburg et al., 2016; Trüeb et al., 2019; Trüeb et al., 2024). As such, by themselves they are of limited usefulness. More recently, a pharmacovigilance study identified the existence of a signal for “post-finasteride syndrome” with finasteride in the FAERS database (Zhong et al., 2025).
A substantial nocebo effect appears to exist in users of 5-ARIs pertaining to PFS-like symptoms (Maksym, Kajdy, & Rabijewski, 2019). A study found that men who were made aware of sexual adverse effects before being treated were much more likely to report them during follow-up (43.6%), compared to men who were not (14.3%) (Mondaini et al., 2007). These data show that the power of suggestion is likely to influence the experience of many individuals using 5-ARIs. A more comprehensive pharmacovigilance study of the FAERS database performed analyses stratified by time period and 5-ARI medication (Gupta et al., 2025b). The study conducted disproportionality analyses for five adverse events related to suicide between 2006 and 2011, 2013 and 2018, and 2019 and 2023. No signals were detected for oral finasteride between 2006 and 2011, but signals emerged in later periods, with increased reporting odds for suicidal ideation between 2013 and 2018 and between 2019 and 2023. Despite oral dutasteride being more efficacious in its action as a 5-ARI, dutasteride showed no significant signals across any time period. The authors concluded that these findings were suggestive of increased awareness of PFS being the cause of heightened reporting of psychiatric adverse events to FAERS, rather than reflecting a true pharmacological effect (Gupta et al., 2025b).
Taken together, all these findings provide limited evidence for the existence of PFS in a small subset of individuals. However, the evidence for persistent long-term adverse effects stemming from finasteride and dutasteride use is tenuous at best.
Minoxidil
Minoxidil is a medication with antihypertensive and vasodilator effects which was originally developed as an oral formulation for high blood pressure. However, it was found to have the unexpected side effect of promoting hair growth, which led to its reformulation as a topical solution and adoption for AGA. Oral minoxidil is also now increasingly being used at lower doses to treat hair loss.
Mechanism of Action of Minoxidil in Hair Loss
The exact means by which minoxidil is involved in promoting hair growth is not fully understood (Gupta et al., 2023; Iyengar & Li, 2025). It is believed that minoxidil functions by improving blood flow to hair follicles, which in turn increases circulation and which may help revitalise shrunken follicles, extend the growth phase of the hair cycle, and encourage thicker, longer hair strands (Zeltzer et al., 2024; Tan et al., 2025). A sulphotransferase enzyme converts minoxidil into its active metabolite, minoxidil sulfate. Differences in sulphotransferase enzyme expression between individuals appear to augment the efficacy of minoxidil (Goren & Naccarato, 2018). Clinical effects on hair growth typically begin after 2 to 4 months of consistent use, with maximal results seen around 6 to 12 months. Clinical response with minoxidil therapy appears to be highly variable. In randomised controlled trials, minoxidil monotherapy has been effective in increasing total hair density, as well as terminal hair density in both male and female AGA compared to controls (Gupta et al., 2022a; Gupta et al., 2024a; Gupta et al., 2024b; Gupta et al., 2025a). In these studies, the efficacy of minoxidil has also been shown to be strongly dose-dependent.
Minoxidil has been evaluated for its therapeutic effects on hair loss in some small studies of transfeminine and transmasculine people (Zaminski et al., 2025; Tang et al., 2025b). These studies have reported positive results, in line with data from the wider population.
Comparison of Different Minoxidil Routes of Administration
The most widely used formulations of minoxidil include oral minoxidil and topical minoxidil. The main difference is that oral minoxidil is absorbed extensively into systemic circulation, whereas topical minoxidil is designed to act locally at the site of application, at which it stimulates hair follicles directly, hence resulting in limited systemic exposure.
Oral minoxidil is metabolised rapidly into minoxidil sulfate in the gastrointestinal tract (Patel, Nessel & Kumar, 2023). Peak plasma concentrations are typically reached within 1 hour. Oral minoxidil has been used at a range of 0.25–7.5 mg/day for AGA in clinical trials. The oral route has an average bioavailability of nearly 100%, whereas the local absorption of topical minoxidil into the scalp is around 1.4%. However, there remains substantial interindividual variation for each. As such, clinical doses of topical minoxidil are much higher (typically a 2 or 5% concentration) in order to compensate. Food does not appear to influence the bioavailability of oral minoxidil (Gupta et al., 2023).
The efficacy of oral and topical minoxidil has been investigated extensively in clinical studies. Higher doses of oral minoxidil have been associated with more favourable outcomes for AGA in terms of hair diameter, total hair density, and terminal hair density, but also with increasing adverse effects (Gupta et al., 2022b). Generally, lower doses have been used in women as compared to men. Oral minoxidil has been investigated at doses of up to 7.5 mg/day in clinical trials in this indication (Sanabria et al., 2024a). Large meta-analyses have found that studies are mixed on whether oral or topical minoxidil, on average, results in better, worse, or equal efficacy (Gupta et al., 2022a; Gupta et al., 2024a; Gupta et al., 2024b; Fazal et al., 2025; Gupta et al., 2025a). However, the dose-dependent effects of oral minoxidil have similarly been found to occur with topical minoxidil (Singh et al., 2022). A possibility is that oral and topical minoxidil may not have always been used at clinically equivalent doses.
The inconsistent differences in efficacy shown between oral and topical minoxidil in clinical studies may be driven by interindividual variation in response due to sulphotransferase enzyme expression, particularly in the scalp (Patel, Nessel & Kumar, 2023). There is growing evidence that in some individuals oral minoxidil may be more efficacious than topical minoxidil and vice versa (Goren et al., 2015; Goren et al., 2016; Gupta et al., 2024b; Gupta et al., 2025a). These data suggest that a subset of individuals who may not respond to one route of administration could see benefit by changing to the other.
Sublingual minoxidil has also been investigated for treating AGA (Sinclair et al., 2020; Bokhari, Jones, & Sinclair, 2021; Sinclair et al., 2025). Another route of administration which is being considered is injectable minoxidil (Needle et al., 2025). However, these routes have received comparatively much less attention and so limited data are available to inform their usage. A randomised controlled trial comparing oral and sublingual minoxidil at a daily dosage of 5 mg found similar efficacy at 24 weeks follow-up, suggesting that sublingual minoxidil may be a useful alternative to oral minoxidil (Sanabria et al., 2024b). Whilst these initial data are promising, further and larger scale studies are likely to be needed before sublingual minoxidil could see the same level of adoption as oral and topical administration.
Safety and Tolerability of Oral and Topical Minoxidil
Minoxidil has generally been shown to be well tolerated in clinical trials. Nevertheless, usage is associated with various adverse effects in some individuals (Gupta et al., 2022b; Gupta et al., 2023; Iyengar & Li, 2025). The adverse effects of minoxidil have been shown to be dependent on the route of administration, as well as being positively dose-dependent.
A retrospective study of users of oral minoxidil investigated the frequency of adverse effects in both men and women receiving a median dose of 1.63 mg/day (Vañó-Galván et al., 2021). The following were found to occur: hypertrichosis (excessive facial/body hair) in 15.1%, lightheadedness in 1.7%, fluid retention in 1.3%, tachycardia in 0.9%, headache in 0.4%, periorbital edema (temporary swelling around the eyes) in 0.3%, and insomnia in 0.2%. The total frequency of adverse effects was 20.4%, which prompted discontinuation in 1.2% of users, overall. Another study reported an overall hypertrichosis incidence of 24%, with the highest rates being found in the sideburns (81%), temples (73%), arms (63%), and upper lip (51%) (Jimenez-Cauhe et al., 2021). By contrast, topical minoxidil is associated with much lower overall rates of hypertrichosis. Most studies have reported incidence rates of between 0 and 5% (Lucky et al., 2004; Blume-Peytavi et al., 2016; Ramos et al., 2020; Penha et al., 2024; Yang et al., 2024). These findings are consistent with a meta-analysis that reported point estimates of incidence rates for hypertrichosis of 10%, 15%, and 33% for oral minoxidil at 0.25 mg/day, 0.5 mg/day, and 1.25 mg/day, respectively, and 0% and 2% for topical minoxidil at a 2% and 5% concentration, respectively (Wiechert et al., 2025). Despite this, the discontinuation rate across all studies was 0.49%. There also seemed to be no statistically significant difference between the rate of discontinuation for oral and topical formulations, suggesting that hypertrichosis appears to be very well tolerated.
A concern associated with the use of oral minoxidil is its potential impact on cardiovascular health (Ibraheim et al., 2023). Since tachycardia can increase myocardial workload and lead to symptoms such as palpitations or chest discomfort, oral minoxidil should be approached cautiously, especially by individuals with underlying cardiovascular issues. Fortunately, the overall risk of severe cardiovascular complications from low-dose oral minoxidil seems to be very low in the general population (Randolph & Tosti, 2021; Vañó-Galván et al., 2021). Meanwhile, skin reactions appear to be relatively common in users of topical minoxidil. This often manifests as scalp eczema and itching, although rates of incidence vary by study (Lucky et al., 2004; Rossi et al., 2012; Penha et al., 2024). The culprit behind this irritating effect appears not to be minoxidil itself, but rather the ingredients in certain formulations such as propylene glycol (Suchonwanit, Thammarucha, & Leerunyakul, 2019). These solvents help deliver minoxidil into the scalp, but are known to cause skin irritation in susceptible individuals. It also appears that, for most people, long-term topical minoxidil therapy may be precluded by non-compliance (Ali Mapar & Omidian, 2007; Shadi, 2023).
The increase in overall body hair growth (i.e., hypertrichosis) is arguably the most consequential side effects for transfeminine people found to occur with minoxidil. As noted above, hypertrichosis is much more common with oral minoxidil than with topical minoxidil. This is a result of the differences in pharmacology between these routes and the extensive systemic absorption that occurs in the case of the former (Desai et al., 2024; Wiechert et al., 2025). In transmasculine people, an increase in body hair growth and diameter could be beneficial. However, these effects are usually not desired by transfeminine people. Consequently, some transfeminine people may prefer to use topical minoxidil over oral minoxidil, despite possible benefits to effectiveness from the latter in some individuals.
Steroidal and Non-Steroidal Antiandrogens
Antiandrogens such as spironolactone and cyproterone acetate are widely employed to reduce or suppress testosterone levels in transfeminine people. Some clinics have also used the non-steroidal antiandrogen bicalutamide. However, these medications have all also been investigated in the treatment of pattern hair loss in cisgender women. After gonadectomy, antiandrogen treatment is often discontinued. Nevertheless, it appears some transfeminine people continue antiandrogen treatment, particularly spironolactone, in order to suppress the effects of non-gonadal androgen production.
Spironolactone
Spironolactone has been studied at oral doses of 25 to 200 mg/day for the treatment of pattern hair loss in women (Wang et al., 2023; Rosenthal et al., 2024). Oral spironolactone has been found to be effective in halting and, in some cases, reversing female AGA (Sinclair, Wewerinke & Jolley, 2005; Burns et al., 2020). Often, it has been paired with other interventions such as minoxidil. A randomised controlled trial found that oral spironolactone at a dose of 80 to 100 mg/day had similar efficacy to minoxidil when used as a single agent therapy (Liang et al., 2022). Spironolactone has also been studied topically (Abdel‐Raouf et al., 2020; Ammar et al., 2022). In the highest quality studies, spironolactone has been found to act additively with minoxidil in improving hair density and hair diameter.
Overall, spironolactone appears to be well tolerated for treating AGA, as well as other androgen sensitive conditions in women (Barbieri et al., 2021; Wang et al., 2023; Martin et al., 2025). Likewise, it may also be useful for some transfeminine people as an adjunct therapy, especially when paired with minoxidil. However, spironolactone has been scarcely studied for male AGA. It could well be the case that other more established therapies, such as dutasteride, would be better for transfeminine people with more extensive hair loss.
Flutamide and Bicalutamide
Flutamide is a potent non-steroidal antiandrogen and antagonist of the androgen receptor which has predominantly been used to treat prostate cancer. Some clinicians have also employed the use of flutamide in treating female AGA, with positive findings (Carmina & Lobo, 2003; Yazdabadi & Sinclair, 2011; Paradisi et al., 2011; Faghihi et al., 2022). In a randomised controlled trial, 500 mg/day flutamide was found to be superior to 100 mg/day spironolactone in treating scalp hair loss (Cusan et al., 1994). However, this may have been at least partially down to the cyclic use of spironolactone, which meant that women randomised to spironolactone were not actually receiving it for the duration of the entire month. In spite of the above findings, flutamide is associated with a high risk of elevated liver enzymes, which can progress to life-threatening organ failure in a very small but clinically significant subset of cases (Ozono et al., 2000; Paradisi et al., 2011; Giorgetti et al., 2017). This appears to have precluded its widespread adoption for female AGA.
Bicalutamide is another non-steroidal antiandrogen which has been considered for female AGA (Perez, Nguyen, & Senna, 2025). Generally, bicalutamide is believed to have a lower risk of liver toxicity than flutamide, making it a much more promising candidate for large-scale adoption in otherwise healthy people (Kolvenbag & Blackledge, 1996; Devjani et al., 2023). A number of retrospective studies have reported encouraging data with 10 to 50 mg/day oral bicalutamide, both in terms of safety and efficacy (Ismali et al., 2020; Fernandez-Nieto et al., 2020; Yoong et al., 2025). Interestingly, a study also found that bicalutamide was associated with improvements in hypertrichosis induced by oral minoxidil (Moussa et al., 2022). Only one randomised controlled trial has been conducted with bicalutamide, in which minoxidil plus 25 mg/day oral bicalutamide was compared to minoxidil plus placebo (da Silva Libório et al., 2025). In this study, there was no additional benefit of bicalutamide on total hair density. This finding is surprising in light of the seemingly additive effects that occur with minoxidil and spironolactone. It is possible that differences in methodology could be responsible for this discrepancy. Another study, although also retrospective, found that 50 mg/day oral bicalutamide was associated with moderately greater improvement in scalp hair loss than was 100 mg/day oral spironolactone (Jha et al., 2024).
Concerns about safety have also historically precluded widespread adoption of bicalutamide in transgender medicine. However, increasing numbers of studies are adding to our knowledge of bicalutamide in transfeminine people (Fuqua, Shi, & Eugster, 2024; Angus et al., 2024).
Other Medications and Future Developments
Despite several decades of research, only two medications have been approved for male AGA by the FDA in the United States. These are minoxidil and finasteride. Even though the use of these agents is increasingly common in men, they remain only partially effective in reversing androgenic hair loss.
Topical Androgen Receptor Antagonists
Two novel topical androgen receptor antagonists of possible interest are clascoterone and pyrilutamide (Saceda-Corralo et al., 2023; Devjani et al., 2023). Unlike oral non-steroidal antiandrogens such as flutamide and bicalutamide, these medications are applied topically so that there is minimal systemic absorption. Clascoterone has shown some limited success in phase 3 clinical trials for treating acne in men and women and has been approved by the FDA for this indication (Hebert et al., 2020). It is hoped that clascoterone could also be effective in treating AGA. An exploratory study found that clascosterone was superior to topical cyproterone acetate and alfatradiol in improving hair shaft diameter and hair follicle density and had comparable efficacy to minoxidil (Cartwright et al., 2019). Phase 3 clinical trials of clascosterone in AGA are currently underway. Pyrilutamide had shown favourable results in phase 2 clinical trials for both male and female AGA, however it failed to outperform placebo in phase 3 clinical trials (Kintor Pharmaceuticals, 2024). Studies are now underway using a higher dose of pyrilutamide over a longer duration of follow-up in the hope that this will show improved results.
Despite the above, these findings are rather disappointing. Notably, clascoterone only marginally outperformed placebo in clinical trials for acne (reduction in symptoms by about 8 to 18% more than placebo) (Hebert et al., 2020). A systematic review and network meta-analysis found that oral spironolactone was substantially more effective for treating acne than topical clascoterone (Basendwh et al., 2024). Clascoterone and pyrilutamide may someday provide another option for treating AGA in transfeminine people. However, since their mechanism of action is not dissimilar to well-established therapies such as 5-ARIs, it seems that this class of medications is unlikely to ever be revolutionary.
Prostaglandin Analogues
Prostaglandin analogues could have some utility in treating AGA. These include latanoprost and bimatoprost. Prostaglandin analogues have mostly been used to treat glaucoma by lowering intraocular pressure but are also believed to prolong the anagen phase and hence cause hair growth in certain susceptible tissues (Valente Duarte de Sousa & Tosti, 2013).
Latanoprost has mostly been studied for alopecia areata within the context of hair loss. However, one small randomised controlled trial found that topical latanoprost outperformed placebo after 24 weeks in increasing hair density in young men with mild AGA (Blume-Peytavi et al., 2012). Bimatoprost has also been evaluated in various clinical trials. In four separate phase 2 clinical trials, bimatoprost mildly to moderately outperformed placebo (Anastassakis, 2022). However, compared to minoxidil, findings were inconsistent. Bimatoprost had similar efficacy in some measures in some studies, but inferior efficacy in others. Overall, prostaglandin analogues appear to have received little subsequent attention. These data are relatively underwhelming by themselves compared to the large amount of literature pertaining to more established AGA therapies.
Mitochondrial Pyruvate Carrier Inhibition
In perhaps one of the more interesting developments in recent years, researchers have identified a topically delivered molecule, called PP-405, that appears to be capable of reactivating dormant follicles by modulating mitochondrial pyruvate carrier (MPC) activity (Brown, 2025). Unlike current therapies that focus on hormone suppression and increased blood flow, PP-405 is a regenerative, stem-cell-focused approach to treating hair loss. The molecule is currently being investigated for male and female AGA.
Recently, a phase 2a clinical trial that randomised 78 men and women to either PP-405 or placebo concluded with positive safety findings (Meara, 2025). However, preliminary results from 4 weeks of treatment at 8 weeks follow-up also showed a rapid and statistically significant clinical response versus placebo. This was despite the study not actually being conducted to show efficacy. The researchers found that 31% of the men with advanced AGA who were treated with the active medication showed a 20% or greater increase in total hair density, compared to 0% of patients in the placebo group. This is particularly notable because current interventions for AGA, such as minoxidil, typically take at least several months of follow-up to show a statistically significant difference from placebo. Most strikingly, PP-405 apparently induced new terminal hair growth from follicles where no hair was previously present.
As of September 2025, PP-405 is in phase 2b clinical trials and is expected to enter phase 3 clinical trials early next year if subsequent clinical findings are promising. With all this said, it should be noted that these are merely early results. More rigorous studies are necessary to determine if PP-405 can be an effective intervention against AGA.
Summary and Conclusions
AGA is a common and distressing condition that has particular relevance for transfeminine people, given the role of hair in gender identity and expression. While feminising hormone therapy appears to at least partially reverse AGA, many individuals appear to experience incomplete regrowth. Limited data suggest that adjunct use of 5-ARIs and/or minoxidil, particularly in the first few years of hormonal transition, may have positive effects but more studies are necessary to confirm this.
The most established treatments for AGA in the wider population are 5-ARIs, including finasteride and dutasteride, and minoxidil. The role of 5-ARIs in women remain less clear. Nevertheless, dutasteride achieves superior outcomes in male AGA to finasteride, whilst having similar safety and tolerability. Hence, wherever possible, it seems reasonable to use the former should the use of a 5α-reductase inhibitor be desired. Minoxidil, whether oral or topical, provides dose-dependent improvements in total and terminal hair density in male and female AGA and acts synergistically with 5-ARIs. However, oral minoxidil is associated with higher rates of hypertrichosis, which may be undesirable for many transfeminine individuals.
Other agents, such as spironolactone and bicalutamide, could also offer additional benefit by antagonising the androgen receptor. Spironolactone is already widely used in transfeminine hormone therapy and shows synergy with minoxidil in studies of female AGA. Bicalutamide is of emerging interest given its relatively favourable safety profile. Novel therapies of benefit to transfeminine people may also become available in the future.
References
Abdel‐Raouf, H., Aly, U. F., Medhat, W., Ahmed, S. S., & Abdel‐Aziz, R. T. (2021). A novel topical combination of minoxidil and spironolactone for androgenetic alopecia: clinical, histopathological, and physicochemical study. Dermatologic Therapy, 34(1), e14678. [DOI:10.1111/dth.14678]
Adenuga, P., Summers, P., & Bergfeld, W. (2012). Hair regrowth in a male patient with extensive androgenetic alopecia on estrogen therapy. Journal of the American Academy of Dermatology, 67(3), e121–e123. [DOI:10.1016/j.jaad.2011.10.017]
Almudimeegh, A., Al-Mutairi, H., Al-Tassan, F., Al-Quraishi, Y., & Nagshabandi, K. N. (2024). Comparison between dutasteride and finasteride in hair regrowth and reversal of miniaturization in male and female androgenetic alopecia: a systematic review. Dermatology Reports, 16(4), 9909. [DOI:10.4081/dr.2024.9909]
Ali, A. K., Heran, B. S., & Etminan, M. (2015). Persistent sexual dysfunction and suicidal ideation in young men treated with low‐dose finasteride: a pharmacovigilance study. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 35(7), 687–695. [DOI:10.1002/phar.1612]
Altendorf, S., Bertolini, M., Le Riche, A., Tosti, A., & Paus, R. (2025). Frontiers in the physiology of male pattern androgenetic alopecia: Beyond the androgen horizon. Physiological Reviews, online ahead of print. [DOI:10.1152/physrev.00005.2024]
Ammar, A. M., Elshahid, A. R., Abdel‐Dayem, H. A., Mohamed, A. A., & Elsaie, M. L. (2022). Dermoscopic evaluation of the efficacy of combination of topical spironolactone 5% and minoxidil 5% solutions in the treatment of androgenetic alopecia: a cross sectional‐comparative study. Journal of Cosmetic Dermatology, 21(11), 5790–5799. [DOI:10.1111/jocd.15328]
Amory, J. K., Wang, C., Swerdloff, R. S., Anawalt, B. D., Matsumoto, A. M., Bremner, W. J., Walker, S. E., Haberer, L. J., & Clark, R. V. (2007). The effect of 5α-reductase inhibition with dutasteride and finasteride on semen parameters and serum hormones in healthy men. The Journal of Clinical Endocrinology & Metabolism, 92(5), 1659–1665. [DOI:10.1210/jc.2006-2203]
Anand/HairLossCure2020. (2024 June 18). Booming Demand: U.S. Sees 200% Surge in Finasteride Prescriptions Over 7 Years. American Hair Loss Association (AHLA). [URL]
Anastassakis, K. (2022). Hormonal and genetic etiology of male androgenetic alopecia. In Anastassakis, K. (Ed.). Androgenetic Alopecia From A to Z: Vol. 1 Basic Science, Diagnosis, Etiology, and Related Disorders (pp. 135–180). Cham: Springer International Publishing. [DOI:10.1007/978-3-030-76111-0_11]
Angus, L. M., Hong, Q. V., Cheung, A. S., & Nolan, B. J. (2024). Effect of bicalutamide on serum total testosterone concentration in transgender adults: a case series. Therapeutic Advances in Endocrinology and Metabolism, 15, 20420188241305022. [DOI:10.1177/20420188241305022]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2021). A systematic review of antiandrogens and feminization in transgender women. Clinical Endocrinology, 94(5), 743–752. [DOI:10.1111/cen.14329]
Asad, N., Naseer, M., & Ghafoor, R. (2024). Efficacy of Topical Finasteride 0.25% With Minoxidil 5% Versus Topical Minoxidil 5% Alone in Treatment of Male Pattern Androgenic Alopecia, Journal of Drugs in Dermatology, 23(11), 1003–1008. [DOI:10.36849/JDD.7826]
Barbieri, J. S., Margolis, D. J., & Mostaghimi, A. (2021). Temporal trends and clinician variability in potassium monitoring of healthy young women treated for acne with spironolactone. JAMA Dermatology, 157(3), 296–300. [DOI:10.1001/jamadermatol.2020.5468]
Basendwh, M. A., Alharbi, A. A., Bukhamsin, S. A., Abdulwahab, R. A., & Alaboud, S. A. (2024). The efficacy of Topical Clascoterone versus systematic spironolactone for treatment of acne vulgaris: A systematic review and network meta-analysis. Plos One, 19(5), e0298155. [DOI:10.1371/journal.pone.0298155]
Blume-Peytavi, U., Lönnfors, S., Hillmann, K., & Bartels, N. G. (2012). A randomized double-blind placebo-controlled pilot study to assess the efficacy of a 24-week topical treatment by latanoprost 0.1% on hair growth and pigmentation in healthy volunteers with androgenetic alopecia. Journal of the American Academy of Dermatology, 66(5), 794–800. [DOI:10.1016/j.jaad.2011.05.026]
Blume-Peytavi, U., Shapiro, J., Messenger, A. G., Hordinsky, M. K., Zhang, P., Quiza, C., Doshi, U., & Olsen, E. A. (2016). Efficacy and Safety of Once-Daily Minoxidil Foam 5% Versus Twice-Daily Minoxidil Solution 2% in Female Pattern Hair Loss: A Phase III, Randomized, Investigator-Blinded Study. Journal of Drugs in Dermatology, 15(7), 883–889. [Google Scholar] [PubMed] [URL]
Bokhari, L., Jones, L. N., & Sinclair, R. D. (2022). Sublingual minoxidil for the treatment of male and female pattern hair loss: a randomized, double‐blind, placebo‐controlled, phase 1B clinical trial. Journal of the European Academy of Dermatology & Venereology, 36(1), e62–e66. [DOI:10.1111/jdv.17623]
Brown, L. (2025 August 12). The Great Unbalding: Could A New Drug Actually Regrow Hair? Intelligencer (New York Magazine). [URL]
Burns, L. J., De Souza, B., Flynn, E., Hagigeorges, D., & Senna, M. M. (2020). Spironolactone for treatment of female pattern hair loss. Journal of the American Academy of Dermatology, 83(1), 276–278. [DOI:10.1016/j.jaad.2020.03.087]
Carmina, E., & Lobo, R. A. (2003). Treatment of hyperandrogenic alopecia in women. Fertility and Sterility, 79(1), 91–95. [DOI:10.1016/S0015-0282(02)04551-X]
Cartwright, M., Mazzetti, A., Moro, L., Rosette, C., Gerloni, M. (2019). 10063. A summary of in vitro, phase I, and phase II studies evaluating the mechanism of action, safety, and efficacy of clascoterone (cortexolone 17α propionate, CB-03-01) in androgenetic alopecia. Journal of the American Academy of Dermatology, 81(4 Suppl 1), AB13. [DOI:10.1016/j.jaad.2019.06.087]
Caruso, D., Abbiati, F., Giatti, S., Romano, S., Fusco, L., Cavaletti, G., & Melcangi, R. C. (2015). Patients treated for male pattern hair with finasteride show, after discontinuation of the drug, altered levels of neuroactive steroids in cerebrospinal fluid and plasma. The Journal of Steroid Biochemistry and Molecular Biology, 146, 74–79. [DOI:10.1016/j.jsbmb.2014.03.012]
Chen, Y., Hysi, P., Maj, C., Heilmann-Heimbach, S., Spector, T. D., Liu, F., & Kayser, M. (2023). Genetic prediction of male pattern baldness based on large independent datasets. European Journal of Human Genetics, 31(3), 321–328. [DOI:10.1038/s41431-022-01201-y]
Cho, Y., Bea, S., Bae, J. H., Kim, D. H., Lee, J. H., & Shin, J. Y. (2024). Cognitive dysfunction following finasteride use: a disproportionality analysis of the global pharmacovigilance database. Expert Opinion on Drug Safety, 23(8), 1027–1033. [DOI:10.1080/14740338.2023.2294926]
Choi, G. S., Sim, W. Y., Kang, H., Huh, C. H., Lee, Y. W., Shantakumar, S., Ho, Y. F., Oh, E. J., Duh, M. S., Cheng, W. Y., Bobbili, P., Thompson-Leduc, P., & Ong, G. (2022). Long-term effectiveness and safety of dutasteride versus finasteride in patients with male androgenic alopecia in South Korea: a multicentre chart review study. Annals of Dermatology, 34(5), 349–359. [DOI:10.5021/ad.22.027]
Cilio, S., Tsampoukas, G., Morgado, A., Ramos, P., & Minhas, S. (2025). Post-finasteride syndrome - a true clinical entity? International Journal of Impotence Research, 37, 426–435. [DOI:10.1038/s41443-025-01025-6]
Clark, R. V., Hermann, D. J., Cunningham, G. R., Wilson, T. H., Morrill, B. B., & Hobbs, S. (2004). Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5α-reductase inhibitor. The Journal of Clinical Endocrinology & Metabolism, 89(5), 2179–2184. [DOI:10.1210/jc.2003-030330]
Cocchetti, C., Castellini, G., Maggi, M., Romani, A., Vignozzi, L., Greenman, Y., den Heijer, M., T’Sjoen, G., & Fisher, A. D. (2023). Effects of hormonal treatment on dermatological outcome in transgender people: a multicentric prospective study (ENIGI). Journal of Endocrinological Investigation, 46(4), 779–786. [DOI:10.1007/s40618-022-01944-x]
Coelingh Bennink, H. J., Prowse, A., Egberts, J. F., Debruyne, F. M., Huhtaniemi, I. T., & Tombal, B. (2024). The loss of estradiol by androgen deprivation in prostate cancer patients shows the importance of estrogens in males. Journal of the Endocrine Society, 8(7), bvae107. [DOI:10.1210/jendso/bvae107]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Corona, G., Tirabassi, G., Santi, D., Maseroli, E., Gacci, M., Dicuio, M., Sforza, A., Mannucci, E., & Maggi, M. (2017). Sexual dysfunction in subjects treated with inhibitors of 5α‐reductase for benign prostatic hyperplasia: a comprehensive review and meta‐analysis. Andrology, 5(4), 671–678. [DOI:10.1111/andr.12353]
Cousen, P., & Messenger, A. (2010). Female pattern hair loss in complete androgen insensitivity syndrome. British Journal of Dermatology, 162(5), 1135–1137. [DOI:10.1111/j.1365-2133.2010.09661.x]
Cusan, L., Dupont, A., Gomez, J. L., Tremblay, R. R., & Labrie, F. (1994). Comparison of flutamide and spironolactone in the treatment of hirsutism: a randomized controlled trial. Fertility and Sterility, 61(2), 281–287. [DOI:10.1016/S0015-0282(16)56518-2]
da Silva Libório, R., da Motta, A. V., Miot, H. A., & Ramos, P. M. (2025). Bicalutamide 25 mg combined with minoxidil 1 mg versus minoxidil 1 mg for female pattern hair loss: A randomized double-blind clinical trial. JAAD International, 19, 48–55. [DOI:10.1016/j.jdin.2024.12.002]
Desai, D. D., Nohria, A., Brinks, A., Needle, C., Shapiro, J., & Sicco, K. I. L. (2024). Minoxidil-induced hypertrichosis: Pathophysiology, clinical implications, and therapeutic strategies. JAAD Reviews, 2, 41–49. [DOI:10.1016/j.jdrv.2024.08.002]
Devjani, S., Ezemma, O., Kelley, K. J., Stratton, E., & Senna, M. (2023). Androgenetic alopecia: therapy update. Drugs, 83(8), 701–715. [DOI:10.1007/s40265-023-01880-x]
Dewhurst, J., & Underhill, R. (1979). The Suppression of Facial Hair Growth in Transsexuals Using Cyproterone Acetate. British Journal of Sexual Medicine, 6, 19–23. [Google Scholar] [PDF]
Dhurat, R. S., & Daruwalla, S. B. (2021). Androgenetic alopecia: Update on etiology. Dermatological Reviews, 2(3), 115–121. [DOI:10.1002/der2.58]
Estill, M. C., Ford, A., Omeira, R., & Rodman, M. (2023). Finasteride and dutasteride for the treatment of male androgenetic alopecia: a review of efficacy and reproductive adverse effects. Georgetown Medical Review, 7(1). [DOI:10.52504/001c.88531]
Faghihi, G., Iraji, F., Siadat, A. H., Saber, M., Jelvan, M., & Hoseyni, M. S. (2022). Comparison between “5% minoxidil plus 2% flutamide” solution vs.“5% minoxidil” solution in the treatment of androgenetic alopecia. Journal of Cosmetic Dermatology, 21(10), 4447–4453. [DOI:10.1111/jocd.14788]
Fazal, F., Malik, B. H., Mumtaz Malik, H., Sabir, B., Mustafa, H., Ahmed, M., Abid, A., Leeza Adil, M., Shafi, U., & Saad, M. (2025). Can oral minoxidil be the game changer in androgenetic alopecia? A comprehensive review and meta-analysis comparing topical and oral minoxidil for treating androgenetic alopecia. Skin Health and Disease, 5(2), 95–101. [DOI:10.1093/skinhd/vzaf009]
Fernandez-Nieto, D., Saceda-Corralo, D., Jimenez-Cauhe, J., Moreno-Arrones, O. M., Rodrigues-Barata, R., Hermosa-Gelbard, A., & Vano-Galvan, S. (2020). Bicalutamide: a potential new oral antiandrogenic drug for female pattern hair loss. Journal of the American Academy of Dermatology, 83(5), e355–e356. [DOI:10.1016/j.jaad.2020.04.054]
Fujimaki, H., Yanagisawa, M., Yoshitake, T., & Sato, A. (2024). Age-related progression of androgenetic alopecia: Statistical analysis of 5372 Japanese men. JAAD International, 17, 84–85. [DOI:10.1016/j.jdin.2024.07.003]
Fuqua, J. S., Shi, E., & Eugster, E. A. (2024). A retrospective review of the use of bicalutamide in transfeminine youth; a single center experience. International Journal of Transgender Health, 25(3), 533–537. [DOI:10.1080/26895269.2023.2294321]
Gao, J. L., Sanz, J., Tan, N., King, D. S., Modest, A. M., & Dommasch, E. D. (2023). Androgenetic alopecia incidence in transgender and gender diverse populations: a retrospective comparative cohort study. Journal of the American Academy of Dermatology, 89(3), 504–510. [DOI:10.1016/j.jaad.2023.01.037]
Gao, J. L., Streed Jr, C. G., Thompson, J., Dommasch, E. D., & Peebles, J. K. (2023). Androgenetic alopecia in transgender and gender diverse populations: a review of therapeutics. Journal of the American Academy of Dermatology, 89(4), 774–783. [DOI:10.1016/j.jaad.2021.08.067]
Giorgetti, R., Di Muzio, M., Giorgetti, A., Girolami, D., Borgia, L., & Tagliabracci, A. (2017). Flutamide-induced hepatotoxicity: ethical and scientific issues. European Review for Medical and Pharmacological Sciences, 21(1 Suppl), 69–77. [Google Scholar] [PubMed]
Gisleskog, P. O., Hermann, D., Hammarlund‐Udenaes, M., & Karlsson, M. O. (1998). A model for the turnover of dihydrotestosterone in the presence of the irreversible 5α‐reductase inhibitors GI198745 and finasteride. Clinical Pharmacology & Therapeutics, 64(6), 636–647. [DOI:10.1016/S0009-9236(98)90054-6]
Goren, A., Shapiro, J., Roberts, J., McCoy, J., Desai, N., Zarrab, Z., Pietrzak, A., & Lotti, T. (2015). Clinical utility and validity of minoxidil response testing in androgenetic alopecia. Dermatologic Therapy, 28(1), 13–16. [DOI:10.1111/dth.12164]
Goren, A., Kovacevic, M., McCoy, J., & Shapiro, J. (2017). 841 Minoxidil dose response study in female pattern hair loss patients determined to be non-responders to 5% topical Minoxidil. Journal of Investigative Dermatology, 137(5 Suppl 1), S144–S144. [DOI:10.1016/j.jid.2017.02.867]
Goren, A., & Naccarato, T. (2018). Minoxidil in the treatment of androgenetic alopecia. Dermatologic Therapy, 31(5), e12686. [DOI:10.1111/dth.12686]
Guo, M., Heran, B., Flannigan, R., Kezouh, A., & Etminan, M. (2016). Persistent sexual dysfunction with finasteride 1 mg taken for hair loss. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 36(11), 1180–1184. [DOI:10.1002/phar.1837]
Gupta, A. K., Venkataraman, M., Talukder, M., & Bamimore, M. A. (2022). Relative efficacy of minoxidil and the 5-α reductase inhibitors in androgenetic alopecia treatment of male patients: a network meta-analysis. JAMA Dermatology, 158(3), 266–274. [DOI:10.1001/jamadermatol.2021.5743]
Gupta, A. K., Hall, D. C., Talukder, M., & Bamimore, M. A. (2022). There is a positive dose-dependent association between low-dose oral minoxidil and its efficacy for androgenetic alopecia: findings from a systematic review with meta-regression analyses. Skin Appendage Disorders, 8(5), 355–361. [DOI:10.1159/000525137]
Gupta, A. K., Talukder, M., Shemar, A., Piraccini, B. M., & Tosti, A. (2023). Low-dose oral minoxidil for alopecia: a comprehensive review. Skin Appendage Disorders, 9(6), 423–437. [DOI:10.1159/000531890]
Gupta, A. K., Bamimore, M. A., Wang, T., & Talukder, M. (2024). The impact of monotherapies for male androgenetic alopecia: A network meta‐analysis study. Journal of Cosmetic Dermatology, 23(9), 2964–2972. [DOI:10.1111/jocd.16362]
Gupta, A. K., Wang, T., Bamimore, M. A., & Talukder, M. (2024). The relative effect of monotherapy with 5‐alpha reductase inhibitors and minoxidil for female pattern hair loss: A network meta‐analysis study. Journal of Cosmetic Dermatology, 23(1), 154–160. [DOI:10.1111/jocd.15910]
Gupta, A. K., Bamimore, M. A., Williams, G., & Talukder, M. (2025). Comparative Efficacy of Minoxidil and 5‐Alpha Reductase Inhibitors Monotherapy for Male Pattern Hair Loss: Network Meta‐Analysis Study of Current Empirical Evidence. Journal of Cosmetic Dermatology, 24(7), e70320. [DOI:10.1111/jocd.70320]
Gupta, A. K., Bamimore, M. A., Williams, G., & Talukder, M. (2025). Finasteride Use: Evaluation of Depression and Suicide Risk. Journal of Cosmetic Dermatology, 24(3), e70102. [DOI:10.1111/jocd.70102]
Gupta, M. A., Vujcic, B., & Gupta, A. K. (2020). Finasteride Use Is Associated with Higher Odds of Obstructive Sleep Apnea: Results from the US Food and Drug Administration Adverse Events Reporting System. Skinmed, 18(3), 146–150. [Google Scholar] [PubMed]
Hebert, A., Thiboutot, D., Gold, L. S., Cartwright, M., Gerloni, M., Fragasso, E., & Mazzetti, A. (2020). Efficacy and safety of topical clascoterone cream, 1%, for treatment in patients with facial acne: two phase 3 randomized clinical trials. JAMA Dermatology, 156(6), 621–630. [DOI:10.1001/jamadermatol.2020.0465]
Hirshburg, J. M., Kelsey, P. A., Therrien, C. A., Gavino, A. C., & Reichenberg, J. S. (2016). Adverse effects and safety of 5-alpha reductase inhibitors (finasteride, dutasteride): a systematic review. The Journal of Clinical and Aesthetic Dermatology, 9(7), 56–62. [Google Scholar] [PubMed] [PubMed Central] [PDF]
Hu, R., Xu, F., Sheng, Y., Qi, S., Han, Y., Miao, Y., Rui, W., & Yang, Q. (2015). Combined treatment with oral finasteride and topical minoxidil in male androgenetic alopecia: a randomized and comparative study in Chinese patients. Dermatologic Therapy, 28(5), 303–308. [DOI:10.1111/dth.12246]
Ibraheim, M. K., Elsensohn, A., Hauschild, C., Hilliard, A., & Dao, H. (2023). Low dose oral minoxidil and the conundrum of cardiovascular complications. Dermatology Online Journal, 29(4). [DOI:10.5070/D329461861]
Imperato-McGinley, J., & Zhu, Y. S. (2002). Androgens and male physiology the syndrome of 5α-reductase-2 deficiency. Molecular and Cellular Endocrinology, 198(1–2), 51–59. [DOI:10.1016/S0303-7207(02)00368-4]
Irwig, M. S., & Kolukula, S. (2011). Persistent sexual side effects of finasteride for male pattern hair loss. The Journal of Sexual Medicine, 8(6), 1747–1753. [DOI:10.1111/j.1743-6109.2011.02255.x]
Irwig, M. S. (2012). Persistent sexual side effects of finasteride: could they be permanent? The Journal of Sexual Medicine, 9(11), 2927–2932. [DOI:10.1111/j.1743-6109.2012.02846.x]
Irwig, M. S. (2014). Persistent sexual and nonsexual adverse effects of finasteride in younger men. Sexual Medicine Reviews, 2(1), 24–35. [DOI:10.1002/smrj.19]
Irwig, M. S. (2021). Is there a role for 5α‐reductase inhibitors in transgender individuals? Andrology, 9(6), 1729–1731. [DOI:10.1111/andr.12881]
Ismail, F. F., Meah, N., de Carvalho, L. T., Bhoyrul, B., Wall, D., & Sinclair, R. (2020). Safety of oral bicalutamide in female pattern hair loss: a retrospective review of 316 patients. Journal of the American Academy of Dermatology, 83(5), 1478–1479. [DOI:10.1016/j.jaad.2020.03.034]
Iyengar, L., & Li, J. (2025). Male and female pattern hair loss. Australian Prescriber, 48(3), 93–97. [DOI:10.18773/austprescr.2025.020]
Izard, J. P., & Siemens, D. R. (2020). Androgen deprivation therapy and mental health: impact on depression and cognition. European Urology Focus, 6(6), 1162–1164. [DOI:10.1016/j.euf.2019.11.010]
Janivara, R., Hazra, U., Pfennig, A., Harlemon, M., Kim, M. S., Eaaswarkhanth, M., Chen, W. C., Ogunbiyi, A., Kachambwa, P., Petersen, L. N., Jalloh, M., Mensah, J. E., Adjei, A. A., Adusei, B., Joffe, M., Gueye, S. M., Aisuodionoe-Shadrach, O. I., Fernandez, P. W., Rohan, T. E., Andrews, C., Rebbeck, T. R., Adebiyi, A. O., Agalliu, I., & Lachance, J. (2025). Uncovering the genetic architecture and evolutionary roots of androgenetic alopecia in African men. Human Genetics and Genomics Advances, 6(3), 100428. [DOI:10.1016/j.xhgg.2025.100428]
Jha, A. K., Zeeshan, M. D., Singh, A., & Singh, A. K. (2024). Efficacy and safety of spironolactone versus bicalutamide in female pattern hair loss: A retrospective comparative study. Australasian Journal of Dermatology, 65(5), 437–443. [DOI:10.1111/ajd.14306]
Jimenez-Cauhe, J., Saceda-Corralo, D., Rodrigues-Barata, R., Hermosa-Gelbard, A., Moreno-Arrones, O. M., Gil-Redondo, R., Ortega-Quijano, D., Fernandez-Nieto, D., Jaen-Olasolo, P., & Vaño-Galvan, S. (2021). Characterization and management of hypertrichosis induced by low-dose oral minoxidil in the treatment of hair loss. Journal of the American Academy of Dermatology, 84(1), 222–223. [DOI:10.1016/j.jaad.2020.08.124]
Keam, S. J., & Scott, L. J. (2008). Dutasteride: a review of its use in the management of prostate disorders. Drugs, 68(4), 463–485. [DOI:10.2165/00003495-200868040-00008]
Khaled, H. N., Allah, A. M. A., Abdelhameed, A. A., & Shehata, W. A. (2020). Role of serum androgens and prostate-specific antigen levels in men with androgenetic alopecia. Egyptian Journal of Dermatology and Venerology, 40(2), 106–111. [DOI:10.4103/ejdv.ejdv_46_19]
Kiguradze, T., Temps, W. H., Yarnold, P. R., Cashy, J., Brannigan, R. E., Nardone, B., Micali, G., West, D. P., & Belknap, S. M. (2017). Persistent erectile dysfunction in men exposed to the 5α-reductase inhibitors, finasteride, or dutasteride. PeerJ, 5, e3020. [DOI:10.7717/peerj.3020]
Kintor Pharmaceuticals. (2024). Annual Results Announcement for the Year Ended 31 December 2023. Kintor Pharmaceuticals. [PDF]
Kolvenbag, G. J., & Blackledge, G. R. (1996). Worldwide activity and safety of bicalutamide: a summary review. Urology, 47(1), 70–79. [DOI:10.1016/S0090-4295(96)80012-4]
Lee, S., Jo, H., Fond, G., Boyer, L., Smith, L., Hajek, A., & Yon, D. K. (2025). Signal detection between 5-alpha reductase inhibitors and the risk of suicidality and depression: an international pharmacovigilance analysis. European Journal of Clinical Pharmacology, 81(8), 1177–1185. [DOI:10.1007/s00228-025-03851-5]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal treatment of transgender women with oral estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Leliefeld, H. H., Debruyne, F. M., & Reisman, Y. (2025). The post-finasteride syndrome: possible etiological mechanisms and symptoms. International Journal of Impotence Research, 37(6), 414–421. [DOI:10.1038/s41443-023-00759-5]
Liang, X., Chang, Y., Wu, H., Liu, Y., Zhao, J., Wang, L., & Zhuo, F. (2022). Efficacy and safety of 5% minoxidil alone, minoxidil plus oral spironolactone, and minoxidil plus microneedling on female pattern hair loss: a prospective, single-center, parallel-group, evaluator blinded, randomized trial. Frontiers in Medicine, 9, 905140. [DOI:10.3389/fmed.2022.905140]
Lucky, A. W., Piacquadio, D. J., Ditre, C. M., Dunlap, F., Kantor, I., Pandya, A. G., Savin, R. C., & Tharp, M. D. (2004). A randomized, placebo-controlled trial of 5% and 2% topical minoxidil solutions in the treatment of female pattern hair loss. Journal of the American Academy of Dermatology, 50(4), 541–553. [DOI:10.1016/j.jaad.2003.06.014]
Maeda, T., Kikuchi, E., Hasegawa, M., Homma, K., Ando, T., Suzuki, K., Kaneko, G., Mizuno, R., Miyajima, A., & Oya, M. (2018). Influence of dutasteride treatment on serum hormone levels and aging male symptoms in patients with benign prostatic enlargement. International Journal of Urology, 25(1), 70–74. [DOI:10.1111/iju.13470]
Maksym, R. B., Kajdy, A., & Rabijewski, M. (2019). Post-finasteride syndrome–does it really exist? The Aging Male, 22(4), 250–259. [DOI:10.1080/13685538.2018.1548589]
Mapar, M. A., & Omidian, M. (2007). Is topical minoxidil solution effective on androgenetic alopecia in routine daily practice? Journal of Dermatological Treatment, 18(5), 268–270. [DOI:10.1080/09546630701383727]
Marks, D. H., & Senna, M. M. (2020). Androgenetic alopecia in gender minority patients. Dermatologic Clinics, 38(2), 239–247 [DOI:10.1016/j.det.2019.10.010]
Martin, A., Chen, L. C., Hsieh, T. Y. J., & Senna, M. M. (2025). Despite progesterone receptor affinity, spironolactone is not associated with meningioma development: A population-based cohort study. Journal of the American Academy of Dermatology, 92(6), 1456–1457. [DOI:10.1016/j.jaad.2025.02.056]
Meara, K. (2025 June 23). Pelage’s PP405 Demonstrates Efficacy in Phase 2a Trial for Androgenetic Alopecia. Dermatology Times. [URL]
Mondaini, N., Gontero, P., Giubilei, G., Lombardi, G., Cai, T., Gavazzi, A., & Bartoletti, R. (2007). Finasteride 5 mg and sexual side effects: how many of these are related to a nocebo phenomenon? The Journal of Sexual Medicine, 4(6), 1708–1712. [DOI:10.1111/j.1743-6109.2007.00563.x]
Moussa, A., Kazmi, A., Bokhari, L., & Sinclair, R. D. (2022). Bicalutamide improves minoxidil-induced hypertrichosis in female pattern hair loss: A retrospective review of 35 patients. Journal of the American Academy of Dermatology, 87(2), 488–490. [DOI:10.1016/j.jaad.2021.10.048]
Needle, C. D., Brinks, A. L., Kearney, C. A., Shapiro, J., & Lo Sicco, K. I. (2025). Key considerations of injectable minoxidil for alopecia. Archives of Dermatological Research, 317(1), 572. [DOI:10.1007/s00403-025-04068-3]
Neubauer, Z., Ong, M. M., & Lipner, S. R. (2025). Similar Risk of Sexual Dysfunction in Androgenetic Alopecia Patients Treated With Dutasteride vs. Finasteride: A Retrospective Cohort Database Study. International Journal of Dermatology, advance online publication. [DOI:10.1111/ijd.17887]
Nguyen, D. D., Marchese, M., Cone, E. B., Paciotti, M., Basaria, S., Bhojani, N., & Trinh, Q. D. (2021). Investigation of suicidality and psychological adverse events in patients treated with finasteride. JAMA Dermatology, 157(1), 35–42. [DOI:10.1001/jamadermatol.2020.3385]
Nguyen, N. H., Taylor, J. M., Huang, K. X., & Lee, J. C. (2025). Estrogen hormone therapy stabilizes lateral hairline in transfeminine patients: Implications for facial feminization surgery. Journal of Plastic, Reconstructive & Aesthetic Surgery, 101, 246–251. [DOI:10.1016/j.bjps.2024.07.044]
Olsen, E. A., Hordinsky, M., Whiting, D., Stough, D., Hobbs, S., Ellis, M. L., Wilson, T., Rittmaster, R. S., & Dutasteride Alopecia Research Team (2006). The importance of dual 5α-reductase inhibition in the treatment of male pattern hair loss: results of a randomized placebo-controlled study of dutasteride versus finasteride. Journal of the American Academy of Dermatology, 55(6), 1014–1023. [DOI:10.1016/j.jaad.2006.05.007]
Ovcharenko, Y., Khobzei, K., & Lortkipanidze, N. (2025). Androgenetic alopecia. In Ovcharenko, Y., Lyakhovitsky, A., & Jafferany, M. (Eds.). Psychotrichology: Psychiatric and Psychosocial Aspects of Hair Diseases (pp. 137–170). Cham: Springer Nature Switzerland. [DOI:10.1007/978-3-031-64083-4_9]
Ozono, S., Okajima, E., Yamaguchi, A., Yoshikawa, M., Iwai, A., Moriya, A., Yoshida, K., Samma, S., Maruyama, Y., & Hirao, Y. (2000). A prospective randomized multicenter study of chlormadinone acetate versus flutamide in total androgen blockade for prostate cancer. Japanese Journal of Clinical Oncology, 30(9), 389–396. [DOI:10.1093/jjco/hyd106]
Paradisi, R., Porcu, E., Fabbri, R., Seracchioli, R., Battaglia, C., & Venturoli, S. (2011). Prospective cohort study on the effects and tolerability of flutamide in patients with female pattern hair loss. Annals of Pharmacotherapy, 45(4), 469–475. [DOI:10.1345/aph.1P600]
Patel, P., Nessel, T. A., & Kumar, D. (2023). Minoxidil. In StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. [Google Scholar] [PubMed] [PubMed Bookshelf]
Penha, M. A., Miot, H. A., Kasprzak, M., & Ramos, P. M. (2024). Oral minoxidil vs topical minoxidil for male androgenetic alopecia: a randomized clinical trial. JAMA Dermatology, 160(6), 600–605. [DOI:10.1001/jamadermatol.2024.0284]
Pereira, A. F. J. R., & Coelho, T. O. D. A. (2020). Post-finasteride syndrome. Anais Brasileiros de Dermatologia, 95(3), 271–277. [DOI:10.1016/j.abd.2020.02.001]
Perez, S. M., Nguyen, B., & Senna, M. M. (2025). Efficacy and safety of bicalutamide in female hair loss: a review of the literature. JAAD Reviews, 4, 61–68. [DOI:10.1016/j.jdrv.2025.03.005]
Pirastu, N., Joshi, P. K., de Vries, P. S., Cornelis, M. C., McKeigue, P. M., Keum, N., Franceschini, N., Colombo, M., Giovannucci, E. L., Spiliopoulou, A., Franke, L., North, K. E., Kraft, P., Morrison, A. C., Esko, T., & Wilson, J. F. (2017). GWAS for male-pattern baldness identifies 71 susceptibility loci explaining 38% of the risk. Nature Communications, 8(1), 1584. [DOI:10.1038/s41467-017-01490-8]
Prince, J. C. J., & Safer, J. D. (2020). Endocrine treatment of transgender individuals: current guidelines and strategies. Expert Review of Endocrinology & Metabolism, 15(6), 395–403. [DOI:10.1080/17446651.2020.1825075]
Randolph, M., & Tosti, A. (2021). Oral minoxidil treatment for hair loss: a review of efficacy and safety. Journal of the American Academy of Dermatology, 84(3), 737–746. [DOI:10.1016/j.jaad.2020.06.1009]
Ramos, P. M., Sinclair, R. D., Kasprzak, M., & Miot, H. A. (2020). Minoxidil 1 mg oral versus minoxidil 5% topical solution for the treatment of female-pattern hair loss: a randomized clinical trial. Journal of the American Academy of Dermatology, 82(1), 252–253. [DOI:10.1016/j.jaad.2019.08.060]
Rosenthal, A., Conde, G., Greco, J. F., & Gharavi, N. M. (2024). Management of androgenic alopecia: a systematic review of the literature. Journal of Cosmetic and Laser Therapy, 26(1–4), 1–16. [DOI:10.1080/14764172.2024.2362126]
Rossi, A., Cantisani, C., Melis, L., Iorio, A., Scali, E., & Calvieri, S. (2012). Minoxidil use in dermatology, side effects and recent patents. Recent Patents on Inflammation & Allergy Drug Discovery, 6(2), 130–136. [DOI:10.2174/187221312800166859]
Rossi, A., & Caro, G. (2024). Efficacy of the association of topical minoxidil and topical finasteride compared to their use in monotherapy in men with androgenetic alopecia: A prospective, randomized, controlled, assessor blinded, 3‐arm, pilot trial. Journal of Cosmetic Dermatology, 23(2), 502–509. [DOI:10.1111/jocd.15953]
Ryu, H. K., Kim, K. M., Yoo, E. A., Sim, W. Y., & Chung, B. C. (2006). Evaluation of androgens in the scalp hair and plasma of patients with male‐pattern baldness before and after finasteride administration. British Journal of Dermatology, 154(4), 730–734. [DOI:10.4103/ijd.ijd_729_23]
Saceda-Corralo, D., Domínguez-Santas, M., Vañó-Galván, S., & Grimalt, R. (2023). What’s new in therapy for male androgenetic alopecia? American Journal of Clinical Dermatology, 24(1), 15–24. [DOI:10.1007/s40257-022-00730-y]
Sadasivam, I. P., Sambandam, R., Kaliyaperumal, D., & Dileep, J. E. (2024). Androgenetic Alopecia in Men: An Update On Genetics. Indian Journal of Dermatology, 69(3), 282. [DOI:10.4103/ijd.ijd_729_23]
Sanabria, B. D., Perdomo, Y. C., Miot, H. A., & Ramos, P. M. (2024). Oral minoxidil 7.5 mg for hair loss increases heart rate with no change in blood pressure in 24 h Holter and 24 h ambulatory blood pressure monitoring. Anais Brasileiros de Dermatologia, 99(5), 734–736. [DOI:10.1016/j.abd.2023.08.016]
Sanabria, B., Miot, H. A., Sinclair, R., Chaves, C., & Müller Ramos, P. (2024). Sublingual Minoxidil 5 mg versus Oral Minoxidil 5 mg for male androgenetic alopecia: A double-blind randomized clinical trial. Journal of the European Academy of Dermatology and Venereology, 39(8), e700–e702. [DOI:10.1111/jdv.20508]
Severi, G., Sinclair, R., Hopper, J. L., English, D. R., McCredie, M. R. E., Boyle, P., & Giles, G. G. (2003). Androgenetic alopecia in men aged 40–69 years: prevalence and risk factors. British Journal of Dermatology, 149(6), 1207–1213. [DOI:10.1111/j.1365-2133.2003.05565.x]
Shadi, Z. (2023). Compliance to topical minoxidil and reasons for discontinuation among patients with androgenetic alopecia. Dermatology and Therapy, 13(5), 1157–1169. [DOI:10.1007/s13555-023-00919-x]
Siebert, A. L., Lapping-Carr, L., & Morgans, A. K. (2020). Neuropsychiatric impact of androgen deprivation therapy in patients with prostate cancer: current evidence and recommendations for the clinician. European Urology Focus, 6(6), 1170–1179. [DOI:10.1016/j.euf.2020.05.014]
Sinclair, R., Wewerinke, M., & Jolley, D. (2005). Treatment of female pattern hair loss with oral antiandrogens. British Journal of Dermatology, 152(3), 466–473. [DOI:10.1111/j.1365-2133.2005.06218.x]
Sinclair, R., Trindade de Carvalho, L., Ferial Ismail, F., & Meah, N. (2020). Treatment of male and female pattern hair loss with sublingual minoxidil: a retrospective case-series of 64 patients. Journal of the European Academy of Dermatology and Venereology, 34(12), e795–e796. [DOI:10.1111/jdv.16616]
Sinclair, R., Sicinska, J., Bokhari, L., & Kasprzak, M. (2025). Sublingual minoxidil increases fibre diameter in male androgenetic alopecia: a proxy for reversal of hair follicle miniaturization. Clinical and Experimental Dermatology, 50(7), 1362–1365. [DOI:10.1093/ced/llaf012]
Singh, S., Patil, A., Kianfar, N., Waśkiel‐Burnat, A., Rudnicka, L., Sinclair, R., & Goldust, M. (2022). Does topical minoxidil at concentrations higher than 5% provide additional clinical benefit? Clinical and Experimental Dermatology, 47(11), 1951–1955. [DOI:10.1111/ced.15338]
Stanczyk, F. Z., Azen, C. G., & Pike, M. C. (2013). Effect of finasteride on serum levels of androstenedione, testosterone and their 5α-reduced metabolites in men at risk for prostate cancer. The Journal of Steroid Biochemistry and Molecular Biology, 138, 10–16. [DOI:10.1016/j.jsbmb.2013.02.015]
Stevenson, M. O., Wixon, N., & Safer, J. D. (2016). Scalp hair regrowth in hormone-treated transgender woman. Transgender Health, 1(1), 202–204. [DOI:10.1089/trgh.2016.0022]
Suchonwanit, P., Srisuwanwattana, P., Chalermroj, N., & Khunkhet, S. (2018). A randomized, double‐blind controlled study of the efficacy and safety of topical solution of 0.25% finasteride admixed with 3% minoxidil vs. 3% minoxidil solution in the treatment of male androgenetic alopecia. Journal of the European Academy of Dermatology and Venereology, 32(12), 2257–2263. [DOI:10.1111/jdv.15171]
Suchonwanit, P., Iamsumang, W., & Rojhirunsakool, S. (2019). Efficacy of topical combination of 0.25% finasteride and 3% minoxidil versus 3% minoxidil solution in female pattern hair loss: a randomized, double-blind, controlled study. American Journal of Clinical Dermatology, 20(1), 147–153. [DOI:10.1007/s40257-018-0387-0]
Suchonwanit, P., Thammarucha, S., & Leerunyakul, K. (2019). Minoxidil and its use in hair disorders: a review. Drug Design, Development and Therapy, 13, 2777–2786. [DOI:10.2147/DDDT.S214907]
T’Sjoen, G., Arcelus, J., Gooren, L., Klink, D. T., & Tangpricha, V. (2019). Endocrinology of transgender medicine. Endocrine Reviews, 40(1), 97–117. [DOI:10.1210/er.2018-00011]
Tan, J. J. Y., Thuya, W. L., Zhu, H., Kristo, J. G., Common, J. E., Wu, C., Ho, P. C. L., & Kang, L. (2025). Comparative analysis of 3D and 2D cell-culturing methods in hair follicle spheroid morphogenesis and drug responsiveness. Biomaterials Advances, 177, 214423. [DOI:10.1016/j.bioadv.2025.214423]
Tang, G. T., Zwickl, S., Sinclair, R., Zajac, J. D., & Cheung, A. S. (2023). Effect of gender-affirming hormone therapy on hair growth: a systematic review of the literature. Clinical and Experimental Dermatology, 48(10), 1117–1127. [DOI:10.1093/ced/llad203]
Tang, G. T., Sinclair, R., Leemaqz, S., & Cheung, A. S. (2025). Scalp hair parameter changes in transgender individuals commencing gender-affirming hormone therapy: A 24-week prospective observational study. Journal of the American Academy of Dermatology, 92(6), 1390–1394. [DOI:10.1016/j.jaad.2025.01.087]
Tang, G. T., Leemaqz, S., Eisman, S., Sinclair, R., & Cheung, A. (2025). Treating testosterone therapy related hair loss in trans people: a clinical trial of sublingual minoxidil. AusPATH 2025 Symposium. [PDF]
Tanglertsampan, C. (2012). Efficacy and safety of 3% minoxidil versus combined 3% minoxidil/0.1% finasteride in male pattern hair loss: a randomized, double-blind, comparative study. Journal of the Medical Association of Thailand, 95(10), 1312–1316. [Google Scholar] [PubMed] [PDF]
Traish, A. M., Hassani, J., Guay, A. T., Zitzmann, M., & Hansen, M. L. (2011). Adverse side effects of 5α-reductase inhibitors therapy: persistent diminished libido and erectile dysfunction and depression in a subset of patients. The Journal of Sexual Medicine, 8(3), 872–884. [DOI:10.1111/j.1743-6109.2010.02157.x]
Traish, A. M., Krakowsky, Y., Doros, G., & Morgentaler, A. (2019). Do 5α-reductase inhibitors raise circulating serum testosterone levels? A comprehensive review and meta-analysis to explaining paradoxical results. Sexual Medicine Reviews, 7(1), 95–114. [DOI:10.1016/j.sxmr.2018.06.002]
Trüeb, R. M., Régnier, A., Dutra Rezende, H., & Gavazzoni Dias, M. F. R. (2019). Post-finasteride syndrome: an induced delusional disorder with the potential of a mass psychogenic illness? Skin Appendage Disorders, 5(5), 320–326. [DOI:10.1159/000497362]
Trüeb, R. M., Luu, N. N. C., Caballero-Uribe, N., Dias, R. G., & Rezende, H. D. (2024). Comment on Current Investigations into the Postfinasteride Syndrome. International Journal of Trichology, 16(1), 6–12. [DOI:10.4103/ijt.ijt_122_21]
Uleri, A., Cornu, J. N., Gobbo, A., Herrmann, T. R., De Nunzio, C., Hashim, H., & Baboudjian, M. (2024). Association of 5α-reductase inhibitors with depression and suicide: a mini systematic review and meta-analysis. European Urology Focus, 10(5), 751–753. [DOI:10.1016/j.euf.2024.04.009]
Upreti, R., Naredo, G., Faqehi, A. M., Hughes, K. A., Stewart, L. H., Walker, B. R., Homer, N. Z., & Andrew, R. (2015). Simultaneous pharmacokinetic and pharmacodynamic analysis of 5α-reductase inhibitors and androgens by liquid chromatography tandem mass spectrometry. Talanta, 131, 728–735. [DOI:10.1016/j.talanta.2014.07.087]
Valente Duarte de Sousa, I. C., & Tosti, A. (2013). New investigational drugs for androgenetic alopecia. Expert Opinion on Investigational Drugs, 22(5), 573–589. [DOI:10.1517/13543784.2013.784743]
Vañó-Galván, S., Pirmez, R., Hermosa-Gelbard, A., Moreno-Arrones, Ó. M., Saceda-Corralo, D., Rodrigues-Barata, R., Jimenez-Cauhe, J., Koh, W. L., Poa, J. E., Jerjen, R., Trindade de Carvalho, L., John, J. M., Salas-Callo, C. I., Vincenzi, C., Yin, L., Lo-Sicco, K., Waskiel-Burnat, A., Starace, M., Zamorano, J. L., Jaén-Olasolo, P., Piraccini, B. M., Rudnicka, L., Shapiro, J., Tosti, A., Sinclair, R., & Bhoyrul, B. (2021). Safety of low-dose oral minoxidil for hair loss: a multicenter study of 1404 patients. Journal of the American Academy of Dermatology, 84(6), 1644–1651. [DOI:10.1016/j.jaad.2021.02.054]
Vierhapper, H., Maier, H., Nowotny, P., & Waldhäusl, W. (2003). Production rates of testosterone and of dihydrotestosterone in female pattern hair loss. Metabolism, 52(7), 927–929. [DOI:10.1016/S0026-0495(03)00060-X]
Vierhapper, H., Nowotny, P., Maier, H., & Waldhausl, W. (2001). Production rates of dihydrotestosterone in healthy men and women and in men with male pattern baldness: determination by stable isotope/dilution and mass spectrometry. The Journal of Clinical Endocrinology & Metabolism, 86(12), 5762–5764. [DOI:10.1210/jcem.86.12.8078]
Wang, C., Du, Y., Bi, L., Lin, X., Zhao, M., & Fan, W. (2023). The efficacy and safety of oral and topical spironolactone in androgenetic alopecia treatment: a systematic review. Clinical, Cosmetic and Investigational Dermatology, 16, 603–612. [DOI:10.2147/CCID.S398950]
Weichert, M., Chen, M., Guo, W., Schrock, N., & Briley, J. (2025). Efficacy and safety of minoxidil therapy: A systematic review and meta-analysis weighing the benefits against the risk of hypertrichosis. JAAD Reviews, 4, 58–60. [DOI:10.1016/j.jdrv.2025.02.013]
Yamana, K., Labrie, F., & Luu-The, V. (2010). Human type 3 5α-reductase is expressed in peripheral tissues at higher levels than types 1 and 2 and its activity is potently inhibited by finasteride and dutasteride. Hormone Molecular Biology and Clinical Investigation, 2(3), 293–299. [DOI:10.1515/HMBCI.2010.035]
Yang, X., Qiao, R., Cheng, W., Lan, X., Li, Y., & Jiang, Y. (2024). Comparative efficacy of 2% minoxidil alone against combination of 2% minoxidil and low-level laser therapy in female pattern hair loss–a randomized controlled trial in Chinese females. Photodiagnosis and Photodynamic Therapy, 45, 103966. [DOI:10.1016/j.pdpdt.2024.103966]
Yazdabadi, A., & Sinclair, R. (2011). Treatment of female pattern hair loss with the androgen receptor antagonist flutamide. Australasian Journal of Dermatology, 52(2), 132–134. [DOI:10.1111/j.1440-0960.2010.00735.x]
Yoong, N., Yii, V., Sinclair, R., & Bhoyrul, B. (2025). Combination of oral minoxidil and bicalutamide for the treatment of female pattern hair loss in adolescents. Journal of the American Academy of Dermatology, 92(5), 1156–1157. [DOI:10.1016/j.jaad.2025.01.056]
Zakhem, G. A., Goldberg, J. E., Motosko, C. C., Cohen, B. E., & Ho, R. S. (2019). Sexual dysfunction in men taking systemic dermatologic medication: a systematic review. Journal of the American Academy of Dermatology, 81(1), 163–172. [DOI:10.1016/j.jaad.2019.03.043]
Zaminski, D., Zampella, J., Shapiro, J., Sicco, K. I. L., & Mazori, D. R. (2025). Use of low-dose oral minoxidil for hair growth in transgender and gender non-binary adult patients: a retrospective cohort study. Journal of the American Academy of Dermatology, advance online publication. [DOI:10.1016/j.jaad.2025.06.029]
Zeltzer, A. A., Keren, A., & GILHAR, A. (2024). Topical Minoxidil Rejuvenates Hair Follicles from Men with Androgenetic Alopecia In Vivo. Acta Dermato-Venereologica, 104, 24213. [DOI:10.2340/actadv.v104.24213]
Zhong, X., Yang, Y., Wei, S., & Liu, Y. (2025). Multidimensional assessment of adverse events of finasteride: a real-world pharmacovigilance analysis based on FDA Adverse Event Reporting System (FAERS) from 2004 to April 2024. PloS One, 20(3), e0309849. [DOI:10.1371/journal.pone.0309849]
Zhou, Z., Song, S., Gao, Z., Wu, J., Ma, J., & Cui, Y. (2019). The efficacy and safety of dutasteride compared with finasteride in treating men with androgenetic alopecia: a systematic review and meta-analysis. Clinical Interventions In Aging, 14, 399–406. [DOI:10.2147/CIA.S192435]
\ No newline at end of file
+A Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People - Transfeminine Science
A Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People
By Sam | First published September 8, 2025 | Last modified September 9, 2025
Abstract / TL;DR
Androgenetic alopecia is a common condition affecting a significant proportion of transfeminine people at onset of hormonal transition. Although several studies have assessed the influence of gender-affirming hormone therapy on scalp hair, there are no robust data to guide optimal dosing regimens. In the wider population, 5α-reductase inhibitors and other treatments, such as minoxidil, have been found to act dose-dependently and additively or synergistically with each other to halt and partially reverse hair loss caused by androgens. Some transfeminine people prefer to add these treatments to their regimens to try to regrow more hair or as prophylaxis against further hair loss. Dutasteride is a superior 5α-reductase inhibitor to finasteride in terms of efficacy and has similar adverse effects. For transfeminine people who wish to use minoxidil, the route of administration should be considered and determined on an individual basis. Other agents, such as spironolactone, may also provide benefit. Research into new therapies which could one day result in new pharmaceutical options for transfeminine people is currently ongoing in the general population.
Introduction
Scalp hair loss, particularly androgenetic alopecia (AGA), is a condition that affects millions of people worldwide and that carries significant psychosocial implications. Hair is a major component of human identity and is often integral to gender expression. As such, hair loss can be particularly distressing for transfeminine people (Marks & Senna, 2020; Gao et al., 2023a; Tang et al., 2023). Over the last decade, there has been a surge in interest in pharmaceutical treatments for treating AGA among both the scientific community and the general population. However, considerably less attention has been devoted to treating AGA in transfeminine people, specifically.
A few studies have evaluated the effects of feminising hormone therapy on scalp hair. A multicentre prospective study found a slight but statistically significant reduction in average Norwood–Hamilton score after 12 months in transfeminine people treated with estradiol and cyproterone acetate (Cocchetti et al., 2023). In another study, duration of estradiol use with or without spironolactone was associated with a −0.07 cm (95% CI: [−0.10, −0.04]) reduction in lateral forehead length per year in the first few years of treatment (Nguyen et al., 2025). Finally, a prospective study demonstrated statistically significant increases in follicular density and total hair count at both the midfrontal and vertex (crown) scalp after 24 weeks in transfeminine people on an unspecified regimen (Tang et al., 2025a). This was accompanied by a reduction in average hair shaft diameter, driven by increases in intermediate and vellus hair density, but with no significant change in terminal hair density. Individual case studies and series showing improvement have also been reported in the literature (Dewhurst & Underhill, 1979; Adenuga, Summers, & Bergfel, 2012; Stevenson, Wixon, & Safer, 2016). Overall, it appears that AGA progression is usually halted and partially reversed due to reduced or suppressed testosterone levels. However, there is a lack of data to inform long-term outcomes. Reversal of AGA may be incomplete in many transfeminine people, particularly in advanced stages, due to irreversible follicular miniaturisation.
The most common therapies for AGA in cisgender people include 5α-reductase inhibitors (5-ARIs) and minoxidil (Gao et al., 2023b; Devjani et al., 2023). Minoxidil use in transfeminine people has been examined by a small study (Zaminski et al., 2025). A study also found that concomitant use of finasteride, dutasteride, and/or minoxidil was associated with a lower hairline compared to treatment with estradiol alone or estradiol and spironolactone alone (Nguyen et al., 2025). However, limitations of these studies include risk of bias due to study design, possible confounding due to secondary factors, and in the case of the former study, lack of a control group. As such, there is a paucity of reliable data to show if these treatments, especially 5-ARIs, provide additional benefit over gender-affirming hormone therapy alone (T’Sjoen et al., 2019; Prince & Safer, 2020; Irwig, 2021; Coleman et al., 2022; Gao et al., 2023b). Despite this, a considerable subset of transfeminine people opt to use 5α-reductase inhibitors and minoxidil (Leinung, Feustel, & Joseph, 2018; Nguyen et al., 2025).
The purpose of this literature review is to critically summarise various pharmaceutical interventions that have been shown to have an acceptable safety profile in the general population and that may therefore be used as adjunct therapy to treat and as prophylaxis against hair loss in transfeminine people if desired. Non-pharmaceutical interventions, such as microneedling and hair transplantation, are outside the scope of this article. Specifically, this review focuses predominantly on 5-ARIs and minoxidil but also discusses androgen receptor antagonists, in addition to some further therapies that may be available in the future.
Etiology of Androgenetic Scalp Hair Loss
Androgenetic alopecia, commonly referred to as male-pattern or female-pattern hair loss, is a polygenic condition characterised by progressive miniaturisation of scalp hair follicles in a pattern-specific distribution (Anastassakis, 2022; Ovcharenko, Khobzei, & Lortkipanidze, 2025). The pathophysiology of male-pattern hair loss is fundamentally androgen-dependent. Specifically, dihydrotestosterone (DHT) binds to the androgen receptors in susceptible hair follicles, in turn initiating a cascade of transcriptional changes that alter the follicular growth cycle. This includes shortening of the anagen (growth) phase, prolongation of the telogen (resting) phase, and eventually, follicular miniaturisation (Dhurat & Daruwalla, 2021). Notably, while androgens are necessary for the development of AGA, they are not sufficient on their own; individuals with high circulating androgens may not develop AGA if their follicles lack the required sensitivity level (Khaled et al. 2020).
The genetic architecture of AGA is complex and involves multiple loci, although the AR gene on the X chromosome is generally thought to be a significant contributor (Sadasivam et al., 2024). Polymorphisms in the AR gene, particularly those affecting receptor sensitivity, have been associated with increased risk of AGA. Additionally, epigenetic factors, including methylation patterns and histone modifications, may also play a role in regulating gene expression relevant to hair follicle cycling. Large genome-wide association studies (GWAS) have identified other loci which modulate follicular response to androgens in the scalp (Pirastu et al., 2017; Chen et al., 2022; Janivara et al., 2025).
Not all scalp follicles are equally sensitive to androgens. Susceptibility is region-specific and genetically determined, with the vertex and frontal scalp being most affected (Severi et al., 2003; Fujimaki et al., 2024). The expression of 5α-reductase, the enzyme responsible for converting testosterone to DHT, is elevated in these regions, which serves to further amplify local androgenic signalling (Dhurat & Daruwalla, 2021). While androgens promote hair growth in areas such as the beard and chest, they paradoxically cause scalp hair loss in genetically predisposed individuals (Anastassakis, 2022; Ovcharenko, Khobzei, & Lortkipanidze, 2025). Miniaturised scalp hair follicles in AGA undergo a progressive transformation. There may be a critical window for intervention in which current therapeutic strategies may reverse or significantly slow progression. Ultimately, the follicles eventually enter a state of dormancy or senescence, rendering them unable to produce cosmetically significant hair. However, the follicles themselves do appear to remain in situ.
Although pattern hair loss is often divided into “male-“ or “female-“ pattern hair loss in the literature, the presentation is often similar. Studies of men with pattern hair loss have consistently shown the role of DHT (Vierhapper et al., 2001; Ryu et al., 2006; Olsen et al., 2006). There are three isoforms of 5α-reductase: type I, II, and III, all of which are thought to contribute to AGA. However, the mechanism of type III 5α-reductase is less well understood. It is notable that men with 5α-reductase type II deficiency do not experience AGA (Imperato-McGinley & Zhu, 2002). Consequently, reducing intracellular DHT concentrations can prevent AGA. However, the involvement of DHT in female AGA is less clear. A study of women with pattern hair loss found that mean average concentrations of testosterone and DHT were higher than in controls without AGA, but still within the female range (Vierhapper et al., 2003). Hence, female concentrations of testosterone and DHT appear to be associated with pattern hair loss in a certain subset of women. A case of a woman with complete androgen insensitivity syndrome (CAIS) with female pattern hair loss has been reported in the literature (Cousen & Messenger, 2010).
5α-Reductase Inhibitors
5α-Reductase inhibitors are a class of medications developed to treat conditions caused by the effects of DHT, such as benign prostatic hyperplasia and androgenetic alopecia. As implied by their name, these medications function by inhibiting 5α-reductase enzyme activity. Whilst not curative, 5-ARIs appear to halt or substantially slow hair loss in cisgender men. Globally, the two most widely used 5-ARIs include finasteride and dutasteride.
Efficacy of Finasteride Compared to Dutasteride in AGA
In the United States, finasteride prescriptions have increased exponentially in recent years, largely driven by its use in the treatment of hair loss (AHLA, 2024). Dutasteride prescriptions are far fewer, estimated in the hundreds of thousands, but also growing due to increased use for AGA. This difference is largely due to finasteride being widely licensed for this indication throughout the world, whilst dutasteride remains off-label in most countries (Altendorf et al., 2025). However, dutasteride is licensed for use in the treatment of AGA in South Korea, Japan and Mexico. Increasing interest is driving further adoption and research into its use.
Despite its use for AGA being mostly off-label, dutasteride is now widely regarded as a more efficacious and hence superior 5-ARI than finasteride. Numerous studies have established that dutasteride results in greater suppression of serum DHT concentrations (i.e., about 70% with finasteride vs 90–95% with dutasteride) (Clark et al., 2004; Olsen et al., 2006; Amory et al., 2007; Upreti et al., 2015). Another study directly comparing scalp tissue concentrations found that dutasteride reduced DHT substantially more than finasteride (mean reduction of about 65% with finasteride versus 90% with dutasteride, though with wide interindividual variation) (Hobo et al., 2023). These differences have been primarily attributed to its broader inhibition of the 5α-reductase enzyme. More specifically, finasteride is a selective inhibitor of type II and III 5α-reductase, whereas dutasteride indiscriminately acts on all three isoforms (Gisleskog et al., 1998; Keam & Scott, 2008; Yamana, Labrie, & Luu-The, 2010). Dutasteride has also been theorised to accumulate inside certain tissues, further enhancing its therapeutic effect.
In accordance with the above, two large network meta-analysis studies found that oral dutasteride is superior to oral and topical finasteride in the treatment of male AGA in terms of both total hair density and terminal hair density (Gupta et al., 2024a; Gupta et al., 2025a). These studies also found the effects of finasteride and dutasteride to be dose-dependent. On average, there was no difference between treatment groups using oral and topical finasteride. A systematic review found that dutasteride was superior to finasteride in some studies in terms of hair thickness (Almudimeegh et al., 2024). However, in contrast to the above findings in the case of male AGA, a network meta-analysis of studies investigating different interventions for female AGA found that clinical trials assessing the effectiveness of dutasteride do not yet exist (Gupta et al., 2024b). Notably, oral finasteride given at a dose of 1 mg/day was not found to be effective in treating female AGA, yet oral finasteride used at a dose of 5 mg/day outperformed all other single-agent interventions. Because of the dose-dependent effects of 5-ARIs, it could well be that dutasteride might be more efficacious than finasteride in the treatment of female AGA, as in male AGA. Hopefully, future clinical trials will shed light on this.
Adverse Effects of 5α-Reductase Inhibitors
5-ARIs are associated with certain adverse effects in a subset of individuals. These include, but are not limited to decreased libido, erectile dysfunction, reduced ejaculate volume and, possibly, fatigue and depression (Gupta, Vujcic, & Gupta, 2020; Choi et al., 2022; Zhong et al., 2025; Cilio et al., 2025). In the case of transfeminine hormone therapy, some of these effects may actually be desirable.
Currently, there are no large cohort studies or randomised controlled trials that have investigated the incidence of certain adverse effects of hormone therapy with 5-ARIs in transfeminine people. Studies of the general cisgender population may provide some limited insight. A meta-analysis of clinical trials found that the odds of hypoactive sexual desire and erectile dysfunction were each approximately 1.5-fold greater, in male users of 5-ARIs versus placebo (Corona et al., 2017). No difference was found between finasteride and dutasteride in their effects on sexual desire. This is in accordance with findings from more recent publications (Zhou et al., 2019; Zahkem et al., 2019; Estill et al., 2023; Neubauer, Ong, & Lipner, 2025). It is possible that 5-ARIs could produce further reduction in sexual function combined with other hormone therapy medications in transfeminine people, but there are no data to confirm or refute this.
Some studies have found that 5-ARIs are associated with slightly increased circulating testosterone levels (Amory et al., 2007; Stanczyk, Azen, & Pike, 2013; Maeda et al., 2018). However, a 2019 meta-analysis of studies of cisgender men concluded that finasteride and dutasteride use did not unequivocally result in statistically significant increases in serum testosterone levels (Traish et al., 2019). The relevance of marginally raised testosterone in individuals receiving gender-affirming hormone therapy is unclear because exogenous estrogen and antiandrogen therapy typically suppresses endogenous testosterone production to concentrations far below the male range. Nevertheless, a retrospective analysis of transfeminine people using oral estradiol and spironolactone did find that finasteride use appeared to have a moderately deleterious effect on testosterone suppression (Leinung, Feustel, & Joseph, 2018). Studies have generally found that spironolactone does not, by itself, actually lower testosterone concentrations in transfeminine people (Angus et al., 2021). As such, these findings might not be applicable to other antigonadotrophic antiandrogens such as GnRH agonists and progestogens including cyproterone acetate. The influence of 5-ARIs on testosterone concentrations in transfeminine people would be an interesting point for more studies to explore in the future.
The possible effect of 5-ARIs on cognitive function, mood, depression, and suicide risk is controversial. Androgen deprivation therapy, in general, results in an increased risk of psychiatric complications (Izard & Siemens, 2020; Siebert, Lapping-Carr, & Morgans, 2020). These effects have been attributed to the depletion of circulating testosterone available for conversion into estrogens and hence can be mitigated with the use of concomitant estradiol administration (Coelingh Bennink et al., 2024). However, in some studies, 5-ARIs have been associated with increased rates of depression despite testosterone levels remaining well within the male range. Several large pharmacovigilance studies have found signals for reduced cognitive function, depression, and suicidality in users of finasteride and dutasteride (Nguyen et al., 2021; Cho et al., 2024; Zhong et al., 2025; Gupta et al., 2025b; Lee at al., 2025). In some of these studies, the signals have been quite strong. These data have prompted some countries to mandate warnings about possible long-term side effects on product labels. The psychiatric effects of 5-ARIs have been hypothesised to be a consequence of this class of drugs also inhibiting the synthesis of neuroactive steroids such as allopregnanolone. These neurosteroids may have antidepressant and anxiolytic effects. In contrast to the findings of pharmacovigilance studies, a 2024 meta-analysis incorporating prospective study data from around 2 million users of 5-ARIs did not find associations for depression (aHR: 1.30, 95% CI: 0.85–2.00) or suicide (aHR 1.30, 95% CI: 0.65–2.61) (Uleri et al., 2024). Subgroup analyses for finasteride and dutasteride yielded similar findings. It should be noted that despite the extremely large sample size, the confidence intervals in the pooled risk estimates were still wide and hence could not rule out marginal risk increases. Overall, these findings may be both concerning and reassuring at the same time. Whilst the involvement of 5-ARIs in mood and depression still remains unclear, the preponderance of evidence strongly suggests that any excess risk is likely to be small.
Post-Finasteride Syndrome
“Post-finasteride syndrome” (PFS) is an even more controversial and poorly understood condition characterised by persistent sexual, neurological, and psychological symptoms that arise during or after the use of finasteride or dutasteride (Cilio et al., 2025; Leliefeld, Debruyne, & Reisman, 2025). PFS has gained increasing attention since it was popularised in the literature in 2012 following a subset of 5-ARI users reporting enduring adverse effects after discontinuation (Irwig, 2012).
Various case reports and studies have been published reporting associations between 5-ARIs and PFS symptoms (Traish et al., 2011; Irwig & Kolukula, 2011; Irwig, 2012; Irwig, 2014; Caruso et al, 2015; Ali, Heran, & Etminan, 2015; Guo et al., 2016; Kiguradze et al., 2017; Pereira & Coelho, 2020). Notably, one apparently well-designed study reported altered levels of neuroactive steroids in cerebrospinal fluid and plasma after discontinuation of finasteride in men who reported suffering from PFS symptoms (Caruso et al, 2015). A retrospective analysis also found that rates of erectile dysfunction were higher in men with cumulatively greater exposure to finasteride and dutasteride (Kiguradze et al., 2017). However, overall these data have been of low-quality, at high risk of bias, may have been confounded by secondary variables, and universally suffer from the lack of a placebo control group to establish causation (Hirshburg et al., 2016; Trüeb et al., 2019; Trüeb et al., 2024). As such, by themselves they are of limited usefulness. More recently, a pharmacovigilance study identified the existence of a signal for “post-finasteride syndrome” with finasteride in the FAERS database (Zhong et al., 2025).
A substantial nocebo effect appears to exist in users of 5-ARIs pertaining to PFS-like symptoms (Maksym, Kajdy, & Rabijewski, 2019). A study found that men who were made aware of sexual adverse effects before being treated were much more likely to report them during follow-up (43.6%), compared to men who were not (14.3%) (Mondaini et al., 2007). These data show that the power of suggestion is likely to influence the experience of many individuals using 5-ARIs. A more comprehensive pharmacovigilance study of the FAERS database performed analyses stratified by time period and 5-ARI medication (Gupta et al., 2025b). The study conducted disproportionality analyses for five adverse events related to suicide between 2006 and 2011, 2013 and 2018, and 2019 and 2023. No signals were detected for oral finasteride between 2006 and 2011, but signals emerged in later periods, with increased reporting odds for suicidal ideation between 2013 and 2018 and between 2019 and 2023. Despite oral dutasteride being more efficacious in its action as a 5-ARI, dutasteride showed no significant signals across any time period. The authors concluded that these findings were suggestive of increased awareness of PFS being the cause of heightened reporting of psychiatric adverse events to FAERS, rather than reflecting a true pharmacological effect (Gupta et al., 2025b).
Taken together, all these findings provide limited evidence for the existence of PFS in a small subset of individuals. However, the evidence for persistent long-term adverse effects stemming from finasteride and dutasteride use is tenuous at best.
Minoxidil
Minoxidil is a medication with antihypertensive and vasodilator effects which was originally developed as an oral formulation for high blood pressure. However, it was found to have the unexpected side effect of promoting hair growth, which led to its reformulation as a topical solution and adoption for AGA. Oral minoxidil is also now increasingly being used at lower doses to treat hair loss.
Mechanism of Action of Minoxidil in Hair Loss
The exact means by which minoxidil is involved in promoting hair growth is not fully understood (Gupta et al., 2023; Iyengar & Li, 2025). It is believed that minoxidil functions by improving blood flow to hair follicles, which in turn increases circulation and which may help revitalise shrunken follicles, extend the growth phase of the hair cycle, and encourage thicker, longer hair strands (Zeltzer et al., 2024; Tan et al., 2025). A sulphotransferase enzyme converts minoxidil into its active metabolite, minoxidil sulfate. Differences in sulphotransferase enzyme expression between individuals appear to augment the efficacy of minoxidil (Goren & Naccarato, 2018). Clinical effects on hair growth typically begin after 2 to 4 months of consistent use, with maximal results seen around 6 to 12 months. Clinical response with minoxidil therapy appears to be highly variable. In randomised controlled trials, minoxidil monotherapy has been effective in increasing total hair density, as well as terminal hair density in both male and female AGA compared to controls (Gupta et al., 2022a; Gupta et al., 2024a; Gupta et al., 2024b; Gupta et al., 2025a). In these studies, the efficacy of minoxidil has also been shown to be strongly dose-dependent.
Minoxidil has been evaluated for its therapeutic effects on hair loss in some small studies of transfeminine and transmasculine people (Zaminski et al., 2025; Tang et al., 2025b). These studies have reported positive results, in line with data from the wider population.
Comparison of Different Minoxidil Routes of Administration
The most widely used formulations of minoxidil include oral minoxidil and topical minoxidil. The main difference is that oral minoxidil is absorbed extensively into systemic circulation, whereas topical minoxidil is designed to act locally at the site of application, at which it stimulates hair follicles directly, hence resulting in limited systemic exposure.
Oral minoxidil is metabolised rapidly into minoxidil sulfate in the gastrointestinal tract (Patel, Nessel & Kumar, 2023). Peak plasma concentrations are typically reached within 1 hour. Oral minoxidil has been used at a range of 0.25–7.5 mg/day for AGA in clinical trials. The oral route has an average bioavailability of nearly 100%, whereas the local absorption of topical minoxidil into the scalp is around 1.4%. However, there remains substantial interindividual variation for each. As such, clinical doses of topical minoxidil are much higher (typically a 2 or 5% concentration) in order to compensate. Food does not appear to influence the bioavailability of oral minoxidil (Gupta et al., 2023).
The efficacy of oral and topical minoxidil has been investigated extensively in clinical studies. Higher doses of oral minoxidil have been associated with more favourable outcomes for AGA in terms of hair diameter, total hair density, and terminal hair density, but also with increasing adverse effects (Gupta et al., 2022b). Generally, lower doses have been used in women as compared to men. Oral minoxidil has been investigated at doses of up to 7.5 mg/day in clinical trials in this indication (Sanabria et al., 2024a). Large meta-analyses have found that studies are mixed on whether oral or topical minoxidil, on average, results in better, worse, or equal efficacy (Gupta et al., 2022a; Gupta et al., 2024a; Gupta et al., 2024b; Fazal et al., 2025; Gupta et al., 2025a). However, the dose-dependent effects of oral minoxidil have similarly been found to occur with topical minoxidil (Singh et al., 2022). A possibility is that oral and topical minoxidil may not have always been used at clinically equivalent doses.
The inconsistent differences in efficacy shown between oral and topical minoxidil in clinical studies may be driven by interindividual variation in response due to sulphotransferase enzyme expression, particularly in the scalp (Patel, Nessel & Kumar, 2023). There is growing evidence that in some individuals oral minoxidil may be more efficacious than topical minoxidil and vice versa (Goren et al., 2015; Goren et al., 2016; Gupta et al., 2024b; Gupta et al., 2025a). These data suggest that a subset of individuals who may not respond to one route of administration could see benefit by changing to the other.
Sublingual minoxidil has also been investigated for treating AGA (Sinclair et al., 2020; Bokhari, Jones, & Sinclair, 2021; Sinclair et al., 2025). Another route of administration which is being considered is injectable minoxidil (Needle et al., 2025). However, these routes have received comparatively much less attention and so limited data are available to inform their usage. A randomised controlled trial comparing oral and sublingual minoxidil at a daily dosage of 5 mg found similar efficacy at 24 weeks follow-up, suggesting that sublingual minoxidil may be a useful alternative to oral minoxidil (Sanabria et al., 2024b). Whilst these initial data are promising, further and larger scale studies are likely to be needed before sublingual minoxidil could see the same level of adoption as oral and topical administration.
Safety and Tolerability of Oral and Topical Minoxidil
Minoxidil has generally been shown to be well tolerated in clinical trials. Nevertheless, usage is associated with various adverse effects in some individuals (Gupta et al., 2022b; Gupta et al., 2023; Iyengar & Li, 2025). The adverse effects of minoxidil have been shown to be dependent on the route of administration, as well as being positively dose-dependent.
A retrospective study of users of oral minoxidil investigated the frequency of adverse effects in both men and women receiving a median dose of 1.63 mg/day (Vañó-Galván et al., 2021). The following were found to occur: hypertrichosis (excessive facial/body hair) in 15.1%, lightheadedness in 1.7%, fluid retention in 1.3%, tachycardia in 0.9%, headache in 0.4%, periorbital edema (temporary swelling around the eyes) in 0.3%, and insomnia in 0.2%. The total frequency of adverse effects was 20.4%, which prompted discontinuation in 1.2% of users, overall. Another study reported an overall hypertrichosis incidence of 24%, with the highest rates being found in the sideburns (81%), temples (73%), arms (63%), and upper lip (51%) (Jimenez-Cauhe et al., 2021). By contrast, topical minoxidil is associated with much lower overall rates of hypertrichosis. Most studies have reported incidence rates of between 0 and 5% (Lucky et al., 2004; Blume-Peytavi et al., 2016; Ramos et al., 2020; Penha et al., 2024; Yang et al., 2024). These findings are consistent with a meta-analysis that reported point estimates of incidence rates for hypertrichosis of 10%, 15%, and 33% for oral minoxidil at 0.25 mg/day, 0.5 mg/day, and 1.25 mg/day, respectively, and 0% and 2% for topical minoxidil at a 2% and 5% concentration, respectively (Wiechert et al., 2025). Despite this, the discontinuation rate across all studies was 0.49%. There also seemed to be no statistically significant difference between the rate of discontinuation for oral and topical formulations, suggesting that hypertrichosis appears to be very well tolerated.
A concern associated with the use of oral minoxidil is its potential impact on cardiovascular health (Ibraheim et al., 2023). Since tachycardia can increase myocardial workload and lead to symptoms such as palpitations or chest discomfort, oral minoxidil should be approached cautiously, especially by individuals with underlying cardiovascular issues. Fortunately, the overall risk of severe cardiovascular complications from low-dose oral minoxidil seems to be very low in the general population (Randolph & Tosti, 2021; Vañó-Galván et al., 2021). Meanwhile, skin reactions appear to be relatively common in users of topical minoxidil. This often manifests as scalp eczema and itching, although rates of incidence vary by study (Lucky et al., 2004; Rossi et al., 2012; Penha et al., 2024). The culprit behind this irritating effect appears not to be minoxidil itself, but rather the ingredients in certain formulations such as propylene glycol (Suchonwanit, Thammarucha, & Leerunyakul, 2019). These solvents help deliver minoxidil into the scalp, but are known to cause skin irritation in susceptible individuals. It also appears that, for most people, long-term topical minoxidil therapy may be precluded by non-compliance (Ali Mapar & Omidian, 2007; Shadi, 2023).
The increase in overall body hair growth (i.e., hypertrichosis) is arguably the most consequential side effects for transfeminine people found to occur with minoxidil. As noted above, hypertrichosis is much more common with oral minoxidil than with topical minoxidil. This is a result of the differences in pharmacology between these routes and the extensive systemic absorption that occurs in the case of the former (Desai et al., 2024; Wiechert et al., 2025). In transmasculine people, an increase in body hair growth and diameter could be beneficial. However, these effects are usually not desired by transfeminine people. Consequently, some transfeminine people may prefer to use topical minoxidil over oral minoxidil, despite possible benefits to effectiveness from the latter in some individuals.
Steroidal and Non-Steroidal Antiandrogens
Antiandrogens such as spironolactone and cyproterone acetate are widely employed to reduce or suppress testosterone levels in transfeminine people. Some clinics have also used the non-steroidal antiandrogen bicalutamide. However, these medications have all also been investigated in the treatment of pattern hair loss in cisgender women. After gonadectomy, antiandrogen treatment is often discontinued. Nevertheless, it appears some transfeminine people continue antiandrogen treatment, particularly spironolactone, in order to suppress the effects of non-gonadal androgen production.
Spironolactone
Spironolactone has been studied at oral doses of 25 to 200 mg/day for the treatment of pattern hair loss in women (Wang et al., 2023; Rosenthal et al., 2024). Oral spironolactone has been found to be effective in halting and, in some cases, reversing female AGA (Sinclair, Wewerinke & Jolley, 2005; Burns et al., 2020). Often, it has been paired with other interventions such as minoxidil. A randomised controlled trial found that oral spironolactone at a dose of 80 to 100 mg/day had similar efficacy to minoxidil when used as a single agent therapy (Liang et al., 2022). Spironolactone has also been studied topically (Abdel‐Raouf et al., 2020; Ammar et al., 2022). In the highest quality studies, spironolactone has been found to act additively with minoxidil in improving hair density and hair diameter.
Overall, spironolactone appears to be well tolerated for treating AGA, as well as other androgen sensitive conditions in women (Barbieri et al., 2021; Wang et al., 2023; Martin et al., 2025). Likewise, it may also be useful for some transfeminine people as an adjunct therapy, especially when paired with minoxidil. However, spironolactone has been scarcely studied for male AGA. It could well be the case that other more established therapies, such as dutasteride, would be better for transfeminine people with more extensive hair loss.
Flutamide and Bicalutamide
Flutamide is a potent non-steroidal antiandrogen and antagonist of the androgen receptor which has predominantly been used to treat prostate cancer. Some clinicians have also employed the use of flutamide in treating female AGA, with positive findings (Carmina & Lobo, 2003; Yazdabadi & Sinclair, 2011; Paradisi et al., 2011; Faghihi et al., 2022). In a randomised controlled trial, 500 mg/day flutamide was found to be superior to 100 mg/day spironolactone in treating scalp hair loss (Cusan et al., 1994). However, this may have been at least partially down to the cyclic use of spironolactone, which meant that women randomised to spironolactone were not actually receiving it for the duration of the entire month. In spite of the above findings, flutamide is associated with a high risk of elevated liver enzymes, which can progress to life-threatening organ failure in a very small but clinically significant subset of cases (Ozono et al., 2000; Paradisi et al., 2011; Giorgetti et al., 2017). This appears to have precluded its widespread adoption for female AGA.
Bicalutamide is another non-steroidal antiandrogen which has been considered for female AGA (Perez, Nguyen, & Senna, 2025). Generally, bicalutamide is believed to have a lower risk of liver toxicity than flutamide, making it a much more promising candidate for large-scale adoption in otherwise healthy people (Kolvenbag & Blackledge, 1996; Devjani et al., 2023). A number of retrospective studies have reported encouraging data with 10 to 50 mg/day oral bicalutamide, both in terms of safety and efficacy (Ismali et al., 2020; Fernandez-Nieto et al., 2020; Yoong et al., 2025). Interestingly, a study also found that bicalutamide was associated with improvements in hypertrichosis induced by oral minoxidil (Moussa et al., 2022). Only one randomised controlled trial has been conducted with bicalutamide, in which minoxidil plus 25 mg/day oral bicalutamide was compared to minoxidil plus placebo (da Silva Libório et al., 2025). In this study, there was no additional benefit of bicalutamide on total hair density. This finding is surprising in light of the seemingly additive effects that occur with minoxidil and spironolactone. It is possible that differences in methodology could be responsible for this discrepancy. Another study, although also retrospective, found that 50 mg/day oral bicalutamide was associated with moderately greater improvement in scalp hair loss than was 100 mg/day oral spironolactone (Jha et al., 2024).
Concerns about safety have also historically precluded widespread adoption of bicalutamide in transgender medicine. However, increasing numbers of studies are adding to our knowledge of bicalutamide in transfeminine people (Fuqua, Shi, & Eugster, 2024; Angus et al., 2024).
Other Medications and Future Developments
Despite several decades of research, only two medications have been approved for male AGA by the FDA in the United States. These are minoxidil and finasteride. Even though the use of these agents is increasingly common in men, they remain only partially effective in reversing androgenic hair loss.
Topical Androgen Receptor Antagonists
Two novel topical androgen receptor antagonists of possible interest are clascoterone and pyrilutamide (Saceda-Corralo et al., 2023; Devjani et al., 2023). Unlike oral non-steroidal antiandrogens such as flutamide and bicalutamide, these medications are applied topically so that there is minimal systemic absorption. Clascoterone has shown some limited success in phase 3 clinical trials for treating acne in men and women and has been approved by the FDA for this indication (Hebert et al., 2020). It is hoped that clascoterone could also be effective in treating AGA. An exploratory study found that clascosterone was superior to topical cyproterone acetate and alfatradiol in improving hair shaft diameter and hair follicle density and had comparable efficacy to minoxidil (Cartwright et al., 2019). Phase 3 clinical trials of clascosterone in AGA are currently underway. Pyrilutamide had shown favourable results in phase 2 clinical trials for both male and female AGA, however it failed to outperform placebo in phase 3 clinical trials (Kintor Pharmaceuticals, 2024). Studies are now underway using a higher dose of pyrilutamide over a longer duration of follow-up in the hope that this will show improved results.
Despite the above, these findings are rather disappointing. Notably, clascoterone only marginally outperformed placebo in clinical trials for acne (reduction in symptoms by about 8 to 18% more than placebo) (Hebert et al., 2020). A systematic review and network meta-analysis found that oral spironolactone was substantially more effective for treating acne than topical clascoterone (Basendwh et al., 2024). Clascoterone and pyrilutamide may someday provide another option for treating AGA in transfeminine people. However, since their mechanism of action is not dissimilar to well-established therapies such as 5-ARIs, it seems that this class of medications is unlikely to ever be revolutionary.
Prostaglandin Analogues
Prostaglandin analogues could have some utility in treating AGA. These include latanoprost and bimatoprost. Prostaglandin analogues have mostly been used to treat glaucoma by lowering intraocular pressure but are also believed to prolong the anagen phase and hence cause hair growth in certain susceptible tissues (Valente Duarte de Sousa & Tosti, 2013).
Latanoprost has mostly been studied for alopecia areata within the context of hair loss. However, one small randomised controlled trial found that topical latanoprost outperformed placebo after 24 weeks in increasing hair density in young men with mild AGA (Blume-Peytavi et al., 2012). Bimatoprost has also been evaluated in various clinical trials. In four separate phase 2 clinical trials, bimatoprost mildly to moderately outperformed placebo (Anastassakis, 2022). However, compared to minoxidil, findings were inconsistent. Bimatoprost had similar efficacy in some measures in some studies, but inferior efficacy in others. Overall, prostaglandin analogues appear to have received little subsequent attention. These data are relatively underwhelming by themselves compared to the large amount of literature pertaining to more established AGA therapies.
Mitochondrial Pyruvate Carrier Inhibition
In perhaps one of the more interesting developments in recent years, researchers have identified a topically delivered molecule, called PP-405, that appears to be capable of reactivating dormant follicles by modulating mitochondrial pyruvate carrier (MPC) activity (Brown, 2025). Unlike current therapies that focus on hormone suppression and increased blood flow, PP-405 is a regenerative, stem-cell-focused approach to treating hair loss. The molecule is currently being investigated for male and female AGA.
Recently, a phase 2a clinical trial that randomised 78 men and women to either PP-405 or placebo concluded with positive safety findings (Meara, 2025). However, preliminary results from 4 weeks of treatment at 8 weeks follow-up also showed a rapid and statistically significant clinical response versus placebo. This was despite the study not actually being conducted to show efficacy. The researchers found that 31% of the men with advanced AGA who were treated with the active medication showed a 20% or greater increase in total hair density, compared to 0% of patients in the placebo group. This is particularly notable because current interventions for AGA, such as minoxidil, typically take at least several months of follow-up to show a statistically significant difference from placebo. Most strikingly, PP-405 apparently induced new terminal hair growth from follicles where no hair was previously present.
As of September 2025, PP-405 is in phase 2b clinical trials and is expected to enter phase 3 clinical trials early next year if subsequent clinical findings are promising. With all this said, it should be noted that these are merely early results. More rigorous studies are necessary to determine if PP-405 can be an effective intervention against AGA.
Summary and Conclusions
AGA is a common and distressing condition that has particular relevance for transfeminine people, given the role of hair in gender identity and expression. While feminising hormone therapy appears to at least partially reverse AGA, many individuals appear to experience incomplete regrowth. Limited data suggest that adjunct use of 5-ARIs and/or minoxidil, particularly in the first few years of hormonal transition, may have positive effects but more studies are necessary to confirm this.
The most established treatments for AGA in the wider population are 5-ARIs, including finasteride and dutasteride, and minoxidil. The role of 5-ARIs in women remain less clear. Nevertheless, dutasteride achieves superior outcomes in male AGA to finasteride, whilst having similar safety and tolerability. Hence, wherever possible, it seems reasonable to use the former should the use of a 5α-reductase inhibitor be desired. Minoxidil, whether oral or topical, provides dose-dependent improvements in total and terminal hair density in male and female AGA and acts synergistically with 5-ARIs. However, oral minoxidil is associated with higher rates of hypertrichosis, which may be undesirable for many transfeminine individuals.
Other agents, such as spironolactone and bicalutamide, could also offer additional benefit by antagonising the androgen receptor. Spironolactone is already widely used in transfeminine hormone therapy and shows synergy with minoxidil in studies of female AGA. Bicalutamide is of emerging interest given its relatively favourable safety profile. Novel therapies of benefit to transfeminine people may also become available in the future.
References
Abdel‐Raouf, H., Aly, U. F., Medhat, W., Ahmed, S. S., & Abdel‐Aziz, R. T. (2021). A novel topical combination of minoxidil and spironolactone for androgenetic alopecia: clinical, histopathological, and physicochemical study. Dermatologic Therapy, 34(1), e14678. [DOI:10.1111/dth.14678]
Adenuga, P., Summers, P., & Bergfeld, W. (2012). Hair regrowth in a male patient with extensive androgenetic alopecia on estrogen therapy. Journal of the American Academy of Dermatology, 67(3), e121–e123. [DOI:10.1016/j.jaad.2011.10.017]
Almudimeegh, A., Al-Mutairi, H., Al-Tassan, F., Al-Quraishi, Y., & Nagshabandi, K. N. (2024). Comparison between dutasteride and finasteride in hair regrowth and reversal of miniaturization in male and female androgenetic alopecia: a systematic review. Dermatology Reports, 16(4), 9909. [DOI:10.4081/dr.2024.9909]
Ali, A. K., Heran, B. S., & Etminan, M. (2015). Persistent sexual dysfunction and suicidal ideation in young men treated with low‐dose finasteride: a pharmacovigilance study. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 35(7), 687–695. [DOI:10.1002/phar.1612]
Altendorf, S., Bertolini, M., Le Riche, A., Tosti, A., & Paus, R. (2025). Frontiers in the physiology of male pattern androgenetic alopecia: Beyond the androgen horizon. Physiological Reviews, online ahead of print. [DOI:10.1152/physrev.00005.2024]
Ammar, A. M., Elshahid, A. R., Abdel‐Dayem, H. A., Mohamed, A. A., & Elsaie, M. L. (2022). Dermoscopic evaluation of the efficacy of combination of topical spironolactone 5% and minoxidil 5% solutions in the treatment of androgenetic alopecia: a cross sectional‐comparative study. Journal of Cosmetic Dermatology, 21(11), 5790–5799. [DOI:10.1111/jocd.15328]
Amory, J. K., Wang, C., Swerdloff, R. S., Anawalt, B. D., Matsumoto, A. M., Bremner, W. J., Walker, S. E., Haberer, L. J., & Clark, R. V. (2007). The effect of 5α-reductase inhibition with dutasteride and finasteride on semen parameters and serum hormones in healthy men. The Journal of Clinical Endocrinology & Metabolism, 92(5), 1659–1665. [DOI:10.1210/jc.2006-2203]
Anand/HairLossCure2020. (2024 June 18). Booming Demand: U.S. Sees 200% Surge in Finasteride Prescriptions Over 7 Years. American Hair Loss Association (AHLA). [URL]
Anastassakis, K. (2022). Hormonal and genetic etiology of male androgenetic alopecia. In Anastassakis, K. (Ed.). Androgenetic Alopecia From A to Z: Vol. 1 Basic Science, Diagnosis, Etiology, and Related Disorders (pp. 135–180). Cham: Springer International Publishing. [DOI:10.1007/978-3-030-76111-0_11]
Angus, L. M., Hong, Q. V., Cheung, A. S., & Nolan, B. J. (2024). Effect of bicalutamide on serum total testosterone concentration in transgender adults: a case series. Therapeutic Advances in Endocrinology and Metabolism, 15, 20420188241305022. [DOI:10.1177/20420188241305022]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2021). A systematic review of antiandrogens and feminization in transgender women. Clinical Endocrinology, 94(5), 743–752. [DOI:10.1111/cen.14329]
Asad, N., Naseer, M., & Ghafoor, R. (2024). Efficacy of Topical Finasteride 0.25% With Minoxidil 5% Versus Topical Minoxidil 5% Alone in Treatment of Male Pattern Androgenic Alopecia, Journal of Drugs in Dermatology, 23(11), 1003–1008. [DOI:10.36849/JDD.7826]
Barbieri, J. S., Margolis, D. J., & Mostaghimi, A. (2021). Temporal trends and clinician variability in potassium monitoring of healthy young women treated for acne with spironolactone. JAMA Dermatology, 157(3), 296–300. [DOI:10.1001/jamadermatol.2020.5468]
Basendwh, M. A., Alharbi, A. A., Bukhamsin, S. A., Abdulwahab, R. A., & Alaboud, S. A. (2024). The efficacy of Topical Clascoterone versus systematic spironolactone for treatment of acne vulgaris: A systematic review and network meta-analysis. Plos One, 19(5), e0298155. [DOI:10.1371/journal.pone.0298155]
Blume-Peytavi, U., Lönnfors, S., Hillmann, K., & Bartels, N. G. (2012). A randomized double-blind placebo-controlled pilot study to assess the efficacy of a 24-week topical treatment by latanoprost 0.1% on hair growth and pigmentation in healthy volunteers with androgenetic alopecia. Journal of the American Academy of Dermatology, 66(5), 794–800. [DOI:10.1016/j.jaad.2011.05.026]
Blume-Peytavi, U., Shapiro, J., Messenger, A. G., Hordinsky, M. K., Zhang, P., Quiza, C., Doshi, U., & Olsen, E. A. (2016). Efficacy and Safety of Once-Daily Minoxidil Foam 5% Versus Twice-Daily Minoxidil Solution 2% in Female Pattern Hair Loss: A Phase III, Randomized, Investigator-Blinded Study. Journal of Drugs in Dermatology, 15(7), 883–889. [Google Scholar] [PubMed] [URL]
Bokhari, L., Jones, L. N., & Sinclair, R. D. (2022). Sublingual minoxidil for the treatment of male and female pattern hair loss: a randomized, double‐blind, placebo‐controlled, phase 1B clinical trial. Journal of the European Academy of Dermatology & Venereology, 36(1), e62–e66. [DOI:10.1111/jdv.17623]
Brown, L. (2025 August 12). The Great Unbalding: Could A New Drug Actually Regrow Hair? Intelligencer (New York Magazine). [URL]
Burns, L. J., De Souza, B., Flynn, E., Hagigeorges, D., & Senna, M. M. (2020). Spironolactone for treatment of female pattern hair loss. Journal of the American Academy of Dermatology, 83(1), 276–278. [DOI:10.1016/j.jaad.2020.03.087]
Carmina, E., & Lobo, R. A. (2003). Treatment of hyperandrogenic alopecia in women. Fertility and Sterility, 79(1), 91–95. [DOI:10.1016/S0015-0282(02)04551-X]
Cartwright, M., Mazzetti, A., Moro, L., Rosette, C., Gerloni, M. (2019). 10063. A summary of in vitro, phase I, and phase II studies evaluating the mechanism of action, safety, and efficacy of clascoterone (cortexolone 17α propionate, CB-03-01) in androgenetic alopecia. Journal of the American Academy of Dermatology, 81(4 Suppl 1), AB13. [DOI:10.1016/j.jaad.2019.06.087]
Caruso, D., Abbiati, F., Giatti, S., Romano, S., Fusco, L., Cavaletti, G., & Melcangi, R. C. (2015). Patients treated for male pattern hair with finasteride show, after discontinuation of the drug, altered levels of neuroactive steroids in cerebrospinal fluid and plasma. The Journal of Steroid Biochemistry and Molecular Biology, 146, 74–79. [DOI:10.1016/j.jsbmb.2014.03.012]
Chen, Y., Hysi, P., Maj, C., Heilmann-Heimbach, S., Spector, T. D., Liu, F., & Kayser, M. (2023). Genetic prediction of male pattern baldness based on large independent datasets. European Journal of Human Genetics, 31(3), 321–328. [DOI:10.1038/s41431-022-01201-y]
Cho, Y., Bea, S., Bae, J. H., Kim, D. H., Lee, J. H., & Shin, J. Y. (2024). Cognitive dysfunction following finasteride use: a disproportionality analysis of the global pharmacovigilance database. Expert Opinion on Drug Safety, 23(8), 1027–1033. [DOI:10.1080/14740338.2023.2294926]
Choi, G. S., Sim, W. Y., Kang, H., Huh, C. H., Lee, Y. W., Shantakumar, S., Ho, Y. F., Oh, E. J., Duh, M. S., Cheng, W. Y., Bobbili, P., Thompson-Leduc, P., & Ong, G. (2022). Long-term effectiveness and safety of dutasteride versus finasteride in patients with male androgenic alopecia in South Korea: a multicentre chart review study. Annals of Dermatology, 34(5), 349–359. [DOI:10.5021/ad.22.027]
Cilio, S., Tsampoukas, G., Morgado, A., Ramos, P., & Minhas, S. (2025). Post-finasteride syndrome - a true clinical entity? International Journal of Impotence Research, 37, 426–435. [DOI:10.1038/s41443-025-01025-6]
Clark, R. V., Hermann, D. J., Cunningham, G. R., Wilson, T. H., Morrill, B. B., & Hobbs, S. (2004). Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5α-reductase inhibitor. The Journal of Clinical Endocrinology & Metabolism, 89(5), 2179–2184. [DOI:10.1210/jc.2003-030330]
Cocchetti, C., Castellini, G., Maggi, M., Romani, A., Vignozzi, L., Greenman, Y., den Heijer, M., T’Sjoen, G., & Fisher, A. D. (2023). Effects of hormonal treatment on dermatological outcome in transgender people: a multicentric prospective study (ENIGI). Journal of Endocrinological Investigation, 46(4), 779–786. [DOI:10.1007/s40618-022-01944-x]
Coelingh Bennink, H. J., Prowse, A., Egberts, J. F., Debruyne, F. M., Huhtaniemi, I. T., & Tombal, B. (2024). The loss of estradiol by androgen deprivation in prostate cancer patients shows the importance of estrogens in males. Journal of the Endocrine Society, 8(7), bvae107. [DOI:10.1210/jendso/bvae107]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Corona, G., Tirabassi, G., Santi, D., Maseroli, E., Gacci, M., Dicuio, M., Sforza, A., Mannucci, E., & Maggi, M. (2017). Sexual dysfunction in subjects treated with inhibitors of 5α‐reductase for benign prostatic hyperplasia: a comprehensive review and meta‐analysis. Andrology, 5(4), 671–678. [DOI:10.1111/andr.12353]
Cousen, P., & Messenger, A. (2010). Female pattern hair loss in complete androgen insensitivity syndrome. British Journal of Dermatology, 162(5), 1135–1137. [DOI:10.1111/j.1365-2133.2010.09661.x]
Cusan, L., Dupont, A., Gomez, J. L., Tremblay, R. R., & Labrie, F. (1994). Comparison of flutamide and spironolactone in the treatment of hirsutism: a randomized controlled trial. Fertility and Sterility, 61(2), 281–287. [DOI:10.1016/S0015-0282(16)56518-2]
da Silva Libório, R., da Motta, A. V., Miot, H. A., & Ramos, P. M. (2025). Bicalutamide 25 mg combined with minoxidil 1 mg versus minoxidil 1 mg for female pattern hair loss: A randomized double-blind clinical trial. JAAD International, 19, 48–55. [DOI:10.1016/j.jdin.2024.12.002]
Desai, D. D., Nohria, A., Brinks, A., Needle, C., Shapiro, J., & Sicco, K. I. L. (2024). Minoxidil-induced hypertrichosis: Pathophysiology, clinical implications, and therapeutic strategies. JAAD Reviews, 2, 41–49. [DOI:10.1016/j.jdrv.2024.08.002]
Devjani, S., Ezemma, O., Kelley, K. J., Stratton, E., & Senna, M. (2023). Androgenetic alopecia: therapy update. Drugs, 83(8), 701–715. [DOI:10.1007/s40265-023-01880-x]
Dewhurst, J., & Underhill, R. (1979). The Suppression of Facial Hair Growth in Transsexuals Using Cyproterone Acetate. British Journal of Sexual Medicine, 6, 19–23. [Google Scholar] [PDF]
Dhurat, R. S., & Daruwalla, S. B. (2021). Androgenetic alopecia: Update on etiology. Dermatological Reviews, 2(3), 115–121. [DOI:10.1002/der2.58]
Estill, M. C., Ford, A., Omeira, R., & Rodman, M. (2023). Finasteride and dutasteride for the treatment of male androgenetic alopecia: a review of efficacy and reproductive adverse effects. Georgetown Medical Review, 7(1). [DOI:10.52504/001c.88531]
Faghihi, G., Iraji, F., Siadat, A. H., Saber, M., Jelvan, M., & Hoseyni, M. S. (2022). Comparison between “5% minoxidil plus 2% flutamide” solution vs.“5% minoxidil” solution in the treatment of androgenetic alopecia. Journal of Cosmetic Dermatology, 21(10), 4447–4453. [DOI:10.1111/jocd.14788]
Fazal, F., Malik, B. H., Mumtaz Malik, H., Sabir, B., Mustafa, H., Ahmed, M., Abid, A., Leeza Adil, M., Shafi, U., & Saad, M. (2025). Can oral minoxidil be the game changer in androgenetic alopecia? A comprehensive review and meta-analysis comparing topical and oral minoxidil for treating androgenetic alopecia. Skin Health and Disease, 5(2), 95–101. [DOI:10.1093/skinhd/vzaf009]
Fernandez-Nieto, D., Saceda-Corralo, D., Jimenez-Cauhe, J., Moreno-Arrones, O. M., Rodrigues-Barata, R., Hermosa-Gelbard, A., & Vano-Galvan, S. (2020). Bicalutamide: a potential new oral antiandrogenic drug for female pattern hair loss. Journal of the American Academy of Dermatology, 83(5), e355–e356. [DOI:10.1016/j.jaad.2020.04.054]
Fujimaki, H., Yanagisawa, M., Yoshitake, T., & Sato, A. (2024). Age-related progression of androgenetic alopecia: Statistical analysis of 5372 Japanese men. JAAD International, 17, 84–85. [DOI:10.1016/j.jdin.2024.07.003]
Fuqua, J. S., Shi, E., & Eugster, E. A. (2024). A retrospective review of the use of bicalutamide in transfeminine youth; a single center experience. International Journal of Transgender Health, 25(3), 533–537. [DOI:10.1080/26895269.2023.2294321]
Gao, J. L., Sanz, J., Tan, N., King, D. S., Modest, A. M., & Dommasch, E. D. (2023). Androgenetic alopecia incidence in transgender and gender diverse populations: a retrospective comparative cohort study. Journal of the American Academy of Dermatology, 89(3), 504–510. [DOI:10.1016/j.jaad.2023.01.037]
Gao, J. L., Streed Jr, C. G., Thompson, J., Dommasch, E. D., & Peebles, J. K. (2023). Androgenetic alopecia in transgender and gender diverse populations: a review of therapeutics. Journal of the American Academy of Dermatology, 89(4), 774–783. [DOI:10.1016/j.jaad.2021.08.067]
Giorgetti, R., Di Muzio, M., Giorgetti, A., Girolami, D., Borgia, L., & Tagliabracci, A. (2017). Flutamide-induced hepatotoxicity: ethical and scientific issues. European Review for Medical and Pharmacological Sciences, 21(1 Suppl), 69–77. [Google Scholar] [PubMed]
Gisleskog, P. O., Hermann, D., Hammarlund‐Udenaes, M., & Karlsson, M. O. (1998). A model for the turnover of dihydrotestosterone in the presence of the irreversible 5α‐reductase inhibitors GI198745 and finasteride. Clinical Pharmacology & Therapeutics, 64(6), 636–647. [DOI:10.1016/S0009-9236(98)90054-6]
Goren, A., Shapiro, J., Roberts, J., McCoy, J., Desai, N., Zarrab, Z., Pietrzak, A., & Lotti, T. (2015). Clinical utility and validity of minoxidil response testing in androgenetic alopecia. Dermatologic Therapy, 28(1), 13–16. [DOI:10.1111/dth.12164]
Goren, A., Kovacevic, M., McCoy, J., & Shapiro, J. (2017). 841 Minoxidil dose response study in female pattern hair loss patients determined to be non-responders to 5% topical Minoxidil. Journal of Investigative Dermatology, 137(5 Suppl 1), S144–S144. [DOI:10.1016/j.jid.2017.02.867]
Goren, A., & Naccarato, T. (2018). Minoxidil in the treatment of androgenetic alopecia. Dermatologic Therapy, 31(5), e12686. [DOI:10.1111/dth.12686]
Guo, M., Heran, B., Flannigan, R., Kezouh, A., & Etminan, M. (2016). Persistent sexual dysfunction with finasteride 1 mg taken for hair loss. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 36(11), 1180–1184. [DOI:10.1002/phar.1837]
Gupta, A. K., Venkataraman, M., Talukder, M., & Bamimore, M. A. (2022). Relative efficacy of minoxidil and the 5-α reductase inhibitors in androgenetic alopecia treatment of male patients: a network meta-analysis. JAMA Dermatology, 158(3), 266–274. [DOI:10.1001/jamadermatol.2021.5743]
Gupta, A. K., Hall, D. C., Talukder, M., & Bamimore, M. A. (2022). There is a positive dose-dependent association between low-dose oral minoxidil and its efficacy for androgenetic alopecia: findings from a systematic review with meta-regression analyses. Skin Appendage Disorders, 8(5), 355–361. [DOI:10.1159/000525137]
Gupta, A. K., Talukder, M., Shemar, A., Piraccini, B. M., & Tosti, A. (2023). Low-dose oral minoxidil for alopecia: a comprehensive review. Skin Appendage Disorders, 9(6), 423–437. [DOI:10.1159/000531890]
Gupta, A. K., Bamimore, M. A., Wang, T., & Talukder, M. (2024). The impact of monotherapies for male androgenetic alopecia: A network meta‐analysis study. Journal of Cosmetic Dermatology, 23(9), 2964–2972. [DOI:10.1111/jocd.16362]
Gupta, A. K., Wang, T., Bamimore, M. A., & Talukder, M. (2024). The relative effect of monotherapy with 5‐alpha reductase inhibitors and minoxidil for female pattern hair loss: A network meta‐analysis study. Journal of Cosmetic Dermatology, 23(1), 154–160. [DOI:10.1111/jocd.15910]
Gupta, A. K., Bamimore, M. A., Williams, G., & Talukder, M. (2025). Comparative Efficacy of Minoxidil and 5‐Alpha Reductase Inhibitors Monotherapy for Male Pattern Hair Loss: Network Meta‐Analysis Study of Current Empirical Evidence. Journal of Cosmetic Dermatology, 24(7), e70320. [DOI:10.1111/jocd.70320]
Gupta, A. K., Bamimore, M. A., Williams, G., & Talukder, M. (2025). Finasteride Use: Evaluation of Depression and Suicide Risk. Journal of Cosmetic Dermatology, 24(3), e70102. [DOI:10.1111/jocd.70102]
Gupta, M. A., Vujcic, B., & Gupta, A. K. (2020). Finasteride Use Is Associated with Higher Odds of Obstructive Sleep Apnea: Results from the US Food and Drug Administration Adverse Events Reporting System. Skinmed, 18(3), 146–150. [Google Scholar] [PubMed]
Hebert, A., Thiboutot, D., Gold, L. S., Cartwright, M., Gerloni, M., Fragasso, E., & Mazzetti, A. (2020). Efficacy and safety of topical clascoterone cream, 1%, for treatment in patients with facial acne: two phase 3 randomized clinical trials. JAMA Dermatology, 156(6), 621–630. [DOI:10.1001/jamadermatol.2020.0465]
Hirshburg, J. M., Kelsey, P. A., Therrien, C. A., Gavino, A. C., & Reichenberg, J. S. (2016). Adverse effects and safety of 5-alpha reductase inhibitors (finasteride, dutasteride): a systematic review. The Journal of Clinical and Aesthetic Dermatology, 9(7), 56–62. [Google Scholar] [PubMed] [PubMed Central] [PDF]
Hobo, Y., Nishikawa, J., Taniguchi Asai, N., Yoneyama, K., Watanabe, Y., Miyashiro, Y., & Fujikata, A. (2023). Evaluation of the therapeutic effects of AGA drugs by measuring finasteride, dutasteride, and dihydrotestosterone in hair. Clinica Chimica Acta, 547, 117456. [DOI:10.1016/j.cca.2023.117456]
Hu, R., Xu, F., Sheng, Y., Qi, S., Han, Y., Miao, Y., Rui, W., & Yang, Q. (2015). Combined treatment with oral finasteride and topical minoxidil in male androgenetic alopecia: a randomized and comparative study in Chinese patients. Dermatologic Therapy, 28(5), 303–308. [DOI:10.1111/dth.12246]
Ibraheim, M. K., Elsensohn, A., Hauschild, C., Hilliard, A., & Dao, H. (2023). Low dose oral minoxidil and the conundrum of cardiovascular complications. Dermatology Online Journal, 29(4). [DOI:10.5070/D329461861]
Imperato-McGinley, J., & Zhu, Y. S. (2002). Androgens and male physiology the syndrome of 5α-reductase-2 deficiency. Molecular and Cellular Endocrinology, 198(1–2), 51–59. [DOI:10.1016/S0303-7207(02)00368-4]
Irwig, M. S., & Kolukula, S. (2011). Persistent sexual side effects of finasteride for male pattern hair loss. The Journal of Sexual Medicine, 8(6), 1747–1753. [DOI:10.1111/j.1743-6109.2011.02255.x]
Irwig, M. S. (2012). Persistent sexual side effects of finasteride: could they be permanent? The Journal of Sexual Medicine, 9(11), 2927–2932. [DOI:10.1111/j.1743-6109.2012.02846.x]
Irwig, M. S. (2014). Persistent sexual and nonsexual adverse effects of finasteride in younger men. Sexual Medicine Reviews, 2(1), 24–35. [DOI:10.1002/smrj.19]
Irwig, M. S. (2021). Is there a role for 5α‐reductase inhibitors in transgender individuals? Andrology, 9(6), 1729–1731. [DOI:10.1111/andr.12881]
Ismail, F. F., Meah, N., de Carvalho, L. T., Bhoyrul, B., Wall, D., & Sinclair, R. (2020). Safety of oral bicalutamide in female pattern hair loss: a retrospective review of 316 patients. Journal of the American Academy of Dermatology, 83(5), 1478–1479. [DOI:10.1016/j.jaad.2020.03.034]
Iyengar, L., & Li, J. (2025). Male and female pattern hair loss. Australian Prescriber, 48(3), 93–97. [DOI:10.18773/austprescr.2025.020]
Izard, J. P., & Siemens, D. R. (2020). Androgen deprivation therapy and mental health: impact on depression and cognition. European Urology Focus, 6(6), 1162–1164. [DOI:10.1016/j.euf.2019.11.010]
Janivara, R., Hazra, U., Pfennig, A., Harlemon, M., Kim, M. S., Eaaswarkhanth, M., Chen, W. C., Ogunbiyi, A., Kachambwa, P., Petersen, L. N., Jalloh, M., Mensah, J. E., Adjei, A. A., Adusei, B., Joffe, M., Gueye, S. M., Aisuodionoe-Shadrach, O. I., Fernandez, P. W., Rohan, T. E., Andrews, C., Rebbeck, T. R., Adebiyi, A. O., Agalliu, I., & Lachance, J. (2025). Uncovering the genetic architecture and evolutionary roots of androgenetic alopecia in African men. Human Genetics and Genomics Advances, 6(3), 100428. [DOI:10.1016/j.xhgg.2025.100428]
Jha, A. K., Zeeshan, M. D., Singh, A., & Singh, A. K. (2024). Efficacy and safety of spironolactone versus bicalutamide in female pattern hair loss: A retrospective comparative study. Australasian Journal of Dermatology, 65(5), 437–443. [DOI:10.1111/ajd.14306]
Jimenez-Cauhe, J., Saceda-Corralo, D., Rodrigues-Barata, R., Hermosa-Gelbard, A., Moreno-Arrones, O. M., Gil-Redondo, R., Ortega-Quijano, D., Fernandez-Nieto, D., Jaen-Olasolo, P., & Vaño-Galvan, S. (2021). Characterization and management of hypertrichosis induced by low-dose oral minoxidil in the treatment of hair loss. Journal of the American Academy of Dermatology, 84(1), 222–223. [DOI:10.1016/j.jaad.2020.08.124]
Keam, S. J., & Scott, L. J. (2008). Dutasteride: a review of its use in the management of prostate disorders. Drugs, 68(4), 463–485. [DOI:10.2165/00003495-200868040-00008]
Khaled, H. N., Allah, A. M. A., Abdelhameed, A. A., & Shehata, W. A. (2020). Role of serum androgens and prostate-specific antigen levels in men with androgenetic alopecia. Egyptian Journal of Dermatology and Venerology, 40(2), 106–111. [DOI:10.4103/ejdv.ejdv_46_19]
Kiguradze, T., Temps, W. H., Yarnold, P. R., Cashy, J., Brannigan, R. E., Nardone, B., Micali, G., West, D. P., & Belknap, S. M. (2017). Persistent erectile dysfunction in men exposed to the 5α-reductase inhibitors, finasteride, or dutasteride. PeerJ, 5, e3020. [DOI:10.7717/peerj.3020]
Kintor Pharmaceuticals. (2024). Annual Results Announcement for the Year Ended 31 December 2023. Kintor Pharmaceuticals. [PDF]
Kolvenbag, G. J., & Blackledge, G. R. (1996). Worldwide activity and safety of bicalutamide: a summary review. Urology, 47(1), 70–79. [DOI:10.1016/S0090-4295(96)80012-4]
Lee, S., Jo, H., Fond, G., Boyer, L., Smith, L., Hajek, A., & Yon, D. K. (2025). Signal detection between 5-alpha reductase inhibitors and the risk of suicidality and depression: an international pharmacovigilance analysis. European Journal of Clinical Pharmacology, 81(8), 1177–1185. [DOI:10.1007/s00228-025-03851-5]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal treatment of transgender women with oral estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Leliefeld, H. H., Debruyne, F. M., & Reisman, Y. (2025). The post-finasteride syndrome: possible etiological mechanisms and symptoms. International Journal of Impotence Research, 37(6), 414–421. [DOI:10.1038/s41443-023-00759-5]
Liang, X., Chang, Y., Wu, H., Liu, Y., Zhao, J., Wang, L., & Zhuo, F. (2022). Efficacy and safety of 5% minoxidil alone, minoxidil plus oral spironolactone, and minoxidil plus microneedling on female pattern hair loss: a prospective, single-center, parallel-group, evaluator blinded, randomized trial. Frontiers in Medicine, 9, 905140. [DOI:10.3389/fmed.2022.905140]
Lucky, A. W., Piacquadio, D. J., Ditre, C. M., Dunlap, F., Kantor, I., Pandya, A. G., Savin, R. C., & Tharp, M. D. (2004). A randomized, placebo-controlled trial of 5% and 2% topical minoxidil solutions in the treatment of female pattern hair loss. Journal of the American Academy of Dermatology, 50(4), 541–553. [DOI:10.1016/j.jaad.2003.06.014]
Maeda, T., Kikuchi, E., Hasegawa, M., Homma, K., Ando, T., Suzuki, K., Kaneko, G., Mizuno, R., Miyajima, A., & Oya, M. (2018). Influence of dutasteride treatment on serum hormone levels and aging male symptoms in patients with benign prostatic enlargement. International Journal of Urology, 25(1), 70–74. [DOI:10.1111/iju.13470]
Maksym, R. B., Kajdy, A., & Rabijewski, M. (2019). Post-finasteride syndrome–does it really exist? The Aging Male, 22(4), 250–259. [DOI:10.1080/13685538.2018.1548589]
Mapar, M. A., & Omidian, M. (2007). Is topical minoxidil solution effective on androgenetic alopecia in routine daily practice? Journal of Dermatological Treatment, 18(5), 268–270. [DOI:10.1080/09546630701383727]
Marks, D. H., & Senna, M. M. (2020). Androgenetic alopecia in gender minority patients. Dermatologic Clinics, 38(2), 239–247 [DOI:10.1016/j.det.2019.10.010]
Martin, A., Chen, L. C., Hsieh, T. Y. J., & Senna, M. M. (2025). Despite progesterone receptor affinity, spironolactone is not associated with meningioma development: A population-based cohort study. Journal of the American Academy of Dermatology, 92(6), 1456–1457. [DOI:10.1016/j.jaad.2025.02.056]
Meara, K. (2025 June 23). Pelage’s PP405 Demonstrates Efficacy in Phase 2a Trial for Androgenetic Alopecia. Dermatology Times. [URL]
Mondaini, N., Gontero, P., Giubilei, G., Lombardi, G., Cai, T., Gavazzi, A., & Bartoletti, R. (2007). Finasteride 5 mg and sexual side effects: how many of these are related to a nocebo phenomenon? The Journal of Sexual Medicine, 4(6), 1708–1712. [DOI:10.1111/j.1743-6109.2007.00563.x]
Moussa, A., Kazmi, A., Bokhari, L., & Sinclair, R. D. (2022). Bicalutamide improves minoxidil-induced hypertrichosis in female pattern hair loss: A retrospective review of 35 patients. Journal of the American Academy of Dermatology, 87(2), 488–490. [DOI:10.1016/j.jaad.2021.10.048]
Needle, C. D., Brinks, A. L., Kearney, C. A., Shapiro, J., & Lo Sicco, K. I. (2025). Key considerations of injectable minoxidil for alopecia. Archives of Dermatological Research, 317(1), 572. [DOI:10.1007/s00403-025-04068-3]
Neubauer, Z., Ong, M. M., & Lipner, S. R. (2025). Similar Risk of Sexual Dysfunction in Androgenetic Alopecia Patients Treated With Dutasteride vs. Finasteride: A Retrospective Cohort Database Study. International Journal of Dermatology, advance online publication. [DOI:10.1111/ijd.17887]
Nguyen, D. D., Marchese, M., Cone, E. B., Paciotti, M., Basaria, S., Bhojani, N., & Trinh, Q. D. (2021). Investigation of suicidality and psychological adverse events in patients treated with finasteride. JAMA Dermatology, 157(1), 35–42. [DOI:10.1001/jamadermatol.2020.3385]
Nguyen, N. H., Taylor, J. M., Huang, K. X., & Lee, J. C. (2025). Estrogen hormone therapy stabilizes lateral hairline in transfeminine patients: Implications for facial feminization surgery. Journal of Plastic, Reconstructive & Aesthetic Surgery, 101, 246–251. [DOI:10.1016/j.bjps.2024.07.044]
Olsen, E. A., Hordinsky, M., Whiting, D., Stough, D., Hobbs, S., Ellis, M. L., Wilson, T., Rittmaster, R. S., & Dutasteride Alopecia Research Team (2006). The importance of dual 5α-reductase inhibition in the treatment of male pattern hair loss: results of a randomized placebo-controlled study of dutasteride versus finasteride. Journal of the American Academy of Dermatology, 55(6), 1014–1023. [DOI:10.1016/j.jaad.2006.05.007]
Ovcharenko, Y., Khobzei, K., & Lortkipanidze, N. (2025). Androgenetic alopecia. In Ovcharenko, Y., Lyakhovitsky, A., & Jafferany, M. (Eds.). Psychotrichology: Psychiatric and Psychosocial Aspects of Hair Diseases (pp. 137–170). Cham: Springer Nature Switzerland. [DOI:10.1007/978-3-031-64083-4_9]
Ozono, S., Okajima, E., Yamaguchi, A., Yoshikawa, M., Iwai, A., Moriya, A., Yoshida, K., Samma, S., Maruyama, Y., & Hirao, Y. (2000). A prospective randomized multicenter study of chlormadinone acetate versus flutamide in total androgen blockade for prostate cancer. Japanese Journal of Clinical Oncology, 30(9), 389–396. [DOI:10.1093/jjco/hyd106]
Paradisi, R., Porcu, E., Fabbri, R., Seracchioli, R., Battaglia, C., & Venturoli, S. (2011). Prospective cohort study on the effects and tolerability of flutamide in patients with female pattern hair loss. Annals of Pharmacotherapy, 45(4), 469–475. [DOI:10.1345/aph.1P600]
Patel, P., Nessel, T. A., & Kumar, D. (2023). Minoxidil. In StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. [Google Scholar] [PubMed] [PubMed Bookshelf]
Penha, M. A., Miot, H. A., Kasprzak, M., & Ramos, P. M. (2024). Oral minoxidil vs topical minoxidil for male androgenetic alopecia: a randomized clinical trial. JAMA Dermatology, 160(6), 600–605. [DOI:10.1001/jamadermatol.2024.0284]
Pereira, A. F. J. R., & Coelho, T. O. D. A. (2020). Post-finasteride syndrome. Anais Brasileiros de Dermatologia, 95(3), 271–277. [DOI:10.1016/j.abd.2020.02.001]
Perez, S. M., Nguyen, B., & Senna, M. M. (2025). Efficacy and safety of bicalutamide in female hair loss: a review of the literature. JAAD Reviews, 4, 61–68. [DOI:10.1016/j.jdrv.2025.03.005]
Pirastu, N., Joshi, P. K., de Vries, P. S., Cornelis, M. C., McKeigue, P. M., Keum, N., Franceschini, N., Colombo, M., Giovannucci, E. L., Spiliopoulou, A., Franke, L., North, K. E., Kraft, P., Morrison, A. C., Esko, T., & Wilson, J. F. (2017). GWAS for male-pattern baldness identifies 71 susceptibility loci explaining 38% of the risk. Nature Communications, 8(1), 1584. [DOI:10.1038/s41467-017-01490-8]
Prince, J. C. J., & Safer, J. D. (2020). Endocrine treatment of transgender individuals: current guidelines and strategies. Expert Review of Endocrinology & Metabolism, 15(6), 395–403. [DOI:10.1080/17446651.2020.1825075]
Randolph, M., & Tosti, A. (2021). Oral minoxidil treatment for hair loss: a review of efficacy and safety. Journal of the American Academy of Dermatology, 84(3), 737–746. [DOI:10.1016/j.jaad.2020.06.1009]
Ramos, P. M., Sinclair, R. D., Kasprzak, M., & Miot, H. A. (2020). Minoxidil 1 mg oral versus minoxidil 5% topical solution for the treatment of female-pattern hair loss: a randomized clinical trial. Journal of the American Academy of Dermatology, 82(1), 252–253. [DOI:10.1016/j.jaad.2019.08.060]
Rosenthal, A., Conde, G., Greco, J. F., & Gharavi, N. M. (2024). Management of androgenic alopecia: a systematic review of the literature. Journal of Cosmetic and Laser Therapy, 26(1–4), 1–16. [DOI:10.1080/14764172.2024.2362126]
Rossi, A., Cantisani, C., Melis, L., Iorio, A., Scali, E., & Calvieri, S. (2012). Minoxidil use in dermatology, side effects and recent patents. Recent Patents on Inflammation & Allergy Drug Discovery, 6(2), 130–136. [DOI:10.2174/187221312800166859]
Rossi, A., & Caro, G. (2024). Efficacy of the association of topical minoxidil and topical finasteride compared to their use in monotherapy in men with androgenetic alopecia: A prospective, randomized, controlled, assessor blinded, 3‐arm, pilot trial. Journal of Cosmetic Dermatology, 23(2), 502–509. [DOI:10.1111/jocd.15953]
Ryu, H. K., Kim, K. M., Yoo, E. A., Sim, W. Y., & Chung, B. C. (2006). Evaluation of androgens in the scalp hair and plasma of patients with male‐pattern baldness before and after finasteride administration. British Journal of Dermatology, 154(4), 730–734. [DOI:10.1111/j.1365-2133.2005.07072.x]
Saceda-Corralo, D., Domínguez-Santas, M., Vañó-Galván, S., & Grimalt, R. (2023). What’s new in therapy for male androgenetic alopecia? American Journal of Clinical Dermatology, 24(1), 15–24. [DOI:10.1007/s40257-022-00730-y]
Sadasivam, I. P., Sambandam, R., Kaliyaperumal, D., & Dileep, J. E. (2024). Androgenetic Alopecia in Men: An Update On Genetics. Indian Journal of Dermatology, 69(3), 282. [DOI:10.4103/ijd.ijd_729_23]
Sanabria, B. D., Perdomo, Y. C., Miot, H. A., & Ramos, P. M. (2024). Oral minoxidil 7.5 mg for hair loss increases heart rate with no change in blood pressure in 24 h Holter and 24 h ambulatory blood pressure monitoring. Anais Brasileiros de Dermatologia, 99(5), 734–736. [DOI:10.1016/j.abd.2023.08.016]
Sanabria, B., Miot, H. A., Sinclair, R., Chaves, C., & Müller Ramos, P. (2024). Sublingual Minoxidil 5 mg versus Oral Minoxidil 5 mg for male androgenetic alopecia: A double-blind randomized clinical trial. Journal of the European Academy of Dermatology and Venereology, 39(8), e700–e702. [DOI:10.1111/jdv.20508]
Severi, G., Sinclair, R., Hopper, J. L., English, D. R., McCredie, M. R. E., Boyle, P., & Giles, G. G. (2003). Androgenetic alopecia in men aged 40–69 years: prevalence and risk factors. British Journal of Dermatology, 149(6), 1207–1213. [DOI:10.1111/j.1365-2133.2003.05565.x]
Shadi, Z. (2023). Compliance to topical minoxidil and reasons for discontinuation among patients with androgenetic alopecia. Dermatology and Therapy, 13(5), 1157–1169. [DOI:10.1007/s13555-023-00919-x]
Siebert, A. L., Lapping-Carr, L., & Morgans, A. K. (2020). Neuropsychiatric impact of androgen deprivation therapy in patients with prostate cancer: current evidence and recommendations for the clinician. European Urology Focus, 6(6), 1170–1179. [DOI:10.1016/j.euf.2020.05.014]
Sinclair, R., Wewerinke, M., & Jolley, D. (2005). Treatment of female pattern hair loss with oral antiandrogens. British Journal of Dermatology, 152(3), 466–473. [DOI:10.1111/j.1365-2133.2005.06218.x]
Sinclair, R., Trindade de Carvalho, L., Ferial Ismail, F., & Meah, N. (2020). Treatment of male and female pattern hair loss with sublingual minoxidil: a retrospective case-series of 64 patients. Journal of the European Academy of Dermatology and Venereology, 34(12), e795–e796. [DOI:10.1111/jdv.16616]
Sinclair, R., Sicinska, J., Bokhari, L., & Kasprzak, M. (2025). Sublingual minoxidil increases fibre diameter in male androgenetic alopecia: a proxy for reversal of hair follicle miniaturization. Clinical and Experimental Dermatology, 50(7), 1362–1365. [DOI:10.1093/ced/llaf012]
Singh, S., Patil, A., Kianfar, N., Waśkiel‐Burnat, A., Rudnicka, L., Sinclair, R., & Goldust, M. (2022). Does topical minoxidil at concentrations higher than 5% provide additional clinical benefit? Clinical and Experimental Dermatology, 47(11), 1951–1955. [DOI:10.1111/ced.15338]
Stanczyk, F. Z., Azen, C. G., & Pike, M. C. (2013). Effect of finasteride on serum levels of androstenedione, testosterone and their 5α-reduced metabolites in men at risk for prostate cancer. The Journal of Steroid Biochemistry and Molecular Biology, 138, 10–16. [DOI:10.1016/j.jsbmb.2013.02.015]
Stevenson, M. O., Wixon, N., & Safer, J. D. (2016). Scalp hair regrowth in hormone-treated transgender woman. Transgender Health, 1(1), 202–204. [DOI:10.1089/trgh.2016.0022]
Suchonwanit, P., Srisuwanwattana, P., Chalermroj, N., & Khunkhet, S. (2018). A randomized, double‐blind controlled study of the efficacy and safety of topical solution of 0.25% finasteride admixed with 3% minoxidil vs. 3% minoxidil solution in the treatment of male androgenetic alopecia. Journal of the European Academy of Dermatology and Venereology, 32(12), 2257–2263. [DOI:10.1111/jdv.15171]
Suchonwanit, P., Iamsumang, W., & Rojhirunsakool, S. (2019). Efficacy of topical combination of 0.25% finasteride and 3% minoxidil versus 3% minoxidil solution in female pattern hair loss: a randomized, double-blind, controlled study. American Journal of Clinical Dermatology, 20(1), 147–153. [DOI:10.1007/s40257-018-0387-0]
Suchonwanit, P., Thammarucha, S., & Leerunyakul, K. (2019). Minoxidil and its use in hair disorders: a review. Drug Design, Development and Therapy, 13, 2777–2786. [DOI:10.2147/DDDT.S214907]
T’Sjoen, G., Arcelus, J., Gooren, L., Klink, D. T., & Tangpricha, V. (2019). Endocrinology of transgender medicine. Endocrine Reviews, 40(1), 97–117. [DOI:10.1210/er.2018-00011]
Tan, J. J. Y., Thuya, W. L., Zhu, H., Kristo, J. G., Common, J. E., Wu, C., Ho, P. C. L., & Kang, L. (2025). Comparative analysis of 3D and 2D cell-culturing methods in hair follicle spheroid morphogenesis and drug responsiveness. Biomaterials Advances, 177, 214423. [DOI:10.1016/j.bioadv.2025.214423]
Tang, G. T., Zwickl, S., Sinclair, R., Zajac, J. D., & Cheung, A. S. (2023). Effect of gender-affirming hormone therapy on hair growth: a systematic review of the literature. Clinical and Experimental Dermatology, 48(10), 1117–1127. [DOI:10.1093/ced/llad203]
Tang, G. T., Sinclair, R., Leemaqz, S., & Cheung, A. S. (2025). Scalp hair parameter changes in transgender individuals commencing gender-affirming hormone therapy: A 24-week prospective observational study. Journal of the American Academy of Dermatology, 92(6), 1390–1394. [DOI:10.1016/j.jaad.2025.01.087]
Tang, G. T., Leemaqz, S., Eisman, S., Sinclair, R., & Cheung, A. (2025). Treating testosterone therapy related hair loss in trans people: a clinical trial of sublingual minoxidil. AusPATH 2025 Symposium. [PDF]
Tanglertsampan, C. (2012). Efficacy and safety of 3% minoxidil versus combined 3% minoxidil/0.1% finasteride in male pattern hair loss: a randomized, double-blind, comparative study. Journal of the Medical Association of Thailand, 95(10), 1312–1316. [Google Scholar] [PubMed] [PDF]
Traish, A. M., Hassani, J., Guay, A. T., Zitzmann, M., & Hansen, M. L. (2011). Adverse side effects of 5α-reductase inhibitors therapy: persistent diminished libido and erectile dysfunction and depression in a subset of patients. The Journal of Sexual Medicine, 8(3), 872–884. [DOI:10.1111/j.1743-6109.2010.02157.x]
Traish, A. M., Krakowsky, Y., Doros, G., & Morgentaler, A. (2019). Do 5α-reductase inhibitors raise circulating serum testosterone levels? A comprehensive review and meta-analysis to explaining paradoxical results. Sexual Medicine Reviews, 7(1), 95–114. [DOI:10.1016/j.sxmr.2018.06.002]
Trüeb, R. M., Régnier, A., Dutra Rezende, H., & Gavazzoni Dias, M. F. R. (2019). Post-finasteride syndrome: an induced delusional disorder with the potential of a mass psychogenic illness? Skin Appendage Disorders, 5(5), 320–326. [DOI:10.1159/000497362]
Trüeb, R. M., Luu, N. N. C., Caballero-Uribe, N., Dias, R. G., & Rezende, H. D. (2024). Comment on Current Investigations into the Postfinasteride Syndrome. International Journal of Trichology, 16(1), 6–12. [DOI:10.4103/ijt.ijt_122_21]
Uleri, A., Cornu, J. N., Gobbo, A., Herrmann, T. R., De Nunzio, C., Hashim, H., & Baboudjian, M. (2024). Association of 5α-reductase inhibitors with depression and suicide: a mini systematic review and meta-analysis. European Urology Focus, 10(5), 751–753. [DOI:10.1016/j.euf.2024.04.009]
Upreti, R., Naredo, G., Faqehi, A. M., Hughes, K. A., Stewart, L. H., Walker, B. R., Homer, N. Z., & Andrew, R. (2015). Simultaneous pharmacokinetic and pharmacodynamic analysis of 5α-reductase inhibitors and androgens by liquid chromatography tandem mass spectrometry. Talanta, 131, 728–735. [DOI:10.1016/j.talanta.2014.07.087]
Valente Duarte de Sousa, I. C., & Tosti, A. (2013). New investigational drugs for androgenetic alopecia. Expert Opinion on Investigational Drugs, 22(5), 573–589. [DOI:10.1517/13543784.2013.784743]
Vañó-Galván, S., Pirmez, R., Hermosa-Gelbard, A., Moreno-Arrones, Ó. M., Saceda-Corralo, D., Rodrigues-Barata, R., Jimenez-Cauhe, J., Koh, W. L., Poa, J. E., Jerjen, R., Trindade de Carvalho, L., John, J. M., Salas-Callo, C. I., Vincenzi, C., Yin, L., Lo-Sicco, K., Waskiel-Burnat, A., Starace, M., Zamorano, J. L., Jaén-Olasolo, P., Piraccini, B. M., Rudnicka, L., Shapiro, J., Tosti, A., Sinclair, R., & Bhoyrul, B. (2021). Safety of low-dose oral minoxidil for hair loss: a multicenter study of 1404 patients. Journal of the American Academy of Dermatology, 84(6), 1644–1651. [DOI:10.1016/j.jaad.2021.02.054]
Vierhapper, H., Maier, H., Nowotny, P., & Waldhäusl, W. (2003). Production rates of testosterone and of dihydrotestosterone in female pattern hair loss. Metabolism, 52(7), 927–929. [DOI:10.1016/S0026-0495(03)00060-X]
Vierhapper, H., Nowotny, P., Maier, H., & Waldhausl, W. (2001). Production rates of dihydrotestosterone in healthy men and women and in men with male pattern baldness: determination by stable isotope/dilution and mass spectrometry. The Journal of Clinical Endocrinology & Metabolism, 86(12), 5762–5764. [DOI:10.1210/jcem.86.12.8078]
Wang, C., Du, Y., Bi, L., Lin, X., Zhao, M., & Fan, W. (2023). The efficacy and safety of oral and topical spironolactone in androgenetic alopecia treatment: a systematic review. Clinical, Cosmetic and Investigational Dermatology, 16, 603–612. [DOI:10.2147/CCID.S398950]
Weichert, M., Chen, M., Guo, W., Schrock, N., & Briley, J. (2025). Efficacy and safety of minoxidil therapy: A systematic review and meta-analysis weighing the benefits against the risk of hypertrichosis. JAAD Reviews, 4, 58–60. [DOI:10.1016/j.jdrv.2025.02.013]
Yamana, K., Labrie, F., & Luu-The, V. (2010). Human type 3 5α-reductase is expressed in peripheral tissues at higher levels than types 1 and 2 and its activity is potently inhibited by finasteride and dutasteride. Hormone Molecular Biology and Clinical Investigation, 2(3), 293–299. [DOI:10.1515/HMBCI.2010.035]
Yang, X., Qiao, R., Cheng, W., Lan, X., Li, Y., & Jiang, Y. (2024). Comparative efficacy of 2% minoxidil alone against combination of 2% minoxidil and low-level laser therapy in female pattern hair loss–a randomized controlled trial in Chinese females. Photodiagnosis and Photodynamic Therapy, 45, 103966. [DOI:10.1016/j.pdpdt.2024.103966]
Yazdabadi, A., & Sinclair, R. (2011). Treatment of female pattern hair loss with the androgen receptor antagonist flutamide. Australasian Journal of Dermatology, 52(2), 132–134. [DOI:10.1111/j.1440-0960.2010.00735.x]
Yoong, N., Yii, V., Sinclair, R., & Bhoyrul, B. (2025). Combination of oral minoxidil and bicalutamide for the treatment of female pattern hair loss in adolescents. Journal of the American Academy of Dermatology, 92(5), 1156–1157. [DOI:10.1016/j.jaad.2025.01.056]
Zakhem, G. A., Goldberg, J. E., Motosko, C. C., Cohen, B. E., & Ho, R. S. (2019). Sexual dysfunction in men taking systemic dermatologic medication: a systematic review. Journal of the American Academy of Dermatology, 81(1), 163–172. [DOI:10.1016/j.jaad.2019.03.043]
Zaminski, D., Zampella, J., Shapiro, J., Sicco, K. I. L., & Mazori, D. R. (2025). Use of low-dose oral minoxidil for hair growth in transgender and gender non-binary adult patients: a retrospective cohort study. Journal of the American Academy of Dermatology, advance online publication. [DOI:10.1016/j.jaad.2025.06.029]
Zeltzer, A. A., Keren, A., & GILHAR, A. (2024). Topical Minoxidil Rejuvenates Hair Follicles from Men with Androgenetic Alopecia In Vivo. Acta Dermato-Venereologica, 104, 24213. [DOI:10.2340/actadv.v104.24213]
Zhong, X., Yang, Y., Wei, S., & Liu, Y. (2025). Multidimensional assessment of adverse events of finasteride: a real-world pharmacovigilance analysis based on FDA Adverse Event Reporting System (FAERS) from 2004 to April 2024. PloS One, 20(3), e0309849. [DOI:10.1371/journal.pone.0309849]
Zhou, Z., Song, S., Gao, Z., Wu, J., Ma, J., & Cui, Y. (2019). The efficacy and safety of dutasteride compared with finasteride in treating men with androgenetic alopecia: a systematic review and meta-analysis. Clinical Interventions In Aging, 14, 399–406. [DOI:10.2147/CIA.S192435]
\ No newline at end of file
diff --git a/transfemscience.org/articles/high-dose-transdermal-e2/index.html b/transfemscience.org/articles/high-dose-transdermal-e2/index.html
index fcbef099..a3c6b2fa 100644
--- a/transfemscience.org/articles/high-dose-transdermal-e2/index.html
+++ b/transfemscience.org/articles/high-dose-transdermal-e2/index.html
@@ -1 +1 @@
-A Review of Studies on Estradiol Levels and Testosterone Suppression with High-Dose Transdermal Estradiol Gel and Ointment in Cisgender Men with Prostate Cancer - Transfeminine Science
A Review of Studies on Estradiol Levels and Testosterone Suppression with High-Dose Transdermal Estradiol Gel and Ointment in Cisgender Men with Prostate Cancer
By Aly | First published June 30, 2019 | Last modified November 2, 2022
Abstract / TL;DR
High-dose estradiol monotherapy, generally used non-orally, is a therapeutic approach for testosterone suppression which is sometimes used in transfeminine people and men with prostate cancer. Limited data are available for this approach with certain forms of estradiol, so considering research in other patient populations besides transfeminine people can provide valuable information. In the 1970s and 1980s, studies were conducted on high-dose transdermal estradiol gel and ointment for the treatment of prostate cancer in men. Estradiol at 3 to 20 mg/day appeared to give 84 to 473 pg/mL estradiol and 105 to 260 ng/dL testosterone in these men. While informative, the findings of the studies are only so useful due to various unfortunate limitations. In any case, it’s apparent that transdermal estradiol gel and related forms don’t seem to be well-suited for achieving the high levels of estradiol that are needed for robust testosterone suppression with high-dose estradiol monotherapy in men with prostate cancer or transfeminine people. High-dose transdermal estradiol patches, or alternatively genital application of transdermal estradiol forms, may be more useful options for this approach on the other hand.
Introduction
High-dose estrogen monotherapy is a means of testosterone suppression which is sometimes used to treat prostate cancer in men. High-dose parenteral estradiol therapy is one particular approach used for this purpose which has less toxicity compared to use of oral and synthetic estrogens. In the late 1970s and early 1980s, high-dose transdermal estradiol gel/ointment was assessed by French researchers in three clinical studies for the treatment of prostate cancer (Steg & Benoit, 1979; Steg et al., 1979; Steg et al., 1980; Steg et al., 1983; Steg & Benoit, 1983; Benoit, 1985). The dosages used in the studies ranged from 3 to 20 mg/day. For comparison, typical doses of transdermal estradiol gel used in transfeminine hormone therapy to achieve physiological estradiol levels range from about 1 to 6 mg/day.
High-dose estradiol monotherapy, generally administered parenterally, is sometimes used as a means of suppressing testosterone levels in transfeminine hormone therapy similarly to the hormonal treatment of prostate cancer. This approach omits the use of antiandrogens and their costs, side effects, and risks, though at the same time incurs risks of higher estradiol exposure. There is limited information available on estradiol levels and testosterone suppression with high-dose estradiol gel and similar forms. Hence, a review of the relevant details from the studies of high-dose estradiol gel for prostate cancer is of value as it can be used to inform the use of high-dose estradiol monotherapy in transfeminine people.
The French studies in question appear to be the only clinical studies of high-dose transdermal estradiol gel/ointment for prostate cancer that have been published. This is in contrast to high-dose transdermal estradiol patches and injectable estradiol esters like polyestradiol phosphate (PEP) and estradiol undecylate (EU), which have been much more widely used in men with prostate cancer.
Data and Excerpts
A Note on Estradiol Delivery Rates
The authors use “γ” (“gamma”) as a unit in their papers. This is a now-deprecated non-SI unit of mass equal to 1 μg (Wiki). That is, “1 γ” means the same thing as “1 μg”. The researchers provided values using this unit as an estimate of the amount of estradiol actually absorbed into the body after taking into account the “cutaneous absorption coefficient” (i.e., transdermal bioavailability) of estradiol. However, when compared to established μg/day values for commercial transdermal estradiol patches and the estradiol levels associated with these patches, the values in the papers are very different from what would be expected. For this reason, and because estimates for transdermal estradiol patches were more rigorously determined and likely to be correct, the delivery rates reported by the authors are probably not accurate. It was important to include them in this article nonetheless however because the researchers modified their transdermal estradiol formulations between their studies, resulting in inconsistencies between the estradiol doses in gel/ointment and their estimated delivery values.
Study 1: Estradiol Gel 3 mg/day versus 6 mg/day
The following three papers pertain to the first study:
Steg, A., Benoit, G., Limouzin-Lamotte, A., Mahoudeau, J., Caillens, M., & Raichvarg, D. (1979). Cancer de la prostate: effets métaboliques des bêta-estradiol par voie percutanée. [Cancer of the prostate: metabolic effect of percutaneous beta-estradiol. / Prostatic carcinoma: metabolic effect of percutaneous beta-estradiol.] La Nouvelle Presse Medicale, 8(46), 3801–3802. [Google Scholar 1] [Google Scholar 2] [Pascal and Francis] [PubMed] [PDF]
Steg, A., Benoit, G., Limouzin-Lamotte, A., Mahoudeau, J., Caillens, M., & Raichvarg, D. (1980). Cancer de la prostate: Effets métaboliques du bêta-estradiol par voie percutanée. [Cancer of the prostate: metabolic effects of percutaneously administered beta-estradiol.] Revue Médicale de la Suisse Romande, 100(11), 895–897. [Google Scholar 1] [Google Scholar 2] [Pascal and Francis] [PubMed] [PDF]
In the study, 21 men with prostate cancer were divided into two groups and treated for 1 month with:
3 mg/day (“300 μg/day”) estradiol in 5 g/day gel applied to the abdomen in one daily dose
6 mg/day (“600 μg/day”) estradiol in 10 g/day gel applied to the abdomen in two daily divided doses
Hormone levels in the study were as follows:
Estradiol levels
Testosterone levels
Pre-treatment
23.2 ± 10.8 pg/mL
Pre-treatment
341 ± 180 ng/dL
E2 gel 3 mg/day
84.2 ± 54.3 pg/mL
E2 gel 3 mg/day
188 ± 125 ng/dL
E2 gel 6 mg/day
184.7 ± 98.46 pg/mL
E2 gel 6 mg/day
105 ± 113 ng/dL
Estrone levels
DHT levels
Pre-treatment
37.6 ± 16.2 pg/mL
Pre-treatment
36 ± 27 ng/dL
E2 gel 3 mg/day
86.7 ± 42.5 pg/mL
E2 gel 3 mg/day
23 ± 14 ng/dL
E2 gel 6 mg/day
150.9 ± 101.4 pg/mL
E2 gel 6 mg/day
12 ± 12 ng/dL
LH levels
FSH levels
Pre-treatment
6.21 ± 6.2 mIU/mL
Pre-treatment
7.56 ± 4.9 mIU/mL
E2 gel 3 mg/day
4.15 ± 3.3 mIU/mL
E2 gel 3 mg/day
6.44 ± 5.0 mIU/mL
E2 gel 6 mg/day
3.41 ± 3.2 mIU/mL
E2 gel 6 mg/day
4.13 ± 5.0 mIU/mL
The researchers stated that the testosterone levels resulting with 6 mg/day estradiol gel (105 ± 113 ng/) were similar to those that are known to occur with 3 mg/day oral diethylstilbestrol. However, this seems to be a somewhat inaccurate statement as 3 mg/day oral diethylstilbestrol has been found to consistently suppress testosterone levels into the castrate range (≤50 ng/dL) in studies (Wiki).
Study 2: Estradiol Ointment 10 mg/day versus Diethylstilbestrol Oral 3 mg/day
The following paper pertains to the second study:
Steg, A., Benoit, G., Maisonneuve, P., Tallet, F., Nahoul, K., Sultan, Y., Raichwarg, D., & Limouzin-Lamotte, M. A. (1983). Étude comparative du diéthylstilboestrol et du 17 bêta-oestradiol per-cutané dans le traitement du cancer de la prostate. [A comparative study of percutaneous 17 beta-estradiol and diethylstilbestrol in the treatment of prostatic cancer.] Annales d’Urologie, 17(4), 197–202. [Google Scholar] [PDF]
In the study, 56 men with prostate cancer were divided into two groups and treated for 3 months with:
10 mg/day (“600 μg/day”) estradiol in “ointment” applied to the abdomen in two daily divided doses
3 mg/day oral diethylstilbestrol in three daily divided doses
Hormone levels in the study were as follows:
Estradiol levels
Testosterone levels
Pre-treatment (E2 group)
30 pg/mL
Pre-treatment (E2 group)
450 ng/dL
E2 ointment 10 mg/day
107 ± 81 pg/mL
E2 ointment 10 mg/day
180 ± 160 ng/dL
Pre-treatment (DES group)
26 pg/mL
Pre-treatment (DES group)
420 ± 130 ng/dL
DES oral 3 mg/day
19 pg/mL
DES oral 3 mg/day
51 ± 0.9 ng/dL
LH levels
FSH levels
Pre-treatment (E2 group)
3.2 mIU/mL
Pre-treatment (E2 group)
4.7 mIU/mL
E2 ointment 10 mg/day
2.0 ± 0.9 mIU/mL
E2 ointment 10 mg/day
1.7 mIU/mL
Pre-treatment (DES group)
4.0 ± 3.2 mIU/mL
Pre-treatment (DES group)
5.8 mIU/mL
DES oral 3 mg/day
1.6 ± 1.2 mIU/mL
DES oral 3 mg/day
1.7 mIU/mL
Some SDs (±) are missing in the table above because not all were provided in the original paper. The SDs that were included above were actually reported in the paper for Study 3.
Some noteworthy translated excerpts from the paper:
This study shows that the clinical effects were more dramatic in the DES group, with a sharper drop in the free plasma testosterone level, than in the E2 group.
E2 has been administered too weakly because it is too low in the commercially used ointment and because application to the thick abdominal skin has a lower absorption coefficient than a thinner skin such as forearms for example. It is possible that a dose equivalent to 15 or 20 mg of gel, applied on the forearms, may allow to lower the testosterone below [100 ng/dL]. The marketing of a more concentrated ointment is therefore desirable, as well as the use of a more efficient absorption site.
The search for new estrogens and new routes of administration is therefore necessary: E2, a natural female hormone, induces no thromboembolic events. It seems to us, therefore, that a larger dose must be proposed to be as effective as 3 mg DES. A gel twice more concentrated is therefore currently under study.
The double-concentration estradiol gel was subsequently reported in Study 3 (see below).
It should be noted that the transdermal estradiol formulation in this study is different from that used in Study 1. Specifically, “6 mg/day estradiol gel” was stated to deliver “600 μg/day estradiol” in Study 1, whereas “10 mg/day estradiol ointment” was stated to deliver “600 μg/day estradiol” in Study 2. These apparent discrepancies might be of relevance to the unexpectedly lower estradiol levels and testosterone suppression in Study 2 compared to Study 1.
Study 3: Estradiol Ointment 20 mg/day versus Bilateral Orchiectomy
The following paper pertains to the third study:
Steg, A., & Benoit, G. (1983). Étude comparative de fortes doses d’oestradiol-17 β administrées par voie per-cutanée et de l’orchidectomie bilatérale dans le traitement du cancer de la prostate. [Prostatic carcinoma. Bilateral orchiectomy versus percutaneous administration of large doses of 17 β-estradiol. A comparative study. / Comparative study of percutaneous administration of large doses of 17β œstradiol and bilateral orchidectomy in prostatic carcinoma.] Annales d’Urologie, 17(5), 286–288. [Google Scholar] [Pascal and Francis] [PDF]
In the study, 38 men with prostate cancer were divided into two groups and treated for 1 month with:
20 mg/day (“1200 μg/day”) estradiol in an ointment applied to the skin in four daily divided doses
Bilateral orchiectomy (i.e., surgical removal of the testes)
Hormone levels in the study were as follows:
Condition
Estradiol levels
Testosterone levels
LH levels
Pre-treatment (E2 group)
52 ± 30 pg/mL
460 ± 230 ng/dL
2.6 ± 0.9 mIU/mL
E2 ointment 20 mg/day
473 ± 375 pg/mL
260 ± 160 ng/dL
1.7 ± 0.8 mIU/mL
Pre-treatment (orchi group)
Not reported
500 ± 160 ng/dL
5.5 ± 6 mIU/mL
Bilateral orchiectomy
Not reported
20 ± 10 ng/dL
19 ± 8 mIU/mL
Some noteworthy translated excerpts from the paper:
The purpose of this study was to determine whether hormonal efficacy [in the treatment of prostate cancer] was enhanced by the administration of large doses of E2. […] The study shows that, whereas orchiectomy lowers plasma testosterone levels dramatically, E2, at [the double dose used in this study compared to our previous study] (1200 [μg] versus 600 [μg]), surprisingly does not further lower it at all, but plasma E2 is substantially increased.
We have found that the administration of E2 causes significant biological changes; E2 is well-absorbed, testosterone is lowered, and LH and FSH are also lowered. But when comparing these effects with those obtained by castration, and with those also observed after administration of 3 mg of diethylstilbestrol (in our previous study), the results are much less brilliant. Indeed: the plasma testosterone is moderately lowered by E2 whereas the fall is much more pronounced with DES and is spectacular with castration; pituitary suppression is comparable with E2 and DES and of course orchiectomy increases LH; and if we compare the results obtained before and after treatment, it is clear that if before treatment the figures are comparable, after treatment the fall is pronounced only with DES and especially with orchiectomy.
The objective of this study was to determine whether doubling the dose of E2 enhances its antiandrogenic effect. [With both a lower dose and a higher dose], E2 is very well-absorbed. [With both doses], the pituitary suppression is identical, but on the other hand, not only is plasma testosterone not lower with [the higher dose], but it seems to be even [higher than with the lower dose]. This difference is also found clinically since with [the higher dose], 10% of patients were improved while with [the lower dose], 30% improvement was obtained.
How can this result be explained? Perhaps this is due to the contrary effects of E2 on androgens. Indeed: E2 on the one hand suppresses pituitary activity, which leads to a fall of testicular testosterone secretion, and at the same time E2 decreases 5α-reductase activity in the prostate; on the other hand, E2 causes an increase in SHBG and competes with free testosterone binding to SHBG and in this way increases the proportion of active free testosterone. In addition, testosterone also increases the level of cytoplasmic androgen receptors.
This work shows that E2 is well-absorbed percutaneously and causes practically no cardiovascular events. On the other hand, it appears that the antiandrogenic effect is significantly less by this route than that obtained by diethylstilbestrol and castration, and that in any case, we cannot improve the efficacy by increasing the dose of E2.
The finding that the 20 mg/day estradiol ointment resulted in less suppression of testosterone levels than the 10 mg/day estradiol ointment despite markedly higher estradiol levels is surprising. However, the researchers’ theoretical explanation of the result seems questionable. Abundant clinical research with high-dose estradiol for prostate cancer (e.g., transdermal patches, injectable polyestradiol phosphate injections, injectable estradiol undecylate) has shown stronger testosterone suppression with high estradiol levels in this range or below and seems to thoroughly contradict their results. Studies have consistently shown that estradiol levels of 200 to 500 pg/mL and above suppress testosterone levels by 90% and greater (to ~50 ng/dL and below) (Wiki; Graphs). Instead of the authors’ interpretation of their unexpected results, it seems possible that the testosterone assays for this study may have been inaccurate or perhaps some other methodological problem may have been responsible.
The concentration and dosage of estradiol ointment in this study were double those of Study 2, yet the estradiol levels measured in this study were almost 5-fold higher than those in Study 2. It’s possible that the higher concentration of ointment used may have resulted in disproportionately greater absorption, as one study found that the smaller the application area of estradiol gel (and hence higher the post-application concentration), the greater the resulting estradiol levels (Graph). On the other hand, it’s possible that the estradiol assays were inaccurate (as also suggested for testosterone above). Lastly, in contrast to the previous two papers, the authors don’t state where the ointment was applied in this study. It was probably the abdomen similarly, but this isn’t certain; they may have used one or more other locations with differing skin permeability (as they discussed the possibility of doing in Study 1). It’d be expected that they’d mention this change though, which perhaps makes the possibility unlikely.
Review Article
A review of hormone therapy for prostate cancer was also published by one of the researchers:
Benoit, G. (1985). Que Penser du Traitement Hormonal du Cancer de la Prostate / Hormonothérapie du Cancer de la Prostate. [Thoughts on the Hormonal Treatment of Prostate Cancer / Hormone Therapy of Prostate Cancer.] Gazette Médicale, 92(5), 33–39. [Google Scholar] [PDF] [Translation]
In the paper, she briefly mentioned her and her colleagues’ studies on high-dose transdermal estradiol gel/ointment for prostate cancer and provided some additional information pertaining to these studies. Here are the relevant translated excerpts from the paper:
In a study in Cochin (16), the efficacy of diethylstilbestrol was 50% of objective responses, but at a cost of 23% of thromboembolic events. These thromboembolic events are similar to the accidents that occur in women taking oral estrogen–progestogens. These are oral synthetic estrogens. In a series of diethylstilbestrol-treated prostate cancers, Steg (16) has shown that this treatment induces an increase in triglycerides and a decrease in coagulation factor VIII. Abbou (1) found, on his behalf, an increase in circulating immune complexes in patients with thrombosis under synthetic estrogen–progestogen therapy.
It therefore seems to us no longer possible to prescribe diethylstilbestrol as first intention. To reduce these cardiovascular events, most authors have tried to use natural estrogens administered intramuscularly, subcutaneously, or percutaneously. Cochin’s experience of using a natural estrogen (17-beta-estradiol) percutaneously has shown that this treatment may be effective, but that it is very difficult to administer: one in two patients is unable to apply the treatment correctly and regularly, which greatly reduces the use of such treatment (15).
As an indication, we report several studies made successively in Cochin (16, 17) with diethylstilbestrol, a natural estrogen (17-beta-estradiol), testicular pulpectomy, and an LHRH analogue administered subcutaneously.
The results confirm that diethylstilbestrol causes thromboembolic events. These different estrogenic treatments promote gynecomastia. Medical castration by LHRH analogues or surgical pulpectomy causes hot flushes. All these treatments reach, in a variable way, libido and sexual potency. This last effect is complex: it is known that castration, in young men, after puberty, does not regularly cause impotence, as shown by the history of castrates. However, it is necessary to warn the patient that each of these treatments can reach his manhood.
Synthetic estrogens, especially diethylstilbestrol, should no longer be used as first-line agents in the treatment of metastatic prostate cancer because of their thromboembolic risk. Natural estrogens do not seem efficient enough to be used regularly. Complete castration performs excessive mutilation. In our opinion, only testicular pulpectomy, LHRH analogues, and antiandrogens remain in competition.
The review also contains the following adapted summary table of data from their studies:
DES
E2 gel/ointment
Orchiectomy
GnRH analogue
Testosterone levels
50 ng/dL
100 ng/dL
20 ng/dL
50 ng/dL
50% partial remission
50%
30%
33%
41%
Cardiovascular events
23%
0%
0%
0%
The gonadotropin-releasing hormone (GnRH) analogue data were from other studies and papers by the same researchers (Steg et al., 1984; Steg et al., 1985a; Steg et al., 1985b). These studies did not assess transdermal estradiol and hence are not otherwise discussed in the present article.
The authors’ comments in this paper raise the additional question of whether inconsistent and suboptimal exposure to estradiol gel/ointment may have influenced the findings of their studies.
Summary and Discussion
In the reviewed clinical studies of high-dose transdermal estradiol gel/ointment in men with prostate cancer, the following results were obtained in terms of changes in measured hormone levels:
Estradiol dose
Estradiol levels
Testosterone levels
Before treatment
23–52 pg/mL
341–460 ng/dL
3 mg/day gel (“300 μg/day”)
84 pg/mL
188 ng/dL
6 mg/day gel (“600 μg/day”)
185 pg/mL
105 ng/dL
10 mg/day ointment (“600 μg/day”)
107 pg/mL
180 ng/dL
20 mg/day ointment (“1200 μg/day”)
473 pg/mL
260 ng/dL
Unfortunately, the data reported in these studies is not as useful as might have been hoped. This is due to inconsistencies with the transdermal estradiol formulations and doses used, reported issues with patient compliance in terms of consistent and correct administration, inconsistencies with the measured estradiol and testosterone levels, and omission of certain important details (e.g., ointment concentrations, gel/ointment compositions and differences, how the estimated estradiol delivery rates were determined, etc.). As a result of these limitations, there may be only so much that can be taken away from the studies.
In any case, these studies do contribute to an impression that transdermal estradiol gel achieves relatively low estradiol levels and is relatively weak in terms of estrogenic strength even when used at high doses. Other studies of high-dose transdermal estradiol gel/cream, for instance at doses of 3 to 8 mg/day, have reported relatively low estradiol levels similarly, for instance 100 to 200 pg/mL on average (e.g., Lauritzen, 1990; Wiki). Based on these studies, transdermal estradiol gel doesn’t seem to be a very effective or affordable way of achieving the estradiol levels that are needed for adequate suppression of testosterone levels with high-dose estradiol monotherapy in transfeminine people.
In contrast to transdermal estradiol gel, transdermal estradiol patches can consistently achieve much higher estradiol levels and testosterone suppression in comparison (Wiki). About 50 to 100 pg/mL estradiol per 100 μg/day estradiol patch appears to be achieved on average, and multiple 100 μg/day patches up to as high as eight at a time appear to result in approximately linear increases in estradiol levels (Aly, 2020; Wiki). The consistent and higher estradiol levels with estradiol patches makes them generally a more favorable form of estradiol for transdermal administration than estradiol gel and related formulations. Additionally, genital application of transdermal estradiol gel and other transdermal estradiol formulations can allow for dramatically higher estradiol levels (e.g., 5- to 8-fold) than application to conventional transdermal skin sites (e.g., forearm, abdomen) (Aly, 2019). This may also provide transdermal estradiol formulations with greater potential for high-dose estradiol monotherapy.
References
Benoit, G. (1985). Que Penser du Traitement Hormonal du Cancer de la Prostate / Hormonothérapie du Cancer de la Prostate. [Thoughts on the Hormonal Treatment of Prostate Cancer / Hormone Therapy of Prostate Cancer.] Gazette Médicale, 92(5), 33–39. [Google Scholar] [PDF] [Translation]
Lauritzen, C. (1990). Clinical use of oestrogens and progestogens. Maturitas, 12(3), 199–214. [DOI:10.1016/0378-5122(90)90004-P]
Steg, A., Benoit, G., Limouzin-Lamotte, A., Mahoudeau, J., Caillens, M., & Raichvarg, D. (1979). Cancer de la prostate: effets métaboliques des bêta-estradiol par voie percutanée. [Cancer of the prostate: metabolic effect of percutaneous beta-estradiol. / Prostatic carcinoma: metabolic effect of percutaneous beta-estradiol.] La Nouvelle Presse Medicale, 8(46), 3801–3802. [Google Scholar 1] [Google Scholar 2] [Pascal and Francis] [PubMed] [PDF]
Steg, A., Benoit, G., Limouzin-Lamotte, A., Mahoudeau, J., Caillens, M., & Raichvarg, D. (1980). Cancer de la prostate: Effets métaboliques du bêta-estradiol par voie percutanée. [Cancer of the prostate: metabolic effects of percutaneously administered beta-estradiol.] Revue Médicale de la Suisse Romande, 100(11), 895–897. [Google Scholar 1] [Google Scholar 2] [Pascal and Francis] [PubMed] [PDF]
Steg, A., Benoit, G., Maisonneuve, P., Tallet, F., Nahoul, K., Sultan, Y., Raichwarg, D., & Limouzin-Lamotte, M. A. (1983). Étude comparative du diéthylstilboestrol et du 17 bêta-oestradiol per-cutané dans le traitement du cancer de la prostate. [A comparative study of percutaneous 17 beta-estradiol and diethylstilbestrol in the treatment of prostatic cancer.] Annales d’Urologie, 17(4), 197–202. [Google Scholar] [PDF]
Steg, A., & Benoit, G. (1983). Étude comparative de fortes doses d’oestradiol-17 β administrées par voie per-cutanée et de l’orchidectomie bilatérale dans le traitement du cancer de la prostate. [Prostatic carcinoma. Bilateral orchiectomy versus percutaneous administration of large doses of 17 β-estradiol. A comparative study. / Comparative study of percutaneous administration of large doses of 17β œstradiol and bilateral orchidectomy in prostatic carcinoma.] Annales d’Urologie, 17(5), 286–288. [Google Scholar] [Pascal and Francis] [PDF]
Steg, A., Chiche, R., Boccon-Gibod, L., Debré, B., Duchier, J., & Schally, A. V. (1984). Traitement du cancer de la prostate par un agoniste de la LH-RH: le D Trp 6 LH-RH: Résultats préliminaires à propos de trente observations. [Treatment of prostatic cancer with an LH-RH agonist: the D Trp6 LH-RH. Preliminary results in 30 cases. / Treatment of prostate cancer with D Trp6 LH-RH, an LH-RH agonist. Preliminary results of thirty cases.] Annales d’Urologie, 18(6), 388–392. [Google Scholar 1] [Google Scholar 2] [PubMed]
Steg, A., Chiche, R., Boccon-Gibod, L., & Debre, B. (1985). Traitement du cancer de la prostate évolué par un agoniste de la gonadoréline, le DTrp6 LH-RH. Quarante et une observations. [Treatment of advanced prostatic cancer with a gonadorelin agonist, dTrp6 LHRH. 41 cases.] La Presse Médicale (Paris, France: 1983), 14(40), 2045–2048. [Google Scholar 1] [Google Scholar 2] [PubMed]
Steg, A., Chiche, R., Boccon-Gibod, L., Debre, B., & Duchier, J. (1985). Traitement du cancer de la prostate par un agoniste de la LH-RH: le D Trp6 LH-RH―résultats préliminaires à propos de trente observations. [Treatment of prostate cancer with D-TRP6 LH-RH, an LH-RH agonist—preliminary results in 30 cases.] La Semaine des Hôpitaux de Paris, 61(10), 615–618. [Google Scholar 1] [Google Scholar 2]
\ No newline at end of file
+A Review of Studies on Estradiol Levels and Testosterone Suppression with High-Dose Transdermal Estradiol Gel and Ointment in Cisgender Men with Prostate Cancer - Transfeminine Science
A Review of Studies on Estradiol Levels and Testosterone Suppression with High-Dose Transdermal Estradiol Gel and Ointment in Cisgender Men with Prostate Cancer
By Aly | First published June 30, 2019 | Last modified November 2, 2022
Abstract / TL;DR
High-dose estradiol monotherapy, generally used non-orally, is a therapeutic approach for testosterone suppression which is sometimes used in transfeminine people and men with prostate cancer. Limited data are available for this approach with certain forms of estradiol, so considering research in other patient populations besides transfeminine people can provide valuable information. In the 1970s and 1980s, studies were conducted on high-dose transdermal estradiol gel and ointment for the treatment of prostate cancer in men. Estradiol at 3 to 20 mg/day appeared to give 84 to 473 pg/mL estradiol and 105 to 260 ng/dL testosterone in these men. While informative, the findings of the studies are only so useful due to various unfortunate limitations. In any case, it’s apparent that transdermal estradiol gel and related forms don’t seem to be well-suited for achieving the high levels of estradiol that are needed for robust testosterone suppression with high-dose estradiol monotherapy in men with prostate cancer or transfeminine people. High-dose transdermal estradiol patches, or alternatively genital application of transdermal estradiol forms, may be more useful options for this approach on the other hand.
Introduction
High-dose estrogen monotherapy is a means of testosterone suppression which is sometimes used to treat prostate cancer in men. High-dose parenteral estradiol therapy is one particular approach used for this purpose which has less toxicity compared to use of oral and synthetic estrogens. In the late 1970s and early 1980s, high-dose transdermal estradiol gel/ointment was assessed by French researchers in three clinical studies for the treatment of prostate cancer (Steg & Benoit, 1979; Steg et al., 1979; Steg et al., 1980; Steg et al., 1983; Steg & Benoit, 1983; Benoit, 1985). The dosages used in the studies ranged from 3 to 20 mg/day. For comparison, typical doses of transdermal estradiol gel used in transfeminine hormone therapy to achieve physiological estradiol levels range from about 1 to 6 mg/day.
High-dose estradiol monotherapy, generally administered parenterally, is sometimes used as a means of suppressing testosterone levels in transfeminine hormone therapy similarly to the hormonal treatment of prostate cancer. This approach omits the use of antiandrogens and their costs, side effects, and risks, though at the same time incurs risks of higher estradiol exposure. There is limited information available on estradiol levels and testosterone suppression with high-dose estradiol gel and similar forms. Hence, a review of the relevant details from the studies of high-dose estradiol gel for prostate cancer is of value as it can be used to inform the use of high-dose estradiol monotherapy in transfeminine people.
The French studies in question appear to be the only clinical studies of high-dose transdermal estradiol gel/ointment for prostate cancer that have been published. This is in contrast to high-dose transdermal estradiol patches and injectable estradiol esters like polyestradiol phosphate (PEP) and estradiol undecylate (EU), which have been much more widely used in men with prostate cancer.
Data and Excerpts
A Note on Estradiol Delivery Rates
The authors use “γ” (“gamma”) as a unit in their papers. This is a now-deprecated non-SI unit of mass equal to 1 μg (Wiki). That is, “1 γ” means the same thing as “1 μg”. The researchers provided values using this unit as an estimate of the amount of estradiol actually absorbed into the body after taking into account the “cutaneous absorption coefficient” (i.e., transdermal bioavailability) of estradiol. However, when compared to established μg/day values for commercial transdermal estradiol patches and the estradiol levels associated with these patches, the values in the papers are very different from what would be expected. For this reason, and because estimates for transdermal estradiol patches were more rigorously determined and likely to be correct, the delivery rates reported by the authors are probably not accurate. It was important to include them in this article nonetheless however because the researchers modified their transdermal estradiol formulations between their studies, resulting in inconsistencies between the estradiol doses in gel/ointment and their estimated delivery values.
Study 1: Estradiol Gel 3 mg/day versus 6 mg/day
The following three papers pertain to the first study:
Steg, A., Benoit, G., Limouzin-Lamotte, A., Mahoudeau, J., Caillens, M., & Raichvarg, D. (1979). Cancer de la prostate: effets métaboliques des bêta-estradiol par voie percutanée. [Cancer of the prostate: metabolic effect of percutaneous beta-estradiol. / Prostatic carcinoma: metabolic effect of percutaneous beta-estradiol.] La Nouvelle Presse Medicale, 8(46), 3801–3802. [Google Scholar 1] [Google Scholar 2] [Pascal and Francis] [PubMed] [PDF]
Steg, A., Benoit, G., Limouzin-Lamotte, A., Mahoudeau, J., Caillens, M., & Raichvarg, D. (1980). Cancer de la prostate: Effets métaboliques du bêta-estradiol par voie percutanée. [Cancer of the prostate: metabolic effects of percutaneously administered beta-estradiol.] Revue Médicale de la Suisse Romande, 100(11), 895–897. [Google Scholar 1] [Google Scholar 2] [Pascal and Francis] [PubMed] [PDF]
In the study, 21 men with prostate cancer were divided into two groups and treated for 1 month with:
3 mg/day (“300 μg/day”) estradiol in 5 g/day gel applied to the abdomen in one daily dose
6 mg/day (“600 μg/day”) estradiol in 10 g/day gel applied to the abdomen in two daily divided doses
Hormone levels in the study were as follows:
Estradiol levels
Testosterone levels
Pre-treatment
23.2 ± 10.8 pg/mL
Pre-treatment
341 ± 180 ng/dL
E2 gel 3 mg/day
84.2 ± 54.3 pg/mL
E2 gel 3 mg/day
188 ± 125 ng/dL
E2 gel 6 mg/day
184.7 ± 98.46 pg/mL
E2 gel 6 mg/day
105 ± 113 ng/dL
Estrone levels
DHT levels
Pre-treatment
37.6 ± 16.2 pg/mL
Pre-treatment
36 ± 27 ng/dL
E2 gel 3 mg/day
86.7 ± 42.5 pg/mL
E2 gel 3 mg/day
23 ± 14 ng/dL
E2 gel 6 mg/day
150.9 ± 101.4 pg/mL
E2 gel 6 mg/day
12 ± 12 ng/dL
LH levels
FSH levels
Pre-treatment
6.21 ± 6.2 mIU/mL
Pre-treatment
7.56 ± 4.9 mIU/mL
E2 gel 3 mg/day
4.15 ± 3.3 mIU/mL
E2 gel 3 mg/day
6.44 ± 5.0 mIU/mL
E2 gel 6 mg/day
3.41 ± 3.2 mIU/mL
E2 gel 6 mg/day
4.13 ± 5.0 mIU/mL
The researchers stated that the testosterone levels resulting with 6 mg/day estradiol gel (105 ± 113 ng/) were similar to those that are known to occur with 3 mg/day oral diethylstilbestrol. However, this seems to be a somewhat inaccurate statement as 3 mg/day oral diethylstilbestrol has been found to consistently suppress testosterone levels into the castrate range (≤50 ng/dL) in studies (Wiki).
Study 2: Estradiol Ointment 10 mg/day versus Diethylstilbestrol Oral 3 mg/day
The following paper pertains to the second study:
Steg, A., Benoit, G., Maisonneuve, P., Tallet, F., Nahoul, K., Sultan, Y., Raichwarg, D., & Limouzin-Lamotte, M. A. (1983). Étude comparative du diéthylstilboestrol et du 17 bêta-oestradiol per-cutané dans le traitement du cancer de la prostate. [A comparative study of percutaneous 17 beta-estradiol and diethylstilbestrol in the treatment of prostatic cancer.] Annales d’Urologie, 17(4), 197–202. [Google Scholar] [PDF]
In the study, 56 men with prostate cancer were divided into two groups and treated for 3 months with:
10 mg/day (“600 μg/day”) estradiol in “ointment” applied to the abdomen in two daily divided doses
3 mg/day oral diethylstilbestrol in three daily divided doses
Hormone levels in the study were as follows:
Estradiol levels
Testosterone levels
Pre-treatment (E2 group)
30 pg/mL
Pre-treatment (E2 group)
450 ng/dL
E2 ointment 10 mg/day
107 ± 81 pg/mL
E2 ointment 10 mg/day
180 ± 160 ng/dL
Pre-treatment (DES group)
26 pg/mL
Pre-treatment (DES group)
420 ± 130 ng/dL
DES oral 3 mg/day
19 pg/mL
DES oral 3 mg/day
51 ± 0.9 ng/dL
LH levels
FSH levels
Pre-treatment (E2 group)
3.2 mIU/mL
Pre-treatment (E2 group)
4.7 mIU/mL
E2 ointment 10 mg/day
2.0 ± 0.9 mIU/mL
E2 ointment 10 mg/day
1.7 mIU/mL
Pre-treatment (DES group)
4.0 ± 3.2 mIU/mL
Pre-treatment (DES group)
5.8 mIU/mL
DES oral 3 mg/day
1.6 ± 1.2 mIU/mL
DES oral 3 mg/day
1.7 mIU/mL
Some SDs (±) are missing in the table above because not all were provided in the original paper. The SDs that were included above were actually reported in the paper for Study 3.
Some noteworthy translated excerpts from the paper:
This study shows that the clinical effects were more dramatic in the DES group, with a sharper drop in the free plasma testosterone level, than in the E2 group.
E2 has been administered too weakly because it is too low in the commercially used ointment and because application to the thick abdominal skin has a lower absorption coefficient than a thinner skin such as forearms for example. It is possible that a dose equivalent to 15 or 20 mg of gel, applied on the forearms, may allow to lower the testosterone below [100 ng/dL]. The marketing of a more concentrated ointment is therefore desirable, as well as the use of a more efficient absorption site.
The search for new estrogens and new routes of administration is therefore necessary: E2, a natural female hormone, induces no thromboembolic events. It seems to us, therefore, that a larger dose must be proposed to be as effective as 3 mg DES. A gel twice more concentrated is therefore currently under study.
The double-concentration estradiol gel was subsequently reported in Study 3 (see below).
It should be noted that the transdermal estradiol formulation in this study is different from that used in Study 1. Specifically, “6 mg/day estradiol gel” was stated to deliver “600 μg/day estradiol” in Study 1, whereas “10 mg/day estradiol ointment” was stated to deliver “600 μg/day estradiol” in Study 2. These apparent discrepancies might be of relevance to the unexpectedly lower estradiol levels and testosterone suppression in Study 2 compared to Study 1.
Study 3: Estradiol Ointment 20 mg/day versus Bilateral Orchiectomy
The following paper pertains to the third study:
Steg, A., & Benoit, G. (1983). Étude comparative de fortes doses d’oestradiol-17 β administrées par voie per-cutanée et de l’orchidectomie bilatérale dans le traitement du cancer de la prostate. [Prostatic carcinoma. Bilateral orchiectomy versus percutaneous administration of large doses of 17 β-estradiol. A comparative study. / Comparative study of percutaneous administration of large doses of 17β œstradiol and bilateral orchidectomy in prostatic carcinoma.] Annales d’Urologie, 17(5), 286–288. [Google Scholar] [Pascal and Francis] [PDF]
In the study, 38 men with prostate cancer were divided into two groups and treated for 1 month with:
20 mg/day (“1200 μg/day”) estradiol in an ointment applied to the skin in four daily divided doses
Bilateral orchiectomy (i.e., surgical removal of the testes)
Hormone levels in the study were as follows:
Condition
Estradiol levels
Testosterone levels
LH levels
Pre-treatment (E2 group)
52 ± 30 pg/mL
460 ± 230 ng/dL
2.6 ± 0.9 mIU/mL
E2 ointment 20 mg/day
473 ± 375 pg/mL
260 ± 160 ng/dL
1.7 ± 0.8 mIU/mL
Pre-treatment (orchi group)
Not reported
500 ± 160 ng/dL
5.5 ± 6 mIU/mL
Bilateral orchiectomy
Not reported
20 ± 10 ng/dL
19 ± 8 mIU/mL
Some noteworthy translated excerpts from the paper:
The purpose of this study was to determine whether hormonal efficacy [in the treatment of prostate cancer] was enhanced by the administration of large doses of E2. […] The study shows that, whereas orchiectomy lowers plasma testosterone levels dramatically, E2, at [the double dose used in this study compared to our previous study] (1200 [μg] versus 600 [μg]), surprisingly does not further lower it at all, but plasma E2 is substantially increased.
We have found that the administration of E2 causes significant biological changes; E2 is well-absorbed, testosterone is lowered, and LH and FSH are also lowered. But when comparing these effects with those obtained by castration, and with those also observed after administration of 3 mg of diethylstilbestrol (in our previous study), the results are much less brilliant. Indeed: the plasma testosterone is moderately lowered by E2 whereas the fall is much more pronounced with DES and is spectacular with castration; pituitary suppression is comparable with E2 and DES and of course orchiectomy increases LH; and if we compare the results obtained before and after treatment, it is clear that if before treatment the figures are comparable, after treatment the fall is pronounced only with DES and especially with orchiectomy.
The objective of this study was to determine whether doubling the dose of E2 enhances its antiandrogenic effect. [With both a lower dose and a higher dose], E2 is very well-absorbed. [With both doses], the pituitary suppression is identical, but on the other hand, not only is plasma testosterone not lower with [the higher dose], but it seems to be even [higher than with the lower dose]. This difference is also found clinically since with [the higher dose], 10% of patients were improved while with [the lower dose], 30% improvement was obtained.
How can this result be explained? Perhaps this is due to the contrary effects of E2 on androgens. Indeed: E2 on the one hand suppresses pituitary activity, which leads to a fall of testicular testosterone secretion, and at the same time E2 decreases 5α-reductase activity in the prostate; on the other hand, E2 causes an increase in SHBG and competes with free testosterone binding to SHBG and in this way increases the proportion of active free testosterone. In addition, testosterone also increases the level of cytoplasmic androgen receptors.
This work shows that E2 is well-absorbed percutaneously and causes practically no cardiovascular events. On the other hand, it appears that the antiandrogenic effect is significantly less by this route than that obtained by diethylstilbestrol and castration, and that in any case, we cannot improve the efficacy by increasing the dose of E2.
The finding that the 20 mg/day estradiol ointment resulted in less suppression of testosterone levels than the 10 mg/day estradiol ointment despite markedly higher estradiol levels is surprising. However, the researchers’ theoretical explanation of the result seems questionable. Abundant clinical research with high-dose estradiol for prostate cancer (e.g., transdermal patches, injectable polyestradiol phosphate injections, injectable estradiol undecylate) has shown stronger testosterone suppression with high estradiol levels in this range or below and seems to thoroughly contradict their results. Studies have consistently shown that estradiol levels of 200 to 500 pg/mL and above suppress testosterone levels by 90% and greater (to ~50 ng/dL and below) (Wiki; Graphs). Instead of the authors’ interpretation of their unexpected results, it seems possible that the testosterone assays for this study may have been inaccurate or perhaps some other methodological problem may have been responsible.
The concentration and dosage of estradiol ointment in this study were double those of Study 2, yet the estradiol levels measured in this study were almost 5-fold higher than those in Study 2. It’s possible that the higher concentration of ointment used may have resulted in disproportionately greater absorption, as one study found that the smaller the application area of estradiol gel (and hence higher the post-application concentration), the greater the resulting estradiol levels (Graph). On the other hand, it’s possible that the estradiol assays were inaccurate (as also suggested for testosterone above). Lastly, in contrast to the previous two papers, the authors don’t state where the ointment was applied in this study. It was probably the abdomen similarly, but this isn’t certain; they may have used one or more other locations with differing skin permeability (as they discussed the possibility of doing in Study 1). It’d be expected that they’d mention this change though, which perhaps makes the possibility unlikely.
Review Article
A review of hormone therapy for prostate cancer was also published by one of the researchers:
Benoit, G. (1985). Que Penser du Traitement Hormonal du Cancer de la Prostate / Hormonothérapie du Cancer de la Prostate. [Thoughts on the Hormonal Treatment of Prostate Cancer / Hormone Therapy of Prostate Cancer.] Gazette Médicale, 92(5), 33–39. [Google Scholar] [PDF] [Translation]
In the paper, she briefly mentioned her and her colleagues’ studies on high-dose transdermal estradiol gel/ointment for prostate cancer and provided some additional information pertaining to these studies. Here are the relevant translated excerpts from the paper:
In a study in Cochin (16), the efficacy of diethylstilbestrol was 50% of objective responses, but at a cost of 23% of thromboembolic events. These thromboembolic events are similar to the accidents that occur in women taking oral estrogen–progestogens. These are oral synthetic estrogens. In a series of diethylstilbestrol-treated prostate cancers, Steg (16) has shown that this treatment induces an increase in triglycerides and a decrease in coagulation factor VIII. Abbou (1) found, on his behalf, an increase in circulating immune complexes in patients with thrombosis under synthetic estrogen–progestogen therapy.
It therefore seems to us no longer possible to prescribe diethylstilbestrol as first intention. To reduce these cardiovascular events, most authors have tried to use natural estrogens administered intramuscularly, subcutaneously, or percutaneously. Cochin’s experience of using a natural estrogen (17-beta-estradiol) percutaneously has shown that this treatment may be effective, but that it is very difficult to administer: one in two patients is unable to apply the treatment correctly and regularly, which greatly reduces the use of such treatment (15).
As an indication, we report several studies made successively in Cochin (16, 17) with diethylstilbestrol, a natural estrogen (17-beta-estradiol), testicular pulpectomy, and an LHRH analogue administered subcutaneously.
The results confirm that diethylstilbestrol causes thromboembolic events. These different estrogenic treatments promote gynecomastia. Medical castration by LHRH analogues or surgical pulpectomy causes hot flushes. All these treatments reach, in a variable way, libido and sexual potency. This last effect is complex: it is known that castration, in young men, after puberty, does not regularly cause impotence, as shown by the history of castrates. However, it is necessary to warn the patient that each of these treatments can reach his manhood.
Synthetic estrogens, especially diethylstilbestrol, should no longer be used as first-line agents in the treatment of metastatic prostate cancer because of their thromboembolic risk. Natural estrogens do not seem efficient enough to be used regularly. Complete castration performs excessive mutilation. In our opinion, only testicular pulpectomy, LHRH analogues, and antiandrogens remain in competition.
The review also contains the following adapted summary table of data from their studies:
DES
E2 gel/ointment
Orchiectomy
GnRH analogue
Testosterone levels
50 ng/dL
100 ng/dL
20 ng/dL
50 ng/dL
50% partial remission
50%
30%
33%
41%
Cardiovascular events
23%
0%
0%
0%
The gonadotropin-releasing hormone (GnRH) analogue data were from other studies and papers by the same researchers (Steg et al., 1984; Steg et al., 1985a; Steg et al., 1985b). These studies did not assess transdermal estradiol and hence are not otherwise discussed in the present article.
The authors’ comments in this paper raise the additional question of whether inconsistent and suboptimal exposure to estradiol gel/ointment may have influenced the findings of their studies.
Summary and Discussion
In the reviewed clinical studies of high-dose transdermal estradiol gel/ointment in men with prostate cancer, the following results were obtained in terms of changes in measured hormone levels:
Estradiol dose
Estradiol levels
Testosterone levels
Before treatment
23–52 pg/mL
341–460 ng/dL
3 mg/day gel (“300 μg/day”)
84 pg/mL
188 ng/dL
6 mg/day gel (“600 μg/day”)
185 pg/mL
105 ng/dL
10 mg/day ointment (“600 μg/day”)
107 pg/mL
180 ng/dL
20 mg/day ointment (“1200 μg/day”)
473 pg/mL
260 ng/dL
Unfortunately, the data reported in these studies is not as useful as might have been hoped. This is due to inconsistencies with the transdermal estradiol formulations and doses used, reported issues with patient compliance in terms of consistent and correct administration, inconsistencies with the measured estradiol and testosterone levels, and omission of certain important details (e.g., ointment concentrations, gel/ointment compositions and differences, how the estimated estradiol delivery rates were determined, etc.). As a result of these limitations, there may be only so much that can be taken away from the studies.
In any case, these studies do contribute to an impression that transdermal estradiol gel achieves relatively low estradiol levels and is relatively weak in terms of estrogenic strength even when used at high doses. Other studies of high-dose transdermal estradiol gel/cream, for instance at doses of 3 to 8 mg/day, have reported relatively low estradiol levels similarly, for instance 100 to 200 pg/mL on average (e.g., Lauritzen, 1990; Wiki). Based on these studies, transdermal estradiol gel doesn’t seem to be a very effective or affordable way of achieving the estradiol levels that are needed for adequate suppression of testosterone levels with high-dose estradiol monotherapy in transfeminine people.
In contrast to transdermal estradiol gel, transdermal estradiol patches can consistently achieve much higher estradiol levels and testosterone suppression in comparison (Wiki). About 50 to 100 pg/mL estradiol per 100 μg/day estradiol patch appears to be achieved on average, and multiple 100 μg/day patches up to as high as eight at a time appear to result in approximately linear increases in estradiol levels (Aly, 2020; Wiki). The consistent and higher estradiol levels with estradiol patches makes them generally a more favorable form of estradiol for transdermal administration than estradiol gel and related formulations. Additionally, genital application of transdermal estradiol gel and other transdermal estradiol formulations can allow for dramatically higher estradiol levels (e.g., 5- to 8-fold) than application to conventional transdermal skin sites (e.g., forearm, abdomen) (Aly, 2019). This may also provide transdermal estradiol formulations with greater potential for high-dose estradiol monotherapy.
References
Aly. (2019). Genital Application via the Scrotum and Neolabia for Greatly Enhanced Absorption of Transdermal Estradiol in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Benoit, G. (1985). Que Penser du Traitement Hormonal du Cancer de la Prostate / Hormonothérapie du Cancer de la Prostate. [Thoughts on the Hormonal Treatment of Prostate Cancer / Hormone Therapy of Prostate Cancer.] Gazette Médicale, 92(5), 33–39. [Google Scholar] [PDF] [Translation]
Lauritzen, C. (1990). Clinical use of oestrogens and progestogens. Maturitas, 12(3), 199–214. [DOI:10.1016/0378-5122(90)90004-P]
Steg, A., Benoit, G., Limouzin-Lamotte, A., Mahoudeau, J., Caillens, M., & Raichvarg, D. (1979). Cancer de la prostate: effets métaboliques des bêta-estradiol par voie percutanée. [Cancer of the prostate: metabolic effect of percutaneous beta-estradiol. / Prostatic carcinoma: metabolic effect of percutaneous beta-estradiol.] La Nouvelle Presse Medicale, 8(46), 3801–3802. [Google Scholar 1] [Google Scholar 2] [Pascal and Francis] [PubMed] [PDF]
Steg, A., Benoit, G., Limouzin-Lamotte, A., Mahoudeau, J., Caillens, M., & Raichvarg, D. (1980). Cancer de la prostate: Effets métaboliques du bêta-estradiol par voie percutanée. [Cancer of the prostate: metabolic effects of percutaneously administered beta-estradiol.] Revue Médicale de la Suisse Romande, 100(11), 895–897. [Google Scholar 1] [Google Scholar 2] [Pascal and Francis] [PubMed] [PDF]
Steg, A., Benoit, G., Maisonneuve, P., Tallet, F., Nahoul, K., Sultan, Y., Raichwarg, D., & Limouzin-Lamotte, M. A. (1983). Étude comparative du diéthylstilboestrol et du 17 bêta-oestradiol per-cutané dans le traitement du cancer de la prostate. [A comparative study of percutaneous 17 beta-estradiol and diethylstilbestrol in the treatment of prostatic cancer.] Annales d’Urologie, 17(4), 197–202. [Google Scholar] [PDF]
Steg, A., & Benoit, G. (1983). Étude comparative de fortes doses d’oestradiol-17 β administrées par voie per-cutanée et de l’orchidectomie bilatérale dans le traitement du cancer de la prostate. [Prostatic carcinoma. Bilateral orchiectomy versus percutaneous administration of large doses of 17 β-estradiol. A comparative study. / Comparative study of percutaneous administration of large doses of 17β œstradiol and bilateral orchidectomy in prostatic carcinoma.] Annales d’Urologie, 17(5), 286–288. [Google Scholar] [Pascal and Francis] [PDF]
Steg, A., Chiche, R., Boccon-Gibod, L., Debré, B., Duchier, J., & Schally, A. V. (1984). Traitement du cancer de la prostate par un agoniste de la LH-RH: le D Trp 6 LH-RH: Résultats préliminaires à propos de trente observations. [Treatment of prostatic cancer with an LH-RH agonist: the D Trp6 LH-RH. Preliminary results in 30 cases. / Treatment of prostate cancer with D Trp6 LH-RH, an LH-RH agonist. Preliminary results of thirty cases.] Annales d’Urologie, 18(6), 388–392. [Google Scholar 1] [Google Scholar 2] [PubMed]
Steg, A., Chiche, R., Boccon-Gibod, L., & Debre, B. (1985). Traitement du cancer de la prostate évolué par un agoniste de la gonadoréline, le DTrp6 LH-RH. Quarante et une observations. [Treatment of advanced prostatic cancer with a gonadorelin agonist, dTrp6 LHRH. 41 cases.] La Presse Médicale (Paris, France: 1983), 14(40), 2045–2048. [Google Scholar 1] [Google Scholar 2] [PubMed]
Steg, A., Chiche, R., Boccon-Gibod, L., Debre, B., & Duchier, J. (1985). Traitement du cancer de la prostate par un agoniste de la LH-RH: le D Trp6 LH-RH―résultats préliminaires à propos de trente observations. [Treatment of prostate cancer with D-TRP6 LH-RH, an LH-RH agonist—preliminary results in 30 cases.] La Semaine des Hôpitaux de Paris, 61(10), 615–618. [Google Scholar 1] [Google Scholar 2]
\ No newline at end of file
diff --git a/transfemscience.org/articles/hormone-levels-female-puberty/index.html b/transfemscience.org/articles/hormone-levels-female-puberty/index.html
index 97d74152..f618f717 100644
--- a/transfemscience.org/articles/hormone-levels-female-puberty/index.html
+++ b/transfemscience.org/articles/hormone-levels-female-puberty/index.html
@@ -1 +1 @@
-Hormone Levels During Normal Puberty in Cisgender Girls - Transfeminine Science
Hormone Levels During Normal Puberty in Cisgender Girls
By Aly | First published April 29, 2020 | Last modified April 7, 2023
Preface
This is a collection of published data on hormone levels throughout normal puberty in cisgender girls. Levels of estrogens (estradiol and estrone), progesterone, and androgens are included. Data are limited to those determined with mass spectrometry-based analytic techniques wherever available.
Estrogen Levels
Esoterix/LabCorp (2020)
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF]:
Table: Estradiol and estrone levels in cisgender girls and women (HPLC–MS/MS):
Estradiol
Estrone
Life stage
Age (years)
Mean (pg/mL)
Range (pg/mL)
Mean (pg/mL)
Range (pg/mL)
Tanner stage 1
<9.2
8.0
5.0–20
13
4.0–29
Tanner stage 2
9.2–13.7
16
10–24
21
10–33
Tanner stage 3
10.0–14.4
25
7.0–60
30
15–43
Tanner stage 4
10.7–15.6
47
21–85
36
16–77
Tanner stage 5
11.8–18.6
110
34–170
61
29–105
Adult follicular
>18.0
?
30–100
?
30–100
Adult luteal
>18.0
?
70–300
?
90–160
Figure: Mean (± ref. range) estradiol and estrone levels in normal female puberty (HPLC–MS/MS):
Frederiksen et al. (2020)
Frederiksen, H., Johannsen, T. H., Andersen, S. E., Albrethsen, J., Landersoe, S. K., Petersen, J. H., Andersen, A. N., Vestergaard, E. T., Schorring, M. E., Linneberg, A., Main, K. M., Andersson, A. M., & Juul, A. (2020). Sex-specific estrogen levels and reference intervals from infancy to late adulthood determined by LC-MS/MS. The Journal of Clinical Endocrinology & Metabolism, 105(3), 754–768. [DOI:10.1210/clinem/dgz196]:
(The graphs below were created from analysis of this study by Sam.)
Figure: Mean (± SD) estradiol and estrone levels in normal female puberty (LC–MS/MS):
Figure: Median (± SD) estradiol and estrone levels in normal female puberty (LC–MS/MS):
The lower estradiol and estrone levels in TS5 relative to TS4 are likely due to sampling error.
Kushnir et al. (2008)
Kushnir, M. M., Rockwood, A. L., Bergquist, J., Varshavsky, M., Roberts, W. L., Yue, B., Bunker, A. M., & Meikle, A. W. (2008). High-sensitivity tandem mass spectrometry assay for serum estrone and estradiol. American Journal of Clinical Pathology, 129(4), 530–539. [DOI:10.1309/LC03BHQ5XJPJYEKG]:
Table: Estradiol and estrone levels in cisgender girls and women (LC–MS/MS):
Estradiol
Estrone
Life stage or age (years)
Ref. range (pg/mL)
Ref. range (pg/mL)
Tanner stage 1
<55
<26
Tanner stage 2
2–133
1–39
Tanner stage 3
12–277
8–117
Tanner stage 4/5
2–259
4–109
Before menarche
1–84
<41
After menarche
3–264
4–113
7–9 years
<35
<25
10–12 years
<87
<42
13–15 years
9–248
8–105
16–17 years
2–266
4–133
Postmenopausal (41–63 years)
2–21
3–32
Figure: Median estradiol and estrone levels around puberty in cisgender girls (LC–MS/MS):
Janfaza, M., Sherman, T. I., Larmore, K. A., Brown-Dawson, J., & Klein, K. O. (2006). Estradiol levels and secretory dynamics in normal girls and boys as determined by an ultrasensitive bioassay: a 10 year experience. Journal of Pediatric Endocrinology and Metabolism, 19(7), 901–910. [DOI:10.1515/JPEM.2006.19.7.901]:
Madsen, A., Bruserud, I. S., Bertelsen, B. E., Roelants, M., Oehme, N. H. B., Viste, K., Bjerknes, R., Almås, B., Rosendahl, K., Mellgren, G., Sagen, J. V., & Juliusson, P. B. (2020). Hormone references for ultrasound breast staging and endocrine profiling to detect female onset of puberty. The Journal of Clinical Endocrinology & Metabolism, 105(12), e4886–e4895. [DOI:10.1210/clinem/dgaa679]:
Table: Estradiol and estrone levels in cisgender girls (LC–MS/MS):
Estradiol (pg/mL)
Estrone (pg/mL)
Tanner stage
n
Median
Range (2.5–97.5%)
Median
Range (2.5–97.5%)
B1
260
1.3
0.24–10
3.8
1.2–10
B2
69
6.5
1.6–56
9.2
3.5–32
B3
63–66
25
1.2–77
21
3.2–52
B4
73–78
48
7.1–219
32
11–108
B5
76–81
41
13–268
36
15–129
Other Studies
Courant, F., Aksglaede, L., Antignac, J. P., Monteau, F., Sorensen, K., Andersson, A. M., Skakkebaek, N. E., Juul, A., & Bizec, B. L. (2010). Assessment of circulating sex steroid levels in prepubertal and pubertal boys and girls by a novel ultrasensitive gas chromatography-tandem mass spectrometry method. The Journal of Clinical Endocrinology & Metabolism, 95(1), 82–92. [DOI:10.1210/jc.2009-1140]
Biro, F. M., Pinney, S. M., Huang, B., Baker, E. R., Walt Chandler, D., & Dorn, L. D. (2014). Hormone changes in peripubertal girls. The Journal of Clinical Endocrinology & Metabolism, 99(10), 3829–3835. [DOI:10.1210/jc.2013-4528]
Ankarberg-Lindgren, C., Dahlgren, J., & Andersson, M. X. (2018). High-sensitivity quantification of serum androstenedione, testosterone, dihydrotestosterone, estrone and estradiol by gas chromatography–tandem mass spectrometry with sex-and puberty-specific reference intervals. The Journal of Steroid Biochemistry and Molecular Biology, 183, 116–124. [DOI:10.1016/j.jsbmb.2018.06.005]
Note: Has estrone sulfate levels in the different Tanner breast stages.
Bae, Y. J., Zeidler, R., Baber, R., Vogel, M., Wirkner, K., Loeffler, M., Ceglarek, U., Kiess, W., Körner, A., Thiery, J., & Kratzsch, J. (2019). Reference intervals of nine steroid hormones over the life-span analyzed by LC-MS/MS: Effect of age, gender, puberty, and oral contraceptives. The Journal of Steroid Biochemistry and Molecular Biology, 193, 105409. [DOI:10.1016/j.jsbmb.2019.105409]
Progesterone Levels
Esoterix/LabCorp (2020)
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF] [Alt] [Alt]:
Table: Progesterone levels in cisgender girls and women (HPLC–MS/MS):
Life stage / age (years)
Range (ng/mL)
<10 years
≤0.26
11 years
≤2.6
12 years
≤8.6
13 years
≤6.9
14 years
≤12.0
15 years
≤10.8
16 years
≤12.9
Adult early follicular (days 1–6)
≤0.17
Adult late follicular (days 7–12)
≤1.4
Adult mid-cycle (days 13–15)
≤15.6
Adult luteal (days 16–28)*
≤25.6
Postmenopausal
≤0.10
Pregnancy first trimester
6.3–45.5
Pregnancy second trimester
15.4–52.1
Pregnancy third trimester
25.0–99.9
Note: Luteal progesterone peaked from 3.5 to 37.5 ng/dL ranging on cycle days 17 to 23.
Progesterone levels in males: ≤0.15 ng/mL (age 1–16 years) and ≤0.11 ng/mL (adult).
Fisher, D. A., Salameh, W., & Furlanetto, R. W. (2007). [The Quest Diagnostics Manual:] Endocrinology: Test Selection and Interpretation, 4th Edition. San Juan Capistrano, California: Quest Diagnostics. [Google Scholar] [Google Books] [WorldCat] [PDF] [Alt PDF] [Alt]:
Table: Progesterone levels in cisgender girls and women (LC–MS/MS):
Life stage / age
Range (ng/mL)
5–9 years
≤0.6
10–13 years
≤10.2
14–17 years
≤11.9
Adult early follicular
≤0.6
Adult late follicular
≤14.5
Adult mid-cycle
≤16.1
Adult luteal
≤31.4
Postmenopausal
≤0.2
Progesterone levels in males: ≤1.2 ng/mL (age 5–17 years) and ≤0.3 ng/mL (adult).
Table: Progesterone levels in cisgender girls and women (RIA):
Life stage
Range (ng/dL)
Tanner stage 1
≤0.33
Tanner stage 2
≤0.55
Tanner stage 3
≤4.5
Tanner stage 4
≤13.0
Tanner stage 5
≤9.5
Adult follicular
0.15–0.70
Adult luteal
2.0–25.0
Adult postmenopausal
≤0.40
Pregnancy first trimester
10.3–44.0
Pregnancy second trimester
19.5–82.5
Pregnancy third trimester
65.0–229.0
Progesterone levels in males: <0.10–1.1 ng/mL (age 1–10 years + puberty) and 0.13–0.97 ng/mL (adult).
Note: RIA gives less accurate and often overestimated values relative to MS-based techniques.
Kühnel (2000)
Kühnel, W. (2000). IMMULITE® and IMMULITE® 2000 Reference Range Compendium, First English Edition. Los Angeles, California: Diagnostic Products Corporation. [Google Scholar] [URL] [PDF 1] [PDF 2]:
Table: Progesterone levels in cisgender girls and women (IA):
Life stage / age (years)
n
Median (ng/mL)
Range (variable) (ng/mL)
7–8 years
24
0.50
0.25–0.99
9–10 years
40
0.55
0.13–1.00
11 years
22
0.66
0.37–0.92
12 years
17
0.78
0.49–1.68
13 years
25
0.72
0.40–1.46
14 years
30
1.20
0.56–12.3
15 years
48
1.40
0.70–13.4
16 years
40
5.0
0.65–14.5
17 years
30
1.40
0.77–11.3
18–19 years
12
2.4
1.27–13.0
Tanner stage 1
28
0.46
0.23–0.77
Tanner stage 2–3
70
0.70
0.36–2.2
Tanner stage 4
30
1.25
0.57–9.5
Tanner stage 5
66
2.4
0.75–14.6
Adult follicular
382
0.47
ND–1.13
Adult mid-follicular (days 5–11)
186
0.43
ND–0.98
Adult mid-cycle
27
1.06
0.48–1.72
Adult luteal
323
8.9
0.95–21
Adult mid-luteal (days 7–8)
54
13.1
6.0–24
Pregnancy first trimester
28
22.2
9.3–33.2
Pregnancy second trimester
10
35.4
29.5–50.0
Pregnancy third trimester
8
102
83.1–160
Abbreviations/etc.: n = tests, not subjects; range = 90%, 95%, or absolute range; ND = not detectable.
Note: IA gives less accurate and often overestimated values relative to MS-based techniques.
Other Studies
Ceglarek, U., Werner, M., Kortz, L., Körner, A., Kiess, W., Thiery, J., & Kratzsch, J. (2010). Preclinical challenges in steroid analysis of human samples. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 505–512. [DOI:10.1016/j.jsbmb.2010.03.039]
Androgen Levels
Esoterix/LabCorp (2020)
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF]:
Table: Total testosterone levels in cisgender girls and women (HPLC–MS/MS):
Life stage
Age (years)
Mean (ng/dL)
Range (ng/dL)
Tanner stage 1
<9.2
4.9
<2.5–10
Tanner stage 2
9.2–13.7
18
7–28
Tanner stage 3
10.0–14.4
25
15–35
Tanner stage 4
10.7–15.6
22
13–32
Tanner stage 5
11.8–18.6
28
20–38
Adult premenopausal
>18.0
?
10–55
Adult postmenopausal
⪆50 years
?
7–40
Table: Dihydrotesterone (DHT) levels in cisgender girls and women (HPLC–MS/MS):
Life stage
Age (years)
Mean (ng/dL)
Range (ng/dL)
Tanner stage 1
<9.2
?
<3
Tanner stage 2
9.2–13.7
8
5–12
Tanner stage 3
10.0–14.4
12
7–19
Tanner stage 4
10.7–15.6
7
4–13
Tanner stage 5
11.8–18.6
9
3–18
Adult
>18.0
?
4–22
Table: Androstenedione levels in cisgender girls and women (HPLC–MS/MS):
Life stage
Age (years)
Mean (ng/dL)
Range (ng/dL)
Tanner stage 1
<9.2
<10
<10–17
Tanner stage 2
9.2–13.7
33
10–72
Tanner stage 3
10.0–14.4
97
50–170
Tanner stage 4
10.7–15.6
105
47–208
Tanner stage 5
11.8–18.6
137
50–224
Adult premenopausal
18–40 years
?
28–230
Adult postmenopausal
⪆50 years
?
<10–93
Table: Dehydroepiandrosterone sulfate (DHEA-S) levels in cisgender girls/women (HPLC–MS/MS):
Life stage
Age (years)
Mean (ng/dL)
Range (ng/dL)
Tanner stage 1
<9.2
40
19–144
Tanner stage 2
9.2–13.7
72
34–129
Tanner stage 3
10.0–14.4
88
32–226
Tanner stage 4
10.7–15.6
120
58–260
Tanner stage 5
11.8–18.6
148
44–248
Adult premenopausal
18–50 years
?
17–372
Adult postmenopausal
⪆50 years
?
<215
ARUP Laboratories (Undated)
ARUP Laboratories. (Undated). Testosterone, Free and Total, Includes Sex Hormone-Binding Globulin (Adult Females, Children, or Individuals on Testosterone-Suppressing Hormone Therapy). ARUP Laboratories. [URL] [Alt]:
Table: Total and free testosterone levels in cisgender girls and women (LC–MS/MS):
Life stage / age
Total: Range (ng/dL)
Free: Range (pg/mL)
Tanner stage 1
2–17 ng/dL
<2.2
Tanner stage 2
5–40 ng/dL
0.4–4.5
Tanner stage 3
10–63 ng/dL
1.3–7.5
Tanner stage 4
11–62 ng/dL
1.1–15.5
Tanner stage 5
11–62 ng/dL
0.8–9.2
1–6 years
<30 ng/dL
<0.6
7–9 years
1–11 ng/dL
0.6–1.8
10–11 years
3–32 ng/dL
0.1–3.5
12–13 years
6–50 ng/dL
0.9–6.8
14–15 years
6–52 ng/dL
1.2–7.5
16–17 years
9–58 ng/dL
1.2–9.9
18–30 years
9–55 ng/dL
0.8–7.4
31–40 years
9–55 ng/dL
1.3–9.2
41–51 years
9–55 ng/dL
1.1–5.8
Postmenopausal
5–32 ng/dL
0.6–3.8
Other Studies
Kushnir, M. M., Rockwood, A. L., Roberts, W. L., Pattison, E. G., Bunker, A. M., Fitzgerald, R. L., & Meikle, A. W. (2006). Performance characteristics of a novel tandem mass spectrometry assay for serum testosterone. Clinical Chemistry, 52(1), 120–128. [DOI:10.1373/clinchem.2005.052167]
Kushnir, M. M., Blamires, T., Rockwood, A. L., Roberts, W. L., Yue, B., Erdogan, E., Bunker, A. M., & Meikle, A. W. (2010). LC-MS/MS assay for androstenedione, dehydroepiandrosterone and testosterone with pediatric and adult reference intervals. Clinical Chemistry, 56(7), 1138–1147. [DOI:10.1373/clinchem.2010.143222]
Prolactin Levels
Kühnel, W. (2000). IMMULITE® and IMMULITE® 2000 Reference Range Compendium, First English Edition. Los Angeles, California: Diagnostic Products Corporation. [Google Scholar] [URL] [PDF 1] [PDF 2]
References
Ankarberg-Lindgren, C., & Norjavaara, E. (2009). Are Estradiol Results Determined by the Tandem Mass Spectrometry Assay Clinically Useful for Children? American Journal of Clinical Pathology, 131(5), 746–750. [DOI:10.1309/ajcpnanprf50lchn]
Ankarberg-Lindgren, C., & Norjavaara, E. (2009). Estradiol in Pediatric Endocrinology. American Journal of Clinical Pathology, 132(6), 978–980. [DOI:10.1309/ajcpa65ouufasoan]
Ankarberg-Lindgren, C., Dahlgren, J., & Andersson, M. X. (2018). High-sensitivity quantification of serum androstenedione, testosterone, dihydrotestosterone, estrone and estradiol by gas chromatography–tandem mass spectrometry with sex-and puberty-specific reference intervals. The Journal of Steroid Biochemistry and Molecular Biology, 183, 116–124. [DOI:10.1016/j.jsbmb.2018.06.005]
ARUP Laboratories. (Undated). Testosterone, Free and Total, Includes Sex Hormone-Binding Globulin (Adult Females, Children, or Individuals on Testosterone-Suppressing Hormone Therapy). ARUP Laboratories. [URL] [Alt]:
Bae, Y. J., Zeidler, R., Baber, R., Vogel, M., Wirkner, K., Loeffler, M., Ceglarek, U., Kiess, W., Körner, A., Thiery, J., & Kratzsch, J. (2019). Reference intervals of nine steroid hormones over the life-span analyzed by LC-MS/MS: Effect of age, gender, puberty, and oral contraceptives. The Journal of Steroid Biochemistry and Molecular Biology, 193, 105409. [DOI:10.1016/j.jsbmb.2019.105409]
Biro, F. M., Pinney, S. M., Huang, B., Baker, E. R., Walt Chandler, D., & Dorn, L. D. (2014). Hormone changes in peripubertal girls. The Journal of Clinical Endocrinology & Metabolism, 99(10), 3829–3835. [DOI:10.1210/jc.2013-4528]
Ceglarek, U., Werner, M., Kortz, L., Körner, A., Kiess, W., Thiery, J., & Kratzsch, J. (2010). Preclinical challenges in steroid analysis of human samples. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 505–512. [DOI:10.1016/j.jsbmb.2010.03.039]
Courant, F., Aksglaede, L., Antignac, J. P., Monteau, F., Sorensen, K., Andersson, A. M., Skakkebaek, N. E., Juul, A., & Bizec, B. L. (2010). Assessment of circulating sex steroid levels in prepubertal and pubertal boys and girls by a novel ultrasensitive gas chromatography-tandem mass spectrometry method. The Journal of Clinical Endocrinology & Metabolism, 95(1), 82–92. [DOI:10.1210/jc.2009-1140]
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF]
Fisher, D. A., Salameh, W., & Furlanetto, R. W. (2007). [The Quest Diagnostics Manual:] Endocrinology: Test Selection and Interpretation, 4th Edition. San Juan Capistrano, California: Quest Diagnostics. [Google Scholar] [Google Books] [WorldCat] [PDF] [Alt PDF]
Frederiksen, H., Johannsen, T. H., Andersen, S. E., Albrethsen, J., Landersoe, S. K., Petersen, J. H., Andersen, A. N., Vestergaard, E. T., Schorring, M. E., Linneberg, A., Main, K. M., Andersson, A. M., & Juul, A. (2020). Sex-specific estrogen levels and reference intervals from infancy to late adulthood determined by LC-MS/MS. The Journal of Clinical Endocrinology & Metabolism, 105(3), 754–768. [DOI:10.1210/clinem/dgz196]
Janfaza, M., Sherman, T. I., Larmore, K. A., Brown-Dawson, J., & Klein, K. O. (2006). Estradiol levels and secretory dynamics in normal girls and boys as determined by an ultrasensitive bioassay: a 10 year experience. Journal of Pediatric Endocrinology and Metabolism, 19(7), 901–910. [DOI:10.1515/JPEM.2006.19.7.901]
Kushnir, M. M., Rockwood, A. L., Roberts, W. L., Pattison, E. G., Bunker, A. M., Fitzgerald, R. L., & Meikle, A. W. (2006). Performance characteristics of a novel tandem mass spectrometry assay for serum testosterone. Clinical Chemistry, 52(1), 120–128. [DOI:10.1373/clinchem.2005.052167]
Kühnel, W. (2000). IMMULITE® and IMMULITE® 2000 Reference Range Compendium, First English Edition. Los Angeles, California: Diagnostic Products Corporation. [Google Scholar] [URL] [PDF 1] [PDF 2]
Kushnir, M. M., Rockwood, A. L., Bergquist, J., Varshavsky, M., Roberts, W. L., Yue, B., Bunker, A. M., & Meikle, A. W. (2008). High-sensitivity tandem mass spectrometry assay for serum estrone and estradiol. American Journal of Clinical Pathology, 129(4), 530–539. [DOI:10.1309/LC03BHQ5XJPJYEKG]
Kushnir, M. M., Blamires, T., Rockwood, A. L., Roberts, W. L., Yue, B., Erdogan, E., Bunker, A. M., & Meikle, A. W. (2010). LC-MS/MS assay for androstenedione, dehydroepiandrosterone and testosterone with pediatric and adult reference intervals. Clinical Chemistry, 56(7), 1138–1147. [DOI:10.1373/clinchem.2010.143222]
Madsen, A., Bruserud, I. S., Bertelsen, B. E., Roelants, M., Oehme, N. H. B., Viste, K., Bjerknes, R., Almås, B., Rosendahl, K., Mellgren, G., Sagen, J. V., & Juliusson, P. B. (2020). Hormone references for ultrasound breast staging and endocrine profiling to detect female onset of puberty. The Journal of Clinical Endocrinology & Metabolism, 105(12), e4886–e4895. [DOI:10.1210/clinem/dgaa679]
Hormone Levels During Normal Puberty in Cisgender Girls
By Aly | First published April 29, 2020 | Last modified April 7, 2023
Preface
This is a collection of published data on hormone levels throughout normal puberty in cisgender girls. Levels of estrogens (estradiol and estrone), progesterone, and androgens are included. Data are limited to those determined with mass spectrometry-based analytic techniques wherever available.
Estrogen Levels
Esoterix/LabCorp (2020)
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF]:
Table: Estradiol and estrone levels in cisgender girls and women (HPLC–MS/MS):
Estradiol
Estrone
Life stage
Age (years)
Mean (pg/mL)
Range (pg/mL)
Mean (pg/mL)
Range (pg/mL)
Tanner stage 1
<9.2
8.0
5.0–20
13
4.0–29
Tanner stage 2
9.2–13.7
16
10–24
21
10–33
Tanner stage 3
10.0–14.4
25
7.0–60
30
15–43
Tanner stage 4
10.7–15.6
47
21–85
36
16–77
Tanner stage 5
11.8–18.6
110
34–170
61
29–105
Adult follicular
>18.0
?
30–100
?
30–100
Adult luteal
>18.0
?
70–300
?
90–160
Figure: Mean (± ref. range) estradiol and estrone levels in normal female puberty (HPLC–MS/MS):
Frederiksen et al. (2020)
Frederiksen, H., Johannsen, T. H., Andersen, S. E., Albrethsen, J., Landersoe, S. K., Petersen, J. H., Andersen, A. N., Vestergaard, E. T., Schorring, M. E., Linneberg, A., Main, K. M., Andersson, A. M., & Juul, A. (2020). Sex-specific estrogen levels and reference intervals from infancy to late adulthood determined by LC-MS/MS. The Journal of Clinical Endocrinology & Metabolism, 105(3), 754–768. [DOI:10.1210/clinem/dgz196]:
(The graphs below were created from analysis of this study by Sam.)
Figure: Mean (± SD) estradiol and estrone levels in normal female puberty (LC–MS/MS):
Figure: Median (± SD) estradiol and estrone levels in normal female puberty (LC–MS/MS):
The lower estradiol and estrone levels in TS5 relative to TS4 are likely due to sampling error.
Kushnir et al. (2008)
Kushnir, M. M., Rockwood, A. L., Bergquist, J., Varshavsky, M., Roberts, W. L., Yue, B., Bunker, A. M., & Meikle, A. W. (2008). High-sensitivity tandem mass spectrometry assay for serum estrone and estradiol. American Journal of Clinical Pathology, 129(4), 530–539. [DOI:10.1309/LC03BHQ5XJPJYEKG]:
Table: Estradiol and estrone levels in cisgender girls and women (LC–MS/MS):
Estradiol
Estrone
Life stage or age (years)
Ref. range (pg/mL)
Ref. range (pg/mL)
Tanner stage 1
<55
<26
Tanner stage 2
2–133
1–39
Tanner stage 3
12–277
8–117
Tanner stage 4/5
2–259
4–109
Before menarche
1–84
<41
After menarche
3–264
4–113
7–9 years
<35
<25
10–12 years
<87
<42
13–15 years
9–248
8–105
16–17 years
2–266
4–133
Postmenopausal (41–63 years)
2–21
3–32
Figure: Median estradiol and estrone levels around puberty in cisgender girls (LC–MS/MS):
Janfaza, M., Sherman, T. I., Larmore, K. A., Brown-Dawson, J., & Klein, K. O. (2006). Estradiol levels and secretory dynamics in normal girls and boys as determined by an ultrasensitive bioassay: a 10 year experience. Journal of Pediatric Endocrinology and Metabolism, 19(7), 901–910. [DOI:10.1515/JPEM.2006.19.7.901]:
Madsen, A., Bruserud, I. S., Bertelsen, B. E., Roelants, M., Oehme, N. H. B., Viste, K., Bjerknes, R., Almås, B., Rosendahl, K., Mellgren, G., Sagen, J. V., & Juliusson, P. B. (2020). Hormone references for ultrasound breast staging and endocrine profiling to detect female onset of puberty. The Journal of Clinical Endocrinology & Metabolism, 105(12), e4886–e4895. [DOI:10.1210/clinem/dgaa679]:
Table: Estradiol and estrone levels in cisgender girls (LC–MS/MS):
Estradiol (pg/mL)
Estrone (pg/mL)
Tanner stage
n
Median
Range (2.5–97.5%)
Median
Range (2.5–97.5%)
B1
260
1.3
0.24–10
3.8
1.2–10
B2
69
6.5
1.6–56
9.2
3.5–32
B3
63–66
25
1.2–77
21
3.2–52
B4
73–78
48
7.1–219
32
11–108
B5
76–81
41
13–268
36
15–129
Other Studies
Courant, F., Aksglaede, L., Antignac, J. P., Monteau, F., Sorensen, K., Andersson, A. M., Skakkebaek, N. E., Juul, A., & Bizec, B. L. (2010). Assessment of circulating sex steroid levels in prepubertal and pubertal boys and girls by a novel ultrasensitive gas chromatography-tandem mass spectrometry method. The Journal of Clinical Endocrinology & Metabolism, 95(1), 82–92. [DOI:10.1210/jc.2009-1140]
Biro, F. M., Pinney, S. M., Huang, B., Baker, E. R., Walt Chandler, D., & Dorn, L. D. (2014). Hormone changes in peripubertal girls. The Journal of Clinical Endocrinology & Metabolism, 99(10), 3829–3835. [DOI:10.1210/jc.2013-4528]
Ankarberg-Lindgren, C., Dahlgren, J., & Andersson, M. X. (2018). High-sensitivity quantification of serum androstenedione, testosterone, dihydrotestosterone, estrone and estradiol by gas chromatography–tandem mass spectrometry with sex-and puberty-specific reference intervals. The Journal of Steroid Biochemistry and Molecular Biology, 183, 116–124. [DOI:10.1016/j.jsbmb.2018.06.005]
Note: Has estrone sulfate levels in the different Tanner breast stages.
Bae, Y. J., Zeidler, R., Baber, R., Vogel, M., Wirkner, K., Loeffler, M., Ceglarek, U., Kiess, W., Körner, A., Thiery, J., & Kratzsch, J. (2019). Reference intervals of nine steroid hormones over the life-span analyzed by LC-MS/MS: Effect of age, gender, puberty, and oral contraceptives. The Journal of Steroid Biochemistry and Molecular Biology, 193, 105409. [DOI:10.1016/j.jsbmb.2019.105409]
Progesterone Levels
Esoterix/LabCorp (2020)
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF] [Alt] [Alt]:
Table: Progesterone levels in cisgender girls and women (HPLC–MS/MS):
Life stage / age (years)
Range (ng/mL)
<10 years
≤0.26
11 years
≤2.6
12 years
≤8.6
13 years
≤6.9
14 years
≤12.0
15 years
≤10.8
16 years
≤12.9
Adult early follicular (days 1–6)
≤0.17
Adult late follicular (days 7–12)
≤1.4
Adult mid-cycle (days 13–15)
≤15.6
Adult luteal (days 16–28)*
≤25.6
Postmenopausal
≤0.10
Pregnancy first trimester
6.3–45.5
Pregnancy second trimester
15.4–52.1
Pregnancy third trimester
25.0–99.9
Note: Luteal progesterone peaked from 3.5 to 37.5 ng/dL ranging on cycle days 17 to 23.
Progesterone levels in males: ≤0.15 ng/mL (age 1–16 years) and ≤0.11 ng/mL (adult).
Fisher, D. A., Salameh, W., & Furlanetto, R. W. (2007). [The Quest Diagnostics Manual:] Endocrinology: Test Selection and Interpretation, 4th Edition. San Juan Capistrano, California: Quest Diagnostics. [Google Scholar] [Google Books] [WorldCat] [PDF] [Alt PDF] [Alt]:
Table: Progesterone levels in cisgender girls and women (LC–MS/MS):
Life stage / age
Range (ng/mL)
5–9 years
≤0.6
10–13 years
≤10.2
14–17 years
≤11.9
Adult early follicular
≤0.6
Adult late follicular
≤14.5
Adult mid-cycle
≤16.1
Adult luteal
≤31.4
Postmenopausal
≤0.2
Progesterone levels in males: ≤1.2 ng/mL (age 5–17 years) and ≤0.3 ng/mL (adult).
Table: Progesterone levels in cisgender girls and women (RIA):
Life stage
Range (ng/dL)
Tanner stage 1
≤0.33
Tanner stage 2
≤0.55
Tanner stage 3
≤4.5
Tanner stage 4
≤13.0
Tanner stage 5
≤9.5
Adult follicular
0.15–0.70
Adult luteal
2.0–25.0
Adult postmenopausal
≤0.40
Pregnancy first trimester
10.3–44.0
Pregnancy second trimester
19.5–82.5
Pregnancy third trimester
65.0–229.0
Progesterone levels in males: <0.10–1.1 ng/mL (age 1–10 years + puberty) and 0.13–0.97 ng/mL (adult).
Note: RIA gives less accurate and often overestimated values relative to MS-based techniques.
Kühnel (2000)
Kühnel, W. (2000). IMMULITE® and IMMULITE® 2000 Reference Range Compendium, First English Edition. Los Angeles, California: Diagnostic Products Corporation. [Google Scholar] [URL] [PDF 1] [PDF 2]:
Table: Progesterone levels in cisgender girls and women (IA):
Life stage / age (years)
n
Median (ng/mL)
Range (variable) (ng/mL)
7–8 years
24
0.50
0.25–0.99
9–10 years
40
0.55
0.13–1.00
11 years
22
0.66
0.37–0.92
12 years
17
0.78
0.49–1.68
13 years
25
0.72
0.40–1.46
14 years
30
1.20
0.56–12.3
15 years
48
1.40
0.70–13.4
16 years
40
5.0
0.65–14.5
17 years
30
1.40
0.77–11.3
18–19 years
12
2.4
1.27–13.0
Tanner stage 1
28
0.46
0.23–0.77
Tanner stage 2–3
70
0.70
0.36–2.2
Tanner stage 4
30
1.25
0.57–9.5
Tanner stage 5
66
2.4
0.75–14.6
Adult follicular
382
0.47
ND–1.13
Adult mid-follicular (days 5–11)
186
0.43
ND–0.98
Adult mid-cycle
27
1.06
0.48–1.72
Adult luteal
323
8.9
0.95–21
Adult mid-luteal (days 7–8)
54
13.1
6.0–24
Pregnancy first trimester
28
22.2
9.3–33.2
Pregnancy second trimester
10
35.4
29.5–50.0
Pregnancy third trimester
8
102
83.1–160
Abbreviations/etc.: n = tests, not subjects; range = 90%, 95%, or absolute range; ND = not detectable.
Note: IA gives less accurate and often overestimated values relative to MS-based techniques.
Other Studies
Ceglarek, U., Werner, M., Kortz, L., Körner, A., Kiess, W., Thiery, J., & Kratzsch, J. (2010). Preclinical challenges in steroid analysis of human samples. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 505–512. [DOI:10.1016/j.jsbmb.2010.03.039]
Androgen Levels
Esoterix/LabCorp (2020)
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF]:
Table: Total testosterone levels in cisgender girls and women (HPLC–MS/MS):
Life stage
Age (years)
Mean (ng/dL)
Range (ng/dL)
Tanner stage 1
<9.2
4.9
<2.5–10
Tanner stage 2
9.2–13.7
18
7–28
Tanner stage 3
10.0–14.4
25
15–35
Tanner stage 4
10.7–15.6
22
13–32
Tanner stage 5
11.8–18.6
28
20–38
Adult premenopausal
>18.0
?
10–55
Adult postmenopausal
⪆50 years
?
7–40
Table: Dihydrotesterone (DHT) levels in cisgender girls and women (HPLC–MS/MS):
Life stage
Age (years)
Mean (ng/dL)
Range (ng/dL)
Tanner stage 1
<9.2
?
<3
Tanner stage 2
9.2–13.7
8
5–12
Tanner stage 3
10.0–14.4
12
7–19
Tanner stage 4
10.7–15.6
7
4–13
Tanner stage 5
11.8–18.6
9
3–18
Adult
>18.0
?
4–22
Table: Androstenedione levels in cisgender girls and women (HPLC–MS/MS):
Life stage
Age (years)
Mean (ng/dL)
Range (ng/dL)
Tanner stage 1
<9.2
<10
<10–17
Tanner stage 2
9.2–13.7
33
10–72
Tanner stage 3
10.0–14.4
97
50–170
Tanner stage 4
10.7–15.6
105
47–208
Tanner stage 5
11.8–18.6
137
50–224
Adult premenopausal
18–40 years
?
28–230
Adult postmenopausal
⪆50 years
?
<10–93
Table: Dehydroepiandrosterone sulfate (DHEA-S) levels in cisgender girls/women (HPLC–MS/MS):
Life stage
Age (years)
Mean (ng/dL)
Range (ng/dL)
Tanner stage 1
<9.2
40
19–144
Tanner stage 2
9.2–13.7
72
34–129
Tanner stage 3
10.0–14.4
88
32–226
Tanner stage 4
10.7–15.6
120
58–260
Tanner stage 5
11.8–18.6
148
44–248
Adult premenopausal
18–50 years
?
17–372
Adult postmenopausal
⪆50 years
?
<215
ARUP Laboratories (Undated)
ARUP Laboratories. (Undated). Testosterone, Free and Total, Includes Sex Hormone-Binding Globulin (Adult Females, Children, or Individuals on Testosterone-Suppressing Hormone Therapy). ARUP Laboratories. [URL] [Alt]:
Table: Total and free testosterone levels in cisgender girls and women (LC–MS/MS):
Life stage / age
Total: Range (ng/dL)
Free: Range (pg/mL)
Tanner stage 1
2–17 ng/dL
<2.2
Tanner stage 2
5–40 ng/dL
0.4–4.5
Tanner stage 3
10–63 ng/dL
1.3–7.5
Tanner stage 4
11–62 ng/dL
1.1–15.5
Tanner stage 5
11–62 ng/dL
0.8–9.2
1–6 years
<30 ng/dL
<0.6
7–9 years
1–11 ng/dL
0.6–1.8
10–11 years
3–32 ng/dL
0.1–3.5
12–13 years
6–50 ng/dL
0.9–6.8
14–15 years
6–52 ng/dL
1.2–7.5
16–17 years
9–58 ng/dL
1.2–9.9
18–30 years
9–55 ng/dL
0.8–7.4
31–40 years
9–55 ng/dL
1.3–9.2
41–51 years
9–55 ng/dL
1.1–5.8
Postmenopausal
5–32 ng/dL
0.6–3.8
Other Studies
Kushnir, M. M., Rockwood, A. L., Roberts, W. L., Pattison, E. G., Bunker, A. M., Fitzgerald, R. L., & Meikle, A. W. (2006). Performance characteristics of a novel tandem mass spectrometry assay for serum testosterone. Clinical Chemistry, 52(1), 120–128. [DOI:10.1373/clinchem.2005.052167]
Kushnir, M. M., Blamires, T., Rockwood, A. L., Roberts, W. L., Yue, B., Erdogan, E., Bunker, A. M., & Meikle, A. W. (2010). LC-MS/MS assay for androstenedione, dehydroepiandrosterone and testosterone with pediatric and adult reference intervals. Clinical Chemistry, 56(7), 1138–1147. [DOI:10.1373/clinchem.2010.143222]
Prolactin Levels
Kühnel, W. (2000). IMMULITE® and IMMULITE® 2000 Reference Range Compendium, First English Edition. Los Angeles, California: Diagnostic Products Corporation. [Google Scholar] [URL] [PDF 1] [PDF 2]
References
Ankarberg-Lindgren, C., & Norjavaara, E. (2009). Are Estradiol Results Determined by the Tandem Mass Spectrometry Assay Clinically Useful for Children? American Journal of Clinical Pathology, 131(5), 746–750. [DOI:10.1309/ajcpnanprf50lchn]
Ankarberg-Lindgren, C., & Norjavaara, E. (2009). Estradiol in Pediatric Endocrinology. American Journal of Clinical Pathology, 132(6), 978–980. [DOI:10.1309/ajcpa65ouufasoan]
Ankarberg-Lindgren, C., Dahlgren, J., & Andersson, M. X. (2018). High-sensitivity quantification of serum androstenedione, testosterone, dihydrotestosterone, estrone and estradiol by gas chromatography–tandem mass spectrometry with sex-and puberty-specific reference intervals. The Journal of Steroid Biochemistry and Molecular Biology, 183, 116–124. [DOI:10.1016/j.jsbmb.2018.06.005]
ARUP Laboratories. (Undated). Testosterone, Free and Total, Includes Sex Hormone-Binding Globulin (Adult Females, Children, or Individuals on Testosterone-Suppressing Hormone Therapy). ARUP Laboratories. [URL] [Alt]:
Bae, Y. J., Zeidler, R., Baber, R., Vogel, M., Wirkner, K., Loeffler, M., Ceglarek, U., Kiess, W., Körner, A., Thiery, J., & Kratzsch, J. (2019). Reference intervals of nine steroid hormones over the life-span analyzed by LC-MS/MS: Effect of age, gender, puberty, and oral contraceptives. The Journal of Steroid Biochemistry and Molecular Biology, 193, 105409. [DOI:10.1016/j.jsbmb.2019.105409]
Biro, F. M., Pinney, S. M., Huang, B., Baker, E. R., Walt Chandler, D., & Dorn, L. D. (2014). Hormone changes in peripubertal girls. The Journal of Clinical Endocrinology & Metabolism, 99(10), 3829–3835. [DOI:10.1210/jc.2013-4528]
Ceglarek, U., Werner, M., Kortz, L., Körner, A., Kiess, W., Thiery, J., & Kratzsch, J. (2010). Preclinical challenges in steroid analysis of human samples. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 505–512. [DOI:10.1016/j.jsbmb.2010.03.039]
Courant, F., Aksglaede, L., Antignac, J. P., Monteau, F., Sorensen, K., Andersson, A. M., Skakkebaek, N. E., Juul, A., & Bizec, B. L. (2010). Assessment of circulating sex steroid levels in prepubertal and pubertal boys and girls by a novel ultrasensitive gas chromatography-tandem mass spectrometry method. The Journal of Clinical Endocrinology & Metabolism, 95(1), 82–92. [DOI:10.1210/jc.2009-1140]
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF]
Fisher, D. A., Salameh, W., & Furlanetto, R. W. (2007). [The Quest Diagnostics Manual:] Endocrinology: Test Selection and Interpretation, 4th Edition. San Juan Capistrano, California: Quest Diagnostics. [Google Scholar] [Google Books] [WorldCat] [PDF] [Alt PDF]
Frederiksen, H., Johannsen, T. H., Andersen, S. E., Albrethsen, J., Landersoe, S. K., Petersen, J. H., Andersen, A. N., Vestergaard, E. T., Schorring, M. E., Linneberg, A., Main, K. M., Andersson, A. M., & Juul, A. (2020). Sex-specific estrogen levels and reference intervals from infancy to late adulthood determined by LC-MS/MS. The Journal of Clinical Endocrinology & Metabolism, 105(3), 754–768. [DOI:10.1210/clinem/dgz196]
Janfaza, M., Sherman, T. I., Larmore, K. A., Brown-Dawson, J., & Klein, K. O. (2006). Estradiol levels and secretory dynamics in normal girls and boys as determined by an ultrasensitive bioassay: a 10 year experience. Journal of Pediatric Endocrinology and Metabolism, 19(7), 901–910. [DOI:10.1515/JPEM.2006.19.7.901]
Kushnir, M. M., Rockwood, A. L., Roberts, W. L., Pattison, E. G., Bunker, A. M., Fitzgerald, R. L., & Meikle, A. W. (2006). Performance characteristics of a novel tandem mass spectrometry assay for serum testosterone. Clinical Chemistry, 52(1), 120–128. [DOI:10.1373/clinchem.2005.052167]
Kühnel, W. (2000). IMMULITE® and IMMULITE® 2000 Reference Range Compendium, First English Edition. Los Angeles, California: Diagnostic Products Corporation. [Google Scholar] [URL] [PDF 1] [PDF 2]
Kushnir, M. M., Rockwood, A. L., Bergquist, J., Varshavsky, M., Roberts, W. L., Yue, B., Bunker, A. M., & Meikle, A. W. (2008). High-sensitivity tandem mass spectrometry assay for serum estrone and estradiol. American Journal of Clinical Pathology, 129(4), 530–539. [DOI:10.1309/LC03BHQ5XJPJYEKG]
Kushnir, M. M., Blamires, T., Rockwood, A. L., Roberts, W. L., Yue, B., Erdogan, E., Bunker, A. M., & Meikle, A. W. (2010). LC-MS/MS assay for androstenedione, dehydroepiandrosterone and testosterone with pediatric and adult reference intervals. Clinical Chemistry, 56(7), 1138–1147. [DOI:10.1373/clinchem.2010.143222]
Madsen, A., Bruserud, I. S., Bertelsen, B. E., Roelants, M., Oehme, N. H. B., Viste, K., Bjerknes, R., Almås, B., Rosendahl, K., Mellgren, G., Sagen, J. V., & Juliusson, P. B. (2020). Hormone references for ultrasound breast staging and endocrine profiling to detect female onset of puberty. The Journal of Clinical Endocrinology & Metabolism, 105(12), e4886–e4895. [DOI:10.1210/clinem/dgaa679]
\ No newline at end of file
diff --git a/transfemscience.org/articles/hormones-fat-metabolism-suppl/index.html b/transfemscience.org/articles/hormones-fat-metabolism-suppl/index.html
index b8fc2e1c..f69dae1c 100644
--- a/transfemscience.org/articles/hormones-fat-metabolism-suppl/index.html
+++ b/transfemscience.org/articles/hormones-fat-metabolism-suppl/index.html
@@ -1 +1 @@
-Sources/Excerpts: On the Impact of Sex Hormones on Fat Metabolism with an Eye Towards Transfeminine Hormone Therapy - Transfeminine Science
Sources/Excerpts: On the Impact of Sex Hormones on Fat Metabolism with an Eye Towards Transfeminine Hormone Therapy
By Lain | First published September 4, 2019 | Last modified October 5, 2020
Preface
These are my raw notes that include a large number of raw quotes from various source materials as well as some thoughts of mine. These are far more detailed than my writeup and again are largely just quotes. This page is a supplement and the main article can be found here.
Pedersen et al. (2004)
Pedersen, S. B., Kristensen, K., Hermann, P. A., Katzenellenbogen, J. A., & Richelsen, B. (2004). Estrogen Controls Lipolysis by Up-Regulating α2A-Adrenergic Receptors Directly in Human Adipose Tissue through the Estrogen Receptor α. Implications for the Female Fat Distribution. The Journal of Clinical Endocrinology & Metabolism, 89(4), 1869–1878. [DOI:10.1210/jc.2003-031327]
Power & Schulkin (1998)
Power, M. L., & Schulkin, J. (2008). Sex differences in fat storage, fat metabolism, and the health risks from obesity: possible evolutionary origins. British Journal of Nutrition, 99(5), 931–940. [DOI:10.1017/s0007114507853347]:
Women have greater adipose stores in thighs and buttocks(8); men tend to be more likely to have significant amounts of abdominal fat, and to be more susceptible to abdominal adiposity(8). Women have larger stores of subcutaneous fat; men are more likely to have visceral fat(11).
Interestingly, not only do men on average have a greater proportion of fat as visceral fat, it would appear that turnover of visceral fat is higher in men compared with women. Men have consistently been shown to have greater rates of both fatty acid release (lipolysis) and fatty acid uptake (lipogenesis) in visceral fat compared with women(6). Adrenergic stimulation increases splanchnic fatty acid release in men but not in women(24). Thus, not only are men more susceptible to excess visceral fat, the effects of visceral fat on health may differ between the sexes as well.
At rest, women shunt more circulating NEFA into re-esterification pathways than do men(32). Women have higher VLDL-TAG production rates than men, but similar circulating concentrations(7). This is further evidence that women have higher rates of re-esterfication and thus reuptake of NEFA into adipose tissue than do men. In the basal condition, women are physiologically adapted to store fat more so than are men.
Women have higher rates of fat uptake into leg fat depots than do men(33). Rates of fatty acid release from abdominal adipose tissue are higher in women than men, but they are lower from gluteal or femoral adipose tissue(6). After feeding, fatty acid uptake is higher in abdominal adipose tissue relative to gluteal or femoral in both men and women. However, in women the majority of fatty acid uptake in abdominal adipose tissue is into subcutaneous fat, while in men a larger proportion goes into visceral fat(6). These findings are consistent with women being more likely to store fat subcutaneously and preferentially in the gluteal and femoral regions compared with men.
Women have higher rates of fat oxidation than men during sustained bouts of increased energy expenditure, such as endurance training. Men are more likely to up regulate glucose and amino acid metabolism during sustained exercise bouts(34,35). The difference is associated with oestrogen. Giving exogenous oestrogen to males decreases carbohydrate and amino acid metabolism during exercise, and increases fat oxidation(36). Thus it would appear that women are more physiologically geared to use fat as a metabolic fuel under conditions of sustained increased demand, while men rely more on glucose and protein metabolism.
Testosterone acts to increase lipolysis, inhibit lipoprotein lipase activity, and decrease TAG accumulation in adipose tissue. Lowering circulating testosterone levels in healthy young men increases total adipose tissue, with the largest percentage increase occurring in subcutaneous adipose tissue; raising circulating testosterone decreases total adipose tissue(37). Oestrogens play multiple roles in the regulation of adipose tissue, in both men and women. Oestradiol has direct effects on adipose tissue, and also acts centrally to affect food intake and energy expenditure. Androgens appear to block proliferation and differentiation of preadipocytes(38). Oestradiol enhances proliferation of preadipocytes from both men and women in vitro(39). The effect was greater in preadipoces from females compared with those from males.
Adipose tissues express both androgen and oestrogen receptors. Visceral fat has higher levels of androgen and oestrogen receptors than does subcutaneous fat, and this is true for both men and women(42). Both the a and b oestrogen receptors are found in adipose tissue(41). In subcutaneous fat, oestradiol acts through the a receptor to up regulate α2A-adrenergic receptors which results in decreased lipolysis. In contrast, oestradiol does not appear to affect the concentration of α2A-adrenergic receptors in adipocytes from visceral fat(41). Subcutaneous adipocytes from premenopausal women have higher α2A-adrenergic receptor density and lower lipolytic activity in response to adrenaline than do subcutaneous adipocytes from men(43).
Adipose tissue serves as an endocrine organ, producing leptin and many other regulatory peptides (Table 1). Adipose tissue is a source of steroids, either stored or metabolically converted from precursors. For example, oestrone is converted to oestradiol and androstendione is converted to testosterone in adipose tissue (Table 1).
Reubinoff et al. (1995)
Reubinoff, B. E., Grubstein, A., Meirow, D., Berry, E., Schenker, J. G., & Brzezinski, A. (1995). Effects of low-dose estrogen oral contraceptives on weight, body composition, and fat distribution in young women. Fertility and Sterility, 63(3), 516–521. [DOI:10.1016/s0015-0282(16)57419-6]:
The levels of the enzyme lipoprotein lipase (LPL) activity in adipocytes serve as a reliable indicator of fat formation and accumulation. Lypolisis, on the other hand, is the enzymatic degradation of lipids within the adipocyte. During their reproductive period of life, women have a higher femoral than abdominal LPL activity (14).
Progesterone stimulates femoral LPL activity, whereas T inhibits it. Estradiol (on a short-term basis) is lipolytic.
Nedungadi & Clegg (2009)
Nedungadi, T. P., & Clegg, D. J. (2009). Sexual Dimorphism in Body Fat Distribution and Risk for Cardiovascular Diseases. Journal of Cardiovascular Translational Research, 2(3), 321–327. [DOI:10.1007/s12265-009-9101-1]:
Lipoprotein lipase (LPL) is the major enzyme involved in the fatty acid uptake and is the key regulator of fat accumulation in various adipose tissues. Higher LPL tissue activity is associated with the development of abdominal visceral obesity. Men have a higher level of LPL activity than premenopausal women.
The lipolytic pathway involves the breakdown of energy stored in the form of triglycerides and is initiated when the energy supply from the metabolic fuels is depleted. There is a higher amount of lipolytic activity in the visceral fat when compared to the subcutaneous fat in premenopausal women as compared to the post menopausal women lacking estrogen [37, 45, 46]. Hence, in women, there is less visceral obesity in comparison to men [34]. Catecholamines trigger lipolysis via α-1, α-2, and α-3 adrenergic receptors, while they inhibit lipolysis via the α-2 adrenergic receptors [35].
In visceral adipose tissue, there is an increase in the expression of androgen receptors in males relative to estrogen receptors. Adipose tissue-specific androgen receptor knockout mice have increased intraadipose tissue estradiol levels, which precedes subcutaneous obesity [60].
Body weight is regulated through coordinated metabolic processes. This includes peripheral signals called “adiposity signals” such as leptin and insulin [59], whose circulating levels are proportional to adipose tissue mass [13, 50]. These adiposity signals interact with the brain to regulate energy homeostasis. There are critical brain regions responsible for mediating these effects, specifically in the hypothalamus [47, 49]. Leptin and insulin have receptors in these critical brain regions and influence release and activity of different neuronal populations known to be involved in body weight regulation.
Leptin is secreted at a higher rate from subcutaneous fat than from visceral fat, thus circulating leptin correlates better with total subcutaneous fat than with total body fat. Insulin secretion is better correlated with visceral fat; thus, its levels better reflect visceral rather than total body adiposity [58]. Male rats are relatively more sensitive to the catabolic action of insulin delivered into the brain [14], whereas female rats are relatively more sensitive to the catabolic action of leptin delivered into the brain [16]. A comparable phenomenon has been reported in a recent study in humans, suggesting that men lose more body weight and body fat and change their waist circumference following intranasal insulin administration when compared to women [61]. Intranasal insulin administration increases insulin concentration of the cerebrospinal fluid and thereby alters brain functions. Therefore, sexually differential sensitivity to the catabolic effects of insulin exists in rodents and humans.
Uniquely, despite having higher circulating leptin levels, females have been demonstrated to be more sensitive to the anorexigenic effects of leptin [15]. Female rats are more responsive than males to the effects of centrally administered leptin to decrease food intake and body weight, and this has been demonstrated to be due to increased leptininduced Stat3 activation in the basal hypothalamus [5, 29]. So, an increase in the Stat3 signaling translates to an increase in the energy expenditure by leptin and also increased SNS regulation.
Among the two major estrogen receptor subtypes, it is believed that ERα mediates the anti-obesity effect in both males and females [41, 43, 55]. Studies with ERα null mice demonstrated obesity and had a massive increase in the adipose tissue [32, 48], whereas ERβ null mice were not obese. Heine et al. reported that male and female mice with a targeted deletion in the ERα subunit have increased adiposity in both sexes.
Clegg et al. (2006)
Clegg, D. J., Brown, L. M., Woods, S. C., & Benoit, S. C. (2006). Gonadal Hormones Determine Sensitivity to Central Leptin and Insulin. Diabetes, 55(4), 978–987. [DOI:10.2337/diabetes.55.04.06.db05-1339]:
These data indicate that estrogen acts within the brain to increase leptin sensitivity, decrease insulin sensitivity, and favor subcutaneous over visceral fat.
Thus, when systemic estrogen is present (intact females and males or OVX females administered subcutaneous estrogen), leptin is catabolic in the brain, whereas when estrogen is low (OVX females and intact males), leptin is relatively ineffective. Conversely, estrogen reduces the sensitivity of the brain to the anorexigenic action of insulin.
An important implication from these findings is that gonadal steroids mediate body fat distribution and interact with the integrated adiposity message conveyed to the brain by leptin and insulin, resulting in differential sensitivity to these signals in males and females.
This implies that the relative amount of androgens and estrogens is a key determinant of the brain’s sensitivity to the catabolic actions of insulin and leptin, with proportionally more estrogen favoring leptin sensitivity and proportionally less estrogen favoring insulin sensitivity. Finally, our data suggest that estrogen’s direct actions in the brain determine body fat distribution.
Note: I would love to see a study of trans women who on GAHT gained or lost weight and correlations with different gene polymorphisms for estrogen receptor alpha and leptin receptors.
Mauvais-Jarvis, Clegg, & Hevener (2013)
Mauvais-Jarvis, F., Clegg, D. J., & Hevener, A. L. (2013). The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews, 34(3), 309–338. [DOI:10.1210/er.2012-1055]:
In postmenopausal women, however, when the ovaries fail to produce E2 and in men—who have naturally low levels of circulating E2—E2 does not function as a circulating hormone; rather, it is synthesized in extragonadal sites such as breast, brain, muscle, bone, and adipose tissue where it acts locally as a paracrine or intracrine factor (8).
EST is a cytosolic enzyme that provides a molecular switch in target cells that inhibits estrogen activity by conjugating a sulfonate group to estrogens, thereby preventing binding to estrogen receptors and enhancing urinary excretion of the hormone.
Early studies of the reproductive actions of estrogens led to the establishment of a paradigm in which classical nuclear ERs acted as ligand-activated transcription factors (11). ER modulation of gene transcription is a highly dynamic process. The ER exists in 2 main forms, ER and ER, each of which has multiple isoforms and exhibit distinct tissue expression patterns and functions (12). The classical “genomic” mechanism of ER action typically occurs within hours, leading to activation or repression of target genes. In this classic signaling pathway, ligandactivated ER dissociates from its chaperone heat-shock protein and binds as a dimer either directly to an estrogen response element (ERE) in target gene promoters or indirectly to activator protein 1 or specificity protein 1 response elements through protein tethering to DNA (13). After binding, these ER dimers interact with cofactors (coactivators or cosuppressors) to regulate gene expression.
Cellular estrogenic action depends on: 1) the ER signaling and sensitivity; 2) the activity of enzymes like aromatase involved in the biosynthesis of E2 from androgenic precursors; and 3) the inactivation of E2 in E2 sulfate (E2-S) by the estrogen sulfotransferase (EST).
Although reproductive functions are mostly mediated via classical nuclear ER acting as ligand-activated transcription factors, a large component of ER actions related to energy metabolism also involves extranuclear ERs, indirectly modulating gene expression or acting independently of nuclear events (18). E2 can activate rapid signals Figure 1. Figure 1. Origin of circulating and tissue estrogens. A, In healthy premenopausal women, estrogen (E2) is produced by the ovaries and functions as a circulating hormone that acts on distant target tissues. Here WAT is represented. B, In postmenopausal women and in men, E2 does not function as a circulating hormone; rather, it is synthesized in extragonadal sites from circulating androgenic precursors such as T, androstenedione (4A), or dehydroepiandrosterone (DHEA). Cellular estrogenic action depends on: 1) the ER signaling and sensitivity; 2) the activity of enzymes like aromatase involved in the biosynthesis of E2 from androgenic precursors; and 3) the inactivation of E2 in E2 sulfate (E2-S) by the estrogen sulfotransferase (EST).
E2 can activate rapid signals that act within seconds or minutes via extranuclear and membrane-associated forms of ERs (19). ER and ER are localized to caveolae where they congregate with other signaling molecules, thereby facilitating interaction and rapid intracellular signaling. These signal proteins include G proteins, growth factor receptors, tyrosine kinases (Src), linker proteins (MNAR), and orphan G protein-coupled receptors. This multiprotein complex provides the necessary interactions for membrane ER to activate growth factor receptors and G proteins. In turn, these E2-induced rapid signals modify protein function via phosphorylation. However, they can also modulate gene expression and thus the production of proteins.
ER deficiency in both male and female mice causes increased body weight and adiposity predominately through reduced energy expenditure and slight increases in food intake.
These effects of E2 are mediated via MC4 receptor because E2 is unable to induce anorexia when the MC4 receptor antagonists Shu 9119 or agouti-related peptide (AgRP) are applied concomitantly with E2 administration in rats (70).
Mice with small hairpin RNA-mediated ERgene silencing as well as transgenic mice in which ER has been selectively deleted from SF1-containing neurons of the VMH, develop reduced sensitivity to E2-induced weight loss, increased visceral fat deposition, and reductions in energy expenditure. All of these results occur without an impact on food intake (60, 78), supporting the notion that ER signaling in VMN neurons plays an important role in regulating physical activity, thermogenesis, and fat distribution.
Collectively, these findings suggest that ER in the brainstem, and specifically the [nucleus tractus solitarius (NTS)], is an additional site mediating the anorexigenic effects of estrogens.
Subcutaneous adipose tissue permits efficient storage of maximal calories per unit volume of tissue.
Estrogens are produced in the adipocytes (via aromatization from androgenic precursors) and increase in proportion to total body adiposity (132, 133).
E2 also suppresses white adipose tissue (WAT) accumulation by decreasing fatty acid and triglyceride synthesis and lipogenesis.
Extensive evidence demonstrates that E2 has direct effects on cultured adipocytes with the overall effect of inhibiting lipogenesis and adipogenesis.
[…] suggesting that the absence of E2 promotes immune cell inflammation. Indeed, circulating levels of proinflammatory cytokines are elevated in women after natural or surgical menopause.
Macrophages are elemental players in innate and adaptive immunity; over the past decade their roles in modulating whole-body metabolism and insulin sensitivity have been topics of increasing interest (295, 296).
[…] EST terminates estrogen activity and represents an important parameter of estrogen output upstream of ER. With regard to metabolism, EST is highly expressed in WAT of male mice, but it is not detectable in WAT of normal cycling female mice (320).
Note: Interesting to see if trans folks on GAHT have different type 2 diabetes probability than normal population.
An obvious explanation for these observations is that EST expression is high in male WAT (320) to protect from excessive estrogen actions. It follows that EST suppression produces WAT estrogen excess leading to inflammation. Consistent with this possibility is the observation that E2 treatment leading to high blood concentrations produces WAT inflammation even in females (193).
Note: Makes you wonder if there is potential for higher WAT inflammation in trans folks – especially whne many have E2 levels higher than folks with estrogen producing genitals.
There are reports suggesting that oral E2 may exacerbate insulin resistance and adipocytokine parameters, worsening cardiovascular risk (329). Transdermal E2, however, has minimal effects on insulin resistance and results in higher adiponectin. This suggests that transdermal E2 may be a preferable treatment compared to oral CEE for obese women with metabolic syndrome.
The authors concluded that in women without diabetes, both oral and transdermal estrogen, with or without progestin, increase lean body mass, reduce abdominal fat, improve insulin resistance, decrease LDL/high-density lipoprotein-cholesterol ratio, and decrease blood pressure.
B. Effect of selective estrogen receptor modulators and aromatase inhibitors on metabolism.
In fact, tamoxifen therapy in a case-control study breast cancer survivor was associated with a 24% increased risk of developing diabetes.
In OVX mice, raloxifene reversed OVX-induced increases in food intake, body weight, fat mass, and hyperleptinemia to an extent similar to that of E2. This suggests that in rodents, raloxifene acts as an ER agonist in hypothalamic neurons and fat.
Sullivan et al (356) looked at the effect of a novel SERM (GSK232802A) on body weight, food intake, physical activity, and metabolic rate in an OVX nonhuman primate model. They observed that GSK232802A produced a 5% decrease in weight and reduced adiposity by suppressing food intake and increasing activity. These results occurred without changes in energy expenditure, suggesting that GSK232802A treatment may counteract postmenopausal obesity (356).
Schiffer et al. (2017)
Schiffer, L., Kempegowda, P., Arlt, W., & O’Reilly, M. W. (2017). MECHANISMS IN ENDOCRINOLOGY: The sexually dimorphic role of androgens in human metabolic disease. European Journal of Endocrinology, 177(3), R125–R143. [DOI:10.1530/eje-17-0124]:
Delineating the specific effects of androgen deprivation therapy in this patient population is clouded by co-administration of relatively large doses of oestrogen. Male-to-female transgender patients on combined estrogen and anti-androgen treatment develop an adverse lipid profile.
Cross-sectional studies analysing age-advanced men, men across different ages and obese vs non-obese men consistently support the association between low T and increased fat mass compared to eugonadal controls (107, 142, 143). BMI negatively correlates with total and free T (142, 144), and waist circumference is negatively associated with total T in men.
T administration in men reduces accumulation of visceral and retroperitoneal fat compared to controls, but not in SC depots; hypogonadal men also have increased visceral fat mass.
Mueller et al. (2011)
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body Composition and Bone Mineral Density in Male-to-Female Transsexuals During Cross-Sex Hormone Therapy Using Gonadotrophin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074]:
When the BMI values were compared, there was a significant increase during cross-sex hormone treatment with oestrogens in the absence of testosterone after 12 months and after 24 months. Moreover, total fat mass increased also during the study period, while total lean mass decreased significantly.
Recently, Lapauw et al. reported that in MtFs treated with female steroid hormones over an average of 8 years, total lean mass was approximately 20% lower whereas total fat mass was about 30% higher in comparison with a male control group (Lapauw et al., 2008). In this study, transsexuals had been treated with either ethinyl oestradiol, oestradiol valerate, or conjugated equine oestrogens combined with cyproterone acetate (Lapauw et al., 2008).
In MtFs treated with ethinyl oestradiol and antiandrogens, a significant increase in all subcutaneous fat depots, with a lesser but proportional and significant increase in the visceral fat depot and a decrease in thigh muscle area, was reported.
With regard to body composition and bone mineral density, there appears to be no difference when GnRH agonists are used for complete androgen deprivation in MtFs. In comparison with commonly used hormone regimens, the complication rates appear to be lower in patients receiving the treatment regimen described here, as reported earlier.
\ No newline at end of file
+Sources/Excerpts: On the Impact of Sex Hormones on Fat Metabolism with an Eye Towards Transfeminine Hormone Therapy - Transfeminine Science
Sources/Excerpts: On the Impact of Sex Hormones on Fat Metabolism with an Eye Towards Transfeminine Hormone Therapy
By Lain | First published September 4, 2019 | Last modified October 5, 2020
Preface
These are my raw notes that include a large number of raw quotes from various source materials as well as some thoughts of mine. These are far more detailed than my writeup and again are largely just quotes. This page is a supplement and the main article can be found here.
Pedersen et al. (2004)
Pedersen, S. B., Kristensen, K., Hermann, P. A., Katzenellenbogen, J. A., & Richelsen, B. (2004). Estrogen Controls Lipolysis by Up-Regulating α2A-Adrenergic Receptors Directly in Human Adipose Tissue through the Estrogen Receptor α. Implications for the Female Fat Distribution. The Journal of Clinical Endocrinology & Metabolism, 89(4), 1869–1878. [DOI:10.1210/jc.2003-031327]
Power & Schulkin (1998)
Power, M. L., & Schulkin, J. (2008). Sex differences in fat storage, fat metabolism, and the health risks from obesity: possible evolutionary origins. British Journal of Nutrition, 99(5), 931–940. [DOI:10.1017/s0007114507853347]:
Women have greater adipose stores in thighs and buttocks(8); men tend to be more likely to have significant amounts of abdominal fat, and to be more susceptible to abdominal adiposity(8). Women have larger stores of subcutaneous fat; men are more likely to have visceral fat(11).
Interestingly, not only do men on average have a greater proportion of fat as visceral fat, it would appear that turnover of visceral fat is higher in men compared with women. Men have consistently been shown to have greater rates of both fatty acid release (lipolysis) and fatty acid uptake (lipogenesis) in visceral fat compared with women(6). Adrenergic stimulation increases splanchnic fatty acid release in men but not in women(24). Thus, not only are men more susceptible to excess visceral fat, the effects of visceral fat on health may differ between the sexes as well.
At rest, women shunt more circulating NEFA into re-esterification pathways than do men(32). Women have higher VLDL-TAG production rates than men, but similar circulating concentrations(7). This is further evidence that women have higher rates of re-esterfication and thus reuptake of NEFA into adipose tissue than do men. In the basal condition, women are physiologically adapted to store fat more so than are men.
Women have higher rates of fat uptake into leg fat depots than do men(33). Rates of fatty acid release from abdominal adipose tissue are higher in women than men, but they are lower from gluteal or femoral adipose tissue(6). After feeding, fatty acid uptake is higher in abdominal adipose tissue relative to gluteal or femoral in both men and women. However, in women the majority of fatty acid uptake in abdominal adipose tissue is into subcutaneous fat, while in men a larger proportion goes into visceral fat(6). These findings are consistent with women being more likely to store fat subcutaneously and preferentially in the gluteal and femoral regions compared with men.
Women have higher rates of fat oxidation than men during sustained bouts of increased energy expenditure, such as endurance training. Men are more likely to up regulate glucose and amino acid metabolism during sustained exercise bouts(34,35). The difference is associated with oestrogen. Giving exogenous oestrogen to males decreases carbohydrate and amino acid metabolism during exercise, and increases fat oxidation(36). Thus it would appear that women are more physiologically geared to use fat as a metabolic fuel under conditions of sustained increased demand, while men rely more on glucose and protein metabolism.
Testosterone acts to increase lipolysis, inhibit lipoprotein lipase activity, and decrease TAG accumulation in adipose tissue. Lowering circulating testosterone levels in healthy young men increases total adipose tissue, with the largest percentage increase occurring in subcutaneous adipose tissue; raising circulating testosterone decreases total adipose tissue(37). Oestrogens play multiple roles in the regulation of adipose tissue, in both men and women. Oestradiol has direct effects on adipose tissue, and also acts centrally to affect food intake and energy expenditure. Androgens appear to block proliferation and differentiation of preadipocytes(38). Oestradiol enhances proliferation of preadipocytes from both men and women in vitro(39). The effect was greater in preadipoces from females compared with those from males.
Adipose tissues express both androgen and oestrogen receptors. Visceral fat has higher levels of androgen and oestrogen receptors than does subcutaneous fat, and this is true for both men and women(42). Both the a and b oestrogen receptors are found in adipose tissue(41). In subcutaneous fat, oestradiol acts through the a receptor to up regulate α2A-adrenergic receptors which results in decreased lipolysis. In contrast, oestradiol does not appear to affect the concentration of α2A-adrenergic receptors in adipocytes from visceral fat(41). Subcutaneous adipocytes from premenopausal women have higher α2A-adrenergic receptor density and lower lipolytic activity in response to adrenaline than do subcutaneous adipocytes from men(43).
Adipose tissue serves as an endocrine organ, producing leptin and many other regulatory peptides (Table 1). Adipose tissue is a source of steroids, either stored or metabolically converted from precursors. For example, oestrone is converted to oestradiol and androstendione is converted to testosterone in adipose tissue (Table 1).
Reubinoff et al. (1995)
Reubinoff, B. E., Grubstein, A., Meirow, D., Berry, E., Schenker, J. G., & Brzezinski, A. (1995). Effects of low-dose estrogen oral contraceptives on weight, body composition, and fat distribution in young women. Fertility and Sterility, 63(3), 516–521. [DOI:10.1016/s0015-0282(16)57419-6]:
The levels of the enzyme lipoprotein lipase (LPL) activity in adipocytes serve as a reliable indicator of fat formation and accumulation. Lypolisis, on the other hand, is the enzymatic degradation of lipids within the adipocyte. During their reproductive period of life, women have a higher femoral than abdominal LPL activity (14).
Progesterone stimulates femoral LPL activity, whereas T inhibits it. Estradiol (on a short-term basis) is lipolytic.
Nedungadi & Clegg (2009)
Nedungadi, T. P., & Clegg, D. J. (2009). Sexual Dimorphism in Body Fat Distribution and Risk for Cardiovascular Diseases. Journal of Cardiovascular Translational Research, 2(3), 321–327. [DOI:10.1007/s12265-009-9101-1]:
Lipoprotein lipase (LPL) is the major enzyme involved in the fatty acid uptake and is the key regulator of fat accumulation in various adipose tissues. Higher LPL tissue activity is associated with the development of abdominal visceral obesity. Men have a higher level of LPL activity than premenopausal women.
The lipolytic pathway involves the breakdown of energy stored in the form of triglycerides and is initiated when the energy supply from the metabolic fuels is depleted. There is a higher amount of lipolytic activity in the visceral fat when compared to the subcutaneous fat in premenopausal women as compared to the post menopausal women lacking estrogen [37, 45, 46]. Hence, in women, there is less visceral obesity in comparison to men [34]. Catecholamines trigger lipolysis via α-1, α-2, and α-3 adrenergic receptors, while they inhibit lipolysis via the α-2 adrenergic receptors [35].
In visceral adipose tissue, there is an increase in the expression of androgen receptors in males relative to estrogen receptors. Adipose tissue-specific androgen receptor knockout mice have increased intraadipose tissue estradiol levels, which precedes subcutaneous obesity [60].
Body weight is regulated through coordinated metabolic processes. This includes peripheral signals called “adiposity signals” such as leptin and insulin [59], whose circulating levels are proportional to adipose tissue mass [13, 50]. These adiposity signals interact with the brain to regulate energy homeostasis. There are critical brain regions responsible for mediating these effects, specifically in the hypothalamus [47, 49]. Leptin and insulin have receptors in these critical brain regions and influence release and activity of different neuronal populations known to be involved in body weight regulation.
Leptin is secreted at a higher rate from subcutaneous fat than from visceral fat, thus circulating leptin correlates better with total subcutaneous fat than with total body fat. Insulin secretion is better correlated with visceral fat; thus, its levels better reflect visceral rather than total body adiposity [58]. Male rats are relatively more sensitive to the catabolic action of insulin delivered into the brain [14], whereas female rats are relatively more sensitive to the catabolic action of leptin delivered into the brain [16]. A comparable phenomenon has been reported in a recent study in humans, suggesting that men lose more body weight and body fat and change their waist circumference following intranasal insulin administration when compared to women [61]. Intranasal insulin administration increases insulin concentration of the cerebrospinal fluid and thereby alters brain functions. Therefore, sexually differential sensitivity to the catabolic effects of insulin exists in rodents and humans.
Uniquely, despite having higher circulating leptin levels, females have been demonstrated to be more sensitive to the anorexigenic effects of leptin [15]. Female rats are more responsive than males to the effects of centrally administered leptin to decrease food intake and body weight, and this has been demonstrated to be due to increased leptininduced Stat3 activation in the basal hypothalamus [5, 29]. So, an increase in the Stat3 signaling translates to an increase in the energy expenditure by leptin and also increased SNS regulation.
Among the two major estrogen receptor subtypes, it is believed that ERα mediates the anti-obesity effect in both males and females [41, 43, 55]. Studies with ERα null mice demonstrated obesity and had a massive increase in the adipose tissue [32, 48], whereas ERβ null mice were not obese. Heine et al. reported that male and female mice with a targeted deletion in the ERα subunit have increased adiposity in both sexes.
Clegg et al. (2006)
Clegg, D. J., Brown, L. M., Woods, S. C., & Benoit, S. C. (2006). Gonadal Hormones Determine Sensitivity to Central Leptin and Insulin. Diabetes, 55(4), 978–987. [DOI:10.2337/diabetes.55.04.06.db05-1339]:
These data indicate that estrogen acts within the brain to increase leptin sensitivity, decrease insulin sensitivity, and favor subcutaneous over visceral fat.
Thus, when systemic estrogen is present (intact females and males or OVX females administered subcutaneous estrogen), leptin is catabolic in the brain, whereas when estrogen is low (OVX females and intact males), leptin is relatively ineffective. Conversely, estrogen reduces the sensitivity of the brain to the anorexigenic action of insulin.
An important implication from these findings is that gonadal steroids mediate body fat distribution and interact with the integrated adiposity message conveyed to the brain by leptin and insulin, resulting in differential sensitivity to these signals in males and females.
This implies that the relative amount of androgens and estrogens is a key determinant of the brain’s sensitivity to the catabolic actions of insulin and leptin, with proportionally more estrogen favoring leptin sensitivity and proportionally less estrogen favoring insulin sensitivity. Finally, our data suggest that estrogen’s direct actions in the brain determine body fat distribution.
Note: I would love to see a study of trans women who on GAHT gained or lost weight and correlations with different gene polymorphisms for estrogen receptor alpha and leptin receptors.
Mauvais-Jarvis, Clegg, & Hevener (2013)
Mauvais-Jarvis, F., Clegg, D. J., & Hevener, A. L. (2013). The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews, 34(3), 309–338. [DOI:10.1210/er.2012-1055]:
In postmenopausal women, however, when the ovaries fail to produce E2 and in men—who have naturally low levels of circulating E2—E2 does not function as a circulating hormone; rather, it is synthesized in extragonadal sites such as breast, brain, muscle, bone, and adipose tissue where it acts locally as a paracrine or intracrine factor (8).
EST is a cytosolic enzyme that provides a molecular switch in target cells that inhibits estrogen activity by conjugating a sulfonate group to estrogens, thereby preventing binding to estrogen receptors and enhancing urinary excretion of the hormone.
Early studies of the reproductive actions of estrogens led to the establishment of a paradigm in which classical nuclear ERs acted as ligand-activated transcription factors (11). ER modulation of gene transcription is a highly dynamic process. The ER exists in 2 main forms, ER and ER, each of which has multiple isoforms and exhibit distinct tissue expression patterns and functions (12). The classical “genomic” mechanism of ER action typically occurs within hours, leading to activation or repression of target genes. In this classic signaling pathway, ligandactivated ER dissociates from its chaperone heat-shock protein and binds as a dimer either directly to an estrogen response element (ERE) in target gene promoters or indirectly to activator protein 1 or specificity protein 1 response elements through protein tethering to DNA (13). After binding, these ER dimers interact with cofactors (coactivators or cosuppressors) to regulate gene expression.
Cellular estrogenic action depends on: 1) the ER signaling and sensitivity; 2) the activity of enzymes like aromatase involved in the biosynthesis of E2 from androgenic precursors; and 3) the inactivation of E2 in E2 sulfate (E2-S) by the estrogen sulfotransferase (EST).
Although reproductive functions are mostly mediated via classical nuclear ER acting as ligand-activated transcription factors, a large component of ER actions related to energy metabolism also involves extranuclear ERs, indirectly modulating gene expression or acting independently of nuclear events (18). E2 can activate rapid signals Figure 1. Figure 1. Origin of circulating and tissue estrogens. A, In healthy premenopausal women, estrogen (E2) is produced by the ovaries and functions as a circulating hormone that acts on distant target tissues. Here WAT is represented. B, In postmenopausal women and in men, E2 does not function as a circulating hormone; rather, it is synthesized in extragonadal sites from circulating androgenic precursors such as T, androstenedione (4A), or dehydroepiandrosterone (DHEA). Cellular estrogenic action depends on: 1) the ER signaling and sensitivity; 2) the activity of enzymes like aromatase involved in the biosynthesis of E2 from androgenic precursors; and 3) the inactivation of E2 in E2 sulfate (E2-S) by the estrogen sulfotransferase (EST).
E2 can activate rapid signals that act within seconds or minutes via extranuclear and membrane-associated forms of ERs (19). ER and ER are localized to caveolae where they congregate with other signaling molecules, thereby facilitating interaction and rapid intracellular signaling. These signal proteins include G proteins, growth factor receptors, tyrosine kinases (Src), linker proteins (MNAR), and orphan G protein-coupled receptors. This multiprotein complex provides the necessary interactions for membrane ER to activate growth factor receptors and G proteins. In turn, these E2-induced rapid signals modify protein function via phosphorylation. However, they can also modulate gene expression and thus the production of proteins.
ER deficiency in both male and female mice causes increased body weight and adiposity predominately through reduced energy expenditure and slight increases in food intake.
These effects of E2 are mediated via MC4 receptor because E2 is unable to induce anorexia when the MC4 receptor antagonists Shu 9119 or agouti-related peptide (AgRP) are applied concomitantly with E2 administration in rats (70).
Mice with small hairpin RNA-mediated ERgene silencing as well as transgenic mice in which ER has been selectively deleted from SF1-containing neurons of the VMH, develop reduced sensitivity to E2-induced weight loss, increased visceral fat deposition, and reductions in energy expenditure. All of these results occur without an impact on food intake (60, 78), supporting the notion that ER signaling in VMN neurons plays an important role in regulating physical activity, thermogenesis, and fat distribution.
Collectively, these findings suggest that ER in the brainstem, and specifically the [nucleus tractus solitarius (NTS)], is an additional site mediating the anorexigenic effects of estrogens.
Subcutaneous adipose tissue permits efficient storage of maximal calories per unit volume of tissue.
Estrogens are produced in the adipocytes (via aromatization from androgenic precursors) and increase in proportion to total body adiposity (132, 133).
E2 also suppresses white adipose tissue (WAT) accumulation by decreasing fatty acid and triglyceride synthesis and lipogenesis.
Extensive evidence demonstrates that E2 has direct effects on cultured adipocytes with the overall effect of inhibiting lipogenesis and adipogenesis.
[…] suggesting that the absence of E2 promotes immune cell inflammation. Indeed, circulating levels of proinflammatory cytokines are elevated in women after natural or surgical menopause.
Macrophages are elemental players in innate and adaptive immunity; over the past decade their roles in modulating whole-body metabolism and insulin sensitivity have been topics of increasing interest (295, 296).
[…] EST terminates estrogen activity and represents an important parameter of estrogen output upstream of ER. With regard to metabolism, EST is highly expressed in WAT of male mice, but it is not detectable in WAT of normal cycling female mice (320).
Note: Interesting to see if trans folks on GAHT have different type 2 diabetes probability than normal population.
An obvious explanation for these observations is that EST expression is high in male WAT (320) to protect from excessive estrogen actions. It follows that EST suppression produces WAT estrogen excess leading to inflammation. Consistent with this possibility is the observation that E2 treatment leading to high blood concentrations produces WAT inflammation even in females (193).
Note: Makes you wonder if there is potential for higher WAT inflammation in trans folks – especially whne many have E2 levels higher than folks with estrogen producing genitals.
There are reports suggesting that oral E2 may exacerbate insulin resistance and adipocytokine parameters, worsening cardiovascular risk (329). Transdermal E2, however, has minimal effects on insulin resistance and results in higher adiponectin. This suggests that transdermal E2 may be a preferable treatment compared to oral CEE for obese women with metabolic syndrome.
The authors concluded that in women without diabetes, both oral and transdermal estrogen, with or without progestin, increase lean body mass, reduce abdominal fat, improve insulin resistance, decrease LDL/high-density lipoprotein-cholesterol ratio, and decrease blood pressure.
B. Effect of selective estrogen receptor modulators and aromatase inhibitors on metabolism.
In fact, tamoxifen therapy in a case-control study breast cancer survivor was associated with a 24% increased risk of developing diabetes.
In OVX mice, raloxifene reversed OVX-induced increases in food intake, body weight, fat mass, and hyperleptinemia to an extent similar to that of E2. This suggests that in rodents, raloxifene acts as an ER agonist in hypothalamic neurons and fat.
Sullivan et al (356) looked at the effect of a novel SERM (GSK232802A) on body weight, food intake, physical activity, and metabolic rate in an OVX nonhuman primate model. They observed that GSK232802A produced a 5% decrease in weight and reduced adiposity by suppressing food intake and increasing activity. These results occurred without changes in energy expenditure, suggesting that GSK232802A treatment may counteract postmenopausal obesity (356).
Schiffer et al. (2017)
Schiffer, L., Kempegowda, P., Arlt, W., & O’Reilly, M. W. (2017). MECHANISMS IN ENDOCRINOLOGY: The sexually dimorphic role of androgens in human metabolic disease. European Journal of Endocrinology, 177(3), R125–R143. [DOI:10.1530/eje-17-0124]:
Delineating the specific effects of androgen deprivation therapy in this patient population is clouded by co-administration of relatively large doses of oestrogen. Male-to-female transgender patients on combined estrogen and anti-androgen treatment develop an adverse lipid profile.
Cross-sectional studies analysing age-advanced men, men across different ages and obese vs non-obese men consistently support the association between low T and increased fat mass compared to eugonadal controls (107, 142, 143). BMI negatively correlates with total and free T (142, 144), and waist circumference is negatively associated with total T in men.
T administration in men reduces accumulation of visceral and retroperitoneal fat compared to controls, but not in SC depots; hypogonadal men also have increased visceral fat mass.
Mueller et al. (2011)
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body Composition and Bone Mineral Density in Male-to-Female Transsexuals During Cross-Sex Hormone Therapy Using Gonadotrophin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074]:
When the BMI values were compared, there was a significant increase during cross-sex hormone treatment with oestrogens in the absence of testosterone after 12 months and after 24 months. Moreover, total fat mass increased also during the study period, while total lean mass decreased significantly.
Recently, Lapauw et al. reported that in MtFs treated with female steroid hormones over an average of 8 years, total lean mass was approximately 20% lower whereas total fat mass was about 30% higher in comparison with a male control group (Lapauw et al., 2008). In this study, transsexuals had been treated with either ethinyl oestradiol, oestradiol valerate, or conjugated equine oestrogens combined with cyproterone acetate (Lapauw et al., 2008).
In MtFs treated with ethinyl oestradiol and antiandrogens, a significant increase in all subcutaneous fat depots, with a lesser but proportional and significant increase in the visceral fat depot and a decrease in thigh muscle area, was reported.
With regard to body composition and bone mineral density, there appears to be no difference when GnRH agonists are used for complete androgen deprivation in MtFs. In comparison with commonly used hormone regimens, the complication rates appear to be lower in patients receiving the treatment regimen described here, as reported earlier.
\ No newline at end of file
diff --git a/transfemscience.org/articles/hormones-fat-metabolism/index.html b/transfemscience.org/articles/hormones-fat-metabolism/index.html
index 166d57a8..c0725b5e 100644
--- a/transfemscience.org/articles/hormones-fat-metabolism/index.html
+++ b/transfemscience.org/articles/hormones-fat-metabolism/index.html
@@ -1 +1 @@
-On the Impact of Sex Hormones on Fat Metabolism with an Eye Towards Transfeminine Hormone Therapy - Transfeminine Science
If you are into the technical details I would urge you after reading through this to look through my notes as they go into even more gory details.
Much like my previous post this post is inspired by my personal journey in gender-affirming hormone therapy (GAHT) and my desire to understand how GAHT will impact me along with my attempts to optimize it for my desired bodily outcome. I have tried to provide an ultra high level summary/abstract, an in-depth writeup of my literature review and finally my raw notes and quotes upon which I based this article on. As always I welcome feedback. I found writing this to be especially challenging both because I struggled to find the right ‘voice’ and technical level in my writeup, and also with translating cis-focused academic literature into trans-inclusive language. Please know that while all mistakes are mine, my intentions are never to offend, erase, or exclude.
Summary
Within the context of transfeminine hormone therapy, it’s often stated that people will have a more feminine fat distribution, gaining fat around their thighs and buttocks (a gynoid fat distribution).
This appears to occur due to both the presence of estrogens as well as the absence of testosterone. Some ways in which estrogens cause this to happen are by directly acting on estrogen receptors in fat deposits in these areas and increasing fat cell growth (adipogenesis) and decreasing fat metabolism in the aforementioned areas (lipolysis).
In addition to directly acting on fat cells, estrogens also act on parts of the brain, specifically the hypothalamus, a center for regulating body fat and body metabolism. In the hypothalamus, estrogen receptors interact with leptin – a hormone associated with body fat regulation – as well with as a number of other hormones and elements that modify the hypothalamus. In general, estrogens effects on the hypothalamus seem to reduce appetite and increase fat metabolism.
These two major actions of estrogens on fat metabolism seem to result in the understanding that estrogens cause subcutaneous fat distributed gynoid, that is resistant to metabolism, while at the same time reducing hunger and increasing overall metabolism.
In keeping with this, there is some research to indicate that transfeminine populations increase their total body fat percentage under GAHT, however, there is some research that shows there is also an increase in total BMI as well.
My review of the literature has made me consider that looking at the genes of transfeminine individuals about to start (or already taking) GAHT could possibly help people understand if they are at risk for disproportionate weight gain due to GAHT and whether other interventional actions could be taken.
The rest of this post will go into much greater detail on how estrogens impact fat metabolism and deposition.
Background
When talking about fat distribution in humans we often talk about a feminine (gynoid) or masculine (android) distribution of fat. Gynoid fat distribution is typically characterized as fat deposited in the thighs and buttocks whereas an android fat distribution is characterized by abdominal fat. In addition a gynoid fat distribution is characterized by having a much higher percentage of subcutaneous fat (fat under the skin) as opposed to visceral fat (fat inside the abdominal cavity – for instance around the organs). This seems to likely be due to the fact that subcutaneous fat depots are among the most efficient ways to store calories per volume of tissue (Clegg et al., 2006). This is an especially important distinction as visceral fat is highly correlated with cardiovascular disease. Following along with that research shows that premenopausal natal women are far less likely to experience cardiovascular disease.
These different fat distributions are ultimately achieved by fat being metabolised (lipolysis) and synthesized (lipogenesis) at different levels in different fat tissues.
There are many ways in which sex hormones mediate lipolysis (fat metabolism) and lipogenesis (fat generation). These effects can be categorized broadly as either direct or peripheral. Direct actions typically mean estrogen or androgen receptor modulation on fat cells (adipocytes) directly whereas peripheral actions are the effects of estrogen or androgen within the brain to cause downstream impact on various aspects of metabolism including rate of fat metabolism, rate of fat uptake and hunger.
As fat is the major store of energy in the body, fat can be viewed as an organ and in fact an endocrine organ (Power & Schulkin, 2008). It produces leptin, a hormone that is central to maintaining body fat percentage as it is part of the leptin/ghrelin/insulin hormone system involved in hunger, and energy homeostasis. Leptin along with insulin are peripheral “adiposity signals” and their circulating levels are relative to the percentage of body fat.
Direct Effects
Direct effects of sex hormones on fat distribution are those which directly modulate fat cells as opposed to peripheral affects which result from downstream modulation of other signaling pathways. Adipose tissue contains both estrogen and androgen receptors with visceral fat having a higher concentration of both receptors (Power & Schulkin, 2008).
Below are some of the direct effects of hormones:
In subcutaneous fat estradiol acts through the estrogen receptor alpha subtype (ERα) to upregulate α2A-adrenergic receptors (increase the density of this receptor in the fat cells) which result in decreased lipolysis whereas estradiol does not seem to impact the concentration of α2A-adrenergic receptors in visceral fat (Power & Schulkin, 2008).
Fatty acid uptake is the incorporation of fatty acids into fat cells. Rates of fatty acid uptake are lower in natal women compared to men, in the buttocks and thighs – and after eating, fatty acid uptake is higher in both natal women and men in the abdomen but with women it is stored largely subcutaneously vs. viscerally (interabdominally) in natal men (Power & Schulkin, 2008).
Estrogen is associated with higher rates of fat metabolism during sustained bouts of increased energy usage whereas natal men are more likely to up regulate glucose and amino acid metabolism. When given exogenous estrogen, natal men decrease carbohydrate and amino acid metabolism and increase fat metabolism (Power & Schulkin, 2008).
Testosterone increases lipolysis (breakdown of fat), inhibits lipoprotein lipase activity (an enzyme that converts triglycerides into fatty acids suitable for cellular metabolism), and decreases triglyceride (TAG – a fatty acid) accumulation in adipose tissue. With this occurring largely in subcutaneous adipose tissue (Power & Schulkin, 2008).
The estrogen receptor α subunit (ERα) appears to be responsible for anti-obesity effects (Nedungadi & Clegg, 2009).
Androgens also appear to block the proliferation and differentiation of preadipocytes – this ties in with them also directing stem cells to create muscle cells (Power & Schulkin, 2008).
Conflictingly somewhat,there is also evidence to support the fact that estradiol suppresses white adipose tissue accumulation by decreasing triglyceride (TAG) and fatty acid synthesis as well as lipogenesis (fat uptake and storage) (Power & Schulkin, 2008).
Peripheral Effects
Peripheral effects of hormones are those mediated largely through the brain. Rather than acting directly on fat cells both androgens and estrogens act on parts of the brain – often the effect is on the hypothalamus – a center for regulating various metabolic activities like fat metabolism, (more broadly called energy homeostasis). Some of the effects hormones may cause include regulating hunger, regulating the rate of various tissues metabolism, and regulating the uptake of fat into tissues.
Leptin and Hormones
Leptin is a hormone secreted from fat and is one of the ways in which hunger is mediated. Increased leptin correlates with decreased hunger and decreased total weight. Leptin is secreted from subcutaneous fat at a higher rate than visceral fat (Power & Schulkin, 2008), thus as estrogen mediates a higher proportion of subcutaneous fat more leptin is found in the body. In addition estrogen in the hypothalamus causes increased sensitivity to leptin which results in decreased hunger (anorexigenic effects) (Nedungadi & Clegg, 2009; Clegg et al., 2006; Mauvais-Jarvis, Clegg, & Hevener, 2013). The brainstem (specifically the NTS – nucleus tractus solitarius) is also a source of anorexigenic effects via the estrogen receptor (Mauvais-Jarvis, Clegg, & Hevener, 2013). In addition leptin can also cause an increase in energy expenditure – via the Stat3 signaling pathway in the hypothalamus (Nedungadi & Clegg, 2009). When estrogen is not present but androgens are, insulin signaling pathways are far more responsible for mediating hunger and energy expenditure (Nedungadi & Clegg, 2009). In natal men, low testosterone is correlated with increased fat mass, probably because neither the insulin pathway for reducing hunger or the leptin one is very active (Schiffer et al., 2017).
Conclusions Related to Transfeminine Hormone Therapy
Obviously any conclusions we wish to draw about how a standard transfeminine hormone therapy regimen of androgen suppression and estrogens addition is speculative and based upon knowledge of how hormones interact with fat. Despite this, I think we can make some statements which are reasonable (and also backed up by observational evidence):
As testosterone decreases, overall body fat increases (because testosterone inhibits fat cell formation and decreases fat uptake overall (Power & Schulkin, 2008).
Subcutaneous fat deposits begin to increase.
This likely occurs by increased lipogenesis mediated by upregulation of α2A-adrenergic receptors and decrease LPL (lipoprotein lipase) in SC fat.
Visceral fat may decrease relative to total body fat possibly because of leptin pathways (Nedungadi & Clegg, 2009; Clegg et al., 2006). While visceral fat may decrease relative to total body fat the rate of turnover of visceral fat decreases, so both the uptake as well as the metabolism of visceral fat decreases (Power & Schulkin, 2008).
The leptin pathway becomes sensitized and the insulin pathway becomes less sensitive.
Total body fat increases, especially subcutaneous fat causing total overall leptin to increase. This may cause a decrease in appetite and weight loss in general.
When undergoing exercise more fat will be burned than amino acids (proteins) and carbohydrates.
However, somewhat at odds with what the actions of transfeminine hormone therapy seem likely to be, the research we have on transgender populations suggests an overall increase in weight:
During GAHT over 12 to 24 months total fat mass increased and total non fat mass decreased (Mueller et al., 2011).
Over an average of 8 years total lean mass (not body fat) was approximately 20% lower whereas total fat mass was about 30% higher in comparison with a control group of cis men (Mueller et al., 2011).
When treated with ethinyl estradiol and antiandrogens subcutaneous fat depots increased significantly and a proportional increase in visceral fat was also observed (Mueller et al., 2011).
There is at least some evidence (Belisle & Love, 1986) to suggest that this maybe due to cyproterone acetate, a commonly prescribed antiandrogen outside of the U.S., causing weight gain. I believe that there is not nearly enough research to draw really solid conclusions as there are so many confounding factors related to weight loss.
In addition I would love to see studies that relate genetics to the effects of GAHT on weight and body mass. For instance it may be possible to identify in advance, genetic polymorphisms that would cause say weight gain under GAHT (due to a specific phenotype of something involved in leptin signaling pathways) and create an intervention prior to or during GAHT.
References
Belisle, S., & Love, E. J. (1986). Clinical efficacy and safety of cyproterone acetate in severe hirsutism: results of a multicentered Canadian study. Fertility and Sterility, 46(6), 1015–1020. [DOI:10.1016/s0015-0282(16)49873-0]
Clegg, D. J., Brown, L. M., Woods, S. C., & Benoit, S. C. (2006). Gonadal Hormones Determine Sensitivity to Central Leptin and Insulin. Diabetes, 55(4), 978–987. [DOI:10.2337/diabetes.55.04.06.db05-1339]
Mauvais-Jarvis, F., Clegg, D. J., & Hevener, A. L. (2013). The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews, 34(3), 309–338. [DOI:10.1210/er.2012-1055]
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body Composition and Bone Mineral Density in Male-to-Female Transsexuals During Cross-Sex Hormone Therapy Using Gonadotrophin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074]
Nedungadi, T. P., & Clegg, D. J. (2009). Sexual Dimorphism in Body Fat Distribution and Risk for Cardiovascular Diseases. Journal of Cardiovascular Translational Research, 2(3), 321–327. [DOI:10.1007/s12265-009-9101-1]
Power, M. L., & Schulkin, J. (2008). Sex differences in fat storage, fat metabolism, and the health risks from obesity: possible evolutionary origins. British Journal of Nutrition, 99(5), 931–940. [DOI:10.1017/s0007114507853347]
Reubinoff, B. E., Grubstein, A., Meirow, D., Berry, E., Schenker, J. G., & Brzezinski, A. (1995). Effects of low-dose estrogen oral contraceptives on weight, body composition, and fat distribution in young women. Fertility and Sterility, 63(3), 516–521. [DOI:10.1016/s0015-0282(16)57419-6]
Schiffer, L., Kempegowda, P., Arlt, W., & O’Reilly, M. W. (2017). MECHANISMS IN ENDOCRINOLOGY: The sexually dimorphic role of androgens in human metabolic disease. European Journal of Endocrinology, 177(3), R125–R143. [DOI:10.1530/eje-17-0124]
\ No newline at end of file
+On the Impact of Sex Hormones on Fat Metabolism with an Eye Towards Transfeminine Hormone Therapy - Transfeminine Science
If you are into the technical details I would urge you after reading through this to look through my notes as they go into even more gory details.
Much like my previous post this post is inspired by my personal journey in gender-affirming hormone therapy (GAHT) and my desire to understand how GAHT will impact me along with my attempts to optimize it for my desired bodily outcome. I have tried to provide an ultra high level summary/abstract, an in-depth writeup of my literature review and finally my raw notes and quotes upon which I based this article on. As always I welcome feedback. I found writing this to be especially challenging both because I struggled to find the right ‘voice’ and technical level in my writeup, and also with translating cis-focused academic literature into trans-inclusive language. Please know that while all mistakes are mine, my intentions are never to offend, erase, or exclude.
Summary
Within the context of transfeminine hormone therapy, it’s often stated that people will have a more feminine fat distribution, gaining fat around their thighs and buttocks (a gynoid fat distribution).
This appears to occur due to both the presence of estrogens as well as the absence of testosterone. Some ways in which estrogens cause this to happen are by directly acting on estrogen receptors in fat deposits in these areas and increasing fat cell growth (adipogenesis) and decreasing fat metabolism in the aforementioned areas (lipolysis).
In addition to directly acting on fat cells, estrogens also act on parts of the brain, specifically the hypothalamus, a center for regulating body fat and body metabolism. In the hypothalamus, estrogen receptors interact with leptin – a hormone associated with body fat regulation – as well with as a number of other hormones and elements that modify the hypothalamus. In general, estrogens effects on the hypothalamus seem to reduce appetite and increase fat metabolism.
These two major actions of estrogens on fat metabolism seem to result in the understanding that estrogens cause subcutaneous fat distributed gynoid, that is resistant to metabolism, while at the same time reducing hunger and increasing overall metabolism.
In keeping with this, there is some research to indicate that transfeminine populations increase their total body fat percentage under GAHT, however, there is some research that shows there is also an increase in total BMI as well.
My review of the literature has made me consider that looking at the genes of transfeminine individuals about to start (or already taking) GAHT could possibly help people understand if they are at risk for disproportionate weight gain due to GAHT and whether other interventional actions could be taken.
The rest of this post will go into much greater detail on how estrogens impact fat metabolism and deposition.
Background
When talking about fat distribution in humans we often talk about a feminine (gynoid) or masculine (android) distribution of fat. Gynoid fat distribution is typically characterized as fat deposited in the thighs and buttocks whereas an android fat distribution is characterized by abdominal fat. In addition a gynoid fat distribution is characterized by having a much higher percentage of subcutaneous fat (fat under the skin) as opposed to visceral fat (fat inside the abdominal cavity – for instance around the organs). This seems to likely be due to the fact that subcutaneous fat depots are among the most efficient ways to store calories per volume of tissue (Clegg et al., 2006). This is an especially important distinction as visceral fat is highly correlated with cardiovascular disease. Following along with that research shows that premenopausal natal women are far less likely to experience cardiovascular disease.
These different fat distributions are ultimately achieved by fat being metabolised (lipolysis) and synthesized (lipogenesis) at different levels in different fat tissues.
There are many ways in which sex hormones mediate lipolysis (fat metabolism) and lipogenesis (fat generation). These effects can be categorized broadly as either direct or peripheral. Direct actions typically mean estrogen or androgen receptor modulation on fat cells (adipocytes) directly whereas peripheral actions are the effects of estrogen or androgen within the brain to cause downstream impact on various aspects of metabolism including rate of fat metabolism, rate of fat uptake and hunger.
As fat is the major store of energy in the body, fat can be viewed as an organ and in fact an endocrine organ (Power & Schulkin, 2008). It produces leptin, a hormone that is central to maintaining body fat percentage as it is part of the leptin/ghrelin/insulin hormone system involved in hunger, and energy homeostasis. Leptin along with insulin are peripheral “adiposity signals” and their circulating levels are relative to the percentage of body fat.
Direct Effects
Direct effects of sex hormones on fat distribution are those which directly modulate fat cells as opposed to peripheral affects which result from downstream modulation of other signaling pathways. Adipose tissue contains both estrogen and androgen receptors with visceral fat having a higher concentration of both receptors (Power & Schulkin, 2008).
Below are some of the direct effects of hormones:
In subcutaneous fat estradiol acts through the estrogen receptor alpha subtype (ERα) to upregulate α2A-adrenergic receptors (increase the density of this receptor in the fat cells) which result in decreased lipolysis whereas estradiol does not seem to impact the concentration of α2A-adrenergic receptors in visceral fat (Power & Schulkin, 2008).
Fatty acid uptake is the incorporation of fatty acids into fat cells. Rates of fatty acid uptake are lower in natal women compared to men, in the buttocks and thighs – and after eating, fatty acid uptake is higher in both natal women and men in the abdomen but with women it is stored largely subcutaneously vs. viscerally (interabdominally) in natal men (Power & Schulkin, 2008).
Estrogen is associated with higher rates of fat metabolism during sustained bouts of increased energy usage whereas natal men are more likely to up regulate glucose and amino acid metabolism. When given exogenous estrogen, natal men decrease carbohydrate and amino acid metabolism and increase fat metabolism (Power & Schulkin, 2008).
Testosterone increases lipolysis (breakdown of fat), inhibits lipoprotein lipase activity (an enzyme that converts triglycerides into fatty acids suitable for cellular metabolism), and decreases triglyceride (TAG – a fatty acid) accumulation in adipose tissue. With this occurring largely in subcutaneous adipose tissue (Power & Schulkin, 2008).
The estrogen receptor α subunit (ERα) appears to be responsible for anti-obesity effects (Nedungadi & Clegg, 2009).
Androgens also appear to block the proliferation and differentiation of preadipocytes – this ties in with them also directing stem cells to create muscle cells (Power & Schulkin, 2008).
Conflictingly somewhat,there is also evidence to support the fact that estradiol suppresses white adipose tissue accumulation by decreasing triglyceride (TAG) and fatty acid synthesis as well as lipogenesis (fat uptake and storage) (Power & Schulkin, 2008).
Peripheral Effects
Peripheral effects of hormones are those mediated largely through the brain. Rather than acting directly on fat cells both androgens and estrogens act on parts of the brain – often the effect is on the hypothalamus – a center for regulating various metabolic activities like fat metabolism, (more broadly called energy homeostasis). Some of the effects hormones may cause include regulating hunger, regulating the rate of various tissues metabolism, and regulating the uptake of fat into tissues.
Leptin and Hormones
Leptin is a hormone secreted from fat and is one of the ways in which hunger is mediated. Increased leptin correlates with decreased hunger and decreased total weight. Leptin is secreted from subcutaneous fat at a higher rate than visceral fat (Power & Schulkin, 2008), thus as estrogen mediates a higher proportion of subcutaneous fat more leptin is found in the body. In addition estrogen in the hypothalamus causes increased sensitivity to leptin which results in decreased hunger (anorexigenic effects) (Nedungadi & Clegg, 2009; Clegg et al., 2006; Mauvais-Jarvis, Clegg, & Hevener, 2013). The brainstem (specifically the NTS – nucleus tractus solitarius) is also a source of anorexigenic effects via the estrogen receptor (Mauvais-Jarvis, Clegg, & Hevener, 2013). In addition leptin can also cause an increase in energy expenditure – via the Stat3 signaling pathway in the hypothalamus (Nedungadi & Clegg, 2009). When estrogen is not present but androgens are, insulin signaling pathways are far more responsible for mediating hunger and energy expenditure (Nedungadi & Clegg, 2009). In natal men, low testosterone is correlated with increased fat mass, probably because neither the insulin pathway for reducing hunger or the leptin one is very active (Schiffer et al., 2017).
Conclusions Related to Transfeminine Hormone Therapy
Obviously any conclusions we wish to draw about how a standard transfeminine hormone therapy regimen of androgen suppression and estrogens addition is speculative and based upon knowledge of how hormones interact with fat. Despite this, I think we can make some statements which are reasonable (and also backed up by observational evidence):
As testosterone decreases, overall body fat increases (because testosterone inhibits fat cell formation and decreases fat uptake overall (Power & Schulkin, 2008).
Subcutaneous fat deposits begin to increase.
This likely occurs by increased lipogenesis mediated by upregulation of α2A-adrenergic receptors and decrease LPL (lipoprotein lipase) in SC fat.
Visceral fat may decrease relative to total body fat possibly because of leptin pathways (Nedungadi & Clegg, 2009; Clegg et al., 2006). While visceral fat may decrease relative to total body fat the rate of turnover of visceral fat decreases, so both the uptake as well as the metabolism of visceral fat decreases (Power & Schulkin, 2008).
The leptin pathway becomes sensitized and the insulin pathway becomes less sensitive.
Total body fat increases, especially subcutaneous fat causing total overall leptin to increase. This may cause a decrease in appetite and weight loss in general.
When undergoing exercise more fat will be burned than amino acids (proteins) and carbohydrates.
However, somewhat at odds with what the actions of transfeminine hormone therapy seem likely to be, the research we have on transgender populations suggests an overall increase in weight:
During GAHT over 12 to 24 months total fat mass increased and total non fat mass decreased (Mueller et al., 2011).
Over an average of 8 years total lean mass (not body fat) was approximately 20% lower whereas total fat mass was about 30% higher in comparison with a control group of cis men (Mueller et al., 2011).
When treated with ethinyl estradiol and antiandrogens subcutaneous fat depots increased significantly and a proportional increase in visceral fat was also observed (Mueller et al., 2011).
There is at least some evidence (Belisle & Love, 1986) to suggest that this maybe due to cyproterone acetate, a commonly prescribed antiandrogen outside of the U.S., causing weight gain. I believe that there is not nearly enough research to draw really solid conclusions as there are so many confounding factors related to weight loss.
In addition I would love to see studies that relate genetics to the effects of GAHT on weight and body mass. For instance it may be possible to identify in advance, genetic polymorphisms that would cause say weight gain under GAHT (due to a specific phenotype of something involved in leptin signaling pathways) and create an intervention prior to or during GAHT.
References
Belisle, S., & Love, E. J. (1986). Clinical efficacy and safety of cyproterone acetate in severe hirsutism: results of a multicentered Canadian study. Fertility and Sterility, 46(6), 1015–1020. [DOI:10.1016/s0015-0282(16)49873-0]
Clegg, D. J., Brown, L. M., Woods, S. C., & Benoit, S. C. (2006). Gonadal Hormones Determine Sensitivity to Central Leptin and Insulin. Diabetes, 55(4), 978–987. [DOI:10.2337/diabetes.55.04.06.db05-1339]
Lain. (2019). Sources/Excerpts: On the Impact of Sex Hormones on Fat Metabolism with an Eye Towards Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Mauvais-Jarvis, F., Clegg, D. J., & Hevener, A. L. (2013). The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews, 34(3), 309–338. [DOI:10.1210/er.2012-1055]
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body Composition and Bone Mineral Density in Male-to-Female Transsexuals During Cross-Sex Hormone Therapy Using Gonadotrophin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074]
Nedungadi, T. P., & Clegg, D. J. (2009). Sexual Dimorphism in Body Fat Distribution and Risk for Cardiovascular Diseases. Journal of Cardiovascular Translational Research, 2(3), 321–327. [DOI:10.1007/s12265-009-9101-1]
Power, M. L., & Schulkin, J. (2008). Sex differences in fat storage, fat metabolism, and the health risks from obesity: possible evolutionary origins. British Journal of Nutrition, 99(5), 931–940. [DOI:10.1017/s0007114507853347]
Reubinoff, B. E., Grubstein, A., Meirow, D., Berry, E., Schenker, J. G., & Brzezinski, A. (1995). Effects of low-dose estrogen oral contraceptives on weight, body composition, and fat distribution in young women. Fertility and Sterility, 63(3), 516–521. [DOI:10.1016/s0015-0282(16)57419-6]
Schiffer, L., Kempegowda, P., Arlt, W., & O’Reilly, M. W. (2017). MECHANISMS IN ENDOCRINOLOGY: The sexually dimorphic role of androgens in human metabolic disease. European Journal of Endocrinology, 177(3), R125–R143. [DOI:10.1530/eje-17-0124]
\ No newline at end of file
diff --git a/transfemscience.org/articles/index.html b/transfemscience.org/articles/index.html
index 619f01e7..b967f98d 100644
--- a/transfemscience.org/articles/index.html
+++ b/transfemscience.org/articles/index.html
@@ -1 +1 @@
-Articles - Transfeminine Science
\ No newline at end of file
diff --git a/transfemscience.org/articles/injectable-e2-meta-analysis/index.html b/transfemscience.org/articles/injectable-e2-meta-analysis/index.html
index 2c88b1d7..bc5e9124 100644
--- a/transfemscience.org/articles/injectable-e2-meta-analysis/index.html
+++ b/transfemscience.org/articles/injectable-e2-meta-analysis/index.html
@@ -1 +1 @@
-An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations - Transfeminine Science
An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations
By Aly | First published July 16, 2021 | Last modified May 8, 2025
Abstract / TL;DR
Injectable estradiol preparations such as estradiol valerate and estradiol cypionate in oil are frequently used as estrogens in transfeminine hormone therapy. However, there is little characterization of these preparations in transfeminine people and dosing recommendations by transgender health guidelines appear to be based on expert opinion rather than on clinical data. To help shed light on the properties of injectable estradiol and to better inform dosing considerations in transfeminine people, an informal meta-analysis of available clinical data on estradiol concentration–time curves with major injectable estradiol formulations was conducted. The included preparations were injectable estradiol benzoate in oil, estradiol valerate in oil, estradiol cypionate both in oil and as a suspension, estradiol enanthate in oil, estradiol undecylate in oil, and polyestradiol phosphate. The literature was searched for clinical concentration–time data with these injectable estradiol esters and these data were collected and analyzed. Meta-analysis consisted of data for each injectable estradiol preparation being processed and fit with pharmacokinetic models. Selected pharmacokinetic parameters were additionally determined and reported. The results of this work were discussed with regard to characteristics of injectable estradiol preparations like curve shapes, durations, estrogenic exposure, and variability between people and studies. Recommendations for injectable estradiol preparations by transgender health guidelines were also explored in light of the present results. Current guidelines recommend doses of these preparations that appear to be highly excessive with injection intervals that are too widely spaced. Based on the findings of the present meta-analysis, recommendations by guidelines should be reassessed. Finally, the fitted curves in this work were incorporated into an interactive web-based injectable estradiol simulator intended for use by transfeminine people and their medical providers to help guide therapeutic decisions.
Introduction
Estradiol is the main estrogen used in transfeminine hormone therapy and is available in a variety of different forms for use by different routes of administration. The most commonly employed forms are oral, sublingual, transdermal, and injectable preparations. Injectable estradiol preparations have been discontinued in many countries and hence are unavailable for use in transfeminine hormone therapy in many parts of the world, for instance in most of Europe (Glintborg et al., 2021). However, they are still used by many transfeminine people particularly in the United States and in the do-it-yourself (DIY) community. The most commonly used forms include estradiol valerate, estradiol cypionate, and estradiol enanthate all in oil. Injectable estradiol preparations have certain advantages over other estradiol forms that make them a popular choice for use in transfeminine hormone therapy. These include often lower cost, capacity to easily achieve higher estradiol levels that can be useful for testosterone suppression, less frequent administration, and theoretically reduced health risks relative to oral estradiol at equivalent doses due to the lack of the first pass with this route (Aly, 2020). The higher estradiol levels with injections are particularly useful for estradiol monotherapy, in which an antiandrogen is not used.
Clinically used injectable estradiol preparations are formulated not as estradiol but as estradiol esters. When injected into muscle or fat in oil solutions or crystalline aqueous suspensions, these estradiol esters form depots at the injection site from which they are slowly released. Subsequent to release, estradiol esters are rapidly metabolized into estradiol and hence act as prodrugs. When estradiol itself is given by intramuscular injection in an aqueous solution or oil solution, it is rapidly absorbed and has a very short duration. Due to having lipophilic esters, most clinically used injectable estradiol esters are more fat-soluble than estradiol (as measured by oil–water partition coefficient (P)) (Table). When these esters are administered as oil solutions by intramuscular or subcutaneous injection, their increased lipophilicity causes them to be released from the injection-site depot more slowly than estradiol and to therefore have longer durations. In the case of fatty acid esters, the longer the chain length of the ester—as in e.g. estradiol valerate (5 carbons) vs. estradiol enanthate (7 carbons) vs. estradiol undecylate (10 carbons)—the greater the fat solubility, the slower the rate of release from the depot, and the longer the time to peak levels and duration (Edkins, 1959; Sinkula, 1978; Chien, 1981; Kuhl, 2005; Kalicharan, 2017; Vhora et al., 2019). The durations of both injectable oil solutions and aqueous suspensions depend on the ester and its particular physicochemical properties, but the characteristics of these preparations are different and they work in distinct ways to produce their depot effects (Enever et al., 1983; Aly, 2019). The durations of oil solutions are dependent on the lipophilicity of the ester as well as oil vehicle, whereas the durations of aqueous suspensions depend on the properties of the ester crystal lattice as well as crystal sizes (Chien, 1981; Enever et al., 1983; Aly, 2019). The polymeric estradiol ester polyestradiol phosphate is more hydrophilic (water-soluble) than estradiol and works differently than other injectable estradiol preparations. Ιt is composed of many estradiol molecules linked together via phosphate esters (on average 13 molecules of estradiol per one molecule of polyestradiol phosphate) and has a prolonged duration due to slow cleavage into estradiol following injection. Estradiol esters are able to substantially prolong the duration of estradiol when used as injectables and these preparations have durations ranging from days to months depending on the ester and how it is formulated (Table).
In order to aid understanding of concentration–time profiles with injectable estradiol preparations, I’ve developed an interactive web-based injectable estradiol simulator for transfeminine people and their medical providers. During work on this simulator, it became apparent that there is substantial variability in estradiol levels and curve shapes between different studies even with the same injectable estradiol ester. The injectable estradiol simulator was originally designed to simulate curves from only a single well-known pharmacokinetic study that directly compared estradiol benzoate, estradiol valerate, and estradiol cypionate in oil (Oriowo et al., 1980 [Graph]). However, due to the considerable differences in estradiol levels and curves across studies, it was decided that relying on only one study for such a project would be untenable. Instead, for the simulations to be reasonably accurate to the available data, many studies would need to be incorporated. Including additional studies would also allow for inclusion of other injectable estradiol esters in the simulator. As a result, the present work—an informal meta-analysis of estradiol curves with injectable estradiol formulations—was conducted for the simulator project.
Methods
A literature search was performed to identify studies reporting clinical estradiol concentration–time data with major injectable estradiol formulations (Table 1). All of these preparations have been used in transfeminine hormone therapy at one time or another in different parts of the world, although only estradiol valerate in oil and estradiol cypionate in oil are widely used today. Some of the injectable preparations included have notably been discontinued. Acceptable data for the search included mean and individual estradiol concentration data and Cmax estradiol levels (mean peak estradiol levels of individual subjects at time Tmax). Databases like PubMed, Google Scholar, and WorldCat were searched using relevant keywords (e.g., estradiol ester names and variations thereof as well as major brand names). Publications with relevant information were catalogued for data collection. Only single-dose data and multi-dose data that allowed estradiol levels to return to baseline between doses (as in e.g. repeated once-monthly combined injectable contraceptives) were included. Studies were included regardless of the hypothalamic–pituitary–gonadal axis (HPG axis) status of the participants. The study selection criteria aimed to maximize data inclusion due to scarcity of data for several preparations. If however there were many studies for a specific preparation, studies with only 1 or 2 subjects were generally skipped due to the limited additional value that they would provide. When data were in figures in papers—as was generally the case—they were extracted from the graphs using WebPlotDigitizer.
Table 1: Major injectable estradiol formulations (ordered roughly from shortest- to longest-acting):
Following their collection, data were processed, aggregated, and modeled. Data were adjusted for endogenous estradiol production and were normalized by dose. Adjustment for endogenous estradiol production was generally done via subtraction of baseline estradiol levels. In a number of cases however, subtraction of trough estradiol levels or of estradiol levels from a control group was required instead. Data were also weighted by sample size. In a handful of instances, certain missing information (e.g., time to peak levels, baseline levels, subject body weights) was filled in with reasonable assumptions to help maximize data inclusion. Data were processed in the form of mean estradiol curve data rather than individual-subject data (except for rare n=1 studies). The combined processed data from all studies for each injectable estradiol preparation were fit via least squares regression to one-, two-, and three-compartmentpharmacokinetic models with first-order absorption and elimination that were obtained from the literature and other sources (e.g., Colburn, 1981; Wagner, 1993; Fisher & Shafer, 2007; Lixoft, 2008; Abuhelwa, Foster, & Upton, 2015; Certara, 2020). These models fit most curves from individual studies very well. Fitting the combined curve fits of all individual studies (as opposed to fitting all of the combined processed data directly) was additionally evaluated for each injectable estradiol preparation, and if it was feasible for the preparation and allowed for better fitting results, was employed instead. Fitting directly to the combined processed data has the effect of weighting individual studies by quantity of time points, whereas fitting the combined curve fits of studies eliminates this. The Akaike information criterion (AIC) was used to help guide model selection for fitting of the preparations. Curve fitting was performed using the Python library Lmfit with the Levenberg–Marquardt algorithm. Cmax concentrations are a different form of data than mean curve estradiol concentration–time data, and for this reason, were not included in the fitting unless data were very limited for a given injectable estradiol preparation. Outlying data were also excluded from fitting in a number of instances and this allowed for improved curve fits with more uniform area-under-the-curve levels. The main criterion used for excluding curves was fit area-under-the-curve levels that deviated considerably from what was typical for the injectable estradiol preparations (generally less than about 50% of the average or greater than about 150% of the average).
A selection of pharmacokinetic parameters were calculated for each injectable estradiol preparation using the single-dose fit curves and compartmental pharmacokinetic analyses. These parameters included maximal or peak concentrations of estradiol after a single dose scaled to 5 mg (Cmax), time to maximal concentrations of estradiol after a single dose (Tmax), total area-under-the-curve concentrations of estradiol after a single dose (AUC0–∞), terminal elimination half-life after a single dose (t1/2), and the terminal 90% life after a single dose (t90%) (calculated as t1/2 × 3.322). In addition, selected pharmacokinetic parameters were calculated for simulated repeated administration of each injectable preparation at steady state with a dose and dose interval of 5 mg once every 7 days using the single-dose fit curves and compartmental pharmacokinetic analyses. These parameters included time to peak concentrations of estradiol (Tmax), peak and trough concentrations of estradiol (Cmax and Cmin, respectively), peak–trough difference (PTD; Cmax – Cmin), peak–trough ratio (PTR; Cmax ÷ Cmin), and integratedmean concentrations of estradiol (Cavg). Simulation of repeated administration was performed by stacking estradiol levels for multiple injections. Cmax and Tmax were defined and calculated in general as peak estradiol level and time to peak level of the fit mean curve as opposed to the mean peak level and mean time to peak level of individual subjects. This is because the latter would not be possible to compute as most studies reported only estradiol mean curve data. Pharmacokinetic parameters were calculated using relevant pharmacokinetic equations and, as a sanity check, were compared against those computed by PKSolver, a Microsoft Excel pharmacokinetics add-in program (Zhang et al., 2010).
Results
The figures in the subsequent sections show the original data from studies adjusted for endogenous estradiol levels and normalized to a common dose as well as the curve fits to the data (or alternatively the curve fits of the fits of the data depending on the preparation) for the included injectable estradiol preparations. Estradiol benzoate, estradiol cypionate in oil, and estradiol cypionate suspension were fit to the fits of all individual studies for these preparations, whereas estradiol enanthate, estradiol undecylate, and polyestradiol phosphate were fit directly to the combined processed data for these esters. In the case of estradiol valerate, the two fitting approaches gave nearly identical curves, and so fitting the combined processed original data was done for simplicity for this preparation. Cmax studies were excluded in the fitting for all preparations except estradiol enanthate, for which available estradiol concentration–time data were otherwise very limited. The data for the injectable estradiol preparations were generally fit best by a three-compartment pharmacokinetic model (Desmos). As a result, and for consistency, this model was used in the fitting of all preparations.
Estradiol Benzoate
Injectable estradiol benzoate has been extensively used in the past in scientific research, most notably in studies elucidating the function and dynamics of the HPG axis. One such use of estradiol benzoate has been the estrogen provocation test, a diagnostic test of HPG axis function. Due to its use in research, substantial estradiol concentration–time data with injectable estradiol benzoate exists. A total of 26 publications and concentration–time data for 355 individual injections were identified (Table 2).
Table 2: Studies of injectable estradiol benzoate (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
A number of studies were excluded from fitting due to much higher or lower area-under-the-curve levels than average. A couple of studies were omitted from the meta-analysis as they only reported total estrogen levels rather than estradiol levels with estradiol benzoate (Akande, 1974; Weiss, Nachtigall, & Ganguly, 1976). Two studies were omitted due partly to being very old and using very early and inaccurate blood tests (Varangot & Cedard, 1957; Ittrich & Pots, 1965 [Graph]). The processed original data and fit of fits curve for estradiol benzoate are shown in Figure 1.
Figure 1: Published estradiol concentration–time curves and fit of fit curves (thick black or white line) with a single intramuscular injection of estradiol benzoate in oil solution over a period of 7 days. Each curve was adjusted for endogenous estradiol levels, normalized to a dose of 5 mg, and fit with a compartmental pharmacokinetic model. Following this, the combined fit curves of the individual studies were fit using the same pharmacokinetic model. The original data from the studies for estradiol benzoate are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Valerate
Studies with curve data on injectable estradiol valerate come from its use in menopausal hormone therapy and other therapeutic indications for estrogens, its use in combined injectable contraceptives, and use in scientific research. A total of 28 publications and concentration–time data for 309 individual injections were identified for estradiol valerate (Table 3).
Table 3: Studies of injectable estradiol valerate (Spreadsheet; Plotly):
Study
na
Subjects
Dose
Reference(s)
S7175
12
Premenopausal women with menstrual migraine (n=10) and amenorrheic/postmenopausal women with history of menstrual migraine (n=2)
Normally cycling transmasculine people not on hormone therapy (n=31), transfeminine people not on hormone therapy (n=14), and gonadally intact transfeminine people on oral estrogen therapy (n=9)
a Total number of injections, not total number of subjects.
A few of these studies were excluded from fitting due generally to much higher or lower area-under-the-curve levels than average or due to being Cmax data. One study was omitted as it only reported estrone levels rather than estradiol levels (Ibrahim, 1996). Another study was not included due to being in pregnant women with concomitant pregnancy termination (Garner & Armstrong, 1977). One last study was omitted due partly to being very old and using very early and inaccurate blood tests (Ittrich & Pots, 1965 [Graph]). The processed original data and fit curve for estradiol valerate are shown in Figure 2.
Figure 2: Published estradiol concentration–time curves and fit curve (thick black or white line) with a single intramuscular injection of estradiol valerate in oil solution over a period of 30 days. Curves were adjusted for endogenous estradiol levels, normalized to a dose of 10 mg, and fit with a compartmental pharmacokinetic model. Fitting of the combined fits of individual studies for this preparation was explored but gave a nearly identical overall curve, so the overall fit curve for the combined processed original data was used for simplicity for this preparation. The original data from the studies for estradiol valerate are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Cypionate Oil
Estradiol cypionate in oil is used in menopausal hormone therapy and for other estrogen indications. However, its use has been more limited relative to other injectable estradiol preparations, like estradiol valerate. Only a handful of studies with relevant data were identified for estradiol cypionate in oil. This included 4 publications and estradiol concentration–time data for 49 individual injections (Table 4).
Table 4: Studies of injectable estradiol cypionate in oil (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
No curves were excluded from fitting in the case of this preparation. The processed original data and fit of fit curves for estradiol cypionate in oil are shown in Figure 3.
Figure 3: Published estradiol concentration–time curves and fit of fit curves (thick black or white line) with a single intramuscular injection of estradiol cypionate in oil solution over a period of 30 days. Each curve was adjusted for endogenous estradiol levels, normalized to a dose of 5 mg, and fit with a compartmental pharmacokinetic model. Following this, the combined fit curves of the individual studies were fit using the same pharmacokinetic model. The original data from the studies for estradiol cypionate in oil are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Cypionate Suspension
Estradiol cypionate suspension has been used exclusively in combined injectable contraceptives. For this reason, many relatively high quality pharmacokinetic studies with this injectable preparation have been conducted. A total of 9 publications and estradiol concentration–time data for 131 individual injections were identified for estradiol cypionate suspension (Table 5).
Table 5: Studies of injectable estradiol cypionate suspension (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects. b By subcutaneous injection rather than intramuscular injection.
One of these studies used subcutaneous injection instead of the usual intramuscular injection but the resulting curve was very similar to the curve for intramuscular injection in the same study (Sierra-Ramírez et al., 2011 [Graph]). Several Cmax studies were excluded from fitting for this preparation. One pharmacokinetic study only measured estradiol cypionate levels rather than estradiol levels and hence was not included (Martins et al., 2019 [Graph]). The processed original data and fit of fit curves for estradiol cypionate suspension are shown in Figure 4.
Figure 4: Published estradiol concentration–time curves and fit of fits curve (thick black or white line) with a single intramuscular (or in one case subcutaneous) injection of a microcrystalline aqueous suspension of estradiol cypionate over a period of 30 days. Each curve was adjusted for endogenous estradiol levels, normalized to a dose of 5 mg, and fit with a compartmental pharmacokinetic model. Following this, the combined fit curves of the individual studies were fit using the same pharmacokinetic model. The original data from the studies for estradiol cypionate suspension are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Enanthate
Estradiol enanthate has been used exclusively in combined injectable contraceptives. Several pharmacokinetic studies have been conducted with it because of this. A total of 7 publications and concentration–time data for 270 individual injections were identified for estradiol enanthate (Table 6).
Table 6: Studies of injectable estradiol enanthate (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
Of the available data, 216 of the injections were from a single study and mainly included only Cmax levels. Wiemeyer et al. (1986) was excluded from fitting due to having unusually high area-under-the-curve levels with a small sample size (n=3). Because of the scarcity of estradiol concentration–time data available for estradiol enanthate, Cmax studies were included in the fitting for this preparation. The processed original data and fit curve for estradiol enanthate are shown in Figure 5.
Figure 5: Published estradiol concentration–time curves and fit curve (thick black or white line) with a single intramuscular injection of estradiol enanthate in oil solution over a period of 30 days. Curves were adjusted for endogenous estradiol levels, normalized to a dose of 10 mg, and fit with a compartmental pharmacokinetic model. The original data from the studies for estradiol enanthate are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Undecylate
Estradiol undecylate was formerly used in the treatment of prostate cancer and in menopausal hormone therapy as well as for other estrogen therapeutic indications. However, it was discontinued many years ago and is no longer used today. Nonetheless, estradiol undecylate is of significant historical interest as an injectable estradiol preparation. A total of 4 publications and estradiol concentration–time data for 7 individual injections were identified for estradiol undecylate (Table 7).
Table 7: Studies of injectable estradiol undecylate (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
Unfortunately, the identified data were of very low quality, with small sample sizes and considerable variations in estradiol levels. Moreover, estradiol undecylate is a very long-acting injectable estradiol ester with a duration measured in months, and the follow up in these studies only went to about 2 weeks post-injection. For these reasons, it was not possible to fit the data for estradiol undecylate in a reasonably accurate way—as suggested by area-under-the-curve estradiol levels that were only around one-third those of the other non-polymeric injectable estradiol esters. Limited multi-dose hormone concentration–time data also exist for estradiol undecylate, but these data could not be incorporated (Jacobi & Altwein, 1979 [Graph]; Jacobi et al., 1980 [Graph]; Derra, 1981 [Graph]). The processed original data and fit curve for estradiol undecylate are shown in Figure 6.
Figure 6: Published estradiol concentration–time curves and fit curve (thick black or white line) with a single intramuscular injection of estradiol undecylate in oil solution over a period of 90 days. Curves were adjusted for endogenous estradiol levels, normalized to a dose of 50 mg, and fit with a compartmental pharmacokinetic model. The original data from the studies for estradiol undecylate are also provided elsewhere (Spreadsheet; Plotly).
Polyestradiol Phosphate
Polyestradiol phosphate has been used primarily in the treatment of prostate cancer but has also been used for estrogen therapeutic indications like treatment of breast cancer and menopausal hormone therapy. While this injectable estradiol preparation has been used widely in the past, it appears to have recently been discontinued. All of the identified studies with estradiol concentration–time data on polyestradiol phosphate were in men with prostate cancer. A total of 11 publications and concentration–time data for 114 individual injections were identified for polyestradiol phosphate (Table 8).
Table 8: Studies of injectable polyestradiol phosphate (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
A few older and strongly outlying studies were excluded from the fitting. The processed original data and fit curve for polyestradiol phosphate are shown in Figure 7.
Figure 7: Published estradiol concentration–time curves and fit curve (thick black or white line) with a single intramuscular injection of an aqueous solution of polyestradiol phosphate over a period of 90 days. The graph was clipped to maximum estradiol levels of 600 pg/mL (~2,200 pmol/L) for better viewability. Curves were adjusted for endogenous estradiol levels, normalized to a dose of 160 mg, and fit with a compartmental pharmacokinetic model. The original data from the studies for polyestradiol phosphate are also provided elsewhere (Spreadsheet; Plotly).
Other Injectable Estradiol Preparations
A number of clinical studies with estradiol concentration–time data for other injectable estradiol preparations were also identified during literature search:
Estradiol (unesterified) in an “aqueous” preparation (type of aqueous preparation unspecified but probably a microcrystalline aqueous suspension) (Jones et al., 1978 [Graph])
These preparations were not included in the present meta-analysis due to their relative obscurity and the limited data available for them. In addition, there were concerns about fitting the used pharmacokinetic models to the formulations with multiple estradiol components and to the microsphere formulations.
No estradiol concentration–time data were identified for certain other injectable estradiol forms of interest, like unesterified estradiol in aqueous solution, estradiol benzoate as a microcrystalline aqueous suspension (Agofollin Depot; Ovocyclin M), or estradiol benzoate butyrate/dihydroxyprogesterone acetophenide in oil (Redimen, Soluna, Unijab) (another lesser-known combined injectable contraceptive).
All Injectable Estradiol Preparations Together
Figure 8 shows the curve fits for all of the injectable estradiol preparations scaled to a single dose of 5 mg (or equivalent) together in the same figure. The dose for polyestradiol phosphate was scaled to be about 6.5 times higher than the other injectable estradiol preparations in order to make it roughly equivalent to them in terms of total estradiol exposure. This was because polyestradiol phosphate was found to produce much lower area-under-the-curve estradiol levels than the other injectable estradiol preparations (see the Discussion section). Estradiol undecylate was not included in Figure 8 as a decent fit curve could not be obtained for it due to the very limited data available for this preparation.
Figure 8: Curve fits of published estradiol concentration–time data with different injectable estradiol preparations by intramuscular injection scaled to equivalent doses and plotted over a period of 20 days in a single combined graph. Polyestradiol phosphate is scaled to a 6.5-fold higher dose that is roughly equivalent to that for the other esters as it gave total estradiol levels that were around 6 or 7 times lower than the other esters at the same dose. An alternative version of this figure without estradiol benzoate and with the x-axis spanning 30 days is also provided (Graph).
Figure 9 shows simulated curves at steady state for repeated administration of all of the injectable estradiol preparations scaled to a dose of 5 mg (or equivalent) once every 7 days. As with the previous figure, the dose for polyestradiol phosphate was scaled to be about 6.5 times higher than the other injectable estradiol preparations and estradiol undecylate was not included in the figure.
Figure 9: Simulated curves at steady state for repeated administration of different injectable estradiol preparations by intramuscular injection scaled to equivalent doses and plotted over three injection cycles. This simulation was based on the fit curves of the published single-dose estradiol concentration–time data reported in this meta-analysis. Polyestradiol phosphate is scaled to a 6.5-fold higher dose that is roughly equivalent to that for the other esters as it gave total estradiol levels that were around 6 or 7 times lower than the other esters at the same dose. An alternative version of this figure without estradiol benzoate is also provided (Graph).
For more simulated estradiol concentration–time curves with repeated injections of these injectable estradiol preparations, please see the accompanying interactive web simulator.
Selected Pharmacokinetic Parameters
The table below shows selected pharmacokinetic parameters for the fit curves of the included injectable estradiol preparations (Table 9). Estradiol undecylate was not included in the table due to the lack of data needed to achieve a decent curve fit for this preparation and the uncertainty of its parameters.
Table 9: Selected pharmacokinetic parameters for estradiol with injectable estradiol preparations following a single 5 mg dose by intramuscular injection:
Estradiol preparation
Tmax (d)
Cmax (pg/mL)
t1/2 (d)
t90% (d)
AUC0–∞ (pg•d/mL)
Estradiol benzoate in oil
0.65
971
1.2
3.9
2410
Estradiol valerate in oil
2.1
295
3.0
9.9
1886
Estradiol cypionate oil
4.3
155
6.7
22.3
2150
Estradiol cypionate suspension
1.2
241
5.1
16.9
2096
Estradiol enanthate in oil
6.5
160
4.6
15.1
2183
Polyestradiol phosphate a
18.0
34
28.4
94.2
2117
a Scaled instead to a single 32.5 mg injection (6.5 times higher dose than with the other esters).
The table below shows selected pharmacokinetic parameters for simulated curves at steady state with repeated administration of the included injectable estradiol preparations (Table 10). As with the previous table, estradiol undecylate was not included.
Table 10: Selected pharmacokinetic parameters for estradiol with injectable estradiol preparations with simulated repeated administration of 5 mg once every 7 days by intramuscular injection:
Estradiol preparation
Tmax (d)
Cmax (pg/mL)
Cmin (pg/mL)
Peak–trough diff. (pg/mL)
Peak–trough ratio
Cavg (pg/mL)
Estradiol benzoate in oil
0.64
990
29
962
35
344
Estradiol valerate in oil
1.9
384
142
242
2.7
269
Estradiol cypionate oil
3.1
339
262
77
1.3
307
Estradiol cypionate suspension
1.0
404
189
214
2.1
299
Estradiol enanthate in oil
4.0
329
288
41
1.1
312
Polyestradiol phosphate a
3.2
304
299
5
1.0
302
a Scaled instead to repeated injections of 32.5 mg every 7 days (6.5 times higher dose than with the other esters).
Terminal half-life (t1/2) is the time for the concentration of estradiol to decrease by 50% after pseudo-equilibrium of distribution has been reached—not the time required for half of an administered dose of the estradiol ester to be eliminated (Toutain & Bousquet-Mélou, 2004). It is calculated using only the terminal portion of a concentration–time curve, without the absorption or distribution phases influencing it (Toutain & Bousquet-Mélou, 2004). Due to flip–flop kinetics with depot injectables and the very short blood half-life of estradiol (~0.5–2 hours), what is being described by the terminal half-life in the case of depot estradiol injectables is not actually elimination of estradiol from blood but rather is the absorption of estradiol from the injection-site depot (Toutain & Bousquet-Mélou, 2004; Yáñez et al., 2011).
Discussion
Data Quality, Limitations, and Variability Between Studies
The accuracies of the curve fits for the different included injectable estradiol preparations are limited by the available data for these preparations. The quantity and quality of data are variable among these preparations. In some cases, such as with estradiol valerate in oil and estradiol cypionate in suspension, the data are overall quite good. In other instances, such as with estradiol cypionate in oil and estradiol enanthate in oil, the available data are more limited. There was undersampling of certain parts of the concentration–time curve with some preparations, for instance estradiol benzoate in oil (the early curve), estradiol enanthate in oil (much of the curve), and polyestradiol phosphate (the late curve). In the case of estradiol undecylate in oil, the available data for this preparation weren’t adequate to achieve a decent curve fit at all. The fit curves and calculated pharmacokinetic parameters of the included injectable estradiol preparations should be interpreted with the imperfect data in mind. For example, the curve shapes and pharmacokinetic parameters for the different preparations should not be taken as precise determinations in most cases but instead as rough estimates that would no doubt change with more and better data. Indeed, the fits and pharmacokinetic parameters were often noticeably sensitive to the influences of individual studies. Modeling decisions, such as the choice of pharmacokinetic model, or whether to fit directly to the combined processed data versus to the fits of individual studies, also yielded significantly different curve fits as well as calculated pharmacokinetic parameters.
Due to scarcity of data for several injectable estradiol preparations, the study selection criteria maximized data inclusion in order to allow for better curve fits at the risk of including potentially less reliable data. As examples, studies were included regardless of the status of the HPG axis of the participants, and Cmax data were included in the fitting if data were very limited. In the case of HPG axis state, studies with cycling women may result in greater error due to more variable levels of endogenous estradiol. Moreover, acute high levels of estradiol can induce a surge in luteinizing hormone levels after several days in gonadally intact women, and this may cause a delayed bump in estradiol levels (Wiki). One of the more overt instances of this can be seen in a study of estradiol benzoate in such women (Shaw, 1978 [Graph]). Many if not most of the included studies with estradiol benzoate involved women with intact HPG axes, whereas studies of this sort were uncommon with the other preparations. In the case of Cmax data, these data when Cmax corresponds to the mean of individual peaks are a different type of data than the peak of the mean curve of all individuals. Cmax levels can differ in both magnitude and timing compared to the mean curve peak (e.g., Oriowo et al., 1980 [Graph]; Rahimy, Ryan, & Hopkins, 1999). This is because for instance not all individuals peak at the same time and this variability in time to peak normally serves to dilute peak levels for the mean curve when compared to individual maximal concentrations. However, Cmax levels are in any case generally in the vicinity of the mean curve peak. While Cmax levels were excluded in the fitting for most injectable estradiol preparations, they were included in the case of estradiol enanthate. This was because the available mean and individual estradiol curve data were very limited for this specific preparation, and inclusion of Cmax data allowed for improved fitting in spite of its limitations. Lastly, some of the included data was once-monthly multi-dose, and research with once-monthly estradiol enanthate-containing combined injectable contraceptives has found that the time to peak levels may shift with repeated long-term use (Schiavon et al., 1988; Garza-Flores, 1994).
There was considerable variability between studies in terms of estradiol levels and concentration–time curve shapes with the same injectable estradiol preparation. The reasons for the large variability across studies are not fully clear. In any case, there are many potential factors that may contribute to this variability. These include preparation- and injection-related factors like formulation (e.g., oil vehicle, other components and excipients, concentration, particle size), injection volume, site of injection (e.g., buttocks, thigh, upper arm), injection technique (e.g., force of injection—and resulting depot droplet dimensions), and syringe dead space. They additionally include various subject- and research-related variables like differing blood-testing methodology, differing sample characteristics (e.g., age, weight, gender, ethnicity, physical activity, HPG axis state), and sampling error (Sinkula, 1978; Chien, 1981; Minto et al., 1997; Larsen & Larsen, 2009; Larsen et al., 2009; Florence, 2010; Larsen, Thing, & Larsen, 2012; Kalicharan, 2017). Older studies, which used potentially less accurate blood tests and tended to have smaller numbers of subjects, seemed to particularly add to the variability between studies. These studies may represent less reliable data than more recent research with larger sample sizes. The exclusion criteria helped to remove outliers for the different injectable estradiol preparations however. This meta-analysis does not take into account the potential factors underlying the variability between studies. To do so would be difficult, as in many cases information on these variables is not provided in individual studies and research quantifying their precise influences and relative importances is limited.
It is in any case known from other studies that different oil vehicles are absorbed at different rates from the injection site (Svendsen & Aaes‐Jørgensen, 1979; Schultz et al., 1998; Larsen et al., 2001) and can result in different concentration–time curve shapes (Ballard, 1978 [Excerpt]; Knudsen, Hansen, & Larsen, 1985). This is thought to be due to differences in oil lipophilicity and depot release rates. Viscosity of oils has also been hypothesized to potentially influence rate of depot escape (Schug, Donath, & Blume, 2012). However, research so far has not supported this hypothesis (Larsen & Larsen, 2009; Larsen, Thing, & Larsen, 2012). Oil vehicles can vary with injectable estradiol preparations even for the same estradiol ester. For instance, pharmaceutical estradiol valerate is formulated in sesame oil, castor oil, or sunflower oil depending on the preparation (Table). It is notable however that these three oils have similar lipophilicities (Table). On the other hand, homebrewed injectable estradiol preparations used by DIY transfeminine people often employ medium-chain triglyceride (MCT) oil as the oil vehicle. This oil (in the proprietary form of Viscoleo) has notably been found to be much more rapidly absorbed than conventional oils like sesame oil and castor oil in animals (Svendsen & Aaes‐Jørgensen, 1979; Schultz et al., 1998; Larsen et al., 2001). In addition, although based on very limited data, MCT oil has been found to give spikier and shorter-lasting depot injectable curves in humans (Knudsen, Hansen, & Larsen, 1985). As such, injectable estradiol preparations using MCT oil as the vehicle may have differing and less favorable concentration–time curve shapes than pharmaceutical injectable estradiol products. Other excipients, like benzyl alcohol, as well as factors like injection site and volume, have additionally been found to influence pharmacokinetic properties with depot injectables (Minto et al., 1997; Kalicharan, Schot, & Vromans, 2016). Excipients besides oil vehicle also vary by formulation (Table).
An implication of the variability between studies is that there is not a single estradiol concentration–time curve for a given injectable estradiol preparation but rather there are many, with these curves determined by variables such as formulation, dose/administration, and subject characteristics, among others. Hence, the curve fits determined in this meta-analysis represent only an estimation of the most typical and hence likely case, but the true curve for a preparation in a given context may be quite different.
Fitting all studies for a given injectable estradiol preparation individually first, and then fitting the fits of these studies, allowed for improved curve fits relative to directly fitting all of the combined processed original data for the preparation. The latter approach has limitations in that it has the effect of inherently weighting individual studies by quantity of time points (resulting in studies with greater time sampling having greater influence on the fit). Additionally, and more problematically, this approach can lead to distortions in curve shape due to different studies sampling different portions of the curve to differing extents in conjunction with systematic differences in curves between these studies. These are problems that fitting the fits of individual studies instead can solve. However, it is not possible to fit all individual studies, as some studies have limited time sampling and curve characterization which precludes fitting them appropriately. Cmax data are an example of this, which on their own cannot be fit properly. As such, it was not possible to fit the fits of the individual studies for all injectable estradiol preparations. Consequently, the fitting approach in this regard was not the same across esters, with some fit instead directly to the combined processed original data (e.g., estradiol enanthate, polyestradiol phosphate).
In spite of the various limitations of this work, aggregated analysis and modeling with injectable estradiol preparations has not previously been done. This informal meta-analysis provides among the most detailed insight into estradiol levels and curve shapes with these preparations available to date.
Durations and Curve Shapes
The curve shapes of non-polymeric injectable estradiol esters in oil relate strongly to lipophilicity. The more lipophilic the ester, the lower the peak levels and the more protracted the estradiol concentration–time curve. Accordingly, estradiol benzoate, one of the least lipophilic estradiol esters, has one of the spikiest curves and shortest durations, whereas more lipophilic estradiol esters, like estradiol cypionate in oil and estradiol enanthate, have comparatively flatter curves with delayed peaks and longer durations.
Duration of Estradiol Valerate
The estradiol concentration–time curve for injectable estradiol valerate in the well-known Oriowo et al. (1980) [Graph] study is notably spikier and shorter-lasting than the overall curve for estradiol valerate in this meta-analysis. On the other hand, the overall curve for injectable estradiol valerate in this meta-analysis was similar to (and considerably influenced by) the curves from several relatively recent and presumably better-quality studies of this injectable estradiol ester (e.g., Göretzlehner et al., 2002; Valle Alvarez, 2011; Schug, Donath, & Blume, 2012). It’s noteworthy that Oriowo et al. (1980) used a peanut oil-based formulation of estradiol valerate that differed from pharmaceutical injectable estradiol valerate preparations, which generally use sesame oil or castor oil as the carrier (as well as other excipients) (Table). This may have influenced the curve shape of estradiol valerate in Oriowo et al. (1980). The study also had a small sample size relative to the more recent studies (n=9 versus n=17, n=32, and n=24×2, respectively). Based on the newer and overall data, estradiol valerate appears to have a curve that is noticeably flatter and more prolonged than that suggested by Oriowo et al. (1980).
Duration of Estradiol Cypionate in Oil versus Estradiol Enanthate
Available estradiol concentration–time data for injectable estradiol cypionate in oil and estradiol enanthate in oil are more limited than with several of the other injectable estradiol preparations, and no direct comparisons of these two preparations exist at present. Based on some of the available literature on these injectable estradiol esters, most notably discussion by Oriowo et al. (1980) and a review of the pharmacokinetics of combined injectable contraceptives (Garza-Flores, 1994 [Graph]), it seemed that the duration of estradiol enanthate in oil was longer than that of estradiol cypionate in oil. However, this was based on limited research from separate and hence indirectly comparative studies of these esters. The estradiol cypionate in oil data from the relevant Garza-Flores (1994) figure was based on Oriowo et al. (1980) [Graph], and there are reasons to be cautious about relying on these data alone. The main concern is that curve shapes with the same injectable estradiol preparation can vary considerably across studies, as the present meta-analysis has shown. The reasons for this have yet to be fully clarified as already discussed, but among other factors may include varying formulations across studies of the same injectable estradiol ester. It is notable in this regard that Oriowo et al. (1980) used a formulation of estradiol cypionate that differs from conventional pharmaceutical estradiol cypionate in oil preparations—specifically, the study used a peanut oil-based formulation (with few other specifics) rather than the cottonseed oil-based preparation employed in marketed pharmaceutical formulations (Table). The study also had a somewhat small sample size (n=10) and may have had significant sampling error. Hence, single studies, perhaps particularly Oriowo et al. (1980), should be interpreted cautiously.
A small but interesting pharmacokinetic study which directly compared injectable testosterone cypionate (n=6) and testosterone enanthate (n=6) both in oil is relevant to the topic in question. This study found that equivalent doses of these testosterone esters using otherwise identical formulations produced virtually identical testosterone concentration–time curves (Schulte-Beerbühl & Nieschlag, 1980 [Graph]). The findings of this study are consistent with the fact that the lipophilicities of testosterone cypionate and testosterone enanthate (as measured by predicted log P) are very similar when directly compared (e.g., 5.1 vs. 5.11 with ALOGPS, 6.29 vs. 6.11 with ChemAxon logP, and 6.4 vs. 6.3 with XLogP3, respectively (Table). This of course is of importance as lipophilicity is thought to be the key factor determining the release kinetics of oil-based depot injectables (Sinkula, 1978; Shah, 2007; Larsen & Larsen, 2009; Larsen, Thing, & Larsen, 2012; Shahiwala, Mehta, & Momin, 2018). Analogously similar lipophilicities can be seen when comparing estradiol cypionate and estradiol enanthate, which employ the same ester moieties (e.g., predicted log P values of 6.47 vs. 6.45 with ALOGPS and 7.1 vs. 7.0 with XLogP3, respectively) (Table). Hence, on a theoretical level, injectable estradiol cypionate and estradiol enanthate, like injectable testosterone cypionate and testosterone enanthate, might be expected to produce very similar curves—at least provided all other variables, such as formulation, are held constant.
The present meta-analysis found that the overall estradiol curve for estradiol cypionate in oil was significantly less spikey and more prolonged than that observed in Oriowo et al. (1980). It is noteworthy in this regard that all of the other studies included for estradiol cypionate in oil specifically employed pharmaceutical Depo-Estradiol and that the overall curve for this preparation appears to be more consistent with its licensed injection interval for use in menopausal hormone therapy (1–5 mg once every 3–4 weeks) (Depo-Estradiol Label). Moreover, this meta-analysis found that injectable estradiol cypionate in oil and estradiol enanthate in oil had fairly similar and comparably flat and prolonged estradiol concentration–time curves. However, estradiol cypionate in oil appeared to peak earlier than estradiol enanthate, while estradiol enanthate was eliminated more rapidly than estradiol cypionate in oil in the terminal portion of the curve. In any case, the available concentration–time data for these preparations are limited, and the present work is not able to determine whether these estradiol esters have truly differing pharmacokinetic properties, as the apparent differences between the curves for these preparations may simply be due to statistical error. Taken together, estradiol cypionate in oil may have a less spikey and longer-lasting curve than that implied by Oriowo et al. (1980), and estradiol cypionate in oil and estradiol enanthate may have more similar curves than has been previously assumed.
Curve Shape of Estradiol Cypionate Suspension
While estradiol cypionate as an aqueous suspension is a relatively long-lasting injectable estradiol preparation similarly to estradiol cypionate in oil and estradiol enanthate in oil, it seems to differ in the shape of its estradiol concentration–time curve from these preparations. Estradiol cypionate as a suspension has a curve that appears to peak significantly earlier than estradiol cypionate in oil and other longer-acting oil-based injectable estradiol preparations. This might relate to the differing mechanisms of depot action and unique properties of injectable aqueous suspensions (Aly, 2019). In line with this notion, injectable medroxyprogesterone acetate suspension (Depo-Provera) also appears to peak rapidly despite having a very long duration (longer durations tending to be associated with delayed peaks in the case of oil-based depot injectables) (Graphs). Although aqueous suspensions generally last longer than oil solutions as injectables (Enever et al., 1983; Aly, 2019), this is not always the case, and estradiol cypionate suspension interestingly seems to be shorter-acting than estradiol cypionate in oil.
Estradiol Exposure and Potency
The average estradiol levels with the non-polymeric injectable estradiol esters when scaled to a dose and dosing interval of 5 mg every 7 days were around 300 pg/mL (~1,100 pmol/L). For comparison, in premenopausal cisgender women, estradiol production is on average about 200 μg/day (or 6 mg per month/cycle) and mean estradiol levels are around 100 pg/mL (~370 pmol/L) (Aly, 2019). After adjusting for the molecular weight of the ester, the estradiol levels for a given dose of non-polymeric injectable estradiol esters are in fairly close agreement with the estradiol levels for an equal quantity of estradiol produced endogenously by the ovaries in premenopausal cisgender women (very roughly around 1.2 mg estradiol per 7 days for injectable estradiol esters and 1.4 mg estradiol per 7 days for ovarian production to achieve average integrated estradiol levels of around 100 pg/mL). The preceding is in accordance with the fact that injectable estradiol valerate has been reported to have approximately 100% bioavailability (with this being less characterized but likely also the case for the other non-polymeric injectable estradiol esters) (Düsterberg & Nishino, 1982; Seibert & Günzel, 1994).
Although non-polymeric injectable estradiol esters have differing estradiol concentration–time curve shapes, they all appear to achieve fairly similar area-under-the-curve levels of estradiol when compared to one another. This is in accordance with the fact that differences in molecular weight and hence estradiol content with the different estradiol esters are fairly minor (all of the assessed non-polymeric esters range from 62 to 76% of that of estradiol in terms of estradiol content, and all but estradiol undecylate are in the range of 69 to 76%) (Table). The appearance of differences in area-under-the-curve levels of estradiol in the present meta-analysis is probably just due to statistical error, and true differences cannot be established by this meta-analysis. An implication of the similar area-under-the-curve estradiol levels with the different non-polymeric injectable estradiol esters is that these preparations can all be expected to deliver a roughly comparable amount of estradiol for the same dose.
On the other hand, the polymeric ester polyestradiol phosphate appears to produce around 6- to 7-fold lower area-under-the-curve and average estradiol levels than non-polymeric estradiol esters. This suggests that the estradiol in polyestradiol phosphate is not 100% bioavailable, and is supported by the fact that this ester is used clinically at substantially higher dosages than other injectable estradiol esters (40–320 mg/month), even for the same indications such as menopausal hormone therapy and treatment of prostate cancer (Wiki; Estradurin Labels). This does not seem to have been previously described in the literature, and the reasons for it are unknown. It seems possible that polyestradiol phosphate may be partially excreted before it can be cleaved into estradiol and thereby rendered partly inactive, in turn necessitating the use of higher doses to achieve the same estradiol levels and therapeutic effect.
Although two given injectable estradiol preparations may produce equivalent total estradiol levels, this does not necessarily mean that they will always have the same estrogenic potency (i.e., strength of effect at a given dose). It is plausible that spikier estradiol concentration–time curves, like with estradiol benzoate, may have overall lower estrogenic potency than more steady curves, like with estradiol enanthate. This is because estrogen receptors for a given tissue should become saturated at a certain point due to the finite quantity of available receptors in the tissue. As a result, high peak estradiol levels with spikier curves may effectively be “wasted” to varying extents in different tissues. On the other hand, more spikey estradiol curves, due to higher peak estradiol levels, might have greater influence on tissues that require high estradiol levels for effect such as the liver (and by extension on coagulation and associated health risks) (Aly, 2020). However, these possibilities are speculative and theoretical. Although some literature exists that is relevant to this issue (e.g., Parkes, 1937; Bradbury, Long, & Durham, 1953), there is very little research in this area. Consequently, it is not currently possible to take into account time-related variations in estradiol levels or differing estradiol curve shapes when assessing the comparative estrogenic potency between injectable estradiol preparations (or between other estradiol forms/routes). It is also noteworthy that these variations depend on injection interval and may be reduced with shorter injection intervals that maintain steadier estradiol levels, which must also be considered.
Variability Between Individuals
There is substantial variation in total estradiol levels and curve shapes between people with the same injectable estradiol preparation. Indicators of interindividual variability such as standard deviation or 95% range have not been included in this meta-analysis at this time due to the large amount of additional time and work this would require (e.g., additional extraction of error bars from all studies and analysis). In any case, individual studies that were included show this marked interindividual variation (e.g., Oriowo et al., 1980; Derra, 1981 [Graph]; Aedo et al., 1985 [Graphs]; Sang et al., 1987 [Graphs]; Rahimy & Ryan, 1999 [Graph]; Valle Alvarez, 2011 [Graph]; Schug, Donath, & Blume, 2012 [Graphs]). Highly variable estradiol levels are already well-established with oral and transdermal estradiol (Kuhl, 2005; Wiki). Less variability might be expected with non-polymeric injectable estradiol esters since these preparations appear to have approximately complete bioavailability. However, it seems that even with injectable forms of estradiol, the variability between people is still quite substantial. An implication of this is that the appropriate dose and dosing interval of an injectable estradiol formulation for a given person will vary considerably. This emphasizes the importance of blood work to ensure that injectable estradiol preparations are neither overdosed—which can increase health risks such as blood clots (Aly, 2020)—nor underdosed—which may result in suboptimal testosterone suppression and therapeutic efficacy.
Insights for Clinical Guidelines and Dosing Recommendations
Clinical guidelines for transgender health (see also Aly (2020)) provide recommendations on doses and dosing intervals of injectable estradiol valerate in oil and estradiol cypionate in oil (Table 11). Dosing recommendations are not given for other injectable estradiol preparations, which are much less commonly used in transgender medicine. The recommended doses for estradiol valerate and estradiol cypionate vary widely depending on the guidelines, whereas the recommended intervals are consistently once every 1 to 2 weeks. The doses for estradiol valerate range from 2 to 20 mg/week or 5 to 80 mg/2 weeks and the doses for estradiol cypionate range from <1 to 10 mg/week or <2 to 80 mg/2 weeks. For reference, the Endocrine Society guidelines and the University of California, San Francisco (UCSF) guidelines are the most major clinical guidelines for transgender hormone therapy at present (Aly, 2020). The Endocrine Society guidelines recommend 5 to 30 mg/2 weeks or 2 to 10 mg/week for either estradiol valerate or estradiol cypionate (Hembree et al., 2017). Conversely, the UCSF guidelines recommend <20 to 40 mg/2 weeks for estradiol valerate and <2 to 5 mg/2 weeks for estradiol cypionate (with the option to divide dose into weekly injections if cyclical side effects occur) (Deutsch, 2016a).
Table 11: Recommended doses and injection intervals of injectable estradiol preparations (specifically estradiol valerate and estradiol cypionate) in transgender medicine clinical guidelinesa:
a Several other guidelines recommend doses and intervals that appear to be taken directly from the Endocrine Society or UCSF guidelines and thus are not listed here but can be found elsewhere (Aly, 2020).
A number of concerns arise when the doses and intervals of injectable estradiol valerate and estradiol cypionate recommended by the major transgender clinical guidelines are considered in the context of the present informal meta-analysis and when they are compared between guidelines. Based on the present work, dosages of injectable preparations recommended by the major transgender clinical guidelines appear to result in estradiol exposure that is markedly higher than that with the recommended dosages for other routes and forms of estradiol (e.g., oral or transdermal). Whereas a dosage of 5 mg/week of any non-polymeric injectable estradiol ester appears to give average estradiol levels of around 300 pg/mL (~1,100 pmol/L), which are already supraphysiological, doses of injectable estradiol valerate or estradiol cypionate recommended by guidelines are as high as 15 to 20 mg per week. The average estradiol concentrations that would be expected to result from such doses per this meta-analysis (e.g., ~600–1,200 pg/mL or 2,200–4,400 pmol/L at 10–20 mg/week) (Figure 10) would vastly exceed the ranges for estradiol levels in transfeminine people advised by the same guidelines (generally about 50–200 pg/mL or ~180–730 pmol/L) (Table). This is not merely theoretical; for example, a study that used 40 mg/week estradiol valerate by intramuscular injection in cisgender women with estrogen deficiency to produce “pseudopregnancy” reported measured estradiol levels of about 2,500 pg/mL (~9,200 pmol/L) at 3 months and 3,100 pg/mL (~11,400 pmol/L) at 6 months of treatment (Ulrich, Pfeifer, & Lauritzen, 1994). Moreover, highly supraphysiological estradiol levels with guideline-based injectable estradiol doses are not unexpected when normal production of estradiol in premenopausal cisgender women is considered (~1.4 mg per week or 6 mg per month/cycle giving mean estradiol levels of ~100 pg/mL or 370 pmol/L) (Aly, 2019). Clinical safety data on high doses of injectable estradiol esters like estradiol valerate and estradiol cypionate are lacking at present, but excessive estrogenic exposure is known to increase the risk of health complications such as blood clots (Aly, 2020). The very high doses of these preparations that are recommended by guidelines should raise considerable reservations about their safety.
Figure 10: Simulated estradiol levels with injectable estradiol valerate at the doses and interval (5–40 mg/2 weeks) preferentially recommended by current major transgender care guidelines. Steady-state estradiol levels are reached by about the second or third injection with this injection interval and levels do not further accumulate. An alternative version of this figure with half-doses at a once-weekly interval (i.e., 2.5–20 mg/week) is also provided (Graph).
The present author elsewhere has listed doses of injectable estradiol preparations that are roughly comparable in terms of total estradiol exposure to doses for other estradiol forms and routes used in transfeminine people (Aly, 2020). These doses range from about 1 to 6 mg per week for “low dose” to “very high dose” therapy with non-polymeric injectable estradiol esters (Graph). This dose range for injectable estradiol is likely to be more appropriate for use in transfeminine people than current recommendations by many guidelines. Although high estradiol levels can be useful in transfeminine hormone therapy when antiandrogens are not used due to their greater efficacy than physiological levels in terms of testosterone suppression, only modestly supraphysiological estradiol levels (e.g., ~200–300 pg/mL or 730–1,100 pmol/L) appear to be required for strong testosterone suppression (Aly, 2019; Langley et al., 2021; Aly, 2020). In relation to this, doses of injectable estradiol need not be excessive.
Some guidelines, such as the Endocrine Society guidelines, recommend the same doses and intervals for both estradiol valerate and estradiol cypionate, whereas other guidelines, such as the UCSF guidelines, recommend different doses for these two injectable estradiol esters. Concerningly, the doses for estradiol valerate and estradiol cypionate recommended by the UCSF guidelines differ by roughly an order of magnitude (<20 to 40 mg/2 weeks for estradiol valerate and <2 to 5 mg/2 weeks for estradiol cypionate). These estradiol esters appear to produce similar average estradiol levels (e.g., around 300 pg/mL or 1,100 pmol/L at a dosage of 5 mg/week) and have concentration–time curve shapes that are not extremely different, with estradiol cypionate being only somewhat flatter and more prolonged than estradiol valerate. As such, it would appear that similar doses should be appropriate for these esters. This is supported by the fact that the same doses of estradiol valerate and estradiol cypionate are used in combined injectable contraceptives in cisgender women (both 5 mg once per month) and that these doses were carefully determined during an intensive clinical development programme for these preparations (Garza-Flores, 1994; Newton, d’Arcangues, & Hall, 1994; Sang, 1994; Toppozada, 1994). This programme notably included dose-ranging and direct-comparison studies. Based on the present analysis, the current recommendations by the UCSF guidelines may result in marked overdosage in the case of estradiol valerate and potential underdosage in the case of estradiol cypionate.
Transgender health guidelines recommend an injection interval for estradiol valerate and estradiol cypionate in oil of once every 1 to 2 weeks. Although an injection interval of 2 weeks seems technically feasible in the case of both of these preparations, such an interval would appear to result in substantial fluctuations in estradiol levels, with high peak levels and low troughs. This is particularly true in the case of the shorter-acting estradiol valerate (Figures 10, 11). Considering the wide fluctuations and unknown effects of this variability, as well as the fact that testosterone suppression when applicable may depend on sustained higher estradiol levels, it may be advisable that a once-weekly interval be preferentially recommended for these preparations. This would achieve steadier estradiol levels and would reduce potential problems due to high or low estradiol levels (Figure 11). Alternatively, a shorter interval of once every 5 days may be used with estradiol valerate to further reduce the variability in estradiol levels that occurs with this preparation (Figure 11). On the other hand, an injection interval of once every 10 days to 2 weeks may be practical and allowable in the case of the longer-acting estradiol cypionate in oil (as well as estradiol enanthate) (Figure 11)—provided that the injection cycles are well-tolerated and testosterone suppression remains adequate. When selecting different injection intervals, doses should be scaled by the interval to maintain equivalent total estradiol exposure (e.g., 3.5 mg/5 days, 5 mg/7 days, 7 mg/10 days, or 10 mg/14 days for high-dose non-polymeric injectable estradiol esters).
Figure 11: Simulated estradiol levels with a high dosage of injectable estradiol valerate or estradiol cypionate in oil at different injection intervals (doses scaled by interval to be equivalent in total estradiol exposure).
With the preceding concerns about the doses and intervals of injectable estradiol preparations recommended by transgender care guidelines considered, the question of how these recommendations were determined arises. Unfortunately, current guidelines do not generally describe how they arrived at their recommendations nor do they usually cite sources to support them. It is notable that the UCSF guidelines recommend doses and intervals for injectable estradiol preparations that are nearly identical to those advised by Christian Hamburger and Harry Benjamin in the late 1960s in the first medical textbook on transgender people (Hamburger & Benjamin, 1969). These authors recommended a dose of 10–40 mg/2 weeks for estradiol valerate and of 2–5 mg/2 weeks for estradiol cypionate (although Benjamin additionally stated that after 4–8 months, the same doses could be used at a longer injection interval of once every 4 weeks). These recommendations were notably made before estradiol blood tests became practicably available and were prior to the advent of modern pharmacokinetic studies. Hence, the recommendations for at least these guidelines appear to be based mainly on past expert opinion and long-standing historical precedent rather than on pharmacokinetic or clinical data. The same is likely to also be true for most other guidelines. High doses with certain injectable estradiol preparations (namely estradiol valerate) were probably originally employed for the purpose of achieving longer durations and more convenient injection intervals. This was notably prior to the risks of excessive estrogenic exposure like blood clots becoming known, and these doses may simply have never been revised.
Among the surveyed guidelines for transgender hormone therapy, only the UCSF guidelines (Deutsch, 2016b) and the Sherbourne Health/Rainbow Health Ontario guidelines (Bourns, 2019) referenced pharmacokinetic literature in their discussion of injectable estradiol. The specific publications cited by these guidelines were Düsterberg & Nishino (1982), Sierra-Ramírez et al. (2011), and Thurman et al. (2013). Although it is favorable to see guidelines considering published pharmacokinetic data for informing use of these preparations, there are a few concerns about the studies that were cited. Düsterberg & Nishino (1982) in its study of injectable estradiol valerate had a very small sample size (n=2), and this study was excluded as an outlier in the present meta-analysis due to unusually high estradiol levels. The findings of Düsterberg & Nishino (1982) also do not seem to have actually been used to guide dosing recommendations in the case of the UCSF guidelines, since if this were the case, the recommended doses should have been much lower. On the other hand, Bourns (2019) cited the same study and recommended injectable estradiol valerate at doses of 3–4 mg/week or 6–8 mg/2 weeks. These doses are well below those recommended by other transgender care guidelines and appear to be more appropriate for use in transfeminine people in light of the present meta-analysis. Sierra-Ramírez et al. (2011) and Thurman et al. (2013), although better-quality studies than Düsterberg & Nishino (1982), described injectable estradiol cypionate suspension rather than estradiol cypionate in oil. The oil-based version of estradiol cypionate is the form normally used in transfeminine hormone therapy, and there are important differences between these estradiol cypionate preparations such that pharmacokinetic studies for the suspension can’t necessarily be generalized to the oil solution. These preparations do in any case produce similar total estradiol levels however and hence doses should be comparable for them.
This meta-analysis is only informal and unpublished research. Nonetheless, based on the results of this work and the preceding discussion, current dosing recommendations for injectable estradiol preparations by most transgender clinical guidelines appear to be highly excessive and likely unsafe, with injection intervals that may additionally be too widely spaced. Transgender care guidelines should consider reassessing these recommendations, and the transgender medical community should make an effort to better characterize the pharmacokinetics and optimal dosing schemes of injectable estradiol preparations in transfeminine people in the future. Since clinical data on these preparations are scarce and will probably remain so in the near-term, use of published pharmacokinetic data may be further considered for guiding dosing recommendations for injectable estradiol. As identified and catalogued by this meta-analysis, there is a wealth of existing data that could be used to better inform transgender care guidelines in terms of the use of injectable estradiol preparations in transfeminine people.
Interactive Web Simulator
This informal meta-analysis of estradiol concentration–time data with injectable estradiol preparations was conducted for the purpose of deriving accurate and representative estradiol curves for incorporation into a web-based injectable estradiol simulator intended for use by transfeminine people and their clinicians. This web app is able to simulate both single-injection curves and repeated-injection curves with these preparations. An informational page for this simulator can be found at the following location:
There are various possibilities for further work on this project in the future. For example, assessment of interindividual variability for estradiol levels with injectable estradiol preparations could be included in the meta-analysis. As another example, it would be fairly straightforward and valuable to expand the meta-analysis as well as simulator to other hormonal preparations such as injectable testosterone preparations and other estradiol routes and forms like oral estradiol, sublingual estradiol, and estradiol pellets. Pharmacokinetic literature for some of these preparations has already been collected by this author. However, these future possibilities would require much additional time and effort to complete.
Special Thanks
A special thank you to Violet and Lila for their indispensable input and guidance on modeling topics during the work on this project. An additional thanks to Violet for deriving a special three-compartment pharmacokinetic model that was used in this work. Please also check out Violet’s own projects Tilia—an effort to empower trans people with tools to manage their hormonal transitions—and TransKit—a work-in-progress pharmacokinetic simulation library specifically tailored for transgender hormone therapy. Lastly, thank you to all the peer reviewers who carefully reviewed this article prior to it being posted.
Updates
Update 1: WPATH SOC8 Guidelines
In September 2022, the World Professional Association for Transgender Health (WPATH) Standards of Care for the Health of Transgender and Gender Diverse People Version 8 (SOC8) were published and made recommendations on transgender hormone therapy for the first time (Coleman et al., 2022). These guidelines are among the most highly regarded and consulted transgender care guidelines. In terms of the recommended doses of hormonal medications for transgender people, the WPATH SOC8 appear to have largely copied the Endocrine Society’s 2017 guidelines on transgender hormone therapy (Hembree et al., 2017). More specifically, in the case of injectable estradiol preparations for transfeminine people, doses of 5–30 mg/2 weeks or 2–10 mg/week estradiol valerate or estradiol cypionate were recommended. There was no discussion of injectable estradiol in the guidelines besides the preceding doses and intervals being included in a table, and no literature citations were included to support these doses. As described in the present work, these recommendations include doses and intervals that appear to be highly excessive, too widely spaced, and are likely unsafe. As such, major transgender care guidelines unfortunately continue to make uncited recommendations for injectable estradiol that are out of step with insights available from abundant published pharmacokinetic data. These recommendations are likely inadvisable, with the possibility of substantial health risks.
Update 2: Literature Mentions
The following publications in the literature have cited or mentioned Transfeminine Science’s injectable estradiol simulator and/or meta-analysis since the project was published in mid-2021:
Hughes et al. (2022)
Hughes, J. H., Woo, K. H., Keizer, R. J., & Goswami, S. (2022). Clinical Decision Support for Precision Dosing: Opportunities for Enhanced Equity and Inclusion in Health Care. Clinical Pharmacology & Therapeutics, 113(3), 565–574. [DOI:10.1002/cpt.2799]:
Lastly, we recommend that developers of [clinical decision support software (CDSS)] for dosing take an iterative and participatory approach to designing systems. By involving stakeholders in the design process, they will develop solutions that best suit users’ needs and are more likely to be adopted and used correctly. This participatory approach should involve interviews and usability testing with clinicians. Formal usability testing and analysis with real end users can improve the quality and usefulness of a system.88 Though patients themselves are not typically the end users of CDSS, their expertise (especially that of marginalized groups and organized patient advocacy organizations) can also inform CDSS developers. As an example, transgender people have compiled their own resources to understanding dosing regimens in the absence of clear clinical guidelines.89 Developers of CDSS could provide a great deal of value to these patient populations, and improve their software’s utility, by working with them to understand their needs from a dosing tool.
89. Aly, W. An interactive web simulator for estradiol levels with injectable estradiol esters. Transfeminine Science <https://transfemscience.org/articles/injectable-e2-simulator-release/> (2021) Accessed November 1, 2022.
Jaafar et al. (2022)
Jaafar, S., Torres-Leguizamon, M., Duplessy, C., & Stambolis-Ruhstorfer, M. (2022). Hormonothérapie injectable et réduction des risques: pratiques, difficultés, santé des personnes trans en France. [Hormone replacement therapy injections and harm reduction: practices, difficulties, and transgender people’s health in France.] Sante Publique, 34(HS2), 109–122. [Google Scholar] [PubMed] [DOI:10.3917/spub.hs2.0109] [Translated]:
With regard to feminizing [substitutive hormone therapy (HS)], there are no specialty injectables based on estrogens in the French pharmacopoeia. This makes it impossible to set up estrogen monotherapies which require high dosages that are more difficult to obtain with specialties with other galenic forms [5]. Faced with this lack of care, some trans women and transfeminine people obtain estradiol-based injectable solutions on the Internet or through other sources [6]. […]
5. Aly. An informal meta-analysis of estradiol curves with injectable estradiol preparations [Internet]. Transfem Sci. 2021 July 16. [Visited on 29/12/2022]. Online : https://transfemscience.org/articles/injectable-e2-meta-analysis/.
Linet (2023)
Linet, T. (2023). Prise en charge endocrinologique d’une personne trans. [Endocrinological care of a trans person.] In Faucher, P., Hassoun, D., & Linet, T. (Eds.). Santé sexuelle et reproductive des personnes LGBT [Sexual and Reproductive Health of LGBT People] (pp. 109–124). Issy-les-Moulineaux, France: Elsevier Masson. [Google Books] [URL] [WorldCat] [Excerpt] [Translated]:
Choice of estrogen.
Estradiol is generally the most prescribed estrogen. It is given orally or sublingually in transfeminine people with no significant cardiovascular risk factors. For others, the percutaneous form (patches, gel) is recommended.
The starting dose is 2 mg of estradiol orally with a step increase of 2 mg every 2 to 3 months until the optimal dose is reached [1]. For the patches, the initial dosage and the increments are 50 or 100 μg, and for the gel 2.5 g. This means that the optimal dose is generally 6 to 8 mg per day for the oral route, 3 to 4 mg for the sublingual route, and 300 to 400 μg for the patches (see table 11.1).
It may happen in consultation that the person does not wish to use the prescribed estrogens and wishes to continue the self-prescription of injectable estrogens. It is then possible to evaluate with them the most suitable dosage using the Transfem Science Injection Simulator (https://transfemscience.org/misc/injectable-e2-simulator/). Risk prevention related to injections (needles) can be done. Associations can help the person find 25 G needles of 40 mm useful this type of treatment.
Rothman et al. (2024)
Rothman, M. S., Ariel, D., Kelley, C., Hamnvik, O. R., Abramowitz, J., Irwig, M. S., Soe, K., Davidge-Pitts, C., Misakian, A. L., Safer, J. D., & Iwamoto, S. J. (2024). The Use of Injectable Estradiol in Transgender and Gender Diverse Adults: A Scoping Review of Dose and Serum Estradiol Levels. Endocrine Practice, 30(9), 870–878. [DOI:10.1016/j.eprac.2024.05.008]:
In recent years, we have noted trends in our clinical practices with TGD adults requesting injectable estradiol, particularly in the United States. The reasons given can vary; it may be due to ease of weekly or every two weeks administration, fatigue of taking daily oral medications and skin reactions to or cost of transdermal preparations. There have been discussions as to the roles of estrone/estradiol ratios in feminization and whether injectable estradiol might lead to more favorable results, however research has not supported a role for estrone in optimizing feminizing outcomes [13]. There is also a belief that higher levels can be attained with injections and may lead to faster and more complete feminization; however, there is a lack of data in the literature to support these conclusions. Such conversations occurring on reddit.com and even some hormone provider websites, are perhaps related to the historical use of high dose injectable estradiol noted above [14]. However, there is a paucity of data to guide clinicians on what dose, type and at what interval estradiol esters should be injected and when levels should be measured to ensure physiologic range estradiol levels. In fact, recent reports and clinical observations have raised concerns that the dosing suggested in guidelines may result in supraphysiological estradiol levels and that higher doses and levels may put patients at elevated risk of thromboembolic events [15-18]. This scoping review examines the available data on levels achieved with various dosages of estradiol injections in TGD adults. We also report on testosterone suppression, route (i.e., SC vs. IM), and type of estradiol ester as well as timing of blood draw relative to dose, where available.
Acknowledgment
[…] [We] thank Aly from Transfemscience for community representation and correspondence.
Toffoli Ribeiro, C., Gois, Í., da Rosa Borges, M., Ferreira, L. G. A., Brandão Vasco, M., Ferreira, J. G., Maia, T. C., & Dias-da-Silva, M. R. (2024). Assessment of parenteral estradiol and dihydroxyprogesterone use among other feminizing regimens for transgender women: insights on satisfaction with breast development from community-based healthcare services. Annals of Medicine, 56(1), 2406458. [DOI:10.1080/07853890.2024.2406458]:
Utilizing a previously published meta-analysis method of estradiol concentration-time data from publicly available information on cisgender women who had used EEn or EEn/DHPA [17], we reanalyzed and integrated data from various studies. […]
[…] The V3C Fitter and Desmos tools, accessible online at https://alyw234237.github.io/injectable-e2-simulator/v3c-fitter/ and https://www.desmos.com/calculator/ndgvp2avhj?lang=pt-BR respectively, were utilized for fitting the three-compartment pharmacokinetic model. […]
Pharmacokinetics of injectable estradiol enanthate
[…] The boxplot graph (Figure 5) illustrates that the median estradiol levels in trans women using EEn/DHPA fell within this population’s expected average range values (100–200pg/mL).
Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. […]
References
[17] Aly. 2021. An informal meta-analysis of estradiol curves with injectable estradiol preparations. Transfeminine Sci. https:// transfemscience.org/articles/injectable-e2-meta-analysis/
Update 3: Herndon et al. (2023)
In March 2023, the following study on injectable estradiol in transfeminine people was published online:
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of Subcutaneous and Intramuscular Estradiol Regimens as part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
The study was a retrospective analysis of individualized injectable estradiol in adult transfeminine people who received hormone therapy at the Mayo Clinic. Doses of injectable estradiol were adjusted by clinical providers based on estradiol levels, testosterone suppression, and feminization goals, and subsequently these clinical data were retrospectively studied by Mayo Clinic researchers. The primary aim of the study was to compare injectable estradiol by intramuscular versus subcutaneous routes. However, other general considerations for injectable estradiol, such as dosing, estradiol levels, testosterone suppression, type of injectable estradiol ester (estradiol valerate vs. estradiol cypionate), and estradiol monotherapy versus concomitant use of antiandrogens, were also assessed. The paper noted that the study was the largest to assess injectable estradiol in transfeminine people to date and was the first to directly compare intramuscular and subcutaneous injectable estradiol routes in transfeminine people.
Injectable estradiol doses were adjusted to achieve estradiol and testosterone levels within therapeutic ranges defined by the Endocrine Society 2017 guidelines (>100 pg/mL [367 pg/mL] for estradiol and <50 ng/dL [<1.7 nmol/L] for testosterone). Estradiol levels were measured exclusively using liquid chromatography–tandem mass spectrometry (LC–MS/MS), while the assay method for measuring testosterone levels was not specified. In terms of when in the injection cycle estradiol levels were measured, the authors stated the following: (1) “Timing of estradiol blood draw in relation to injection was not protocolized” and (2) “In our practice, although estradiol concentrations were generally checked midway through the injection cycle, we were unable to document with certainty the timing of the estradiol lab testing which may have influenced the results and/or the outliers”. Only the most recent blood test for each individual was analyzed, with the results of earlier tests discarded. Doses were analyzed in per-week amounts, regardless of dosing frequency (either once weekly or once every two weeks).
There were a total of 130 transfeminine people on injectable estradiol who were analyzed in the study. Of these individuals, 56 received intramuscular (i.m.) injections and 74 received subcutaneous (s.c.) injections. The median duration of therapy for injectable estradiol was 3.0 years for both routes. The vast majority of people used weekly injections (91.1% for i.m., 98.6% for s.c.), whereas the small remainder (n=6) injected once every 2 weeks. Similarly, the great majority used injectable estradiol valerate (89.3% for i.m., 86.5% for s.c.), while a small subset (n=16) used injectable estradiol cypionate. The authors did not state the injectable vehicles, but they can be confidently assumed to have both been in oil. The treatment-individualized doses per week of injectable estradiol were median 4 mg (interquartile range (IQR) 3–5.15 mg; range 1–8 mg) for the i.m. route and median 3.75 mg (IQR 3–4 mg; range 1–8 mg) for the s.c. route, with the differences in doses between groups being slightly but significantly different (p = 0.005). For the small number of people on two-week injection cycles, the doses for the combined i.m. and s.c. groups were median 8.5 mg (range 6–16 mg) every 2 weeks. Estradiol levels with injectable estradiol were median 189.5 pg/mL (IQR 126.8–252.5 or 122.5–257 pg/mL; range ~33–575 pg/mL] for i.m. and median 196 pg/mL (IQR 125.3–298.5 pg/mL; range ~23–581 pg/mL) for s.c., with the differences between groups not being significantly different (p = 0.70). The percentages of individuals with estradiol levels in target range (>100 pg/mL) were 78.6% for i.m. and 82.4% for s.c. The estradiol doses and levels of individual patients for each route were also provided in the paper (Graph). It can be seen that more individuals clustered into the higher range of doses with i.m. than with s.c. injections.
In the case of estradiol valerate versus estradiol cypionate, dose per week for i.m. with estradiol valerate was median 4 mg (IQR 3–5.45 mg) and with estradiol cypionate was median 4 mg (IQR 2.25–5 mg). In the case of s.c., dose per week with estradiol valerate was median 4 mg (IQR 3–4 mg) and with estradiol cypionate was median 3 mg (IQR 2–3 mg). The doses between estradiol valerate and estradiol cypionate were not significantly different in the case of i.m. (p = 0.51), but were significantly different in the case of s.c. (p = 0.025). Estradiol levels with the two different injectable estradiol esters were not provided.
On multiple regression analysis, injectable estradiol dose was significantly positively associated with estradiol levels (p < 0.001) following adjustment for several variables (injection route, body mass index (BMI), antiandrogen use, gonadectomy status). Each 1 mg per week in dose was associated with estradiol levels that were increased by (estimate ± standard error) 57.42 ± 10.46 pg/mL. No other variable, including notably BMI, was significantly associated with estradiol levels. According to the authors, the dose-dependently higher estradiol levels with injectable estradiol suggested the need to start at lower doses that are gradually increased in conjunction with close monitoring of hormone levels.
Testosterone levels in those with gonads were 11 ng/dL (IQR 0–19.8 ng/dL) for i.m. and 11 ng/dL (0–20 ng/dL) for s.c., with levels not significantly different between groups (p = 0.92). Adequate testosterone suppression (<50 ng/dL) in those with gonads was achieved in 84.6% with i.m. and 86.6% with s.c. In the small subset of individuals on injections every two weeks (n=6), 100% of individuals achieved target estradiol and testosterone levels. A majority of individuals on injectable estradiol in the study concomitantly used an antiandrogen, with this usually being spironolactone or finasteride. In a minority of individuals, injectable estradiol monotherapy, without concomitant use of an antiandrogen, was employed and hormone levels were measured (n=17). In this subgroup, estradiol levels were median 220 pg/mL (IQR 180–264 pg/mL) at a dose per week of median 4 mg (IQR 3–6 mg), with estradiol levels noticeably higher than in the overall group. In terms of hormone levels for those on injectable estradiol monotherapy, 100% achieved therapeutic estradiol levels (>100 pg/mL) and 88.2% achieved target testosterone levels (<50 ng/dL). The authors noted that most individuals on injectable estradiol monotherapy were able to adequately suppress testosterone, but that higher doses and levels of estradiol may be needed for testosterone suppression in this context.
Herndon et al. (2023) noted that existing recommendations for injectable estradiol in transfeminine people suggest doses of 2 to 10 mg per week or 5 to 30 mg every 2 weeks, referencing the Endocrine Society 2017 guidelines (Hembree et al., 2017) and UCSF 2016 guidelines (Deutsch, 2016a). They also noted that the UCSF 2016 guidelines recommended lower doses of estradiol cypionate than estradiol valerate, which they attributed to pharmacokinetic differences between the esters (citing Oriowo et al. (1980) for this claim). However, the authors noted that the differences between estradiol valerate and estradiol cypionate doses they observed were small, and questioned the clinical relevance of the differences. The authors also tactfully critiqued dosing recommendations by existing guidelines, and suggested their own data to guide dosing instead, with the following relevant excerpts:
Prior studies used for development of guidelines for parenteral doses are suboptimal given their small sample sizes or pre-specificized [gender-affirming hormone therapy (GAHT)] protocols with no adjustment of estradiol doses or no information on hormone concentrations achieved. [Discussion of Deutsch, Bhakri, & Kubicek (2015) and Mueller et al. (2011) …]
Overall, the studies used to support the current dosing recommendation guidelines for parenteral estradiol dosing are limited, incomplete with regards to hormone concentrations achieved, and do not provide SC as an available option. The doses of estradiol used in this study (with either SC or IM approach), were successful in achieving serum estradiol concentrations at the cisgender female range. Most importantly, as compared to current available guidelines and consensus statements [1, 2], these doses of estradiol valerate are less than half of what is recommended for both initial and maintenance dosing and achieved suppression of testosterone.
Lower doses of parenteral injections than previously described in clinical practice guidelines achieved therapeutic estradiol concentrations. Our data can serve as a dosing guide for initial and maintenance use of parenteral estradiol, which is different than what has been previously described.
Herndon et al. (2023) concluded that injectable estradiol by both i.m. and s.c. routes is effective in achieving therapeutic estradiol levels in transfeminine people. They noted that there were not meaningful differences between i.m. and s.c. in terms of dose, although i.m. may require slightly higher doses than s.c. to achieve therapeutic estradiol levels. The authors stated that doses of injectable estradiol to achieve therapeutic estradiol levels in transfeminine people were lower than previously recommended by guidelines and other publications. They concluded that clinical use of injectable estradiol in transfeminine people should include discussion of both i.m. and s.c. routes and invidiualization by patient. Finally, they called for more clinical studies on injectable estradiol in transfeminine people to evaluate clinical outcomes, feminization, and additional risks and benefits of this route compared to other routes.
The findings of Herndon et al. (2023) are pleasingly consistent with the results of the present meta-analysis. Based on the findings of this meta-analysis, assuming a linear relationship between dose and estradiol levels, estradiol levels with non-polymeric injectable estradiol esters, like estradiol valerate and estradiol cypionate in oil via intramuscular injection, increase by around 60 pg/mL on average for each 1 mg per week in dose (with Herndon et al. (2023) finding a value of 57 pg/mL per 1 mg using a multiple linear regression model). In relation to this, mean integrated estradiol levels of around 250 pg/mL on average would be expected at a dosage of 4 mg once per week. Accordingly, Herndon et al. (2023) found median estradiol levels of 190 to 196 pg/mL at per-week median doses of 3.75 to 4 mg. Similarly, the present work recommended injectable estradiol doses with non-polymeric esters of 1 to 6 mg per week (to achieve mean integrated estradiol levels of roughly 50–300 pg/mL), which is comparable to the range of about 2 to 6 mg per week used in most transfeminine people in Herndon et al. (2023) (to achieve estradiol levels of at least 100 pg/mL, along with adequate testosterone suppression). Additionally, it was noted in this meta-analysis—based on clinical research in cisgender men with prostate cancer—that only modestly supraphysiological estradiol levels, of no more than approximately 200 to 300 pg/mL, are likely to be needed for strong testosterone suppression in transfeminine people. This has likewise been confirmed with solid clinical data in transfeminine people by Herndon et al. (2023), with 88% of those on injectable estradiol monotherapy having testosterone levels of <50 ng/dL at a median injectable estradiol dose of 4 mg/week and with median estradiol levels of 220 pg/mL. It is the opinion of the present author that Herndon et al. (2023) is a very important and formative study, with clinical implications that go far beyond merely supporting the s.c. use of injectable estradiol. The study represents the first major step in the published literature to correcting the dosing and intervals of injectable estradiol in transgender care guidelines and in transgender health generally. I commend the researchers for their work.
Update 4: Rothman et al. (2024a) and Rothman et al. (2024b)
In February 2024, the following short review on injectable estradiol dosing in transfeminine people by Micol Rothman and colleagues was published online:
Rothman, M. S., Hamnvik, O. P. R., Davidge-Pitts, C., Safer, J. D., Ariel, D., Tangpricha, V., Abramowitz, J., Soe, K., Sarvaideo, J., Kelley, C., Irwig, M. S., & Iwamoto, S. J. (2024). Revisiting Injectable Estrogen Dosing Recommendations for Gender-Affirming Hormone Therapy. Transgender Health, 9(6), 463–465. [DOI:10.1089/trgh.2023.0209]
Here is the abstract of the paper:
Injectable estrogens are options for gender-affirming hormone therapy per guidelines, which suggest intramuscular dosages of 5–30 mg every 2 weeks or 2–10 mg weekly with estradiol cypionate or valerate interchangeably. Data among transgender and gender-diverse patients are limited due to local unavailability and concerns around laboratory assay variability and estradiol (E2) level fluctuation. We note a concerning trend where patients are prescribed high-dose injections based on the guidelines leading to serum E2 levels well above the range recommended in the same guidelines. Our review indicates that 5 mg weekly or lower should be prescribed when initiating injectable estrogens to avoid supraphysiologic E2 levels.
Then, in May 2024, the following longer and more comprehensive review on injectable estradiol dosing in transfeminine people by Rothman and most of the same other academics was published online:
Rothman, M. S., Ariel, D., Kelley, C., Hamnvik, O. R., Abramowitz, J., Irwig, M. S., Soe, K., Davidge-Pitts, C., Misakian, A. L., Safer, J. D., & Iwamoto, S. J. (2024). The Use of Injectable Estradiol in Transgender and Gender Diverse Adults: A Scoping Review of Dose and Serum Estradiol Levels. Endocrine Practice, 30(9), 870–878. [DOI:10.1016/j.eprac.2024.05.008]
Here is the abstract of this paper:
Objective: Feminizing gender-affirming hormone therapy is the mainstay of treatment for many transgender and gender diverse people. Injectable estradiol preparations are recommended by the World Professional Association for Transgender Health Standards of Care 8 and the Endocrine Society guidelines. Many patients prefer this route of administration, but few studies have rigorously assessed optimal dosing or route.
Methods: We performed a scoping review of the available data on estradiol levels achieved with various dosages of estradiol injections in transgender and gender diverse adults on feminizing gender-affirming hormone therapy. We also report on testosterone suppression, route (ie, subcutaneous vs intramuscular), and type of injectable estradiol ester as well as timing of blood draw relative to the most recent dose, where available.
Results: The data we reviewed suggest that the current guidelines, which recommend starting doses 2 to 10 mg weekly or 5 to 30 mg every 2 weeks of estradiol cypionate or valerate, are too high and likely lead to patients having supraphysiologic levels across much of their injection cycle.
Conclusions: The optimal starting dose for injectable estradiol remains unclear and whether it should differ for cypionate and valerate. Based on the data available, we suggest that clinicians start injectable estradiol cypionate or valerate via subcutaneous or intramuscular injections at a dose ≤5 mg weekly and then titrate accordingly to keep levels within guideline-recommended range. Future studies should assess timing of injections and subsequent levels more precisely across the injection cycle and between esters.
This paper notably also cited the present Transfeminine Science article as raising concerns about guideline-based dosing for injectable estradiol and potential health complications from these doses.
Aside from Micol Rothman herself, these reviews were also authored by other well-known experts in transgender health. For instance, two of the coauthors, Joshua Safer and Michael Irwig, were authors for the WPATH SOC8 hormone therapy chapter (WPATH SOC8 Full Contributor List). Additionally, Safer was one of the authors for the Endocrine Society’s transgender hormone therapy guidelines (Hembree et al., 2017). As such, it would appear that transgender medicine has finally started to seriously correct injectable estradiol dosing. This is a very important development. Now, the appropriate dosing and intervals of injectable estradiol will need to be more precisely established and the corrections will need to make their way into updated transgender hormone therapy guidelines and general clinical practice.
A letter to the editor commented on and concorded with Rothman and colleagues’ second literature review:
Patel, K. T., & Tangpricha, V. (2024). Parenteral Estradiol for Transgender Women: Time to adjust the dose. Endocrine Practice, 30(9), 893–894. [DOI:10.1016/j.eprac.2024.07.005]
Update 5: Kariyawasam et al. (2024)
In March 2024, the following study of estradiol levels with different routes of estradiol in transfeminine people, including injectable estradiol, was published:
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2024). Comparison of Estrone/Estradiol Ratio and Levels in Transfeminine Individuals on Different Routes of Estradiol. Transgender Health, ahead of print. [DOI:10.1089/trgh.2023.0138]
The study stratified injectable estradiol doses into different dosing levels, accounted for timing of blood draws, and compared injectable estradiol to other estradiol routes. The other routes included oral estradiol, sublingual estradiol, and transdermal estradiol. The form of injectable estradiol used was estradiol valerate in dose groups including ≤4 mg/week (“low-dose”), >4 mg/week to ≤8 mg/week (“medium-dose”), and >8 mg/week (“high-dose”). In the study, this injectable estradiol regimen resulted in supraphysiological estradiol levels in the medium- to high-dose groups (>4 mg/week) and dramatically higher estradiol levels than with the other estradiol routes (Data). Median estradiol levels were reported in a subsequent paper as follows: “Figure 2 from the paper shows estradiol levels across the 3 groups. Although exact numbers are not given in this figure, we learned through correspondence with the authors that the low dose injection group [n=8] had a median level of 202.7 ± SD 232.6 pg/mL, the medium group [n=22] 465.2 ± SD 466.3 pg/mL, and the high group [n=3] 574.4 ± SD147.3 pg/mL (converted from SI units)” (Rothman et al., 2024b). Although the sample sizes for the different dose groups were small, this study, along with Herndon et al. (2023), provides some of the best clinical data on estradiol levels with injectable estradiol in transfeminine people that have so far been published.
Update 6: Patel et al. (2024)
In June 2024, the following open-access review discussing injectable estradiol in transfeminine people and calling for updated transgender health guidelines was published:
Patel, R., Korenman, S., Weimer, A., & Grock, S. (2024). A Call for Updates to Hormone Therapy Guidelines for Gender-Diverse Adults Assigned Male at Birth. Cureus, 16(6), e62262. [DOI:10.7759/cureus.62262] [PDF]
The following quote is the relevant excerpt on injectable estradiol from the review:
The current guideline-based dosing recommendations for estradiol vary considerably, which is problematic for clinicians and patients who rely on guidelines to initiate treatment. Most notably, the conversion rates between parenteral estradiol valerate and estradiol cypionate vary drastically between the UCSF Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People (UCSF Guidelines) and The Endocrine Society Clinical Practice Guidelines for Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons (the Endocrine Society Guidelines). The UCSF Guidelines indicate the conversion between estradiol valerate and cypionate to be as high as a 4:1 ratio [2], while the Endocrine Society Guidelines provide no dosing differentiations [1]. Herndon and colleagues demonstrated that the conversion between estradiol cypionate and estradiol valerate is closer to 1:1 [4]. Further equivalence studies are needed to clarify ideal dosing conversions.
The Endocrine Society Guidelines recommend titrating estradiol to 100-200 pg/mL [1]. The UCSF Guidelines recommend 2-4 mg daily as the starting dose for oral estradiol and 5 mg weekly for parenteral estradiol valerate [2]. The Endocrine Society Guidelines suggest oral estradiol 2-6 mg daily and parenteral estradiol 2- 10 mg weekly [1]. However, Chantrapanichkul et al. found that intramuscular injections of estradiol valerate greater than 5 mg weekly led to mean estradiol concentrations well above 200 pg/mL, while 4-5 mg of oral estradiol daily only led to minimum desired concentrations [5]. Similarly, Herndon et al. found that subcutaneous estradiol at a median dose of 3.75 mg per week led to a median estradiol level of 196 pg/mL [4]. Thus, current guideline-based dosing may lead providers to choose doses of injectable estradiol that would result in supratherapeutic serum estradiol levels. In light of these recent publications, it is clear that guideline-based dosing for estradiol needs updating. In our clinical experience, parenteral estradiol valerate at doses of 2-4 mg weekly typically leads to physiologic estradiol levels. Estradiol cypionate should likely be dosed in a 1:1 ratio with estradiol valerate until future data are obtained.
Lastly, while estradiol valerate and cypionate are only FDA-approved for intramuscular administration, many patients prefer subcutaneous administration. There are small studies that suggest the pharmacokinetics of intramuscular and subcutaneous estradiol are similar [4]. While the UCSF Guidelines comment on the use of subcutaneous estradiol, other guidelines should be updated to include this option for patients [2].
Update 7: Toffoli Ribeiro et al. (2024)
Toffoli Ribeiro, C., Gois, Í., da Rosa Borges, M., Ferreira, L. G. A., Brandão Vasco, M., Ferreira, J. G., Maia, T. C., & Dias-da-Silva, M. R. (2024). Assessment of parenteral estradiol and dihydroxyprogesterone use among other feminizing regimens for transgender women: insights on satisfaction with breast development from community-based healthcare services. Annals of Medicine, 56(1), 2406458. [DOI:10.1080/07853890.2024.2406458]:
This study examines the effects of a commonly used injectable hormone combination, specifically estradiol enanthate with dihydroxyprogesterone acetophenide (EEn/DHPA), […] Our research focused on a retrospective longitudinal study involving a large cohort of transwomen evaluated between 2020 and 2022, comprising 101 participants. We assessed serum levels of estradiol (E2), testosterone (T), luteinizing hormone (LH), and follicle-stimulating hormone (FSH), comparing the EEn/DHPA hormonal regimen with other combined estrogen-progestogen (CEP) therapies. […] Our findings indicated that participants using the EEn/DHPA regimen exhibited significantly higher serum E2 levels (mean: 186 pg/mL ± 32 pg/mL) than those using other therapies (62 ± 7 pg/mL), along with lower FSH levels, but no significant differences in T and LH levels. […] These results suggest that an injectable, low-cost EEn/DHPA administered every three weeks could serve as an alternative feminizing regimen, particularly considering the extensive long-term experience of the local transgender community. Further longitudinal studies on the efficacy of feminizing-body effects and endovascular risks of various parenteral CEP types are warranted to improve primary healthcare provision for transgender persons.
Introduction
Injectable combined estrogens with progestogens (CEP) have long been widely used in Brazil and other Latin American countries, predominantly among ciswomen as an injectable contraceptive and by Brazilian transgender women and travestis as GAHT [8]. Despite the absence of recognition by the Endocrine Society as an alternative hormonal regimen due to concerns regarding thrombogenicity and challenges in routine monitoring through blood testing, the prevalent use of CEP necessitates evaluating its regimen recommendations. This has led our research to delve deeper into understanding CEP regimens, considering the experiences of travestis amidst distinct sociocultural lifestyles and limited access to public endocrinological care services [15,16]. Hence, our objective is to elucidate our observations in monitoring trans individuals utilizing CEP regimens by evaluating hormone levels […] within a cohort of transwomen employing the most common injectable CEP, namely estradiol enanthate with dihydroxyprogesterone acetophenide (EEn/DHPA) and comparing these observations with other GAHT regimens.
Subjects and methods
Estradiol enanthate pharmacokinetics curve
Utilizing a previously published meta-analysis method of estradiol concentration-time data from publicly available information on cisgender women who had used EEn or EEn/DHPA [17], we reanalyzed and integrated data from various studies. A unified single-dose curve for 30 days was created. We employed least squares regression for studies with four or more concentration-time data points (solid lines). We manually adjusted other studies with three data points to fit into a single-dose curve.
Each study’s data were adjusted for baseline estradiol levels or endogenous estradiol production and then normalized by 10 mg. The V3C Fitter and Desmos tools, accessible online at https://alyw234237.github.io/injectable-e2-simulator/v3c-fitter/ and https://www.desmos.com/calculator/ndgvp2avhj?lang=pt-BR respectively, were utilized for fitting the three-compartment pharmacokinetic model. Estradiol levels from transgender women on EEn/DHPA in this study were presented using a box plot graph featuring percentiles at 10, 25, 50, 75, and 90.
Results
Hormonal levels during the follow-up of feminizing regimens
Scatter plot graphs depicted the measurement of sex hormones (Figure 2). Serum estradiol levels in the EEn/ DHPA group (mean: 186.4pg/mL ± 32.8pg/mL) were significantly higher than those in the group using other therapies (62.2±6.9pg/mL) (Figure 2(A)). Within the EEn/DHPA group, serum FSH levels were significantly lower compared to the other group (Others) (Figure 2(B)). However, no significant difference was found between the groups concerning testosterone (Figure 2(C)) and LH (Figure 2(D)) levels.
Pharmacokinetics of injectable estradiol enanthate
Serum estradiol levels in trans women using EEn/DHPA reached the target levels for this population during hormone therapy, a trend not observed in participants using other feminizing hormone therapies (Table 1). The boxplot graph (Figure 5) illustrates that the median estradiol levels in trans women using EEn/DHPA fell within this population’s expected average range values (100–200pg/mL).
Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. For studies with four or more concentration-time data points (solid lines) and the fit of combined data (thick black line), least squares regression to a three-compartment pharmacokinetic model was employed. A single-dose curve was manually adjusted for studies with three data points (dashed lines). Data from each study were adjusted for endogenous estradiol production via baseline or trough estradiol levels subtraction and normalized by 10mg. The graph illustrates estradiol levels from the transwoman cohort in a boxplot. The shaded area represents the optimal target range for estradiol levels in transwomen under hormone therapy. The boxplot graph displays the percentiles 10, 25, 50, 75, and 90 for estradiol levels of transwomen under EEn/DHPA in this study (N=53).
Discussion
Our study represents a pioneering contribution to the literature by demonstrating that Brazilian trans women undergoing EEn/DHPA therapy achieved estradiol levels comparable to those observed during the follicular phase in cisgender women. […]
Our study further noted that DHPA demonstrates comparable efficacy to cyproterone or other anti-androgens in achieving optimal LH pituitary suppression and reducing testosterone levels. EEn/ DHPA, an affordable injectable contraceptive widely accessible in South American countries, presents a promising avenue for attaining target hormone levels among transfeminine individuals.
Additionally, our investigation, which reviewed pharmacokinetic data, supports the potential implementation of EEn/DHPA in a 21-day regimen to sustain optimal estradiol levels. While alternative medications exist to inhibit testosterone production and action, their availability varies based on regional healthcare provider systems. […]
EEn/DHPA, commonly used as a long-lasting injectable contraceptive [21–23], has found application in feminizing hormone therapy for transfeminine people, notably in travestis in Brazil [7,8,24,25]. […]
In conclusion, our long-term cohort study suggests that administering parenteral estradiol enanthate with dihydroxyprogesterone acetophenide every three weeks could serve as a practical option for feminizing hormone regimens in transgender women. Nonetheless, adopting an individualized approach that takes into account each individual’s goals, response to prior hormone therapies, and medical history is crucial. This personalized approach is central to improving healthcare provision and ensuring optimal outcomes in bodily changes. By continuing to explore and refine hormone therapy regimens, we can better support the health and well-being of transgender individuals on their gender-affirming journey.
References
[17] Aly. 2021. An informal meta-analysis of estradiol curves with injectable estradiol preparations. Transfeminine Sci. https:// transfemscience.org/articles/injectable-e2-meta-analysis/
Update 8: Misakian et al. (2025)
Misakian, A. L., Kelley, C. E., Sullivan, E. A., Chang, J. J., Singh, G., Kokosa, S., Avila, J., Cooper, H., Liang, J. W., Botzheim, B., Quint, M., Jeevananthan, A., Chi, E., Harmer, M., Hiatt, L., Kowalewski, M., Steinberg, B., Tausinga, T., Tanner, H., Ho, T. F., … Ariel, D. (2025). Injectable Estradiol Use in Transgender and Gender-Diverse Individuals throughout the United States. The Journal of Clinical Endocrinology & Metabolism, dgaf015. [DOI:10.1210/clinem/dgaf015]:
Context: Guidelines for use of injectable estradiol esters (valerate [EV] and cypionate [EC]) among transgender and gender-diverse (TGD) individuals designated male at birth vary considerably, with many providers noting supraphysiologic serum estradiol concentrations based on current dosing recommendations.
Objectives: This work aimed to 1. determine the dose of injectable estradiol (subcutaneous [SC] and intramuscular [IM]) needed to reach guideline-recommended estradiol concentrations for TGD adults using EC/EV; 2. describe the relationship between estradiol concentration relative to timing/dose of last estradiol injection and other covariates; and 3. determine dosing differences between IM/SC EV/EC.
Methods: A cross-sectional retrospective study was conducted across 6 US medical centers including TGD adults on same-dose injectable estradiol for more than 75 days, with confirmed timing of estradiol concentration relative to last injection, from January 1, 2019 to December 31, 2023. Descriptive statistics were used to describe patient characteristics and weighted linear mixed models to evaluate relationship between various covariates and estradiol concentration.
Results: Data from 562 patients were included. Among those injecting every 7 days who reached the guideline-recommended estradiol concentration (n = 131, 27.5%), the median estradiol dose was 4.0 mg (interquartile range, 3.0-5.0 mg). Among all patients, the majority reached supraphysiologic estradiol concentrations (>200 pg/mL [>734 pmol/L]) while dose and timing in the injection cycle were significant covariates for the estradiol concentration. There were no significant dosing differences between IM/SC EV/EC.
Conclusion: Injectable estradiol esters effectively reach guideline-recommended estradiol concentrations but at lower doses than previously recommended. Estradiol concentrations are best interpreted relative to timing of last injection. Route of administration and type of ester do not significantly affect estradiol concentrations.
[…]
And a letter to the editor commenting on the paper:
Milano, C., & Harper, J. (2025). Comments on Injectable Estradiol Use in Transgender and Gender-Diverse Individuals in the US. The Journal of Clinical Endocrinology & Metabolism, dgaf134. [DOI:10.1210/clinem/dgaf134]
Update 9: Slack et al. (2025)
Slack, D. J., Di Via Ioschpe, A., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., Karim, S., & Safer, J. D. (2025). Examining the Influence of the Route of Administration and Dose of Estradiol on Serum Estradiol and Testosterone Levels in Feminizing Gender-Affirming Hormone Therapy. Endocrine Practice, 31(1), 19–27. [DOI:10.1016/j.eprac.2024.10.002]:
Introduction: […] This study investigates the effect of route of administration (ROA) and dose of estradiol on estradiol (E2) and testosterone (T) levels in transfeminine individuals.
Methods: We conducted a chart review of 573 patients with an active prescription for estradiol for feminizing GAHT and serum hormone levels available.
Results: […] Intramuscular estradiol was associated with lower T and higher E2 compared to oral and transdermal ROAs (P < .001), with many achieving target hormone levels even at low doses.
Conclusions: […] The intramuscular ROA appears to be the most potent delivery of estradiol with impact on serum hormone levels with doses on the low end of guideline-suggested ranges.
[…]
Update 10: Carlson et al. (2025)
Carlson, S. M., Dominguez, C., Jeevananthan, A., & Crowley, M. J. (2025). Follow-Up Estradiol Levels Based on Regimen Formulation With Guideline-Concordant Gender-Affirming Hormone Therapy. Journal of the Endocrine Society, 9(3), bvae205. [DOI:10.1210/jendso/bvae205]:
Context: Endocrine Society guidelines for dosing of feminizing gender-affirming hormone therapy (GAHT) have remained essentially unchanged since 2009. The Endocrine Society recommends periodic monitoring of serum estradiol levels, with the goal of maintaining levels in the premenopausal cisgender female range (100-200 pg/mL). However, it is not clear whether guideline-concordant dosing consistently produces guideline-recommended levels across common estradiol formulation types (oral pills, parenteral injections, transdermal patches).
Objective: All transgender and nonbinary patients receiving estradiol-based GAHT between October 2015 and March 2023 were reviewed at a single center, with the goal of determining the frequency with which guideline-concordant dosing with different estradiol formulations led to guideline-recommended estradiol levels.
Methods: Demographics, GAHT regimen, and estradiol levels were obtained via chart review, and data were analyzed descriptively.
Results: The analytic population included n = 35 individuals, including n = 9 prescribed oral estradiol pills, n = 11 prescribed parenteral injections, and n = 15 prescribed transdermal patches. With guideline-concordant doses of oral estradiol (mean 2.8 mg daily), the mean follow-up level was 168 pg/mL; 32% of follow-up levels were subtherapeutic and 14% were supratherapeutic. With guideline-concordant doses of parenteral estradiol (mean 5.8 mg weekly), the mean midpoint follow-up level was 342 pg/mL; 91% of midpoint follow-up levels were supratherapeutic. With guideline-concordant doses of transdermal estradiol (mean 0.09 mg/day), the mean follow-up level was 81.5 pg/mL; 70% of follow-up levels were subtherapeutic.
Conclusion: Supratherapeutic follow-up estradiol levels were common with guideline-concordant parenteral estradiol doses, as were subtherapeutic follow-up levels with guideline-concordant transdermal doses. These findings may suggest the need for revision of guideline-recommended estradiol doses for these formulations
[…]
Update 11: Kanin et al. (2025)
Kanin, M., Slack, M., Patel, R., Chen, K. T., Jackson, N., Williams, K. C., & Grock, S. (2025). Injectable Estradiol Dosing Regimens in Transgender and Nonbinary Adults Listed as Male at Birth. Journal of the Endocrine Society, bvaf004. [DOI:10.1210/jendso/bvaf004]:
Context: Many transgender and nonbinary (TGNB) individuals assigned male at birth (AMAB) seek hormone therapy to achieve physical and emotional changes. Standard therapy includes estradiol, with or without an antiandrogen. Our clinical observations suggest that currently recommended injectable estradiol dosing may lead to supratherapeutic estradiol levels.
Objective: We sought to evaluate whether lower-than-recommended doses of injectable estradiol were effective in achieving serum estradiol and testosterone goals.
Methods: We conducted a retrospective cohort study to evaluate injectable estradiol dosing in treatment-naive AMAB individuals initiating hormone therapy. Data from a single provider at an academic center from January 2017 to March 2023 were analyzed. A total of 29 patients were eligible for inclusion. The primary variables of estradiol dosage, serum estradiol, and testosterone levels were analyzed over 15 months.
Results: The average estradiol dose decreased from 4.3 to 3.7 mg weekly (P < .001) during the study period with a final on-treatment estradiol level of 248 pg/mL. All individuals achieved a testosterone level of less than 50 ng/dL during the study period. The average initial on-treatment testosterone level was not significantly different from average final on-treatment measurement of 24.0 mg/dL (P = .95). […]
Conclusion: Lower doses of injectable estradiol can achieve therapeutic estradiol levels with excellent testosterone suppression. […]
[…]
This study had been previously published as a conference abstract:
Kanin, M., Slack, M., Patel, R., Chen, K. T., Jackson, N., Williams, K., & Grock, S. (2024). 8309 Injectable Estradiol Dosing Regimens; A Retrospective Review of Hormone Therapy for Gender-Diverse Adults Assigned Male at Birth. Journal of the Endocrine Society, 8(Suppl 1), bvae163-1706. [DOI:10.1210/jendso/bvae163.1706]
Abuhelwa, A. Y., Foster, D. J., & Upton, R. N. (2015). ADVAN-style analytical solutions for common pharmacokinetic models. Journal of Pharmacological and Toxicological Methods, 73, 42–48. [DOI:10.1016/j.vascn.2015.03.004]
Aedo, A. R., Landgren, B. M., Johannisson, E., & Diczfalusy, E. (1985). Pharmacokinetic and pharmacodynamic investigations with monthly injectable contraceptive preparations. Contraception, 31(5), 453–469. [DOI:10.1016/0010-7824(85)90081-2]
Aisaka, K., Ando, S., Kokubo, K., Yoshida, K., & Mori, H. (1986). いわゆる潜在性高prolactin血症患者におけるprolactin分泌予備能の検討. [Studies on Prolactin Secreting Capacity in the Ovulatory Infertile Patients with Transient Hyperprolactinemia.] 日本内分泌学会雑誌 / Nihon Naibunpi Gakkai Zasshi / Folia Endocrinologica Japonica, 62(5), 662–671. [DOI:10.1507/endocrine1927.62.5_662]
Akande, E. O. (1974). The effect of oestrogen on plasma levels of luteinizing hormone in euthyroid and thyrotoxic postmenopausal women. The Journal of Obstetrics and Gynaecology of the British Commonwealth / BJOG, 81(10), 795–803. [DOI:10.1111/j.1471-0528.1974.tb00383.x]
Ballard, B. E. (1978). An Overview of Prolonged Action Drug Dosage Forms. In Robinson, J. R. (Ed.). Sustained and Controlled Release Drug Delivery Systems (pp. 1–69). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books]
Behre, H. M., Oberpenning, F., & Nieschlag, E. (1990). Comparative pharmacokinetics of androgen preparations: application of computer analysis and simulation. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 1st Edition (pp. 115–135). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-662-00814-0_6]
Behre, H. M., & Nieschlag, E. (1998). Comparative pharmacokinetics of testosterone esters. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 329–348). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_11]
Behre, H. M., Wang, C., Handelsman, D. J., & Nieschlag, E. (2004). Pharmacology of testosterone preparations. Testosterone: Action · Deficiency · Substitution, 3rd Edition (pp. 405–444). Cambridge/New York: Cambridge University Press. [DOI:10.1017/CBO9780511545221.015] [PDF]
Behre, H. M., & Nieschlag, E. (2012). Testosterone preparations for clinical use in males. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Testosterone: Action · Deficiency · Substitution, 4th Edition (pp. 309–335). Cambridge/New York: Cambridge University Press. [DOI:10.1017/CBO9781139003353.016]
Bider, D., Ben-Rafael, Z., Shalev, J., Goldenberg, M., Mashiach, S., & Blankstein, J. (1989). Pituitary and ovarian suppression rate after high dosage of gonadotropin-releasing hormone agonist. Fertility and Sterility, 51(4), 578–581. [DOI:10.1016/S0015-0282(16)60602-7]
Blackwell, R. E., Boots, L. R., & Potter Jr, H. D. (1982). Evaluation of Delestrogen and Parlodel as a luteolytic agent in humans. Fertility and Sterility, 37(2), 213–217. [DOI:10.1016/S0015-0282(16)46042-5]
Bourns, A. (2019). Guidelines for Gender-Affirming Primary Care with Trans and Non-Binary Patients, 4th Edition. Toronto: Rainbow Health Ontario/Sherbourne Health. [URL] [PDF]
Bradbury, J. T., Long, R. C., & Durham, W. C. (1953). Progesterone and estrogen requirements to induce and maintain decidua. Fertility and Sterility, 4(1), 63–75. [DOI:10.1016/S0015-0282(16)31145-1]
Braun, P., Wildt, L., & Leyendecker, G. (1983). The effect of danazol on gonadotropin secretion during the follicular phase of the menstrual cycle. Fertility and Sterility, 40(1), 37–44. [DOI:10.1016/S0015-0282(16)47174-8]
Buckman, M. T., Johnson, J., Ellis, H., Srivastava, L., & Peake, G. T. (1980). Differential lipemic and proteinemic response to oral ethinyl estradiol and parenteral estradiol cypionate. Metabolism, 29(9), 803–805. [DOI:10.1016/0026-0495(80)90117-1]
Callen-Lorde Community Health Center. (2018). Protocols for the Provision of Hormone Therapy. New York City: Callen-Lorde Community Health Center. [URL] [PDF]
Canales, E. S., Cabezas, A., Vázquez-Matute, L., & Zárate, A. (1978). Induction of ovulation with clomiphene and estradiol benzoate in anovulatory women refractory to clomiphene alone. Fertility and Sterility, 29(5), 496–499. [DOI:10.1016/S0015-0282(16)43271-1]
Canales, E. S., Fonseca, M. E., Mason, M., & Zárate, A. (1981). Feedback effect of estradiol on follicle-stimulating hormone and prolactin secretion during the puerperium. International Journal of Gynecology & Obstetrics, 19(1), 79–81. [DOI:10.1016/0020-7292(81)90043-6]
Cano, A., Gimeno, F., Fuente, T., Parrilla, J. J., & Abad, L. (1986). The positive feedback of estradiol on gonadotropin secretion in women with perimenopausal dysfunctional uterine bleeding. European Journal of Obstetrics & Gynecology and Reproductive Biology, 22(5–6), 353–358. [DOI:10.1016/0028-2243(86)90125-5]
Carlson, S. M., Dominguez, C., Jeevananthan, A., & Crowley, M. J. (2025). Follow-Up Estradiol Levels Based on Regimen Formulation With Guideline-Concordant Gender-Affirming Hormone Therapy. Journal of the Endocrine Society, 9(3), bvae205. [DOI:10.1210/jendso/bvae205]
Cavanaugh, T., Hopwood, R., Gonzalez, A., & Thompson, J. (2015). The Medical Care of Transgender Persons. Boston: Fenway Health. [URL] [PDF]
Chantrapanichkul, P., Stevenson, M. O., Suppakitjanusant, P., Goodman, M., & Tangpricha, V. (2021). Serum Hormone Concentrations in Transgender Individuals Receiving Gender-Affirming Hormone Therapy: A Longitudinal Retrospective Cohort Study. Endocrine Practice, 27(1), 27–33. [DOI:10.4158/EP-2020-0414] [Table]
Chien, Y. W. (1981). Long-acting parenteral drug formulations. Journal of Parenteral Science and Technology / PDA Journal of Pharmaceutical Science and Technology, 35(3), 106–139. [Google Scholar] [URL] [PDF]
Cirrincione, L. R., Winston McPherson, G., Rongitsch, J., Sadilkova, K., Drees, J. C., Krasowski, M. D., Dickerson, J. A., & Greene, D. N. (2021). Sublingual estradiol is associated with higher estrone concentrations than transdermal or injectable preparations in transgender women and gender nonbinary adults. LGBT Health, 8(2), 125–132. [DOI:10.1089/lgbt.2020.0249] [Table]
Colburn, W. A. (1981). Simultaneous pharmacokinetic and pharmacodynamic modeling. Journal of Pharmacokinetics and Biopharmaceutics, 9(3), 367–388. [DOI:10.1007/BF01059272]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Dahl, M., Feldman, J. L., Goldberg, J., & Jaberi, A. (2015). Endocrine Therapy for Transgender Adults in British Columbia: Suggested Guidelines. Physical Aspects of Transgender Endocrine Therapy. Vancouver: Vancouver Coastal Health. [Google Scholar] [PDF]
Davidson, A., Franicevich, J., Freeman, M., Lin, R., Martinez, L., Monihan, M., Porch, M., Samuel, L., Stukalin, R., Vormohr, J., & Zevin, B. (2013). Protocols for Hormonal Reassignment of Gender. San Francisco: San Francisco Department of Public Health/Tom Waddell Health Center. [Google Scholar] [PDF]
Depo®-Estradiol Estradiol Cypionate Label. U.S. Food and Drug Administration/Pharmacia & Upjohn (Pfizer). [URL] [PDF]
Derra, C. (1981). Hormonprofile unter Östrogen- und Antiandrogentherapie bei Patienten mit Prostatakarzinom: Östradiolundecylat versus Cyproteronacetat. [Hormone Profiles under Estrogen and Antiandrogen Therapy in Patients with Prostate Cancer: Estradiol Undecylate versus Cyproterone Acetate.] (Doctoral dissertation, University of Mainz.) [Google Scholar] [WorldCat] [PDF] [Translation]
Deutsch, M. (2014). Medical Transition. In Erickson-Schroth, L. (Ed.). Trans Bodies, Trans Selves: A Resource for the Transgender Community, 1st Edition (pp. 241–264). Oxford: Oxford University Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Deutsch, M. B., Bhakri, V., & Kubicek, K. (2015). Effects of Cross-Sex Hormone Treatment on Transgender Women and Men. Obstetrics & Gynecology, 125(3), 605–610. [DOI:10.1097/AOG.0000000000000692]
Deutsch, M. B. (Ed.). (2016). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition. San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Düsterberg, B., & Nishino, Y. (1982). Pharmacokinetic and pharmacological features of oestradiol valerate. Maturitas, 4(4), 315–324. [DOI:10.1016/0378-5122(82)90064-0]
Düsterberg, B., & Wendt, H. (1983). Plasma levels of dehydroepiandrosterone and 17β-estradiol after intramuscular administration of Gynodian-Depot® in 3 women. Hormones, 17(2), 84–89. [DOI:10.1159/000179680]
Düsterberg, B., Schmidt-Gollwitzer, M., & Hümpel, M. (1985). Pharmacokinetics and biotransformation of estradiol valerate in ovariectomized women. Hormone Research in Paediatrics, 21(3), 145–154. [DOI:10.1159/000180039]
Edkins, R. P. (1959). The Modification of the Duration of Drug Action: Pharmaceutical Considerations. Journal of Pharmacy and Pharmacology, 11(Suppl 1), 54T–66T. [DOI:10.1111/j.2042-7158.1959.tb10412.x]
Enever, R. P., Fotherby, K., Naderi, S., & Lewis, G. A. (1983). Long-acting contraceptive agents: The influence of physicochemical properties of some esters of norethisterone upon the plasma levels of free norethisterone. Steroids, 41(3), 381–396. [DOI:10.1016/0039-128X(83)90109-5]
Eriksson, O., Bäckström, T., Stridsberg, M., Hammarlund-Udenaes, M., & Naessén, T. (2006). Differential response to estrogen challenge test in women with and without premenstrual dysphoria. Psychoneuroendocrinology, 31(4), 415–427. [DOI:10.1016/j.psyneuen.2005.10.004]
Espino y Sosa, S., Cortés Fuentes, M., Gómez Rico, J. A., & Cortés Bonilla, M. (2019). Non-polymeric Microspheres for the Therapeutic Use of Estrogens: An Innovative Technology. In Khan, W. A. (Ed.). Estrogen. London: IntechOpen. [DOI:10.5772/intechopen.82553]
Fisher, D., & Shafer, S. (2007). Fisher/Shafer NONMEM Workshop Pharmacokinetic and Pharmacodynamic Analysis with NONMEM. Basic Concepts. [PDF]
Florence, A. T. (2010). Looking at Formulations. In Florence, A. T. An Introduction to Clinical Pharmaceutics (pp. 69–100). London/Chicago: Pharmaceutical Press. [Google Scholar] [Google Books]
Fotherby, K., Benagiano, G., Toppozada, H. K., Abdel-Rahman, A., Navaroli, F., Arce, B., Ramos-Cordero, R., Gual, C., Landgren, B. M., & Johannisson, E. (1982). A preliminary pharmacological trial of the monthly injectable contraceptive Cycloprovera. Contraception, 25(3), 261–272. [DOI:10.1016/0010-7824(82)90049-X]
Futterweit, W., Gabrilove, J., & Smith, H. (1984). Testicular steroidogenic response to human chorionic gonadotropin of fifteen male transsexuals on chronic estrogen treatment. Metabolism, 33(10), 936–942. [DOI:10.1016/0026-0495(84)90248-8] [Figure]
Garner, P. R., & Armstrong, D. T. (1977). The effect of human chorionic gonadotropin and estradiol-17β on the maintenance of the human corpus luteum of early pregnancy. American Journal of Obstetrics and Gynecology, 128(5), 469–475. [DOI:10.1016/0002-9378(77)90026-6]
Garza-Flores, J., Rodriguez, V., Perez-Palacios, G., Virutamasen, P., Tang-Keow, P., Konsayreepong, R., Kovacs, L., Koloszar, S., & Hall, P. E. (1987). A multicentered pharmacokinetic, pharmacodynamic study of once-a-month injectable contraceptives. I. Different doses of HRP112 and of DepoProvera. Contraception, 36(4), 441–457. [DOI:10.1016/0010-7824(87)90093-X]
Garza-Flores, J., Alba, V. M., Cravioto, M. C., Hernandez, L., Perez-Palacios, G., Alvarado, G., Rivera, R., Recio, R., & Bassol, S. (1989). Estrogen-progestogen once-a-month injectable contraceptives and serum prolactin. Contraception, 39(5), 519–529. [DOI:10.1016/0010-7824(89)90107-8]
Garza-Flores, J., Fatinikun, T., Hernandez, L., Ramos, I., Cardenas, M., & Menjivar, M. (1991). A pilot study on the assessment of a progesterone/estradiol sustained release as once-a-month-injectable contraceptive. Contraception, 44(1), 45–59. [DOI:10.1016/0010-7824(91)90105-O]
Garza-Flores, J. (1994). Pharmacokinetics of once-a-month injectable contraceptives. Contraception, 49(4), 347–359. [DOI:10.1016/0010-7824(94)90032-9]
Geppert, G. (1975). Untersuchungen zur Pharmakokinetik von Östradiol-17β, Östradiol-Benzoat, Östradiol-Valerianat und Östradiol-Undezylat bei der Frau: Der Verlauf der Konzentrationen von Östradiol-17β, Östron, LH und FSH im Serum. [Studies on the Pharmacokinetics of Estradiol-17β, Estradiol Benzoate, Estradiol Valerate, and Estradiol Undecylate in Women: The Course of the Relationships Between Estradiol-17β, Estrone, LH, and FSH in Serum.] (Doctoral dissertation, University of Bonn.) [Google Scholar] [WorldCat] [PDF] [Translation]
Glintborg, D., T’Sjoen, G., Ravn, P., & Andersen, M. S. (2021). Management of endocrine disease: Optimal feminizing hormone treatment in transgender people. European Journal of Endocrinology, 185(2), R49–R63. [DOI:10.1530/EJE-21-0059]
Goh, H. H., & Ratnam, S. S. (1988). The LH surge in humans: its mechanism and sex difference. Gynecological Endocrinology, 2(2), 165–182. [DOI:10.3109/09513598809023624]
Goh, H. H., & Ratnam, S. S. (1990). Effect of estrogens on prolactin secretion in transsexual subjects. Archives of Sexual Behavior, 19(5), 507–516. [DOI:10.1007/BF02442351]
Goh, V. H. H., & Lee, K. O. (1998). Does a positive oestrogen feedback on the hypothalamic-pituitary axis exist concurrently with a defective testosterone feedback in Klinefelter’s syndrome? Hormone Research in Paediatrics, 50(3), 160–165. [DOI:10.1159/000023266]
Goodman, R. E., Anderson, D. C., Bu’Lock, D. E., Sheffield, B., Lynch, S. S., & Butt, W. R. (1985). Study of the effect of estradiol on gonadotrophin levels in untreated male-to-female transsexuals. Archives of Sexual Behavior, 14(2), 141–146. [DOI:10.1007/BF01541659]
Gooren, L. J. G., Rao, B. R., Van Kessel, H., & Harmsen-Louman, W. (1984). Estrogen positive feedback on LH secretion in transsexuality. Psychoneuroendocrinology, 9(3), 249–259. [DOI:10.1016/0306-4530(84)90004-0]
Gooren, L. (2005). Hormone treatment of the adult transsexual patient. Hormone Research in Paediatrics, 64(Suppl 2), 31–36. [DOI:10.1159/000087751]
Göretzlehner, G., Ackermann, W., Angelow, K., Bergmann, G., Bieck, E., Golbs, S., & Kliem, O. (2002). Pharmakokinetik von Estron, Estradiol, FSH, LH und Prolaktin nach intramuskulärer Applikation von 5 mg Estradiolvalerat. [Pharmacokinetics of estradiol valerate in postmenopausal women after intramuscular administration.] Journal für Menopause, 9(2), 46–49. [Google Scholar] [URL] [PDF] [Translation]
Gorton, N., Jaffe, J. M., Thompson, J., Menkin, D., Nesteby, A., Dunn, D., Baker, K. K., Harbatkin, D., Do, T., Radix, A., Meacher, P., Goldstein, Z., Carpenter, W., Caine, M., Henn, S., Murayama, R., Feldmann, J., & Zayas, S. (2019). TransLine Gender Affirming Hormone Therapy Prescriber Guidelines. San Francisco: Lyon-Martin Health Services/TransLine. [URL] [PDF]
Gunnarsson, P. O., & Norlén, B. J. (1988). Clinical pharmacology of polyestradiol phosphate. The Prostate, 13(4), 299–304. [DOI:10.1002/pros.2990130405]
Hamburger, C., & Benjamin, H. (1969). Endocrine Treatment of Male and Female Transsexualism / Appendix for the Practicing Physician: Suggestions and Guidelines for the Management of Transsexuals. In Green, R., & Money, J. (Eds.). Transsexualism and Sex Reassignment (pp. 291–307). Baltimore: John Hopkins University Press. [Google Scholar] [Google Books] [PDF]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658]
Henriksson, P., Carlström, K., Pousette, A., Gunnarsson, P. O., Johansson, C. J., Eriksson, B., Altersgård-Brorsson, A. K., Nordle, O., & Stege, R. (1999). Time for revival of estrogens in the treatment of advanced prostatic carcinoma? Pharmacokinetics, and endocrine and clinical effects, of a parenteral estrogen regimen. The Prostate, 40(2), 76–82. [DOI:10.1002/(SICI)1097-0045(19990701)40:2<76::AID-PROS2>3.0.CO;2-Q]
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of Subcutaneous and Intramuscular Estradiol Regimens as part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
Hughes, J. H., Woo, K. H., Keizer, R. J., & Goswami, S. (2022). Clinical Decision Support for Precision Dosing: Opportunities for Enhanced Equity and Inclusion in Health Care. Clinical Pharmacology & Therapeutics, 113(3), 565–574. [DOI:10.1002/cpt.2799]:
Ibrahim, S. (1996/1998). Pharmakokinetische Untersuchungen mit Östradiolvalerat und Hydroxyprogesteroncaproat in Depotform nach einmaliger Applikation bei 24 postmenopausalen Frauen. [Pharmacokinetic studies with estradiol valerate and hydroxyprogesterone caproate in depot form after a single application in 24 postmenopausal women.] (Doctoral dissertation, Dresden University of Technology.) [Google Scholar] [WorldCat] [Partial PDF]
Ittrich, G., & Pots, P. (1965). Östrogenbestimmungen in Blut und Urin nach Verabreichung von Östrogenen. [Estrogen determinations in blood and urine after administration of estrogens.] In Kraatz, H. (Ed.). International Symposium der Gynäkologischen Endokrinologie vom 15. bis 18. Mai 1963. / Abhandlungen der Deutschen Akademie der Wissenschaften zu Berlin, Klasse für Medizin, 1965(1), 53–56. [ISSN:0568-4250] [WorldCat 1] [WorldCat 2] [WorldCat 3] [PDF] [Translation]
Jaafar, S., Torres-Leguizamon, M., Duplessy, C., & Stambolis-Ruhstorfer, M. (2022). Hormonothérapie injectable et réduction des risques: pratiques, difficultés, santé des personnes trans en France. [Hormone replacement therapy injections and harm reduction: practices, difficulties, and transgender people’s health in France.] Sante Publique, 34(HS2), 109–122. [Google Scholar] [PubMed] [DOI:10.3917/spub.hs2.0109]
Jacobi, G. H., & Altwein, J. E. (1979). Bromocriptin als Palliativtherapie beim fortgeschrittenen Prostatakarzinom. [Bromocriptine for palliation of advanced prostatic carcinoma. Experimental and clinical profile of a drug.] Urologia Internationalis, 34(4), 266–290. [DOI:10.1159/000280272]
Jacobi, G. H., Altwein, J. E., Kurth, K. H., Basting, R., & Hohenfellner, R. (1980). Treatment of advanced prostatic cancer with parenteral cyproterone acetate: a phase III randomised trial. British Journal of Urology, 52(3), 208–215. [DOI:10.1111/j.1464-410X.1980.tb02961.x]
Jacobi, G. R. (1982). Experimental Rationale for the Investigation of Antiprolactins as Palliative Treatment for Prostate Cancer. In Jacobi, G. H., & Hohenfellner, R. (Eds.). Prostate Cancer (International Perspectives in Urology, Volume 3) (pp. 419–431). Baltimore/London: Williams & Wilkins. [Google Scholar] [Google Books]
Jilma, B., Eichler, H. G., Breiteneder, H., Wolzt, M., Aringer, M., Graninger, W., Röhrer, C., Veitl, M., & Wagner, O. F. (1994). Effects of 17β-estradiol on circulating adhesion molecules. The Journal of Clinical Endocrinology and Metabolism, 79(6), 1619–1624. [DOI:10.1210/jcem.79.6.7527406]
Johansson, C. J., & Gunnarsson, P. O. (2000). Pharmacodynamic model of testosterone suppression after intramuscular depot estrogen therapy in prostate cancer. The Prostate, 44(1), 26–30. [DOI:10.1002/1097-0045(20000615)44:1<26::AID-PROS4>3.0.CO;2-P]
Jones, T. M., Fang, V. S., Landau, R. L., & Rosenfield, R. (1978). Direct inhibition of Leydig cell function by estradiol. The Journal of Clinical Endocrinology and Metabolism, 47(6), 1368–1373. [DOI:10.1210/jcem-47-6-1368]
Jönsson, G., Olsson, A. M., Luttrop, W., Cekan, Z., Purvis, K., & Diczfalusy, E. (1976). Treatment of prostatic carcinoma with various types of estrogen derivatives. In Munson, P. L., Diczfalusy, E., Glover, J., Olson, R. E., Harris, R. S., Thimann, K. V., Loraine, J. A., & Wool, I. G. (Eds.). Vitamins & Hormones, Volume 33 (pp. 351–376). New York/San Francisco/London: Academic Press. [DOI:10.1016/S0083-6729(08)60965-6]
Kalicharan, R. W., Schot, P., & Vromans, H. (2016). Fundamental understanding of drug absorption from a parenteral oil depot. European Journal of Pharmaceutical Sciences, 83, 19–27. [DOI:10.1016/j.ejps.2015.12.011]
Kalicharan, R. W. (2017). New Insights into Drug Absorption from Oil Depots. (Doctoral dissertation, Utrecht University.) [Google Scholar] [URL] [PDF]
Kanin, M., Slack, M., Patel, R., Chen, K. T., Jackson, N., Williams, K., & Grock, S. (2024). 8309 Injectable Estradiol Dosing Regimens; A Retrospective Review of Hormone Therapy for Gender-Diverse Adults Assigned Male at Birth. Journal of the Endocrine Society, 8(Suppl 1), bvae163-1706. [DOI:10.1210/jendso/bvae163.1706]
Kanin, M., Slack, M., Patel, R., Chen, K. T., Jackson, N., Williams, K. C., & Grock, S. (2025). Injectable Estradiol Dosing Regimens in Transgender and Nonbinary Adults Listed as Male at Birth. Journal of the Endocrine Society, bvaf004. [DOI:10.1210/jendso/bvaf004]
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2024). Comparison of Estrone/Estradiol Ratio and Levels in Transfeminine Individuals on Different Routes of Estradiol. Transgender Health, ahead of print. [DOI:10.1089/trgh.2023.0138] [Data]
Kemeter, P., Bernaschek, G., Altmann, G., & Feichtinger, W. (1984). The effect of 2 mg estradiol-17β plus 1 mg estriol, sequentially combined with 1 mg norethisteroneacetate, on LH, FSH, estradiol-17β, progesterone, testosterone and prolactin after ovarectomy. Archives of Gynecology, 234(3), 219–229. [DOI:10.1007/BF00570759]
Kerdelhué, B., Andrews, M. C., Zhao, Y., Scholler, R., & Jones Jr, H. W. (2006). Short term changes in melatonin and cortisol serum levels after a single administration of estrogen to menopausal women. Neuroendocrinology Letters, 27(5), 659–664. [Google Scholar] [URL] [PDF]
Keye, W. R., & Jaffe, R. B. (1975). Strength-duration characteristics of estrogen effects on gonadotropin response to gonadotropin-releasing hormone in women. I. Effects of varying duration of estradiol administration. The Journal of Clinical Endocrinology and Metabolism, 41(6), 1003–1008. [DOI:10.1210/jcem-41-6-1003]
Knudsen, P., Hansen, L. B., & Larsen, N. E. (1985). Pharmacokinetic implications of different oil vehicles used in depot neuroleptic treatment. Acta Psychiatrica Scandinavica, 72(S322), 7–10. [DOI:10.1111/j.1600-0447.1985.tb08535.x]
Kronawitter, D., Gooren, L. J., Zollver, H., Oppelt, P. G., Beckmann, M. W., Dittrich, R., & Mueller, A. (2009). Effects of transdermal testosterone or oral dydrogesterone on hypoactive sexual desire disorder in transsexual women: results of a pilot study. European Journal of Endocrinology, 161(2), 363–368. [DOI:10.1530/eje-09-0265] [Table]
Kuhl, H. (1986). Hormonsubstitution durch Injektionspräparate und Hautimplantate. [Hormone substitution by injectable preparations and skin implants.] Der Gynäkologe, 19(4), 241–247. [Google Scholar] [PubMed] [PDF] [Translation]
Kuhl, H. (1990). Pharmacokinetics of oestrogens and progestogens. Maturitas, 12(3), 171–197. [DOI:10.1016/0378-5122(90)90003-O]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
LaBudde, J. A., Craig, W. Y., & Spratt, D. I. (2020). Initial Evaluation of Safety and Efficacy of Administration of Estradiol (E2) by Subcutaneous (SC) Injections to Male-to-Female (MTF) Transgender Patients. Fertility and Sterility, 114(3), e91–e91. [DOI:10.1016/j.fertnstert.2020.08.277]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., … & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/S0140-6736(21)00100-8]
Larsen, S. W., Rinvar, E., Svendsen, O., Lykkesfeldt, J., Friis, G. J., & Larsen, C. (2001). Determination of the disappearance rate of iodine-125 labelled oils from the injection site after intramuscular and subcutaneous administration to pigs. International Journal of Pharmaceutics, 230(1–2), 67–75. [DOI:10.1016/S0378-5173(01)00860-2]
Larsen, S. W., & Larsen, C. (2009). Critical factors influencing the in vivo performance of long-acting lipophilic solutions—impact on in vitro release method design. The AAPS Journal, 11(4), 762–770. [DOI:10.1208/s12248-009-9153-9]
Larsen, C., Larsen, S. W., Jensen, H., Yaghmur, A., & Østergaard, J. (2009). Role of in vitro release models in formulation development and quality control of parenteral depots. Expert Opinion on Drug Delivery, 6(12), 1283–1295. [DOI:10.1517/17425240903307431]
Larsen, S. W., Thing, M. A., & Larsen, C. (2012). Oily (lipophilic) solutions and suspensions. In Wright, J. C., & Burgess, D. J. (Eds.). Long Acting Injections and Implants (Advances in Delivery Science and Technology) (pp. 113–135). Boston: Springer. [DOI:10.1007/978-1-4614-0554-2_7]
Le, A., Huang, K. J., & Cirrincione, L. R. (2022). Regulation of drug-metabolizing enzymes by sex-related hormones: clinical implications for transgender medicine. Trends in Pharmacological Sciences, 43(7), 582–592. [DOI:10.1016/j.tips.2022.03.006]
Leinonen, P., Hammond, G. L., Lukkarinen, O., & Vihko, R. (1979). Serum sex hormone binding globulin and testosterone binding after estradiol administration, castration, and their combination in men with prostatic carcinoma. Investigative Urology, 17(1), 24–27. [Google Scholar] [PubMed]
Leinonen, P. (1980). Estrone and estradiol concentrations in the testis and spermatic and peripheral venous blood of elderly men: the influence of estrogen treatment. Journal of Steroid Biochemistry, 13(7), 737–742. [DOI:10.1016/0022-4731(80)90225-3]
Leyendecker, G., Geppert, G., Nocke, W., & Ufer, J. (1975). Untersuchungen zur Pharmakokinetik von Östradiol-17β, Östradiol-Benzoat, Östradiol-Valerianat und Östradiol-Undezylat bei der Frau: Der Verlauf der Konzentrationen von Östradiol-17β, Östron, LH und FSH im Serum. [Estradiol-17β, Estrone, LH and FSH in Serum After Administration of Estradiol-17β, Estradiol-Benzoate, Estradiol-Valeriate and Estradiol-Undecylate in the Female.] Geburtshilfe und Frauenheilkunde, 35(5), 370–374. [Google Scholar] [PubMed] [PDF] [Translation]
Leyendecker, G., Wildt, L., Gips, H., Nocke, W., & Plotz, E. J. (1976). Experimental studies on the positive feedback effect of progesterone, 17α-hydroxyprogesterone and 20α-dihydroprogesterone on the pituitary release of LH and FSH in the human female. Archiv für Gynäkologie, 221(1), 29–45. [DOI:10.1007/BF00667679]
Lichten, E. M., Lichten, J. B., Whitty, A., & Pieper, D. (1996). The confirmation of a biochemical marker for women’s hormonal migraine: The depo‐estradiol challenge test. Headache: The Journal of Head and Face Pain, 36(6), 367–371. [DOI:10.1046/j.1526-4610.1996.3606367.x]
Linet, T. (2023). Prise en charge endocrinologique d’une personne trans. [Endocrinological care of a trans person.] In Faucher, P., Hassoun, D., & Linet, T. (Eds.). Santé sexuelle et reproductive des personnes LGBT [Sexual and Reproductive Health of LGBT People] (pp. 109–124). Issy-les-Moulineaux, France: Elsevier Masson. [Google Books] [URL] [WorldCat] [Excerpt]
Lixsoft. (2008). Mathematical Expressions of the Pharmacokinetic and Pharmacodynamic Models Implemented in the Monolix Software. [PDF]
Martinez, G. (1995). Estradiol and Progesterone Serum Levels in Women Using the Once-a-month Injectable Contraceptive Perlutal. / Benagiano, G., Bianchi, P., von Kesserü, E., Castañeda, A., Correa, J. E., & Martínez, G. (1995). Session 19 what is new about injectable contraceptives? Advances in Contraception, 11(1), 39–42. [DOI:10.1007/BF02436100]
Martins, R. S., Antunes, N. J., Comerlatti, G., Caraccio, G., Moreno, R. A., Frecentese, F., Caliendo, G., & De Nucci, G. (2019). Quantification of estradiol cypionate in plasma by liquid chromatography coupled with tandem mass spectrometry: Application in a pharmacokinetic study in healthy female volunteers. Journal of Pharmaceutical and Biomedical Analysis, 170, 273–278. [DOI:10.1016/j.jpba.2019.03.053]
Messinis, I. E., & Templeton, A. (1987). Effect of high dose exogenous oestrogen on midcycle luteinizing hormone surge in human spontaneous cycles. Clinical Endocrinology, 27(4), 453–459. [DOI:10.1111/j.1365-2265.1987.tb01173.x]
Messinis, I. E., & Templeton, A. A. (1987). Disparate effects of endogenous and exogenous oestradiol on luteal phase function in women. Reproduction, 79(2), 549–554. [DOI:10.1530/jrf.0.0790549]
Milano, C., & Harper, J. (2025). Comments on Injectable Estradiol Use in Transgender and Gender-Diverse Individuals in the US. The Journal of Clinical Endocrinology & Metabolism, dgaf134. [DOI:10.1210/clinem/dgaf134]
Minto, C. F., Howe, C., Wishart, S., Conway, A. J., & Handelsman, D. J. (1997). Pharmacokinetics and pharmacodynamics of nandrolone esters in oil vehicle: effects of ester, injection site and injection volume. Journal of Pharmacology and Experimental Therapeutics, 281(1), 93–102. [URL]
Misakian, A. L., Kelley, C. E., Sullivan, E. A., Chang, J. J., Singh, G., Kokosa, S., Avila, J., Cooper, H., Liang, J. W., Botzheim, B., Quint, M., Jeevananthan, A., Chi, E., Harmer, M., Hiatt, L., Kowalewski, M., Steinberg, B., Tausinga, T., Tanner, H., Ho, T. F., … Ariel, D. (2025). Injectable Estradiol Use in Transgender and Gender-Diverse Individuals throughout the United States. The Journal of Clinical Endocrinology & Metabolism, dgaf015. [DOI:10.1210/clinem/dgaf015]
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body composition and bone mineral density in male-to-female transsexuals during cross-sex hormone therapy using gonadotrophin-releasing hormone agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074] [Table]
Newton, J. R., d’Arcangues, C., & Hall, P. E. (1994). A review of ‘once-a-month’ combined injectable contraceptives. Journal of Obstetrics and Gynaecology, 14(Suppl 1), S1–S34. [DOI:10.3109/01443619409027641]
Nelson, M. D., Szczepaniak, L. S., Wei, J., Szczepaniak, E., Sánchez, F. J., Vilain, E., Stern, J. H., Bergman, R. N., Bairey Merz, C. N., & Clegg, D. J. (2016). Transwomen and the Metabolic Syndrome: Is Orchiectomy Protective? Transgender Health, 1(1), 165–171. [DOI:10.1089/trgh.2016.0016] [Table]
Nieschlag, E., & Behre, H. M. (2010). Testosterone therapy. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Andrology (pp. 437–455). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-540-78355-8_21] [PDF]
Norlén, B. J., Fritjofsson, Å., Grönquist, L., Gunnarsson, P. O., Johansson, S. Å., & Plym-Forshell, G. (1987). Plasma concentrations of estradiol and testosterone in single-drug polyestradiol phosphate therapy for prostatic cancer. European Urology, 13, 193–197. [DOI:10.1159/000472772]
Olson-Kennedy, J., Rosenthal, S. M., Hastings, J., & Wesp, L. (2016). Health considerations for gender non-conforming children and transgender adolescents. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 186–199). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Oriowo, M. A., Landgren, B. M., Stenström, B., & Diczfalusy, E. (1980). A comparison of the pharmacokinetic properties of three estradiol esters. Contraception, 21(4), 415–424. [DOI:10.1016/S0010-7824(80)80018-7]
Parkes, A. S. (1937). Relative duration of action of various esters of oestrone, oestradiol and oestriol. Biochemical Journal, 31(4), 579–585. [DOI:10.1042/bj0310579]
Patel, R., Korenman, S., Weimer, A., & Grock, S. (2024). A Call for Updates to Hormone Therapy Guidelines for Gender-Diverse Adults Assigned Male at Birth. Cureus, 16(6), e62262. [DOI:10.7759/cureus.62262] [PDF]
Patel, K. T., & Tangpricha, V. (2024). Parenteral Estradiol for Transgender Women: Time to adjust the dose. Endocrine Practice, 30(9), 893–894. [DOI:10.1016/j.eprac.2024.07.005]
Presl, J., Horejsi, J., Štroufová, A., & Herzmann, J. (1976). Sexual maturation in girls and the development of estrogen-induced gonadotropic hormone release. Annales de Biologie Animale, Biochimie, Biophysique, 16(3), 377–383. [DOI:10.1051/rnd:19760314]
Rahimy, M. H., & Ryan, K. K. (1999). Lunelle monthly contraceptive injection (medroxyprogesterone acetate and estradiol cypionate injectable suspension): assessment of return of ovulation after three monthly injections in surgically sterile women. Contraception, 60(4), 189–200. [DOI:10.1016/s0010-7824(99)00081-5]
Rahimy, M. H., Ryan, K. K., & Hopkins, N. K. (1999). Lunelle™ monthly contraceptive injection (medroxyprogesterone acetate and estradiol cypionate injectable suspension): steady-state pharmacokinetics of MPA and E2 in surgically sterile women. Contraception, 60(4), 209–214. [DOI:10.1016/S0010-7824(99)00086-4]
Rauramo, L., Punnonen, R., Kaihola, H. L., & Grönroos, M. (1980). Serum oestrone, oestradiol and oestriol concentrations in castrated women during intramuscular oestradiolvalerate and oestradiolbenzoate-oestradiolphenylpropionate therapy. Maturitas, 2(1), 53–58. [DOI:10.1016/0378-5122(80)90060-2]
Rauramo, L., Punnonen, R., & Grönroos, M. (1981). Serum concentrations of oestrone, oestradiol and oestriol during various oestrogen treatments. Maturitas, 3(2), 183–186. [DOI:10.1016/0378-5122(81)90010-4]
Recio, R., Garza-Flores, J., Schiavon, R., Reyes, A., Diaz-Sanchez, V., Valles, V., Luz de la Cruz, D., Oropeza, G., & Perez-Palacios, G. (1986). Pharmacodynamic assessment of dihydroxyprogesterone acetophenide plus estradiol enanthate as a monthly injectable contraceptive. Contraception, 33(6), 579–589. [DOI:10.1016/0010-7824(86)90046-6]
Reimann, I. W., Britzelmeier, C., Haber, P., Wollmann, H., Antonin, K. H., & Bieck, P. R. (1987). Influence of Oestradiol on Alpha2-Adrenoceptor Binding Sites on Intact Platelets of Young Male Volunteers. European Journal of Clinical Pharmacology, 33(2), 147–150. [DOI:10.1007/BF00544558]
Rosenfield, R. L., Fang, V. S., Dupon, C., Kim, M. H., & Refetoff, S. (1973). The effects of low doses of depot estradiol and testosterone in teenagers with ovarian failure and Turner’s syndrome. The Journal of Clinical Endocrinology and Metabolism, 37(4), 574–580. [DOI:10.1210/jcem-37-4-574]
Rosenfield, R. L., & Fang, V. S. (1974). The effects of prolonged physiologic estradiol therapy on the maturation of hypogonadal teen-agers. The Journal of Pediatrics, 85(6), 830–837. [DOI:10.1016/S0022-3476(74)80355-0]
Rothman, M. S., Ariel, D., Kelley, C., Hamnvik, O. R., Abramowitz, J., Irwig, M. S., Soe, K., Davidge-Pitts, C., Misakian, A. L., Safer, J. D., & Iwamoto, S. J. (2024). The Use of Injectable Estradiol in Transgender and Gender Diverse Adults: A Scoping Review of Dose and Serum Estradiol Levels. Endocrine Practice, 30(9), 870–878. [DOI:10.1016/j.eprac.2024.05.008]
Rothman, M. S., Hamnvik, O. P. R., Davidge-Pitts, C., Safer, J. D., Ariel, D., Tangpricha, V., Abramowitz, J., Soe, K., Sarvaideo, J., Kelley, C., Irwig, M. S., & Iwamoto, S. J. (2024). Revisiting Injectable Estrogen Dosing Recommendations for Gender-Affirming Hormone Therapy. Transgender Health, 9(6), 463–465. [DOI:10.1089/trgh.2023.0209]
Sang, G. W., Ge, J. L., Liu, X. H., Shao, Q. X., Zhao, X. J., & Mao, S. M. (1987). 不同剂量庚炔诺酮单独或配伍戊酸雌二醇后的药代动力学及药效学. [Pharmacokinetics and pharmacodynamics of different doses of norethisterone enanthate alone and in combination with estradiol valerate.] 中国 临 床药理 学杂志 / Chinese Journal of Clinical Pharmacology, 3(1), 7–18. [Google Scholar] [CNKI] [DOI:10.13699/j.cnki.1001-6821.1987.01.002] [PDF]
Sang, G. W. (1994). Pharmacodynamic effects of once-a-month combined injectable contraceptives. Contraception, 49(4), 361–385. [DOI:10.1016/0010-7824(94)90033-7]
Schiavon, R., Benavides, S., Oropeza, G., Garza-Flores, J., Recio, R., Díaz-Sanchez, V., & Pérez-Palacios, G. (1988). Serum estrogens and ovulation return in chronic users of a once-a-month injectable contraceptive. Contraception, 37(6), 591–598. [DOI:10.1016/0010-7824(88)90005-4]
Schug, B. S., Donath, F., & Blume, H. H. (2012). Bioavailability and pharmacodynamics of two 10-mg estradiol valerate depot formulations following IM single dose administration in healthy postmenopausal volunteers. International Journal of Clinical Pharmacology and Therapeutics, 50(2), 100–117. [Google Scholar] [DOI:10.5414/cp201589] [PDF]
Schulte-Beerbühl, M., & Nieschlag, E. (1980). Comparison of testosterone, dihydrotestosterone, luteinizing hormone, and follicle-stimulating hormone in serum after injection of testosterone enanthate or testosterone cypionate. Fertility and Sterility, 33(2), 201–203. [DOI:10.1016/S0015-0282(16)44543-7]
Schultz, K., Møllgaard, B., Fisher, A. N., Illum, L., & Larsen, C. (1998). Intramuscular rate of disappearance of oily vehicles in rabbits investigated by gamma-scintigraphy. International Journal of Pharmaceutics, 169(1), 121–126. [DOI:10.1016/S0378-5173(98)00121-5]
Schweikert, H. U., Weissbach, L., Leyendecker, G., Schwinger, E., Wartenberg, H., & Krück, F. (1982). Clinical, endocrinological, and cytological characterization of two 46, XX males. The Journal of Clinical Endocrinology & Metabolism, 54(4), 745–752. [DOI:10.1210/jcem-54-4-745]
Seibert, B., & Günzel, P. (1994). Animal toxicity studies performed for risk assessment of the once-a-month injectable contraceptive Mesigyna®. Contraception, 49(4), 303–333. [DOI:10.1016/0010-7824(94)90030-2]
Shah, J. C. (2007). Controlled Release – Small Molecules. In Stella, V. J., Borchardt, R. T., Hageman, M. J., Oliyai, R., Maag, H., & Tilley, J. W. (Eds.). Prodrugs: Challenges and Rewards, Part 1 (Biotechnology: Pharmaceutical Aspects, Volume V) (pp. 357–377). New York: Springer. [DOI:10.1007/978-0-387-49785-3_9]
Sharula, Chekir, C., Emi, Y., Arai, F., Kikuchi, Y., Sasaki, A., Matsuda, M., Shimizu, K., Tabuchi, K., Kamada, Y., Hiramatsu, Y., & Nakatsuka, M. (2012). Altered arterial stiffness in male‐to‐female transsexuals undergoing hormonal treatment. Journal of Obstetrics and Gynaecology Research, 38(6), 932–940. [DOI:10.1111/j.1447-0756.2011.01815.x] [Data]
Shaw, R. W., Butt, W. R., London, D. R., & Marshall, J. C. (1975). The oestrogen provocation test: a method of assessing the hypothalamic‐pituitary axis in patients with amenorrhoea. Clinical Endocrinology, 4(3), 267–276. [DOI:10.1111/j.1365-2265.1975.tb01534.x]
Shaw, R. W., Butt, W. R., & London, D. R. (1975). The effect of oestrogen pretreatment on subsequent response to luteinizing hormone releasing hormone in normal women. Clinical Endocrinology, 4(3), 297–304. [DOI:10.1111/j.1365-2265.1975.tb01537.x]
Shaw, R. W. (1978). Neuroendocrinology of the menstrual cycle in humans. Clinics in Endocrinology and Metabolism, 7(3), 531–559. [DOI:10.1016/S0300-595X(78)80008-5]
Shahiwala, A., Mehta, T. A., & Momin, M. M. (2018). Parenteral drug delivery systems. In Misra, A., & Shahiwala, A. (Eds.). In-Vitro and In-Vivo Tools in Drug Delivery Research for Optimum Clinical Outcomes (pp. 283–318). Boca Raton: CRC Press. [DOI:10.1201/b22448-9] [Google Books]
Sherwin, B. B., & Gelfand, M. M. (1987). Individual differences in mood with menopausal replacement therapy: Possible role of sex hormone-binding globulin. Journal of Psychosomatic Obstetrics & Gynecology, 6(2), 121–131. [DOI:10.3109/01674828709016773]
Sherwin, B. B., Gelfand, M. M., Schucher, R., & Gabor, J. (1987). Postmenopausal estrogen and androgen replacement and lipoprotein lipid concentrations. American Journal of Obstetrics and Gynecology, 156(2), 414–419. [DOI:10.1016/0002-9378(87)90295-X]
Sherwin, B. B. (1988). Affective changes with estrogen and androgen replacement therapy in surgically menopausal women. Journal of Affective Disorders, 14(2), 177–187. [DOI:10.1016/0165-0327(88)90061-4]
Sierra-Ramírez, J. A., Lara-Ricalde, R., Lujan, M., Velázquez-Ramírez, N., Godínez-Victoria, M., Hernádez-Munguía, I. A., Padilla, A., & Garza-Flores, J. (2011). Comparative pharmacokinetics and pharmacodynamics after subcutaneous and intramuscular administration of medroxyprogesterone acetate (25 mg) and estradiol cypionate (5 mg). Contraception, 84(6), 565–570. [DOI:10.1016/j.contraception.2011.03.014]
Sinkula, A. A. (1978). Methods to Achieve Sustained Drug Delivery. The Chemical Approach. In Robinson, J. R. (Ed.). Sustained and Controlled Release Drug Delivery Systems (pp. 411–555). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books] [PDF]
Slack, D. J., Di Via Ioschpe, A., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., Karim, S., & Safer, J. D. (2025). Examining the Influence of the Route of Administration and Dose of Estradiol on Serum Estradiol and Testosterone Levels in Feminizing Gender-Affirming Hormone Therapy. Endocrine Practice, 31(1), 19–27. [DOI:10.1016/j.eprac.2024.10.002]
Somerville, B. W. (1971). The Role of Oestradiol in Menstrual Migraine. In Somerville, B. W. The Influence of Progesterone and Oestradiol on Migraine (Doctoral dissertation, University of New South Wales) (pp. 93–104). [Google Scholar] [URL] [WorldCat] [PDF]
Somerville, B. W. (1972). The Role of Estradiol Withdrawal in the Etiology of Menstrual Migraine. Neurology, 22(4), 355–365. [DOI:10.1212/WNL.22.4.355]
Somerville, B. W. (1972). The influence of progesterone and estradiol upon migraine. Headache: The Journal of Head and Face Pain, 12(3), 93–102. [DOI:10.1111/j.1526-4610.1972.hed1203093.x]
Somerville, B. W. (1972). The Influence of Hormones Upon Migraine in Women. Medical Journal of Australia, 2(S2), 6–11. [DOI:10.5694/j.1326-5377.1972.tb93039.x]
Somerville, B. W. (1975). Estrogen‐withdrawal migraine: I. Duration of exposure required and attempted prophylaxis by premenstrual estrogen administration. Neurology, 25(3), 239–244. [DOI:10.1212/wnl.25.3.239]
Spack, N. P. (2013). Management of transgenderism. JAMA, 309(5), 478–484. [DOI:10.1001/jama.2012.165234]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., & Pousette, A. (1988). Single drug polyestradiol phosphate therapy in prostatic cancer. American Journal of Clinical Oncology, 11(Suppl 2), S101–S103. [DOI:10.1097/00000421-198801102-00024] [PDF]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & von Schoultz, B. (1989). Single‐drug parenteral estrogen treatment in prostatic cancer: A study of two maintenance‐dose regimens. The Prostate, 14(2), 183–188. [DOI:10.1002/pros.2990140211]
Stege, R., Gunnarsson, P. O., Johansson, C. J., Olsson, P., Pousette, Å., & Carlström, K. (1996). Pharmacokinetics and testosterone suppression of a single dose of polyestradiol phosphate (Estradurin®) in prostatic cancer patients. The Prostate, 28(5), 307–310. [DOI:10.1002/(SICI)1097-0045(199605)28:5<307::AID-PROS6>3.0.CO;2-8]
Sumioki, H. (1987). ヒト排卵機構とGonadotropin分泌調節における “β-Endorphinおよびβ-Lipotropin” の役割に関する研究. [The Role of “β-Endorphin & β-Lipotropin” on the Gonadotropin Regulation in the Mechanism of Human Ovulation.] Folia Endocrinologica Japonica, 63(10), 1289–1307. [DOI:10.1507/endocrine1927.63.10_1289]
Svendsen, O., & Aaes‐Jørgensen, T. (1979). Studies on the fate of vegetable oil after intramuscular injection into experimental animals. Acta Pharmacologica et Toxicologica, 45(5), 352–378. [DOI:10.1111/j.1600-0773.1979.tb02404.x]
Tassinari, R., & Maranghi, F. (2021). Rodent Model of Gender-Affirming Hormone Therapies as Specific Tool for Identifying Susceptibility and Vulnerability of Transgender People and Future Applications for Risk Assessment. International Journal of Environmental Research and Public Health, 18(23), 12640. [DOI:10.3390/ijerph182312640]
Thurman, A., Kimble, T., Hall, P., Schwartz, J. L., & Archer, D. F. (2013). Medroxyprogesterone acetate and estradiol cypionate injectable suspension (Cyclofem) monthly contraceptive injection: steady-state pharmacokinetics. Contraception, 87(6), 738–743. [DOI:10.1016/j.contraception.2012.11.010]
Toffoli Ribeiro, C., Gois, Í., da Rosa Borges, M., Ferreira, L. G. A., Brandão Vasco, M., Ferreira, J. G., Maia, T. C., & Dias-da-Silva, M. R. (2024). Assessment of parenteral estradiol and dihydroxyprogesterone use among other feminizing regimens for transgender women: insights on satisfaction with breast development from community-based healthcare services. Annals of Medicine, 56(1), 2406458. [DOI:10.1080/07853890.2024.2406458]
Toppozada, M. K. (1994). Existing once-a-month combined injectable contraceptives. Contraception, 49(4), 293–301. [DOI:10.1016/0010-7824(94)90029-9]
Toutain, P. L., & Bousquet-Mélou, A. (2004). Plasma terminal half-life. Journal of Veterinary Pharmacology and Therapeutics, 27(6), 427–439. [DOI:10.1111/j.1365-2885.2004.00600.x]
Trans Care BC. (2021). Gender-affirming Care for Trans, Two-Spirit, and Gender Diverse Patients in BC: A Primary Care Toolkit. Vancouver: Provincial Health Services Authority/Trans Care BC. [URL] [PDF]
Travaglini, P., Ambrosi, B., Beck-Peccoz, P., Elli, R., Rondena, M., Bara, R., & Weber, G. (1978). Hypothalamic-pituitary-ovarian function in hyperprolactinemic women. Journal of Endocrinological Investigation, 1(1), 39–45. [DOI:10.1007/BF03346769]
Travaglini, P., Elli, R., Ambrosi, B., Ballabio, M., Moriondo, P., & Faglia, G. (1979). Serum LH increase after estradiol and progesterone administration in hyperprolactinemic women. Journal of Endocrinological Investigation, 2(4), 407–411. [DOI:10.1007/BF03349341]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine position statement “assessment and hormonal management in adolescent and adult trans people, with attention for sexual function and satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
Ulrich, U., Pfeifer, T., & Lauritzen, C. (1994). Rapid Increase in Lumbar Spine Bone Density in Osteopenic Women by High-Dose Intramuscular Estrogen-Progestogen Injections. Hormone and Metabolic Research, 26(9), 428–431. [DOI:10.1055/s-2007-1001723]
Välimäki, M., Pelkonen, R., Salaspuro, M., Härkönen, M., Hirvonen, E., & Ylikahri, R. (1984). Sex hormones in amenorrheic women with alcoholic liver disease. The Journal of Clinical Endocrinology and Metabolism, 59(1), 133–138. [DOI:10.1210/jcem-59-1-133]
Valle Alvarez, D. C. (2011). Efecto de una Dosis de 50 mg de Enantato de Noretisterona y 5 mg de Valerato de Estradiol en los Niveles de Testosterona Total en Hombres Mexicanos Sanos. [Effect of a Dose of 50 mg of Norethisterone Enanthate and 5 mg of Estradiol Valerate on Total Testosterone Levels in Healthy Mexican Men.] (Masters thesis, National Polytechnic Institute of Mexico.) [Google Scholar] [URL] [PDF] [Translation]
Varangot, J., & Cedard, L. (1957). Modifications des Œstrogènes Sanguins Après Administration Intramusculaire de Benzoate d’Œstradiol. [Changes in Serum Estrogens After Intramuscular Administration of Estradiol Benzoate.] Comptes Rendus des Séances de la Société de Biologie et de ses Filiales, 151(10), 1707–1712. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF] [Translation]
Vermeulen, A. (1977). Transport and Distribution of Androgens at Different Ages. In Martini, L., & Motta, M. (Eds.). Androgens and Antiandrogens (pp. 53–65). New York: Raven Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [Excerpt]
Vhora, I., Bardoliwala, D., Ranamalla, S. R., & Javia, A. (2019). Parenteral controlled and prolonged drug delivery systems: therapeutic needs and formulation strategies. In Misra A., & Shahiwala, A. (Eds.). Novel Drug Delivery Technologies (pp. 183–260). Singapore: Springer. [DOI:10.1007/978-981-13-3642-3_7]
Vizziello, G., D’Amato, G., Trentadue, R., & Fanizza, G. (1993). Studio dinamico del blocco ipofisario indotto dalla triptorelina, mediante test all’estradiolo benzoato. [Estradiol benzoate test in the study of pituitary block induced by triptorelin.] Minerva Ginecologica, 45(4), 185–189. [Google Scholar] [PubMed] [PDF] [Translation]
Wagner, J. G. (1993). Pharmacokinetics for the Pharmaceutical Scientist. Lancaster/Basel: Technomic. [DOI:10.1201/9780203743652] [Google Books]
Weiss, G., Nachtigall, L. E., & Ganguly, M. (1976). Induction of an LH surge with estradiol benzoate. A clinical test of pituitary-hypothalamic axis competence. Obstetrics and Gynecology, 47(4), 415–418. [URL]
White, M. C., Rosenstock, J., Anapliotou, M., Mashiter, K., & Joplin, G. F. (1981). Heterogeneity of prolactin responses to oestradiol benzoate in women with prolactinomas. The Lancet, 317(8235), 1394–1396. [DOI:10.1016/S0140-6736(81)92571-X]
Wiemeyer, J. C., Fernandez, M., Moguilevsky, J. A., & Sagasta, C. L. (1986). Pharmacokinetic Studies of Estradiol Enantate in Menopausic Women. Arzneimittel-Forschung, 36(11), 1674–1677. [Google Scholar] [PubMed] [PDF]
Wiemeyer, J. C., Fernandez, M., Sagasta, C. L., & Moguilevsky, J. A. (1987). Estudos farmacocinéticos do enantato de estradiol em mulheres na menopausa. [Pharmacokinetic studies of estradiol enantate in menopausic women.] Jornal Brasileiro de Ginecologia, 97(9), 497–501. [Google Scholar] [LILACS] [PDF]
Yan, J., Pan, J., Chang, Y., & Kang, J. (1987). The effect of monthly injectable contraceptive megestrol acetate compound on pituitary-ovarian function. 上海第二医科大学学报(英文版) / Journal of Shanghai Second Medical University / Medical Bulletin of Shanghai Jiaotong University, 1(2), 7–12. [Google Scholar] [CNKI] [PDF]
Yáñez, J. A., Remsberg, C. M., Sayre, C. L., Forrest, M. L., & Davies, N. M. (2011). Flip-flop pharmacokinetics–delivering a reversal of disposition: challenges and opportunities during drug development. Therapeutic Delivery, 2(5), 643–672. [DOI:10.4155/tde.11.19]
Zhang, Y., Huo, M., Zhou, J., & Xie, S. (2010). PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Computer Methods and Programs in Biomedicine, 99(3), 306–314. [DOI:10.1016/j.cmpb.2010.01.007]
Zhou, X. F., Shao, Q. X., Han, X. J., Weng, L. J., & Sang, G. W. (1998). Pharmacokinetics of medroxyprogesterone acetate after single and multiple injection of Cyclofem® in Chinese women. Contraception, 57(6), 405–411. [DOI:10.1016/S0010-7824(98)00048-1]
\ No newline at end of file
+An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations - Transfeminine Science
An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations
By Aly | First published July 16, 2021 | Last modified May 8, 2025
Abstract / TL;DR
Injectable estradiol preparations such as estradiol valerate and estradiol cypionate in oil are frequently used as estrogens in transfeminine hormone therapy. However, there is little characterization of these preparations in transfeminine people and dosing recommendations by transgender health guidelines appear to be based on expert opinion rather than on clinical data. To help shed light on the properties of injectable estradiol and to better inform dosing considerations in transfeminine people, an informal meta-analysis of available clinical data on estradiol concentration–time curves with major injectable estradiol formulations was conducted. The included preparations were injectable estradiol benzoate in oil, estradiol valerate in oil, estradiol cypionate both in oil and as a suspension, estradiol enanthate in oil, estradiol undecylate in oil, and polyestradiol phosphate. The literature was searched for clinical concentration–time data with these injectable estradiol esters and these data were collected and analyzed. Meta-analysis consisted of data for each injectable estradiol preparation being processed and fit with pharmacokinetic models. Selected pharmacokinetic parameters were additionally determined and reported. The results of this work were discussed with regard to characteristics of injectable estradiol preparations like curve shapes, durations, estrogenic exposure, and variability between people and studies. Recommendations for injectable estradiol preparations by transgender health guidelines were also explored in light of the present results. Current guidelines recommend doses of these preparations that appear to be highly excessive with injection intervals that are too widely spaced. Based on the findings of the present meta-analysis, recommendations by guidelines should be reassessed. Finally, the fitted curves in this work were incorporated into an interactive web-based injectable estradiol simulator intended for use by transfeminine people and their medical providers to help guide therapeutic decisions.
Introduction
Estradiol is the main estrogen used in transfeminine hormone therapy and is available in a variety of different forms for use by different routes of administration. The most commonly employed forms are oral, sublingual, transdermal, and injectable preparations. Injectable estradiol preparations have been discontinued in many countries and hence are unavailable for use in transfeminine hormone therapy in many parts of the world, for instance in most of Europe (Glintborg et al., 2021). However, they are still used by many transfeminine people particularly in the United States and in the do-it-yourself (DIY) community. The most commonly used forms include estradiol valerate, estradiol cypionate, and estradiol enanthate all in oil. Injectable estradiol preparations have certain advantages over other estradiol forms that make them a popular choice for use in transfeminine hormone therapy. These include often lower cost, capacity to easily achieve higher estradiol levels that can be useful for testosterone suppression, less frequent administration, and theoretically reduced health risks relative to oral estradiol at equivalent doses due to the lack of the first pass with this route (Aly, 2020). The higher estradiol levels with injections are particularly useful for estradiol monotherapy, in which an antiandrogen is not used.
Clinically used injectable estradiol preparations are formulated not as estradiol but as estradiol esters. When injected into muscle or fat in oil solutions or crystalline aqueous suspensions, these estradiol esters form depots at the injection site from which they are slowly released. Subsequent to release, estradiol esters are rapidly metabolized into estradiol and hence act as prodrugs. When estradiol itself is given by intramuscular injection in an aqueous solution or oil solution, it is rapidly absorbed and has a very short duration. Due to having lipophilic esters, most clinically used injectable estradiol esters are more fat-soluble than estradiol (as measured by oil–water partition coefficient (P)) (Table). When these esters are administered as oil solutions by intramuscular or subcutaneous injection, their increased lipophilicity causes them to be released from the injection-site depot more slowly than estradiol and to therefore have longer durations. In the case of fatty acid esters, the longer the chain length of the ester—as in e.g. estradiol valerate (5 carbons) vs. estradiol enanthate (7 carbons) vs. estradiol undecylate (10 carbons)—the greater the fat solubility, the slower the rate of release from the depot, and the longer the time to peak levels and duration (Edkins, 1959; Sinkula, 1978; Chien, 1981; Kuhl, 2005; Kalicharan, 2017; Vhora et al., 2019). The durations of both injectable oil solutions and aqueous suspensions depend on the ester and its particular physicochemical properties, but the characteristics of these preparations are different and they work in distinct ways to produce their depot effects (Enever et al., 1983; Aly, 2019). The durations of oil solutions are dependent on the lipophilicity of the ester as well as oil vehicle, whereas the durations of aqueous suspensions depend on the properties of the ester crystal lattice as well as crystal sizes (Chien, 1981; Enever et al., 1983; Aly, 2019). The polymeric estradiol ester polyestradiol phosphate is more hydrophilic (water-soluble) than estradiol and works differently than other injectable estradiol preparations. Ιt is composed of many estradiol molecules linked together via phosphate esters (on average 13 molecules of estradiol per one molecule of polyestradiol phosphate) and has a prolonged duration due to slow cleavage into estradiol following injection. Estradiol esters are able to substantially prolong the duration of estradiol when used as injectables and these preparations have durations ranging from days to months depending on the ester and how it is formulated (Table).
In order to aid understanding of concentration–time profiles with injectable estradiol preparations, I’ve developed an interactive web-based injectable estradiol simulator for transfeminine people and their medical providers. During work on this simulator, it became apparent that there is substantial variability in estradiol levels and curve shapes between different studies even with the same injectable estradiol ester. The injectable estradiol simulator was originally designed to simulate curves from only a single well-known pharmacokinetic study that directly compared estradiol benzoate, estradiol valerate, and estradiol cypionate in oil (Oriowo et al., 1980 [Graph]). However, due to the considerable differences in estradiol levels and curves across studies, it was decided that relying on only one study for such a project would be untenable. Instead, for the simulations to be reasonably accurate to the available data, many studies would need to be incorporated. Including additional studies would also allow for inclusion of other injectable estradiol esters in the simulator. As a result, the present work—an informal meta-analysis of estradiol curves with injectable estradiol formulations—was conducted for the simulator project.
Methods
A literature search was performed to identify studies reporting clinical estradiol concentration–time data with major injectable estradiol formulations (Table 1). All of these preparations have been used in transfeminine hormone therapy at one time or another in different parts of the world, although only estradiol valerate in oil and estradiol cypionate in oil are widely used today. Some of the injectable preparations included have notably been discontinued. Acceptable data for the search included mean and individual estradiol concentration data and Cmax estradiol levels (mean peak estradiol levels of individual subjects at time Tmax). Databases like PubMed, Google Scholar, and WorldCat were searched using relevant keywords (e.g., estradiol ester names and variations thereof as well as major brand names). Publications with relevant information were catalogued for data collection. Only single-dose data and multi-dose data that allowed estradiol levels to return to baseline between doses (as in e.g. repeated once-monthly combined injectable contraceptives) were included. Studies were included regardless of the hypothalamic–pituitary–gonadal axis (HPG axis) status of the participants. The study selection criteria aimed to maximize data inclusion due to scarcity of data for several preparations. If however there were many studies for a specific preparation, studies with only 1 or 2 subjects were generally skipped due to the limited additional value that they would provide. When data were in figures in papers—as was generally the case—they were extracted from the graphs using WebPlotDigitizer.
Table 1: Major injectable estradiol formulations (ordered roughly from shortest- to longest-acting):
Following their collection, data were processed, aggregated, and modeled. Data were adjusted for endogenous estradiol production and were normalized by dose. Adjustment for endogenous estradiol production was generally done via subtraction of baseline estradiol levels. In a number of cases however, subtraction of trough estradiol levels or of estradiol levels from a control group was required instead. Data were also weighted by sample size. In a handful of instances, certain missing information (e.g., time to peak levels, baseline levels, subject body weights) was filled in with reasonable assumptions to help maximize data inclusion. Data were processed in the form of mean estradiol curve data rather than individual-subject data (except for rare n=1 studies). The combined processed data from all studies for each injectable estradiol preparation were fit via least squares regression to one-, two-, and three-compartmentpharmacokinetic models with first-order absorption and elimination that were obtained from the literature and other sources (e.g., Colburn, 1981; Wagner, 1993; Fisher & Shafer, 2007; Lixoft, 2008; Abuhelwa, Foster, & Upton, 2015; Certara, 2020). These models fit most curves from individual studies very well. Fitting the combined curve fits of all individual studies (as opposed to fitting all of the combined processed data directly) was additionally evaluated for each injectable estradiol preparation, and if it was feasible for the preparation and allowed for better fitting results, was employed instead. Fitting directly to the combined processed data has the effect of weighting individual studies by quantity of time points, whereas fitting the combined curve fits of studies eliminates this. The Akaike information criterion (AIC) was used to help guide model selection for fitting of the preparations. Curve fitting was performed using the Python library Lmfit with the Levenberg–Marquardt algorithm. Cmax concentrations are a different form of data than mean curve estradiol concentration–time data, and for this reason, were not included in the fitting unless data were very limited for a given injectable estradiol preparation. Outlying data were also excluded from fitting in a number of instances and this allowed for improved curve fits with more uniform area-under-the-curve levels. The main criterion used for excluding curves was fit area-under-the-curve levels that deviated considerably from what was typical for the injectable estradiol preparations (generally less than about 50% of the average or greater than about 150% of the average).
A selection of pharmacokinetic parameters were calculated for each injectable estradiol preparation using the single-dose fit curves and compartmental pharmacokinetic analyses. These parameters included maximal or peak concentrations of estradiol after a single dose scaled to 5 mg (Cmax), time to maximal concentrations of estradiol after a single dose (Tmax), total area-under-the-curve concentrations of estradiol after a single dose (AUC0–∞), terminal elimination half-life after a single dose (t1/2), and the terminal 90% life after a single dose (t90%) (calculated as t1/2 × 3.322). In addition, selected pharmacokinetic parameters were calculated for simulated repeated administration of each injectable preparation at steady state with a dose and dose interval of 5 mg once every 7 days using the single-dose fit curves and compartmental pharmacokinetic analyses. These parameters included time to peak concentrations of estradiol (Tmax), peak and trough concentrations of estradiol (Cmax and Cmin, respectively), peak–trough difference (PTD; Cmax – Cmin), peak–trough ratio (PTR; Cmax ÷ Cmin), and integratedmean concentrations of estradiol (Cavg). Simulation of repeated administration was performed by stacking estradiol levels for multiple injections. Cmax and Tmax were defined and calculated in general as peak estradiol level and time to peak level of the fit mean curve as opposed to the mean peak level and mean time to peak level of individual subjects. This is because the latter would not be possible to compute as most studies reported only estradiol mean curve data. Pharmacokinetic parameters were calculated using relevant pharmacokinetic equations and, as a sanity check, were compared against those computed by PKSolver, a Microsoft Excel pharmacokinetics add-in program (Zhang et al., 2010).
Results
The figures in the subsequent sections show the original data from studies adjusted for endogenous estradiol levels and normalized to a common dose as well as the curve fits to the data (or alternatively the curve fits of the fits of the data depending on the preparation) for the included injectable estradiol preparations. Estradiol benzoate, estradiol cypionate in oil, and estradiol cypionate suspension were fit to the fits of all individual studies for these preparations, whereas estradiol enanthate, estradiol undecylate, and polyestradiol phosphate were fit directly to the combined processed data for these esters. In the case of estradiol valerate, the two fitting approaches gave nearly identical curves, and so fitting the combined processed original data was done for simplicity for this preparation. Cmax studies were excluded in the fitting for all preparations except estradiol enanthate, for which available estradiol concentration–time data were otherwise very limited. The data for the injectable estradiol preparations were generally fit best by a three-compartment pharmacokinetic model (Desmos). As a result, and for consistency, this model was used in the fitting of all preparations.
Estradiol Benzoate
Injectable estradiol benzoate has been extensively used in the past in scientific research, most notably in studies elucidating the function and dynamics of the HPG axis. One such use of estradiol benzoate has been the estrogen provocation test, a diagnostic test of HPG axis function. Due to its use in research, substantial estradiol concentration–time data with injectable estradiol benzoate exists. A total of 26 publications and concentration–time data for 355 individual injections were identified (Table 2).
Table 2: Studies of injectable estradiol benzoate (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
A number of studies were excluded from fitting due to much higher or lower area-under-the-curve levels than average. A couple of studies were omitted from the meta-analysis as they only reported total estrogen levels rather than estradiol levels with estradiol benzoate (Akande, 1974; Weiss, Nachtigall, & Ganguly, 1976). Two studies were omitted due partly to being very old and using very early and inaccurate blood tests (Varangot & Cedard, 1957; Ittrich & Pots, 1965 [Graph]). The processed original data and fit of fits curve for estradiol benzoate are shown in Figure 1.
Figure 1: Published estradiol concentration–time curves and fit of fit curves (thick black or white line) with a single intramuscular injection of estradiol benzoate in oil solution over a period of 7 days. Each curve was adjusted for endogenous estradiol levels, normalized to a dose of 5 mg, and fit with a compartmental pharmacokinetic model. Following this, the combined fit curves of the individual studies were fit using the same pharmacokinetic model. The original data from the studies for estradiol benzoate are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Valerate
Studies with curve data on injectable estradiol valerate come from its use in menopausal hormone therapy and other therapeutic indications for estrogens, its use in combined injectable contraceptives, and use in scientific research. A total of 28 publications and concentration–time data for 309 individual injections were identified for estradiol valerate (Table 3).
Table 3: Studies of injectable estradiol valerate (Spreadsheet; Plotly):
Study
na
Subjects
Dose
Reference(s)
S7175
12
Premenopausal women with menstrual migraine (n=10) and amenorrheic/postmenopausal women with history of menstrual migraine (n=2)
Normally cycling transmasculine people not on hormone therapy (n=31), transfeminine people not on hormone therapy (n=14), and gonadally intact transfeminine people on oral estrogen therapy (n=9)
a Total number of injections, not total number of subjects.
A few of these studies were excluded from fitting due generally to much higher or lower area-under-the-curve levels than average or due to being Cmax data. One study was omitted as it only reported estrone levels rather than estradiol levels (Ibrahim, 1996). Another study was not included due to being in pregnant women with concomitant pregnancy termination (Garner & Armstrong, 1977). One last study was omitted due partly to being very old and using very early and inaccurate blood tests (Ittrich & Pots, 1965 [Graph]). The processed original data and fit curve for estradiol valerate are shown in Figure 2.
Figure 2: Published estradiol concentration–time curves and fit curve (thick black or white line) with a single intramuscular injection of estradiol valerate in oil solution over a period of 30 days. Curves were adjusted for endogenous estradiol levels, normalized to a dose of 10 mg, and fit with a compartmental pharmacokinetic model. Fitting of the combined fits of individual studies for this preparation was explored but gave a nearly identical overall curve, so the overall fit curve for the combined processed original data was used for simplicity for this preparation. The original data from the studies for estradiol valerate are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Cypionate Oil
Estradiol cypionate in oil is used in menopausal hormone therapy and for other estrogen indications. However, its use has been more limited relative to other injectable estradiol preparations, like estradiol valerate. Only a handful of studies with relevant data were identified for estradiol cypionate in oil. This included 4 publications and estradiol concentration–time data for 49 individual injections (Table 4).
Table 4: Studies of injectable estradiol cypionate in oil (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
No curves were excluded from fitting in the case of this preparation. The processed original data and fit of fit curves for estradiol cypionate in oil are shown in Figure 3.
Figure 3: Published estradiol concentration–time curves and fit of fit curves (thick black or white line) with a single intramuscular injection of estradiol cypionate in oil solution over a period of 30 days. Each curve was adjusted for endogenous estradiol levels, normalized to a dose of 5 mg, and fit with a compartmental pharmacokinetic model. Following this, the combined fit curves of the individual studies were fit using the same pharmacokinetic model. The original data from the studies for estradiol cypionate in oil are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Cypionate Suspension
Estradiol cypionate suspension has been used exclusively in combined injectable contraceptives. For this reason, many relatively high quality pharmacokinetic studies with this injectable preparation have been conducted. A total of 9 publications and estradiol concentration–time data for 131 individual injections were identified for estradiol cypionate suspension (Table 5).
Table 5: Studies of injectable estradiol cypionate suspension (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects. b By subcutaneous injection rather than intramuscular injection.
One of these studies used subcutaneous injection instead of the usual intramuscular injection but the resulting curve was very similar to the curve for intramuscular injection in the same study (Sierra-Ramírez et al., 2011 [Graph]). Several Cmax studies were excluded from fitting for this preparation. One pharmacokinetic study only measured estradiol cypionate levels rather than estradiol levels and hence was not included (Martins et al., 2019 [Graph]). The processed original data and fit of fit curves for estradiol cypionate suspension are shown in Figure 4.
Figure 4: Published estradiol concentration–time curves and fit of fits curve (thick black or white line) with a single intramuscular (or in one case subcutaneous) injection of a microcrystalline aqueous suspension of estradiol cypionate over a period of 30 days. Each curve was adjusted for endogenous estradiol levels, normalized to a dose of 5 mg, and fit with a compartmental pharmacokinetic model. Following this, the combined fit curves of the individual studies were fit using the same pharmacokinetic model. The original data from the studies for estradiol cypionate suspension are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Enanthate
Estradiol enanthate has been used exclusively in combined injectable contraceptives. Several pharmacokinetic studies have been conducted with it because of this. A total of 7 publications and concentration–time data for 270 individual injections were identified for estradiol enanthate (Table 6).
Table 6: Studies of injectable estradiol enanthate (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
Of the available data, 216 of the injections were from a single study and mainly included only Cmax levels. Wiemeyer et al. (1986) was excluded from fitting due to having unusually high area-under-the-curve levels with a small sample size (n=3). Because of the scarcity of estradiol concentration–time data available for estradiol enanthate, Cmax studies were included in the fitting for this preparation. The processed original data and fit curve for estradiol enanthate are shown in Figure 5.
Figure 5: Published estradiol concentration–time curves and fit curve (thick black or white line) with a single intramuscular injection of estradiol enanthate in oil solution over a period of 30 days. Curves were adjusted for endogenous estradiol levels, normalized to a dose of 10 mg, and fit with a compartmental pharmacokinetic model. The original data from the studies for estradiol enanthate are also provided elsewhere (Spreadsheet; Plotly).
Estradiol Undecylate
Estradiol undecylate was formerly used in the treatment of prostate cancer and in menopausal hormone therapy as well as for other estrogen therapeutic indications. However, it was discontinued many years ago and is no longer used today. Nonetheless, estradiol undecylate is of significant historical interest as an injectable estradiol preparation. A total of 4 publications and estradiol concentration–time data for 7 individual injections were identified for estradiol undecylate (Table 7).
Table 7: Studies of injectable estradiol undecylate (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
Unfortunately, the identified data were of very low quality, with small sample sizes and considerable variations in estradiol levels. Moreover, estradiol undecylate is a very long-acting injectable estradiol ester with a duration measured in months, and the follow up in these studies only went to about 2 weeks post-injection. For these reasons, it was not possible to fit the data for estradiol undecylate in a reasonably accurate way—as suggested by area-under-the-curve estradiol levels that were only around one-third those of the other non-polymeric injectable estradiol esters. Limited multi-dose hormone concentration–time data also exist for estradiol undecylate, but these data could not be incorporated (Jacobi & Altwein, 1979 [Graph]; Jacobi et al., 1980 [Graph]; Derra, 1981 [Graph]). The processed original data and fit curve for estradiol undecylate are shown in Figure 6.
Figure 6: Published estradiol concentration–time curves and fit curve (thick black or white line) with a single intramuscular injection of estradiol undecylate in oil solution over a period of 90 days. Curves were adjusted for endogenous estradiol levels, normalized to a dose of 50 mg, and fit with a compartmental pharmacokinetic model. The original data from the studies for estradiol undecylate are also provided elsewhere (Spreadsheet; Plotly).
Polyestradiol Phosphate
Polyestradiol phosphate has been used primarily in the treatment of prostate cancer but has also been used for estrogen therapeutic indications like treatment of breast cancer and menopausal hormone therapy. While this injectable estradiol preparation has been used widely in the past, it appears to have recently been discontinued. All of the identified studies with estradiol concentration–time data on polyestradiol phosphate were in men with prostate cancer. A total of 11 publications and concentration–time data for 114 individual injections were identified for polyestradiol phosphate (Table 8).
Table 8: Studies of injectable polyestradiol phosphate (Spreadsheet; Plotly):
a Total number of injections, not total number of subjects.
A few older and strongly outlying studies were excluded from the fitting. The processed original data and fit curve for polyestradiol phosphate are shown in Figure 7.
Figure 7: Published estradiol concentration–time curves and fit curve (thick black or white line) with a single intramuscular injection of an aqueous solution of polyestradiol phosphate over a period of 90 days. The graph was clipped to maximum estradiol levels of 600 pg/mL (~2,200 pmol/L) for better viewability. Curves were adjusted for endogenous estradiol levels, normalized to a dose of 160 mg, and fit with a compartmental pharmacokinetic model. The original data from the studies for polyestradiol phosphate are also provided elsewhere (Spreadsheet; Plotly).
Other Injectable Estradiol Preparations
A number of clinical studies with estradiol concentration–time data for other injectable estradiol preparations were also identified during literature search:
Estradiol (unesterified) in an “aqueous” preparation (type of aqueous preparation unspecified but probably a microcrystalline aqueous suspension) (Jones et al., 1978 [Graph])
These preparations were not included in the present meta-analysis due to their relative obscurity and the limited data available for them. In addition, there were concerns about fitting the used pharmacokinetic models to the formulations with multiple estradiol components and to the microsphere formulations.
No estradiol concentration–time data were identified for certain other injectable estradiol forms of interest, like unesterified estradiol in aqueous solution, estradiol benzoate as a microcrystalline aqueous suspension (Agofollin Depot; Ovocyclin M), or estradiol benzoate butyrate/dihydroxyprogesterone acetophenide in oil (Redimen, Soluna, Unijab) (another lesser-known combined injectable contraceptive).
All Injectable Estradiol Preparations Together
Figure 8 shows the curve fits for all of the injectable estradiol preparations scaled to a single dose of 5 mg (or equivalent) together in the same figure. The dose for polyestradiol phosphate was scaled to be about 6.5 times higher than the other injectable estradiol preparations in order to make it roughly equivalent to them in terms of total estradiol exposure. This was because polyestradiol phosphate was found to produce much lower area-under-the-curve estradiol levels than the other injectable estradiol preparations (see the Discussion section). Estradiol undecylate was not included in Figure 8 as a decent fit curve could not be obtained for it due to the very limited data available for this preparation.
Figure 8: Curve fits of published estradiol concentration–time data with different injectable estradiol preparations by intramuscular injection scaled to equivalent doses and plotted over a period of 20 days in a single combined graph. Polyestradiol phosphate is scaled to a 6.5-fold higher dose that is roughly equivalent to that for the other esters as it gave total estradiol levels that were around 6 or 7 times lower than the other esters at the same dose. An alternative version of this figure without estradiol benzoate and with the x-axis spanning 30 days is also provided (Graph).
Figure 9 shows simulated curves at steady state for repeated administration of all of the injectable estradiol preparations scaled to a dose of 5 mg (or equivalent) once every 7 days. As with the previous figure, the dose for polyestradiol phosphate was scaled to be about 6.5 times higher than the other injectable estradiol preparations and estradiol undecylate was not included in the figure.
Figure 9: Simulated curves at steady state for repeated administration of different injectable estradiol preparations by intramuscular injection scaled to equivalent doses and plotted over three injection cycles. This simulation was based on the fit curves of the published single-dose estradiol concentration–time data reported in this meta-analysis. Polyestradiol phosphate is scaled to a 6.5-fold higher dose that is roughly equivalent to that for the other esters as it gave total estradiol levels that were around 6 or 7 times lower than the other esters at the same dose. An alternative version of this figure without estradiol benzoate is also provided (Graph).
For more simulated estradiol concentration–time curves with repeated injections of these injectable estradiol preparations, please see the accompanying interactive web simulator.
Selected Pharmacokinetic Parameters
The table below shows selected pharmacokinetic parameters for the fit curves of the included injectable estradiol preparations (Table 9). Estradiol undecylate was not included in the table due to the lack of data needed to achieve a decent curve fit for this preparation and the uncertainty of its parameters.
Table 9: Selected pharmacokinetic parameters for estradiol with injectable estradiol preparations following a single 5 mg dose by intramuscular injection:
Estradiol preparation
Tmax (d)
Cmax (pg/mL)
t1/2 (d)
t90% (d)
AUC0–∞ (pg•d/mL)
Estradiol benzoate in oil
0.65
971
1.2
3.9
2410
Estradiol valerate in oil
2.1
295
3.0
9.9
1886
Estradiol cypionate oil
4.3
155
6.7
22.3
2150
Estradiol cypionate suspension
1.2
241
5.1
16.9
2096
Estradiol enanthate in oil
6.5
160
4.6
15.1
2183
Polyestradiol phosphate a
18.0
34
28.4
94.2
2117
a Scaled instead to a single 32.5 mg injection (6.5 times higher dose than with the other esters).
The table below shows selected pharmacokinetic parameters for simulated curves at steady state with repeated administration of the included injectable estradiol preparations (Table 10). As with the previous table, estradiol undecylate was not included.
Table 10: Selected pharmacokinetic parameters for estradiol with injectable estradiol preparations with simulated repeated administration of 5 mg once every 7 days by intramuscular injection:
Estradiol preparation
Tmax (d)
Cmax (pg/mL)
Cmin (pg/mL)
Peak–trough diff. (pg/mL)
Peak–trough ratio
Cavg (pg/mL)
Estradiol benzoate in oil
0.64
990
29
962
35
344
Estradiol valerate in oil
1.9
384
142
242
2.7
269
Estradiol cypionate oil
3.1
339
262
77
1.3
307
Estradiol cypionate suspension
1.0
404
189
214
2.1
299
Estradiol enanthate in oil
4.0
329
288
41
1.1
312
Polyestradiol phosphate a
3.2
304
299
5
1.0
302
a Scaled instead to repeated injections of 32.5 mg every 7 days (6.5 times higher dose than with the other esters).
Terminal half-life (t1/2) is the time for the concentration of estradiol to decrease by 50% after pseudo-equilibrium of distribution has been reached—not the time required for half of an administered dose of the estradiol ester to be eliminated (Toutain & Bousquet-Mélou, 2004). It is calculated using only the terminal portion of a concentration–time curve, without the absorption or distribution phases influencing it (Toutain & Bousquet-Mélou, 2004). Due to flip–flop kinetics with depot injectables and the very short blood half-life of estradiol (~0.5–2 hours), what is being described by the terminal half-life in the case of depot estradiol injectables is not actually elimination of estradiol from blood but rather is the absorption of estradiol from the injection-site depot (Toutain & Bousquet-Mélou, 2004; Yáñez et al., 2011).
Discussion
Data Quality, Limitations, and Variability Between Studies
The accuracies of the curve fits for the different included injectable estradiol preparations are limited by the available data for these preparations. The quantity and quality of data are variable among these preparations. In some cases, such as with estradiol valerate in oil and estradiol cypionate in suspension, the data are overall quite good. In other instances, such as with estradiol cypionate in oil and estradiol enanthate in oil, the available data are more limited. There was undersampling of certain parts of the concentration–time curve with some preparations, for instance estradiol benzoate in oil (the early curve), estradiol enanthate in oil (much of the curve), and polyestradiol phosphate (the late curve). In the case of estradiol undecylate in oil, the available data for this preparation weren’t adequate to achieve a decent curve fit at all. The fit curves and calculated pharmacokinetic parameters of the included injectable estradiol preparations should be interpreted with the imperfect data in mind. For example, the curve shapes and pharmacokinetic parameters for the different preparations should not be taken as precise determinations in most cases but instead as rough estimates that would no doubt change with more and better data. Indeed, the fits and pharmacokinetic parameters were often noticeably sensitive to the influences of individual studies. Modeling decisions, such as the choice of pharmacokinetic model, or whether to fit directly to the combined processed data versus to the fits of individual studies, also yielded significantly different curve fits as well as calculated pharmacokinetic parameters.
Due to scarcity of data for several injectable estradiol preparations, the study selection criteria maximized data inclusion in order to allow for better curve fits at the risk of including potentially less reliable data. As examples, studies were included regardless of the status of the HPG axis of the participants, and Cmax data were included in the fitting if data were very limited. In the case of HPG axis state, studies with cycling women may result in greater error due to more variable levels of endogenous estradiol. Moreover, acute high levels of estradiol can induce a surge in luteinizing hormone levels after several days in gonadally intact women, and this may cause a delayed bump in estradiol levels (Wiki). One of the more overt instances of this can be seen in a study of estradiol benzoate in such women (Shaw, 1978 [Graph]). Many if not most of the included studies with estradiol benzoate involved women with intact HPG axes, whereas studies of this sort were uncommon with the other preparations. In the case of Cmax data, these data when Cmax corresponds to the mean of individual peaks are a different type of data than the peak of the mean curve of all individuals. Cmax levels can differ in both magnitude and timing compared to the mean curve peak (e.g., Oriowo et al., 1980 [Graph]; Rahimy, Ryan, & Hopkins, 1999). This is because for instance not all individuals peak at the same time and this variability in time to peak normally serves to dilute peak levels for the mean curve when compared to individual maximal concentrations. However, Cmax levels are in any case generally in the vicinity of the mean curve peak. While Cmax levels were excluded in the fitting for most injectable estradiol preparations, they were included in the case of estradiol enanthate. This was because the available mean and individual estradiol curve data were very limited for this specific preparation, and inclusion of Cmax data allowed for improved fitting in spite of its limitations. Lastly, some of the included data was once-monthly multi-dose, and research with once-monthly estradiol enanthate-containing combined injectable contraceptives has found that the time to peak levels may shift with repeated long-term use (Schiavon et al., 1988; Garza-Flores, 1994).
There was considerable variability between studies in terms of estradiol levels and concentration–time curve shapes with the same injectable estradiol preparation. The reasons for the large variability across studies are not fully clear. In any case, there are many potential factors that may contribute to this variability. These include preparation- and injection-related factors like formulation (e.g., oil vehicle, other components and excipients, concentration, particle size), injection volume, site of injection (e.g., buttocks, thigh, upper arm), injection technique (e.g., force of injection—and resulting depot droplet dimensions), and syringe dead space. They additionally include various subject- and research-related variables like differing blood-testing methodology, differing sample characteristics (e.g., age, weight, gender, ethnicity, physical activity, HPG axis state), and sampling error (Sinkula, 1978; Chien, 1981; Minto et al., 1997; Larsen & Larsen, 2009; Larsen et al., 2009; Florence, 2010; Larsen, Thing, & Larsen, 2012; Kalicharan, 2017). Older studies, which used potentially less accurate blood tests and tended to have smaller numbers of subjects, seemed to particularly add to the variability between studies. These studies may represent less reliable data than more recent research with larger sample sizes. The exclusion criteria helped to remove outliers for the different injectable estradiol preparations however. This meta-analysis does not take into account the potential factors underlying the variability between studies. To do so would be difficult, as in many cases information on these variables is not provided in individual studies and research quantifying their precise influences and relative importances is limited.
It is in any case known from other studies that different oil vehicles are absorbed at different rates from the injection site (Svendsen & Aaes‐Jørgensen, 1979; Schultz et al., 1998; Larsen et al., 2001) and can result in different concentration–time curve shapes (Ballard, 1978 [Excerpt]; Knudsen, Hansen, & Larsen, 1985). This is thought to be due to differences in oil lipophilicity and depot release rates. Viscosity of oils has also been hypothesized to potentially influence rate of depot escape (Schug, Donath, & Blume, 2012). However, research so far has not supported this hypothesis (Larsen & Larsen, 2009; Larsen, Thing, & Larsen, 2012). Oil vehicles can vary with injectable estradiol preparations even for the same estradiol ester. For instance, pharmaceutical estradiol valerate is formulated in sesame oil, castor oil, or sunflower oil depending on the preparation (Table). It is notable however that these three oils have similar lipophilicities (Table). On the other hand, homebrewed injectable estradiol preparations used by DIY transfeminine people often employ medium-chain triglyceride (MCT) oil as the oil vehicle. This oil (in the proprietary form of Viscoleo) has notably been found to be much more rapidly absorbed than conventional oils like sesame oil and castor oil in animals (Svendsen & Aaes‐Jørgensen, 1979; Schultz et al., 1998; Larsen et al., 2001). In addition, although based on very limited data, MCT oil has been found to give spikier and shorter-lasting depot injectable curves in humans (Knudsen, Hansen, & Larsen, 1985). As such, injectable estradiol preparations using MCT oil as the vehicle may have differing and less favorable concentration–time curve shapes than pharmaceutical injectable estradiol products. Other excipients, like benzyl alcohol, as well as factors like injection site and volume, have additionally been found to influence pharmacokinetic properties with depot injectables (Minto et al., 1997; Kalicharan, Schot, & Vromans, 2016). Excipients besides oil vehicle also vary by formulation (Table).
An implication of the variability between studies is that there is not a single estradiol concentration–time curve for a given injectable estradiol preparation but rather there are many, with these curves determined by variables such as formulation, dose/administration, and subject characteristics, among others. Hence, the curve fits determined in this meta-analysis represent only an estimation of the most typical and hence likely case, but the true curve for a preparation in a given context may be quite different.
Fitting all studies for a given injectable estradiol preparation individually first, and then fitting the fits of these studies, allowed for improved curve fits relative to directly fitting all of the combined processed original data for the preparation. The latter approach has limitations in that it has the effect of inherently weighting individual studies by quantity of time points (resulting in studies with greater time sampling having greater influence on the fit). Additionally, and more problematically, this approach can lead to distortions in curve shape due to different studies sampling different portions of the curve to differing extents in conjunction with systematic differences in curves between these studies. These are problems that fitting the fits of individual studies instead can solve. However, it is not possible to fit all individual studies, as some studies have limited time sampling and curve characterization which precludes fitting them appropriately. Cmax data are an example of this, which on their own cannot be fit properly. As such, it was not possible to fit the fits of the individual studies for all injectable estradiol preparations. Consequently, the fitting approach in this regard was not the same across esters, with some fit instead directly to the combined processed original data (e.g., estradiol enanthate, polyestradiol phosphate).
In spite of the various limitations of this work, aggregated analysis and modeling with injectable estradiol preparations has not previously been done. This informal meta-analysis provides among the most detailed insight into estradiol levels and curve shapes with these preparations available to date.
Durations and Curve Shapes
The curve shapes of non-polymeric injectable estradiol esters in oil relate strongly to lipophilicity. The more lipophilic the ester, the lower the peak levels and the more protracted the estradiol concentration–time curve. Accordingly, estradiol benzoate, one of the least lipophilic estradiol esters, has one of the spikiest curves and shortest durations, whereas more lipophilic estradiol esters, like estradiol cypionate in oil and estradiol enanthate, have comparatively flatter curves with delayed peaks and longer durations.
Duration of Estradiol Valerate
The estradiol concentration–time curve for injectable estradiol valerate in the well-known Oriowo et al. (1980) [Graph] study is notably spikier and shorter-lasting than the overall curve for estradiol valerate in this meta-analysis. On the other hand, the overall curve for injectable estradiol valerate in this meta-analysis was similar to (and considerably influenced by) the curves from several relatively recent and presumably better-quality studies of this injectable estradiol ester (e.g., Göretzlehner et al., 2002; Valle Alvarez, 2011; Schug, Donath, & Blume, 2012). It’s noteworthy that Oriowo et al. (1980) used a peanut oil-based formulation of estradiol valerate that differed from pharmaceutical injectable estradiol valerate preparations, which generally use sesame oil or castor oil as the carrier (as well as other excipients) (Table). This may have influenced the curve shape of estradiol valerate in Oriowo et al. (1980). The study also had a small sample size relative to the more recent studies (n=9 versus n=17, n=32, and n=24×2, respectively). Based on the newer and overall data, estradiol valerate appears to have a curve that is noticeably flatter and more prolonged than that suggested by Oriowo et al. (1980).
Duration of Estradiol Cypionate in Oil versus Estradiol Enanthate
Available estradiol concentration–time data for injectable estradiol cypionate in oil and estradiol enanthate in oil are more limited than with several of the other injectable estradiol preparations, and no direct comparisons of these two preparations exist at present. Based on some of the available literature on these injectable estradiol esters, most notably discussion by Oriowo et al. (1980) and a review of the pharmacokinetics of combined injectable contraceptives (Garza-Flores, 1994 [Graph]), it seemed that the duration of estradiol enanthate in oil was longer than that of estradiol cypionate in oil. However, this was based on limited research from separate and hence indirectly comparative studies of these esters. The estradiol cypionate in oil data from the relevant Garza-Flores (1994) figure was based on Oriowo et al. (1980) [Graph], and there are reasons to be cautious about relying on these data alone. The main concern is that curve shapes with the same injectable estradiol preparation can vary considerably across studies, as the present meta-analysis has shown. The reasons for this have yet to be fully clarified as already discussed, but among other factors may include varying formulations across studies of the same injectable estradiol ester. It is notable in this regard that Oriowo et al. (1980) used a formulation of estradiol cypionate that differs from conventional pharmaceutical estradiol cypionate in oil preparations—specifically, the study used a peanut oil-based formulation (with few other specifics) rather than the cottonseed oil-based preparation employed in marketed pharmaceutical formulations (Table). The study also had a somewhat small sample size (n=10) and may have had significant sampling error. Hence, single studies, perhaps particularly Oriowo et al. (1980), should be interpreted cautiously.
A small but interesting pharmacokinetic study which directly compared injectable testosterone cypionate (n=6) and testosterone enanthate (n=6) both in oil is relevant to the topic in question. This study found that equivalent doses of these testosterone esters using otherwise identical formulations produced virtually identical testosterone concentration–time curves (Schulte-Beerbühl & Nieschlag, 1980 [Graph]). The findings of this study are consistent with the fact that the lipophilicities of testosterone cypionate and testosterone enanthate (as measured by predicted log P) are very similar when directly compared (e.g., 5.1 vs. 5.11 with ALOGPS, 6.29 vs. 6.11 with ChemAxon logP, and 6.4 vs. 6.3 with XLogP3, respectively (Table). This of course is of importance as lipophilicity is thought to be the key factor determining the release kinetics of oil-based depot injectables (Sinkula, 1978; Shah, 2007; Larsen & Larsen, 2009; Larsen, Thing, & Larsen, 2012; Shahiwala, Mehta, & Momin, 2018). Analogously similar lipophilicities can be seen when comparing estradiol cypionate and estradiol enanthate, which employ the same ester moieties (e.g., predicted log P values of 6.47 vs. 6.45 with ALOGPS and 7.1 vs. 7.0 with XLogP3, respectively) (Table). Hence, on a theoretical level, injectable estradiol cypionate and estradiol enanthate, like injectable testosterone cypionate and testosterone enanthate, might be expected to produce very similar curves—at least provided all other variables, such as formulation, are held constant.
The present meta-analysis found that the overall estradiol curve for estradiol cypionate in oil was significantly less spikey and more prolonged than that observed in Oriowo et al. (1980). It is noteworthy in this regard that all of the other studies included for estradiol cypionate in oil specifically employed pharmaceutical Depo-Estradiol and that the overall curve for this preparation appears to be more consistent with its licensed injection interval for use in menopausal hormone therapy (1–5 mg once every 3–4 weeks) (Depo-Estradiol Label). Moreover, this meta-analysis found that injectable estradiol cypionate in oil and estradiol enanthate in oil had fairly similar and comparably flat and prolonged estradiol concentration–time curves. However, estradiol cypionate in oil appeared to peak earlier than estradiol enanthate, while estradiol enanthate was eliminated more rapidly than estradiol cypionate in oil in the terminal portion of the curve. In any case, the available concentration–time data for these preparations are limited, and the present work is not able to determine whether these estradiol esters have truly differing pharmacokinetic properties, as the apparent differences between the curves for these preparations may simply be due to statistical error. Taken together, estradiol cypionate in oil may have a less spikey and longer-lasting curve than that implied by Oriowo et al. (1980), and estradiol cypionate in oil and estradiol enanthate may have more similar curves than has been previously assumed.
Curve Shape of Estradiol Cypionate Suspension
While estradiol cypionate as an aqueous suspension is a relatively long-lasting injectable estradiol preparation similarly to estradiol cypionate in oil and estradiol enanthate in oil, it seems to differ in the shape of its estradiol concentration–time curve from these preparations. Estradiol cypionate as a suspension has a curve that appears to peak significantly earlier than estradiol cypionate in oil and other longer-acting oil-based injectable estradiol preparations. This might relate to the differing mechanisms of depot action and unique properties of injectable aqueous suspensions (Aly, 2019). In line with this notion, injectable medroxyprogesterone acetate suspension (Depo-Provera) also appears to peak rapidly despite having a very long duration (longer durations tending to be associated with delayed peaks in the case of oil-based depot injectables) (Graphs). Although aqueous suspensions generally last longer than oil solutions as injectables (Enever et al., 1983; Aly, 2019), this is not always the case, and estradiol cypionate suspension interestingly seems to be shorter-acting than estradiol cypionate in oil.
Estradiol Exposure and Potency
The average estradiol levels with the non-polymeric injectable estradiol esters when scaled to a dose and dosing interval of 5 mg every 7 days were around 300 pg/mL (~1,100 pmol/L). For comparison, in premenopausal cisgender women, estradiol production is on average about 200 μg/day (or 6 mg per month/cycle) and mean estradiol levels are around 100 pg/mL (~370 pmol/L) (Aly, 2019). After adjusting for the molecular weight of the ester, the estradiol levels for a given dose of non-polymeric injectable estradiol esters are in fairly close agreement with the estradiol levels for an equal quantity of estradiol produced endogenously by the ovaries in premenopausal cisgender women (very roughly around 1.2 mg estradiol per 7 days for injectable estradiol esters and 1.4 mg estradiol per 7 days for ovarian production to achieve average integrated estradiol levels of around 100 pg/mL). The preceding is in accordance with the fact that injectable estradiol valerate has been reported to have approximately 100% bioavailability (with this being less characterized but likely also the case for the other non-polymeric injectable estradiol esters) (Düsterberg & Nishino, 1982; Seibert & Günzel, 1994).
Although non-polymeric injectable estradiol esters have differing estradiol concentration–time curve shapes, they all appear to achieve fairly similar area-under-the-curve levels of estradiol when compared to one another. This is in accordance with the fact that differences in molecular weight and hence estradiol content with the different estradiol esters are fairly minor (all of the assessed non-polymeric esters range from 62 to 76% of that of estradiol in terms of estradiol content, and all but estradiol undecylate are in the range of 69 to 76%) (Table). The appearance of differences in area-under-the-curve levels of estradiol in the present meta-analysis is probably just due to statistical error, and true differences cannot be established by this meta-analysis. An implication of the similar area-under-the-curve estradiol levels with the different non-polymeric injectable estradiol esters is that these preparations can all be expected to deliver a roughly comparable amount of estradiol for the same dose.
On the other hand, the polymeric ester polyestradiol phosphate appears to produce around 6- to 7-fold lower area-under-the-curve and average estradiol levels than non-polymeric estradiol esters. This suggests that the estradiol in polyestradiol phosphate is not 100% bioavailable, and is supported by the fact that this ester is used clinically at substantially higher dosages than other injectable estradiol esters (40–320 mg/month), even for the same indications such as menopausal hormone therapy and treatment of prostate cancer (Wiki; Estradurin Labels). This does not seem to have been previously described in the literature, and the reasons for it are unknown. It seems possible that polyestradiol phosphate may be partially excreted before it can be cleaved into estradiol and thereby rendered partly inactive, in turn necessitating the use of higher doses to achieve the same estradiol levels and therapeutic effect.
Although two given injectable estradiol preparations may produce equivalent total estradiol levels, this does not necessarily mean that they will always have the same estrogenic potency (i.e., strength of effect at a given dose). It is plausible that spikier estradiol concentration–time curves, like with estradiol benzoate, may have overall lower estrogenic potency than more steady curves, like with estradiol enanthate. This is because estrogen receptors for a given tissue should become saturated at a certain point due to the finite quantity of available receptors in the tissue. As a result, high peak estradiol levels with spikier curves may effectively be “wasted” to varying extents in different tissues. On the other hand, more spikey estradiol curves, due to higher peak estradiol levels, might have greater influence on tissues that require high estradiol levels for effect such as the liver (and by extension on coagulation and associated health risks) (Aly, 2020). However, these possibilities are speculative and theoretical. Although some literature exists that is relevant to this issue (e.g., Parkes, 1937; Bradbury, Long, & Durham, 1953), there is very little research in this area. Consequently, it is not currently possible to take into account time-related variations in estradiol levels or differing estradiol curve shapes when assessing the comparative estrogenic potency between injectable estradiol preparations (or between other estradiol forms/routes). It is also noteworthy that these variations depend on injection interval and may be reduced with shorter injection intervals that maintain steadier estradiol levels, which must also be considered.
Variability Between Individuals
There is substantial variation in total estradiol levels and curve shapes between people with the same injectable estradiol preparation. Indicators of interindividual variability such as standard deviation or 95% range have not been included in this meta-analysis at this time due to the large amount of additional time and work this would require (e.g., additional extraction of error bars from all studies and analysis). In any case, individual studies that were included show this marked interindividual variation (e.g., Oriowo et al., 1980; Derra, 1981 [Graph]; Aedo et al., 1985 [Graphs]; Sang et al., 1987 [Graphs]; Rahimy & Ryan, 1999 [Graph]; Valle Alvarez, 2011 [Graph]; Schug, Donath, & Blume, 2012 [Graphs]). Highly variable estradiol levels are already well-established with oral and transdermal estradiol (Kuhl, 2005; Wiki). Less variability might be expected with non-polymeric injectable estradiol esters since these preparations appear to have approximately complete bioavailability. However, it seems that even with injectable forms of estradiol, the variability between people is still quite substantial. An implication of this is that the appropriate dose and dosing interval of an injectable estradiol formulation for a given person will vary considerably. This emphasizes the importance of blood work to ensure that injectable estradiol preparations are neither overdosed—which can increase health risks such as blood clots (Aly, 2020)—nor underdosed—which may result in suboptimal testosterone suppression and therapeutic efficacy.
Insights for Clinical Guidelines and Dosing Recommendations
Clinical guidelines for transgender health (see also Aly (2020)) provide recommendations on doses and dosing intervals of injectable estradiol valerate in oil and estradiol cypionate in oil (Table 11). Dosing recommendations are not given for other injectable estradiol preparations, which are much less commonly used in transgender medicine. The recommended doses for estradiol valerate and estradiol cypionate vary widely depending on the guidelines, whereas the recommended intervals are consistently once every 1 to 2 weeks. The doses for estradiol valerate range from 2 to 20 mg/week or 5 to 80 mg/2 weeks and the doses for estradiol cypionate range from <1 to 10 mg/week or <2 to 80 mg/2 weeks. For reference, the Endocrine Society guidelines and the University of California, San Francisco (UCSF) guidelines are the most major clinical guidelines for transgender hormone therapy at present (Aly, 2020). The Endocrine Society guidelines recommend 5 to 30 mg/2 weeks or 2 to 10 mg/week for either estradiol valerate or estradiol cypionate (Hembree et al., 2017). Conversely, the UCSF guidelines recommend <20 to 40 mg/2 weeks for estradiol valerate and <2 to 5 mg/2 weeks for estradiol cypionate (with the option to divide dose into weekly injections if cyclical side effects occur) (Deutsch, 2016a).
Table 11: Recommended doses and injection intervals of injectable estradiol preparations (specifically estradiol valerate and estradiol cypionate) in transgender medicine clinical guidelinesa:
a Several other guidelines recommend doses and intervals that appear to be taken directly from the Endocrine Society or UCSF guidelines and thus are not listed here but can be found elsewhere (Aly, 2020).
A number of concerns arise when the doses and intervals of injectable estradiol valerate and estradiol cypionate recommended by the major transgender clinical guidelines are considered in the context of the present informal meta-analysis and when they are compared between guidelines. Based on the present work, dosages of injectable preparations recommended by the major transgender clinical guidelines appear to result in estradiol exposure that is markedly higher than that with the recommended dosages for other routes and forms of estradiol (e.g., oral or transdermal). Whereas a dosage of 5 mg/week of any non-polymeric injectable estradiol ester appears to give average estradiol levels of around 300 pg/mL (~1,100 pmol/L), which are already supraphysiological, doses of injectable estradiol valerate or estradiol cypionate recommended by guidelines are as high as 15 to 20 mg per week. The average estradiol concentrations that would be expected to result from such doses per this meta-analysis (e.g., ~600–1,200 pg/mL or 2,200–4,400 pmol/L at 10–20 mg/week) (Figure 10) would vastly exceed the ranges for estradiol levels in transfeminine people advised by the same guidelines (generally about 50–200 pg/mL or ~180–730 pmol/L) (Table). This is not merely theoretical; for example, a study that used 40 mg/week estradiol valerate by intramuscular injection in cisgender women with estrogen deficiency to produce “pseudopregnancy” reported measured estradiol levels of about 2,500 pg/mL (~9,200 pmol/L) at 3 months and 3,100 pg/mL (~11,400 pmol/L) at 6 months of treatment (Ulrich, Pfeifer, & Lauritzen, 1994). Moreover, highly supraphysiological estradiol levels with guideline-based injectable estradiol doses are not unexpected when normal production of estradiol in premenopausal cisgender women is considered (~1.4 mg per week or 6 mg per month/cycle giving mean estradiol levels of ~100 pg/mL or 370 pmol/L) (Aly, 2019). Clinical safety data on high doses of injectable estradiol esters like estradiol valerate and estradiol cypionate are lacking at present, but excessive estrogenic exposure is known to increase the risk of health complications such as blood clots (Aly, 2020). The very high doses of these preparations that are recommended by guidelines should raise considerable reservations about their safety.
Figure 10: Simulated estradiol levels with injectable estradiol valerate at the doses and interval (5–40 mg/2 weeks) preferentially recommended by current major transgender care guidelines. Steady-state estradiol levels are reached by about the second or third injection with this injection interval and levels do not further accumulate. An alternative version of this figure with half-doses at a once-weekly interval (i.e., 2.5–20 mg/week) is also provided (Graph).
The present author elsewhere has listed doses of injectable estradiol preparations that are roughly comparable in terms of total estradiol exposure to doses for other estradiol forms and routes used in transfeminine people (Aly, 2020). These doses range from about 1 to 6 mg per week for “low dose” to “very high dose” therapy with non-polymeric injectable estradiol esters (Graph). This dose range for injectable estradiol is likely to be more appropriate for use in transfeminine people than current recommendations by many guidelines. Although high estradiol levels can be useful in transfeminine hormone therapy when antiandrogens are not used due to their greater efficacy than physiological levels in terms of testosterone suppression, only modestly supraphysiological estradiol levels (e.g., ~200–300 pg/mL or 730–1,100 pmol/L) appear to be required for strong testosterone suppression (Aly, 2019; Langley et al., 2021; Aly, 2020). In relation to this, doses of injectable estradiol need not be excessive.
Some guidelines, such as the Endocrine Society guidelines, recommend the same doses and intervals for both estradiol valerate and estradiol cypionate, whereas other guidelines, such as the UCSF guidelines, recommend different doses for these two injectable estradiol esters. Concerningly, the doses for estradiol valerate and estradiol cypionate recommended by the UCSF guidelines differ by roughly an order of magnitude (<20 to 40 mg/2 weeks for estradiol valerate and <2 to 5 mg/2 weeks for estradiol cypionate). These estradiol esters appear to produce similar average estradiol levels (e.g., around 300 pg/mL or 1,100 pmol/L at a dosage of 5 mg/week) and have concentration–time curve shapes that are not extremely different, with estradiol cypionate being only somewhat flatter and more prolonged than estradiol valerate. As such, it would appear that similar doses should be appropriate for these esters. This is supported by the fact that the same doses of estradiol valerate and estradiol cypionate are used in combined injectable contraceptives in cisgender women (both 5 mg once per month) and that these doses were carefully determined during an intensive clinical development programme for these preparations (Garza-Flores, 1994; Newton, d’Arcangues, & Hall, 1994; Sang, 1994; Toppozada, 1994). This programme notably included dose-ranging and direct-comparison studies. Based on the present analysis, the current recommendations by the UCSF guidelines may result in marked overdosage in the case of estradiol valerate and potential underdosage in the case of estradiol cypionate.
Transgender health guidelines recommend an injection interval for estradiol valerate and estradiol cypionate in oil of once every 1 to 2 weeks. Although an injection interval of 2 weeks seems technically feasible in the case of both of these preparations, such an interval would appear to result in substantial fluctuations in estradiol levels, with high peak levels and low troughs. This is particularly true in the case of the shorter-acting estradiol valerate (Figures 10, 11). Considering the wide fluctuations and unknown effects of this variability, as well as the fact that testosterone suppression when applicable may depend on sustained higher estradiol levels, it may be advisable that a once-weekly interval be preferentially recommended for these preparations. This would achieve steadier estradiol levels and would reduce potential problems due to high or low estradiol levels (Figure 11). Alternatively, a shorter interval of once every 5 days may be used with estradiol valerate to further reduce the variability in estradiol levels that occurs with this preparation (Figure 11). On the other hand, an injection interval of once every 10 days to 2 weeks may be practical and allowable in the case of the longer-acting estradiol cypionate in oil (as well as estradiol enanthate) (Figure 11)—provided that the injection cycles are well-tolerated and testosterone suppression remains adequate. When selecting different injection intervals, doses should be scaled by the interval to maintain equivalent total estradiol exposure (e.g., 3.5 mg/5 days, 5 mg/7 days, 7 mg/10 days, or 10 mg/14 days for high-dose non-polymeric injectable estradiol esters).
Figure 11: Simulated estradiol levels with a high dosage of injectable estradiol valerate or estradiol cypionate in oil at different injection intervals (doses scaled by interval to be equivalent in total estradiol exposure).
With the preceding concerns about the doses and intervals of injectable estradiol preparations recommended by transgender care guidelines considered, the question of how these recommendations were determined arises. Unfortunately, current guidelines do not generally describe how they arrived at their recommendations nor do they usually cite sources to support them. It is notable that the UCSF guidelines recommend doses and intervals for injectable estradiol preparations that are nearly identical to those advised by Christian Hamburger and Harry Benjamin in the late 1960s in the first medical textbook on transgender people (Hamburger & Benjamin, 1969). These authors recommended a dose of 10–40 mg/2 weeks for estradiol valerate and of 2–5 mg/2 weeks for estradiol cypionate (although Benjamin additionally stated that after 4–8 months, the same doses could be used at a longer injection interval of once every 4 weeks). These recommendations were notably made before estradiol blood tests became practicably available and were prior to the advent of modern pharmacokinetic studies. Hence, the recommendations for at least these guidelines appear to be based mainly on past expert opinion and long-standing historical precedent rather than on pharmacokinetic or clinical data. The same is likely to also be true for most other guidelines. High doses with certain injectable estradiol preparations (namely estradiol valerate) were probably originally employed for the purpose of achieving longer durations and more convenient injection intervals. This was notably prior to the risks of excessive estrogenic exposure like blood clots becoming known, and these doses may simply have never been revised.
Among the surveyed guidelines for transgender hormone therapy, only the UCSF guidelines (Deutsch, 2016b) and the Sherbourne Health/Rainbow Health Ontario guidelines (Bourns, 2019) referenced pharmacokinetic literature in their discussion of injectable estradiol. The specific publications cited by these guidelines were Düsterberg & Nishino (1982), Sierra-Ramírez et al. (2011), and Thurman et al. (2013). Although it is favorable to see guidelines considering published pharmacokinetic data for informing use of these preparations, there are a few concerns about the studies that were cited. Düsterberg & Nishino (1982) in its study of injectable estradiol valerate had a very small sample size (n=2), and this study was excluded as an outlier in the present meta-analysis due to unusually high estradiol levels. The findings of Düsterberg & Nishino (1982) also do not seem to have actually been used to guide dosing recommendations in the case of the UCSF guidelines, since if this were the case, the recommended doses should have been much lower. On the other hand, Bourns (2019) cited the same study and recommended injectable estradiol valerate at doses of 3–4 mg/week or 6–8 mg/2 weeks. These doses are well below those recommended by other transgender care guidelines and appear to be more appropriate for use in transfeminine people in light of the present meta-analysis. Sierra-Ramírez et al. (2011) and Thurman et al. (2013), although better-quality studies than Düsterberg & Nishino (1982), described injectable estradiol cypionate suspension rather than estradiol cypionate in oil. The oil-based version of estradiol cypionate is the form normally used in transfeminine hormone therapy, and there are important differences between these estradiol cypionate preparations such that pharmacokinetic studies for the suspension can’t necessarily be generalized to the oil solution. These preparations do in any case produce similar total estradiol levels however and hence doses should be comparable for them.
This meta-analysis is only informal and unpublished research. Nonetheless, based on the results of this work and the preceding discussion, current dosing recommendations for injectable estradiol preparations by most transgender clinical guidelines appear to be highly excessive and likely unsafe, with injection intervals that may additionally be too widely spaced. Transgender care guidelines should consider reassessing these recommendations, and the transgender medical community should make an effort to better characterize the pharmacokinetics and optimal dosing schemes of injectable estradiol preparations in transfeminine people in the future. Since clinical data on these preparations are scarce and will probably remain so in the near-term, use of published pharmacokinetic data may be further considered for guiding dosing recommendations for injectable estradiol. As identified and catalogued by this meta-analysis, there is a wealth of existing data that could be used to better inform transgender care guidelines in terms of the use of injectable estradiol preparations in transfeminine people.
Interactive Web Simulator
This informal meta-analysis of estradiol concentration–time data with injectable estradiol preparations was conducted for the purpose of deriving accurate and representative estradiol curves for incorporation into a web-based injectable estradiol simulator intended for use by transfeminine people and their clinicians. This web app is able to simulate both single-injection curves and repeated-injection curves with these preparations. An informational page for this simulator can be found at the following location:
There are various possibilities for further work on this project in the future. For example, assessment of interindividual variability for estradiol levels with injectable estradiol preparations could be included in the meta-analysis. As another example, it would be fairly straightforward and valuable to expand the meta-analysis as well as simulator to other hormonal preparations such as injectable testosterone preparations and other estradiol routes and forms like oral estradiol, sublingual estradiol, and estradiol pellets. Pharmacokinetic literature for some of these preparations has already been collected by this author. However, these future possibilities would require much additional time and effort to complete.
Special Thanks
A special thank you to Violet and Lila for their indispensable input and guidance on modeling topics during the work on this project. An additional thanks to Violet for deriving a special three-compartment pharmacokinetic model that was used in this work. Please also check out Violet’s own projects Tilia—an effort to empower trans people with tools to manage their hormonal transitions—and TransKit—a work-in-progress pharmacokinetic simulation library specifically tailored for transgender hormone therapy. Lastly, thank you to all the peer reviewers who carefully reviewed this article prior to it being posted.
Updates
Update 1: WPATH SOC8 Guidelines
In September 2022, the World Professional Association for Transgender Health (WPATH) Standards of Care for the Health of Transgender and Gender Diverse People Version 8 (SOC8) were published and made recommendations on transgender hormone therapy for the first time (Coleman et al., 2022). These guidelines are among the most highly regarded and consulted transgender care guidelines. In terms of the recommended doses of hormonal medications for transgender people, the WPATH SOC8 appear to have largely copied the Endocrine Society’s 2017 guidelines on transgender hormone therapy (Hembree et al., 2017). More specifically, in the case of injectable estradiol preparations for transfeminine people, doses of 5–30 mg/2 weeks or 2–10 mg/week estradiol valerate or estradiol cypionate were recommended. There was no discussion of injectable estradiol in the guidelines besides the preceding doses and intervals being included in a table, and no literature citations were included to support these doses. As described in the present work, these recommendations include doses and intervals that appear to be highly excessive, too widely spaced, and are likely unsafe. As such, major transgender care guidelines unfortunately continue to make uncited recommendations for injectable estradiol that are out of step with insights available from abundant published pharmacokinetic data. These recommendations are likely inadvisable, with the possibility of substantial health risks.
Update 2: Literature Mentions
The following publications in the literature have cited or mentioned Transfeminine Science’s injectable estradiol simulator and/or meta-analysis since the project was published in mid-2021:
Hughes et al. (2022)
Hughes, J. H., Woo, K. H., Keizer, R. J., & Goswami, S. (2022). Clinical Decision Support for Precision Dosing: Opportunities for Enhanced Equity and Inclusion in Health Care. Clinical Pharmacology & Therapeutics, 113(3), 565–574. [DOI:10.1002/cpt.2799]:
Lastly, we recommend that developers of [clinical decision support software (CDSS)] for dosing take an iterative and participatory approach to designing systems. By involving stakeholders in the design process, they will develop solutions that best suit users’ needs and are more likely to be adopted and used correctly. This participatory approach should involve interviews and usability testing with clinicians. Formal usability testing and analysis with real end users can improve the quality and usefulness of a system.88 Though patients themselves are not typically the end users of CDSS, their expertise (especially that of marginalized groups and organized patient advocacy organizations) can also inform CDSS developers. As an example, transgender people have compiled their own resources to understanding dosing regimens in the absence of clear clinical guidelines.89 Developers of CDSS could provide a great deal of value to these patient populations, and improve their software’s utility, by working with them to understand their needs from a dosing tool.
89. Aly, W. An interactive web simulator for estradiol levels with injectable estradiol esters. Transfeminine Science <https://transfemscience.org/articles/injectable-e2-simulator-release/> (2021) Accessed November 1, 2022.
Jaafar et al. (2022)
Jaafar, S., Torres-Leguizamon, M., Duplessy, C., & Stambolis-Ruhstorfer, M. (2022). Hormonothérapie injectable et réduction des risques: pratiques, difficultés, santé des personnes trans en France. [Hormone replacement therapy injections and harm reduction: practices, difficulties, and transgender people’s health in France.] Sante Publique, 34(HS2), 109–122. [Google Scholar] [PubMed] [DOI:10.3917/spub.hs2.0109] [Translated]:
With regard to feminizing [substitutive hormone therapy (HS)], there are no specialty injectables based on estrogens in the French pharmacopoeia. This makes it impossible to set up estrogen monotherapies which require high dosages that are more difficult to obtain with specialties with other galenic forms [5]. Faced with this lack of care, some trans women and transfeminine people obtain estradiol-based injectable solutions on the Internet or through other sources [6]. […]
5. Aly. An informal meta-analysis of estradiol curves with injectable estradiol preparations [Internet]. Transfem Sci. 2021 July 16. [Visited on 29/12/2022]. Online : https://transfemscience.org/articles/injectable-e2-meta-analysis/.
Linet (2023)
Linet, T. (2023). Prise en charge endocrinologique d’une personne trans. [Endocrinological care of a trans person.] In Faucher, P., Hassoun, D., & Linet, T. (Eds.). Santé sexuelle et reproductive des personnes LGBT [Sexual and Reproductive Health of LGBT People] (pp. 109–124). Issy-les-Moulineaux, France: Elsevier Masson. [Google Books] [URL] [WorldCat] [Excerpt] [Translated]:
Choice of estrogen.
Estradiol is generally the most prescribed estrogen. It is given orally or sublingually in transfeminine people with no significant cardiovascular risk factors. For others, the percutaneous form (patches, gel) is recommended.
The starting dose is 2 mg of estradiol orally with a step increase of 2 mg every 2 to 3 months until the optimal dose is reached [1]. For the patches, the initial dosage and the increments are 50 or 100 μg, and for the gel 2.5 g. This means that the optimal dose is generally 6 to 8 mg per day for the oral route, 3 to 4 mg for the sublingual route, and 300 to 400 μg for the patches (see table 11.1).
It may happen in consultation that the person does not wish to use the prescribed estrogens and wishes to continue the self-prescription of injectable estrogens. It is then possible to evaluate with them the most suitable dosage using the Transfem Science Injection Simulator (https://transfemscience.org/misc/injectable-e2-simulator/). Risk prevention related to injections (needles) can be done. Associations can help the person find 25 G needles of 40 mm useful this type of treatment.
Rothman et al. (2024)
Rothman, M. S., Ariel, D., Kelley, C., Hamnvik, O. R., Abramowitz, J., Irwig, M. S., Soe, K., Davidge-Pitts, C., Misakian, A. L., Safer, J. D., & Iwamoto, S. J. (2024). The Use of Injectable Estradiol in Transgender and Gender Diverse Adults: A Scoping Review of Dose and Serum Estradiol Levels. Endocrine Practice, 30(9), 870–878. [DOI:10.1016/j.eprac.2024.05.008]:
In recent years, we have noted trends in our clinical practices with TGD adults requesting injectable estradiol, particularly in the United States. The reasons given can vary; it may be due to ease of weekly or every two weeks administration, fatigue of taking daily oral medications and skin reactions to or cost of transdermal preparations. There have been discussions as to the roles of estrone/estradiol ratios in feminization and whether injectable estradiol might lead to more favorable results, however research has not supported a role for estrone in optimizing feminizing outcomes [13]. There is also a belief that higher levels can be attained with injections and may lead to faster and more complete feminization; however, there is a lack of data in the literature to support these conclusions. Such conversations occurring on reddit.com and even some hormone provider websites, are perhaps related to the historical use of high dose injectable estradiol noted above [14]. However, there is a paucity of data to guide clinicians on what dose, type and at what interval estradiol esters should be injected and when levels should be measured to ensure physiologic range estradiol levels. In fact, recent reports and clinical observations have raised concerns that the dosing suggested in guidelines may result in supraphysiological estradiol levels and that higher doses and levels may put patients at elevated risk of thromboembolic events [15-18]. This scoping review examines the available data on levels achieved with various dosages of estradiol injections in TGD adults. We also report on testosterone suppression, route (i.e., SC vs. IM), and type of estradiol ester as well as timing of blood draw relative to dose, where available.
Acknowledgment
[…] [We] thank Aly from Transfemscience for community representation and correspondence.
Toffoli Ribeiro, C., Gois, Í., da Rosa Borges, M., Ferreira, L. G. A., Brandão Vasco, M., Ferreira, J. G., Maia, T. C., & Dias-da-Silva, M. R. (2024). Assessment of parenteral estradiol and dihydroxyprogesterone use among other feminizing regimens for transgender women: insights on satisfaction with breast development from community-based healthcare services. Annals of Medicine, 56(1), 2406458. [DOI:10.1080/07853890.2024.2406458]:
Utilizing a previously published meta-analysis method of estradiol concentration-time data from publicly available information on cisgender women who had used EEn or EEn/DHPA [17], we reanalyzed and integrated data from various studies. […]
[…] The V3C Fitter and Desmos tools, accessible online at https://alyw234237.github.io/injectable-e2-simulator/v3c-fitter/ and https://www.desmos.com/calculator/ndgvp2avhj?lang=pt-BR respectively, were utilized for fitting the three-compartment pharmacokinetic model. […]
Pharmacokinetics of injectable estradiol enanthate
[…] The boxplot graph (Figure 5) illustrates that the median estradiol levels in trans women using EEn/DHPA fell within this population’s expected average range values (100–200pg/mL).
Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. […]
References
[17] Aly. 2021. An informal meta-analysis of estradiol curves with injectable estradiol preparations. Transfeminine Sci. https:// transfemscience.org/articles/injectable-e2-meta-analysis/
Update 3: Herndon et al. (2023)
In March 2023, the following study on injectable estradiol in transfeminine people was published online:
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of Subcutaneous and Intramuscular Estradiol Regimens as part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
The study was a retrospective analysis of individualized injectable estradiol in adult transfeminine people who received hormone therapy at the Mayo Clinic. Doses of injectable estradiol were adjusted by clinical providers based on estradiol levels, testosterone suppression, and feminization goals, and subsequently these clinical data were retrospectively studied by Mayo Clinic researchers. The primary aim of the study was to compare injectable estradiol by intramuscular versus subcutaneous routes. However, other general considerations for injectable estradiol, such as dosing, estradiol levels, testosterone suppression, type of injectable estradiol ester (estradiol valerate vs. estradiol cypionate), and estradiol monotherapy versus concomitant use of antiandrogens, were also assessed. The paper noted that the study was the largest to assess injectable estradiol in transfeminine people to date and was the first to directly compare intramuscular and subcutaneous injectable estradiol routes in transfeminine people.
Injectable estradiol doses were adjusted to achieve estradiol and testosterone levels within therapeutic ranges defined by the Endocrine Society 2017 guidelines (>100 pg/mL [367 pg/mL] for estradiol and <50 ng/dL [<1.7 nmol/L] for testosterone). Estradiol levels were measured exclusively using liquid chromatography–tandem mass spectrometry (LC–MS/MS), while the assay method for measuring testosterone levels was not specified. In terms of when in the injection cycle estradiol levels were measured, the authors stated the following: (1) “Timing of estradiol blood draw in relation to injection was not protocolized” and (2) “In our practice, although estradiol concentrations were generally checked midway through the injection cycle, we were unable to document with certainty the timing of the estradiol lab testing which may have influenced the results and/or the outliers”. Only the most recent blood test for each individual was analyzed, with the results of earlier tests discarded. Doses were analyzed in per-week amounts, regardless of dosing frequency (either once weekly or once every two weeks).
There were a total of 130 transfeminine people on injectable estradiol who were analyzed in the study. Of these individuals, 56 received intramuscular (i.m.) injections and 74 received subcutaneous (s.c.) injections. The median duration of therapy for injectable estradiol was 3.0 years for both routes. The vast majority of people used weekly injections (91.1% for i.m., 98.6% for s.c.), whereas the small remainder (n=6) injected once every 2 weeks. Similarly, the great majority used injectable estradiol valerate (89.3% for i.m., 86.5% for s.c.), while a small subset (n=16) used injectable estradiol cypionate. The authors did not state the injectable vehicles, but they can be confidently assumed to have both been in oil. The treatment-individualized doses per week of injectable estradiol were median 4 mg (interquartile range (IQR) 3–5.15 mg; range 1–8 mg) for the i.m. route and median 3.75 mg (IQR 3–4 mg; range 1–8 mg) for the s.c. route, with the differences in doses between groups being slightly but significantly different (p = 0.005). For the small number of people on two-week injection cycles, the doses for the combined i.m. and s.c. groups were median 8.5 mg (range 6–16 mg) every 2 weeks. Estradiol levels with injectable estradiol were median 189.5 pg/mL (IQR 126.8–252.5 or 122.5–257 pg/mL; range ~33–575 pg/mL] for i.m. and median 196 pg/mL (IQR 125.3–298.5 pg/mL; range ~23–581 pg/mL) for s.c., with the differences between groups not being significantly different (p = 0.70). The percentages of individuals with estradiol levels in target range (>100 pg/mL) were 78.6% for i.m. and 82.4% for s.c. The estradiol doses and levels of individual patients for each route were also provided in the paper (Graph). It can be seen that more individuals clustered into the higher range of doses with i.m. than with s.c. injections.
In the case of estradiol valerate versus estradiol cypionate, dose per week for i.m. with estradiol valerate was median 4 mg (IQR 3–5.45 mg) and with estradiol cypionate was median 4 mg (IQR 2.25–5 mg). In the case of s.c., dose per week with estradiol valerate was median 4 mg (IQR 3–4 mg) and with estradiol cypionate was median 3 mg (IQR 2–3 mg). The doses between estradiol valerate and estradiol cypionate were not significantly different in the case of i.m. (p = 0.51), but were significantly different in the case of s.c. (p = 0.025). Estradiol levels with the two different injectable estradiol esters were not provided.
On multiple regression analysis, injectable estradiol dose was significantly positively associated with estradiol levels (p < 0.001) following adjustment for several variables (injection route, body mass index (BMI), antiandrogen use, gonadectomy status). Each 1 mg per week in dose was associated with estradiol levels that were increased by (estimate ± standard error) 57.42 ± 10.46 pg/mL. No other variable, including notably BMI, was significantly associated with estradiol levels. According to the authors, the dose-dependently higher estradiol levels with injectable estradiol suggested the need to start at lower doses that are gradually increased in conjunction with close monitoring of hormone levels.
Testosterone levels in those with gonads were 11 ng/dL (IQR 0–19.8 ng/dL) for i.m. and 11 ng/dL (0–20 ng/dL) for s.c., with levels not significantly different between groups (p = 0.92). Adequate testosterone suppression (<50 ng/dL) in those with gonads was achieved in 84.6% with i.m. and 86.6% with s.c. In the small subset of individuals on injections every two weeks (n=6), 100% of individuals achieved target estradiol and testosterone levels. A majority of individuals on injectable estradiol in the study concomitantly used an antiandrogen, with this usually being spironolactone or finasteride. In a minority of individuals, injectable estradiol monotherapy, without concomitant use of an antiandrogen, was employed and hormone levels were measured (n=17). In this subgroup, estradiol levels were median 220 pg/mL (IQR 180–264 pg/mL) at a dose per week of median 4 mg (IQR 3–6 mg), with estradiol levels noticeably higher than in the overall group. In terms of hormone levels for those on injectable estradiol monotherapy, 100% achieved therapeutic estradiol levels (>100 pg/mL) and 88.2% achieved target testosterone levels (<50 ng/dL). The authors noted that most individuals on injectable estradiol monotherapy were able to adequately suppress testosterone, but that higher doses and levels of estradiol may be needed for testosterone suppression in this context.
Herndon et al. (2023) noted that existing recommendations for injectable estradiol in transfeminine people suggest doses of 2 to 10 mg per week or 5 to 30 mg every 2 weeks, referencing the Endocrine Society 2017 guidelines (Hembree et al., 2017) and UCSF 2016 guidelines (Deutsch, 2016a). They also noted that the UCSF 2016 guidelines recommended lower doses of estradiol cypionate than estradiol valerate, which they attributed to pharmacokinetic differences between the esters (citing Oriowo et al. (1980) for this claim). However, the authors noted that the differences between estradiol valerate and estradiol cypionate doses they observed were small, and questioned the clinical relevance of the differences. The authors also tactfully critiqued dosing recommendations by existing guidelines, and suggested their own data to guide dosing instead, with the following relevant excerpts:
Prior studies used for development of guidelines for parenteral doses are suboptimal given their small sample sizes or pre-specificized [gender-affirming hormone therapy (GAHT)] protocols with no adjustment of estradiol doses or no information on hormone concentrations achieved. [Discussion of Deutsch, Bhakri, & Kubicek (2015) and Mueller et al. (2011) …]
Overall, the studies used to support the current dosing recommendation guidelines for parenteral estradiol dosing are limited, incomplete with regards to hormone concentrations achieved, and do not provide SC as an available option. The doses of estradiol used in this study (with either SC or IM approach), were successful in achieving serum estradiol concentrations at the cisgender female range. Most importantly, as compared to current available guidelines and consensus statements [1, 2], these doses of estradiol valerate are less than half of what is recommended for both initial and maintenance dosing and achieved suppression of testosterone.
Lower doses of parenteral injections than previously described in clinical practice guidelines achieved therapeutic estradiol concentrations. Our data can serve as a dosing guide for initial and maintenance use of parenteral estradiol, which is different than what has been previously described.
Herndon et al. (2023) concluded that injectable estradiol by both i.m. and s.c. routes is effective in achieving therapeutic estradiol levels in transfeminine people. They noted that there were not meaningful differences between i.m. and s.c. in terms of dose, although i.m. may require slightly higher doses than s.c. to achieve therapeutic estradiol levels. The authors stated that doses of injectable estradiol to achieve therapeutic estradiol levels in transfeminine people were lower than previously recommended by guidelines and other publications. They concluded that clinical use of injectable estradiol in transfeminine people should include discussion of both i.m. and s.c. routes and invidiualization by patient. Finally, they called for more clinical studies on injectable estradiol in transfeminine people to evaluate clinical outcomes, feminization, and additional risks and benefits of this route compared to other routes.
The findings of Herndon et al. (2023) are pleasingly consistent with the results of the present meta-analysis. Based on the findings of this meta-analysis, assuming a linear relationship between dose and estradiol levels, estradiol levels with non-polymeric injectable estradiol esters, like estradiol valerate and estradiol cypionate in oil via intramuscular injection, increase by around 60 pg/mL on average for each 1 mg per week in dose (with Herndon et al. (2023) finding a value of 57 pg/mL per 1 mg using a multiple linear regression model). In relation to this, mean integrated estradiol levels of around 250 pg/mL on average would be expected at a dosage of 4 mg once per week. Accordingly, Herndon et al. (2023) found median estradiol levels of 190 to 196 pg/mL at per-week median doses of 3.75 to 4 mg. Similarly, the present work recommended injectable estradiol doses with non-polymeric esters of 1 to 6 mg per week (to achieve mean integrated estradiol levels of roughly 50–300 pg/mL), which is comparable to the range of about 2 to 6 mg per week used in most transfeminine people in Herndon et al. (2023) (to achieve estradiol levels of at least 100 pg/mL, along with adequate testosterone suppression). Additionally, it was noted in this meta-analysis—based on clinical research in cisgender men with prostate cancer—that only modestly supraphysiological estradiol levels, of no more than approximately 200 to 300 pg/mL, are likely to be needed for strong testosterone suppression in transfeminine people. This has likewise been confirmed with solid clinical data in transfeminine people by Herndon et al. (2023), with 88% of those on injectable estradiol monotherapy having testosterone levels of <50 ng/dL at a median injectable estradiol dose of 4 mg/week and with median estradiol levels of 220 pg/mL. It is the opinion of the present author that Herndon et al. (2023) is a very important and formative study, with clinical implications that go far beyond merely supporting the s.c. use of injectable estradiol. The study represents the first major step in the published literature to correcting the dosing and intervals of injectable estradiol in transgender care guidelines and in transgender health generally. I commend the researchers for their work.
Update 4: Rothman et al. (2024a) and Rothman et al. (2024b)
In February 2024, the following short review on injectable estradiol dosing in transfeminine people by Micol Rothman and colleagues was published online:
Rothman, M. S., Hamnvik, O. P. R., Davidge-Pitts, C., Safer, J. D., Ariel, D., Tangpricha, V., Abramowitz, J., Soe, K., Sarvaideo, J., Kelley, C., Irwig, M. S., & Iwamoto, S. J. (2024). Revisiting Injectable Estrogen Dosing Recommendations for Gender-Affirming Hormone Therapy. Transgender Health, 9(6), 463–465. [DOI:10.1089/trgh.2023.0209]
Here is the abstract of the paper:
Injectable estrogens are options for gender-affirming hormone therapy per guidelines, which suggest intramuscular dosages of 5–30 mg every 2 weeks or 2–10 mg weekly with estradiol cypionate or valerate interchangeably. Data among transgender and gender-diverse patients are limited due to local unavailability and concerns around laboratory assay variability and estradiol (E2) level fluctuation. We note a concerning trend where patients are prescribed high-dose injections based on the guidelines leading to serum E2 levels well above the range recommended in the same guidelines. Our review indicates that 5 mg weekly or lower should be prescribed when initiating injectable estrogens to avoid supraphysiologic E2 levels.
Then, in May 2024, the following longer and more comprehensive review on injectable estradiol dosing in transfeminine people by Rothman and most of the same other academics was published online:
Rothman, M. S., Ariel, D., Kelley, C., Hamnvik, O. R., Abramowitz, J., Irwig, M. S., Soe, K., Davidge-Pitts, C., Misakian, A. L., Safer, J. D., & Iwamoto, S. J. (2024). The Use of Injectable Estradiol in Transgender and Gender Diverse Adults: A Scoping Review of Dose and Serum Estradiol Levels. Endocrine Practice, 30(9), 870–878. [DOI:10.1016/j.eprac.2024.05.008]
Here is the abstract of this paper:
Objective: Feminizing gender-affirming hormone therapy is the mainstay of treatment for many transgender and gender diverse people. Injectable estradiol preparations are recommended by the World Professional Association for Transgender Health Standards of Care 8 and the Endocrine Society guidelines. Many patients prefer this route of administration, but few studies have rigorously assessed optimal dosing or route.
Methods: We performed a scoping review of the available data on estradiol levels achieved with various dosages of estradiol injections in transgender and gender diverse adults on feminizing gender-affirming hormone therapy. We also report on testosterone suppression, route (ie, subcutaneous vs intramuscular), and type of injectable estradiol ester as well as timing of blood draw relative to the most recent dose, where available.
Results: The data we reviewed suggest that the current guidelines, which recommend starting doses 2 to 10 mg weekly or 5 to 30 mg every 2 weeks of estradiol cypionate or valerate, are too high and likely lead to patients having supraphysiologic levels across much of their injection cycle.
Conclusions: The optimal starting dose for injectable estradiol remains unclear and whether it should differ for cypionate and valerate. Based on the data available, we suggest that clinicians start injectable estradiol cypionate or valerate via subcutaneous or intramuscular injections at a dose ≤5 mg weekly and then titrate accordingly to keep levels within guideline-recommended range. Future studies should assess timing of injections and subsequent levels more precisely across the injection cycle and between esters.
This paper notably also cited the present Transfeminine Science article as raising concerns about guideline-based dosing for injectable estradiol and potential health complications from these doses.
Aside from Micol Rothman herself, these reviews were also authored by other well-known experts in transgender health. For instance, two of the coauthors, Joshua Safer and Michael Irwig, were authors for the WPATH SOC8 hormone therapy chapter (WPATH SOC8 Full Contributor List). Additionally, Safer was one of the authors for the Endocrine Society’s transgender hormone therapy guidelines (Hembree et al., 2017). As such, it would appear that transgender medicine has finally started to seriously correct injectable estradiol dosing. This is a very important development. Now, the appropriate dosing and intervals of injectable estradiol will need to be more precisely established and the corrections will need to make their way into updated transgender hormone therapy guidelines and general clinical practice.
A letter to the editor commented on and concorded with Rothman and colleagues’ second literature review:
Patel, K. T., & Tangpricha, V. (2024). Parenteral Estradiol for Transgender Women: Time to adjust the dose. Endocrine Practice, 30(9), 893–894. [DOI:10.1016/j.eprac.2024.07.005]
Update 5: Kariyawasam et al. (2024)
In March 2024, the following study of estradiol levels with different routes of estradiol in transfeminine people, including injectable estradiol, was published:
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2024). Comparison of Estrone/Estradiol Ratio and Levels in Transfeminine Individuals on Different Routes of Estradiol. Transgender Health, ahead of print. [DOI:10.1089/trgh.2023.0138]
The study stratified injectable estradiol doses into different dosing levels, accounted for timing of blood draws, and compared injectable estradiol to other estradiol routes. The other routes included oral estradiol, sublingual estradiol, and transdermal estradiol. The form of injectable estradiol used was estradiol valerate in dose groups including ≤4 mg/week (“low-dose”), >4 mg/week to ≤8 mg/week (“medium-dose”), and >8 mg/week (“high-dose”). In the study, this injectable estradiol regimen resulted in supraphysiological estradiol levels in the medium- to high-dose groups (>4 mg/week) and dramatically higher estradiol levels than with the other estradiol routes (Data). Median estradiol levels were reported in a subsequent paper as follows: “Figure 2 from the paper shows estradiol levels across the 3 groups. Although exact numbers are not given in this figure, we learned through correspondence with the authors that the low dose injection group [n=8] had a median level of 202.7 ± SD 232.6 pg/mL, the medium group [n=22] 465.2 ± SD 466.3 pg/mL, and the high group [n=3] 574.4 ± SD147.3 pg/mL (converted from SI units)” (Rothman et al., 2024b). Although the sample sizes for the different dose groups were small, this study, along with Herndon et al. (2023), provides some of the best clinical data on estradiol levels with injectable estradiol in transfeminine people that have so far been published.
Update 6: Patel et al. (2024)
In June 2024, the following open-access review discussing injectable estradiol in transfeminine people and calling for updated transgender health guidelines was published:
Patel, R., Korenman, S., Weimer, A., & Grock, S. (2024). A Call for Updates to Hormone Therapy Guidelines for Gender-Diverse Adults Assigned Male at Birth. Cureus, 16(6), e62262. [DOI:10.7759/cureus.62262] [PDF]
The following quote is the relevant excerpt on injectable estradiol from the review:
The current guideline-based dosing recommendations for estradiol vary considerably, which is problematic for clinicians and patients who rely on guidelines to initiate treatment. Most notably, the conversion rates between parenteral estradiol valerate and estradiol cypionate vary drastically between the UCSF Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People (UCSF Guidelines) and The Endocrine Society Clinical Practice Guidelines for Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons (the Endocrine Society Guidelines). The UCSF Guidelines indicate the conversion between estradiol valerate and cypionate to be as high as a 4:1 ratio [2], while the Endocrine Society Guidelines provide no dosing differentiations [1]. Herndon and colleagues demonstrated that the conversion between estradiol cypionate and estradiol valerate is closer to 1:1 [4]. Further equivalence studies are needed to clarify ideal dosing conversions.
The Endocrine Society Guidelines recommend titrating estradiol to 100-200 pg/mL [1]. The UCSF Guidelines recommend 2-4 mg daily as the starting dose for oral estradiol and 5 mg weekly for parenteral estradiol valerate [2]. The Endocrine Society Guidelines suggest oral estradiol 2-6 mg daily and parenteral estradiol 2- 10 mg weekly [1]. However, Chantrapanichkul et al. found that intramuscular injections of estradiol valerate greater than 5 mg weekly led to mean estradiol concentrations well above 200 pg/mL, while 4-5 mg of oral estradiol daily only led to minimum desired concentrations [5]. Similarly, Herndon et al. found that subcutaneous estradiol at a median dose of 3.75 mg per week led to a median estradiol level of 196 pg/mL [4]. Thus, current guideline-based dosing may lead providers to choose doses of injectable estradiol that would result in supratherapeutic serum estradiol levels. In light of these recent publications, it is clear that guideline-based dosing for estradiol needs updating. In our clinical experience, parenteral estradiol valerate at doses of 2-4 mg weekly typically leads to physiologic estradiol levels. Estradiol cypionate should likely be dosed in a 1:1 ratio with estradiol valerate until future data are obtained.
Lastly, while estradiol valerate and cypionate are only FDA-approved for intramuscular administration, many patients prefer subcutaneous administration. There are small studies that suggest the pharmacokinetics of intramuscular and subcutaneous estradiol are similar [4]. While the UCSF Guidelines comment on the use of subcutaneous estradiol, other guidelines should be updated to include this option for patients [2].
Update 7: Toffoli Ribeiro et al. (2024)
Toffoli Ribeiro, C., Gois, Í., da Rosa Borges, M., Ferreira, L. G. A., Brandão Vasco, M., Ferreira, J. G., Maia, T. C., & Dias-da-Silva, M. R. (2024). Assessment of parenteral estradiol and dihydroxyprogesterone use among other feminizing regimens for transgender women: insights on satisfaction with breast development from community-based healthcare services. Annals of Medicine, 56(1), 2406458. [DOI:10.1080/07853890.2024.2406458]:
This study examines the effects of a commonly used injectable hormone combination, specifically estradiol enanthate with dihydroxyprogesterone acetophenide (EEn/DHPA), […] Our research focused on a retrospective longitudinal study involving a large cohort of transwomen evaluated between 2020 and 2022, comprising 101 participants. We assessed serum levels of estradiol (E2), testosterone (T), luteinizing hormone (LH), and follicle-stimulating hormone (FSH), comparing the EEn/DHPA hormonal regimen with other combined estrogen-progestogen (CEP) therapies. […] Our findings indicated that participants using the EEn/DHPA regimen exhibited significantly higher serum E2 levels (mean: 186 pg/mL ± 32 pg/mL) than those using other therapies (62 ± 7 pg/mL), along with lower FSH levels, but no significant differences in T and LH levels. […] These results suggest that an injectable, low-cost EEn/DHPA administered every three weeks could serve as an alternative feminizing regimen, particularly considering the extensive long-term experience of the local transgender community. Further longitudinal studies on the efficacy of feminizing-body effects and endovascular risks of various parenteral CEP types are warranted to improve primary healthcare provision for transgender persons.
Introduction
Injectable combined estrogens with progestogens (CEP) have long been widely used in Brazil and other Latin American countries, predominantly among ciswomen as an injectable contraceptive and by Brazilian transgender women and travestis as GAHT [8]. Despite the absence of recognition by the Endocrine Society as an alternative hormonal regimen due to concerns regarding thrombogenicity and challenges in routine monitoring through blood testing, the prevalent use of CEP necessitates evaluating its regimen recommendations. This has led our research to delve deeper into understanding CEP regimens, considering the experiences of travestis amidst distinct sociocultural lifestyles and limited access to public endocrinological care services [15,16]. Hence, our objective is to elucidate our observations in monitoring trans individuals utilizing CEP regimens by evaluating hormone levels […] within a cohort of transwomen employing the most common injectable CEP, namely estradiol enanthate with dihydroxyprogesterone acetophenide (EEn/DHPA) and comparing these observations with other GAHT regimens.
Subjects and methods
Estradiol enanthate pharmacokinetics curve
Utilizing a previously published meta-analysis method of estradiol concentration-time data from publicly available information on cisgender women who had used EEn or EEn/DHPA [17], we reanalyzed and integrated data from various studies. A unified single-dose curve for 30 days was created. We employed least squares regression for studies with four or more concentration-time data points (solid lines). We manually adjusted other studies with three data points to fit into a single-dose curve.
Each study’s data were adjusted for baseline estradiol levels or endogenous estradiol production and then normalized by 10 mg. The V3C Fitter and Desmos tools, accessible online at https://alyw234237.github.io/injectable-e2-simulator/v3c-fitter/ and https://www.desmos.com/calculator/ndgvp2avhj?lang=pt-BR respectively, were utilized for fitting the three-compartment pharmacokinetic model. Estradiol levels from transgender women on EEn/DHPA in this study were presented using a box plot graph featuring percentiles at 10, 25, 50, 75, and 90.
Results
Hormonal levels during the follow-up of feminizing regimens
Scatter plot graphs depicted the measurement of sex hormones (Figure 2). Serum estradiol levels in the EEn/ DHPA group (mean: 186.4pg/mL ± 32.8pg/mL) were significantly higher than those in the group using other therapies (62.2±6.9pg/mL) (Figure 2(A)). Within the EEn/DHPA group, serum FSH levels were significantly lower compared to the other group (Others) (Figure 2(B)). However, no significant difference was found between the groups concerning testosterone (Figure 2(C)) and LH (Figure 2(D)) levels.
Pharmacokinetics of injectable estradiol enanthate
Serum estradiol levels in trans women using EEn/DHPA reached the target levels for this population during hormone therapy, a trend not observed in participants using other feminizing hormone therapies (Table 1). The boxplot graph (Figure 5) illustrates that the median estradiol levels in trans women using EEn/DHPA fell within this population’s expected average range values (100–200pg/mL).
Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. For studies with four or more concentration-time data points (solid lines) and the fit of combined data (thick black line), least squares regression to a three-compartment pharmacokinetic model was employed. A single-dose curve was manually adjusted for studies with three data points (dashed lines). Data from each study were adjusted for endogenous estradiol production via baseline or trough estradiol levels subtraction and normalized by 10mg. The graph illustrates estradiol levels from the transwoman cohort in a boxplot. The shaded area represents the optimal target range for estradiol levels in transwomen under hormone therapy. The boxplot graph displays the percentiles 10, 25, 50, 75, and 90 for estradiol levels of transwomen under EEn/DHPA in this study (N=53).
Discussion
Our study represents a pioneering contribution to the literature by demonstrating that Brazilian trans women undergoing EEn/DHPA therapy achieved estradiol levels comparable to those observed during the follicular phase in cisgender women. […]
Our study further noted that DHPA demonstrates comparable efficacy to cyproterone or other anti-androgens in achieving optimal LH pituitary suppression and reducing testosterone levels. EEn/ DHPA, an affordable injectable contraceptive widely accessible in South American countries, presents a promising avenue for attaining target hormone levels among transfeminine individuals.
Additionally, our investigation, which reviewed pharmacokinetic data, supports the potential implementation of EEn/DHPA in a 21-day regimen to sustain optimal estradiol levels. While alternative medications exist to inhibit testosterone production and action, their availability varies based on regional healthcare provider systems. […]
EEn/DHPA, commonly used as a long-lasting injectable contraceptive [21–23], has found application in feminizing hormone therapy for transfeminine people, notably in travestis in Brazil [7,8,24,25]. […]
In conclusion, our long-term cohort study suggests that administering parenteral estradiol enanthate with dihydroxyprogesterone acetophenide every three weeks could serve as a practical option for feminizing hormone regimens in transgender women. Nonetheless, adopting an individualized approach that takes into account each individual’s goals, response to prior hormone therapies, and medical history is crucial. This personalized approach is central to improving healthcare provision and ensuring optimal outcomes in bodily changes. By continuing to explore and refine hormone therapy regimens, we can better support the health and well-being of transgender individuals on their gender-affirming journey.
References
[17] Aly. 2021. An informal meta-analysis of estradiol curves with injectable estradiol preparations. Transfeminine Sci. https:// transfemscience.org/articles/injectable-e2-meta-analysis/
Update 8: Misakian et al. (2025)
Misakian, A. L., Kelley, C. E., Sullivan, E. A., Chang, J. J., Singh, G., Kokosa, S., Avila, J., Cooper, H., Liang, J. W., Botzheim, B., Quint, M., Jeevananthan, A., Chi, E., Harmer, M., Hiatt, L., Kowalewski, M., Steinberg, B., Tausinga, T., Tanner, H., Ho, T. F., … Ariel, D. (2025). Injectable Estradiol Use in Transgender and Gender-Diverse Individuals throughout the United States. The Journal of Clinical Endocrinology & Metabolism, dgaf015. [DOI:10.1210/clinem/dgaf015]:
Context: Guidelines for use of injectable estradiol esters (valerate [EV] and cypionate [EC]) among transgender and gender-diverse (TGD) individuals designated male at birth vary considerably, with many providers noting supraphysiologic serum estradiol concentrations based on current dosing recommendations.
Objectives: This work aimed to 1. determine the dose of injectable estradiol (subcutaneous [SC] and intramuscular [IM]) needed to reach guideline-recommended estradiol concentrations for TGD adults using EC/EV; 2. describe the relationship between estradiol concentration relative to timing/dose of last estradiol injection and other covariates; and 3. determine dosing differences between IM/SC EV/EC.
Methods: A cross-sectional retrospective study was conducted across 6 US medical centers including TGD adults on same-dose injectable estradiol for more than 75 days, with confirmed timing of estradiol concentration relative to last injection, from January 1, 2019 to December 31, 2023. Descriptive statistics were used to describe patient characteristics and weighted linear mixed models to evaluate relationship between various covariates and estradiol concentration.
Results: Data from 562 patients were included. Among those injecting every 7 days who reached the guideline-recommended estradiol concentration (n = 131, 27.5%), the median estradiol dose was 4.0 mg (interquartile range, 3.0-5.0 mg). Among all patients, the majority reached supraphysiologic estradiol concentrations (>200 pg/mL [>734 pmol/L]) while dose and timing in the injection cycle were significant covariates for the estradiol concentration. There were no significant dosing differences between IM/SC EV/EC.
Conclusion: Injectable estradiol esters effectively reach guideline-recommended estradiol concentrations but at lower doses than previously recommended. Estradiol concentrations are best interpreted relative to timing of last injection. Route of administration and type of ester do not significantly affect estradiol concentrations.
[…]
And a letter to the editor commenting on the paper:
Milano, C., & Harper, J. (2025). Comments on Injectable Estradiol Use in Transgender and Gender-Diverse Individuals in the US. The Journal of Clinical Endocrinology & Metabolism, dgaf134. [DOI:10.1210/clinem/dgaf134]
Update 9: Slack et al. (2025)
Slack, D. J., Di Via Ioschpe, A., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., Karim, S., & Safer, J. D. (2025). Examining the Influence of the Route of Administration and Dose of Estradiol on Serum Estradiol and Testosterone Levels in Feminizing Gender-Affirming Hormone Therapy. Endocrine Practice, 31(1), 19–27. [DOI:10.1016/j.eprac.2024.10.002]:
Introduction: […] This study investigates the effect of route of administration (ROA) and dose of estradiol on estradiol (E2) and testosterone (T) levels in transfeminine individuals.
Methods: We conducted a chart review of 573 patients with an active prescription for estradiol for feminizing GAHT and serum hormone levels available.
Results: […] Intramuscular estradiol was associated with lower T and higher E2 compared to oral and transdermal ROAs (P < .001), with many achieving target hormone levels even at low doses.
Conclusions: […] The intramuscular ROA appears to be the most potent delivery of estradiol with impact on serum hormone levels with doses on the low end of guideline-suggested ranges.
[…]
Update 10: Carlson et al. (2025)
Carlson, S. M., Dominguez, C., Jeevananthan, A., & Crowley, M. J. (2025). Follow-Up Estradiol Levels Based on Regimen Formulation With Guideline-Concordant Gender-Affirming Hormone Therapy. Journal of the Endocrine Society, 9(3), bvae205. [DOI:10.1210/jendso/bvae205]:
Context: Endocrine Society guidelines for dosing of feminizing gender-affirming hormone therapy (GAHT) have remained essentially unchanged since 2009. The Endocrine Society recommends periodic monitoring of serum estradiol levels, with the goal of maintaining levels in the premenopausal cisgender female range (100-200 pg/mL). However, it is not clear whether guideline-concordant dosing consistently produces guideline-recommended levels across common estradiol formulation types (oral pills, parenteral injections, transdermal patches).
Objective: All transgender and nonbinary patients receiving estradiol-based GAHT between October 2015 and March 2023 were reviewed at a single center, with the goal of determining the frequency with which guideline-concordant dosing with different estradiol formulations led to guideline-recommended estradiol levels.
Methods: Demographics, GAHT regimen, and estradiol levels were obtained via chart review, and data were analyzed descriptively.
Results: The analytic population included n = 35 individuals, including n = 9 prescribed oral estradiol pills, n = 11 prescribed parenteral injections, and n = 15 prescribed transdermal patches. With guideline-concordant doses of oral estradiol (mean 2.8 mg daily), the mean follow-up level was 168 pg/mL; 32% of follow-up levels were subtherapeutic and 14% were supratherapeutic. With guideline-concordant doses of parenteral estradiol (mean 5.8 mg weekly), the mean midpoint follow-up level was 342 pg/mL; 91% of midpoint follow-up levels were supratherapeutic. With guideline-concordant doses of transdermal estradiol (mean 0.09 mg/day), the mean follow-up level was 81.5 pg/mL; 70% of follow-up levels were subtherapeutic.
Conclusion: Supratherapeutic follow-up estradiol levels were common with guideline-concordant parenteral estradiol doses, as were subtherapeutic follow-up levels with guideline-concordant transdermal doses. These findings may suggest the need for revision of guideline-recommended estradiol doses for these formulations
[…]
Update 11: Kanin et al. (2025)
Kanin, M., Slack, M., Patel, R., Chen, K. T., Jackson, N., Williams, K. C., & Grock, S. (2025). Injectable Estradiol Dosing Regimens in Transgender and Nonbinary Adults Listed as Male at Birth. Journal of the Endocrine Society, bvaf004. [DOI:10.1210/jendso/bvaf004]:
Context: Many transgender and nonbinary (TGNB) individuals assigned male at birth (AMAB) seek hormone therapy to achieve physical and emotional changes. Standard therapy includes estradiol, with or without an antiandrogen. Our clinical observations suggest that currently recommended injectable estradiol dosing may lead to supratherapeutic estradiol levels.
Objective: We sought to evaluate whether lower-than-recommended doses of injectable estradiol were effective in achieving serum estradiol and testosterone goals.
Methods: We conducted a retrospective cohort study to evaluate injectable estradiol dosing in treatment-naive AMAB individuals initiating hormone therapy. Data from a single provider at an academic center from January 2017 to March 2023 were analyzed. A total of 29 patients were eligible for inclusion. The primary variables of estradiol dosage, serum estradiol, and testosterone levels were analyzed over 15 months.
Results: The average estradiol dose decreased from 4.3 to 3.7 mg weekly (P < .001) during the study period with a final on-treatment estradiol level of 248 pg/mL. All individuals achieved a testosterone level of less than 50 ng/dL during the study period. The average initial on-treatment testosterone level was not significantly different from average final on-treatment measurement of 24.0 mg/dL (P = .95). […]
Conclusion: Lower doses of injectable estradiol can achieve therapeutic estradiol levels with excellent testosterone suppression. […]
[…]
This study had been previously published as a conference abstract:
Kanin, M., Slack, M., Patel, R., Chen, K. T., Jackson, N., Williams, K., & Grock, S. (2024). 8309 Injectable Estradiol Dosing Regimens; A Retrospective Review of Hormone Therapy for Gender-Diverse Adults Assigned Male at Birth. Journal of the Endocrine Society, 8(Suppl 1), bvae163-1706. [DOI:10.1210/jendso/bvae163.1706]
Abuhelwa, A. Y., Foster, D. J., & Upton, R. N. (2015). ADVAN-style analytical solutions for common pharmacokinetic models. Journal of Pharmacological and Toxicological Methods, 73, 42–48. [DOI:10.1016/j.vascn.2015.03.004]
Aedo, A. R., Landgren, B. M., Johannisson, E., & Diczfalusy, E. (1985). Pharmacokinetic and pharmacodynamic investigations with monthly injectable contraceptive preparations. Contraception, 31(5), 453–469. [DOI:10.1016/0010-7824(85)90081-2]
Aisaka, K., Ando, S., Kokubo, K., Yoshida, K., & Mori, H. (1986). いわゆる潜在性高prolactin血症患者におけるprolactin分泌予備能の検討. [Studies on Prolactin Secreting Capacity in the Ovulatory Infertile Patients with Transient Hyperprolactinemia.] 日本内分泌学会雑誌 / Nihon Naibunpi Gakkai Zasshi / Folia Endocrinologica Japonica, 62(5), 662–671. [DOI:10.1507/endocrine1927.62.5_662]
Akande, E. O. (1974). The effect of oestrogen on plasma levels of luteinizing hormone in euthyroid and thyrotoxic postmenopausal women. The Journal of Obstetrics and Gynaecology of the British Commonwealth / BJOG, 81(10), 795–803. [DOI:10.1111/j.1471-0528.1974.tb00383.x]
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). Injectable Aqueous Suspensions of Sex Hormones and How Depot Injectables Work. Transfeminine Science. [URL]
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Aly. (2020). Clinical Guidelines with Information on Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
Aly. (2021). An Interactive Web Simulator for Estradiol Levels with Injectable Estradiol Esters. Transfeminine Science. [URL]
Ballard, B. E. (1978). An Overview of Prolonged Action Drug Dosage Forms. In Robinson, J. R. (Ed.). Sustained and Controlled Release Drug Delivery Systems (pp. 1–69). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books]
Behre, H. M., Oberpenning, F., & Nieschlag, E. (1990). Comparative pharmacokinetics of androgen preparations: application of computer analysis and simulation. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 1st Edition (pp. 115–135). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-662-00814-0_6]
Behre, H. M., & Nieschlag, E. (1998). Comparative pharmacokinetics of testosterone esters. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 329–348). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_11]
Behre, H. M., Wang, C., Handelsman, D. J., & Nieschlag, E. (2004). Pharmacology of testosterone preparations. Testosterone: Action · Deficiency · Substitution, 3rd Edition (pp. 405–444). Cambridge/New York: Cambridge University Press. [DOI:10.1017/CBO9780511545221.015] [PDF]
Behre, H. M., & Nieschlag, E. (2012). Testosterone preparations for clinical use in males. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Testosterone: Action · Deficiency · Substitution, 4th Edition (pp. 309–335). Cambridge/New York: Cambridge University Press. [DOI:10.1017/CBO9781139003353.016]
Bider, D., Ben-Rafael, Z., Shalev, J., Goldenberg, M., Mashiach, S., & Blankstein, J. (1989). Pituitary and ovarian suppression rate after high dosage of gonadotropin-releasing hormone agonist. Fertility and Sterility, 51(4), 578–581. [DOI:10.1016/S0015-0282(16)60602-7]
Blackwell, R. E., Boots, L. R., & Potter Jr, H. D. (1982). Evaluation of Delestrogen and Parlodel as a luteolytic agent in humans. Fertility and Sterility, 37(2), 213–217. [DOI:10.1016/S0015-0282(16)46042-5]
Bourns, A. (2019). Guidelines for Gender-Affirming Primary Care with Trans and Non-Binary Patients, 4th Edition. Toronto: Rainbow Health Ontario/Sherbourne Health. [URL] [PDF]
Bradbury, J. T., Long, R. C., & Durham, W. C. (1953). Progesterone and estrogen requirements to induce and maintain decidua. Fertility and Sterility, 4(1), 63–75. [DOI:10.1016/S0015-0282(16)31145-1]
Braun, P., Wildt, L., & Leyendecker, G. (1983). The effect of danazol on gonadotropin secretion during the follicular phase of the menstrual cycle. Fertility and Sterility, 40(1), 37–44. [DOI:10.1016/S0015-0282(16)47174-8]
Buckman, M. T., Johnson, J., Ellis, H., Srivastava, L., & Peake, G. T. (1980). Differential lipemic and proteinemic response to oral ethinyl estradiol and parenteral estradiol cypionate. Metabolism, 29(9), 803–805. [DOI:10.1016/0026-0495(80)90117-1]
Callen-Lorde Community Health Center. (2018). Protocols for the Provision of Hormone Therapy. New York City: Callen-Lorde Community Health Center. [URL] [PDF]
Canales, E. S., Cabezas, A., Vázquez-Matute, L., & Zárate, A. (1978). Induction of ovulation with clomiphene and estradiol benzoate in anovulatory women refractory to clomiphene alone. Fertility and Sterility, 29(5), 496–499. [DOI:10.1016/S0015-0282(16)43271-1]
Canales, E. S., Fonseca, M. E., Mason, M., & Zárate, A. (1981). Feedback effect of estradiol on follicle-stimulating hormone and prolactin secretion during the puerperium. International Journal of Gynecology & Obstetrics, 19(1), 79–81. [DOI:10.1016/0020-7292(81)90043-6]
Cano, A., Gimeno, F., Fuente, T., Parrilla, J. J., & Abad, L. (1986). The positive feedback of estradiol on gonadotropin secretion in women with perimenopausal dysfunctional uterine bleeding. European Journal of Obstetrics & Gynecology and Reproductive Biology, 22(5–6), 353–358. [DOI:10.1016/0028-2243(86)90125-5]
Carlson, S. M., Dominguez, C., Jeevananthan, A., & Crowley, M. J. (2025). Follow-Up Estradiol Levels Based on Regimen Formulation With Guideline-Concordant Gender-Affirming Hormone Therapy. Journal of the Endocrine Society, 9(3), bvae205. [DOI:10.1210/jendso/bvae205]
Cavanaugh, T., Hopwood, R., Gonzalez, A., & Thompson, J. (2015). The Medical Care of Transgender Persons. Boston: Fenway Health. [URL] [PDF]
Chantrapanichkul, P., Stevenson, M. O., Suppakitjanusant, P., Goodman, M., & Tangpricha, V. (2021). Serum Hormone Concentrations in Transgender Individuals Receiving Gender-Affirming Hormone Therapy: A Longitudinal Retrospective Cohort Study. Endocrine Practice, 27(1), 27–33. [DOI:10.4158/EP-2020-0414] [Table]
Chien, Y. W. (1981). Long-acting parenteral drug formulations. Journal of Parenteral Science and Technology / PDA Journal of Pharmaceutical Science and Technology, 35(3), 106–139. [Google Scholar] [URL] [PDF]
Cirrincione, L. R., Winston McPherson, G., Rongitsch, J., Sadilkova, K., Drees, J. C., Krasowski, M. D., Dickerson, J. A., & Greene, D. N. (2021). Sublingual estradiol is associated with higher estrone concentrations than transdermal or injectable preparations in transgender women and gender nonbinary adults. LGBT Health, 8(2), 125–132. [DOI:10.1089/lgbt.2020.0249] [Table]
Colburn, W. A. (1981). Simultaneous pharmacokinetic and pharmacodynamic modeling. Journal of Pharmacokinetics and Biopharmaceutics, 9(3), 367–388. [DOI:10.1007/BF01059272]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Dahl, M., Feldman, J. L., Goldberg, J., & Jaberi, A. (2015). Endocrine Therapy for Transgender Adults in British Columbia: Suggested Guidelines. Physical Aspects of Transgender Endocrine Therapy. Vancouver: Vancouver Coastal Health. [Google Scholar] [PDF]
Davidson, A., Franicevich, J., Freeman, M., Lin, R., Martinez, L., Monihan, M., Porch, M., Samuel, L., Stukalin, R., Vormohr, J., & Zevin, B. (2013). Protocols for Hormonal Reassignment of Gender. San Francisco: San Francisco Department of Public Health/Tom Waddell Health Center. [Google Scholar] [PDF]
Depo®-Estradiol Estradiol Cypionate Label. U.S. Food and Drug Administration/Pharmacia & Upjohn (Pfizer). [URL] [PDF]
Derra, C. (1981). Hormonprofile unter Östrogen- und Antiandrogentherapie bei Patienten mit Prostatakarzinom: Östradiolundecylat versus Cyproteronacetat. [Hormone Profiles under Estrogen and Antiandrogen Therapy in Patients with Prostate Cancer: Estradiol Undecylate versus Cyproterone Acetate.] (Doctoral dissertation, University of Mainz.) [Google Scholar] [WorldCat] [PDF] [Translation]
Deutsch, M. (2014). Medical Transition. In Erickson-Schroth, L. (Ed.). Trans Bodies, Trans Selves: A Resource for the Transgender Community, 1st Edition (pp. 241–264). Oxford: Oxford University Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Deutsch, M. B., Bhakri, V., & Kubicek, K. (2015). Effects of Cross-Sex Hormone Treatment on Transgender Women and Men. Obstetrics & Gynecology, 125(3), 605–610. [DOI:10.1097/AOG.0000000000000692]
Deutsch, M. B. (Ed.). (2016). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition. San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Düsterberg, B., & Nishino, Y. (1982). Pharmacokinetic and pharmacological features of oestradiol valerate. Maturitas, 4(4), 315–324. [DOI:10.1016/0378-5122(82)90064-0]
Düsterberg, B., & Wendt, H. (1983). Plasma levels of dehydroepiandrosterone and 17β-estradiol after intramuscular administration of Gynodian-Depot® in 3 women. Hormones, 17(2), 84–89. [DOI:10.1159/000179680]
Düsterberg, B., Schmidt-Gollwitzer, M., & Hümpel, M. (1985). Pharmacokinetics and biotransformation of estradiol valerate in ovariectomized women. Hormone Research in Paediatrics, 21(3), 145–154. [DOI:10.1159/000180039]
Edkins, R. P. (1959). The Modification of the Duration of Drug Action: Pharmaceutical Considerations. Journal of Pharmacy and Pharmacology, 11(Suppl 1), 54T–66T. [DOI:10.1111/j.2042-7158.1959.tb10412.x]
Enever, R. P., Fotherby, K., Naderi, S., & Lewis, G. A. (1983). Long-acting contraceptive agents: The influence of physicochemical properties of some esters of norethisterone upon the plasma levels of free norethisterone. Steroids, 41(3), 381–396. [DOI:10.1016/0039-128X(83)90109-5]
Eriksson, O., Bäckström, T., Stridsberg, M., Hammarlund-Udenaes, M., & Naessén, T. (2006). Differential response to estrogen challenge test in women with and without premenstrual dysphoria. Psychoneuroendocrinology, 31(4), 415–427. [DOI:10.1016/j.psyneuen.2005.10.004]
Espino y Sosa, S., Cortés Fuentes, M., Gómez Rico, J. A., & Cortés Bonilla, M. (2019). Non-polymeric Microspheres for the Therapeutic Use of Estrogens: An Innovative Technology. In Khan, W. A. (Ed.). Estrogen. London: IntechOpen. [DOI:10.5772/intechopen.82553]
Fisher, D., & Shafer, S. (2007). Fisher/Shafer NONMEM Workshop Pharmacokinetic and Pharmacodynamic Analysis with NONMEM. Basic Concepts. [PDF]
Florence, A. T. (2010). Looking at Formulations. In Florence, A. T. An Introduction to Clinical Pharmaceutics (pp. 69–100). London/Chicago: Pharmaceutical Press. [Google Scholar] [Google Books]
Fotherby, K., Benagiano, G., Toppozada, H. K., Abdel-Rahman, A., Navaroli, F., Arce, B., Ramos-Cordero, R., Gual, C., Landgren, B. M., & Johannisson, E. (1982). A preliminary pharmacological trial of the monthly injectable contraceptive Cycloprovera. Contraception, 25(3), 261–272. [DOI:10.1016/0010-7824(82)90049-X]
Futterweit, W., Gabrilove, J., & Smith, H. (1984). Testicular steroidogenic response to human chorionic gonadotropin of fifteen male transsexuals on chronic estrogen treatment. Metabolism, 33(10), 936–942. [DOI:10.1016/0026-0495(84)90248-8] [Figure]
Garner, P. R., & Armstrong, D. T. (1977). The effect of human chorionic gonadotropin and estradiol-17β on the maintenance of the human corpus luteum of early pregnancy. American Journal of Obstetrics and Gynecology, 128(5), 469–475. [DOI:10.1016/0002-9378(77)90026-6]
Garza-Flores, J., Rodriguez, V., Perez-Palacios, G., Virutamasen, P., Tang-Keow, P., Konsayreepong, R., Kovacs, L., Koloszar, S., & Hall, P. E. (1987). A multicentered pharmacokinetic, pharmacodynamic study of once-a-month injectable contraceptives. I. Different doses of HRP112 and of DepoProvera. Contraception, 36(4), 441–457. [DOI:10.1016/0010-7824(87)90093-X]
Garza-Flores, J., Alba, V. M., Cravioto, M. C., Hernandez, L., Perez-Palacios, G., Alvarado, G., Rivera, R., Recio, R., & Bassol, S. (1989). Estrogen-progestogen once-a-month injectable contraceptives and serum prolactin. Contraception, 39(5), 519–529. [DOI:10.1016/0010-7824(89)90107-8]
Garza-Flores, J., Fatinikun, T., Hernandez, L., Ramos, I., Cardenas, M., & Menjivar, M. (1991). A pilot study on the assessment of a progesterone/estradiol sustained release as once-a-month-injectable contraceptive. Contraception, 44(1), 45–59. [DOI:10.1016/0010-7824(91)90105-O]
Garza-Flores, J. (1994). Pharmacokinetics of once-a-month injectable contraceptives. Contraception, 49(4), 347–359. [DOI:10.1016/0010-7824(94)90032-9]
Geppert, G. (1975). Untersuchungen zur Pharmakokinetik von Östradiol-17β, Östradiol-Benzoat, Östradiol-Valerianat und Östradiol-Undezylat bei der Frau: Der Verlauf der Konzentrationen von Östradiol-17β, Östron, LH und FSH im Serum. [Studies on the Pharmacokinetics of Estradiol-17β, Estradiol Benzoate, Estradiol Valerate, and Estradiol Undecylate in Women: The Course of the Relationships Between Estradiol-17β, Estrone, LH, and FSH in Serum.] (Doctoral dissertation, University of Bonn.) [Google Scholar] [WorldCat] [PDF] [Translation]
Glintborg, D., T’Sjoen, G., Ravn, P., & Andersen, M. S. (2021). Management of endocrine disease: Optimal feminizing hormone treatment in transgender people. European Journal of Endocrinology, 185(2), R49–R63. [DOI:10.1530/EJE-21-0059]
Goh, H. H., & Ratnam, S. S. (1988). The LH surge in humans: its mechanism and sex difference. Gynecological Endocrinology, 2(2), 165–182. [DOI:10.3109/09513598809023624]
Goh, H. H., & Ratnam, S. S. (1990). Effect of estrogens on prolactin secretion in transsexual subjects. Archives of Sexual Behavior, 19(5), 507–516. [DOI:10.1007/BF02442351]
Goh, V. H. H., & Lee, K. O. (1998). Does a positive oestrogen feedback on the hypothalamic-pituitary axis exist concurrently with a defective testosterone feedback in Klinefelter’s syndrome? Hormone Research in Paediatrics, 50(3), 160–165. [DOI:10.1159/000023266]
Goodman, R. E., Anderson, D. C., Bu’Lock, D. E., Sheffield, B., Lynch, S. S., & Butt, W. R. (1985). Study of the effect of estradiol on gonadotrophin levels in untreated male-to-female transsexuals. Archives of Sexual Behavior, 14(2), 141–146. [DOI:10.1007/BF01541659]
Gooren, L. J. G., Rao, B. R., Van Kessel, H., & Harmsen-Louman, W. (1984). Estrogen positive feedback on LH secretion in transsexuality. Psychoneuroendocrinology, 9(3), 249–259. [DOI:10.1016/0306-4530(84)90004-0]
Gooren, L. (2005). Hormone treatment of the adult transsexual patient. Hormone Research in Paediatrics, 64(Suppl 2), 31–36. [DOI:10.1159/000087751]
Göretzlehner, G., Ackermann, W., Angelow, K., Bergmann, G., Bieck, E., Golbs, S., & Kliem, O. (2002). Pharmakokinetik von Estron, Estradiol, FSH, LH und Prolaktin nach intramuskulärer Applikation von 5 mg Estradiolvalerat. [Pharmacokinetics of estradiol valerate in postmenopausal women after intramuscular administration.] Journal für Menopause, 9(2), 46–49. [Google Scholar] [URL] [PDF] [Translation]
Gorton, N., Jaffe, J. M., Thompson, J., Menkin, D., Nesteby, A., Dunn, D., Baker, K. K., Harbatkin, D., Do, T., Radix, A., Meacher, P., Goldstein, Z., Carpenter, W., Caine, M., Henn, S., Murayama, R., Feldmann, J., & Zayas, S. (2019). TransLine Gender Affirming Hormone Therapy Prescriber Guidelines. San Francisco: Lyon-Martin Health Services/TransLine. [URL] [PDF]
Gunnarsson, P. O., & Norlén, B. J. (1988). Clinical pharmacology of polyestradiol phosphate. The Prostate, 13(4), 299–304. [DOI:10.1002/pros.2990130405]
Hamburger, C., & Benjamin, H. (1969). Endocrine Treatment of Male and Female Transsexualism / Appendix for the Practicing Physician: Suggestions and Guidelines for the Management of Transsexuals. In Green, R., & Money, J. (Eds.). Transsexualism and Sex Reassignment (pp. 291–307). Baltimore: John Hopkins University Press. [Google Scholar] [Google Books] [PDF]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658]
Henriksson, P., Carlström, K., Pousette, A., Gunnarsson, P. O., Johansson, C. J., Eriksson, B., Altersgård-Brorsson, A. K., Nordle, O., & Stege, R. (1999). Time for revival of estrogens in the treatment of advanced prostatic carcinoma? Pharmacokinetics, and endocrine and clinical effects, of a parenteral estrogen regimen. The Prostate, 40(2), 76–82. [DOI:10.1002/(SICI)1097-0045(19990701)40:2<76::AID-PROS2>3.0.CO;2-Q]
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of Subcutaneous and Intramuscular Estradiol Regimens as part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
Hughes, J. H., Woo, K. H., Keizer, R. J., & Goswami, S. (2022). Clinical Decision Support for Precision Dosing: Opportunities for Enhanced Equity and Inclusion in Health Care. Clinical Pharmacology & Therapeutics, 113(3), 565–574. [DOI:10.1002/cpt.2799]:
Ibrahim, S. (1996/1998). Pharmakokinetische Untersuchungen mit Östradiolvalerat und Hydroxyprogesteroncaproat in Depotform nach einmaliger Applikation bei 24 postmenopausalen Frauen. [Pharmacokinetic studies with estradiol valerate and hydroxyprogesterone caproate in depot form after a single application in 24 postmenopausal women.] (Doctoral dissertation, Dresden University of Technology.) [Google Scholar] [WorldCat] [Partial PDF]
Ittrich, G., & Pots, P. (1965). Östrogenbestimmungen in Blut und Urin nach Verabreichung von Östrogenen. [Estrogen determinations in blood and urine after administration of estrogens.] In Kraatz, H. (Ed.). International Symposium der Gynäkologischen Endokrinologie vom 15. bis 18. Mai 1963. / Abhandlungen der Deutschen Akademie der Wissenschaften zu Berlin, Klasse für Medizin, 1965(1), 53–56. [ISSN:0568-4250] [WorldCat 1] [WorldCat 2] [WorldCat 3] [PDF] [Translation]
Jaafar, S., Torres-Leguizamon, M., Duplessy, C., & Stambolis-Ruhstorfer, M. (2022). Hormonothérapie injectable et réduction des risques: pratiques, difficultés, santé des personnes trans en France. [Hormone replacement therapy injections and harm reduction: practices, difficulties, and transgender people’s health in France.] Sante Publique, 34(HS2), 109–122. [Google Scholar] [PubMed] [DOI:10.3917/spub.hs2.0109]
Jacobi, G. H., & Altwein, J. E. (1979). Bromocriptin als Palliativtherapie beim fortgeschrittenen Prostatakarzinom. [Bromocriptine for palliation of advanced prostatic carcinoma. Experimental and clinical profile of a drug.] Urologia Internationalis, 34(4), 266–290. [DOI:10.1159/000280272]
Jacobi, G. H., Altwein, J. E., Kurth, K. H., Basting, R., & Hohenfellner, R. (1980). Treatment of advanced prostatic cancer with parenteral cyproterone acetate: a phase III randomised trial. British Journal of Urology, 52(3), 208–215. [DOI:10.1111/j.1464-410X.1980.tb02961.x]
Jacobi, G. R. (1982). Experimental Rationale for the Investigation of Antiprolactins as Palliative Treatment for Prostate Cancer. In Jacobi, G. H., & Hohenfellner, R. (Eds.). Prostate Cancer (International Perspectives in Urology, Volume 3) (pp. 419–431). Baltimore/London: Williams & Wilkins. [Google Scholar] [Google Books]
Jilma, B., Eichler, H. G., Breiteneder, H., Wolzt, M., Aringer, M., Graninger, W., Röhrer, C., Veitl, M., & Wagner, O. F. (1994). Effects of 17β-estradiol on circulating adhesion molecules. The Journal of Clinical Endocrinology and Metabolism, 79(6), 1619–1624. [DOI:10.1210/jcem.79.6.7527406]
Johansson, C. J., & Gunnarsson, P. O. (2000). Pharmacodynamic model of testosterone suppression after intramuscular depot estrogen therapy in prostate cancer. The Prostate, 44(1), 26–30. [DOI:10.1002/1097-0045(20000615)44:1<26::AID-PROS4>3.0.CO;2-P]
Jones, T. M., Fang, V. S., Landau, R. L., & Rosenfield, R. (1978). Direct inhibition of Leydig cell function by estradiol. The Journal of Clinical Endocrinology and Metabolism, 47(6), 1368–1373. [DOI:10.1210/jcem-47-6-1368]
Jönsson, G., Olsson, A. M., Luttrop, W., Cekan, Z., Purvis, K., & Diczfalusy, E. (1976). Treatment of prostatic carcinoma with various types of estrogen derivatives. In Munson, P. L., Diczfalusy, E., Glover, J., Olson, R. E., Harris, R. S., Thimann, K. V., Loraine, J. A., & Wool, I. G. (Eds.). Vitamins & Hormones, Volume 33 (pp. 351–376). New York/San Francisco/London: Academic Press. [DOI:10.1016/S0083-6729(08)60965-6]
Kalicharan, R. W., Schot, P., & Vromans, H. (2016). Fundamental understanding of drug absorption from a parenteral oil depot. European Journal of Pharmaceutical Sciences, 83, 19–27. [DOI:10.1016/j.ejps.2015.12.011]
Kalicharan, R. W. (2017). New Insights into Drug Absorption from Oil Depots. (Doctoral dissertation, Utrecht University.) [Google Scholar] [URL] [PDF]
Kanin, M., Slack, M., Patel, R., Chen, K. T., Jackson, N., Williams, K., & Grock, S. (2024). 8309 Injectable Estradiol Dosing Regimens; A Retrospective Review of Hormone Therapy for Gender-Diverse Adults Assigned Male at Birth. Journal of the Endocrine Society, 8(Suppl 1), bvae163-1706. [DOI:10.1210/jendso/bvae163.1706]
Kanin, M., Slack, M., Patel, R., Chen, K. T., Jackson, N., Williams, K. C., & Grock, S. (2025). Injectable Estradiol Dosing Regimens in Transgender and Nonbinary Adults Listed as Male at Birth. Journal of the Endocrine Society, bvaf004. [DOI:10.1210/jendso/bvaf004]
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2024). Comparison of Estrone/Estradiol Ratio and Levels in Transfeminine Individuals on Different Routes of Estradiol. Transgender Health, ahead of print. [DOI:10.1089/trgh.2023.0138] [Data]
Kemeter, P., Bernaschek, G., Altmann, G., & Feichtinger, W. (1984). The effect of 2 mg estradiol-17β plus 1 mg estriol, sequentially combined with 1 mg norethisteroneacetate, on LH, FSH, estradiol-17β, progesterone, testosterone and prolactin after ovarectomy. Archives of Gynecology, 234(3), 219–229. [DOI:10.1007/BF00570759]
Kerdelhué, B., Andrews, M. C., Zhao, Y., Scholler, R., & Jones Jr, H. W. (2006). Short term changes in melatonin and cortisol serum levels after a single administration of estrogen to menopausal women. Neuroendocrinology Letters, 27(5), 659–664. [Google Scholar] [URL] [PDF]
Keye, W. R., & Jaffe, R. B. (1975). Strength-duration characteristics of estrogen effects on gonadotropin response to gonadotropin-releasing hormone in women. I. Effects of varying duration of estradiol administration. The Journal of Clinical Endocrinology and Metabolism, 41(6), 1003–1008. [DOI:10.1210/jcem-41-6-1003]
Knudsen, P., Hansen, L. B., & Larsen, N. E. (1985). Pharmacokinetic implications of different oil vehicles used in depot neuroleptic treatment. Acta Psychiatrica Scandinavica, 72(S322), 7–10. [DOI:10.1111/j.1600-0447.1985.tb08535.x]
Kronawitter, D., Gooren, L. J., Zollver, H., Oppelt, P. G., Beckmann, M. W., Dittrich, R., & Mueller, A. (2009). Effects of transdermal testosterone or oral dydrogesterone on hypoactive sexual desire disorder in transsexual women: results of a pilot study. European Journal of Endocrinology, 161(2), 363–368. [DOI:10.1530/eje-09-0265] [Table]
Kuhl, H. (1986). Hormonsubstitution durch Injektionspräparate und Hautimplantate. [Hormone substitution by injectable preparations and skin implants.] Der Gynäkologe, 19(4), 241–247. [Google Scholar] [PubMed] [PDF] [Translation]
Kuhl, H. (1990). Pharmacokinetics of oestrogens and progestogens. Maturitas, 12(3), 171–197. [DOI:10.1016/0378-5122(90)90003-O]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
LaBudde, J. A., Craig, W. Y., & Spratt, D. I. (2020). Initial Evaluation of Safety and Efficacy of Administration of Estradiol (E2) by Subcutaneous (SC) Injections to Male-to-Female (MTF) Transgender Patients. Fertility and Sterility, 114(3), e91–e91. [DOI:10.1016/j.fertnstert.2020.08.277]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., … & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/S0140-6736(21)00100-8]
Larsen, S. W., Rinvar, E., Svendsen, O., Lykkesfeldt, J., Friis, G. J., & Larsen, C. (2001). Determination of the disappearance rate of iodine-125 labelled oils from the injection site after intramuscular and subcutaneous administration to pigs. International Journal of Pharmaceutics, 230(1–2), 67–75. [DOI:10.1016/S0378-5173(01)00860-2]
Larsen, S. W., & Larsen, C. (2009). Critical factors influencing the in vivo performance of long-acting lipophilic solutions—impact on in vitro release method design. The AAPS Journal, 11(4), 762–770. [DOI:10.1208/s12248-009-9153-9]
Larsen, C., Larsen, S. W., Jensen, H., Yaghmur, A., & Østergaard, J. (2009). Role of in vitro release models in formulation development and quality control of parenteral depots. Expert Opinion on Drug Delivery, 6(12), 1283–1295. [DOI:10.1517/17425240903307431]
Larsen, S. W., Thing, M. A., & Larsen, C. (2012). Oily (lipophilic) solutions and suspensions. In Wright, J. C., & Burgess, D. J. (Eds.). Long Acting Injections and Implants (Advances in Delivery Science and Technology) (pp. 113–135). Boston: Springer. [DOI:10.1007/978-1-4614-0554-2_7]
Le, A., Huang, K. J., & Cirrincione, L. R. (2022). Regulation of drug-metabolizing enzymes by sex-related hormones: clinical implications for transgender medicine. Trends in Pharmacological Sciences, 43(7), 582–592. [DOI:10.1016/j.tips.2022.03.006]
Leinonen, P., Hammond, G. L., Lukkarinen, O., & Vihko, R. (1979). Serum sex hormone binding globulin and testosterone binding after estradiol administration, castration, and their combination in men with prostatic carcinoma. Investigative Urology, 17(1), 24–27. [Google Scholar] [PubMed]
Leinonen, P. (1980). Estrone and estradiol concentrations in the testis and spermatic and peripheral venous blood of elderly men: the influence of estrogen treatment. Journal of Steroid Biochemistry, 13(7), 737–742. [DOI:10.1016/0022-4731(80)90225-3]
Leyendecker, G., Geppert, G., Nocke, W., & Ufer, J. (1975). Untersuchungen zur Pharmakokinetik von Östradiol-17β, Östradiol-Benzoat, Östradiol-Valerianat und Östradiol-Undezylat bei der Frau: Der Verlauf der Konzentrationen von Östradiol-17β, Östron, LH und FSH im Serum. [Estradiol-17β, Estrone, LH and FSH in Serum After Administration of Estradiol-17β, Estradiol-Benzoate, Estradiol-Valeriate and Estradiol-Undecylate in the Female.] Geburtshilfe und Frauenheilkunde, 35(5), 370–374. [Google Scholar] [PubMed] [PDF] [Translation]
Leyendecker, G., Wildt, L., Gips, H., Nocke, W., & Plotz, E. J. (1976). Experimental studies on the positive feedback effect of progesterone, 17α-hydroxyprogesterone and 20α-dihydroprogesterone on the pituitary release of LH and FSH in the human female. Archiv für Gynäkologie, 221(1), 29–45. [DOI:10.1007/BF00667679]
Lichten, E. M., Lichten, J. B., Whitty, A., & Pieper, D. (1996). The confirmation of a biochemical marker for women’s hormonal migraine: The depo‐estradiol challenge test. Headache: The Journal of Head and Face Pain, 36(6), 367–371. [DOI:10.1046/j.1526-4610.1996.3606367.x]
Linet, T. (2023). Prise en charge endocrinologique d’une personne trans. [Endocrinological care of a trans person.] In Faucher, P., Hassoun, D., & Linet, T. (Eds.). Santé sexuelle et reproductive des personnes LGBT [Sexual and Reproductive Health of LGBT People] (pp. 109–124). Issy-les-Moulineaux, France: Elsevier Masson. [Google Books] [URL] [WorldCat] [Excerpt]
Lixsoft. (2008). Mathematical Expressions of the Pharmacokinetic and Pharmacodynamic Models Implemented in the Monolix Software. [PDF]
Martinez, G. (1995). Estradiol and Progesterone Serum Levels in Women Using the Once-a-month Injectable Contraceptive Perlutal. / Benagiano, G., Bianchi, P., von Kesserü, E., Castañeda, A., Correa, J. E., & Martínez, G. (1995). Session 19 what is new about injectable contraceptives? Advances in Contraception, 11(1), 39–42. [DOI:10.1007/BF02436100]
Martins, R. S., Antunes, N. J., Comerlatti, G., Caraccio, G., Moreno, R. A., Frecentese, F., Caliendo, G., & De Nucci, G. (2019). Quantification of estradiol cypionate in plasma by liquid chromatography coupled with tandem mass spectrometry: Application in a pharmacokinetic study in healthy female volunteers. Journal of Pharmaceutical and Biomedical Analysis, 170, 273–278. [DOI:10.1016/j.jpba.2019.03.053]
Messinis, I. E., & Templeton, A. (1987). Effect of high dose exogenous oestrogen on midcycle luteinizing hormone surge in human spontaneous cycles. Clinical Endocrinology, 27(4), 453–459. [DOI:10.1111/j.1365-2265.1987.tb01173.x]
Messinis, I. E., & Templeton, A. A. (1987). Disparate effects of endogenous and exogenous oestradiol on luteal phase function in women. Reproduction, 79(2), 549–554. [DOI:10.1530/jrf.0.0790549]
Milano, C., & Harper, J. (2025). Comments on Injectable Estradiol Use in Transgender and Gender-Diverse Individuals in the US. The Journal of Clinical Endocrinology & Metabolism, dgaf134. [DOI:10.1210/clinem/dgaf134]
Minto, C. F., Howe, C., Wishart, S., Conway, A. J., & Handelsman, D. J. (1997). Pharmacokinetics and pharmacodynamics of nandrolone esters in oil vehicle: effects of ester, injection site and injection volume. Journal of Pharmacology and Experimental Therapeutics, 281(1), 93–102. [URL]
Misakian, A. L., Kelley, C. E., Sullivan, E. A., Chang, J. J., Singh, G., Kokosa, S., Avila, J., Cooper, H., Liang, J. W., Botzheim, B., Quint, M., Jeevananthan, A., Chi, E., Harmer, M., Hiatt, L., Kowalewski, M., Steinberg, B., Tausinga, T., Tanner, H., Ho, T. F., … Ariel, D. (2025). Injectable Estradiol Use in Transgender and Gender-Diverse Individuals throughout the United States. The Journal of Clinical Endocrinology & Metabolism, dgaf015. [DOI:10.1210/clinem/dgaf015]
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body composition and bone mineral density in male-to-female transsexuals during cross-sex hormone therapy using gonadotrophin-releasing hormone agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074] [Table]
Newton, J. R., d’Arcangues, C., & Hall, P. E. (1994). A review of ‘once-a-month’ combined injectable contraceptives. Journal of Obstetrics and Gynaecology, 14(Suppl 1), S1–S34. [DOI:10.3109/01443619409027641]
Nelson, M. D., Szczepaniak, L. S., Wei, J., Szczepaniak, E., Sánchez, F. J., Vilain, E., Stern, J. H., Bergman, R. N., Bairey Merz, C. N., & Clegg, D. J. (2016). Transwomen and the Metabolic Syndrome: Is Orchiectomy Protective? Transgender Health, 1(1), 165–171. [DOI:10.1089/trgh.2016.0016] [Table]
Nieschlag, E., & Behre, H. M. (2010). Testosterone therapy. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Andrology (pp. 437–455). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-540-78355-8_21] [PDF]
Norlén, B. J., Fritjofsson, Å., Grönquist, L., Gunnarsson, P. O., Johansson, S. Å., & Plym-Forshell, G. (1987). Plasma concentrations of estradiol and testosterone in single-drug polyestradiol phosphate therapy for prostatic cancer. European Urology, 13, 193–197. [DOI:10.1159/000472772]
Olson-Kennedy, J., Rosenthal, S. M., Hastings, J., & Wesp, L. (2016). Health considerations for gender non-conforming children and transgender adolescents. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 186–199). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Oriowo, M. A., Landgren, B. M., Stenström, B., & Diczfalusy, E. (1980). A comparison of the pharmacokinetic properties of three estradiol esters. Contraception, 21(4), 415–424. [DOI:10.1016/S0010-7824(80)80018-7]
Parkes, A. S. (1937). Relative duration of action of various esters of oestrone, oestradiol and oestriol. Biochemical Journal, 31(4), 579–585. [DOI:10.1042/bj0310579]
Patel, R., Korenman, S., Weimer, A., & Grock, S. (2024). A Call for Updates to Hormone Therapy Guidelines for Gender-Diverse Adults Assigned Male at Birth. Cureus, 16(6), e62262. [DOI:10.7759/cureus.62262] [PDF]
Patel, K. T., & Tangpricha, V. (2024). Parenteral Estradiol for Transgender Women: Time to adjust the dose. Endocrine Practice, 30(9), 893–894. [DOI:10.1016/j.eprac.2024.07.005]
Presl, J., Horejsi, J., Štroufová, A., & Herzmann, J. (1976). Sexual maturation in girls and the development of estrogen-induced gonadotropic hormone release. Annales de Biologie Animale, Biochimie, Biophysique, 16(3), 377–383. [DOI:10.1051/rnd:19760314]
Rahimy, M. H., & Ryan, K. K. (1999). Lunelle monthly contraceptive injection (medroxyprogesterone acetate and estradiol cypionate injectable suspension): assessment of return of ovulation after three monthly injections in surgically sterile women. Contraception, 60(4), 189–200. [DOI:10.1016/s0010-7824(99)00081-5]
Rahimy, M. H., Ryan, K. K., & Hopkins, N. K. (1999). Lunelle™ monthly contraceptive injection (medroxyprogesterone acetate and estradiol cypionate injectable suspension): steady-state pharmacokinetics of MPA and E2 in surgically sterile women. Contraception, 60(4), 209–214. [DOI:10.1016/S0010-7824(99)00086-4]
Rauramo, L., Punnonen, R., Kaihola, H. L., & Grönroos, M. (1980). Serum oestrone, oestradiol and oestriol concentrations in castrated women during intramuscular oestradiolvalerate and oestradiolbenzoate-oestradiolphenylpropionate therapy. Maturitas, 2(1), 53–58. [DOI:10.1016/0378-5122(80)90060-2]
Rauramo, L., Punnonen, R., & Grönroos, M. (1981). Serum concentrations of oestrone, oestradiol and oestriol during various oestrogen treatments. Maturitas, 3(2), 183–186. [DOI:10.1016/0378-5122(81)90010-4]
Recio, R., Garza-Flores, J., Schiavon, R., Reyes, A., Diaz-Sanchez, V., Valles, V., Luz de la Cruz, D., Oropeza, G., & Perez-Palacios, G. (1986). Pharmacodynamic assessment of dihydroxyprogesterone acetophenide plus estradiol enanthate as a monthly injectable contraceptive. Contraception, 33(6), 579–589. [DOI:10.1016/0010-7824(86)90046-6]
Reimann, I. W., Britzelmeier, C., Haber, P., Wollmann, H., Antonin, K. H., & Bieck, P. R. (1987). Influence of Oestradiol on Alpha2-Adrenoceptor Binding Sites on Intact Platelets of Young Male Volunteers. European Journal of Clinical Pharmacology, 33(2), 147–150. [DOI:10.1007/BF00544558]
Rosenfield, R. L., Fang, V. S., Dupon, C., Kim, M. H., & Refetoff, S. (1973). The effects of low doses of depot estradiol and testosterone in teenagers with ovarian failure and Turner’s syndrome. The Journal of Clinical Endocrinology and Metabolism, 37(4), 574–580. [DOI:10.1210/jcem-37-4-574]
Rosenfield, R. L., & Fang, V. S. (1974). The effects of prolonged physiologic estradiol therapy on the maturation of hypogonadal teen-agers. The Journal of Pediatrics, 85(6), 830–837. [DOI:10.1016/S0022-3476(74)80355-0]
Rothman, M. S., Ariel, D., Kelley, C., Hamnvik, O. R., Abramowitz, J., Irwig, M. S., Soe, K., Davidge-Pitts, C., Misakian, A. L., Safer, J. D., & Iwamoto, S. J. (2024). The Use of Injectable Estradiol in Transgender and Gender Diverse Adults: A Scoping Review of Dose and Serum Estradiol Levels. Endocrine Practice, 30(9), 870–878. [DOI:10.1016/j.eprac.2024.05.008]
Rothman, M. S., Hamnvik, O. P. R., Davidge-Pitts, C., Safer, J. D., Ariel, D., Tangpricha, V., Abramowitz, J., Soe, K., Sarvaideo, J., Kelley, C., Irwig, M. S., & Iwamoto, S. J. (2024). Revisiting Injectable Estrogen Dosing Recommendations for Gender-Affirming Hormone Therapy. Transgender Health, 9(6), 463–465. [DOI:10.1089/trgh.2023.0209]
Sang, G. W., Ge, J. L., Liu, X. H., Shao, Q. X., Zhao, X. J., & Mao, S. M. (1987). 不同剂量庚炔诺酮单独或配伍戊酸雌二醇后的药代动力学及药效学. [Pharmacokinetics and pharmacodynamics of different doses of norethisterone enanthate alone and in combination with estradiol valerate.] 中国 临 床药理 学杂志 / Chinese Journal of Clinical Pharmacology, 3(1), 7–18. [Google Scholar] [CNKI] [DOI:10.13699/j.cnki.1001-6821.1987.01.002] [PDF]
Sang, G. W. (1994). Pharmacodynamic effects of once-a-month combined injectable contraceptives. Contraception, 49(4), 361–385. [DOI:10.1016/0010-7824(94)90033-7]
Schiavon, R., Benavides, S., Oropeza, G., Garza-Flores, J., Recio, R., Díaz-Sanchez, V., & Pérez-Palacios, G. (1988). Serum estrogens and ovulation return in chronic users of a once-a-month injectable contraceptive. Contraception, 37(6), 591–598. [DOI:10.1016/0010-7824(88)90005-4]
Schug, B. S., Donath, F., & Blume, H. H. (2012). Bioavailability and pharmacodynamics of two 10-mg estradiol valerate depot formulations following IM single dose administration in healthy postmenopausal volunteers. International Journal of Clinical Pharmacology and Therapeutics, 50(2), 100–117. [Google Scholar] [DOI:10.5414/cp201589] [PDF]
Schulte-Beerbühl, M., & Nieschlag, E. (1980). Comparison of testosterone, dihydrotestosterone, luteinizing hormone, and follicle-stimulating hormone in serum after injection of testosterone enanthate or testosterone cypionate. Fertility and Sterility, 33(2), 201–203. [DOI:10.1016/S0015-0282(16)44543-7]
Schultz, K., Møllgaard, B., Fisher, A. N., Illum, L., & Larsen, C. (1998). Intramuscular rate of disappearance of oily vehicles in rabbits investigated by gamma-scintigraphy. International Journal of Pharmaceutics, 169(1), 121–126. [DOI:10.1016/S0378-5173(98)00121-5]
Schweikert, H. U., Weissbach, L., Leyendecker, G., Schwinger, E., Wartenberg, H., & Krück, F. (1982). Clinical, endocrinological, and cytological characterization of two 46, XX males. The Journal of Clinical Endocrinology & Metabolism, 54(4), 745–752. [DOI:10.1210/jcem-54-4-745]
Seibert, B., & Günzel, P. (1994). Animal toxicity studies performed for risk assessment of the once-a-month injectable contraceptive Mesigyna®. Contraception, 49(4), 303–333. [DOI:10.1016/0010-7824(94)90030-2]
Shah, J. C. (2007). Controlled Release – Small Molecules. In Stella, V. J., Borchardt, R. T., Hageman, M. J., Oliyai, R., Maag, H., & Tilley, J. W. (Eds.). Prodrugs: Challenges and Rewards, Part 1 (Biotechnology: Pharmaceutical Aspects, Volume V) (pp. 357–377). New York: Springer. [DOI:10.1007/978-0-387-49785-3_9]
Sharula, Chekir, C., Emi, Y., Arai, F., Kikuchi, Y., Sasaki, A., Matsuda, M., Shimizu, K., Tabuchi, K., Kamada, Y., Hiramatsu, Y., & Nakatsuka, M. (2012). Altered arterial stiffness in male‐to‐female transsexuals undergoing hormonal treatment. Journal of Obstetrics and Gynaecology Research, 38(6), 932–940. [DOI:10.1111/j.1447-0756.2011.01815.x] [Data]
Shaw, R. W., Butt, W. R., London, D. R., & Marshall, J. C. (1975). The oestrogen provocation test: a method of assessing the hypothalamic‐pituitary axis in patients with amenorrhoea. Clinical Endocrinology, 4(3), 267–276. [DOI:10.1111/j.1365-2265.1975.tb01534.x]
Shaw, R. W., Butt, W. R., & London, D. R. (1975). The effect of oestrogen pretreatment on subsequent response to luteinizing hormone releasing hormone in normal women. Clinical Endocrinology, 4(3), 297–304. [DOI:10.1111/j.1365-2265.1975.tb01537.x]
Shaw, R. W. (1978). Neuroendocrinology of the menstrual cycle in humans. Clinics in Endocrinology and Metabolism, 7(3), 531–559. [DOI:10.1016/S0300-595X(78)80008-5]
Shahiwala, A., Mehta, T. A., & Momin, M. M. (2018). Parenteral drug delivery systems. In Misra, A., & Shahiwala, A. (Eds.). In-Vitro and In-Vivo Tools in Drug Delivery Research for Optimum Clinical Outcomes (pp. 283–318). Boca Raton: CRC Press. [DOI:10.1201/b22448-9] [Google Books]
Sherwin, B. B., & Gelfand, M. M. (1987). Individual differences in mood with menopausal replacement therapy: Possible role of sex hormone-binding globulin. Journal of Psychosomatic Obstetrics & Gynecology, 6(2), 121–131. [DOI:10.3109/01674828709016773]
Sherwin, B. B., Gelfand, M. M., Schucher, R., & Gabor, J. (1987). Postmenopausal estrogen and androgen replacement and lipoprotein lipid concentrations. American Journal of Obstetrics and Gynecology, 156(2), 414–419. [DOI:10.1016/0002-9378(87)90295-X]
Sherwin, B. B. (1988). Affective changes with estrogen and androgen replacement therapy in surgically menopausal women. Journal of Affective Disorders, 14(2), 177–187. [DOI:10.1016/0165-0327(88)90061-4]
Sierra-Ramírez, J. A., Lara-Ricalde, R., Lujan, M., Velázquez-Ramírez, N., Godínez-Victoria, M., Hernádez-Munguía, I. A., Padilla, A., & Garza-Flores, J. (2011). Comparative pharmacokinetics and pharmacodynamics after subcutaneous and intramuscular administration of medroxyprogesterone acetate (25 mg) and estradiol cypionate (5 mg). Contraception, 84(6), 565–570. [DOI:10.1016/j.contraception.2011.03.014]
Sinkula, A. A. (1978). Methods to Achieve Sustained Drug Delivery. The Chemical Approach. In Robinson, J. R. (Ed.). Sustained and Controlled Release Drug Delivery Systems (pp. 411–555). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books] [PDF]
Slack, D. J., Di Via Ioschpe, A., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., Karim, S., & Safer, J. D. (2025). Examining the Influence of the Route of Administration and Dose of Estradiol on Serum Estradiol and Testosterone Levels in Feminizing Gender-Affirming Hormone Therapy. Endocrine Practice, 31(1), 19–27. [DOI:10.1016/j.eprac.2024.10.002]
Somerville, B. W. (1971). The Role of Oestradiol in Menstrual Migraine. In Somerville, B. W. The Influence of Progesterone and Oestradiol on Migraine (Doctoral dissertation, University of New South Wales) (pp. 93–104). [Google Scholar] [URL] [WorldCat] [PDF]
Somerville, B. W. (1972). The Role of Estradiol Withdrawal in the Etiology of Menstrual Migraine. Neurology, 22(4), 355–365. [DOI:10.1212/WNL.22.4.355]
Somerville, B. W. (1972). The influence of progesterone and estradiol upon migraine. Headache: The Journal of Head and Face Pain, 12(3), 93–102. [DOI:10.1111/j.1526-4610.1972.hed1203093.x]
Somerville, B. W. (1972). The Influence of Hormones Upon Migraine in Women. Medical Journal of Australia, 2(S2), 6–11. [DOI:10.5694/j.1326-5377.1972.tb93039.x]
Somerville, B. W. (1975). Estrogen‐withdrawal migraine: I. Duration of exposure required and attempted prophylaxis by premenstrual estrogen administration. Neurology, 25(3), 239–244. [DOI:10.1212/wnl.25.3.239]
Spack, N. P. (2013). Management of transgenderism. JAMA, 309(5), 478–484. [DOI:10.1001/jama.2012.165234]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., & Pousette, A. (1988). Single drug polyestradiol phosphate therapy in prostatic cancer. American Journal of Clinical Oncology, 11(Suppl 2), S101–S103. [DOI:10.1097/00000421-198801102-00024] [PDF]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & von Schoultz, B. (1989). Single‐drug parenteral estrogen treatment in prostatic cancer: A study of two maintenance‐dose regimens. The Prostate, 14(2), 183–188. [DOI:10.1002/pros.2990140211]
Stege, R., Gunnarsson, P. O., Johansson, C. J., Olsson, P., Pousette, Å., & Carlström, K. (1996). Pharmacokinetics and testosterone suppression of a single dose of polyestradiol phosphate (Estradurin®) in prostatic cancer patients. The Prostate, 28(5), 307–310. [DOI:10.1002/(SICI)1097-0045(199605)28:5<307::AID-PROS6>3.0.CO;2-8]
Sumioki, H. (1987). ヒト排卵機構とGonadotropin分泌調節における “β-Endorphinおよびβ-Lipotropin” の役割に関する研究. [The Role of “β-Endorphin & β-Lipotropin” on the Gonadotropin Regulation in the Mechanism of Human Ovulation.] Folia Endocrinologica Japonica, 63(10), 1289–1307. [DOI:10.1507/endocrine1927.63.10_1289]
Svendsen, O., & Aaes‐Jørgensen, T. (1979). Studies on the fate of vegetable oil after intramuscular injection into experimental animals. Acta Pharmacologica et Toxicologica, 45(5), 352–378. [DOI:10.1111/j.1600-0773.1979.tb02404.x]
Tassinari, R., & Maranghi, F. (2021). Rodent Model of Gender-Affirming Hormone Therapies as Specific Tool for Identifying Susceptibility and Vulnerability of Transgender People and Future Applications for Risk Assessment. International Journal of Environmental Research and Public Health, 18(23), 12640. [DOI:10.3390/ijerph182312640]
Thurman, A., Kimble, T., Hall, P., Schwartz, J. L., & Archer, D. F. (2013). Medroxyprogesterone acetate and estradiol cypionate injectable suspension (Cyclofem) monthly contraceptive injection: steady-state pharmacokinetics. Contraception, 87(6), 738–743. [DOI:10.1016/j.contraception.2012.11.010]
Toffoli Ribeiro, C., Gois, Í., da Rosa Borges, M., Ferreira, L. G. A., Brandão Vasco, M., Ferreira, J. G., Maia, T. C., & Dias-da-Silva, M. R. (2024). Assessment of parenteral estradiol and dihydroxyprogesterone use among other feminizing regimens for transgender women: insights on satisfaction with breast development from community-based healthcare services. Annals of Medicine, 56(1), 2406458. [DOI:10.1080/07853890.2024.2406458]
Toppozada, M. K. (1994). Existing once-a-month combined injectable contraceptives. Contraception, 49(4), 293–301. [DOI:10.1016/0010-7824(94)90029-9]
Toutain, P. L., & Bousquet-Mélou, A. (2004). Plasma terminal half-life. Journal of Veterinary Pharmacology and Therapeutics, 27(6), 427–439. [DOI:10.1111/j.1365-2885.2004.00600.x]
Trans Care BC. (2021). Gender-affirming Care for Trans, Two-Spirit, and Gender Diverse Patients in BC: A Primary Care Toolkit. Vancouver: Provincial Health Services Authority/Trans Care BC. [URL] [PDF]
Travaglini, P., Ambrosi, B., Beck-Peccoz, P., Elli, R., Rondena, M., Bara, R., & Weber, G. (1978). Hypothalamic-pituitary-ovarian function in hyperprolactinemic women. Journal of Endocrinological Investigation, 1(1), 39–45. [DOI:10.1007/BF03346769]
Travaglini, P., Elli, R., Ambrosi, B., Ballabio, M., Moriondo, P., & Faglia, G. (1979). Serum LH increase after estradiol and progesterone administration in hyperprolactinemic women. Journal of Endocrinological Investigation, 2(4), 407–411. [DOI:10.1007/BF03349341]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine position statement “assessment and hormonal management in adolescent and adult trans people, with attention for sexual function and satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
Ulrich, U., Pfeifer, T., & Lauritzen, C. (1994). Rapid Increase in Lumbar Spine Bone Density in Osteopenic Women by High-Dose Intramuscular Estrogen-Progestogen Injections. Hormone and Metabolic Research, 26(9), 428–431. [DOI:10.1055/s-2007-1001723]
Välimäki, M., Pelkonen, R., Salaspuro, M., Härkönen, M., Hirvonen, E., & Ylikahri, R. (1984). Sex hormones in amenorrheic women with alcoholic liver disease. The Journal of Clinical Endocrinology and Metabolism, 59(1), 133–138. [DOI:10.1210/jcem-59-1-133]
Valle Alvarez, D. C. (2011). Efecto de una Dosis de 50 mg de Enantato de Noretisterona y 5 mg de Valerato de Estradiol en los Niveles de Testosterona Total en Hombres Mexicanos Sanos. [Effect of a Dose of 50 mg of Norethisterone Enanthate and 5 mg of Estradiol Valerate on Total Testosterone Levels in Healthy Mexican Men.] (Masters thesis, National Polytechnic Institute of Mexico.) [Google Scholar] [URL] [PDF] [Translation]
Varangot, J., & Cedard, L. (1957). Modifications des Œstrogènes Sanguins Après Administration Intramusculaire de Benzoate d’Œstradiol. [Changes in Serum Estrogens After Intramuscular Administration of Estradiol Benzoate.] Comptes Rendus des Séances de la Société de Biologie et de ses Filiales, 151(10), 1707–1712. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF] [Translation]
Vermeulen, A. (1977). Transport and Distribution of Androgens at Different Ages. In Martini, L., & Motta, M. (Eds.). Androgens and Antiandrogens (pp. 53–65). New York: Raven Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [Excerpt]
Vhora, I., Bardoliwala, D., Ranamalla, S. R., & Javia, A. (2019). Parenteral controlled and prolonged drug delivery systems: therapeutic needs and formulation strategies. In Misra A., & Shahiwala, A. (Eds.). Novel Drug Delivery Technologies (pp. 183–260). Singapore: Springer. [DOI:10.1007/978-981-13-3642-3_7]
Vizziello, G., D’Amato, G., Trentadue, R., & Fanizza, G. (1993). Studio dinamico del blocco ipofisario indotto dalla triptorelina, mediante test all’estradiolo benzoato. [Estradiol benzoate test in the study of pituitary block induced by triptorelin.] Minerva Ginecologica, 45(4), 185–189. [Google Scholar] [PubMed] [PDF] [Translation]
Wagner, J. G. (1993). Pharmacokinetics for the Pharmaceutical Scientist. Lancaster/Basel: Technomic. [DOI:10.1201/9780203743652] [Google Books]
Weiss, G., Nachtigall, L. E., & Ganguly, M. (1976). Induction of an LH surge with estradiol benzoate. A clinical test of pituitary-hypothalamic axis competence. Obstetrics and Gynecology, 47(4), 415–418. [URL]
White, M. C., Rosenstock, J., Anapliotou, M., Mashiter, K., & Joplin, G. F. (1981). Heterogeneity of prolactin responses to oestradiol benzoate in women with prolactinomas. The Lancet, 317(8235), 1394–1396. [DOI:10.1016/S0140-6736(81)92571-X]
Wiemeyer, J. C., Fernandez, M., Moguilevsky, J. A., & Sagasta, C. L. (1986). Pharmacokinetic Studies of Estradiol Enantate in Menopausic Women. Arzneimittel-Forschung, 36(11), 1674–1677. [Google Scholar] [PubMed] [PDF]
Wiemeyer, J. C., Fernandez, M., Sagasta, C. L., & Moguilevsky, J. A. (1987). Estudos farmacocinéticos do enantato de estradiol em mulheres na menopausa. [Pharmacokinetic studies of estradiol enantate in menopausic women.] Jornal Brasileiro de Ginecologia, 97(9), 497–501. [Google Scholar] [LILACS] [PDF]
Yan, J., Pan, J., Chang, Y., & Kang, J. (1987). The effect of monthly injectable contraceptive megestrol acetate compound on pituitary-ovarian function. 上海第二医科大学学报(英文版) / Journal of Shanghai Second Medical University / Medical Bulletin of Shanghai Jiaotong University, 1(2), 7–12. [Google Scholar] [CNKI] [PDF]
Yáñez, J. A., Remsberg, C. M., Sayre, C. L., Forrest, M. L., & Davies, N. M. (2011). Flip-flop pharmacokinetics–delivering a reversal of disposition: challenges and opportunities during drug development. Therapeutic Delivery, 2(5), 643–672. [DOI:10.4155/tde.11.19]
Zhang, Y., Huo, M., Zhou, J., & Xie, S. (2010). PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Computer Methods and Programs in Biomedicine, 99(3), 306–314. [DOI:10.1016/j.cmpb.2010.01.007]
Zhou, X. F., Shao, Q. X., Han, X. J., Weng, L. J., & Sang, G. W. (1998). Pharmacokinetics of medroxyprogesterone acetate after single and multiple injection of Cyclofem® in Chinese women. Contraception, 57(6), 405–411. [DOI:10.1016/S0010-7824(98)00048-1]
\ No newline at end of file
diff --git a/transfemscience.org/articles/injectable-e2-simulator-release/index.html b/transfemscience.org/articles/injectable-e2-simulator-release/index.html
index af5beede..5340492d 100644
--- a/transfemscience.org/articles/injectable-e2-simulator-release/index.html
+++ b/transfemscience.org/articles/injectable-e2-simulator-release/index.html
@@ -1 +1 @@
-An Interactive Web Simulator for Estradiol Levels with Injectable Estradiol Esters - Transfeminine Science
Estradiol is frequently used in injectable form in transfeminine hormone therapy. Injectable estradiol is employed in the form of estradiol esters such as estradiol valerate and estradiol cypionate, which are prodrugs of estradiol that are slowly released from a depot formed at the injection site. These esters are most commonly formulated as oil solutions and are administered via intramuscular or subcutaneous injection. Injectable estradiol is a popular choice among transfeminine people as well as some clinical providers as it has a number of advantages over other estradiol routes and forms. For instance, it allows for easy and inexpensive attainment of higher estradiol levels that can be useful in transfeminine people for achieving better testosterone suppression. This is particularly true in the case of estradiol monotherapy, a therapeutic approach in which an antiandrogen isn’t employed.
Clinically used injectable estradiol preparations were developed many decades ago and are not as commonly used in medicine as estradiol preparations like oral and transdermal estradiol. In fact, injectable estradiol has been discontinued in many countries in favor of non-injectable preparations. In relation to the preceding, research and review material on the pharmacokinetics of these preparations are limited and are scattered throughout the scientific literature. For most of the published concentration–time curves of circulating estradiol with injectable estradiol esters, only a single injection has been administered and the different doses that have been employed have been few. The scarce and obscure information on the pharmacokinetics of these formulations presents challenges for transfeminine people and their clinicians when it comes to understanding the estradiol levels that may result with injectable estradiol preparations. This is particularly true in relation to repeated injections of injectable estradiol formulations at varying doses and injection intervals, which is how these preparations are used in transfeminine hormone therapy. A proper understanding of the estradiol levels with injectable estradiol is important for transfeminine people for avoiding estradiol levels that are too low—which can result in inadequate testosterone suppression and therapeutic efficacy—while also avoiding estradiol levels that are too high—which may produce unnecessary side effects and risks (e.g., Aly, 2020).
To help with overcoming these obstacles, I’ve developed an interactive web app for simulating estradiol levels with injectable estradiol preparations. This simulator can be found at the following page:
Here is a screenshot of the simulator that shows what it looks like and what it can do:
The app simulates estradiol levels with a selection of major injectable estradiol preparations. These preparations include injectable estradiol benzoate (EB) in oil, estradiol valerate (EV) in oil, estradiol cypionate (EC) in oil and as a microcrystallineaqueous suspension, estradiol enanthate (EEn) in oil, estradiol undecylate (EU) in oil, and polyestradiol phosphate (PEP). Options are available in the simulator for specifying injectable estradiol dose (mg), single versus repeated injections, injection interval (days, weeks, or months), units for estradiol concentrations (pg/mL or pmol/L), x-axis maximum value (or time interval to graph) (days), and y-axis max value (or estradiol concentration interval to graph) (pg/mL or pmol/L). One preparation can be simulated at a time or all of the supported injectable estradiol preparations can be graphed together at the same time. When all injectable preparations are simulated at once, the legend can be interacted with to hide or show individual preparations.
The estradiol curves produced by the app are simulations based on available data from published studies with the supported injectable estradiol preparations. The accuracy of the curves is limited by the quality and quantity of these data. In other words, the curves are only estimates, and true estradiol levels with a given preparation may be different than what is shown. It is notable in this regard that estradiol curves with a given injectable estradiol preparation vary considerably between studies, with different levels and curve shapes apparent. There are many potential factors which may contribute to this variability, such as formulation, injection specifics (like injection site, volume, and technique), the type and calibration of blood test used, differing subject characteristics (like age, weight, etc.), and research matters like sampling error. The simulator is not able to take into account these potential variables as data on their influences are scarce and not well-defined. An assumption of the simulator is that estradiol levels and curve shapes scale linearly with dose, which may or may not actually be the case. Lastly, it must be made clear that the estradiol curves correspond to the averages of many people, and individual estradiol levels and curve shapes vary substantially even with the same injectable estradiol preparation. For these varied reasons, the simulator cannot tell a given person what their exact estradiol levels with a given injectable estradiol regimen will be. It can only be used as a guide to roughly estimate where one’s estradiol levels most plausibly could be. In relation to this, estradiol levels, as well as testosterone suppression, should be monitored and verified with blood work to ensure that they are in desired ranges.
A literature review and informal meta-analysis of available estradiol concentration–time data with injectable estradiol preparations was conducted to determine the appropriate estradiol curves for the different estradiol esters included in the simulator. Data were collected from the literature, processed, and modeled using pharmacokinetic models. This work can be found at the following page:
The meta-analysis was not able to derive a reasonable curve for injectable estradiol undecylate due to lack of adequate published data for this ester for modeling. Because of the historical and theoretical importance of estradiol undecylate as an injectable estradiol ester however, it was desirable to nonetheless construct a curve of some form for estradiol undecylate so that it could be included in the simulator. In order to do this, a curve was instead fit to a well-known study for injectable testosterone undecanoate (testosterone undecylate; TU) (Behre et al., 1999) and area-under-the-curve estradiol levels were scaled to be appropriate for those with a given dose of estradiol undecylate based on data with other injectable estradiol preparations. This approach is reasonable as estradiol undecylate and testosterone undecanoate have fairly similar fat solubilities (Table) due to being very similar in chemical structure and as fat solubility is the key property dictating the release rates and curve shapes of these preparations. Accordingly, the resulting curve for estradiol undecylate roughly accords with the reported clinical durations of this ester (Table). In any case, it should be cautioned that the estradiol undecylate curve is not based on real data for this estradiol ester and is only hypothetical or “just for fun”.
The simulator and the curves for the different injectable preparations included may be updated in the future with improvements and new features. Extension of the simulator to other hormonal preparations like injectable testosterone, sublingual estradiol, and estradiol pellets would be fairly straightforward and could be done in the future. However, it would require additional meta-analysis and much further work.
A special thank you to Violet and Lila for their indispensable input and guidance on modeling topics during the work on this project. An additional thanks to Violet for deriving a special three-compartment pharmacokinetic model that was used in the simulator. Please also check out Violet’s own work-in-progress TransKit and Tilia projects—pharmacokinetic tools tailored for transgender hormone therapy.
Updates
Update 1: New Advanced Simulator
Since the release of the injectable estradiol simulator, a more advanced version of the simulator with additional options and functionality has been developed. This advanced simulator was created by Luna via modification of Aly’s original simulator code. It uses the same data (i.e. injectable estradiol curves) as the original simulator, but has the following new features: (1) simulate multiple traces at once; (2) stop after X doses (dose limit); (3) start trace at steady state; and (4) show cis woman menstrual cycle (median, 5th percentile, and 95th percentile estradiol levels; data from Abbott (2009)). The advanced injectable estradiol simulator was released on October 5, 2022 and can be found at the following page:
Here is a screenshot of the advanced simulator and its capabilities:
Update 2: Literature Mentions
Since Transfeminine Science’s injectable estradiol simulator was released in mid-2021, it has been mentioned and cited in the scientific literature in a number of publications (see Aly, 2021).
Behre, H. M., Abshagen, K., Oettel, M., Hubler, D., & Nieschlag, E. (1999). Intramuscular injection of testosterone undecanoate for the treatment of male hypogonadism: phase I studies. European Journal of Endocrinology, 140(5), 414–419. [DOI:10.1530/eje.0.1400414]
\ No newline at end of file
+An Interactive Web Simulator for Estradiol Levels with Injectable Estradiol Esters - Transfeminine Science
Estradiol is frequently used in injectable form in transfeminine hormone therapy. Injectable estradiol is employed in the form of estradiol esters such as estradiol valerate and estradiol cypionate, which are prodrugs of estradiol that are slowly released from a depot formed at the injection site. These esters are most commonly formulated as oil solutions and are administered via intramuscular or subcutaneous injection. Injectable estradiol is a popular choice among transfeminine people as well as some clinical providers as it has a number of advantages over other estradiol routes and forms. For instance, it allows for easy and inexpensive attainment of higher estradiol levels that can be useful in transfeminine people for achieving better testosterone suppression. This is particularly true in the case of estradiol monotherapy, a therapeutic approach in which an antiandrogen isn’t employed.
Clinically used injectable estradiol preparations were developed many decades ago and are not as commonly used in medicine as estradiol preparations like oral and transdermal estradiol. In fact, injectable estradiol has been discontinued in many countries in favor of non-injectable preparations. In relation to the preceding, research and review material on the pharmacokinetics of these preparations are limited and are scattered throughout the scientific literature. For most of the published concentration–time curves of circulating estradiol with injectable estradiol esters, only a single injection has been administered and the different doses that have been employed have been few. The scarce and obscure information on the pharmacokinetics of these formulations presents challenges for transfeminine people and their clinicians when it comes to understanding the estradiol levels that may result with injectable estradiol preparations. This is particularly true in relation to repeated injections of injectable estradiol formulations at varying doses and injection intervals, which is how these preparations are used in transfeminine hormone therapy. A proper understanding of the estradiol levels with injectable estradiol is important for transfeminine people for avoiding estradiol levels that are too low—which can result in inadequate testosterone suppression and therapeutic efficacy—while also avoiding estradiol levels that are too high—which may produce unnecessary side effects and risks (e.g., Aly, 2020).
To help with overcoming these obstacles, I’ve developed an interactive web app for simulating estradiol levels with injectable estradiol preparations. This simulator can be found at the following page:
Here is a screenshot of the simulator that shows what it looks like and what it can do:
The app simulates estradiol levels with a selection of major injectable estradiol preparations. These preparations include injectable estradiol benzoate (EB) in oil, estradiol valerate (EV) in oil, estradiol cypionate (EC) in oil and as a microcrystallineaqueous suspension, estradiol enanthate (EEn) in oil, estradiol undecylate (EU) in oil, and polyestradiol phosphate (PEP). Options are available in the simulator for specifying injectable estradiol dose (mg), single versus repeated injections, injection interval (days, weeks, or months), units for estradiol concentrations (pg/mL or pmol/L), x-axis maximum value (or time interval to graph) (days), and y-axis max value (or estradiol concentration interval to graph) (pg/mL or pmol/L). One preparation can be simulated at a time or all of the supported injectable estradiol preparations can be graphed together at the same time. When all injectable preparations are simulated at once, the legend can be interacted with to hide or show individual preparations.
The estradiol curves produced by the app are simulations based on available data from published studies with the supported injectable estradiol preparations. The accuracy of the curves is limited by the quality and quantity of these data. In other words, the curves are only estimates, and true estradiol levels with a given preparation may be different than what is shown. It is notable in this regard that estradiol curves with a given injectable estradiol preparation vary considerably between studies, with different levels and curve shapes apparent. There are many potential factors which may contribute to this variability, such as formulation, injection specifics (like injection site, volume, and technique), the type and calibration of blood test used, differing subject characteristics (like age, weight, etc.), and research matters like sampling error. The simulator is not able to take into account these potential variables as data on their influences are scarce and not well-defined. An assumption of the simulator is that estradiol levels and curve shapes scale linearly with dose, which may or may not actually be the case. Lastly, it must be made clear that the estradiol curves correspond to the averages of many people, and individual estradiol levels and curve shapes vary substantially even with the same injectable estradiol preparation. For these varied reasons, the simulator cannot tell a given person what their exact estradiol levels with a given injectable estradiol regimen will be. It can only be used as a guide to roughly estimate where one’s estradiol levels most plausibly could be. In relation to this, estradiol levels, as well as testosterone suppression, should be monitored and verified with blood work to ensure that they are in desired ranges.
A literature review and informal meta-analysis of available estradiol concentration–time data with injectable estradiol preparations was conducted to determine the appropriate estradiol curves for the different estradiol esters included in the simulator. Data were collected from the literature, processed, and modeled using pharmacokinetic models. This work can be found at the following page:
The meta-analysis was not able to derive a reasonable curve for injectable estradiol undecylate due to lack of adequate published data for this ester for modeling. Because of the historical and theoretical importance of estradiol undecylate as an injectable estradiol ester however, it was desirable to nonetheless construct a curve of some form for estradiol undecylate so that it could be included in the simulator. In order to do this, a curve was instead fit to a well-known study for injectable testosterone undecanoate (testosterone undecylate; TU) (Behre et al., 1999) and area-under-the-curve estradiol levels were scaled to be appropriate for those with a given dose of estradiol undecylate based on data with other injectable estradiol preparations. This approach is reasonable as estradiol undecylate and testosterone undecanoate have fairly similar fat solubilities (Table) due to being very similar in chemical structure and as fat solubility is the key property dictating the release rates and curve shapes of these preparations. Accordingly, the resulting curve for estradiol undecylate roughly accords with the reported clinical durations of this ester (Table). In any case, it should be cautioned that the estradiol undecylate curve is not based on real data for this estradiol ester and is only hypothetical or “just for fun”.
The simulator and the curves for the different injectable preparations included may be updated in the future with improvements and new features. Extension of the simulator to other hormonal preparations like injectable testosterone, sublingual estradiol, and estradiol pellets would be fairly straightforward and could be done in the future. However, it would require additional meta-analysis and much further work.
A special thank you to Violet and Lila for their indispensable input and guidance on modeling topics during the work on this project. An additional thanks to Violet for deriving a special three-compartment pharmacokinetic model that was used in the simulator. Please also check out Violet’s own work-in-progress TransKit and Tilia projects—pharmacokinetic tools tailored for transgender hormone therapy.
Updates
Update 1: New Advanced Simulator
Since the release of the injectable estradiol simulator, a more advanced version of the simulator with additional options and functionality has been developed. This advanced simulator was created by Luna via modification of Aly’s original simulator code. It uses the same data (i.e. injectable estradiol curves) as the original simulator, but has the following new features: (1) simulate multiple traces at once; (2) stop after X doses (dose limit); (3) start trace at steady state; and (4) show cis woman menstrual cycle (median, 5th percentile, and 95th percentile estradiol levels; data from Abbott (2009)). The advanced injectable estradiol simulator was released on October 5, 2022 and can be found at the following page:
Here is a screenshot of the advanced simulator and its capabilities:
Update 2: Literature Mentions
Since Transfeminine Science’s injectable estradiol simulator was released in mid-2021, it has been mentioned and cited in the scientific literature in a number of publications (see Aly, 2021).
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
Aly. (2021). An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations. Transfeminine Science. [URL]
Behre, H. M., Abshagen, K., Oettel, M., Hubler, D., & Nieschlag, E. (1999). Intramuscular injection of testosterone undecanoate for the treatment of male hypogonadism: phase I studies. European Journal of Endocrinology, 140(5), 414–419. [DOI:10.1530/eje.0.1400414]
\ No newline at end of file
diff --git a/transfemscience.org/articles/nandrolone/index.html b/transfemscience.org/articles/nandrolone/index.html
index 01a58717..0c7e4ec6 100644
--- a/transfemscience.org/articles/nandrolone/index.html
+++ b/transfemscience.org/articles/nandrolone/index.html
@@ -1 +1 @@
-Nandrolone as a Potential Alternative Androgen with Reduced Androgenic Side Effects for Transfeminine and Transmasculine People - Transfeminine Science
Nandrolone as a Potential Alternative Androgen with Reduced Androgenic Side Effects for Transfeminine and Transmasculine People
By Aly | First published March 20, 2020 | Last modified March 23, 2023
Abstract / TL;DR
Nandrolone, or 19-nortestosterone, is a unique androgen and anabolic steroid which is used in the form of injectable ester prodrugs like nandrolone decanoate. It is closely related to testosterone structurally but has a number of important pharmacological differences in comparison. These differences include inactivation by 5α-reductase rather than potentiation, less or no conversion into estradiol, and much greater progestogenic activity. The inactivation of nandrolone by 5α-reductase results in nandrolone theoretically having much lower androgenic strength than testosterone in skin, hair follicles, and the prostate, among other tissues. Consequently, nandrolone may have less propensity for oily skin, acne, facial/body hair growth, scalp hair loss, and prostate problems than testosterone. On the other hand, nandrolone is theoretically expected to have full androgenic efficacy in most other tissues, including bone, fat, and muscle (and hence of masculine skeletal, fat, and muscular changes). This is also likely to be the case when it comes to voice, bottom growth, and sexual function. The reduced androgenic impact of nandrolone in skin and hair follicles relative to testosterone make it a favorable potential alternative option for androgen therapy both for transfeminine people and for some transmasculine people.
Introduction
Nandrolone, also known chemically as 19-nortestosterone (19-NT), is an androgen and anabolic steroid (AAS) similarly to testosterone (T). It is used as a medication by intramuscular or subcutaneous injection in the form of longer-acting esters like nandrolone decanoate (ND; Deca-Durabolin) and nandrolone phenylpropionate (NPP; Durabolin), which are converted into nandrolone in the body. This is analogous to longer-acting testosterone esters like testosterone enanthate (TE) and testosterone undecanoate (TU) and their conversion in the body into testosterone. Nandrolone is used in medicine for a variety of indications that benefit from anabolic effects in tissues like muscle, bone, and kidneys. Examples include wasting syndromes, osteoporosis, kidney disease, and anemia. It is used not only in men but also in women, for instance to prevent and treat postmenopausal osteoporosis in women who can’t tolerate estrogens or in whom estrogens can’t be taken. Nandrolone has additionally been advocated for use in androgen replacement therapy for women (Davis, 1999).
Nandrolone is closely related to testosterone in terms of chemical structure, pharmacodynamic actions, and disposition in the body. At the same time however, nandrolone has a key difference from testosterone that makes it a very favorable alternative option for use as an androgen in transgender hormone therapy. This is the case not only for transfeminine people but also for transmasculine people. This property of nandrolone is that it is inactivated by 5α-reductase in tissues that express this enzyme. This is in contrast to testosterone, which is potentiated in such tissues via conversion into dihydrotestosterone (DHT). Because of this difference, nandrolone has normal androgenic effects in most of the body but a greatly reduced potential for androgenic effects in skin and hair follicles relative to testosterone. As a result, nandrolone has a variably reduced potential for androgenic skin and hair side effects such as oily skin, acne, facial and body hair growth, and scalp hair loss.
Because of its favorable profile relative to testosterone, nandrolone has been advocated for use in androgen replacement therapy in cisgender men (Wu & Kovac, 2016; Pan & Kovac, 2016). It has also been studied for use in male hormonal contraception as an alternative and replacement for testosterone (Knuth et al., 1986; Nieschlag, 2010; Nieschlag & Behre, 2012). Nandrolone is advantageous for cisgender men not only due to preservation of scalp hair but also due to its lower stimulation of the prostate gland. As a result of its more favorable profile in terms of skin and hair follicles, nandrolone is an underappreciated alternative possibility relative to testosterone for use not only in cisgender people but also in transfeminine and transmasculine people.
Chemistry of Nandrolone
Nandrolone is chemically known as 19-nortestosterone. It is very close to testosterone in terms of chemical structure, the only difference between the two compounds being that the C19 methyl group of testosterone has been removed in the case of nandrolone. Nandrolone is not a synthetic compound; it occurs naturally in the human body in trace amounts as an intermediate in the conversion of testosterone to estradiol by the enzyme aromatase (Bricout & White, 2004). As such, although nandrolone couldn’t be said to be “bioidentical”, it’s quite similar to testosterone.
Figure: Chemical structures of testosterone and nandrolone (19-nortestosterone). The structural difference between testosterone and nandrolone is at the C19 position and is highlighted by the circled areas.
Nandrolone is an agonist of the androgen receptor (AR) similarly to testosterone. It shows higher affinity for the AR than does testosterone and has almost no binding affinity for sex hormone-binding globulin (SHBG) however. For these reasons, nandrolone has about 2.5- to 5-fold higher potency than testosterone in rats on measures of general AR agonistic strength, such as stimulation of muscle growth (Sundaram et al., 1995; Tóth & Zakár, 1986; Wiki; Winters, 1976). It is likewise a more potent AAS than testosterone in humans, and is used medically at lower doses in comparison (Wiki).
Testosterone is a substrate for 5α-reductase in tissues that express this enzyme like skin, hair follicles, prostate gland, and seminal vesicles, among others. It is converted by 5α-reductase into dihydrotestosterone (DHT), an androgen with much higher AR affinity and AR agonistic potency than that of testosterone (Wiki). Consequently, it is estimated that the effects of testosterone are potentiated by 2- to 3-fold via conversion into DHT in tissues that express 5α-reductase (Sundaram et al., 1995). The role of DHT in the effects of testosterone is demonstrated by a rare intersex condition known as 5α-reductase type 2 deficiency, as well as by 5α-reductase inhibitors (5α-RIs) like finasteride and dutasteride (Wiki; Wiki). Men with 5α-reductase type 2 deficiency are reported to have reduced facial hair, a female-like pattern of body hair (with terminal hair largely restricted to the underarms and lower pubic triangle), and no scalp hair recession in the temples nor scalp hair loss in general. Conversely, other aspects of male secondary sexual development, like voice deepening, muscle growth, bone changes, and pubertal penile enlargement, are all normal in the condition. In addition, no feminization or gynecomastia occurs. Hence, adults with 5α-reductase type 2 deficiency have a masculine appearance (Imperato-McGinley & Peterson, 1976; Peterson et al., 1977; Photos). In published photographs of individuals with the condition however, features like lack of body hair and rounded feminine hairlines can be noted.
5α-Reductase inhibitors have similar effects relative to the profile of 5α-reductase type 2 deficiency. For this reason, they are used in the treatment and prevention of scalp hair loss in men and off-label for hirsutism (excessive facial/body hair growth) in women. They greatly slow the rate of scalp hair loss over time in men. For example, a large 5-year randomized controlled trial showed that in men with diagnosed scalp hair loss, 100% of those who received placebo had significant further scalp hair loss whereas only 35% of those who received finasteride had further scalp hair loss (Propecia label). Low-quality evidence suggests that finasteride has similar effectiveness to antiandrogens like spironolactone and flutamide in the treatment of hirsutism in women (Barrionuevo et al., 2018; van Zuuren et al., 2015). Dutasteride has similar or greater effectiveness than finasteride for scalp hair loss in men (Arif et al., 2017), whereas it hasn’t yet been studied in the treatment of hirsutism (Wiki).
5α-Reductase is the key to the favorable differences between testosterone and nandrolone. Both testosterone and nandrolone are substrates for 5α-reductase and have similar affinity for this enzyme (Tóth & Zakár, 1986). But whereas testosterone is potentiated via conversion into DHT in tissues that express 5α-reductase, nandrolone is inactivated by 5α-reductase in these tissues (Kicman, 2008; Tóth, 2009). This is because the 5α-reduced metabolite of nandrolone, 5α-dihydronandrolone (DHN), is a weaker AR agonist with much lower affinity for the AR than nandrolone (Tóth & Zakár, 1986; Kumar et al., 1999). The following table shows AR and SHBG relative binding affinities (RBA) of testosterone, nandrolone, trestolone, and their 5α-reduced forms (Kumar et al., 1999):
Steroid
AR RBA
SHBG RBA
Testosterone
100%
100%
5α-Dihydrotestosterone
290%
340%
Nandrolone
230%
5%
5α-Dihydronandrolone
10%
5%
Trestolone
380%
6%
5α-Dihydrotrestolone
30%
2%
In rodents, although nandrolone has about 3-fold higher anabolic or muscle-stimulating potency than testosterone, its androgenic potency in the prostate gland and seminal vesicles is only about 20 to 40% of that of testosterone (Sundaram et al., 1995; Tóth & Zakár, 1986; Wiki). This works out to an approximate 11:1 dissociation between the effects of nandrolone in tissues with little or no 5α-reductase (muscle) versus with high 5α-reductase (prostate gland, seminal vesicles) in rodents (Wiki). The exact ratio of potency between these tissues in humans is unknown, and extrapolation from rodents should be cautioned against. In any case, an analogous dissociation of some level can be anticipated. As such, nandrolone can be expected to have high AR agonistic strength in most of the body, where 5α-reductase has minimal expression (e.g., fat, muscle, bone), but much lower potency in tissues with high 5α-reductase expression, like skin, hair follicles, the prostate gland, and the seminal vesicles.
It’s important to be clear that nandrolone is not simply akin to taking testosterone with a 5α-reductase inhibitor like dutasteride to block 5α-reductase. This is because nandrolone isn’t merely not potentiated by 5α-reductase, it’s weakened by 5α-reductase. For this reason, 5α-reductase inhibitors actually increase the androgenic strength of nandrolone in tissues that express 5α-reductase like the prostate gland in rodents (Sundaram et al., 1995). (And, for this reason, nandrolone should not be taken with a 5α-reductase inhibitor, in contrast to testosterone.) In other words, nandrolone goes beyond testosterone plus a 5α-reductase inhibitor in terms of the dissociation of its potency between tissues that express 5α-reductase and tissues that do not. As a result, nandrolone has much lower androgenic strength in such tissues than does the combination of testosterone plus a 5α-reductase inhibitor. Nandrolone also appears to have lower strength in tissues that express 5α-reductase than do most other AAS, for instance synthetic DHT derivatives like oxandrolone and stanozolol (Tóth & Zakár, 1986). This is because these AAS are not substrates for 5α-reductase and hence are neither potentiated nor weakened by 5α-reductase, similarly to testosterone with a 5α-reductase inhibitor. The inactivation of nandrolone by 5α-reductase makes it very unique among AAS and AR agonists. Only nandrolone and a few of its derivatives like normethandrone and norethandrolone have this very special property.
Testosterone and nandrolone may also be non-identical in their androgenic actions and effects for reasons other than metabolism. This is because different AR agonists including strong androgens like testosterone and DHT, weak androgens like dehydroepiandrosterone (DHEA) and androstenedione (A4), and predominantly anabolic androgens like nandrolone, oxandrolone, and stanozolol, have been shown to produce differential AR-mediated changes in gene expression (Holterhus, Piefke, & Hiort, 2002; Kicman, 2008; Hiort, 2013). However, nandrolone was more similar in its profile to testosterone than the synthetic AAS. In any case, the clinical implications of these differences in relation to therapeutic use of testosterone versus nandrolone, if any, are unknown.
In relation to the fact that nandrolone is an intermediate in the aromatase-mediated reaction that converts testosterone into estradiol, both testosterone and nandrolone are transformed into estradiol, and hence estradiol is their specific estrogen metabolite. Paradoxically however, whereas it has the same potential for 5α-reduction as testosterone, nandrolone as a medication is aromatized into estradiol virtually not at all (Handelsman, 2016). In accordance, nandrolone suppresses estradiol levels in men and results in low estradiol levels even at very high doses (Friedl, 1991; Hobbs et al., 1993; Hobbs, Jones, & Plymate, 1996; Behre et al., 2001). The following table gives an example of estradiol (E2) levels in men with high-dose testosterone versus high-dose nandrolone (Friedl, 1991):
Drug and dose
E2 before
E2 after
Change
Testosterone enanthate 300 mg/week i.m.
32 ± 4.6 pg/mL
79 ± 9.3 pg/mL
+147%
Nandrolone decanoate 300 mg/week i.m.
40 ± 6.0 pg/mL
14 ± 1.6 pg/mL
–65%
Testosterone and nandrolone themselves don’t have significant affinity for the estrogen receptors nor intrinsic estrogenic activity outside of aromatization to estradiol (Kuiper et al., 1997; Table). Although androgens also help to maintain bone mineral density and AAS like nandrolone are used to treat osteoporosis in postmenopausal women (Geusens, 1995; Ebeling, 2010), estrogens are comparatively much more important for maintenance of bone mineral density and prevention of osteoporosis in both men and women (Wiki). Studies of high-dose nandrolone and androstanolone (DHT) have shown that these non-aromatizable AAS are not adequate for maintenance of bone mineral density in men, whereas the aromatizable testosterone is effective for such purposes (Ronald et al., 1998; Crawford et al., 2003; Lemmey et al., 2013; Sartorius, Ly, & Handelsman, 2014; Čeponis et al., 2017). Non-aromatizable AAS are likewise inferior to estrogen and estrogen–progestogen therapy for maintaining bone mineral density in postmenopausal women (Lauritzen, 1984 [Graph]; Hassager et al., 1991).
Because of its minimal conversion into estradiol, nandrolone should be used as an androgen in transmasculine people only in combination with low-dose estradiol to replace the lost estradiol that would otherwise be provided by testosterone. Estradiol levels in men are on average around 30 pg/mL, and estradiol levels of at least 20 to 30 pg/mL appear to be necessary for maintenance of bone density in men (Doran et al., 2001). A low-dose transdermal estradiol patch, for instance 14 to 25 μg/day, is likely to be adequate for achieving such levels. Alternatively, another option like transdermal estradiol gel (e.g., 0.75–1 mg/day) or low-dose oral estradiol (e.g., 0.5–1 mg/day) may be used instead.
Nandrolone likewise has progestogenic activity. However, the progestogenic activity of nandrolone is greatly enhanced relative to that of testosterone. The affinity of nandrolone for the PR is 5- to 20-fold higher than that of testosterone and its progestogenic potency is correspondingly improved (Wiki; Wiki; Bardin & Janne, 1986). Despite this however, the progestogenic potency of nandrolone is still relatively low. For instance, its affinity for the PR is only about 10 to 20% of that of progesterone and the nandrolone-derived progestin norethisterone (also known as 17α-ethynyl-19-nortestosterone). At the same time however, nandrolone is used at relatively large doses compared to norethisterone. This may help to compensate for its lower progestogenic potency and may allow for significant progestogenic effects at typical clinical doses (Camerino & Sciaky, 1975).
The following table shows human and rabbit PR RBAs and relative progestogenic activity (as measured by in-vivo induction of uteroglobin synthesis in rabbit uterus) of progesterone and several AAS including testosterone and nandrolone (Jänne et al., 1978; Bardin & Janne, 1986):
Steroid
hPR RBA
rbPR RBA
Progestogenic activity (rabbit)
Progesterone
100%
100%
100%
Testosterone
1.6%
1.6%
0.9%
Dihydrotestosterone
3.3%
3.3%
1.9%
Nandrolone
8.9%
12%
5.2%
Methyltestosterone
4.6%
3.4%
0.6%
Norethandrolone
130%
70%
115%
The development of nandrolone was an intermediate step in the eventual development of the 19-nortestosterone group of progestins, which are widely used in hormonal birth control. These include progestins like norethisterone and levonorgestrel, among many others.
Advantages of Nandrolone Over Testosterone
Because of its substantially lower androgenic strength in tissues that have high 5α-reductase expression relative to tissues that have minimal or no 5α-reductase expression, nandrolone is likely to have normal androgenic effects in muscle, bone, fat, and most other tissues but much weaker androgenic strength in skin and hair follicles than equivalent doses of testosterone. As a result of this, although nandrolone has high capacity for producing most aspects of masculinization, it has a lower potential for many of the undesirable androgenic skin and hair side effects of testosterone, including oily skin, acne, facial and body hair growth, and scalp hair loss. This may allow for unique advantages of nandrolone over testosterone in transfeminine and transmasculine hormone therapy.
The benefits of nandrolone are expected to partially differ in transfeminine people relative to transmasculine people however. This is due to the different doses of nandrolone used in these individuals, namely low, female replacement doses in transfeminine people and full, male replacement doses in transmasculine people. As an example, transmasculine people taking full male androgen replacement doses should probably expect no less oily skin or acne with nandrolone than with testosterone. This is because very low levels of testosterone are necessary for such effects, as evidenced by the fact that acne prevalence and severity is similar in females compared to males (Skroza et al., 2018) despite the fact that testosterone levels are 20-fold lower in women than in men (Styne & Grumbach, 2016). Conversely, transfeminine people taking nandrolone at doses equivalent to the effect of testosterone levels in the normal female range may experience less or possibly no oily skin or acne with nandrolone compared to use of testosterone instead. This is due to the equivalent testosterone levels being far closer to the biological threshold for manifestation of such effects.
Nandrolone can be anticipated to have far less potential for scalp hair loss relative to testosterone in transmasculine people (Wu & Kovac, 2016; Pan & Kovac, 2016). This is related to the lack of scalp hair loss in men with 5α-reductase type 2 deficiency and the high effectiveness of 5α-reductase inhibitors for scalp hair loss. However, nandrolone is theoretically even better than testosterone plus a 5α-reductase inhibitor when it comes to preservation of scalp hair. This is because again, nandrolone is not simply not potentiated, but is inactivated in tissues that express 5α-reductase. Nandrolone replacement of testosterone represents a highly promising treatment for scalp hair loss in men that offers potentially superior effectiveness to 5α-reductase inhibitors. Moreover, since nandrolone isn’t an inhibitor of 5α-reductase, it doesn’t interfere with 5α-reduced neurosteroid synthesis. Hence, nandrolone doesn’t have the possible psychiatric side effects that have been associated with 5α-reductase inhibitors, for instance depression (Irwig, 2015; Kuhl & Wiegratz, 2017; Wiki).
On the basis of 5α-reductase type 2 deficiency, nandrolone may be expected to produce reduced facial and particularly body hair growth relative to testosterone in transmasculine people. Essentially no body or facial hair growth is likely to occur with nandrolone at doses equivalent to female-range testosterone levels in transfeminine people, and even at supraphysiological levels, facial and body hair growth is likely to be less than with testosterone. However, facial and body hirsutism have nonetheless been reported with nandrolone at postmenopausal osteoporosis doses (Need et al., 1989; Passeri et al., 1993; Geusens, 1995; Flicker et al., 1997).
For transmasculine people who want the benefits of nandrolone except the lesser facial hair growth, topical testosterone to the beard area can potentially be used in conjunction with nandrolone injections to restore full facial hair growth. Another possibility for achieving effects of both testosterone and nandrolone in transmasculine people could be to start on testosterone and use it for a few years, allowing for facial/body hair growth and hairline masculinization, and then switch from testosterone to nandrolone for long-term therapy, allowing for preservation of scalp hair.
One of the reasons that nandrolone is attractive for androgen replacement therapy in cisgender men is that it has very weak relative effects in the prostate gland compared to testosterone. This is due to inactivation of nandrolone in the prostate gland by 5α-reductase similarly to other tissues that express this enzyme. It is anticipated that long-term androgen replacement therapy with nandrolone might allow for a reduced risk of enlarged prostate and prostate cancer compared to testosterone in men (Wu & Kovac, 2016; Pan & Kovac, 2016). As transfeminine people also have prostate glands, the weak effects of nandrolone in the prostate are potentially of relevance to us as well. However, due to estrogen therapy and androgen deprivation, the prostate is atrophied in transfeminine people and prostate cancer is very rare (Wiki). In relation to this, the favorable prostate profile of nandrolone may be of little significance for transfeminine people.
Although nandrolone is quite promising for transgender hormone therapy, it is important to be clear that there are currently no quality clinical studies comparing it to testosterone and showing that it is less androgenic in terms of skin and hair effects (even if there is strong theoretical basis for this notion, particularly in the case of scalp hair loss). Nor are there any studies demonstrating with certainty that it is as effective as testosterone in terms of inducing general masculinization. Moreover, the appropriate dosages of nandrolone for use in androgen replacement in women and men are uncertain. Finally, nandrolone has been little-studied for androgen replacement and its long-term tolerability and health safety for this use haven’t been properly characterized. For these reasons, androgen replacement with nandrolone in cisgender men and transmasculine people is an experimental therapy. Additional research is needed to properly characterize nandrolone for this indication.
Androgen Replacement in Transfeminine People
Some believe that testosterone is important in women for mood, well-being, energy, sexual desire and function, general health, and for other reasons. Androgen levels and activity may be low in many transfeminine people due to hormone therapy and/or gonadectomy, raising concerns about androgen deficiency. However, there is little or no change in androgen levels with natural menopause in cisgender women and only a small decrease in androgen levels with ovariectomy in women (Liu & Handelsman, 1998). Moreover, there is inadequate evidence to support claims of benefit with androgen replacement therapy in women at present, and there are no well-supported benefits of female-range doses of testosterone (Wiki). For these reasons, androgen replacement therapy in women is controversial (Liu & Handelsman, 1998).
Testosterone does stimulate sexual desire in women, but only at levels that are above the normal female range (>50 ng/dL) (Cappelletti & Wallen, 2016; Wiki). And even with high testosterone levels of 80 to 150 ng/dL, stimulation of sexual desire in women is modest (Cappelletti & Wallen, 2016; Wiki). It is likely that estradiol rather than testosterone (or progesterone) is the key sex hormone for sexual desire in women (Cappelletti & Wallen, 2016; Aly, 2020).
Androgens can have detrimental effects in transfeminine people. They can cause androgenic and masculinizing effects like oily skin, acne, seborrhea, facial/body hair growth, scalp hair loss, and voice deepening, among others. In addition, androgens oppose the effects of estrogens in the breasts, and may inhibit breast development even with low-level exposure or physiological levels in females (Dimitrakakis et al., 2003; Peters et al., 2011; Sas et al., 2014; Barbieri, 2017).
For these reasons, transfeminine people don’t necessarily require androgen replacement, and it shouldn’t be routinely used out of concern of androgen deficiency. In any case, some transfeminine people insist on androgen replacement, and others desire supraphysiological levels of androgens for purposes like stimulating libido or helping to build and maintain muscle mass. There is some evidence that even physiological levels of testosterone may help to support muscle mass in women (Huang & Basaria, 2017). Moreover, in transfeminine people who are fully hormonally transitioned, breast development is complete, and hence inhibition of the breasts by androgens is no longer a concern. Another potentially useful indication of androgens in transfeminine people is to counteract cellulite (Wiki; Gruber et al., 2002; Avram, 2004). Since nandrolone has full androgenic potential in fat tissue, it may be similarly effective as testosterone for this purpose. However, there are no good studies of androgens for treatment of cellulite at this time.
Although androgen replacement therapy isn’t recommended for cisgender women or transfeminine people because of a lack of evidence of benefit and potential adverse effects, nandrolone is a very favorable alternative to testosterone for such purposes in those who nonetheless opt for such therapy. This is due to its improved skin and hair profile and hence a probable lower risk of undesirable effects.
Additional Topics on Nandrolone
This section is for additional topics on nandrolone as an androgen in transfeminine and transmasculine people, including its availability, dosage, androgenic/masculinizing effects besides skin and hair (e.g., voice deepening, bottom growth/clitoral enlargement, sexual effects), and liver safety.
Availability of Nandrolone
Nandrolone esters remain widely available for medical use throughout the world (Drugs.com). However, the availability of AAS, including nandrolone esters, has become increasingly limited over time. The shorter-acting ester nandrolone phenylpropionate is no longer available in the United States or many other countries, and the more favorable longer-acting nandrolone decanoate was discontinued in the United States in 2019. However, nandrolone decanoate remains available from compounding pharmacies in the United States, for instance AnazaoHealth. It is only available from this particular pharmacy at a very high concentration of 200 mg/mL however, which although a potentially workable concentration for use in transmasculine people, is likely completely impractical for use in transfeminine people. Compounding pharmacies might be able to do custom concentrations upon request though.
Another potential means of delivering nandrolone is via an orally active nandrolone androgen prohormone like 19-nordehydroepiandrosterone (19-nor-DHEA). This compound is converted into nandrolone analogously to the use of DHEA as a prohormone of testosterone (Wiki). It is sold as an over-the-counter supplement from certain online vendors, similarly to DHEA. 19-Nor-DHEA may be a more suitable option for transfeminine people due to the lower doses needed and less suitable for transmasculine people due to the much higher doses required.
Dosage of Nandrolone
The dosage of nandrolone for use in androgen replacement therapy in women and men hasn’t been established. Hence, we don’t completely know what the appropriate dose is for use in transfeminine and transmasculine hormone therapy. In any case, it is possible for us to estimate.
One review recommended a clinical dose range in males of 50–400 mg/2–4 weeks for testosterone enanthate or cypionate but 50–100/3–4 weeks for nandrolone decanoate (Hickson et al., 1989). This is also in line with several-fold higher potency of nandrolone relative to testosterone.
Dosage for Transmasculine People
A typical dosage of testosterone enanthate for use in androgen replacement therapy in cisgender and transgender men is 50 to 100 mg once per week by intramuscular injection (Wiki; Wiki). Hence, an appropriate dosage of nandrolone decanoate, assuming 3-fold greater potency than testosterone, might be about 16.7 to 33.3 mg once per week by intramuscular injection (ignoring the small differences in molecular weight between testosterone enanthate and nandrolone decanoate). However, it must be emphasized that these doses are merely educated guesses. It’s notable that these doses are quite similar to those used in postmenopausal osteoporosis in women (about 12.5 to 25 mg per week total). Hence, although such doses have certainly been associated with masculinizing effects in women, they might be suboptimal in terms of masculinization for transmasculine people. Consequently, it’s possible that a higher dose, like 50 mg once per week, might be more appropriate. In addition to weekly use, nandrolone decanoate has a duration suitable for use once every 2 weeks (Graphs).
Nandrolone hexyloxyphenylpropionate (NHPP; brand name Anadur) was studied as a male hormonal contraceptive by the World Health Organization and others at a dose of 200 mg/3 weeks i.m. in combination with depot medroxyprogesterone acetate (Knuth et al., 1986; Nieschlag, 2010; Nieschlag & Behre, 2012). It was used as a replacement and alternative for testosterone and was given at a dosage of about 66.7 mg once per week. However, this was an intentionally supraphysiological dosage which was selected to achieve a male contraceptive effect, as suggested by suppression of gonadotropins to undetectable levels (Knuth et al., 1985). Hence, although safe and well-tolerated in limited studies, it might be a high dosage for transmasculine hormone therapy. Very high doses of nandrolone can reproduce the full spectrum of androgenic effects of testosterone, and hence doses of nandrolone that are too high may serve to nullify its advantages over testosterone (van der Vies, 1985).
Dosage for Transfeminine People
In postmenopausal women, nandrolone decanoate has been used for general androgen replacement at a dose of 25 to 50 mg once every 6 to 12 weeks (about 2–8 mg/week or 0.29–1.14 mg/day), and for prevention and treatment of osteoporosis at a dose of 50 mg once every 2 to 4 weeks (about 12.5–25 mg/week or 1.8–3.6 mg/day) (Wiki; Table). However, these doses of nandrolone decanoate are likely to be highly excessive for physiological androgen replacement in women and transfeminine people and are probably also spaced too widely in terms of dosing interval. This is based on the following: (1) normal production of testosterone in premenopausal women is about 150 μg/day; (2) nandrolone is more potent as an androgen receptor agonist than testosterone; and (3) pharmacokinetic concentration–time curves for nandrolone decanoate suggest that an appropriate dosing interval is once every 1 to 2 weeks (Graphs). Indeed, nandrolone decanoate at 50 mg/2–4 weeks has been found to produce significant masculinizing effects in women with long-term use (Geusens, 1995; Wiki). Since nandrolone has high theoretical masculinizing potential in most tissues in the body, like muscle, bone, fat, and vocal tissue, it’s very important that transfeminine people don’t take doses that are too large.
Androgenic and Masculinizing Effects Besides Skin and Hair
Nandrolone has sometimes been described as “non-masculinizing” or “minimally virilizing”, but this is misleading and inaccurate. Nandrolone has high theoretical potential to produce masculinizing skeletal, fat, and muscular effects, like growth of muscles, widening of shoulders, masculine enlargement, elongation, and shaping of bones, and masculine distribution of fat. It also has high capacity to produce voice deepening (the vocal tissue notably being part muscle). The only clear differences in masculinizing effect between nandrolone and testosterone are that nandrolone has a greatly reduced theoretical potential for androgenic effect in skin and hair follicles. Succinctly, nandrolone could be thought of as testosterone with minimization of the undesirable skin and hair effects.
Voice Deepening with Nandrolone
The vocal tissue, consisting of the vocal cords and the larynx, are part muscle, and 5α-reductase is expressed minimally or not at all in this tissue (Bhagavan, 2002; Kicman, 2008). Accordingly, voice deepening is normal in individuals with 5α-reductase type 2 deficiency (Imperato-McGinley & Zhu, 2002). As such, nandrolone would be expected to produce voice deepening similarly to testosterone. In accordance, voice deepening has been reported with nandrolone in women at doses used in postmenopausal osteoporosis (Gerritsma et al., 1994; Geusens, 1995; Frisoli et al., 2005). There are case reports of severe and complete masculinization of the voice in women treated with excessive doses of nandrolone (Damsté, 1964; Brodnitz, 1971; Baker, 1999). Voice changes have also been seen with nandrolone derivatives like normethandrone (Feldman et al., 1960).
Bottom Growth with Nandrolone
DHT is required for normal male sexual differentiation of the genitals during prenatal development. This is evidenced by 5α-reductase type 2 deficiency, in which the genitals are ambiguous but overall more female-like at birth and in childhood (Wiki). Although DHT is critical for prenatal genital masculinization, the same may not be true for pubertal development of the penis (Hiort, 2013). Individuals with 5α-reductase type 2 deficiency undergo masculinization of the genitals at puberty such that a small and near-functional penis develops (Peterson et al., 1977; Marks, 2004; Hiort, 2013; Photos). This is so striking that males with 5α-reductase type 2 deficiency in the Dominican Republic are referred to as “guevedoces”, which is said to literally mean “penis at 12”.
As 5α-reductase may not be critical for penile enlargement at puberty, nandrolone may allow for no less bottom growth (clitoral enlargement) in transmasculine people than does testosterone. Indeed, nandrolone has been reported to produce clitoral enlargement as a side effect in women and penile growth in prepubertal boys (Camerino & Sala, 1960; Camerino & Sciaky, 1975). Similar findings have been made for derivatives of nandrolone like normethandrone and norethandrolone (Feldman et al., 1960; Roche, Towns, & Wettenhall, 1963; Kirschvink et al., 1963; Prunty et al., 1958).
Since this article was published, a review suggesting nandrolone as a potential component of transgender hormone therapy for non-binary individuals has been published:
Cocchetti, C., Ristori, J., Romani, A., Maggi, M., & Fisher, A. D. (2020). Hormonal Treatment Strategies Tailored to Non-Binary Transgender Individuals. Journal of Clinical Medicine, 9(6), 1609. [DOI:10.3390/jcm9061609]
The review includes the following excerpts on nandrolone:
Other options may include nandrolone, an anabolic steroid administered via intramuscular injection, which is not as optimal a substrate for 5α-reductase as testosterone, but it has a stronger effect compared to the testosterone on target tissues devoid of 5α-reductase activity (e.g., muscular tissue) [15]. Indeed, nandrolone can be theoretically used in non-binary [assigned female at birth (AFAB)] individuals requesting masculinization of body shape (i.e., increased muscle mass) with a limited increase in facial and body hair. Regarding the safety profile of this compound, data are limited by the fact that most observations come from the setting of androgenic-anabolic steroid (AAS) abuse [16,17], thus their applicability to appropriate medical therapy is limited [18]. In this setting, concerns about cardiomyopathy and coronary artery disease risk emerged [19], although associated with the administration of nandrolone at extremely higher dosages [20]. Furthermore, nandrolone use does not seem associated to hepatotoxicity, since, as an injectable oil, it is not subject to first-pass hepatic metabolism.
Moreover, some AFAB transgender individuals can benefit from testosterone therapy combined with 5α-reductase inhibitors or from treatment with nandrolone (an androgenic compound less prone to 5α reduction) in case they wish only a partial virilization (i.e., voice deepening and lean mass increase without facial and body hair increase).
In addition to the above review, a second review suggesting nandrolone as a potential option for hormone therapy in non-binary individuals has been published (as a preprint):
Vetri, M., Cataldi, A., Naselli, A., & Vetri, A. (2021). Transsexualism Ethiology and Medical Management: Between Scientific Evidence and Personal Experiences. Preprints, 2021030172. [DOI:10.20944/preprints202103.0172.v1]:
With the following relevant excerpt:
In [female-to-males] (FtMs) requesting partial masculinization, it may be possible to reduce the dose of testosterone or substitute it with nandrolone, an anabolic steroid administered via intramuscular injection. So, we must accept alternative hormonal treatment regimens, other than those reported in current guidelines, for such nonbinary transgender individuals, to try to improve their psychological well-being and quality of life [40].
However, this preprint article appears to have never ended up being accepted for publication.
Update 3: Cocchetti et al. (2020) and Iuliano et al. (2021)
Further articles have mentioned nandrolone as a hypothetical option for transgender people, for instance Cocchetti et al. (2020):
Another hypothetic option [for management of hypoactive sexual desire disorder in transgender women] may include nandrolone, an androgenic compound administered via intramuscular injection, less prone to 5-alpha reduction anabolic steroid leading to limited virilizing dermatological undesired effects [72]. However, data on its efficacy on sexual desire are not yet available.
Actually, the classic view of gender binarism and [gender-affirming hormone therapy (GAHT)] should be reconsidered because of the increasing prevalence of non-binary individuals experiencing [gender dysphoria (GD)]. Therefore, new therapeutic strategies should be also considered [17]. Since there is a lack in standardized hormonal treatment protocols for non-binary [assigned female at birth (AFAB)], GAHT goals should be adjusted according to patients’ needs in order to improve the self-perception and the quality of life [18]. For instance, different testosterone doses and/or other androgen preparations (i.e., nandrolone) are suggested to modulate the requested body changes [18], even if serious ethical concerns on some drugs exist.
References
Anderson, R. A., Martin, C. W., Kung, A. W., Everington, D., Pun, T. C., Tan, K. C., Bancroft, J., Sundaram, K., Moo-Young, A. J., & Baird, D. T. (1999). 7α-Methyl-19-Nortestosterone Maintains Sexual Behavior and Mood in Hypogonadal Men. The Journal of Clinical Endocrinology & Metabolism, 84(10), 3556–3562. [DOI:10.1210/jcem.84.10.6028]
Anderson, R. A., Wallace, A. M., Sattar, N., Kumar, N., & Sundaram, K. (2003). Evidence for Tissue Selectivity of the Synthetic Androgen 7α-Methyl-19-Nortestosterone in Hypogonadal Men. The Journal of Clinical Endocrinology & Metabolism, 88(6), 2784–2793. [DOI:10.1210/jc.2002-021960]
Arif, T., Dorjay, K., Adil, M., & Sami, M. (2017). Dutasteride in Androgenetic Alopecia: An Update. Current Clinical Pharmacology, 12(1), 31–35. [DOI:10.2174/1574884712666170310111125]
Avram, M. M. (2004). Cellulite: a review of its physiology and treatment. Journal of Cosmetic and Laser Therapy, 6(4), 181–185. [DOI:10.1080/14764170410003057]
Azzouni, F., Godoy, A., Li, Y., & Mohler, J. (2012). The 5 Alpha-Reductase Isozyme Family: A Review of Basic Biology and Their Role in Human Diseases. Advances in Urology, 2012 (Personalized Cancer Therapy for Urological Cancers: From Bench to Bedside and Back), 530121. [DOI:10.1155/2012/530121]
Baker, J. (1999). A report on alterations to the speaking and singing voices of four women following hormonal therapy with virilizing agents. Journal of Voice, 13(4), 496–507. [DOI:10.1016/s0892-1997(99)80005-8]
Barbieri, R. L. (2019). Breast. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 248–255.e3). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/B978-0-323-47912-7.00010-X]
Bardin, C. W., & Janne, O. A. (1986). Steroids of One Class Can Mimic, Inhibit and Potentiate the Biological Effects of Other Steroid Classes When Administered at High Doses. In Gregoire, A. T., & Blye, R. P. (Eds.). Contraceptive Steroids: Pharmacology and Safety (pp. 123–143). Boston, MA: Springer US. [DOI:10.1007/978-1-4613-2241-2_6]
Barrionuevo, P., Nabhan, M., Altayar, O., Wang, Z., Erwin, P. J., Asi, N., Martin, K. A., & Murad, M. H. (2018). Treatment Options for Hirsutism: A Systematic Review and Network Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism, 103(4), 1258–1264. [DOI:10.1210/jc.2017-02052]
Bässler, R. (1970). The Morphology of Hormone Induced Structural Changes in the Female Breast. In Altmann, H.-W., et al. (Eds.). Current Topics in Pathology: Ergebnisse der Pathology, Volume 53 (pp. 1–89). Heidelberg: Springer Berlin. [DOI:10.1007/978-3-662-30514-0_1]
Behre, H. M., Nashan, D., Hubert, W., & Nieschlag, E. (1992). Depot gonadotropin-releasing hormone agonist blunts the androgen-induced suppression of spermatogenesis in a clinical trial of male contraception. The Journal of Clinical Endocrinology & Metabolism, 74(1), 84–90. [DOI:10.1210/jcem.74.1.1727833]
Behre, H., Kliesch, S., Lemcke, B., von Eckardstein, S., & Nieschlag, E. (2001). Suppression of spermatogenesis to azoospermia by combined administration of GnRH antagonist and 19-nortestosterone cannot be maintained by this non-aromatizable androgen alone. Human Reproduction, 16(12), 2570–2577. [DOI:10.1093/humrep/16.12.2570]
Bhagavan, N. V. (2002). Endocrine Metabolism V: Reproductive System. In Bhagavan, N. V. Medical Biochemistry, 4th Edition (pp. 781–801). San Diego: Harcourt/Academic Press. [Google Books] [WorldCat]
Bourg, R. (1950). Activité sécrétrice des glandes mammaires; consécutive a des doses massives de testostérone chez la femme après castration. [Secretory activity of the mammary glands; sequellae of massive testosterone doses in women following castration.] Annales d’Endocrinologie, 11(3), 254–260. [Google Scholar] [PubMed] [PDF]
Bricout, V., & Wright, F. (2004). Update on nandrolone and norsteroids: how endogenous or xenobiotic are these substances? European Journal of Applied Physiology, 92(1–2), 1–12. [DOI:10.1007/s00421-004-1051-3]
Brodnitz, F. S. (1971). Hormones and the human voice. Bulletin of the New York Academy of Medicine, 47(2), 183–191. [Google Scholar] [PubMed] [PubMed Central]
Buvat, J., Maggi, M., Guay, A., & Torres, L. O. (2013). Testosterone Deficiency in Men: Systematic Review and Standard Operating Procedures for Diagnosis and Treatment. The Journal of Sexual Medicine, 10(1), 245–284. [DOI:10.1111/j.1743-6109.2012.02783.x]
Camerino, B., & Sala, G. (1960). Anabolic Steroids. In Jucker, E. (Ed.). Fortschritte der Arzneimittelforschung / Progress in Drug Research / Progrès des Recherches Pharmaceutiques, Volume 2 (pp. 71–134). Basel: Birkhäuser Basel. [DOI:10.1007/978-3-0348-7038-2_2]
Camerino, B., & Sciaky, R. (1975). Structure and effects of anabolic steroids. Pharmacology & Therapeutics. Part B: General and Systematic Pharmacology, 1(2), 233–275. [DOI:10.1016/0306-039x(75)90007-0]
Cappelletti, M., & Wallen, K. (2016). Increasing women’s sexual desire: The comparative effectiveness of estrogens and androgens. Hormones and Behavior, 78, 178–193. [DOI:10.1016/j.yhbeh.2015.11.003]
Čeponis, J., Wang, C., Swerdloff, R. S., & Liu, P. Y. (2017). Anabolic and Metabolic Effects of Testosterone and Other Androgens: Direct Effects and Role of Testosterone Metabolic Products. In Simoni, M., & Huhtaniemi, I. T. (Eds.). Endocrinology of the Testis and Male Reproduction (pp. 373–394). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-44441-3_11]
Cocchetti, C., Ristori, J., Romani, A., Maggi, M., & Fisher, A. D. (2020). Hormonal Treatment Strategies Tailored to Non-Binary Transgender Individuals. Journal of Clinical Medicine, 9(6), 1609. [DOI:10.3390/jcm9061609]
Cocchetti, C., Ristori, J., Mazzoli, F., Vignozzi, L., Maggi, M., & Fisher, A. D. (2020). Management of hypoactive sexual desire disorder in transgender women: a guide for clinicians. International journal of Impotence Research, 33(7), 703–709. [DOI:10.1038/s41443-021-00409-8]
Cooke, P. S., Nanjappa, M. K., Ko, C., Prins, G. S., & Hess, R. A. (2017). Estrogens in Male Physiology. Physiological Reviews, 97(3), 995–1043. [DOI:10.1152/physrev.00018.2016]
Crawford, B. A., Liu, P. Y., Kean, M. T., Bleasel, J. F., & Handelsman, D. J. (2003). Randomized Placebo-Controlled Trial of Androgen Effects on Muscle and Bone in Men Requiring Long-Term Systemic Glucocorticoid Treatment. The Journal of Clinical Endocrinology & Metabolism, 88(7), 3167–3176. [DOI:10.1210/jc.2002-021827]
Damsté, P. (1964). Virilization of the Voice due to Anabolic Stereoids. Folia Phoniatrica et Logopaedica, 16(1), 10–18. [DOI:10.1159/000262980]
Davis, S. (1999). The therapeutic use of androgens in women. The Journal of Steroid Biochemistry and Molecular Biology, 69(1–6), 177–184. [DOI:10.1016/s0960-0760(99)00054-0]
Dimitrakakis, C., Zhou, J., Wang, J., Belanger, A., LaBrie, F., Cheng, C., Powell, D., & Bondy, C. (2003). A physiologic role for testosterone in limiting estrogenic stimulation of the breast. Menopause, 10(4), 292–298. [DOI:10.1097/01.gme.0000055522.67459.89]
Doran, P. M., Riggs, B. L., Atkinson, E. J., & Khosla, S. (2001). Effects of Raloxifene, a Selective Estrogen Receptor Modulator, on Bone Turnover Markers and Serum Sex Steroid and Lipid Levels in Elderly Men. Journal of Bone and Mineral Research, 16(11), 2118–2125. [DOI:10.1359/jbmr.2001.16.11.2118]
Drugs.com. International > Nandrolone. Drugs.com. [URL]
Ebeling, P. R. (2010). Androgens and osteoporosis. Current Opinion in Endocrinology, Diabetes & Obesity, 17(3), 284–292. [DOI:10.1097/med.0b013e328339658c]
Feldman, E. B., Carter, A. C., Kossa, J. L., McCarrick, J. F., & Schwartz, H. L. (1960). Endocrinologic and Metabolic Effects of 17α-Methyl-19-nortestosterone in Women. The Journal of Clinical Endocrinology & Metabolism, 20(6), 842–857. [DOI:10.1210/jcem-20-6-842]
Flicker, L., Hopper, J. L., Larkins, R. G., Lichtenstein, M., Buirski, G., & Wark, J. D. (1997). Nandrolone decanoate and intranasal calcitonin as therapy in established osteoporosis. Osteoporosis International, 7(1), 29–35. [DOI:10.1007/bf01623456]
Friedl, K. E., Jones, R. E., Hannan, C. J., & Plymate, S. R. (1989). The Administration of Pharmacological Doses of Testosterone or 19-Nortestosterone to Normal Men is Not Associated with Increased Insulin Secretion or Impaired Glucose Tolerance. The Journal of Clinical Endocrinology & Metabolism, 68(5), 971–975. [DOI:10.1210/jcem-68-5-971]
Friedl, K. E., Dettori, J. R., Hannan, C. J., Patience, T. H., & Plymate, S. R. (1991). Comparison of the effects of high dose testosterone and 19-nortestosterone to a replacement dose of testosterone on strength and body composition in normal men. The Journal of Steroid Biochemistry and Molecular Biology, 40(4–6), 607–612, IN5–IN6. [DOI:10.1016/0960-0760(91)90283-b]
Frisoli, A., Chaves, P. H., Pinheiro, M. M., & Szejnfeld, V. L. (2005). The Effect of Nandrolone Decanoate on Bone Mineral Density, Muscle Mass, and Hemoglobin Levels in Elderly Women With Osteoporosis: A Double-Blind, Randomized, Placebo-Controlled Clinical Trial. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 60(5), 648–653. [DOI:10.1093/gerona/60.5.648]
Gerritsma, E. J., Brocaar, M. P., Hakkesteegt, M. M., & Birkenhäger, J. C. (1994). Virilization of the voice in post-menopausal women due to the anabolic steroid nandrolone decanoate (Decadurabolin). The effects of medication for one year. Clinical Otolaryngology, 19(1), 79–84. [DOI:10.1111/j.1365-2273.1994.tb01153.x]
Geusens, P. (1995). Nandrolone decanoate: Pharmacological properties and therapeutic use in osteoporosis. Clinical Rheumatology, 14(S3), 32–39. [DOI:10.1007/bf02210686]
Gold, J., Batterham, M., Rekers, H., Harms, M., Geurts, T., Helmyr, P., Silva de Mendonca, J., Falleiros Carvalho, L., Panos, G., Pinchera, A., Aiuti, F., Lee, C., Horban, A., Gatell, J., Phanuphak, P., Prasithsirikul, W., Gazzard, B., Bloch, M., & Danner, S. (2006). Effects of nandrolone decanoate compared with placebo or testosterone on HIV-associated wasting. HIV Medicine, 7(3), 146–155. [DOI:10.1111/j.1468-1293.2006.00358.x]
Gruber, C. J., Wieser, F., Gruber, I. M., Ferlitsch, K., Gruber, D. M., & Huber, J. C. (2002). Current concepts in aesthetic endocrinology. Gynecological Endocrinology, 16(6), 431–441. [DOI:10.1080/gye.16.6.431.441]
Hamdy, R. C., Moore, S. W., Whalen, K. E., & Landy, C. (1998). Nandrolone Decanoate for Men With Osteoporosis. American Journal of Therapeutics, 5(2), 89–96. [DOI:10.1097/00045391-199803000-00006]
Handelsman, D. J. (2016). Androgen Physiology, Pharmacology, and Abuse. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition, Volume 2 (pp. 2368–2393.e16). Philadelphia: Saunders/Elsevier. [Google Books] [DOI:10.1016/B978-0-323-18907-1.00138-4]
Hassager, C., Jensen, L., Johansen, J., Riis, B., Melkko, J., Pødenphant, J., Risteli, L., Christiansen, C., & Risteli, J. (1991). The carboxy-terminal propeptide of type I procollagen in serum as a marker of bone formation: The effect of nandrolone decanoate and female sex hormones. Metabolism, 40(2), 205–208. [DOI:10.1016/0026-0495(91)90176-w]
Hickson, R. C., Ball, K. L., & Falduto, M. T. (1989). Adverse Effects of Anabolic Steroids. Medical Toxicology and Adverse Drug Experience, 4(4), 254–271. [DOI:10.1007/bf03259912]
Hiort, O. (2013). The differential role of androgens in early human sex development. BMC Medicine, 11(1), 152. [DOI:10.1186/1741-7015-11-152]
Hirshburg, J. M., Kelsey, P. A., Therrien, C. A., Gavino, A. C., & Reichenberg, J. S. (2016). Adverse Effects and Safety of 5-alpha Reductase Inhibitors (Finasteride, Dutasteride): A Systematic Review. The Journal of Clinical and Aesthetic Dermatology, 9(7), 56–62. [Google Scholar] [PubMed] [PubMed Central]
Hobbs, C. J., Plymate, S. R., Rosen, C. J., & Adler, R. A. (1993). Testosterone administration increases insulin-like growth factor-I levels in normal men. The Journal of Clinical Endocrinology & Metabolism, 77(3), 776–779. [DOI:10.1210/jcem.77.3.7690364]
Hobbs, C. J., Jones, R. E., & Plymate, S. R. (1996). Nandrolone, a 19-nortestosterone, enhances insulin-independent glucose uptake in normal men. The Journal of Clinical Endocrinology & Metabolism, 81(4), 1582–1585. [DOI:10.1210/jcem.81.4.8636371]
Holterhus, P., Piefke, S., & Hiort, O. (2002). Anabolic steroids, testosterone-precursors and virilizing androgens induce distinct activation profiles of androgen responsive promoter constructs. The Journal of Steroid Biochemistry and Molecular Biology, 82(4–5), 269–275. [DOI:10.1016/s0960-0760(02)00220-0]
Huang, G., & Basaria, S. (2017). The Case for Androgens in Menopausal Women: When and How? In Pal, L., & Sayegh, R. A. (Eds.). Essentials of Menopause Management: A Case-Based Approach (pp. 173–196). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-42451-4_10]
Imperato-McGinley, J., & Peterson, R. E. (1976). Male pseudohermaphroditism: The complexities of male phenotypic development. The American Journal of Medicine, 61(2), 251–272. [DOI:10.1016/0002-9343(76)90175-3]
Imperato-McGinley, J., & Zhu, Y. (2002). Androgens and male physiology the syndrome of 5α-reductase-2 deficiency. Molecular and Cellular Endocrinology, 198(1–2), 51–59. [DOI:10.1016/s0303-7207(02)00368-4]
Irwig, M. S. (2015). Safety concerns regarding 5α reductase inhibitors for the treatment of androgenetic alopecia. Current Opinion in Endocrinology, Diabetes & Obesity, 22(3), 248–253. [DOI:10.1097/med.0000000000000158]
Iuliano, S., Izzo, G., Zagari, M. C., Vergine, M., Brunetti, F. S., Brunetti, A., Di Luigi, L., & Aversa, A. (2021). Endocrine management of transgender adults: a clinical approach. Sexes, 2(1), 104–118. [DOI:10.3390/sexes2010009]
Jänne, O., Hemminki, S., Isomaa, V., Kokko, E., Torkkeli, H., Torkkeli, T., & Vierikko, P. (1978). Progestational Activity of Natural and Synthetic Androgens. International Journal of Andrology, 1(s2a) [Endocrine Approach to Male Contraception: Transactions of the Fifth Annual Workshop on the Testis, Held at Geilo, Norway on April 2-5, 1978], 162–174. [DOI:10.1111/j.1365-2605.1978.tb00015.x]
Kicman, A. T. (2008). Pharmacology of anabolic steroids. British Journal of Pharmacology, 154(3), 502–521. [DOI:10.1038/bjp.2008.165]
Kirschvink, J. F. (1963). Studies of Anabolic Steroids. American Journal of Diseases of Children, 106(4), 368–368. [DOI:10.1001/archpedi.1963.02080050370005]
Knuth, U. A., Behre, H., Belkien, L., Bents, H., & Nieschlag, E. (1985). Clinical trial of 19-nortestosterone-hexoxyphenylpropionate (Anadur) for male fertility regulation. Fertility and Sterility, 44(6), 814–821. [DOI:10.1016/s0015-0282(16)49043-6]
Knuth, U. A., Behre, H., Belkien, L., Bents, H., & Nieschlag, E. (1986). 19-Nortestosterone for male fertility regulation. In Zatuchni, G. I., Goldsmith, A., Spieler, J. M., & Sciarra, J. J. (Eds.). Male Contraception: Advances and Future Prospects: Proceedings of an International Workshop on Male Contraception, Advances and Future Prospects, May 28-31, 1985, Geneva, Switzerland (pp. 320–328). Philadelphia: Harper and Row. [Google Scholar] [Google Books] [WorldCat] [PDF]
Knuth, U. A., Yeung, C., & Nieschlag, E. (1989). Combination of 19-nortestosterone-hexyloxyphenylpropionate (Anadur) and depot-medroxyprogesterone-acetate (Clinovir) for male contraception. Fertility and Sterility, 51(6), 1011–1018. [DOI:10.1016/s0015-0282(16)60735-5]
Kuhl, H., & Wiegratz, I. (2017). Das Post-Finasterid-Syndrom. [Post Finasteride Syndrome.] Gynäkologische Endokrinologie, 15(2), 153–163. [DOI:10.1007/s10304-017-0126-2]
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Häggblad, J., Nilsson, S., & Gustafsson, J. (1997). Comparison of the Ligand Binding Specificity and Transcript Tissue Distribution of Estrogen Receptors α and β. Endocrinology, 138(3), 863–870. [DOI:10.1210/endo.138.3.4979]
Kumar, N., Crozat, A., Li, F., Catterall, J., Bardin, C., & Sundaram, K. (1999). 7α-methyl-19-nortestosterone, a synthetic androgen with high potency: structure-activity comparisons with other androgens. The Journal of Steroid Biochemistry and Molecular Biology, 71(5–6), 213–222. [DOI:10.1016/s0960-0760(99)00143-0]
Lauritzen, C. (1984). Pre- and Postmenopause. In Platt, D. (Ed.). Geriatrics 3: Gynecology · Orthopaedics · Anesthesiology · Surgery · Otorhinolaryngology · Ophthalmology · Dermatology (pp. 2–45). Berlin/Heidelberg: Springer Berlin Heidelberg. [DOI:10.1007/978-3-642-68976-5_1]
Lemmey, A. B., Elamanchi, S. R., Marcora, S. M., Casanova, F., & Maddison, P. J. (2013). Efficacy of nandrolone decanoate in treating rheumatoid cachexia in male rheumatoid arthritis patients. In Matsuno, H. (Ed.). Innovative Rheumatology (pp. 271–285). Rijeka, Croatia: InTech. [DOI:10.5772/53236]
Liu, P. Y., & Handelsman, D. J. (1998). Androgen therapy in non-gonadal disease. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 473–512). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_17]
Marks, L. S. (2004). 5α-reductase: history and clinical importance. Reviews in Urology, 6(Suppl 9), S11–S21. [Google Scholar] [PubMed] [PubMed Central]
Mella, J. M., Perret, M. C., Manzotti, M., Catalano, H. N., & Guyatt, G. (2010). Efficacy and Safety of Finasteride Therapy for Androgenetic Alopecia. Archives of Dermatology, 146(10), 1141–1150. [DOI:10.1001/archdermatol.2010.256]
Need, A. (1989). Cross-over study of fat-corrected forearm mineral content during nandrolone decanoate therapy for osteoporosis. Bone, 10(1), 3–6. [DOI:10.1016/8756-3282(89)90139-7]
Neff, M. S., Goldberg, J., Slifkin, R. F., Eiser, A. R., Calamia, V., Kaplan, M., Baez, A., Gupta, S., & Mattoo, N. (1981). A Comparison of Androgens for Anemia in Patients on Hemodialysis. New England Journal of Medicine, 304(15), 871–875. [DOI:10.1056/nejm198104093041504]
Nieschlag, E. (2010). Clinical trials in male hormonal contraception. Contraception, 82(5), 457–470. [DOI:10.1016/j.contraception.2010.03.020]
Nieschlag, E., & Behre, H. M. (2012). The essential role of testosterone in hormonal male contraception. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Testosterone: Action · Deficiency · Substitution, 4th Edition (pp. 470–493). Cambridge/New York: Cambridge University Press. [DOI:10.1017/cbo9781139003353.023]
Nieschlag, E., Kumar, N., & Sitruk-Ware, R. (2013). 7α-Methyl-19-nortestosterone (MENTR): the Population Council’s contribution to research on male contraception and treatment of hypogonadism. Contraception, 87(3), 288–295. [DOI:10.1016/j.contraception.2012.08.036]
Pan, M. M., & Kovac, J. R. (2016). Beyond testosterone cypionate: evidence behind the use of nandrolone in male health and wellness. Translational Andrology and Urology, 5(2), 213–219. [DOI:10.21037/tau.2016.03.03]
Passeri, M., Pedrazzoni, M., Pioli, G., Butturini, L., Ruys, A., & Cortenraad, M. (1993). Effects of nandrolone decanoate on bone mass in established osteoporosis. Maturitas, 17(3), 211–219. [DOI:10.1016/0378-5122(93)90049-n]
Peters, A. A., Ingman, W. V., Tilley, W. D., & Butler, L. M. (2011). Differential Effects of Exogenous Androgen and an Androgen Receptor Antagonist in the Peri- and Postpubertal Murine Mammary Gland. Endocrinology, 152(10), 3728–3737. [DOI:10.1210/en.2011-1133]
Peterson, R. E., Imperato-McGinley, J., Gautier, T., & Sturla, E. (1977). Male pseudohermaphroditism due to steroid 5α-reductase deficiency. The American Journal of Medicine, 62(2), 170–191. [DOI:10.1016/0002-9343(77)90313-8]
Propecia Finasteride Label. U.S. Food and Drug Administration. [URL] [PDF]
Prunty, F. T., Brooks, R. V., Clayton, B. E., & McSwiney, R. R. (1958). Some Effects of 17α-Ethyl-19-Nortestosterone in Man. Proceedings of the Royal Society of Medicine, 51(7), 557–558. [Google Scholar] [PubMed] [PubMed Central]
Roche, A., Towns, J. W., & Wettenhall, H. (1963). Influence of norethandrolone on the stature of short children. The Journal of Pediatrics, 63(5), 967–976. [DOI:10.1016/s0022-3476(63)80228-0]
Russell, N., & Grossmann, M. (2019). MECHANISMS IN ENDOCRINOLOGY: Estradiol as a male hormone. European Journal of Endocrinology, 181(1), R23–R43. [DOI:10.1530/eje-18-1000]
Sardar, P., Jha, A., Roy, D., Majumdar, U., Guha, P., Roy, S., Banerjee, R., Banerjee, A. K., & Bandyopadhyay, D. (2010). Therapeutic Effects of Nandrolone and Testosterone in Adult Male HIV Patients With AIDS Wasting Syndrome (AWS): A Randomized, Double-Blind, Placebo-Controlled Trial. HIV Clinical Trials, 11(4), 220–229. [DOI:10.1310/hct1104-220]
Sartorius, G. A., Ly, L. P., & Handelsman, D. J. (2014). Male Sexual Function Can Be Maintained Without Aromatization: Randomized Placebo‐Controlled Trial of Dihydrotestosterone (DHT) in Healthy, Older Men for 24 Months. The Journal of Sexual Medicine, 11(10), 2562–2570. [DOI:10.1111/jsm.12550]
Sas, T., Gault, E., Zeger Bardsley, M., Menke, L., Freriks, K., Perry, R., Otten, B., de Muinck Keizer-Schrama, S., Timmers, H., Wit, J., Ross, J., & Donaldson, M. (2014). Safety and Efficacy of Oxandrolone in Growth Hormone-Treated Girls with Turner Syndrome: Evidence from Recent Studies and Recommendations for Use. Hormone Research in Paediatrics, 81(5), 289–297. [DOI:10.1159/000358195]
Schürmeyer, T., Belkien, L., Knuth, U., & Nieschlag, E. (1984). Reversible azoospermia induced by the anabolic steroid 19-nortestosterone. The Lancet, 323(8374), 417–420. [DOI:10.1016/s0140-6736(84)91752-5]
Simpson, E. R., & Jones, M. E. (2007). Of Mice and Men: The Many Guises of Estrogens. In Korach, K. S., & Wintermantel, T. (Eds.). Tissue-Specific Estrogen Action: Novel Mechanisms, Novel Ligands, Novel Therapies (Ernst Schering Foundation Symposium Proceedings, Volume 2006/1) (pp. 45–68). Berlin/Heidelberg: Springer. [DOI:10.1007/2789_2006_016]
Skroza, N., Tolino, E., Mambrin, A., Zuber, S., Balduzzi, V., Marchesiello, A., Bernardini, N., Proietti, I., & Potenza, C. (2018). Adult Acne Versus Adolescent Acne: A Retrospective Study of 1,167 Patients. The Journal of Clinical and Aesthetic Dermatology, 11(1), 21–25. [Google Scholar] [PubMed] [PubMed Central] [URL]
Styne, D. M., & Grumbach, M. M. (2016). Physiology and Disorders of Puberty. In Melmed, S., Polonsky, K. S., Larsen, P. R., Kronenberg, & H. M. (Eds.). Williams Textbook of Endocrinology, 13th Edition (pp. 1074–1218). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-29738-7.00025-3]
Sundaram, K., & Kumar, N. (2000). 7alpha-Methyl-19-nortestosterone (MENT): the optimal androgen for male contraception and replacement therapy. International Journal of Andrology, 23(S2), 13–15. [DOI:10.1046/j.1365-2605.2000.00004.x]
Sundaram, K., Kumar, N., & Bardin, C. W. (1994). 7α-Methyl-19-nortestosterone: An Ideal Androgen for Replacement Therapy. In Bardin, W. C. (Ed.). Proceedings of the 1992 Laurentian Hormone Conference (Recent Progress in Hormone Research, Volume 49) (pp. 373–376). San Diego: Academic Press. [DOI:10.1016/b978-0-12-571149-4.50027-1]
Sundaram, K., Kumar, N., Monder, C., & Bardin, C. (1995). Different patterns of metabolism determine the relative anabolic activity of 19-norandrogens. The Journal of Steroid Biochemistry and Molecular Biology, 53(1–6), 253–257. [DOI:10.1016/0960-0760(95)00056-6]
Suvisaari, J. (2000). 7α-Methyl-19-Nortestosterone (MENT): Pharmacokinetics and Antigonadotropic Effects in Men. (Doctoral dissertation, University of Helsinki.) Helsinki: University of Helsinki. [Google Scholar] [URL] [PDF]
Thirumalai, A., Ceponis, J., Amory, J. K., Swerdloff, R., Surampudi, V., Liu, P. Y., Bremner, W. J., Harvey, E., Blithe, D. L., Lee, M. S., Hull, L., Wang, C., & Page, S. T. (2018). Effects of 28 Days of Oral Dimethandrolone Undecanoate in Healthy Men: A Prototype Male Pill. The Journal of Clinical Endocrinology & Metabolism, 104(2), 423–432. [DOI:10.1210/jc.2018-01452]
Tóth, M. (2009). Mioanabolikus Szteroidok és Szelektív Androgénreceptormodulátorok: Hatásmechanizmus és Terápiás Perspektívák. [Myoanabolic Steroids and Selective Androgen Receptor Modulators: Mechanism of Action and Perspectives. Orvosi Hetilap, 150(45), 2051–2059. [DOI:10.1556/oh.2009.28739] [Translation]
Tóth, M., & Zakár, T. (1986). Classification of Anabolic Steroids Using the Method of Competitive Metabolism. Experimental and Clinical Endocrinology & Diabetes, 87(2), 125–132. [DOI:10.1055/s-0029-1210533]
Trost, L., Saitz, T. R., & Hellstrom, W. J. (2013). Side Effects of 5‐Alpha Reductase Inhibitors: A Comprehensive Review. Sexual Medicine Reviews, 1(1), 24–41. [DOI:10.1002/smrj.3]
van der Vies, J. (1985). Implications of basic pharmacology in the therapy with esters of nandrolone. Acta Endocrinologica, 110(3 Suppl a), S38–S44. [DOI:10.1530/acta.0.109s0038]
van Zuuren, E. J., Fedorowicz, Z., Carter, B., & Pandis, N. (2015). Interventions for hirsutism (excluding laser and photoepilation therapy alone). Cochrane Database of Systematic Reviews, 2015, CD010334. [DOI:10.1002/14651858.cd010334.pub2]
Vetri, M., Cataldi, A., Naselli, A., & Vetri, A. (2021). Transsexualism Ethiology and Medical Management: Between Scientific Evidence and Personal Experiences. Preprints, 2021030172. [DOI:10.20944/preprints202103.0172.v1]
Wibowo, E., Schellhammer, P., & Wassersug, R. J. (2011). Role of Estrogen in Normal Male Function: Clinical Implications for Patients With Prostate Cancer on Androgen Deprivation Therapy. Journal of Urology, 185(1), 17–23. [DOI:10.1016/j.juro.2010.08.094]
Wibowo, E., & Wassersug, R. J. (2013). The effect of estrogen on the sexual interest of castrated males: Implications to prostate cancer patients on androgen-deprivation therapy. Critical Reviews in Oncology/Hematology, 87(3), 224–238. [DOI:10.1016/j.critrevonc.2013.01.006]
Winters, S. J. (1990). Androgens: Endocrine Physiology and Pharmacology. In Lin, G. C., & Erinoff, L. (Eds.). Anabolic Steroid Abuse (National Institute on Drug Abuse Research Monograph Series, Volume 102) (pp. 113–130). Rockville, Maryland: National Institute on Drug Abuse/U.S. Department of Health and Human Services. [Google Scholar] [PubMed] [Google Books] [PDF]
World Health Organization & Task Force on Methods for the Regulation of Male Fertility. (1993). Comparison of two androgens plus depot-medroxyprogesterone acetate for suppression to azoospermia in Indonesian men. Fertility and Sterility, 60(6), 1062–1068. [DOI:10.1016/s0015-0282(16)56411-5]
Wu, C., & Kovac, J. R. (2016). Novel Uses for the Anabolic Androgenic Steroids Nandrolone and Oxandrolone in the Management of Male Health. Current Urology Reports, 17(10), 72. [DOI:10.1007/s11934-016-0629-8]
Wu, S., Yuen, F., Swerdloff, R. S., Pak, Y., Thirumalai, A., Liu, P. Y., Amory, J. K., Bai, F., Hull, L., Blithe, D. L., Anawalt, B. D., Parman, T., Kim, K., Lee, M. S., Bremner, W. J., Page, S. T., & Wang, C. (2018). Safety and Pharmacokinetics of Single-Dose Novel Oral Androgen 11β-Methyl-19-Nortestosterone-17β-Dodecylcarbonate in Men. The Journal of Clinical Endocrinology & Metabolism, 104(3), 629–638. [DOI:10.1210/jc.2018-01528]
Yuen, F., Thirumalai, A., Pham, C., Swerdloff, R. S., Anawalt, B. D., Liu, P. Y., Amory, J. K., Bremner, W. J., Dart, C., Wu, H., Hull, L., Blithe, D. L., Long, J., Wang, C., & Page, S. T. (2020). Daily Oral Administration of the Novel Androgen 11β-MNTDC Markedly Suppresses Serum Gonadotropins in Healthy Men. The Journal of Clinical Endocrinology & Metabolism, 105(3), e835–e847. [DOI:10.1210/clinem/dgaa032]
\ No newline at end of file
+Nandrolone as a Potential Alternative Androgen with Reduced Androgenic Side Effects for Transfeminine and Transmasculine People - Transfeminine Science
Nandrolone as a Potential Alternative Androgen with Reduced Androgenic Side Effects for Transfeminine and Transmasculine People
By Aly | First published March 20, 2020 | Last modified March 23, 2023
Abstract / TL;DR
Nandrolone, or 19-nortestosterone, is a unique androgen and anabolic steroid which is used in the form of injectable ester prodrugs like nandrolone decanoate. It is closely related to testosterone structurally but has a number of important pharmacological differences in comparison. These differences include inactivation by 5α-reductase rather than potentiation, less or no conversion into estradiol, and much greater progestogenic activity. The inactivation of nandrolone by 5α-reductase results in nandrolone theoretically having much lower androgenic strength than testosterone in skin, hair follicles, and the prostate, among other tissues. Consequently, nandrolone may have less propensity for oily skin, acne, facial/body hair growth, scalp hair loss, and prostate problems than testosterone. On the other hand, nandrolone is theoretically expected to have full androgenic efficacy in most other tissues, including bone, fat, and muscle (and hence of masculine skeletal, fat, and muscular changes). This is also likely to be the case when it comes to voice, bottom growth, and sexual function. The reduced androgenic impact of nandrolone in skin and hair follicles relative to testosterone make it a favorable potential alternative option for androgen therapy both for transfeminine people and for some transmasculine people.
Introduction
Nandrolone, also known chemically as 19-nortestosterone (19-NT), is an androgen and anabolic steroid (AAS) similarly to testosterone (T). It is used as a medication by intramuscular or subcutaneous injection in the form of longer-acting esters like nandrolone decanoate (ND; Deca-Durabolin) and nandrolone phenylpropionate (NPP; Durabolin), which are converted into nandrolone in the body. This is analogous to longer-acting testosterone esters like testosterone enanthate (TE) and testosterone undecanoate (TU) and their conversion in the body into testosterone. Nandrolone is used in medicine for a variety of indications that benefit from anabolic effects in tissues like muscle, bone, and kidneys. Examples include wasting syndromes, osteoporosis, kidney disease, and anemia. It is used not only in men but also in women, for instance to prevent and treat postmenopausal osteoporosis in women who can’t tolerate estrogens or in whom estrogens can’t be taken. Nandrolone has additionally been advocated for use in androgen replacement therapy for women (Davis, 1999).
Nandrolone is closely related to testosterone in terms of chemical structure, pharmacodynamic actions, and disposition in the body. At the same time however, nandrolone has a key difference from testosterone that makes it a very favorable alternative option for use as an androgen in transgender hormone therapy. This is the case not only for transfeminine people but also for transmasculine people. This property of nandrolone is that it is inactivated by 5α-reductase in tissues that express this enzyme. This is in contrast to testosterone, which is potentiated in such tissues via conversion into dihydrotestosterone (DHT). Because of this difference, nandrolone has normal androgenic effects in most of the body but a greatly reduced potential for androgenic effects in skin and hair follicles relative to testosterone. As a result, nandrolone has a variably reduced potential for androgenic skin and hair side effects such as oily skin, acne, facial and body hair growth, and scalp hair loss.
Because of its favorable profile relative to testosterone, nandrolone has been advocated for use in androgen replacement therapy in cisgender men (Wu & Kovac, 2016; Pan & Kovac, 2016). It has also been studied for use in male hormonal contraception as an alternative and replacement for testosterone (Knuth et al., 1986; Nieschlag, 2010; Nieschlag & Behre, 2012). Nandrolone is advantageous for cisgender men not only due to preservation of scalp hair but also due to its lower stimulation of the prostate gland. As a result of its more favorable profile in terms of skin and hair follicles, nandrolone is an underappreciated alternative possibility relative to testosterone for use not only in cisgender people but also in transfeminine and transmasculine people.
Chemistry of Nandrolone
Nandrolone is chemically known as 19-nortestosterone. It is very close to testosterone in terms of chemical structure, the only difference between the two compounds being that the C19 methyl group of testosterone has been removed in the case of nandrolone. Nandrolone is not a synthetic compound; it occurs naturally in the human body in trace amounts as an intermediate in the conversion of testosterone to estradiol by the enzyme aromatase (Bricout & White, 2004). As such, although nandrolone couldn’t be said to be “bioidentical”, it’s quite similar to testosterone.
Figure: Chemical structures of testosterone and nandrolone (19-nortestosterone). The structural difference between testosterone and nandrolone is at the C19 position and is highlighted by the circled areas.
Nandrolone is an agonist of the androgen receptor (AR) similarly to testosterone. It shows higher affinity for the AR than does testosterone and has almost no binding affinity for sex hormone-binding globulin (SHBG) however. For these reasons, nandrolone has about 2.5- to 5-fold higher potency than testosterone in rats on measures of general AR agonistic strength, such as stimulation of muscle growth (Sundaram et al., 1995; Tóth & Zakár, 1986; Wiki; Winters, 1976). It is likewise a more potent AAS than testosterone in humans, and is used medically at lower doses in comparison (Wiki).
Testosterone is a substrate for 5α-reductase in tissues that express this enzyme like skin, hair follicles, prostate gland, and seminal vesicles, among others. It is converted by 5α-reductase into dihydrotestosterone (DHT), an androgen with much higher AR affinity and AR agonistic potency than that of testosterone (Wiki). Consequently, it is estimated that the effects of testosterone are potentiated by 2- to 3-fold via conversion into DHT in tissues that express 5α-reductase (Sundaram et al., 1995). The role of DHT in the effects of testosterone is demonstrated by a rare intersex condition known as 5α-reductase type 2 deficiency, as well as by 5α-reductase inhibitors (5α-RIs) like finasteride and dutasteride (Wiki; Wiki). Men with 5α-reductase type 2 deficiency are reported to have reduced facial hair, a female-like pattern of body hair (with terminal hair largely restricted to the underarms and lower pubic triangle), and no scalp hair recession in the temples nor scalp hair loss in general. Conversely, other aspects of male secondary sexual development, like voice deepening, muscle growth, bone changes, and pubertal penile enlargement, are all normal in the condition. In addition, no feminization or gynecomastia occurs. Hence, adults with 5α-reductase type 2 deficiency have a masculine appearance (Imperato-McGinley & Peterson, 1976; Peterson et al., 1977; Photos). In published photographs of individuals with the condition however, features like lack of body hair and rounded feminine hairlines can be noted.
5α-Reductase inhibitors have similar effects relative to the profile of 5α-reductase type 2 deficiency. For this reason, they are used in the treatment and prevention of scalp hair loss in men and off-label for hirsutism (excessive facial/body hair growth) in women. They greatly slow the rate of scalp hair loss over time in men. For example, a large 5-year randomized controlled trial showed that in men with diagnosed scalp hair loss, 100% of those who received placebo had significant further scalp hair loss whereas only 35% of those who received finasteride had further scalp hair loss (Propecia label). Low-quality evidence suggests that finasteride has similar effectiveness to antiandrogens like spironolactone and flutamide in the treatment of hirsutism in women (Barrionuevo et al., 2018; van Zuuren et al., 2015). Dutasteride has similar or greater effectiveness than finasteride for scalp hair loss in men (Arif et al., 2017), whereas it hasn’t yet been studied in the treatment of hirsutism (Wiki).
5α-Reductase is the key to the favorable differences between testosterone and nandrolone. Both testosterone and nandrolone are substrates for 5α-reductase and have similar affinity for this enzyme (Tóth & Zakár, 1986). But whereas testosterone is potentiated via conversion into DHT in tissues that express 5α-reductase, nandrolone is inactivated by 5α-reductase in these tissues (Kicman, 2008; Tóth, 2009). This is because the 5α-reduced metabolite of nandrolone, 5α-dihydronandrolone (DHN), is a weaker AR agonist with much lower affinity for the AR than nandrolone (Tóth & Zakár, 1986; Kumar et al., 1999). The following table shows AR and SHBG relative binding affinities (RBA) of testosterone, nandrolone, trestolone, and their 5α-reduced forms (Kumar et al., 1999):
Steroid
AR RBA
SHBG RBA
Testosterone
100%
100%
5α-Dihydrotestosterone
290%
340%
Nandrolone
230%
5%
5α-Dihydronandrolone
10%
5%
Trestolone
380%
6%
5α-Dihydrotrestolone
30%
2%
In rodents, although nandrolone has about 3-fold higher anabolic or muscle-stimulating potency than testosterone, its androgenic potency in the prostate gland and seminal vesicles is only about 20 to 40% of that of testosterone (Sundaram et al., 1995; Tóth & Zakár, 1986; Wiki). This works out to an approximate 11:1 dissociation between the effects of nandrolone in tissues with little or no 5α-reductase (muscle) versus with high 5α-reductase (prostate gland, seminal vesicles) in rodents (Wiki). The exact ratio of potency between these tissues in humans is unknown, and extrapolation from rodents should be cautioned against. In any case, an analogous dissociation of some level can be anticipated. As such, nandrolone can be expected to have high AR agonistic strength in most of the body, where 5α-reductase has minimal expression (e.g., fat, muscle, bone), but much lower potency in tissues with high 5α-reductase expression, like skin, hair follicles, the prostate gland, and the seminal vesicles.
It’s important to be clear that nandrolone is not simply akin to taking testosterone with a 5α-reductase inhibitor like dutasteride to block 5α-reductase. This is because nandrolone isn’t merely not potentiated by 5α-reductase, it’s weakened by 5α-reductase. For this reason, 5α-reductase inhibitors actually increase the androgenic strength of nandrolone in tissues that express 5α-reductase like the prostate gland in rodents (Sundaram et al., 1995). (And, for this reason, nandrolone should not be taken with a 5α-reductase inhibitor, in contrast to testosterone.) In other words, nandrolone goes beyond testosterone plus a 5α-reductase inhibitor in terms of the dissociation of its potency between tissues that express 5α-reductase and tissues that do not. As a result, nandrolone has much lower androgenic strength in such tissues than does the combination of testosterone plus a 5α-reductase inhibitor. Nandrolone also appears to have lower strength in tissues that express 5α-reductase than do most other AAS, for instance synthetic DHT derivatives like oxandrolone and stanozolol (Tóth & Zakár, 1986). This is because these AAS are not substrates for 5α-reductase and hence are neither potentiated nor weakened by 5α-reductase, similarly to testosterone with a 5α-reductase inhibitor. The inactivation of nandrolone by 5α-reductase makes it very unique among AAS and AR agonists. Only nandrolone and a few of its derivatives like normethandrone and norethandrolone have this very special property.
Testosterone and nandrolone may also be non-identical in their androgenic actions and effects for reasons other than metabolism. This is because different AR agonists including strong androgens like testosterone and DHT, weak androgens like dehydroepiandrosterone (DHEA) and androstenedione (A4), and predominantly anabolic androgens like nandrolone, oxandrolone, and stanozolol, have been shown to produce differential AR-mediated changes in gene expression (Holterhus, Piefke, & Hiort, 2002; Kicman, 2008; Hiort, 2013). However, nandrolone was more similar in its profile to testosterone than the synthetic AAS. In any case, the clinical implications of these differences in relation to therapeutic use of testosterone versus nandrolone, if any, are unknown.
In relation to the fact that nandrolone is an intermediate in the aromatase-mediated reaction that converts testosterone into estradiol, both testosterone and nandrolone are transformed into estradiol, and hence estradiol is their specific estrogen metabolite. Paradoxically however, whereas it has the same potential for 5α-reduction as testosterone, nandrolone as a medication is aromatized into estradiol virtually not at all (Handelsman, 2016). In accordance, nandrolone suppresses estradiol levels in men and results in low estradiol levels even at very high doses (Friedl, 1991; Hobbs et al., 1993; Hobbs, Jones, & Plymate, 1996; Behre et al., 2001). The following table gives an example of estradiol (E2) levels in men with high-dose testosterone versus high-dose nandrolone (Friedl, 1991):
Drug and dose
E2 before
E2 after
Change
Testosterone enanthate 300 mg/week i.m.
32 ± 4.6 pg/mL
79 ± 9.3 pg/mL
+147%
Nandrolone decanoate 300 mg/week i.m.
40 ± 6.0 pg/mL
14 ± 1.6 pg/mL
–65%
Testosterone and nandrolone themselves don’t have significant affinity for the estrogen receptors nor intrinsic estrogenic activity outside of aromatization to estradiol (Kuiper et al., 1997; Table). Although androgens also help to maintain bone mineral density and AAS like nandrolone are used to treat osteoporosis in postmenopausal women (Geusens, 1995; Ebeling, 2010), estrogens are comparatively much more important for maintenance of bone mineral density and prevention of osteoporosis in both men and women (Wiki). Studies of high-dose nandrolone and androstanolone (DHT) have shown that these non-aromatizable AAS are not adequate for maintenance of bone mineral density in men, whereas the aromatizable testosterone is effective for such purposes (Ronald et al., 1998; Crawford et al., 2003; Lemmey et al., 2013; Sartorius, Ly, & Handelsman, 2014; Čeponis et al., 2017). Non-aromatizable AAS are likewise inferior to estrogen and estrogen–progestogen therapy for maintaining bone mineral density in postmenopausal women (Lauritzen, 1984 [Graph]; Hassager et al., 1991).
Because of its minimal conversion into estradiol, nandrolone should be used as an androgen in transmasculine people only in combination with low-dose estradiol to replace the lost estradiol that would otherwise be provided by testosterone. Estradiol levels in men are on average around 30 pg/mL, and estradiol levels of at least 20 to 30 pg/mL appear to be necessary for maintenance of bone density in men (Doran et al., 2001). A low-dose transdermal estradiol patch, for instance 14 to 25 μg/day, is likely to be adequate for achieving such levels. Alternatively, another option like transdermal estradiol gel (e.g., 0.75–1 mg/day) or low-dose oral estradiol (e.g., 0.5–1 mg/day) may be used instead.
Nandrolone likewise has progestogenic activity. However, the progestogenic activity of nandrolone is greatly enhanced relative to that of testosterone. The affinity of nandrolone for the PR is 5- to 20-fold higher than that of testosterone and its progestogenic potency is correspondingly improved (Wiki; Wiki; Bardin & Janne, 1986). Despite this however, the progestogenic potency of nandrolone is still relatively low. For instance, its affinity for the PR is only about 10 to 20% of that of progesterone and the nandrolone-derived progestin norethisterone (also known as 17α-ethynyl-19-nortestosterone). At the same time however, nandrolone is used at relatively large doses compared to norethisterone. This may help to compensate for its lower progestogenic potency and may allow for significant progestogenic effects at typical clinical doses (Camerino & Sciaky, 1975).
The following table shows human and rabbit PR RBAs and relative progestogenic activity (as measured by in-vivo induction of uteroglobin synthesis in rabbit uterus) of progesterone and several AAS including testosterone and nandrolone (Jänne et al., 1978; Bardin & Janne, 1986):
Steroid
hPR RBA
rbPR RBA
Progestogenic activity (rabbit)
Progesterone
100%
100%
100%
Testosterone
1.6%
1.6%
0.9%
Dihydrotestosterone
3.3%
3.3%
1.9%
Nandrolone
8.9%
12%
5.2%
Methyltestosterone
4.6%
3.4%
0.6%
Norethandrolone
130%
70%
115%
The development of nandrolone was an intermediate step in the eventual development of the 19-nortestosterone group of progestins, which are widely used in hormonal birth control. These include progestins like norethisterone and levonorgestrel, among many others.
Advantages of Nandrolone Over Testosterone
Because of its substantially lower androgenic strength in tissues that have high 5α-reductase expression relative to tissues that have minimal or no 5α-reductase expression, nandrolone is likely to have normal androgenic effects in muscle, bone, fat, and most other tissues but much weaker androgenic strength in skin and hair follicles than equivalent doses of testosterone. As a result of this, although nandrolone has high capacity for producing most aspects of masculinization, it has a lower potential for many of the undesirable androgenic skin and hair side effects of testosterone, including oily skin, acne, facial and body hair growth, and scalp hair loss. This may allow for unique advantages of nandrolone over testosterone in transfeminine and transmasculine hormone therapy.
The benefits of nandrolone are expected to partially differ in transfeminine people relative to transmasculine people however. This is due to the different doses of nandrolone used in these individuals, namely low, female replacement doses in transfeminine people and full, male replacement doses in transmasculine people. As an example, transmasculine people taking full male androgen replacement doses should probably expect no less oily skin or acne with nandrolone than with testosterone. This is because very low levels of testosterone are necessary for such effects, as evidenced by the fact that acne prevalence and severity is similar in females compared to males (Skroza et al., 2018) despite the fact that testosterone levels are 20-fold lower in women than in men (Styne & Grumbach, 2016). Conversely, transfeminine people taking nandrolone at doses equivalent to the effect of testosterone levels in the normal female range may experience less or possibly no oily skin or acne with nandrolone compared to use of testosterone instead. This is due to the equivalent testosterone levels being far closer to the biological threshold for manifestation of such effects.
Nandrolone can be anticipated to have far less potential for scalp hair loss relative to testosterone in transmasculine people (Wu & Kovac, 2016; Pan & Kovac, 2016). This is related to the lack of scalp hair loss in men with 5α-reductase type 2 deficiency and the high effectiveness of 5α-reductase inhibitors for scalp hair loss. However, nandrolone is theoretically even better than testosterone plus a 5α-reductase inhibitor when it comes to preservation of scalp hair. This is because again, nandrolone is not simply not potentiated, but is inactivated in tissues that express 5α-reductase. Nandrolone replacement of testosterone represents a highly promising treatment for scalp hair loss in men that offers potentially superior effectiveness to 5α-reductase inhibitors. Moreover, since nandrolone isn’t an inhibitor of 5α-reductase, it doesn’t interfere with 5α-reduced neurosteroid synthesis. Hence, nandrolone doesn’t have the possible psychiatric side effects that have been associated with 5α-reductase inhibitors, for instance depression (Irwig, 2015; Kuhl & Wiegratz, 2017; Wiki).
On the basis of 5α-reductase type 2 deficiency, nandrolone may be expected to produce reduced facial and particularly body hair growth relative to testosterone in transmasculine people. Essentially no body or facial hair growth is likely to occur with nandrolone at doses equivalent to female-range testosterone levels in transfeminine people, and even at supraphysiological levels, facial and body hair growth is likely to be less than with testosterone. However, facial and body hirsutism have nonetheless been reported with nandrolone at postmenopausal osteoporosis doses (Need et al., 1989; Passeri et al., 1993; Geusens, 1995; Flicker et al., 1997).
For transmasculine people who want the benefits of nandrolone except the lesser facial hair growth, topical testosterone to the beard area can potentially be used in conjunction with nandrolone injections to restore full facial hair growth. Another possibility for achieving effects of both testosterone and nandrolone in transmasculine people could be to start on testosterone and use it for a few years, allowing for facial/body hair growth and hairline masculinization, and then switch from testosterone to nandrolone for long-term therapy, allowing for preservation of scalp hair.
One of the reasons that nandrolone is attractive for androgen replacement therapy in cisgender men is that it has very weak relative effects in the prostate gland compared to testosterone. This is due to inactivation of nandrolone in the prostate gland by 5α-reductase similarly to other tissues that express this enzyme. It is anticipated that long-term androgen replacement therapy with nandrolone might allow for a reduced risk of enlarged prostate and prostate cancer compared to testosterone in men (Wu & Kovac, 2016; Pan & Kovac, 2016). As transfeminine people also have prostate glands, the weak effects of nandrolone in the prostate are potentially of relevance to us as well. However, due to estrogen therapy and androgen deprivation, the prostate is atrophied in transfeminine people and prostate cancer is very rare (Wiki). In relation to this, the favorable prostate profile of nandrolone may be of little significance for transfeminine people.
Although nandrolone is quite promising for transgender hormone therapy, it is important to be clear that there are currently no quality clinical studies comparing it to testosterone and showing that it is less androgenic in terms of skin and hair effects (even if there is strong theoretical basis for this notion, particularly in the case of scalp hair loss). Nor are there any studies demonstrating with certainty that it is as effective as testosterone in terms of inducing general masculinization. Moreover, the appropriate dosages of nandrolone for use in androgen replacement in women and men are uncertain. Finally, nandrolone has been little-studied for androgen replacement and its long-term tolerability and health safety for this use haven’t been properly characterized. For these reasons, androgen replacement with nandrolone in cisgender men and transmasculine people is an experimental therapy. Additional research is needed to properly characterize nandrolone for this indication.
Androgen Replacement in Transfeminine People
Some believe that testosterone is important in women for mood, well-being, energy, sexual desire and function, general health, and for other reasons. Androgen levels and activity may be low in many transfeminine people due to hormone therapy and/or gonadectomy, raising concerns about androgen deficiency. However, there is little or no change in androgen levels with natural menopause in cisgender women and only a small decrease in androgen levels with ovariectomy in women (Liu & Handelsman, 1998). Moreover, there is inadequate evidence to support claims of benefit with androgen replacement therapy in women at present, and there are no well-supported benefits of female-range doses of testosterone (Wiki). For these reasons, androgen replacement therapy in women is controversial (Liu & Handelsman, 1998).
Testosterone does stimulate sexual desire in women, but only at levels that are above the normal female range (>50 ng/dL) (Cappelletti & Wallen, 2016; Wiki). And even with high testosterone levels of 80 to 150 ng/dL, stimulation of sexual desire in women is modest (Cappelletti & Wallen, 2016; Wiki). It is likely that estradiol rather than testosterone (or progesterone) is the key sex hormone for sexual desire in women (Cappelletti & Wallen, 2016; Aly, 2020).
Androgens can have detrimental effects in transfeminine people. They can cause androgenic and masculinizing effects like oily skin, acne, seborrhea, facial/body hair growth, scalp hair loss, and voice deepening, among others. In addition, androgens oppose the effects of estrogens in the breasts, and may inhibit breast development even with low-level exposure or physiological levels in females (Dimitrakakis et al., 2003; Peters et al., 2011; Sas et al., 2014; Barbieri, 2017).
For these reasons, transfeminine people don’t necessarily require androgen replacement, and it shouldn’t be routinely used out of concern of androgen deficiency. In any case, some transfeminine people insist on androgen replacement, and others desire supraphysiological levels of androgens for purposes like stimulating libido or helping to build and maintain muscle mass. There is some evidence that even physiological levels of testosterone may help to support muscle mass in women (Huang & Basaria, 2017). Moreover, in transfeminine people who are fully hormonally transitioned, breast development is complete, and hence inhibition of the breasts by androgens is no longer a concern. Another potentially useful indication of androgens in transfeminine people is to counteract cellulite (Wiki; Gruber et al., 2002; Avram, 2004). Since nandrolone has full androgenic potential in fat tissue, it may be similarly effective as testosterone for this purpose. However, there are no good studies of androgens for treatment of cellulite at this time.
Although androgen replacement therapy isn’t recommended for cisgender women or transfeminine people because of a lack of evidence of benefit and potential adverse effects, nandrolone is a very favorable alternative to testosterone for such purposes in those who nonetheless opt for such therapy. This is due to its improved skin and hair profile and hence a probable lower risk of undesirable effects.
Additional Topics on Nandrolone
This section is for additional topics on nandrolone as an androgen in transfeminine and transmasculine people, including its availability, dosage, androgenic/masculinizing effects besides skin and hair (e.g., voice deepening, bottom growth/clitoral enlargement, sexual effects), and liver safety.
Availability of Nandrolone
Nandrolone esters remain widely available for medical use throughout the world (Drugs.com). However, the availability of AAS, including nandrolone esters, has become increasingly limited over time. The shorter-acting ester nandrolone phenylpropionate is no longer available in the United States or many other countries, and the more favorable longer-acting nandrolone decanoate was discontinued in the United States in 2019. However, nandrolone decanoate remains available from compounding pharmacies in the United States, for instance AnazaoHealth. It is only available from this particular pharmacy at a very high concentration of 200 mg/mL however, which although a potentially workable concentration for use in transmasculine people, is likely completely impractical for use in transfeminine people. Compounding pharmacies might be able to do custom concentrations upon request though.
Another potential means of delivering nandrolone is via an orally active nandrolone androgen prohormone like 19-nordehydroepiandrosterone (19-nor-DHEA). This compound is converted into nandrolone analogously to the use of DHEA as a prohormone of testosterone (Wiki). It is sold as an over-the-counter supplement from certain online vendors, similarly to DHEA. 19-Nor-DHEA may be a more suitable option for transfeminine people due to the lower doses needed and less suitable for transmasculine people due to the much higher doses required.
Dosage of Nandrolone
The dosage of nandrolone for use in androgen replacement therapy in women and men hasn’t been established. Hence, we don’t completely know what the appropriate dose is for use in transfeminine and transmasculine hormone therapy. In any case, it is possible for us to estimate.
One review recommended a clinical dose range in males of 50–400 mg/2–4 weeks for testosterone enanthate or cypionate but 50–100/3–4 weeks for nandrolone decanoate (Hickson et al., 1989). This is also in line with several-fold higher potency of nandrolone relative to testosterone.
Dosage for Transmasculine People
A typical dosage of testosterone enanthate for use in androgen replacement therapy in cisgender and transgender men is 50 to 100 mg once per week by intramuscular injection (Wiki; Wiki). Hence, an appropriate dosage of nandrolone decanoate, assuming 3-fold greater potency than testosterone, might be about 16.7 to 33.3 mg once per week by intramuscular injection (ignoring the small differences in molecular weight between testosterone enanthate and nandrolone decanoate). However, it must be emphasized that these doses are merely educated guesses. It’s notable that these doses are quite similar to those used in postmenopausal osteoporosis in women (about 12.5 to 25 mg per week total). Hence, although such doses have certainly been associated with masculinizing effects in women, they might be suboptimal in terms of masculinization for transmasculine people. Consequently, it’s possible that a higher dose, like 50 mg once per week, might be more appropriate. In addition to weekly use, nandrolone decanoate has a duration suitable for use once every 2 weeks (Graphs).
Nandrolone hexyloxyphenylpropionate (NHPP; brand name Anadur) was studied as a male hormonal contraceptive by the World Health Organization and others at a dose of 200 mg/3 weeks i.m. in combination with depot medroxyprogesterone acetate (Knuth et al., 1986; Nieschlag, 2010; Nieschlag & Behre, 2012). It was used as a replacement and alternative for testosterone and was given at a dosage of about 66.7 mg once per week. However, this was an intentionally supraphysiological dosage which was selected to achieve a male contraceptive effect, as suggested by suppression of gonadotropins to undetectable levels (Knuth et al., 1985). Hence, although safe and well-tolerated in limited studies, it might be a high dosage for transmasculine hormone therapy. Very high doses of nandrolone can reproduce the full spectrum of androgenic effects of testosterone, and hence doses of nandrolone that are too high may serve to nullify its advantages over testosterone (van der Vies, 1985).
Dosage for Transfeminine People
In postmenopausal women, nandrolone decanoate has been used for general androgen replacement at a dose of 25 to 50 mg once every 6 to 12 weeks (about 2–8 mg/week or 0.29–1.14 mg/day), and for prevention and treatment of osteoporosis at a dose of 50 mg once every 2 to 4 weeks (about 12.5–25 mg/week or 1.8–3.6 mg/day) (Wiki; Table). However, these doses of nandrolone decanoate are likely to be highly excessive for physiological androgen replacement in women and transfeminine people and are probably also spaced too widely in terms of dosing interval. This is based on the following: (1) normal production of testosterone in premenopausal women is about 150 μg/day; (2) nandrolone is more potent as an androgen receptor agonist than testosterone; and (3) pharmacokinetic concentration–time curves for nandrolone decanoate suggest that an appropriate dosing interval is once every 1 to 2 weeks (Graphs). Indeed, nandrolone decanoate at 50 mg/2–4 weeks has been found to produce significant masculinizing effects in women with long-term use (Geusens, 1995; Wiki). Since nandrolone has high theoretical masculinizing potential in most tissues in the body, like muscle, bone, fat, and vocal tissue, it’s very important that transfeminine people don’t take doses that are too large.
Androgenic and Masculinizing Effects Besides Skin and Hair
Nandrolone has sometimes been described as “non-masculinizing” or “minimally virilizing”, but this is misleading and inaccurate. Nandrolone has high theoretical potential to produce masculinizing skeletal, fat, and muscular effects, like growth of muscles, widening of shoulders, masculine enlargement, elongation, and shaping of bones, and masculine distribution of fat. It also has high capacity to produce voice deepening (the vocal tissue notably being part muscle). The only clear differences in masculinizing effect between nandrolone and testosterone are that nandrolone has a greatly reduced theoretical potential for androgenic effect in skin and hair follicles. Succinctly, nandrolone could be thought of as testosterone with minimization of the undesirable skin and hair effects.
Voice Deepening with Nandrolone
The vocal tissue, consisting of the vocal cords and the larynx, are part muscle, and 5α-reductase is expressed minimally or not at all in this tissue (Bhagavan, 2002; Kicman, 2008). Accordingly, voice deepening is normal in individuals with 5α-reductase type 2 deficiency (Imperato-McGinley & Zhu, 2002). As such, nandrolone would be expected to produce voice deepening similarly to testosterone. In accordance, voice deepening has been reported with nandrolone in women at doses used in postmenopausal osteoporosis (Gerritsma et al., 1994; Geusens, 1995; Frisoli et al., 2005). There are case reports of severe and complete masculinization of the voice in women treated with excessive doses of nandrolone (Damsté, 1964; Brodnitz, 1971; Baker, 1999). Voice changes have also been seen with nandrolone derivatives like normethandrone (Feldman et al., 1960).
Bottom Growth with Nandrolone
DHT is required for normal male sexual differentiation of the genitals during prenatal development. This is evidenced by 5α-reductase type 2 deficiency, in which the genitals are ambiguous but overall more female-like at birth and in childhood (Wiki). Although DHT is critical for prenatal genital masculinization, the same may not be true for pubertal development of the penis (Hiort, 2013). Individuals with 5α-reductase type 2 deficiency undergo masculinization of the genitals at puberty such that a small and near-functional penis develops (Peterson et al., 1977; Marks, 2004; Hiort, 2013; Photos). This is so striking that males with 5α-reductase type 2 deficiency in the Dominican Republic are referred to as “guevedoces”, which is said to literally mean “penis at 12”.
As 5α-reductase may not be critical for penile enlargement at puberty, nandrolone may allow for no less bottom growth (clitoral enlargement) in transmasculine people than does testosterone. Indeed, nandrolone has been reported to produce clitoral enlargement as a side effect in women and penile growth in prepubertal boys (Camerino & Sala, 1960; Camerino & Sciaky, 1975). Similar findings have been made for derivatives of nandrolone like normethandrone and norethandrolone (Feldman et al., 1960; Roche, Towns, & Wettenhall, 1963; Kirschvink et al., 1963; Prunty et al., 1958).
Since this article was published, a review suggesting nandrolone as a potential component of transgender hormone therapy for non-binary individuals has been published:
Cocchetti, C., Ristori, J., Romani, A., Maggi, M., & Fisher, A. D. (2020). Hormonal Treatment Strategies Tailored to Non-Binary Transgender Individuals. Journal of Clinical Medicine, 9(6), 1609. [DOI:10.3390/jcm9061609]
The review includes the following excerpts on nandrolone:
Other options may include nandrolone, an anabolic steroid administered via intramuscular injection, which is not as optimal a substrate for 5α-reductase as testosterone, but it has a stronger effect compared to the testosterone on target tissues devoid of 5α-reductase activity (e.g., muscular tissue) [15]. Indeed, nandrolone can be theoretically used in non-binary [assigned female at birth (AFAB)] individuals requesting masculinization of body shape (i.e., increased muscle mass) with a limited increase in facial and body hair. Regarding the safety profile of this compound, data are limited by the fact that most observations come from the setting of androgenic-anabolic steroid (AAS) abuse [16,17], thus their applicability to appropriate medical therapy is limited [18]. In this setting, concerns about cardiomyopathy and coronary artery disease risk emerged [19], although associated with the administration of nandrolone at extremely higher dosages [20]. Furthermore, nandrolone use does not seem associated to hepatotoxicity, since, as an injectable oil, it is not subject to first-pass hepatic metabolism.
Moreover, some AFAB transgender individuals can benefit from testosterone therapy combined with 5α-reductase inhibitors or from treatment with nandrolone (an androgenic compound less prone to 5α reduction) in case they wish only a partial virilization (i.e., voice deepening and lean mass increase without facial and body hair increase).
In addition to the above review, a second review suggesting nandrolone as a potential option for hormone therapy in non-binary individuals has been published (as a preprint):
Vetri, M., Cataldi, A., Naselli, A., & Vetri, A. (2021). Transsexualism Ethiology and Medical Management: Between Scientific Evidence and Personal Experiences. Preprints, 2021030172. [DOI:10.20944/preprints202103.0172.v1]:
With the following relevant excerpt:
In [female-to-males] (FtMs) requesting partial masculinization, it may be possible to reduce the dose of testosterone or substitute it with nandrolone, an anabolic steroid administered via intramuscular injection. So, we must accept alternative hormonal treatment regimens, other than those reported in current guidelines, for such nonbinary transgender individuals, to try to improve their psychological well-being and quality of life [40].
However, this preprint article appears to have never ended up being accepted for publication.
Update 3: Cocchetti et al. (2020) and Iuliano et al. (2021)
Further articles have mentioned nandrolone as a hypothetical option for transgender people, for instance Cocchetti et al. (2020):
Another hypothetic option [for management of hypoactive sexual desire disorder in transgender women] may include nandrolone, an androgenic compound administered via intramuscular injection, less prone to 5-alpha reduction anabolic steroid leading to limited virilizing dermatological undesired effects [72]. However, data on its efficacy on sexual desire are not yet available.
Actually, the classic view of gender binarism and [gender-affirming hormone therapy (GAHT)] should be reconsidered because of the increasing prevalence of non-binary individuals experiencing [gender dysphoria (GD)]. Therefore, new therapeutic strategies should be also considered [17]. Since there is a lack in standardized hormonal treatment protocols for non-binary [assigned female at birth (AFAB)], GAHT goals should be adjusted according to patients’ needs in order to improve the self-perception and the quality of life [18]. For instance, different testosterone doses and/or other androgen preparations (i.e., nandrolone) are suggested to modulate the requested body changes [18], even if serious ethical concerns on some drugs exist.
References
Anderson, R. A., Martin, C. W., Kung, A. W., Everington, D., Pun, T. C., Tan, K. C., Bancroft, J., Sundaram, K., Moo-Young, A. J., & Baird, D. T. (1999). 7α-Methyl-19-Nortestosterone Maintains Sexual Behavior and Mood in Hypogonadal Men. The Journal of Clinical Endocrinology & Metabolism, 84(10), 3556–3562. [DOI:10.1210/jcem.84.10.6028]
Anderson, R. A., Wallace, A. M., Sattar, N., Kumar, N., & Sundaram, K. (2003). Evidence for Tissue Selectivity of the Synthetic Androgen 7α-Methyl-19-Nortestosterone in Hypogonadal Men. The Journal of Clinical Endocrinology & Metabolism, 88(6), 2784–2793. [DOI:10.1210/jc.2002-021960]
Arif, T., Dorjay, K., Adil, M., & Sami, M. (2017). Dutasteride in Androgenetic Alopecia: An Update. Current Clinical Pharmacology, 12(1), 31–35. [DOI:10.2174/1574884712666170310111125]
Avram, M. M. (2004). Cellulite: a review of its physiology and treatment. Journal of Cosmetic and Laser Therapy, 6(4), 181–185. [DOI:10.1080/14764170410003057]
Azzouni, F., Godoy, A., Li, Y., & Mohler, J. (2012). The 5 Alpha-Reductase Isozyme Family: A Review of Basic Biology and Their Role in Human Diseases. Advances in Urology, 2012 (Personalized Cancer Therapy for Urological Cancers: From Bench to Bedside and Back), 530121. [DOI:10.1155/2012/530121]
Baker, J. (1999). A report on alterations to the speaking and singing voices of four women following hormonal therapy with virilizing agents. Journal of Voice, 13(4), 496–507. [DOI:10.1016/s0892-1997(99)80005-8]
Barbieri, R. L. (2019). Breast. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 248–255.e3). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/B978-0-323-47912-7.00010-X]
Bardin, C. W., & Janne, O. A. (1986). Steroids of One Class Can Mimic, Inhibit and Potentiate the Biological Effects of Other Steroid Classes When Administered at High Doses. In Gregoire, A. T., & Blye, R. P. (Eds.). Contraceptive Steroids: Pharmacology and Safety (pp. 123–143). Boston, MA: Springer US. [DOI:10.1007/978-1-4613-2241-2_6]
Barrionuevo, P., Nabhan, M., Altayar, O., Wang, Z., Erwin, P. J., Asi, N., Martin, K. A., & Murad, M. H. (2018). Treatment Options for Hirsutism: A Systematic Review and Network Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism, 103(4), 1258–1264. [DOI:10.1210/jc.2017-02052]
Bässler, R. (1970). The Morphology of Hormone Induced Structural Changes in the Female Breast. In Altmann, H.-W., et al. (Eds.). Current Topics in Pathology: Ergebnisse der Pathology, Volume 53 (pp. 1–89). Heidelberg: Springer Berlin. [DOI:10.1007/978-3-662-30514-0_1]
Behre, H. M., Nashan, D., Hubert, W., & Nieschlag, E. (1992). Depot gonadotropin-releasing hormone agonist blunts the androgen-induced suppression of spermatogenesis in a clinical trial of male contraception. The Journal of Clinical Endocrinology & Metabolism, 74(1), 84–90. [DOI:10.1210/jcem.74.1.1727833]
Behre, H., Kliesch, S., Lemcke, B., von Eckardstein, S., & Nieschlag, E. (2001). Suppression of spermatogenesis to azoospermia by combined administration of GnRH antagonist and 19-nortestosterone cannot be maintained by this non-aromatizable androgen alone. Human Reproduction, 16(12), 2570–2577. [DOI:10.1093/humrep/16.12.2570]
Bhagavan, N. V. (2002). Endocrine Metabolism V: Reproductive System. In Bhagavan, N. V. Medical Biochemistry, 4th Edition (pp. 781–801). San Diego: Harcourt/Academic Press. [Google Books] [WorldCat]
Bourg, R. (1950). Activité sécrétrice des glandes mammaires; consécutive a des doses massives de testostérone chez la femme après castration. [Secretory activity of the mammary glands; sequellae of massive testosterone doses in women following castration.] Annales d’Endocrinologie, 11(3), 254–260. [Google Scholar] [PubMed] [PDF]
Bricout, V., & Wright, F. (2004). Update on nandrolone and norsteroids: how endogenous or xenobiotic are these substances? European Journal of Applied Physiology, 92(1–2), 1–12. [DOI:10.1007/s00421-004-1051-3]
Brodnitz, F. S. (1971). Hormones and the human voice. Bulletin of the New York Academy of Medicine, 47(2), 183–191. [Google Scholar] [PubMed] [PubMed Central]
Buvat, J., Maggi, M., Guay, A., & Torres, L. O. (2013). Testosterone Deficiency in Men: Systematic Review and Standard Operating Procedures for Diagnosis and Treatment. The Journal of Sexual Medicine, 10(1), 245–284. [DOI:10.1111/j.1743-6109.2012.02783.x]
Camerino, B., & Sala, G. (1960). Anabolic Steroids. In Jucker, E. (Ed.). Fortschritte der Arzneimittelforschung / Progress in Drug Research / Progrès des Recherches Pharmaceutiques, Volume 2 (pp. 71–134). Basel: Birkhäuser Basel. [DOI:10.1007/978-3-0348-7038-2_2]
Camerino, B., & Sciaky, R. (1975). Structure and effects of anabolic steroids. Pharmacology & Therapeutics. Part B: General and Systematic Pharmacology, 1(2), 233–275. [DOI:10.1016/0306-039x(75)90007-0]
Cappelletti, M., & Wallen, K. (2016). Increasing women’s sexual desire: The comparative effectiveness of estrogens and androgens. Hormones and Behavior, 78, 178–193. [DOI:10.1016/j.yhbeh.2015.11.003]
Čeponis, J., Wang, C., Swerdloff, R. S., & Liu, P. Y. (2017). Anabolic and Metabolic Effects of Testosterone and Other Androgens: Direct Effects and Role of Testosterone Metabolic Products. In Simoni, M., & Huhtaniemi, I. T. (Eds.). Endocrinology of the Testis and Male Reproduction (pp. 373–394). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-44441-3_11]
Cocchetti, C., Ristori, J., Romani, A., Maggi, M., & Fisher, A. D. (2020). Hormonal Treatment Strategies Tailored to Non-Binary Transgender Individuals. Journal of Clinical Medicine, 9(6), 1609. [DOI:10.3390/jcm9061609]
Cocchetti, C., Ristori, J., Mazzoli, F., Vignozzi, L., Maggi, M., & Fisher, A. D. (2020). Management of hypoactive sexual desire disorder in transgender women: a guide for clinicians. International journal of Impotence Research, 33(7), 703–709. [DOI:10.1038/s41443-021-00409-8]
Cooke, P. S., Nanjappa, M. K., Ko, C., Prins, G. S., & Hess, R. A. (2017). Estrogens in Male Physiology. Physiological Reviews, 97(3), 995–1043. [DOI:10.1152/physrev.00018.2016]
Crawford, B. A., Liu, P. Y., Kean, M. T., Bleasel, J. F., & Handelsman, D. J. (2003). Randomized Placebo-Controlled Trial of Androgen Effects on Muscle and Bone in Men Requiring Long-Term Systemic Glucocorticoid Treatment. The Journal of Clinical Endocrinology & Metabolism, 88(7), 3167–3176. [DOI:10.1210/jc.2002-021827]
Damsté, P. (1964). Virilization of the Voice due to Anabolic Stereoids. Folia Phoniatrica et Logopaedica, 16(1), 10–18. [DOI:10.1159/000262980]
Davis, S. (1999). The therapeutic use of androgens in women. The Journal of Steroid Biochemistry and Molecular Biology, 69(1–6), 177–184. [DOI:10.1016/s0960-0760(99)00054-0]
Dimitrakakis, C., Zhou, J., Wang, J., Belanger, A., LaBrie, F., Cheng, C., Powell, D., & Bondy, C. (2003). A physiologic role for testosterone in limiting estrogenic stimulation of the breast. Menopause, 10(4), 292–298. [DOI:10.1097/01.gme.0000055522.67459.89]
Doran, P. M., Riggs, B. L., Atkinson, E. J., & Khosla, S. (2001). Effects of Raloxifene, a Selective Estrogen Receptor Modulator, on Bone Turnover Markers and Serum Sex Steroid and Lipid Levels in Elderly Men. Journal of Bone and Mineral Research, 16(11), 2118–2125. [DOI:10.1359/jbmr.2001.16.11.2118]
Drugs.com. International > Nandrolone. Drugs.com. [URL]
Ebeling, P. R. (2010). Androgens and osteoporosis. Current Opinion in Endocrinology, Diabetes & Obesity, 17(3), 284–292. [DOI:10.1097/med.0b013e328339658c]
Feldman, E. B., Carter, A. C., Kossa, J. L., McCarrick, J. F., & Schwartz, H. L. (1960). Endocrinologic and Metabolic Effects of 17α-Methyl-19-nortestosterone in Women. The Journal of Clinical Endocrinology & Metabolism, 20(6), 842–857. [DOI:10.1210/jcem-20-6-842]
Flicker, L., Hopper, J. L., Larkins, R. G., Lichtenstein, M., Buirski, G., & Wark, J. D. (1997). Nandrolone decanoate and intranasal calcitonin as therapy in established osteoporosis. Osteoporosis International, 7(1), 29–35. [DOI:10.1007/bf01623456]
Friedl, K. E., Jones, R. E., Hannan, C. J., & Plymate, S. R. (1989). The Administration of Pharmacological Doses of Testosterone or 19-Nortestosterone to Normal Men is Not Associated with Increased Insulin Secretion or Impaired Glucose Tolerance. The Journal of Clinical Endocrinology & Metabolism, 68(5), 971–975. [DOI:10.1210/jcem-68-5-971]
Friedl, K. E., Dettori, J. R., Hannan, C. J., Patience, T. H., & Plymate, S. R. (1991). Comparison of the effects of high dose testosterone and 19-nortestosterone to a replacement dose of testosterone on strength and body composition in normal men. The Journal of Steroid Biochemistry and Molecular Biology, 40(4–6), 607–612, IN5–IN6. [DOI:10.1016/0960-0760(91)90283-b]
Frisoli, A., Chaves, P. H., Pinheiro, M. M., & Szejnfeld, V. L. (2005). The Effect of Nandrolone Decanoate on Bone Mineral Density, Muscle Mass, and Hemoglobin Levels in Elderly Women With Osteoporosis: A Double-Blind, Randomized, Placebo-Controlled Clinical Trial. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 60(5), 648–653. [DOI:10.1093/gerona/60.5.648]
Gerritsma, E. J., Brocaar, M. P., Hakkesteegt, M. M., & Birkenhäger, J. C. (1994). Virilization of the voice in post-menopausal women due to the anabolic steroid nandrolone decanoate (Decadurabolin). The effects of medication for one year. Clinical Otolaryngology, 19(1), 79–84. [DOI:10.1111/j.1365-2273.1994.tb01153.x]
Geusens, P. (1995). Nandrolone decanoate: Pharmacological properties and therapeutic use in osteoporosis. Clinical Rheumatology, 14(S3), 32–39. [DOI:10.1007/bf02210686]
Gold, J., Batterham, M., Rekers, H., Harms, M., Geurts, T., Helmyr, P., Silva de Mendonca, J., Falleiros Carvalho, L., Panos, G., Pinchera, A., Aiuti, F., Lee, C., Horban, A., Gatell, J., Phanuphak, P., Prasithsirikul, W., Gazzard, B., Bloch, M., & Danner, S. (2006). Effects of nandrolone decanoate compared with placebo or testosterone on HIV-associated wasting. HIV Medicine, 7(3), 146–155. [DOI:10.1111/j.1468-1293.2006.00358.x]
Gruber, C. J., Wieser, F., Gruber, I. M., Ferlitsch, K., Gruber, D. M., & Huber, J. C. (2002). Current concepts in aesthetic endocrinology. Gynecological Endocrinology, 16(6), 431–441. [DOI:10.1080/gye.16.6.431.441]
Hamdy, R. C., Moore, S. W., Whalen, K. E., & Landy, C. (1998). Nandrolone Decanoate for Men With Osteoporosis. American Journal of Therapeutics, 5(2), 89–96. [DOI:10.1097/00045391-199803000-00006]
Handelsman, D. J. (2016). Androgen Physiology, Pharmacology, and Abuse. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition, Volume 2 (pp. 2368–2393.e16). Philadelphia: Saunders/Elsevier. [Google Books] [DOI:10.1016/B978-0-323-18907-1.00138-4]
Hassager, C., Jensen, L., Johansen, J., Riis, B., Melkko, J., Pødenphant, J., Risteli, L., Christiansen, C., & Risteli, J. (1991). The carboxy-terminal propeptide of type I procollagen in serum as a marker of bone formation: The effect of nandrolone decanoate and female sex hormones. Metabolism, 40(2), 205–208. [DOI:10.1016/0026-0495(91)90176-w]
Hickson, R. C., Ball, K. L., & Falduto, M. T. (1989). Adverse Effects of Anabolic Steroids. Medical Toxicology and Adverse Drug Experience, 4(4), 254–271. [DOI:10.1007/bf03259912]
Hiort, O. (2013). The differential role of androgens in early human sex development. BMC Medicine, 11(1), 152. [DOI:10.1186/1741-7015-11-152]
Hirshburg, J. M., Kelsey, P. A., Therrien, C. A., Gavino, A. C., & Reichenberg, J. S. (2016). Adverse Effects and Safety of 5-alpha Reductase Inhibitors (Finasteride, Dutasteride): A Systematic Review. The Journal of Clinical and Aesthetic Dermatology, 9(7), 56–62. [Google Scholar] [PubMed] [PubMed Central]
Hobbs, C. J., Plymate, S. R., Rosen, C. J., & Adler, R. A. (1993). Testosterone administration increases insulin-like growth factor-I levels in normal men. The Journal of Clinical Endocrinology & Metabolism, 77(3), 776–779. [DOI:10.1210/jcem.77.3.7690364]
Hobbs, C. J., Jones, R. E., & Plymate, S. R. (1996). Nandrolone, a 19-nortestosterone, enhances insulin-independent glucose uptake in normal men. The Journal of Clinical Endocrinology & Metabolism, 81(4), 1582–1585. [DOI:10.1210/jcem.81.4.8636371]
Holterhus, P., Piefke, S., & Hiort, O. (2002). Anabolic steroids, testosterone-precursors and virilizing androgens induce distinct activation profiles of androgen responsive promoter constructs. The Journal of Steroid Biochemistry and Molecular Biology, 82(4–5), 269–275. [DOI:10.1016/s0960-0760(02)00220-0]
Huang, G., & Basaria, S. (2017). The Case for Androgens in Menopausal Women: When and How? In Pal, L., & Sayegh, R. A. (Eds.). Essentials of Menopause Management: A Case-Based Approach (pp. 173–196). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-42451-4_10]
Imperato-McGinley, J., & Peterson, R. E. (1976). Male pseudohermaphroditism: The complexities of male phenotypic development. The American Journal of Medicine, 61(2), 251–272. [DOI:10.1016/0002-9343(76)90175-3]
Imperato-McGinley, J., & Zhu, Y. (2002). Androgens and male physiology the syndrome of 5α-reductase-2 deficiency. Molecular and Cellular Endocrinology, 198(1–2), 51–59. [DOI:10.1016/s0303-7207(02)00368-4]
Irwig, M. S. (2015). Safety concerns regarding 5α reductase inhibitors for the treatment of androgenetic alopecia. Current Opinion in Endocrinology, Diabetes & Obesity, 22(3), 248–253. [DOI:10.1097/med.0000000000000158]
Iuliano, S., Izzo, G., Zagari, M. C., Vergine, M., Brunetti, F. S., Brunetti, A., Di Luigi, L., & Aversa, A. (2021). Endocrine management of transgender adults: a clinical approach. Sexes, 2(1), 104–118. [DOI:10.3390/sexes2010009]
Jänne, O., Hemminki, S., Isomaa, V., Kokko, E., Torkkeli, H., Torkkeli, T., & Vierikko, P. (1978). Progestational Activity of Natural and Synthetic Androgens. International Journal of Andrology, 1(s2a) [Endocrine Approach to Male Contraception: Transactions of the Fifth Annual Workshop on the Testis, Held at Geilo, Norway on April 2-5, 1978], 162–174. [DOI:10.1111/j.1365-2605.1978.tb00015.x]
Kicman, A. T. (2008). Pharmacology of anabolic steroids. British Journal of Pharmacology, 154(3), 502–521. [DOI:10.1038/bjp.2008.165]
Kirschvink, J. F. (1963). Studies of Anabolic Steroids. American Journal of Diseases of Children, 106(4), 368–368. [DOI:10.1001/archpedi.1963.02080050370005]
Knuth, U. A., Behre, H., Belkien, L., Bents, H., & Nieschlag, E. (1985). Clinical trial of 19-nortestosterone-hexoxyphenylpropionate (Anadur) for male fertility regulation. Fertility and Sterility, 44(6), 814–821. [DOI:10.1016/s0015-0282(16)49043-6]
Knuth, U. A., Behre, H., Belkien, L., Bents, H., & Nieschlag, E. (1986). 19-Nortestosterone for male fertility regulation. In Zatuchni, G. I., Goldsmith, A., Spieler, J. M., & Sciarra, J. J. (Eds.). Male Contraception: Advances and Future Prospects: Proceedings of an International Workshop on Male Contraception, Advances and Future Prospects, May 28-31, 1985, Geneva, Switzerland (pp. 320–328). Philadelphia: Harper and Row. [Google Scholar] [Google Books] [WorldCat] [PDF]
Knuth, U. A., Yeung, C., & Nieschlag, E. (1989). Combination of 19-nortestosterone-hexyloxyphenylpropionate (Anadur) and depot-medroxyprogesterone-acetate (Clinovir) for male contraception. Fertility and Sterility, 51(6), 1011–1018. [DOI:10.1016/s0015-0282(16)60735-5]
Kuhl, H., & Wiegratz, I. (2017). Das Post-Finasterid-Syndrom. [Post Finasteride Syndrome.] Gynäkologische Endokrinologie, 15(2), 153–163. [DOI:10.1007/s10304-017-0126-2]
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Häggblad, J., Nilsson, S., & Gustafsson, J. (1997). Comparison of the Ligand Binding Specificity and Transcript Tissue Distribution of Estrogen Receptors α and β. Endocrinology, 138(3), 863–870. [DOI:10.1210/endo.138.3.4979]
Kumar, N., Crozat, A., Li, F., Catterall, J., Bardin, C., & Sundaram, K. (1999). 7α-methyl-19-nortestosterone, a synthetic androgen with high potency: structure-activity comparisons with other androgens. The Journal of Steroid Biochemistry and Molecular Biology, 71(5–6), 213–222. [DOI:10.1016/s0960-0760(99)00143-0]
Lauritzen, C. (1984). Pre- and Postmenopause. In Platt, D. (Ed.). Geriatrics 3: Gynecology · Orthopaedics · Anesthesiology · Surgery · Otorhinolaryngology · Ophthalmology · Dermatology (pp. 2–45). Berlin/Heidelberg: Springer Berlin Heidelberg. [DOI:10.1007/978-3-642-68976-5_1]
Lemmey, A. B., Elamanchi, S. R., Marcora, S. M., Casanova, F., & Maddison, P. J. (2013). Efficacy of nandrolone decanoate in treating rheumatoid cachexia in male rheumatoid arthritis patients. In Matsuno, H. (Ed.). Innovative Rheumatology (pp. 271–285). Rijeka, Croatia: InTech. [DOI:10.5772/53236]
Liu, P. Y., & Handelsman, D. J. (1998). Androgen therapy in non-gonadal disease. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 473–512). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_17]
Marks, L. S. (2004). 5α-reductase: history and clinical importance. Reviews in Urology, 6(Suppl 9), S11–S21. [Google Scholar] [PubMed] [PubMed Central]
Mella, J. M., Perret, M. C., Manzotti, M., Catalano, H. N., & Guyatt, G. (2010). Efficacy and Safety of Finasteride Therapy for Androgenetic Alopecia. Archives of Dermatology, 146(10), 1141–1150. [DOI:10.1001/archdermatol.2010.256]
Need, A. (1989). Cross-over study of fat-corrected forearm mineral content during nandrolone decanoate therapy for osteoporosis. Bone, 10(1), 3–6. [DOI:10.1016/8756-3282(89)90139-7]
Neff, M. S., Goldberg, J., Slifkin, R. F., Eiser, A. R., Calamia, V., Kaplan, M., Baez, A., Gupta, S., & Mattoo, N. (1981). A Comparison of Androgens for Anemia in Patients on Hemodialysis. New England Journal of Medicine, 304(15), 871–875. [DOI:10.1056/nejm198104093041504]
Nieschlag, E. (2010). Clinical trials in male hormonal contraception. Contraception, 82(5), 457–470. [DOI:10.1016/j.contraception.2010.03.020]
Nieschlag, E., & Behre, H. M. (2012). The essential role of testosterone in hormonal male contraception. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Testosterone: Action · Deficiency · Substitution, 4th Edition (pp. 470–493). Cambridge/New York: Cambridge University Press. [DOI:10.1017/cbo9781139003353.023]
Nieschlag, E., Kumar, N., & Sitruk-Ware, R. (2013). 7α-Methyl-19-nortestosterone (MENTR): the Population Council’s contribution to research on male contraception and treatment of hypogonadism. Contraception, 87(3), 288–295. [DOI:10.1016/j.contraception.2012.08.036]
Pan, M. M., & Kovac, J. R. (2016). Beyond testosterone cypionate: evidence behind the use of nandrolone in male health and wellness. Translational Andrology and Urology, 5(2), 213–219. [DOI:10.21037/tau.2016.03.03]
Passeri, M., Pedrazzoni, M., Pioli, G., Butturini, L., Ruys, A., & Cortenraad, M. (1993). Effects of nandrolone decanoate on bone mass in established osteoporosis. Maturitas, 17(3), 211–219. [DOI:10.1016/0378-5122(93)90049-n]
Peters, A. A., Ingman, W. V., Tilley, W. D., & Butler, L. M. (2011). Differential Effects of Exogenous Androgen and an Androgen Receptor Antagonist in the Peri- and Postpubertal Murine Mammary Gland. Endocrinology, 152(10), 3728–3737. [DOI:10.1210/en.2011-1133]
Peterson, R. E., Imperato-McGinley, J., Gautier, T., & Sturla, E. (1977). Male pseudohermaphroditism due to steroid 5α-reductase deficiency. The American Journal of Medicine, 62(2), 170–191. [DOI:10.1016/0002-9343(77)90313-8]
Propecia Finasteride Label. U.S. Food and Drug Administration. [URL] [PDF]
Prunty, F. T., Brooks, R. V., Clayton, B. E., & McSwiney, R. R. (1958). Some Effects of 17α-Ethyl-19-Nortestosterone in Man. Proceedings of the Royal Society of Medicine, 51(7), 557–558. [Google Scholar] [PubMed] [PubMed Central]
Roche, A., Towns, J. W., & Wettenhall, H. (1963). Influence of norethandrolone on the stature of short children. The Journal of Pediatrics, 63(5), 967–976. [DOI:10.1016/s0022-3476(63)80228-0]
Russell, N., & Grossmann, M. (2019). MECHANISMS IN ENDOCRINOLOGY: Estradiol as a male hormone. European Journal of Endocrinology, 181(1), R23–R43. [DOI:10.1530/eje-18-1000]
Sardar, P., Jha, A., Roy, D., Majumdar, U., Guha, P., Roy, S., Banerjee, R., Banerjee, A. K., & Bandyopadhyay, D. (2010). Therapeutic Effects of Nandrolone and Testosterone in Adult Male HIV Patients With AIDS Wasting Syndrome (AWS): A Randomized, Double-Blind, Placebo-Controlled Trial. HIV Clinical Trials, 11(4), 220–229. [DOI:10.1310/hct1104-220]
Sartorius, G. A., Ly, L. P., & Handelsman, D. J. (2014). Male Sexual Function Can Be Maintained Without Aromatization: Randomized Placebo‐Controlled Trial of Dihydrotestosterone (DHT) in Healthy, Older Men for 24 Months. The Journal of Sexual Medicine, 11(10), 2562–2570. [DOI:10.1111/jsm.12550]
Sas, T., Gault, E., Zeger Bardsley, M., Menke, L., Freriks, K., Perry, R., Otten, B., de Muinck Keizer-Schrama, S., Timmers, H., Wit, J., Ross, J., & Donaldson, M. (2014). Safety and Efficacy of Oxandrolone in Growth Hormone-Treated Girls with Turner Syndrome: Evidence from Recent Studies and Recommendations for Use. Hormone Research in Paediatrics, 81(5), 289–297. [DOI:10.1159/000358195]
Schürmeyer, T., Belkien, L., Knuth, U., & Nieschlag, E. (1984). Reversible azoospermia induced by the anabolic steroid 19-nortestosterone. The Lancet, 323(8374), 417–420. [DOI:10.1016/s0140-6736(84)91752-5]
Simpson, E. R., & Jones, M. E. (2007). Of Mice and Men: The Many Guises of Estrogens. In Korach, K. S., & Wintermantel, T. (Eds.). Tissue-Specific Estrogen Action: Novel Mechanisms, Novel Ligands, Novel Therapies (Ernst Schering Foundation Symposium Proceedings, Volume 2006/1) (pp. 45–68). Berlin/Heidelberg: Springer. [DOI:10.1007/2789_2006_016]
Skroza, N., Tolino, E., Mambrin, A., Zuber, S., Balduzzi, V., Marchesiello, A., Bernardini, N., Proietti, I., & Potenza, C. (2018). Adult Acne Versus Adolescent Acne: A Retrospective Study of 1,167 Patients. The Journal of Clinical and Aesthetic Dermatology, 11(1), 21–25. [Google Scholar] [PubMed] [PubMed Central] [URL]
Styne, D. M., & Grumbach, M. M. (2016). Physiology and Disorders of Puberty. In Melmed, S., Polonsky, K. S., Larsen, P. R., Kronenberg, & H. M. (Eds.). Williams Textbook of Endocrinology, 13th Edition (pp. 1074–1218). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-29738-7.00025-3]
Sundaram, K., & Kumar, N. (2000). 7alpha-Methyl-19-nortestosterone (MENT): the optimal androgen for male contraception and replacement therapy. International Journal of Andrology, 23(S2), 13–15. [DOI:10.1046/j.1365-2605.2000.00004.x]
Sundaram, K., Kumar, N., & Bardin, C. W. (1994). 7α-Methyl-19-nortestosterone: An Ideal Androgen for Replacement Therapy. In Bardin, W. C. (Ed.). Proceedings of the 1992 Laurentian Hormone Conference (Recent Progress in Hormone Research, Volume 49) (pp. 373–376). San Diego: Academic Press. [DOI:10.1016/b978-0-12-571149-4.50027-1]
Sundaram, K., Kumar, N., Monder, C., & Bardin, C. (1995). Different patterns of metabolism determine the relative anabolic activity of 19-norandrogens. The Journal of Steroid Biochemistry and Molecular Biology, 53(1–6), 253–257. [DOI:10.1016/0960-0760(95)00056-6]
Suvisaari, J. (2000). 7α-Methyl-19-Nortestosterone (MENT): Pharmacokinetics and Antigonadotropic Effects in Men. (Doctoral dissertation, University of Helsinki.) Helsinki: University of Helsinki. [Google Scholar] [URL] [PDF]
Thirumalai, A., Ceponis, J., Amory, J. K., Swerdloff, R., Surampudi, V., Liu, P. Y., Bremner, W. J., Harvey, E., Blithe, D. L., Lee, M. S., Hull, L., Wang, C., & Page, S. T. (2018). Effects of 28 Days of Oral Dimethandrolone Undecanoate in Healthy Men: A Prototype Male Pill. The Journal of Clinical Endocrinology & Metabolism, 104(2), 423–432. [DOI:10.1210/jc.2018-01452]
Tóth, M. (2009). Mioanabolikus Szteroidok és Szelektív Androgénreceptormodulátorok: Hatásmechanizmus és Terápiás Perspektívák. [Myoanabolic Steroids and Selective Androgen Receptor Modulators: Mechanism of Action and Perspectives. Orvosi Hetilap, 150(45), 2051–2059. [DOI:10.1556/oh.2009.28739] [Translation]
Tóth, M., & Zakár, T. (1986). Classification of Anabolic Steroids Using the Method of Competitive Metabolism. Experimental and Clinical Endocrinology & Diabetes, 87(2), 125–132. [DOI:10.1055/s-0029-1210533]
Trost, L., Saitz, T. R., & Hellstrom, W. J. (2013). Side Effects of 5‐Alpha Reductase Inhibitors: A Comprehensive Review. Sexual Medicine Reviews, 1(1), 24–41. [DOI:10.1002/smrj.3]
van der Vies, J. (1985). Implications of basic pharmacology in the therapy with esters of nandrolone. Acta Endocrinologica, 110(3 Suppl a), S38–S44. [DOI:10.1530/acta.0.109s0038]
van Zuuren, E. J., Fedorowicz, Z., Carter, B., & Pandis, N. (2015). Interventions for hirsutism (excluding laser and photoepilation therapy alone). Cochrane Database of Systematic Reviews, 2015, CD010334. [DOI:10.1002/14651858.cd010334.pub2]
Vetri, M., Cataldi, A., Naselli, A., & Vetri, A. (2021). Transsexualism Ethiology and Medical Management: Between Scientific Evidence and Personal Experiences. Preprints, 2021030172. [DOI:10.20944/preprints202103.0172.v1]
Wibowo, E., Schellhammer, P., & Wassersug, R. J. (2011). Role of Estrogen in Normal Male Function: Clinical Implications for Patients With Prostate Cancer on Androgen Deprivation Therapy. Journal of Urology, 185(1), 17–23. [DOI:10.1016/j.juro.2010.08.094]
Wibowo, E., & Wassersug, R. J. (2013). The effect of estrogen on the sexual interest of castrated males: Implications to prostate cancer patients on androgen-deprivation therapy. Critical Reviews in Oncology/Hematology, 87(3), 224–238. [DOI:10.1016/j.critrevonc.2013.01.006]
Winters, S. J. (1990). Androgens: Endocrine Physiology and Pharmacology. In Lin, G. C., & Erinoff, L. (Eds.). Anabolic Steroid Abuse (National Institute on Drug Abuse Research Monograph Series, Volume 102) (pp. 113–130). Rockville, Maryland: National Institute on Drug Abuse/U.S. Department of Health and Human Services. [Google Scholar] [PubMed] [Google Books] [PDF]
World Health Organization & Task Force on Methods for the Regulation of Male Fertility. (1993). Comparison of two androgens plus depot-medroxyprogesterone acetate for suppression to azoospermia in Indonesian men. Fertility and Sterility, 60(6), 1062–1068. [DOI:10.1016/s0015-0282(16)56411-5]
Wu, C., & Kovac, J. R. (2016). Novel Uses for the Anabolic Androgenic Steroids Nandrolone and Oxandrolone in the Management of Male Health. Current Urology Reports, 17(10), 72. [DOI:10.1007/s11934-016-0629-8]
Wu, S., Yuen, F., Swerdloff, R. S., Pak, Y., Thirumalai, A., Liu, P. Y., Amory, J. K., Bai, F., Hull, L., Blithe, D. L., Anawalt, B. D., Parman, T., Kim, K., Lee, M. S., Bremner, W. J., Page, S. T., & Wang, C. (2018). Safety and Pharmacokinetics of Single-Dose Novel Oral Androgen 11β-Methyl-19-Nortestosterone-17β-Dodecylcarbonate in Men. The Journal of Clinical Endocrinology & Metabolism, 104(3), 629–638. [DOI:10.1210/jc.2018-01528]
Yuen, F., Thirumalai, A., Pham, C., Swerdloff, R. S., Anawalt, B. D., Liu, P. Y., Amory, J. K., Bremner, W. J., Dart, C., Wu, H., Hull, L., Blithe, D. L., Long, J., Wang, C., & Page, S. T. (2020). Daily Oral Administration of the Novel Androgen 11β-MNTDC Markedly Suppresses Serum Gonadotropins in Healthy Men. The Journal of Clinical Endocrinology & Metabolism, 105(3), e835–e847. [DOI:10.1210/clinem/dgaa032]
\ No newline at end of file
diff --git a/transfemscience.org/articles/nonbinary-transfem-overview/index.html b/transfemscience.org/articles/nonbinary-transfem-overview/index.html
index 490d73dc..50a779fb 100644
--- a/transfemscience.org/articles/nonbinary-transfem-overview/index.html
+++ b/transfemscience.org/articles/nonbinary-transfem-overview/index.html
@@ -1 +1 @@
-An Exploration of Possibilities for Hormone Therapy in Non-Binary Transfeminine People - Transfeminine Science
Transfeminine people who are non-binary often desire different and more partial feminizing and demasculinizing changes compared to what occurs with conventional transfeminine hormone therapy. For example, non-binary transfeminine people may seek an intermediate physical and hormonal state between what is typical for males and females, may desire substantial or full feminization and demasculinization without breast development, may want to have a more sexually neutral or androgynous appearance, or may wish for feminization but concomitant preservation of more male-typical sexual desire and capacity. These partial goals for transfeminine hormone therapy are being increasingly encountered, but therapeutic methods for achieving them have not yet been studied or established in transgender medicine. Nonetheless, there are a variety of hormonal approaches by which such outcomes could theoretically be achieved. Demasculinization can be produced via partial to full testosterone suppression or blockade using varied doses of antiandrogens, progestogens, and/or gonadotropin-releasing hormone modulators. Feminization and other estrogenic effects with less or no breast development may be produced with selective estrogen receptor modulators or low doses of estradiol. Certain other hormonal agents can be used to achieve various specific effects, like 5α-reductase inhibitors to reduce body hair growth and scalp hair loss, topical androgens applied to the breasts to reduce breast development, and the androgen and anabolic steroid nandrolone decanoate as an alternative to testosterone with less effect in skin and hair follicles. Besides hormonal means, breast removal surgery can be sought to negate breast development. Health risks, particularly those related to androgen deprivation without adequate estrogenic replacement, are a concern with partial approaches to hormone therapy. One such risk, bone density loss, may be reduced with calcium and vitamin D supplementation, bisphosphonates, and weight-bearing exercise. There is increasing interest in partial approaches to hormone therapy in transgender health, so hopefully they will become better characterized and more available and utilized in transgender people in the future.
Introduction
“Non-binary” is a term that refers to transgender people who do not identify within the gender binary. Despite the fact that they do not identify as either male or female, many non-binary transmasculine and transfeminine people pursue hormone therapy just like transgender men and women. While some non-binary individuals opt for a full hormonal transition similarly to most binary-identified transgender people, many non-binary people would prefer only a partial hormonal transition. This could be to achieve an intermediate area between masculine and feminine characteristics, to achieve a more sexually neutral appearance, or to induce some but not all aspects of masculinization or feminization.
There are also individuals who seek hormonal feminization and/or demasculinization but do not actually identify as transgender. These cisgender-identified individuals often refer to themselves as “femboys”. Communities of these individuals exist on social media sites like Reddit (e.g., r/FemboyTransition). Feminization-inclined cisgender people who pursue hormonal transition often have similar preferences as transfeminine non-binary people—one of the most common of which is feminization without breast development. Sometimes these initially cisgender-identified hormonally transitioning individuals end up progressing to a transgender identity with time.
Partial approaches to hormonal transition and even widespread identification as non-binary are fairly recent developments. There is very little written on non-conventional approaches to hormone therapy of this sort in the published literature. Moreover, there are no available standards or guidelines for such therapy at this time. A number of recent reviews have started to discuss possibilities for non-binary hormone therapy however (Richards et al., 2016; Seal, 2017; Bass et al., 2018; Cocchetti et al., 2020). There is currently a discordance between the number of people who desire non-conventional hormonal transition and the clinical establishment of such therapy. Consequently, an exploration of the possibilities from a theoretical standpoint would be of value and is the aim of this review.
As a disclaimer, the ideas in this article are experimental and preliminary. No studies with the goal of partial hormonal transition in transgender people have been conducted as of present and there is no data or evidence in non-binary people to inform the use of such approaches. Instead, we can only extrapolate from theory and research in other groups of people at this time. Examples of these other groups in the case of transfeminine non-binary hormone therapy include cisgender men undergoing hormone therapy for prostate cancer, cisgender men going through treatment for gynecomastia (male breast development), and transgender women undergoing hormone therapy. For these reasons, the present discussion is exploratory and should not be taken as therapeutic recommendations.
Conventional Transfeminine Hormone Therapy
The goal of therapy in conventional hormone therapy for transgender women is to produce the maximum degrees of demasculinization and feminization—including breast development—that are possible. This is achieved by suppressing testosterone levels and increasing estradiol levels such that they are both within normal adult female ranges. Alternatively, the actions of testosterone can be blocked instead of full suppression of testosterone levels. The relevant hormonal changes are accomplished through the administration of hormonal medications including estrogens, progestogens, and/or antiandrogens. Estrogens produce feminization, including breast development, while testosterone suppression causes demasculinization—as well as disinhibits feminization. Estrogens, progestogens, and antiandrogens can all contribute to testosterone suppression.
The therapeutic goals of a subset of non-binary transfeminine people are equivalent to those of transgender women and hence are compatible with the effects of conventional transfeminine hormone therapy. That is, the maximum possible feminization, including breast development, and demasculinization are the aims of therapy. Non-binary transfeminine people with these preferences can simply use conventional transfeminine hormone therapy for their hormonal transition as opposed to more experimental and non-conventional partial approaches.
For a comprehensive introduction to conventional transfeminine hormone therapy, see this article:
The above article is intended to provide everything one needs to know to achieve a basic understanding of the subject. If you are new to the topic of transgender hormone therapy, it is highly recommended reading prior to continuing in the current article. The introduction covers the sex hormones, their effects, specific hormonal medications used, routes, and dosages for this type of hormone therapy. Much of this information is also applicable to non-conventional transfeminine hormone therapy.
Partial Transfeminine Hormone Therapy
Some non-binary transfeminine people (as well as cisgender-identified individuals seeking hormonal feminization) desire only partial feminization and/or demasculinization. Depending on the specific aims, this can be more complicated and require more thought than conventional transfeminine hormone therapy. The following goals of partial transfeminine hormone therapy may be encountered:
An intermediate physical and hormonal state between male and female
A more sexually neutral or androgynous appearance that is not necessarily masculine or feminine
Substantial or maximal feminization and demasculinization with little or no breast development
Substantial or maximal feminization and demasculinization with minimal or no loss of sexual desire, sexual function (i.e., erectile and orgasmic capacity), and/or fertility
The first of these goals is fairly straightforward in that it can entail what is essentially conventional transfeminine hormone therapy using lower medication doses. This will result in partial testosterone suppression and a mixture of both androgens and estrogens as major active sex hormones. The second goal involves deprivation of both androgens and estrogens. While possible, this can have negative consequences as sex hormones are important for maintaining certain aspects of health and well-being. There are potential ways to avoid or mitigate such consequences however. The third and fourth goals are also technically possible but are more difficult to achieve and are likely to require more specialized and potentially complex hormonal approaches.
Suppression and Blockade of Testosterone
If the goal of non-binary transfeminine hormone therapy is simply to achieve an androgynous appearance with minimal or no feminization, this can be achieved via deprivation of testosterone without concomitant administration of an estrogen. There are multiple ways to achieve androgen deprivation or testosterone suppression in people assigned male at birth. These include high-dose progestogen therapy, medical and surgical castration with GnRH agonists/antagonists or gonadectomy, high-dose androgen receptor antagonist therapy, and a few other other possibilities. In this section, I’ll discuss androgen deprivation largely from the standpoint of efficacy. There are problems with androgen deprivation alone in terms of tolerability and safety due to the co-consequence of estrogen deficiency however, which I’ll discuss in the subsequent section.
Testosterone Suppression with High Doses of Progestogens
Androgen deprivation can be achieved with high doses of progestogens, which suppress testosterone levels by up to 50 to 70% (Aly, 2019). This is a substantial decrease in testosterone levels, but is not quite into the female range. Androgen receptor antagonists can additionally be included to block the remaining testosterone that is not suppressed if desired. For these purposes, low-dose cyproterone acetate (e.g., 5.0–12.5 mg/day) (Aly, 2019) plus bicalutamide (e.g., 12.5–50 mg/day) or spironolactone (e.g., 200–400 mg/day) is likely to be an effective regimen. As an alternative to cyproterone acetate, high doses of other progestogens, such as just about any other progestin, or alternatively rectal progesterone (Aly, 2018), can be used instead.
Testosterone Suppression with Medical or Surgical Castration
GnRH agonists and antagonists are another option for testosterone suppression. These medications suppress testosterone levels by about 95%, or into the normal female range or male castrate range (<50 ng/dL). However, GnRH agonists and antagonists are very expensive, although there may be some viable options for obtaining them more cheaply (e.g., certain online pharmacies) (Aly, 2019).
Gonadectomy, or surgical removal of the gonads, can be performed as a more permanent alternative to GnRH agonists and antagonists. However, this procedure is expensive (a few thousand dollars USD), requires minor surgery, and can be more difficult to obtain. Most surgeons require letters from gender therapists and real-life experience; informed-consent surgeons do exist however. Gonadectomy is also irreversible, notably resulting in permanent loss of testes and sterility. In any case, gonadectomy is far less expensive and more convenient than GnRH agonists and antagonists in the long run.
Testosterone Blockade with Antiandrogens
Androgen receptor antagonists like bicalutamide and spironolactone act by directly binding to the androgen receptor and displacing androgens like testosterone and DHT from the receptor, thereby preventing its activation by these androgens. This is in contrast to therapies that act by suppressing androgen production and levels.
High-dose bicalutamide monotherapy (e.g., 150–300 mg/day) is a possible approach for androgen deprivation therapy. However, bicalutamide monotherapy increases testosterone and hence estradiol levels. The testosterone will be blocked by bicalutamide and will not have effects, but estradiol is increased to a concentration range that allows for marked or full feminization, including breast development. In addition, bicalutamide alone, even at very high doses, might not be enough to completely block male-range testosterone. With these considerations, if the goal is full demasculinization with no feminization or breast development, bicalutamide monotherapy is not something that, at least alone, can achieve this.
High-dose bicalutamide is expensive and potentially cost-prohibitive. High-dose spironolactone monotherapy is not a good option for this route as it is a relatively weak antiandrogen and likely falls far short of being able to handle male-range levels of testosterone (at least 200 mg/day appears to be required to fully block female testosterone levels) (Aly, 2018; Wiki). Concomitant partial suppression of testosterone and estrogen levels via additional use of a progestogen (e.g., cyproterone acetate) may be a more feasible option than an androgen receptor antagonist alone.
Some potentially major advantages of high-dose bicalutamide monotherapy are that in contrast to marked or full suppression of testosterone levels, bicalutamide monotherapy largely preserves sexual desire and erectile function and likely does not result in infertility.
Other Options for Testosterone Suppression
Another option is only partial demasculinization, which can be achieved essentially by using lower dosages of the medications discussed above (e.g., cyproterone acetate, bicalutamide). If desired, 5α-reductase inhibitors can be added in this context to more substantially decrease scalp hair loss and body hair growth. Note that if testosterone is more fully suppressed or blocked however, there is likely to be little or no benefit with 5α-reductase inhibitors.
Yet another possibility could be to incorporate low-dose nandrolone decanoate, an androgen receptor agonist and anabolic–androgenic steroid (AAS) with much less masculinizing/androgenic effect in skin and hair follicles (Aly, 2020). This AAS will help to suppress and replace testosterone levels. Nandrolone decanoate might also have the benefit of helping to maintain sexual desire and function. However, nandrolone decanoate was recently discontinued in the United States. Oxandrolone is another, similar AAS, but has been associated with liver toxicity.
Estrogen Deficiency and Replacement
While androgen deprivation therapy is effective for achieving the desired changes—specifically demasculinization without feminization—it is not recommended by itself. This is because estradiol is produced from testosterone and hence androgen deprivation results in estrogen deficiency as well. Estrogens are essential for maintaining bone density in both men and women, and without them, a person will quickly lose bone mass, eventually develop osteoporosis, and be at a high risk for bone fractures. Skeletal and postural disfigurement may also eventually occur (Figure; Figure). In addition, the person is likely to experience other menopause-like symptoms, such as hot flashes, mood and sleep problems, sexual dysfunction (e.g., low sexual desire, erectile dysfunction), and accelerated aging of the skin (Wiki). An increased risk of weight gain, type 2 diabetes, cardiovascular disease, and dementia may be associated with sex hormone deficiency as well. As such, extended deprivation of both androgens and estrogens with no estrogenic supplementation is not advisable.
With that said, a couple of clarifications should be made. Due to preservation of estradiol levels, high-dose bicalutamide monotherapy has minimal to no risk of bone density loss or most other menopausal symptoms. In addition, the low-dose cyproterone acetate plus low-dose bicalutamide option may have less of a risk of menopausal symptoms and possibly osteoporosis as well. This is because high-dose progestogens (of which “low-dose” cyproterone acetate certainly qualifies) can help treat certain menopausal symptoms such as hot flashes and possibly bone density loss, and also because some estradiol will be preserved (since testosterone will only be suppressed by 70 to 80% rather than more fully). With that said however, in the latter case, it is probably best not to take any risks.
Selective Estrogen Receptor Modulators (SERMs)
Instead of only androgen and estrogen deprivation, the inclusion of selective estrogen receptor modulators (SERMs), so-called partial estrogens, can be employed. These medications are partial agonists of the estrogen receptor, and have mixed estrogenic and antiestrogenic effects depending on the tissue. For example, the SERM raloxifene has estrogenic effects in bone, fat tissue, and the liver, but antiestrogenic effects in the breasts. In general, SERMs reduce bone density loss and osteoporosis risk while not causing breast development (and actually blocking it). A full list of SERMs can be found here. However, practically speaking, only raloxifene (Evista), tamoxifen (Nolvadex), and toremifene (Fareston) are available, inexpensive, and commonly used. For an overview of the estrogenic and antiestrogenic effects of the different SERMs in different tissues, see here. In general, SERMs have a fairly similar pattern of effects. Although we have some idea of the differential tissue effects of SERMs, in many cases we do not know how they behave in specific tissues. For example, only a single clinical study has shown that a SERM, specifically raloxifene, has estrogenic effects in fat tissue (Francucci et al., 2014). In addition, it is less clear how SERMs behave in, for example, skin, or in most of the brain.
SERMs also have various side effects. For instance, SERMs commonly produce hot flashes as an adverse effect. However, the fairly recently introduced combination of bazedoxifene/conjugated estrogens (Duavee) has been found to reduce the incidence of hot flashes in postmenopausal women (Duavee label). It is still on-patent and hence is expensive however. In any case, SERMs are also likely to produce other menopause-like symptoms. Additionally, SERMs have estrogenic effects in the liver and therefore influence production of coagulation factors and decrease production of insulin-like growth factor-1 (IGF-1), among other potentially undesirable changes. Due to the increase in coagulation with SERMs, they have a notable risk of blood clots and cardiovascular complications like stroke (Aly, 2020). Some SERMs, like tamoxifen, also have unique off-target actions and risks, for instance rare liver toxicity. Raloxifene is a more selective and probably safer SERM than tamoxifen.
SERMs are effective for maintaining bone density. However, they are, unfortunately, only partially estrogenic in bone and hence are submaximally effective for such purposes—they are significantly more effective than no treatment at all but are not as effective as estrogens (Dane et al., 2007; Zirilli et al., 2009; Birzniece et al., 2012; Vestergaard, 2012). Indeed, SERMs have actually been found to significantly antagonize the effects of estradiol on bone, for instance on bone density in premenopausal women (Powles et al., 1996; Burshell et al., 1999) and on bone maturation and growth plate closure in cisgender girls with precocious puberty (Passone et al., 2015). One study in elderly men suggested that 60 mg/day raloxifene is maximally equivalent in terms of bone density effect to estradiol levels of about 26 pg/mL (Doran et al., 2001; Palacios et al., 2020). Below this estradiol level, raloxifene was estrogenic on bone density, whereas above this level, it was antiestrogenic on bone density (Doran et al., 2001; Palacios et al., 2020). As such, although SERMs increase bone density in the context of very low estradiol levels, they are not as effective as estrogens in terms of maintaining bone density and they may still allow for significantly decreased bone density when added to androgen deprivation in non-binary transfeminine people.
Low-Dose Estrogen Supplementation
An alternative to SERMs for avoiding estrogen deficiency is low-dose estrogen therapy. A dosage of oral estradiol of about 1 to 2 mg/day or estradiol levels of about 30 to 50 pg/mL (via another route, for instance transdermal patches) is all that is needed for complete or near-complete prevention of bone density loss (Barbieri, 1992; Roux, 1997; Hadji, Colli, & Regidor, 2019). Moreover, estradiol has a better tolerability and safety profile than SERMs, with a much lower risk of blood clots (Aly, 2020).
A problem with estrogen therapy however is that in the absence of androgens, estrogens even at low levels will induce substantial feminization and breast development. Estradiol levels in normal cisgender girls gradually increase from around 5 to 10 pg/mL at the start of puberty to 50 or 60 pg/mL by late puberty, and these low levels produce full developent of the female secondary sex characteristics (Aly, 2020). Similarly, cisgender females with complete androgen insensitivity syndrome (CAIS) have estradiol levels of only about 35 pg/mL on average yet have complete feminization and excellent breast development (Aly, 2020; Table). Hence, the addition of low-dose estradiol to androgen deprivation would likely be a full transition. Very low doses of estradiol, for instance 0.5 mg/day oral estradiol or a 14 μg/day estradiol patch, achieving estradiol levels of only maybe 20 pg/mL, may be feasible and may result in less feminization. But, while effective for improving bone density (Dane et al., 2007; Birzniece et al., 2012), such doses/levels would not fully protect against bone density loss and other menopause-like symptoms and would likely still produce at least partial feminization. It is notable that even GnRH agonists/antagonists and gonadectomy alone—which reduce estradiol levels to around 10 pg/mL—have a rate of mild gynecomastia of as high as 15% (Di Lorenzo et al., 2005).
Interestingly, spironolactone was found at 100 mg/day to fully prevent GnRH agonist-induced bone density loss in women in a small randomized controlled trial (Moghetti et al., 1999). The authors hypothesized that this was due to its antimineralocorticoid activity, as aldosterone is negatively correlated with bone density (Moghetti et al., 1999). However, in another study, 100 mg/day spironolactone did not prevent bone density loss caused by high-dose progestogen therapy in the form of 5 mg/day lynestrenol in women (Preželj & Kocijančič, 1994; Preželj & Kocijančič, 1999). Hence, spironolactone should not be relied upon for preservation of bone density.
Onset and Reversibility of Bone Loss
Certain medications used in premenopausal women suppress gonadal sex hormone production and are associated with decreased bone density. These therapies can provide insight on the risk of bone density loss that may occur in non-binary transfeminine people deprived of sex hormones. Examples of such medications include progestogen-only birth control, which partially suppresses estradiol levels (to around 20–50 pg/mL) (Hadji, Colli, & Regidor, 2019), and GnRH agonists/antagonists, which partially to fully suppress estradiol levels depending on the medication and dose. Minimal or no bone density loss occurs with estradiol levels of 30 to 50 pg/mL, whereas significant bone density loss occurs with estradiol levels of 20 to 30 pg/mL (Hadji, Colli, & Regidor, 2019). Reassuringly however, bone density has been found to substantially or fully recover within a few years following discontinuation of progestogen-only birth control in young premenopausal women (Nelson, 2010). Along similar lines, therapy with the GnRH antagonist elagolix is considered to be acceptably safe in premenopausal women for up to 2 years at a dose that results in partial suppression of estradiol levels (to about 40 pg/mL) and for up to 6 months at a dose that results in maximal suppression of estradiol levels (to about 10 pg/mL) (Wiki). As such, a limited period of sex hormone deprivation—for instance as a trial of non-binary transfeminine hormone therapy—may be reasonably safe in terms of bone health. Long-term therapy should include adequate measures to avoid bone density loss however.
Prevention of Breast Development
Estrogen Blockade and Suppression
If the goal is to produce full demasculinization and some or full feminization with the sole exception of breast development, there are a number of ways to possibly achieve this. Androgen deprivation without estrogen supplementation will achieve demasculinization without any feminization or breast development (except for bicalutamide monotherapy of course). However, it is not recommended for reasons described above and would not provide feminization. SERMs are an option; in addition to their capacity to treat osteoporosis, they are used to treat gynecomastia in men, and are capable of fully blocking gynecomastia induced by estrogens when used at sufficient doses (Fentiman, 2018). However, SERMs may allow for only partial feminization rather than full. Aromatase inhibitors, in contrast to SERMs, have no apparent place in this form of hormone therapy, as they are, surprisingly, poorly effective for prevention of gynecomastia (Fagerlund et al., 2015; Bedognetti et al., 2010).
Selective Estrogen Receptor Modulators and Testosterone Elevation
SERMs can be used to prevent breast development, as previously described. A problem with the use of SERMs for this purpose however is that when they are used in people assigned male at birth in whom the gonads are intact and testosterone levels are not suppressed, they will induce substantial increases in gonadal testosterone production and circulating testosterone levels. In men with hypogonadism and consequent low testosterone levels, the SERMs clomifene (20–50 mg/day) and enclomifene (12.5–25 mg/day) increase testosterone levels from about 200–300 ng/dL to about 450–600 ng/dL on average (a change of about 2.0- to 2.5-fold, with an absolute increase of 250–400 ng/dL in this patient population) (Bach, Najari, & Kashanian, 2016; Trost & Khera, 2014). Because they are so effective at increasing testosterone levels, SERMs are used to treat male hypogonadism as an alternative to exogenous testosterone administration. Worse still, SERMs appear to cause even greater increases in testosterone levels in non-hypogonadal men. One study found that 50 mg/day clomifene increased testosterone levels by about 850 ng/dL in healthy younger men and by about 500 ng/dL in elderly men (Trost & Khera, 2014). Similarly, strong increases in testosterone levels have been observed with partial suppression of estradiol levels via aromatase inhibition in young and older men (T’Sjoen et al., 2005; Raven et al., 2006; de Ronde et al., 2009).
If testosterone levels are suppressed, increases in testosterone levels with SERMs will, depending on the degree of testosterone suppression, be less applicable (e.g., with high-dose progestogen therapy) or not applicable at all (e.g., with medical/surgical castration). However, if a SERM is combined with, say, bicalutamide alone, the situation may become even worse. This is because bicalutamide itself produces considerable increases in testosterone levels similarly to SERMs. In elderly men with prostate cancer, bicalutamide monotherapy induces a 1.5- to 2.0-fold rise in testosterone levels, increasing them from about 300–400 ng/dL to about 500–600 ng/dL (an absolute change of about 150–250 ng/dL) (Wiki). In healthy younger men, bicalutamide may increase testosterone levels into the upper end of the normal male range (potentially into the range of around 900–1,200 ng/dL) (Wiki).
As bicalutamide is a competitive antagonist of the androgen receptor, its efficacy is fundamentally both dose-dependent and dependent on testosterone levels. Consequently, in combination with a SERM, it is possible that testosterone levels will become too high for bicalutamide to block. Moreover, endogenous androgens and estrogens are together responsible for maintaining normal homeostatic negative feedback on the hypothalamic–pituitary–gonadal axis (HPG axis) in people assigned male at birth. It seems logical that with little to suppress the axis, gonadal production and hence circulating levels of testosterone and estradiol may simply continue to rise until they overwhelm bicalutamide and/or the SERM it is combined with and restore negative feedback on the HPG axis. For these reasons, it is possible that the combination of bicalutamide and a SERM alone might not be a practical option for non-conventional feminizing hormone therapy.
With all of that said however, the combination of bicalutamide and tamoxifen has been assessed in various studies in men with prostate cancer (PubMed), and increases in testosterone levels have, rather surprisingly, not been a problem in these studies. In terms of the findings, bicalutamide and tamoxifen together do, as expected, increase total testosterone levels. However, the rise in total testosterone levels is not much different from that which occurs with bicalutamide alone. Moreover, free testosterone levels are either increased to a certain degree or are not actually raised at all (Boccardo et al., 2005; Saltzstein et al., 2005; Fradet et al., 2007). This is thought to be due to the fact that SERMs have potent estrogenic effects in the liver and result in increased production of sex hormone-binding globulin (SHBG), consequently reducing the fraction of free and hence bioactive testosterone in the circulation. This serves to offset the biological influence of the increase in total testosterone levels. In accordance, and reassuringly, unfavorable changes in markers of androgen receptor signaling, like higher prostate-specific antigen (PSA) levels, have not been observed relative to bicalutamide alone in the studies.
It is not clear why studies of bicalutamide plus tamoxifen have observed increases in total testosterone levels that are not that different from those of bicalutamide alone. Whatever the reason, these studies suggest that the combination of bicalutamide and tamoxifen (or certain other SERMs) might actually be feasible still for non-conventional feminizing hormone therapy. With that said however, elderly men are a different patient population than non-binary transfeminine people. Older men have diminished increases in testosterone levels with bicalutamide and SERMs compared to healthy young men. In relation to this, the combination might not be as favorable for younger people assigned male at birth.
Tamoxifen very well may be exchangeable with raloxifene for use in combination with bicalutamide. However, it should be noted that in contrast to tamoxifen, raloxifene has never been studied in combination with bicalutamide. Or, at least, not in gonadally intact men; one study of bicalutamide with raloxifene in castrated men with prostate cancer does exist, but that does not provide much in the way of useful information (Ho et al., 2017). Nor has raloxifene actually been properly studied for prevention of gynecomastia. A single retrospective chart review reported that it was effective for pubertal gynecomastia in boys (Lawrence et al., 2004). But that is all the data we have. Conversely, there are many high-quality studies of tamoxifen for prevention of gynecomastia, including in combination with bicalutamide.
In any case, used by themselves in men, raloxifene has been found to result in lower increases in testosterone levels than tamoxifen or toremifene (Tsourdi et al., 2009). As such, bicalutamide and raloxifene together may indeed be similar in terms of testosterone levels relative to the combination of bicalutamide and tamoxifen. This might just be due to raloxifene having lower efficacy as a SERM than tamoxifen or toremifene at the relevant clinical doses however (Tsourdi et al., 2009).
Unfortunately, pharmaceutical topical DHT is only available today in France (Drugs.com). Some compounding pharmacies in certain countries might provide topical DHT preparations. However, DHT does not seem to be available from any compounding pharmacies in the United States. In contrast to DHT, testosterone readily converts into estradiol via aromatization and can actually induce some gynecomastia due to excessive estrogenic exposure. As such, unlike non-aromatizable androgens like DHT, use of testosterone for this purpose is likely inappropriate. There are few or no other options for topical androgens besides testosterone and DHT, so the practicality of this approach is limited.
In contrast to SERMs, topical androgens may not be fully effective for preventing breast development. Whereas SERMs can abolish breast development caused by estrogens, sufficient exposure to estrogens can still mediate significant breast development even in the context of strong androgenic exposure. In addition, topical application of androgens to the breasts is likely to cause local body hair growth and other local androgenic effects, like masculine skin changes, oily skin, and acne. For many transfeminine individuals, such changes would probably be unacceptable. Lastly, there is a risk of systemic distribution of the topically applied androgen (Kuhn et al., 1983a), and hence of elevated circulating androgen levels and androgenic effects elsewhere in the body. This risk would be lessened in combination with an androgen receptor antagonist like bicalutamide. However, androgen receptor antagonists also have the potential to block the local effects of the topical androgen.
Breast Removal Surgery and Breast Irradiation
Two non-medication-based alternatives for prevention of breast development are prophylactic surgical breast removal and prophylactic breast irradiation.
If there is no excess skin, mastectomy, or breast removal surgery, can remove the breasts without leaving obvious scars, as was the case in this young transgender man. Mastectomy is a highly effective means of preventing breast development. Of course, it requires surgery however.
Exposure of the breasts to radiation inhibits subsequent breast development (Photos). Irradiation of the breasts is an inexpensive, easy, and effective technique that is commonly used as prophylaxis against gynecomastia in men with prostate cancer treated with estrogens or high-dose bicalutamide monotherapy (Viani, da Silva, & Stefano, 2012). It is less effective than SERMs however and generally only reduces the severity of gynecomastia rather than fully prevents it (Viani, da Silva, & Stefano, 2012). More concerningly, there is a theoretical increased risk of breast cancer with exposure of the breasts to radiation (Aksnessæther et al., 2018). Research has found a 100-fold higher incidence of breast cancer in young women whose breasts were exposed to radiation during childhood as a consequence of radiotherapy for cancer when compared to other young women (Zacharin, 2010). On the other hand, limited available evidence so far suggests minimal if any increase in breast cancer incidence in elderly men on androgen deprivation and/or estrogen therapy treated with breast irradiation to prevent gynecomastia (Aksnessæther et al., 2018; Viani, da Silva, & Stefano, 2012). However, cancer radiotherapy and other forms of radiation exposure increase the risk of breast cancer in men, particularly those exposed at a young age (Niewoehner, 2008; Giordano, 2005). In addition to theoretical cancer risk, heart and lung issues have been associated with breast irradiation in elderly men with prostate cancer (Tunio et al., 2012; Viani, da Silva, & Stefano, 2012). Due to these health risks, breast irradiation for prevention of breast development is an inadvisable option.
An obvious drawback of breast development prevention with both surgical breast removal and prophylactic breast irradiation is that they are irreversible. If the person ever changes their mind about not wanting breasts or eventually decides to fully transition (a not uncommon occurrence), there is no going back on the choice to permanently negate breast development.
Degree, Onset, and Reversibility of Breast Development
For reasons that are not entirely clear, it is notable that transfeminine people tend to have poor or suboptimal breast development relative to cisgender women (Wierckx, Gooren, & T’Sjoen, 2014; de Blok et al., 2018; Reisman, Goldstein, & Safer, 2019). Likewise, in generally elderly men with prostate cancer, high-dose bicalutamide monotherapy and high-dose estrogen therapy both cause high rates of gynecomastia but produce only mild-to-moderate gynecomastia in 90% of cases (Wiki; Ockrim et al., 2003). (Whether their advanced age is a factor here or not is uncertain but could be involved however.) Hence, any person who was assigned male at birth should, generally speaking or on average, not necessarily expect a marked degree of breast development. Small breasts should generally be anticipated as the most likely outcome. There are always exceptions however, with a subset of transfeminine people experiencing substantial breast development. Hence, the degree of breast development one experiences is a matter of chance, and caution should be advised.
There are a few things to be aware of about breast development. One is that it happens slowly and is not something that becomes substantial overnight. Another is that will not continue to progress if hormones are withdrawn. And finally, it seems to be at least partially reversible if medications are discontinued within a certain amount of time (e.g., one year) (Mancino, Young, & Bland, 2018; Kanakis et al., 2019). However, a study found that bicalutamide monotherapy-induced gynecomastia outcome was worse if tamoxifen was introduced within a month of symptom onset rather than used from the start of therapy (Serretta et al. 2012). In any case, for the preceding reasons, a given individual could self-monitor their breast development, and, if it becomes too much for their liking, alter their medication regimen as desired in order to prevent further or reverse existing breast growth. Hence, breast development may not be something that should be feared excessively.
Updates
Update 1: Xu et al. (2021)
In June 2021, the following review on SERMs for non-binary transfeminine people was published:
Xu, J. Y., O’Connell, M. A., Notini, L., Cheung, A. S., Zwickl, S., & Pang, K. C. (2021). Selective Estrogen Receptor Modulators: A Potential Option For Non-Binary Gender-Affirming Hormonal Care? Frontiers in Endocrinology, 12, 701364. [DOI:10.3389/fendo.2021.701364]
The article is the first literature of its kind. In relation to the lack of evidence for the efficacy and safety of SERMs in non-binary transfeminine hormone therapy at present, it stated that SERMs should not be routinely recommended to non-binary transfeminine people. However, it was cautiously permissive of their use in non-binary transfeminine people who request them and provide informed consent. The review concluded that SERMs are a theoretically attractive potential option for non-binary transfeminine hormone therapy and called for clinical research on them for these purposes.
Update 2: van Dijken et al. (2022) and Cocchetti et al. (2022)
In March 2022, one of the first studies on tailored hormone therapy for non-binary people was published:
van Dijken, J. B., Steensma, T. D., Wensing-Kruger, S. A., Heijer, M. D., & Dreijerink, K. M. (2022). Tailored Gender-Affirming Hormone Treatment in Nonbinary Transgender Individuals: A Retrospective Study in a Referral Center Cohort. Transgender Health, 8(3), 220–225. [DOI:10.1089/trgh.2021.0032]
Cocchetti, C., Romani, A., Collet, S., Greenman, Y., Schreiner, T., Wiepjes, C., den Heijer, M., T’Sjoen, G., & Fisher, A. D. (2022). The ENIGI (European Network for the Investigation of Gender Incongruence) Study: Overview of Acquired Endocrine Knowledge and Future Perspectives. Journal of Clinical Medicine, 11(7), 1784. [DOI:10.3390/jcm11071784]
Here are some excerpts from the review:
The main future perspectives [of the ENIGI] should include the evaluation of the efficacy and safety of non-standardized hormonal treatment in non-binary trans people.
Regarding GAHT protocols, all participants were treated in line with the World Professional Association for Transgender Health (WPATH) recommendations [2]. Originally, the binary conception of gender was also extended to the transgender experience, limiting hormonal treatment protocols to obtaining a full virilization or feminization/de-virilization. In line with that, trans AMAB (assigned male at birth) people received both anti-androgens and estrogens aimed at obtaining estradiol and testosterone levels within the adult cisgender women’s range.
Trans AFAB (assigned female at birth) people received—still in a binary perspective of gender—T treatment to obtain full virilization, following the same principles of hormone replacement treatment in hypogonadal cisgender men.
Despite growing evidence based on data from many participants, there are still many questions that need to be answered. First of all, to date the ENIGI collaboration has evaluated only the efficacy and safety profile of standardized GAHT in trans people requesting full de-/masculinization or de-/feminization. However, non-binary transgender people represent a growing body of those who refer to gender clinics, and requests for non-standardized GAHT are increasing [42,43]. In line with this, some authors hypothesized both pharmacological and non-pharmacological strategies to respond to the different requests of non-binary people [44,45]. Given the complete lack of data on the efficacy and safety of these treatments, the ENIGI collaboration should focus more on this issue in the near future, and given the large number of participants, individualized variations in hormone treatment are now welcomed.
Supplementary Material
An interesting anecdotal report of hormone therapy with bicalutamide 50 mg/day, cyproterone acetate 12.5 mg/day, and raloxifene 60 mg/day in a formerly self-identified femboy can be found here (PDF).
References
Aksnessæther, B. Y., Solberg, A., Klepp, O. H., Myklebust, T. Å., Skovlund, E., Hoff, S. R., Vatten, L. J., & Lund, J. (2018). Does Prophylactic Radiation Therapy to Avoid Gynecomastia in Patients With Prostate Cancer Increase the Risk of Breast Cancer? International Journal of Radiation Oncology*Biology*Physics, 101(1), 211–216. [DOI:10.1016/j.ijrobp.2018.01.096]
Bach, P. V., Najari, B. B., & Kashanian, J. A. (2016). Adjunct Management of Male Hypogonadism. Current Sexual Health Reports, 8(4), 231–239. [DOI:10.1007/s11930-016-0089-7]
Barbieri, R. L. (1992). Hormone treatment of endometriosis: The estrogenthreshold hypothesis. American Journal of Obstetrics and Gynecology, 166(2), 740–745. [DOI:10.1016/0002-9378(92)91706-g]
Bass, M., Gonzalez, L. J., Colip, L., Sharon, N., & Conklin, J. (2018). Rethinking gender: The nonbinary approach. American Journal of Health-System Pharmacy, 75(22), 1821–1823. [DOI:10.2146/ajhp180236]
Bedognetti, D., Rubagotti, A., Zoppoli, G., & Boccardo, F. (2010). Gynaecomastia: The Anastrozole Paradox. Journal of Pediatric Endocrinology and Metabolism, 23(1–2), 205–206. [DOI:10.1515/jpem.2010.23.1-2.205]
Benveniste, O., Simon, A., & Herson, S. (2001). Successful Percutaneous Dihydrotestosterone Treatment of Gynecomastia Occurring during Highly Active Antiretroviral Therapy: Four Cases and a Review of the Literature. Clinical Infectious Diseases, 33(6), 891–893. [DOI:10.1086/322637]
Birzniece, V., Meinhardt, U. J., Gibney, J., Johannsson, G., Armstrong, N., Baxter, R. C., & Ho, K. K. (2012). Differential Effects of Raloxifene and Estrogen on Body Composition in Growth Hormone-Replaced Hypopituitary Women. The Journal of Clinical Endocrinology & Metabolism, 97(3), 1005–1012. [DOI:10.1210/jc.2011-2837]
Boccardo, F., Rubagotti, A., Battaglia, M., Di Tonno, P., Selvaggi, F., Conti, G., Comeri, G., Bertaccini, A., Martorana, G., Galassi, P., Zattoni, F., Macchiarella, A., Siragusa, A., Muscas, G., Durand, F., Potenzoni, D., Manganelli, A., Ferraris, V., & Montefiore, F. (2005). Evaluation of Tamoxifen and Anastrozole in the Prevention of Gynecomastia and Breast Pain Induced by Bicalutamide Monotherapy of Prostate Cancer. Journal of Clinical Oncology, 23(4), 808–815. [DOI:10.1200/jco.2005.12.013]
Burshell, A. L., Anderson, S. J., Leib, E. S., Johnston, C. C., & Costantino, J. P. (1999). The effect of tamoxifen on pre and postmenopausal bone. Journal of Bone and Mineral Research, 14(Suppl 1), S158–S158. [Google Scholar]
Caron, P., Khoury, J. M., Bennet, A., & Louvet, J. P. (1987). Treatment of pubertal gynecomastia with percutaneous dihydrotestosterone gel - long-term clinical outcome. La Semaine des Hôpitaux, 63, 2167–2170. [Google Scholar]
Carswell, J. M., & Roberts, S. A. (2017). Induction and Maintenance of Amenorrhea in Transmasculine and Nonbinary Adolescents. Transgender Health, 2(1), 195–201. [DOI:10.1089/trgh.2017.0021]
Chen, L., Ko, N., & Chen, K. (2019). Medical Treatment for Osteoporosis: From Molecular to Clinical Opinions. International Journal of Molecular Sciences, 20(9), 2213. [DOI:10.3390/ijms20092213]
Cocchetti, C., Ristori, J., Romani, A., Maggi, M., & Fisher, A. D. (2020). Hormonal Treatment Strategies Tailored to Non-Binary Transgender Individuals. Journal of Clinical Medicine, 9(6), 1609. [DOI:10.3390/jcm9061609]
Cocchetti, C., Romani, A., Collet, S., Greenman, Y., Schreiner, T., Wiepjes, C., den Heijer, M., T’Sjoen, G., & Fisher, A. D. (2022). The ENIGI (European Network for the Investigation of Gender Incongruence) Study: Overview of Acquired Endocrine Knowledge and Future Perspectives. Journal of Clinical Medicine, 11(7), 1784. [DOI:10.3390/jcm11071784]
De Corrado, G., Vetri, M., Padova, G., & Pezzino, V. (1998). Effect of prolonged treatment with percutaneous dihydrotestosterone (DHT) in persistent pubertal gynaecomastia (PPG). International Journal of Andrology, 21, 40–40. [Google Scholar]
Dane, C., Dane, B., Cetin, A., & Erginbas, M. (2007). Comparison of the effects of raloxifene and low-dose hormone replacement therapy on bone mineral density and bone turnover in the treatment of postmenopausal osteoporosis. Gynecological Endocrinology, 23(7), 398–403. [DOI:10.1080/09513590701414907]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de G. Buff Passone, C., Kuperman, H., Cabral de Menezes-Filho, H., Spassapan Oliveira Esteves, L., Lana Obata Giroto, R., & Damiani, D. (2015). Tamoxifen Improves Final Height Prediction in Girls with McCune-Albright Syndrome: A Long Follow-Up. Hormone Research in Paediatrics, 84(3), 184–189. [DOI:10.1159/000435881]
de Ronde, W., ten Kulve, J., Woerdeman, J., Kaufman, J., & de Jong, F. H. (2009). Effects of oestradiol on gonadotrophin levels in normal and castrated men. Clinical Endocrinology, 71(6), 874–879. [DOI:10.1111/j.1365-2265.2009.03573.x]
Di Lorenzo, G., Autorino, R., Perdonà, S., & De Placido, S. (2005). Management of gynaecomastia in patients with prostate cancer: a systematic review. The Lancet Oncology, 6(12), 972–979. [DOI:10.1016/s1470-2045(05)70464-2]
Doran, P. M., Riggs, B. L., Atkinson, E. J., & Khosla, S. (2001). Effects of Raloxifene, a Selective Estrogen Receptor Modulator, on Bone Turnover Markers and Serum Sex Steroid and Lipid Levels in Elderly Men. Journal of Bone and Mineral Research, 16(11), 2118–2125. [DOI:10.1359/jbmr.2001.16.11.2118]
Eberle, A. J., & Keenan, B. S. (1985). Treatment of persistent pubertal gynecomastia with dehydrotestosterone heptanoate (DHT-hp). Pediatric Research, 19(4), 185A–185A (abstract no. 448). [DOI:10.1203/00006450-198504000-00478]
Eberle, A. J., Sparrow, J. T., & Keenan, B. S. (1986). Treatment of persistent pubertal gynecomastia with dihydrotestosterone heptanoate. The Journal of Pediatrics, 109(1), 144–149. [DOI:10.1016/s0022-3476(86)80596-0]
Fagerlund, A., Cormio, L., Palangi, L., Lewin, R., Santanelli di Pompeo, F., Elander, A., & Selvaggi, G. (2015). Gynecomastia in Patients with Prostate Cancer: A Systematic Review. PLOS ONE, 10(8), e0136094. [DOI:10.1371/journal.pone.0136094]
Fentiman, I. S. (2018). Managing Male Mammary Maladies. European Journal of Breast Health, 14(1), 5–9. [DOI:10.5152/ejbh.2017.3841]
Fradet, Y., Egerdie, B., Andersen, M., Tammela, T. L., Nachabe, M., Armstrong, J., Morris, T., & Navani, S. (2007). Tamoxifen as Prophylaxis for Prevention of Gynaecomastia and Breast Pain Associated with Bicalutamide 150mg Monotherapy in Patients with Prostate Cancer: A Randomised, Placebo-Controlled, Dose–Response Study. European Urology, 52(1), 106–115. [DOI:10.1016/j.eururo.2007.01.031]
Francucci, C. M., Pantaleo, D., Iori, N., Camilletti, A., Massi, F., & Boscaro, M. (2005). Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women. Journal of Endocrinological Investigation, 28(9), 623–631. [DOI:10.1007/bf03347261]
Giordano, S. H. (2005). A Review of the Diagnosis and Management of Male Breast Cancer. The Oncologist, 10(7), 471–479. [DOI:10.1634/theoncologist.10-7-471]
Hadji, P., Colli, E., & Regidor, P. (2019). Bone health in estrogen-free contraception. Osteoporosis International, 30(12), 2391–2400. [DOI:10.1007/s00198-019-05103-6]
Heresová, J., Hampl, R., Kuska, L., & Stárka, L. (1981). Podávání nandrolondekanoátu nemocným s gynekomastií [Nandrolone decanoate administered to patients with Gynaecomastia]. Casopis Lekaru Ceskych, 120(52), 1602–1605. [Google Scholar] [PubMed]
Heresová, J., & Vrzáňová, M. (2003). Gynekomasti. Interní Medicína Pro Praxi, 5(1), 6–9. [URL] [PDF]
Ho, T. H., Nunez-Nateras, R., Hou, Y., Bryce, A. H., Northfelt, D. W., Dueck, A. C., Wong, B., Stanton, M. L., Joseph, R. W., & Castle, E. P. (2017). A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer. Clinical Genitourinary Cancer, 15(2), 196–202.e1. [DOI:10.1016/j.clgc.2016.08.026]
Kanakis, G. A., Nordkap, L., Bang, A. K., Calogero, A. E., Bártfai, G., Corona, G., Forti, G., Toppari, J., Goulis, D. G., & Jørgensen, N. (2019). EAA clinical practice guidelines—gynecomastia evaluation and management. Andrology, 7(6), 778–793. [DOI:10.1111/andr.12636]
Keenan, B. S., Fagan, C. J., & Richards, G. E. (1989). Adolescent gynecomastia in lean and obese subjects - radiographic characteristics and response to therapy with dihydrotestosterone enanthate (DHT-EN). Clinical Research, 37(1), A50–A50. [Google Scholar]
Kuhn, J. M., Laudat, M. H., Rieu, M., Roca, R., Wolf, L. M., Luton, J. P., & Bricaire, H. (1982). Sex steroid studies and dihydrotestosterone (DHT) treatment in idiopathic and hypogonadic gynecomastia. Journal of Steroid Biochemistry and Molecular Biology17(3), R81–R81 (abstract no. 242). [Google Scholar]
Kuhn, J. M., Laudat, M. H., Roca, R., Dugue, M. A., Luton, J. P., & Bricaire, H. (1983). Gynécomasties: effet du traitement prolongé par la dihydrotestostérone par voie per-cutanée [Gynecomastia: effect of prolonged treatment with dihydrotestosterone by the percutaneous route]. Presse Medicale, 12(1), 21–25. [Google Scholar 1] [Google Scholar 2] [PubMed]
Kuhn, J., Roca, R., Laudat, M., Rieu, M., Luton, J., & Bricaire, H. (1983). Studies on the treatment of idiopathic gynaecomastia with percutaneous dihydrotestosterone. Clinical Endocrinology, 19(4), 513–520. [DOI:10.1111/j.1365-2265.1983.tb00026.x]
Lawrence, S. E., Arnold Faught, K., Vethamuthu, J., & Lawson, M. L. (2004). Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. The Journal of Pediatrics, 145(1), 71–76. [DOI:10.1016/j.jpeds.2004.03.057]
Mancino, A. T., Young, Z. T., & Bland, K. I. (2018). Gynecomastia. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 104–115.e5). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00007-6]
Moghetti, P., Castello, R., Zamberlan, N., Rossini, M., Gatti, D., Negri, C., Tosi, F., Muggeo, M., & Adami, S. (1999). Spironolactone, But Not Flutamide, Administration Prevents Bone Loss in Hyperandrogenic Women Treated with Gonadotropin-Releasing Hormone Agonist. The Journal of Clinical Endocrinology & Metabolism, 84(4), 1250–1254. [DOI:10.1210/jcem.84.4.5606]
Moser, C., & Devereux, M. (2019). The Medical Management of Gender Dysphoric, Gender Fluid, Gender Nonconforming, Gender Queer, Nonbinary, and Transgender Patients: One Clinic’s Approach. Current Sexual Health Reports, 11(4), 421–429. [DOI:10.1007/s11930-019-00221-y]
Naroji, S., Shimy, K., Delot, E., & Gomez-Lobo, V. (2021). 59. Tamoxifen for Puberty Induction and Bone Mineral Density Protection in a Gender Non Binary Patient With 46, XY Complete Gonadal Dysgenesis: A Case Report. Journal of Pediatric and Adolescent Gynecology, 34(2), 262–262 (abstract no. 59). [DOI:10.1016/j.jpag.2021.02.063]
Nelson, A. L. (2010). DMPA: battered and bruised but still needed and used in the USA. Expert Review of Obstetrics & Gynecology, 5(6), 673–686. [DOI:10.1586/eog.10.60]
Niewoehner, C. B., & Schorer, A. E. (2008). Gynaecomastia and breast cancer in men. BMJ, 336(7646), 709–713. [DOI:10.1136/bmj.39511.493391.be]
Ockrim, J., Lalani, E., Laniado, M., Carter, S. S., & Abel, P. (2003). Transdermal Estradiol Therapy for Advanced Prostate Cancer—Forward to the Past? Journal of Urology, 169(5), 1735–1737. [DOI:10.1097/01.ju.0000061024.75334.40]
Palacios, S., Regidor, P., Colli, E., Skouby, S. O., Apter, D., Roemer, T., Egarter, C., Nappi, R. E., Skřivánek, A., Jakimiuk, A. J., Weyers, S., Ács, N., Elia, D., Gemzell Danielsson, K., & Bitzer, J. (2020). Oestrogen-free oral contraception with a 4 mg drospirenone-only pill: new data and a review of the literature. The European Journal of Contraception & Reproductive Health Care, 25(3), 221–227. [DOI:10.1080/13625187.2020.1743828]
Pang, K. C., Notini, L., McDougall, R., Gillam, L., Savulescu, J., Wilkinson, D., Clark, B. A., Olson-Kennedy, J., Telfer, M. M., & Lantos, J. D. (2020). Long-term Puberty Suppression for a Nonbinary Teenager. Pediatrics, 145(2), e20191606. [DOI:10.1542/peds.2019-1606]
Powles, T. J., Hickish, T., Kanis, J. A., Tidy, A., & Ashley, S. (1996). Effect of tamoxifen on bone mineral density measured by dual-energy x-ray absorptiometry in healthy premenopausal and postmenopausal women. Journal of Clinical Oncology, 14(1), 78–84. [DOI:10.1200/jco.1996.14.1.78]
Preželj, J., & Kocijančič, A. (1994). Antiandrogen Treatment with Spironolactone and Linestrenol Decreases Bone Mineral Density in Eumenorrhoeic Women with Androgen Excess. Hormone and Metabolic Research, 26(1), 46–48. [DOI:10.1055/s-2007-1000771]
Prezelj, J., & Kocijancic, A. (1999). Comment on Spironolactone, But Not Flutamide, Administration Prevents Bone Loss in Hyperandrogenic Women Treated with Gonadotropin-Releasing Hormone Agonist. The Journal of Clinical Endocrinology & Metabolism, 84(12), 4747–4751. [DOI:10.1210/jcem.84.12.4747a]
Raven, G., de Jong, F. H., Kaufman, J., & de Ronde, W. (2006). In Men, Peripheral Estradiol Levels Directly Reflect the Action of Estrogens at the Hypothalamo-Pituitary Level to Inhibit Gonadotropin Secretion. The Journal of Clinical Endocrinology & Metabolism, 91(9), 3324–3328. [DOI:10.1210/jc.2006-0462]
Reisman, T., Goldstein, Z., & Safer, J. D. (2019). A Review of Breast Development in Cisgender Women and Implications for Transgender Women. Endocrine Practice, 25(12), 1338–1345. [DOI:10.4158/ep-2019-0183]
Richards, C., Bouman, W. P., Seal, L., Barker, M. J., Nieder, T. O., & T’Sjoen, G. (2016). Non-binary or genderqueer genders. International Review of Psychiatry, 28(1), 95–102. [DOI:10.3109/09540261.2015.1106446]
Rizzoli, R. (2018). Postmenopausal osteoporosis: Assessment and management. Best Practice & Research Clinical Endocrinology & Metabolism, 32(5), 739–757. [DOI:10.1016/j.beem.2018.09.005]
Roux, C. (1997). Estrogen therapy in postmenopausal osteoporosis. What we know and what we don’t. Revue du Rhumatisme (English Ed.), 64(6), 402–409. [Google Scholar] [PubMed] [PDF]
Saltzstein, D., Sieber, P., Morris, T., & Gallo, J. (2005). Prevention and management of bicalutamide-induced gynecomastia and breast pain: randomized endocrinologic and clinical studies with tamoxifen and anastrozole. Prostate Cancer and Prostatic Diseases, 8(1), 75–83. [DOI:10.1038/sj.pcan.4500782]
Seal, L. (2017). Adult Endocrinology. In Richards, C., Bouman, W. P., & Barker, M.-J. (Eds.). Genderqueer and Non-Binary Genders (Critical and Applied Approaches in Sexuality, Gender and Identity) (pp. 183–223). London: Palgrave Macmillan UK. [DOI:10.1057/978-1-137-51053-2_10]
Serretta, V., Altieri, V., Morgia, G., Nicolosi, F., De Grande, G., Mazza, R., Melloni, D., Allegro, R., Ferraù, F., & Gebbia, V. (2012). A Randomized Trial Comparing Tamoxifen Therapy vs. Tamoxifen Prophylaxis in Bicalutamide-Induced Gynecomastia. Clinical Genitourinary Cancer, 10(3), 174–179. [DOI:10.1016/j.clgc.2012.03.002]
T’Sjoen, G. G., Giagulli, V. A., Delva, H., Crabbe, P., De Bacquer, D., & Kaufman, J. (2005). Comparative Assessment in Young and Elderly Men of the Gonadotropin Response to Aromatase Inhibition. The Journal of Clinical Endocrinology & Metabolism, 90(10), 5717–5722. [DOI:10.1210/jc.2005-0982]
Ting, A. C., Chow, L. W., & Leung, Y. (2000). Comparison of Tamoxifen with Danazol in the Management of Idiopathic Gynecomastia. The American Surgeon, 66(1), 38–40. [DOI:10.1177/000313480006600108]
Trost, L. W., & Khera, M. (2014). Alternative Treatment Modalities for the Hypogonadal Patient. Current Urology Reports, 15(7), 417. [DOI:10.1007/s11934-014-0417-2]
Tsourdi, E., Kourtis, A., Farmakiotis, D., Katsikis, I., Salmas, M., & Panidis, D. (2009). The effect of selective estrogen receptor modulator administration on the hypothalamic-pituitary-testicular axis in men with idiopathic oligozoospermia. Fertility and Sterility, 91(4), 1427–1430. [DOI:10.1016/j.fertnstert.2008.06.002]
Tunio, M., Al-Asiri, M., Al-Amro, A., Bayoumi, Y., & Fareed, M. (2012). Optimal Prophylactic and Definitive Therapy for Bicalutamide-Induced Gynecomastia: Results of a Meta-analysis. Current Oncology, 19(4), 280–288. [DOI:10.3747/co.19.993]
van Dijken, J. B., Steensma, T. D., Wensing-Kruger, S. A., Heijer, M. D., & Dreijerink, K. M. (2022). Tailored Gender-Affirming Hormone Treatment in Nonbinary Transgender Individuals: A Retrospective Study in a Referral Center Cohort. Transgender Health, 8(3), 220–225. [DOI:10.1089/trgh.2021.0032]
Vestergaard, P. (2012). Raloxifene for the treatment of postmenopausal osteoporosis. International Journal of Clinical Rheumatology, 7(3), 261–269. [Google Scholar] [DOI:10.2217/ijr.12.19] [PDF]
Viani, G. A., Bernardes da Silva, L. G., & Stefano, E. J. (2012). Prevention of Gynecomastia and Breast Pain Caused by Androgen Deprivation Therapy in Prostate Cancer: Tamoxifen or Radiotherapy? International Journal of Radiation Oncology*Biology*Physics, 83(4), e519–e524. [DOI:10.1016/j.ijrobp.2012.01.036]
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical Review: Breast Development in Trans Women Receiving Cross-Sex Hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]
Xu, J. Y., O’Connell, M. A., Notini, L., Cheung, A. S., Zwickl, S., & Pang, K. C. (2021). Selective Estrogen Receptor Modulators: A Potential Option For Non-Binary Gender-Affirming Hormonal Care? Frontiers in Endocrinology, 12, 701364. [DOI:10.3389/fendo.2021.701364]
Zacharin, M. (2009). Disorders of ovarian function in childhood and adolescence: evolving needs of the growing child. An endocrine perspective. BJOG: An International Journal of Obstetrics & Gynaecology, 117(2), 156–162. [DOI:10.1111/j.1471-0528.2009.02399.x]
Zirilli, L., Maffei, L., Meunier, P., Chavassieux, P., Carani, C., & Rochira, V. (2009). The effects of long-term raloxifene and estradiol treatments on bone in a patient with congenital aromatase deficiency. Bone, 45(5), 827–832. [DOI:10.1016/j.bone.2009.03.672]
\ No newline at end of file
+An Exploration of Possibilities for Hormone Therapy in Non-Binary Transfeminine People - Transfeminine Science
Transfeminine people who are non-binary often desire different and more partial feminizing and demasculinizing changes compared to what occurs with conventional transfeminine hormone therapy. For example, non-binary transfeminine people may seek an intermediate physical and hormonal state between what is typical for males and females, may desire substantial or full feminization and demasculinization without breast development, may want to have a more sexually neutral or androgynous appearance, or may wish for feminization but concomitant preservation of more male-typical sexual desire and capacity. These partial goals for transfeminine hormone therapy are being increasingly encountered, but therapeutic methods for achieving them have not yet been studied or established in transgender medicine. Nonetheless, there are a variety of hormonal approaches by which such outcomes could theoretically be achieved. Demasculinization can be produced via partial to full testosterone suppression or blockade using varied doses of antiandrogens, progestogens, and/or gonadotropin-releasing hormone modulators. Feminization and other estrogenic effects with less or no breast development may be produced with selective estrogen receptor modulators or low doses of estradiol. Certain other hormonal agents can be used to achieve various specific effects, like 5α-reductase inhibitors to reduce body hair growth and scalp hair loss, topical androgens applied to the breasts to reduce breast development, and the androgen and anabolic steroid nandrolone decanoate as an alternative to testosterone with less effect in skin and hair follicles. Besides hormonal means, breast removal surgery can be sought to negate breast development. Health risks, particularly those related to androgen deprivation without adequate estrogenic replacement, are a concern with partial approaches to hormone therapy. One such risk, bone density loss, may be reduced with calcium and vitamin D supplementation, bisphosphonates, and weight-bearing exercise. There is increasing interest in partial approaches to hormone therapy in transgender health, so hopefully they will become better characterized and more available and utilized in transgender people in the future.
Introduction
“Non-binary” is a term that refers to transgender people who do not identify within the gender binary. Despite the fact that they do not identify as either male or female, many non-binary transmasculine and transfeminine people pursue hormone therapy just like transgender men and women. While some non-binary individuals opt for a full hormonal transition similarly to most binary-identified transgender people, many non-binary people would prefer only a partial hormonal transition. This could be to achieve an intermediate area between masculine and feminine characteristics, to achieve a more sexually neutral appearance, or to induce some but not all aspects of masculinization or feminization.
There are also individuals who seek hormonal feminization and/or demasculinization but do not actually identify as transgender. These cisgender-identified individuals often refer to themselves as “femboys”. Communities of these individuals exist on social media sites like Reddit (e.g., r/FemboyTransition). Feminization-inclined cisgender people who pursue hormonal transition often have similar preferences as transfeminine non-binary people—one of the most common of which is feminization without breast development. Sometimes these initially cisgender-identified hormonally transitioning individuals end up progressing to a transgender identity with time.
Partial approaches to hormonal transition and even widespread identification as non-binary are fairly recent developments. There is very little written on non-conventional approaches to hormone therapy of this sort in the published literature. Moreover, there are no available standards or guidelines for such therapy at this time. A number of recent reviews have started to discuss possibilities for non-binary hormone therapy however (Richards et al., 2016; Seal, 2017; Bass et al., 2018; Cocchetti et al., 2020). There is currently a discordance between the number of people who desire non-conventional hormonal transition and the clinical establishment of such therapy. Consequently, an exploration of the possibilities from a theoretical standpoint would be of value and is the aim of this review.
As a disclaimer, the ideas in this article are experimental and preliminary. No studies with the goal of partial hormonal transition in transgender people have been conducted as of present and there is no data or evidence in non-binary people to inform the use of such approaches. Instead, we can only extrapolate from theory and research in other groups of people at this time. Examples of these other groups in the case of transfeminine non-binary hormone therapy include cisgender men undergoing hormone therapy for prostate cancer, cisgender men going through treatment for gynecomastia (male breast development), and transgender women undergoing hormone therapy. For these reasons, the present discussion is exploratory and should not be taken as therapeutic recommendations.
Conventional Transfeminine Hormone Therapy
The goal of therapy in conventional hormone therapy for transgender women is to produce the maximum degrees of demasculinization and feminization—including breast development—that are possible. This is achieved by suppressing testosterone levels and increasing estradiol levels such that they are both within normal adult female ranges. Alternatively, the actions of testosterone can be blocked instead of full suppression of testosterone levels. The relevant hormonal changes are accomplished through the administration of hormonal medications including estrogens, progestogens, and/or antiandrogens. Estrogens produce feminization, including breast development, while testosterone suppression causes demasculinization—as well as disinhibits feminization. Estrogens, progestogens, and antiandrogens can all contribute to testosterone suppression.
The therapeutic goals of a subset of non-binary transfeminine people are equivalent to those of transgender women and hence are compatible with the effects of conventional transfeminine hormone therapy. That is, the maximum possible feminization, including breast development, and demasculinization are the aims of therapy. Non-binary transfeminine people with these preferences can simply use conventional transfeminine hormone therapy for their hormonal transition as opposed to more experimental and non-conventional partial approaches.
For a comprehensive introduction to conventional transfeminine hormone therapy, see this article:
The above article is intended to provide everything one needs to know to achieve a basic understanding of the subject. If you are new to the topic of transgender hormone therapy, it is highly recommended reading prior to continuing in the current article. The introduction covers the sex hormones, their effects, specific hormonal medications used, routes, and dosages for this type of hormone therapy. Much of this information is also applicable to non-conventional transfeminine hormone therapy.
Partial Transfeminine Hormone Therapy
Some non-binary transfeminine people (as well as cisgender-identified individuals seeking hormonal feminization) desire only partial feminization and/or demasculinization. Depending on the specific aims, this can be more complicated and require more thought than conventional transfeminine hormone therapy. The following goals of partial transfeminine hormone therapy may be encountered:
An intermediate physical and hormonal state between male and female
A more sexually neutral or androgynous appearance that is not necessarily masculine or feminine
Substantial or maximal feminization and demasculinization with little or no breast development
Substantial or maximal feminization and demasculinization with minimal or no loss of sexual desire, sexual function (i.e., erectile and orgasmic capacity), and/or fertility
The first of these goals is fairly straightforward in that it can entail what is essentially conventional transfeminine hormone therapy using lower medication doses. This will result in partial testosterone suppression and a mixture of both androgens and estrogens as major active sex hormones. The second goal involves deprivation of both androgens and estrogens. While possible, this can have negative consequences as sex hormones are important for maintaining certain aspects of health and well-being. There are potential ways to avoid or mitigate such consequences however. The third and fourth goals are also technically possible but are more difficult to achieve and are likely to require more specialized and potentially complex hormonal approaches.
Suppression and Blockade of Testosterone
If the goal of non-binary transfeminine hormone therapy is simply to achieve an androgynous appearance with minimal or no feminization, this can be achieved via deprivation of testosterone without concomitant administration of an estrogen. There are multiple ways to achieve androgen deprivation or testosterone suppression in people assigned male at birth. These include high-dose progestogen therapy, medical and surgical castration with GnRH agonists/antagonists or gonadectomy, high-dose androgen receptor antagonist therapy, and a few other other possibilities. In this section, I’ll discuss androgen deprivation largely from the standpoint of efficacy. There are problems with androgen deprivation alone in terms of tolerability and safety due to the co-consequence of estrogen deficiency however, which I’ll discuss in the subsequent section.
Testosterone Suppression with High Doses of Progestogens
Androgen deprivation can be achieved with high doses of progestogens, which suppress testosterone levels by up to 50 to 70% (Aly, 2019). This is a substantial decrease in testosterone levels, but is not quite into the female range. Androgen receptor antagonists can additionally be included to block the remaining testosterone that is not suppressed if desired. For these purposes, low-dose cyproterone acetate (e.g., 5.0–12.5 mg/day) (Aly, 2019) plus bicalutamide (e.g., 12.5–50 mg/day) or spironolactone (e.g., 200–400 mg/day) is likely to be an effective regimen. As an alternative to cyproterone acetate, high doses of other progestogens, such as just about any other progestin, or alternatively rectal progesterone (Aly, 2018), can be used instead.
Testosterone Suppression with Medical or Surgical Castration
GnRH agonists and antagonists are another option for testosterone suppression. These medications suppress testosterone levels by about 95%, or into the normal female range or male castrate range (<50 ng/dL). However, GnRH agonists and antagonists are very expensive, although there may be some viable options for obtaining them more cheaply (e.g., certain online pharmacies) (Aly, 2019).
Gonadectomy, or surgical removal of the gonads, can be performed as a more permanent alternative to GnRH agonists and antagonists. However, this procedure is expensive (a few thousand dollars USD), requires minor surgery, and can be more difficult to obtain. Most surgeons require letters from gender therapists and real-life experience; informed-consent surgeons do exist however. Gonadectomy is also irreversible, notably resulting in permanent loss of testes and sterility. In any case, gonadectomy is far less expensive and more convenient than GnRH agonists and antagonists in the long run.
Testosterone Blockade with Antiandrogens
Androgen receptor antagonists like bicalutamide and spironolactone act by directly binding to the androgen receptor and displacing androgens like testosterone and DHT from the receptor, thereby preventing its activation by these androgens. This is in contrast to therapies that act by suppressing androgen production and levels.
High-dose bicalutamide monotherapy (e.g., 150–300 mg/day) is a possible approach for androgen deprivation therapy. However, bicalutamide monotherapy increases testosterone and hence estradiol levels. The testosterone will be blocked by bicalutamide and will not have effects, but estradiol is increased to a concentration range that allows for marked or full feminization, including breast development. In addition, bicalutamide alone, even at very high doses, might not be enough to completely block male-range testosterone. With these considerations, if the goal is full demasculinization with no feminization or breast development, bicalutamide monotherapy is not something that, at least alone, can achieve this.
High-dose bicalutamide is expensive and potentially cost-prohibitive. High-dose spironolactone monotherapy is not a good option for this route as it is a relatively weak antiandrogen and likely falls far short of being able to handle male-range levels of testosterone (at least 200 mg/day appears to be required to fully block female testosterone levels) (Aly, 2018; Wiki). Concomitant partial suppression of testosterone and estrogen levels via additional use of a progestogen (e.g., cyproterone acetate) may be a more feasible option than an androgen receptor antagonist alone.
Some potentially major advantages of high-dose bicalutamide monotherapy are that in contrast to marked or full suppression of testosterone levels, bicalutamide monotherapy largely preserves sexual desire and erectile function and likely does not result in infertility.
Other Options for Testosterone Suppression
Another option is only partial demasculinization, which can be achieved essentially by using lower dosages of the medications discussed above (e.g., cyproterone acetate, bicalutamide). If desired, 5α-reductase inhibitors can be added in this context to more substantially decrease scalp hair loss and body hair growth. Note that if testosterone is more fully suppressed or blocked however, there is likely to be little or no benefit with 5α-reductase inhibitors.
Yet another possibility could be to incorporate low-dose nandrolone decanoate, an androgen receptor agonist and anabolic–androgenic steroid (AAS) with much less masculinizing/androgenic effect in skin and hair follicles (Aly, 2020). This AAS will help to suppress and replace testosterone levels. Nandrolone decanoate might also have the benefit of helping to maintain sexual desire and function. However, nandrolone decanoate was recently discontinued in the United States. Oxandrolone is another, similar AAS, but has been associated with liver toxicity.
Estrogen Deficiency and Replacement
While androgen deprivation therapy is effective for achieving the desired changes—specifically demasculinization without feminization—it is not recommended by itself. This is because estradiol is produced from testosterone and hence androgen deprivation results in estrogen deficiency as well. Estrogens are essential for maintaining bone density in both men and women, and without them, a person will quickly lose bone mass, eventually develop osteoporosis, and be at a high risk for bone fractures. Skeletal and postural disfigurement may also eventually occur (Figure; Figure). In addition, the person is likely to experience other menopause-like symptoms, such as hot flashes, mood and sleep problems, sexual dysfunction (e.g., low sexual desire, erectile dysfunction), and accelerated aging of the skin (Wiki). An increased risk of weight gain, type 2 diabetes, cardiovascular disease, and dementia may be associated with sex hormone deficiency as well. As such, extended deprivation of both androgens and estrogens with no estrogenic supplementation is not advisable.
With that said, a couple of clarifications should be made. Due to preservation of estradiol levels, high-dose bicalutamide monotherapy has minimal to no risk of bone density loss or most other menopausal symptoms. In addition, the low-dose cyproterone acetate plus low-dose bicalutamide option may have less of a risk of menopausal symptoms and possibly osteoporosis as well. This is because high-dose progestogens (of which “low-dose” cyproterone acetate certainly qualifies) can help treat certain menopausal symptoms such as hot flashes and possibly bone density loss, and also because some estradiol will be preserved (since testosterone will only be suppressed by 70 to 80% rather than more fully). With that said however, in the latter case, it is probably best not to take any risks.
Selective Estrogen Receptor Modulators (SERMs)
Instead of only androgen and estrogen deprivation, the inclusion of selective estrogen receptor modulators (SERMs), so-called partial estrogens, can be employed. These medications are partial agonists of the estrogen receptor, and have mixed estrogenic and antiestrogenic effects depending on the tissue. For example, the SERM raloxifene has estrogenic effects in bone, fat tissue, and the liver, but antiestrogenic effects in the breasts. In general, SERMs reduce bone density loss and osteoporosis risk while not causing breast development (and actually blocking it). A full list of SERMs can be found here. However, practically speaking, only raloxifene (Evista), tamoxifen (Nolvadex), and toremifene (Fareston) are available, inexpensive, and commonly used. For an overview of the estrogenic and antiestrogenic effects of the different SERMs in different tissues, see here. In general, SERMs have a fairly similar pattern of effects. Although we have some idea of the differential tissue effects of SERMs, in many cases we do not know how they behave in specific tissues. For example, only a single clinical study has shown that a SERM, specifically raloxifene, has estrogenic effects in fat tissue (Francucci et al., 2014). In addition, it is less clear how SERMs behave in, for example, skin, or in most of the brain.
SERMs also have various side effects. For instance, SERMs commonly produce hot flashes as an adverse effect. However, the fairly recently introduced combination of bazedoxifene/conjugated estrogens (Duavee) has been found to reduce the incidence of hot flashes in postmenopausal women (Duavee label). It is still on-patent and hence is expensive however. In any case, SERMs are also likely to produce other menopause-like symptoms. Additionally, SERMs have estrogenic effects in the liver and therefore influence production of coagulation factors and decrease production of insulin-like growth factor-1 (IGF-1), among other potentially undesirable changes. Due to the increase in coagulation with SERMs, they have a notable risk of blood clots and cardiovascular complications like stroke (Aly, 2020). Some SERMs, like tamoxifen, also have unique off-target actions and risks, for instance rare liver toxicity. Raloxifene is a more selective and probably safer SERM than tamoxifen.
SERMs are effective for maintaining bone density. However, they are, unfortunately, only partially estrogenic in bone and hence are submaximally effective for such purposes—they are significantly more effective than no treatment at all but are not as effective as estrogens (Dane et al., 2007; Zirilli et al., 2009; Birzniece et al., 2012; Vestergaard, 2012). Indeed, SERMs have actually been found to significantly antagonize the effects of estradiol on bone, for instance on bone density in premenopausal women (Powles et al., 1996; Burshell et al., 1999) and on bone maturation and growth plate closure in cisgender girls with precocious puberty (Passone et al., 2015). One study in elderly men suggested that 60 mg/day raloxifene is maximally equivalent in terms of bone density effect to estradiol levels of about 26 pg/mL (Doran et al., 2001; Palacios et al., 2020). Below this estradiol level, raloxifene was estrogenic on bone density, whereas above this level, it was antiestrogenic on bone density (Doran et al., 2001; Palacios et al., 2020). As such, although SERMs increase bone density in the context of very low estradiol levels, they are not as effective as estrogens in terms of maintaining bone density and they may still allow for significantly decreased bone density when added to androgen deprivation in non-binary transfeminine people.
Low-Dose Estrogen Supplementation
An alternative to SERMs for avoiding estrogen deficiency is low-dose estrogen therapy. A dosage of oral estradiol of about 1 to 2 mg/day or estradiol levels of about 30 to 50 pg/mL (via another route, for instance transdermal patches) is all that is needed for complete or near-complete prevention of bone density loss (Barbieri, 1992; Roux, 1997; Hadji, Colli, & Regidor, 2019). Moreover, estradiol has a better tolerability and safety profile than SERMs, with a much lower risk of blood clots (Aly, 2020).
A problem with estrogen therapy however is that in the absence of androgens, estrogens even at low levels will induce substantial feminization and breast development. Estradiol levels in normal cisgender girls gradually increase from around 5 to 10 pg/mL at the start of puberty to 50 or 60 pg/mL by late puberty, and these low levels produce full developent of the female secondary sex characteristics (Aly, 2020). Similarly, cisgender females with complete androgen insensitivity syndrome (CAIS) have estradiol levels of only about 35 pg/mL on average yet have complete feminization and excellent breast development (Aly, 2020; Table). Hence, the addition of low-dose estradiol to androgen deprivation would likely be a full transition. Very low doses of estradiol, for instance 0.5 mg/day oral estradiol or a 14 μg/day estradiol patch, achieving estradiol levels of only maybe 20 pg/mL, may be feasible and may result in less feminization. But, while effective for improving bone density (Dane et al., 2007; Birzniece et al., 2012), such doses/levels would not fully protect against bone density loss and other menopause-like symptoms and would likely still produce at least partial feminization. It is notable that even GnRH agonists/antagonists and gonadectomy alone—which reduce estradiol levels to around 10 pg/mL—have a rate of mild gynecomastia of as high as 15% (Di Lorenzo et al., 2005).
Interestingly, spironolactone was found at 100 mg/day to fully prevent GnRH agonist-induced bone density loss in women in a small randomized controlled trial (Moghetti et al., 1999). The authors hypothesized that this was due to its antimineralocorticoid activity, as aldosterone is negatively correlated with bone density (Moghetti et al., 1999). However, in another study, 100 mg/day spironolactone did not prevent bone density loss caused by high-dose progestogen therapy in the form of 5 mg/day lynestrenol in women (Preželj & Kocijančič, 1994; Preželj & Kocijančič, 1999). Hence, spironolactone should not be relied upon for preservation of bone density.
Onset and Reversibility of Bone Loss
Certain medications used in premenopausal women suppress gonadal sex hormone production and are associated with decreased bone density. These therapies can provide insight on the risk of bone density loss that may occur in non-binary transfeminine people deprived of sex hormones. Examples of such medications include progestogen-only birth control, which partially suppresses estradiol levels (to around 20–50 pg/mL) (Hadji, Colli, & Regidor, 2019), and GnRH agonists/antagonists, which partially to fully suppress estradiol levels depending on the medication and dose. Minimal or no bone density loss occurs with estradiol levels of 30 to 50 pg/mL, whereas significant bone density loss occurs with estradiol levels of 20 to 30 pg/mL (Hadji, Colli, & Regidor, 2019). Reassuringly however, bone density has been found to substantially or fully recover within a few years following discontinuation of progestogen-only birth control in young premenopausal women (Nelson, 2010). Along similar lines, therapy with the GnRH antagonist elagolix is considered to be acceptably safe in premenopausal women for up to 2 years at a dose that results in partial suppression of estradiol levels (to about 40 pg/mL) and for up to 6 months at a dose that results in maximal suppression of estradiol levels (to about 10 pg/mL) (Wiki). As such, a limited period of sex hormone deprivation—for instance as a trial of non-binary transfeminine hormone therapy—may be reasonably safe in terms of bone health. Long-term therapy should include adequate measures to avoid bone density loss however.
Prevention of Breast Development
Estrogen Blockade and Suppression
If the goal is to produce full demasculinization and some or full feminization with the sole exception of breast development, there are a number of ways to possibly achieve this. Androgen deprivation without estrogen supplementation will achieve demasculinization without any feminization or breast development (except for bicalutamide monotherapy of course). However, it is not recommended for reasons described above and would not provide feminization. SERMs are an option; in addition to their capacity to treat osteoporosis, they are used to treat gynecomastia in men, and are capable of fully blocking gynecomastia induced by estrogens when used at sufficient doses (Fentiman, 2018). However, SERMs may allow for only partial feminization rather than full. Aromatase inhibitors, in contrast to SERMs, have no apparent place in this form of hormone therapy, as they are, surprisingly, poorly effective for prevention of gynecomastia (Fagerlund et al., 2015; Bedognetti et al., 2010).
Selective Estrogen Receptor Modulators and Testosterone Elevation
SERMs can be used to prevent breast development, as previously described. A problem with the use of SERMs for this purpose however is that when they are used in people assigned male at birth in whom the gonads are intact and testosterone levels are not suppressed, they will induce substantial increases in gonadal testosterone production and circulating testosterone levels. In men with hypogonadism and consequent low testosterone levels, the SERMs clomifene (20–50 mg/day) and enclomifene (12.5–25 mg/day) increase testosterone levels from about 200–300 ng/dL to about 450–600 ng/dL on average (a change of about 2.0- to 2.5-fold, with an absolute increase of 250–400 ng/dL in this patient population) (Bach, Najari, & Kashanian, 2016; Trost & Khera, 2014). Because they are so effective at increasing testosterone levels, SERMs are used to treat male hypogonadism as an alternative to exogenous testosterone administration. Worse still, SERMs appear to cause even greater increases in testosterone levels in non-hypogonadal men. One study found that 50 mg/day clomifene increased testosterone levels by about 850 ng/dL in healthy younger men and by about 500 ng/dL in elderly men (Trost & Khera, 2014). Similarly, strong increases in testosterone levels have been observed with partial suppression of estradiol levels via aromatase inhibition in young and older men (T’Sjoen et al., 2005; Raven et al., 2006; de Ronde et al., 2009).
If testosterone levels are suppressed, increases in testosterone levels with SERMs will, depending on the degree of testosterone suppression, be less applicable (e.g., with high-dose progestogen therapy) or not applicable at all (e.g., with medical/surgical castration). However, if a SERM is combined with, say, bicalutamide alone, the situation may become even worse. This is because bicalutamide itself produces considerable increases in testosterone levels similarly to SERMs. In elderly men with prostate cancer, bicalutamide monotherapy induces a 1.5- to 2.0-fold rise in testosterone levels, increasing them from about 300–400 ng/dL to about 500–600 ng/dL (an absolute change of about 150–250 ng/dL) (Wiki). In healthy younger men, bicalutamide may increase testosterone levels into the upper end of the normal male range (potentially into the range of around 900–1,200 ng/dL) (Wiki).
As bicalutamide is a competitive antagonist of the androgen receptor, its efficacy is fundamentally both dose-dependent and dependent on testosterone levels. Consequently, in combination with a SERM, it is possible that testosterone levels will become too high for bicalutamide to block. Moreover, endogenous androgens and estrogens are together responsible for maintaining normal homeostatic negative feedback on the hypothalamic–pituitary–gonadal axis (HPG axis) in people assigned male at birth. It seems logical that with little to suppress the axis, gonadal production and hence circulating levels of testosterone and estradiol may simply continue to rise until they overwhelm bicalutamide and/or the SERM it is combined with and restore negative feedback on the HPG axis. For these reasons, it is possible that the combination of bicalutamide and a SERM alone might not be a practical option for non-conventional feminizing hormone therapy.
With all of that said however, the combination of bicalutamide and tamoxifen has been assessed in various studies in men with prostate cancer (PubMed), and increases in testosterone levels have, rather surprisingly, not been a problem in these studies. In terms of the findings, bicalutamide and tamoxifen together do, as expected, increase total testosterone levels. However, the rise in total testosterone levels is not much different from that which occurs with bicalutamide alone. Moreover, free testosterone levels are either increased to a certain degree or are not actually raised at all (Boccardo et al., 2005; Saltzstein et al., 2005; Fradet et al., 2007). This is thought to be due to the fact that SERMs have potent estrogenic effects in the liver and result in increased production of sex hormone-binding globulin (SHBG), consequently reducing the fraction of free and hence bioactive testosterone in the circulation. This serves to offset the biological influence of the increase in total testosterone levels. In accordance, and reassuringly, unfavorable changes in markers of androgen receptor signaling, like higher prostate-specific antigen (PSA) levels, have not been observed relative to bicalutamide alone in the studies.
It is not clear why studies of bicalutamide plus tamoxifen have observed increases in total testosterone levels that are not that different from those of bicalutamide alone. Whatever the reason, these studies suggest that the combination of bicalutamide and tamoxifen (or certain other SERMs) might actually be feasible still for non-conventional feminizing hormone therapy. With that said however, elderly men are a different patient population than non-binary transfeminine people. Older men have diminished increases in testosterone levels with bicalutamide and SERMs compared to healthy young men. In relation to this, the combination might not be as favorable for younger people assigned male at birth.
Tamoxifen very well may be exchangeable with raloxifene for use in combination with bicalutamide. However, it should be noted that in contrast to tamoxifen, raloxifene has never been studied in combination with bicalutamide. Or, at least, not in gonadally intact men; one study of bicalutamide with raloxifene in castrated men with prostate cancer does exist, but that does not provide much in the way of useful information (Ho et al., 2017). Nor has raloxifene actually been properly studied for prevention of gynecomastia. A single retrospective chart review reported that it was effective for pubertal gynecomastia in boys (Lawrence et al., 2004). But that is all the data we have. Conversely, there are many high-quality studies of tamoxifen for prevention of gynecomastia, including in combination with bicalutamide.
In any case, used by themselves in men, raloxifene has been found to result in lower increases in testosterone levels than tamoxifen or toremifene (Tsourdi et al., 2009). As such, bicalutamide and raloxifene together may indeed be similar in terms of testosterone levels relative to the combination of bicalutamide and tamoxifen. This might just be due to raloxifene having lower efficacy as a SERM than tamoxifen or toremifene at the relevant clinical doses however (Tsourdi et al., 2009).
Unfortunately, pharmaceutical topical DHT is only available today in France (Drugs.com). Some compounding pharmacies in certain countries might provide topical DHT preparations. However, DHT does not seem to be available from any compounding pharmacies in the United States. In contrast to DHT, testosterone readily converts into estradiol via aromatization and can actually induce some gynecomastia due to excessive estrogenic exposure. As such, unlike non-aromatizable androgens like DHT, use of testosterone for this purpose is likely inappropriate. There are few or no other options for topical androgens besides testosterone and DHT, so the practicality of this approach is limited.
In contrast to SERMs, topical androgens may not be fully effective for preventing breast development. Whereas SERMs can abolish breast development caused by estrogens, sufficient exposure to estrogens can still mediate significant breast development even in the context of strong androgenic exposure. In addition, topical application of androgens to the breasts is likely to cause local body hair growth and other local androgenic effects, like masculine skin changes, oily skin, and acne. For many transfeminine individuals, such changes would probably be unacceptable. Lastly, there is a risk of systemic distribution of the topically applied androgen (Kuhn et al., 1983a), and hence of elevated circulating androgen levels and androgenic effects elsewhere in the body. This risk would be lessened in combination with an androgen receptor antagonist like bicalutamide. However, androgen receptor antagonists also have the potential to block the local effects of the topical androgen.
Breast Removal Surgery and Breast Irradiation
Two non-medication-based alternatives for prevention of breast development are prophylactic surgical breast removal and prophylactic breast irradiation.
If there is no excess skin, mastectomy, or breast removal surgery, can remove the breasts without leaving obvious scars, as was the case in this young transgender man. Mastectomy is a highly effective means of preventing breast development. Of course, it requires surgery however.
Exposure of the breasts to radiation inhibits subsequent breast development (Photos). Irradiation of the breasts is an inexpensive, easy, and effective technique that is commonly used as prophylaxis against gynecomastia in men with prostate cancer treated with estrogens or high-dose bicalutamide monotherapy (Viani, da Silva, & Stefano, 2012). It is less effective than SERMs however and generally only reduces the severity of gynecomastia rather than fully prevents it (Viani, da Silva, & Stefano, 2012). More concerningly, there is a theoretical increased risk of breast cancer with exposure of the breasts to radiation (Aksnessæther et al., 2018). Research has found a 100-fold higher incidence of breast cancer in young women whose breasts were exposed to radiation during childhood as a consequence of radiotherapy for cancer when compared to other young women (Zacharin, 2010). On the other hand, limited available evidence so far suggests minimal if any increase in breast cancer incidence in elderly men on androgen deprivation and/or estrogen therapy treated with breast irradiation to prevent gynecomastia (Aksnessæther et al., 2018; Viani, da Silva, & Stefano, 2012). However, cancer radiotherapy and other forms of radiation exposure increase the risk of breast cancer in men, particularly those exposed at a young age (Niewoehner, 2008; Giordano, 2005). In addition to theoretical cancer risk, heart and lung issues have been associated with breast irradiation in elderly men with prostate cancer (Tunio et al., 2012; Viani, da Silva, & Stefano, 2012). Due to these health risks, breast irradiation for prevention of breast development is an inadvisable option.
An obvious drawback of breast development prevention with both surgical breast removal and prophylactic breast irradiation is that they are irreversible. If the person ever changes their mind about not wanting breasts or eventually decides to fully transition (a not uncommon occurrence), there is no going back on the choice to permanently negate breast development.
Degree, Onset, and Reversibility of Breast Development
For reasons that are not entirely clear, it is notable that transfeminine people tend to have poor or suboptimal breast development relative to cisgender women (Wierckx, Gooren, & T’Sjoen, 2014; de Blok et al., 2018; Reisman, Goldstein, & Safer, 2019). Likewise, in generally elderly men with prostate cancer, high-dose bicalutamide monotherapy and high-dose estrogen therapy both cause high rates of gynecomastia but produce only mild-to-moderate gynecomastia in 90% of cases (Wiki; Ockrim et al., 2003). (Whether their advanced age is a factor here or not is uncertain but could be involved however.) Hence, any person who was assigned male at birth should, generally speaking or on average, not necessarily expect a marked degree of breast development. Small breasts should generally be anticipated as the most likely outcome. There are always exceptions however, with a subset of transfeminine people experiencing substantial breast development. Hence, the degree of breast development one experiences is a matter of chance, and caution should be advised.
There are a few things to be aware of about breast development. One is that it happens slowly and is not something that becomes substantial overnight. Another is that will not continue to progress if hormones are withdrawn. And finally, it seems to be at least partially reversible if medications are discontinued within a certain amount of time (e.g., one year) (Mancino, Young, & Bland, 2018; Kanakis et al., 2019). However, a study found that bicalutamide monotherapy-induced gynecomastia outcome was worse if tamoxifen was introduced within a month of symptom onset rather than used from the start of therapy (Serretta et al. 2012). In any case, for the preceding reasons, a given individual could self-monitor their breast development, and, if it becomes too much for their liking, alter their medication regimen as desired in order to prevent further or reverse existing breast growth. Hence, breast development may not be something that should be feared excessively.
Updates
Update 1: Xu et al. (2021)
In June 2021, the following review on SERMs for non-binary transfeminine people was published:
Xu, J. Y., O’Connell, M. A., Notini, L., Cheung, A. S., Zwickl, S., & Pang, K. C. (2021). Selective Estrogen Receptor Modulators: A Potential Option For Non-Binary Gender-Affirming Hormonal Care? Frontiers in Endocrinology, 12, 701364. [DOI:10.3389/fendo.2021.701364]
The article is the first literature of its kind. In relation to the lack of evidence for the efficacy and safety of SERMs in non-binary transfeminine hormone therapy at present, it stated that SERMs should not be routinely recommended to non-binary transfeminine people. However, it was cautiously permissive of their use in non-binary transfeminine people who request them and provide informed consent. The review concluded that SERMs are a theoretically attractive potential option for non-binary transfeminine hormone therapy and called for clinical research on them for these purposes.
Update 2: van Dijken et al. (2022) and Cocchetti et al. (2022)
In March 2022, one of the first studies on tailored hormone therapy for non-binary people was published:
van Dijken, J. B., Steensma, T. D., Wensing-Kruger, S. A., Heijer, M. D., & Dreijerink, K. M. (2022). Tailored Gender-Affirming Hormone Treatment in Nonbinary Transgender Individuals: A Retrospective Study in a Referral Center Cohort. Transgender Health, 8(3), 220–225. [DOI:10.1089/trgh.2021.0032]
Cocchetti, C., Romani, A., Collet, S., Greenman, Y., Schreiner, T., Wiepjes, C., den Heijer, M., T’Sjoen, G., & Fisher, A. D. (2022). The ENIGI (European Network for the Investigation of Gender Incongruence) Study: Overview of Acquired Endocrine Knowledge and Future Perspectives. Journal of Clinical Medicine, 11(7), 1784. [DOI:10.3390/jcm11071784]
Here are some excerpts from the review:
The main future perspectives [of the ENIGI] should include the evaluation of the efficacy and safety of non-standardized hormonal treatment in non-binary trans people.
Regarding GAHT protocols, all participants were treated in line with the World Professional Association for Transgender Health (WPATH) recommendations [2]. Originally, the binary conception of gender was also extended to the transgender experience, limiting hormonal treatment protocols to obtaining a full virilization or feminization/de-virilization. In line with that, trans AMAB (assigned male at birth) people received both anti-androgens and estrogens aimed at obtaining estradiol and testosterone levels within the adult cisgender women’s range.
Trans AFAB (assigned female at birth) people received—still in a binary perspective of gender—T treatment to obtain full virilization, following the same principles of hormone replacement treatment in hypogonadal cisgender men.
Despite growing evidence based on data from many participants, there are still many questions that need to be answered. First of all, to date the ENIGI collaboration has evaluated only the efficacy and safety profile of standardized GAHT in trans people requesting full de-/masculinization or de-/feminization. However, non-binary transgender people represent a growing body of those who refer to gender clinics, and requests for non-standardized GAHT are increasing [42,43]. In line with this, some authors hypothesized both pharmacological and non-pharmacological strategies to respond to the different requests of non-binary people [44,45]. Given the complete lack of data on the efficacy and safety of these treatments, the ENIGI collaboration should focus more on this issue in the near future, and given the large number of participants, individualized variations in hormone treatment are now welcomed.
Supplementary Material
An interesting anecdotal report of hormone therapy with bicalutamide 50 mg/day, cyproterone acetate 12.5 mg/day, and raloxifene 60 mg/day in a formerly self-identified femboy can be found here (PDF).
References
Aksnessæther, B. Y., Solberg, A., Klepp, O. H., Myklebust, T. Å., Skovlund, E., Hoff, S. R., Vatten, L. J., & Lund, J. (2018). Does Prophylactic Radiation Therapy to Avoid Gynecomastia in Patients With Prostate Cancer Increase the Risk of Breast Cancer? International Journal of Radiation Oncology*Biology*Physics, 101(1), 211–216. [DOI:10.1016/j.ijrobp.2018.01.096]
Aly. (2018). A Review of Studies on Spironolactone and Testosterone Suppression in Cisgender Men, Cisgender Women, and Transfeminine People. Transfeminine Science. [URL]
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2018). Buserelin, a Gonadotropin-Releasing Hormone Agonist, is Inexpensively Available as a Nasal Spray From Online Pharmacies. Transfeminine Science. [URL]
Aly. (2019). Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
Aly. (2020). Hormone Levels During Normal Puberty in Cisgender Girls. Transfeminine Science. [URL]
Aly. (2020). Nandrolone as a Potential Alternative Androgen with Reduced Androgenic Side Effects for Transfeminine and Transmasculine People. Transfeminine Science. [URL]
Bach, P. V., Najari, B. B., & Kashanian, J. A. (2016). Adjunct Management of Male Hypogonadism. Current Sexual Health Reports, 8(4), 231–239. [DOI:10.1007/s11930-016-0089-7]
Barbieri, R. L. (1992). Hormone treatment of endometriosis: The estrogenthreshold hypothesis. American Journal of Obstetrics and Gynecology, 166(2), 740–745. [DOI:10.1016/0002-9378(92)91706-g]
Bass, M., Gonzalez, L. J., Colip, L., Sharon, N., & Conklin, J. (2018). Rethinking gender: The nonbinary approach. American Journal of Health-System Pharmacy, 75(22), 1821–1823. [DOI:10.2146/ajhp180236]
Bedognetti, D., Rubagotti, A., Zoppoli, G., & Boccardo, F. (2010). Gynaecomastia: The Anastrozole Paradox. Journal of Pediatric Endocrinology and Metabolism, 23(1–2), 205–206. [DOI:10.1515/jpem.2010.23.1-2.205]
Benveniste, O., Simon, A., & Herson, S. (2001). Successful Percutaneous Dihydrotestosterone Treatment of Gynecomastia Occurring during Highly Active Antiretroviral Therapy: Four Cases and a Review of the Literature. Clinical Infectious Diseases, 33(6), 891–893. [DOI:10.1086/322637]
Birzniece, V., Meinhardt, U. J., Gibney, J., Johannsson, G., Armstrong, N., Baxter, R. C., & Ho, K. K. (2012). Differential Effects of Raloxifene and Estrogen on Body Composition in Growth Hormone-Replaced Hypopituitary Women. The Journal of Clinical Endocrinology & Metabolism, 97(3), 1005–1012. [DOI:10.1210/jc.2011-2837]
Boccardo, F., Rubagotti, A., Battaglia, M., Di Tonno, P., Selvaggi, F., Conti, G., Comeri, G., Bertaccini, A., Martorana, G., Galassi, P., Zattoni, F., Macchiarella, A., Siragusa, A., Muscas, G., Durand, F., Potenzoni, D., Manganelli, A., Ferraris, V., & Montefiore, F. (2005). Evaluation of Tamoxifen and Anastrozole in the Prevention of Gynecomastia and Breast Pain Induced by Bicalutamide Monotherapy of Prostate Cancer. Journal of Clinical Oncology, 23(4), 808–815. [DOI:10.1200/jco.2005.12.013]
Burshell, A. L., Anderson, S. J., Leib, E. S., Johnston, C. C., & Costantino, J. P. (1999). The effect of tamoxifen on pre and postmenopausal bone. Journal of Bone and Mineral Research, 14(Suppl 1), S158–S158. [Google Scholar]
Caron, P., Khoury, J. M., Bennet, A., & Louvet, J. P. (1987). Treatment of pubertal gynecomastia with percutaneous dihydrotestosterone gel - long-term clinical outcome. La Semaine des Hôpitaux, 63, 2167–2170. [Google Scholar]
Carswell, J. M., & Roberts, S. A. (2017). Induction and Maintenance of Amenorrhea in Transmasculine and Nonbinary Adolescents. Transgender Health, 2(1), 195–201. [DOI:10.1089/trgh.2017.0021]
Chen, L., Ko, N., & Chen, K. (2019). Medical Treatment for Osteoporosis: From Molecular to Clinical Opinions. International Journal of Molecular Sciences, 20(9), 2213. [DOI:10.3390/ijms20092213]
Cocchetti, C., Ristori, J., Romani, A., Maggi, M., & Fisher, A. D. (2020). Hormonal Treatment Strategies Tailored to Non-Binary Transgender Individuals. Journal of Clinical Medicine, 9(6), 1609. [DOI:10.3390/jcm9061609]
Cocchetti, C., Romani, A., Collet, S., Greenman, Y., Schreiner, T., Wiepjes, C., den Heijer, M., T’Sjoen, G., & Fisher, A. D. (2022). The ENIGI (European Network for the Investigation of Gender Incongruence) Study: Overview of Acquired Endocrine Knowledge and Future Perspectives. Journal of Clinical Medicine, 11(7), 1784. [DOI:10.3390/jcm11071784]
De Corrado, G., Vetri, M., Padova, G., & Pezzino, V. (1998). Effect of prolonged treatment with percutaneous dihydrotestosterone (DHT) in persistent pubertal gynaecomastia (PPG). International Journal of Andrology, 21, 40–40. [Google Scholar]
Dane, C., Dane, B., Cetin, A., & Erginbas, M. (2007). Comparison of the effects of raloxifene and low-dose hormone replacement therapy on bone mineral density and bone turnover in the treatment of postmenopausal osteoporosis. Gynecological Endocrinology, 23(7), 398–403. [DOI:10.1080/09513590701414907]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de G. Buff Passone, C., Kuperman, H., Cabral de Menezes-Filho, H., Spassapan Oliveira Esteves, L., Lana Obata Giroto, R., & Damiani, D. (2015). Tamoxifen Improves Final Height Prediction in Girls with McCune-Albright Syndrome: A Long Follow-Up. Hormone Research in Paediatrics, 84(3), 184–189. [DOI:10.1159/000435881]
de Ronde, W., ten Kulve, J., Woerdeman, J., Kaufman, J., & de Jong, F. H. (2009). Effects of oestradiol on gonadotrophin levels in normal and castrated men. Clinical Endocrinology, 71(6), 874–879. [DOI:10.1111/j.1365-2265.2009.03573.x]
Di Lorenzo, G., Autorino, R., Perdonà, S., & De Placido, S. (2005). Management of gynaecomastia in patients with prostate cancer: a systematic review. The Lancet Oncology, 6(12), 972–979. [DOI:10.1016/s1470-2045(05)70464-2]
Doran, P. M., Riggs, B. L., Atkinson, E. J., & Khosla, S. (2001). Effects of Raloxifene, a Selective Estrogen Receptor Modulator, on Bone Turnover Markers and Serum Sex Steroid and Lipid Levels in Elderly Men. Journal of Bone and Mineral Research, 16(11), 2118–2125. [DOI:10.1359/jbmr.2001.16.11.2118]
Eberle, A. J., & Keenan, B. S. (1985). Treatment of persistent pubertal gynecomastia with dehydrotestosterone heptanoate (DHT-hp). Pediatric Research, 19(4), 185A–185A (abstract no. 448). [DOI:10.1203/00006450-198504000-00478]
Eberle, A. J., Sparrow, J. T., & Keenan, B. S. (1986). Treatment of persistent pubertal gynecomastia with dihydrotestosterone heptanoate. The Journal of Pediatrics, 109(1), 144–149. [DOI:10.1016/s0022-3476(86)80596-0]
Fagerlund, A., Cormio, L., Palangi, L., Lewin, R., Santanelli di Pompeo, F., Elander, A., & Selvaggi, G. (2015). Gynecomastia in Patients with Prostate Cancer: A Systematic Review. PLOS ONE, 10(8), e0136094. [DOI:10.1371/journal.pone.0136094]
Fentiman, I. S. (2018). Managing Male Mammary Maladies. European Journal of Breast Health, 14(1), 5–9. [DOI:10.5152/ejbh.2017.3841]
Fradet, Y., Egerdie, B., Andersen, M., Tammela, T. L., Nachabe, M., Armstrong, J., Morris, T., & Navani, S. (2007). Tamoxifen as Prophylaxis for Prevention of Gynaecomastia and Breast Pain Associated with Bicalutamide 150mg Monotherapy in Patients with Prostate Cancer: A Randomised, Placebo-Controlled, Dose–Response Study. European Urology, 52(1), 106–115. [DOI:10.1016/j.eururo.2007.01.031]
Francucci, C. M., Pantaleo, D., Iori, N., Camilletti, A., Massi, F., & Boscaro, M. (2005). Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women. Journal of Endocrinological Investigation, 28(9), 623–631. [DOI:10.1007/bf03347261]
Giordano, S. H. (2005). A Review of the Diagnosis and Management of Male Breast Cancer. The Oncologist, 10(7), 471–479. [DOI:10.1634/theoncologist.10-7-471]
Hadji, P., Colli, E., & Regidor, P. (2019). Bone health in estrogen-free contraception. Osteoporosis International, 30(12), 2391–2400. [DOI:10.1007/s00198-019-05103-6]
Heresová, J., Hampl, R., Kuska, L., & Stárka, L. (1981). Podávání nandrolondekanoátu nemocným s gynekomastií [Nandrolone decanoate administered to patients with Gynaecomastia]. Casopis Lekaru Ceskych, 120(52), 1602–1605. [Google Scholar] [PubMed]
Heresová, J., & Vrzáňová, M. (2003). Gynekomasti. Interní Medicína Pro Praxi, 5(1), 6–9. [URL] [PDF]
Ho, T. H., Nunez-Nateras, R., Hou, Y., Bryce, A. H., Northfelt, D. W., Dueck, A. C., Wong, B., Stanton, M. L., Joseph, R. W., & Castle, E. P. (2017). A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer. Clinical Genitourinary Cancer, 15(2), 196–202.e1. [DOI:10.1016/j.clgc.2016.08.026]
Kanakis, G. A., Nordkap, L., Bang, A. K., Calogero, A. E., Bártfai, G., Corona, G., Forti, G., Toppari, J., Goulis, D. G., & Jørgensen, N. (2019). EAA clinical practice guidelines—gynecomastia evaluation and management. Andrology, 7(6), 778–793. [DOI:10.1111/andr.12636]
Keenan, B. S., Fagan, C. J., & Richards, G. E. (1989). Adolescent gynecomastia in lean and obese subjects - radiographic characteristics and response to therapy with dihydrotestosterone enanthate (DHT-EN). Clinical Research, 37(1), A50–A50. [Google Scholar]
Kuhn, J. M., Laudat, M. H., Rieu, M., Roca, R., Wolf, L. M., Luton, J. P., & Bricaire, H. (1982). Sex steroid studies and dihydrotestosterone (DHT) treatment in idiopathic and hypogonadic gynecomastia. Journal of Steroid Biochemistry and Molecular Biology17(3), R81–R81 (abstract no. 242). [Google Scholar]
Kuhn, J. M., Laudat, M. H., Roca, R., Dugue, M. A., Luton, J. P., & Bricaire, H. (1983). Gynécomasties: effet du traitement prolongé par la dihydrotestostérone par voie per-cutanée [Gynecomastia: effect of prolonged treatment with dihydrotestosterone by the percutaneous route]. Presse Medicale, 12(1), 21–25. [Google Scholar 1] [Google Scholar 2] [PubMed]
Kuhn, J., Roca, R., Laudat, M., Rieu, M., Luton, J., & Bricaire, H. (1983). Studies on the treatment of idiopathic gynaecomastia with percutaneous dihydrotestosterone. Clinical Endocrinology, 19(4), 513–520. [DOI:10.1111/j.1365-2265.1983.tb00026.x]
Lawrence, S. E., Arnold Faught, K., Vethamuthu, J., & Lawson, M. L. (2004). Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. The Journal of Pediatrics, 145(1), 71–76. [DOI:10.1016/j.jpeds.2004.03.057]
Mancino, A. T., Young, Z. T., & Bland, K. I. (2018). Gynecomastia. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 104–115.e5). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00007-6]
Moghetti, P., Castello, R., Zamberlan, N., Rossini, M., Gatti, D., Negri, C., Tosi, F., Muggeo, M., & Adami, S. (1999). Spironolactone, But Not Flutamide, Administration Prevents Bone Loss in Hyperandrogenic Women Treated with Gonadotropin-Releasing Hormone Agonist. The Journal of Clinical Endocrinology & Metabolism, 84(4), 1250–1254. [DOI:10.1210/jcem.84.4.5606]
Moser, C., & Devereux, M. (2019). The Medical Management of Gender Dysphoric, Gender Fluid, Gender Nonconforming, Gender Queer, Nonbinary, and Transgender Patients: One Clinic’s Approach. Current Sexual Health Reports, 11(4), 421–429. [DOI:10.1007/s11930-019-00221-y]
Naroji, S., Shimy, K., Delot, E., & Gomez-Lobo, V. (2021). 59. Tamoxifen for Puberty Induction and Bone Mineral Density Protection in a Gender Non Binary Patient With 46, XY Complete Gonadal Dysgenesis: A Case Report. Journal of Pediatric and Adolescent Gynecology, 34(2), 262–262 (abstract no. 59). [DOI:10.1016/j.jpag.2021.02.063]
Nelson, A. L. (2010). DMPA: battered and bruised but still needed and used in the USA. Expert Review of Obstetrics & Gynecology, 5(6), 673–686. [DOI:10.1586/eog.10.60]
Niewoehner, C. B., & Schorer, A. E. (2008). Gynaecomastia and breast cancer in men. BMJ, 336(7646), 709–713. [DOI:10.1136/bmj.39511.493391.be]
Ockrim, J., Lalani, E., Laniado, M., Carter, S. S., & Abel, P. (2003). Transdermal Estradiol Therapy for Advanced Prostate Cancer—Forward to the Past? Journal of Urology, 169(5), 1735–1737. [DOI:10.1097/01.ju.0000061024.75334.40]
Palacios, S., Regidor, P., Colli, E., Skouby, S. O., Apter, D., Roemer, T., Egarter, C., Nappi, R. E., Skřivánek, A., Jakimiuk, A. J., Weyers, S., Ács, N., Elia, D., Gemzell Danielsson, K., & Bitzer, J. (2020). Oestrogen-free oral contraception with a 4 mg drospirenone-only pill: new data and a review of the literature. The European Journal of Contraception & Reproductive Health Care, 25(3), 221–227. [DOI:10.1080/13625187.2020.1743828]
Pang, K. C., Notini, L., McDougall, R., Gillam, L., Savulescu, J., Wilkinson, D., Clark, B. A., Olson-Kennedy, J., Telfer, M. M., & Lantos, J. D. (2020). Long-term Puberty Suppression for a Nonbinary Teenager. Pediatrics, 145(2), e20191606. [DOI:10.1542/peds.2019-1606]
Powles, T. J., Hickish, T., Kanis, J. A., Tidy, A., & Ashley, S. (1996). Effect of tamoxifen on bone mineral density measured by dual-energy x-ray absorptiometry in healthy premenopausal and postmenopausal women. Journal of Clinical Oncology, 14(1), 78–84. [DOI:10.1200/jco.1996.14.1.78]
Preželj, J., & Kocijančič, A. (1994). Antiandrogen Treatment with Spironolactone and Linestrenol Decreases Bone Mineral Density in Eumenorrhoeic Women with Androgen Excess. Hormone and Metabolic Research, 26(1), 46–48. [DOI:10.1055/s-2007-1000771]
Prezelj, J., & Kocijancic, A. (1999). Comment on Spironolactone, But Not Flutamide, Administration Prevents Bone Loss in Hyperandrogenic Women Treated with Gonadotropin-Releasing Hormone Agonist. The Journal of Clinical Endocrinology & Metabolism, 84(12), 4747–4751. [DOI:10.1210/jcem.84.12.4747a]
Raven, G., de Jong, F. H., Kaufman, J., & de Ronde, W. (2006). In Men, Peripheral Estradiol Levels Directly Reflect the Action of Estrogens at the Hypothalamo-Pituitary Level to Inhibit Gonadotropin Secretion. The Journal of Clinical Endocrinology & Metabolism, 91(9), 3324–3328. [DOI:10.1210/jc.2006-0462]
Reisman, T., Goldstein, Z., & Safer, J. D. (2019). A Review of Breast Development in Cisgender Women and Implications for Transgender Women. Endocrine Practice, 25(12), 1338–1345. [DOI:10.4158/ep-2019-0183]
Richards, C., Bouman, W. P., Seal, L., Barker, M. J., Nieder, T. O., & T’Sjoen, G. (2016). Non-binary or genderqueer genders. International Review of Psychiatry, 28(1), 95–102. [DOI:10.3109/09540261.2015.1106446]
Rizzoli, R. (2018). Postmenopausal osteoporosis: Assessment and management. Best Practice & Research Clinical Endocrinology & Metabolism, 32(5), 739–757. [DOI:10.1016/j.beem.2018.09.005]
Roux, C. (1997). Estrogen therapy in postmenopausal osteoporosis. What we know and what we don’t. Revue du Rhumatisme (English Ed.), 64(6), 402–409. [Google Scholar] [PubMed] [PDF]
Saltzstein, D., Sieber, P., Morris, T., & Gallo, J. (2005). Prevention and management of bicalutamide-induced gynecomastia and breast pain: randomized endocrinologic and clinical studies with tamoxifen and anastrozole. Prostate Cancer and Prostatic Diseases, 8(1), 75–83. [DOI:10.1038/sj.pcan.4500782]
Seal, L. (2017). Adult Endocrinology. In Richards, C., Bouman, W. P., & Barker, M.-J. (Eds.). Genderqueer and Non-Binary Genders (Critical and Applied Approaches in Sexuality, Gender and Identity) (pp. 183–223). London: Palgrave Macmillan UK. [DOI:10.1057/978-1-137-51053-2_10]
Serretta, V., Altieri, V., Morgia, G., Nicolosi, F., De Grande, G., Mazza, R., Melloni, D., Allegro, R., Ferraù, F., & Gebbia, V. (2012). A Randomized Trial Comparing Tamoxifen Therapy vs. Tamoxifen Prophylaxis in Bicalutamide-Induced Gynecomastia. Clinical Genitourinary Cancer, 10(3), 174–179. [DOI:10.1016/j.clgc.2012.03.002]
T’Sjoen, G. G., Giagulli, V. A., Delva, H., Crabbe, P., De Bacquer, D., & Kaufman, J. (2005). Comparative Assessment in Young and Elderly Men of the Gonadotropin Response to Aromatase Inhibition. The Journal of Clinical Endocrinology & Metabolism, 90(10), 5717–5722. [DOI:10.1210/jc.2005-0982]
Ting, A. C., Chow, L. W., & Leung, Y. (2000). Comparison of Tamoxifen with Danazol in the Management of Idiopathic Gynecomastia. The American Surgeon, 66(1), 38–40. [DOI:10.1177/000313480006600108]
Trost, L. W., & Khera, M. (2014). Alternative Treatment Modalities for the Hypogonadal Patient. Current Urology Reports, 15(7), 417. [DOI:10.1007/s11934-014-0417-2]
Tsourdi, E., Kourtis, A., Farmakiotis, D., Katsikis, I., Salmas, M., & Panidis, D. (2009). The effect of selective estrogen receptor modulator administration on the hypothalamic-pituitary-testicular axis in men with idiopathic oligozoospermia. Fertility and Sterility, 91(4), 1427–1430. [DOI:10.1016/j.fertnstert.2008.06.002]
Tunio, M., Al-Asiri, M., Al-Amro, A., Bayoumi, Y., & Fareed, M. (2012). Optimal Prophylactic and Definitive Therapy for Bicalutamide-Induced Gynecomastia: Results of a Meta-analysis. Current Oncology, 19(4), 280–288. [DOI:10.3747/co.19.993]
van Dijken, J. B., Steensma, T. D., Wensing-Kruger, S. A., Heijer, M. D., & Dreijerink, K. M. (2022). Tailored Gender-Affirming Hormone Treatment in Nonbinary Transgender Individuals: A Retrospective Study in a Referral Center Cohort. Transgender Health, 8(3), 220–225. [DOI:10.1089/trgh.2021.0032]
Vestergaard, P. (2012). Raloxifene for the treatment of postmenopausal osteoporosis. International Journal of Clinical Rheumatology, 7(3), 261–269. [Google Scholar] [DOI:10.2217/ijr.12.19] [PDF]
Viani, G. A., Bernardes da Silva, L. G., & Stefano, E. J. (2012). Prevention of Gynecomastia and Breast Pain Caused by Androgen Deprivation Therapy in Prostate Cancer: Tamoxifen or Radiotherapy? International Journal of Radiation Oncology*Biology*Physics, 83(4), e519–e524. [DOI:10.1016/j.ijrobp.2012.01.036]
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical Review: Breast Development in Trans Women Receiving Cross-Sex Hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]
Xu, J. Y., O’Connell, M. A., Notini, L., Cheung, A. S., Zwickl, S., & Pang, K. C. (2021). Selective Estrogen Receptor Modulators: A Potential Option For Non-Binary Gender-Affirming Hormonal Care? Frontiers in Endocrinology, 12, 701364. [DOI:10.3389/fendo.2021.701364]
Zacharin, M. (2009). Disorders of ovarian function in childhood and adolescence: evolving needs of the growing child. An endocrine perspective. BJOG: An International Journal of Obstetrics & Gynaecology, 117(2), 156–162. [DOI:10.1111/j.1471-0528.2009.02399.x]
Zirilli, L., Maffei, L., Meunier, P., Chavassieux, P., Carani, C., & Rochira, V. (2009). The effects of long-term raloxifene and estradiol treatments on bone in a patient with congenital aromatase deficiency. Bone, 45(5), 827–832. [DOI:10.1016/j.bone.2009.03.672]
\ No newline at end of file
diff --git a/transfemscience.org/articles/oral-e2-leinung-2018/index.html b/transfemscience.org/articles/oral-e2-leinung-2018/index.html
index db25e073..f7ee222d 100644
--- a/transfemscience.org/articles/oral-e2-leinung-2018/index.html
+++ b/transfemscience.org/articles/oral-e2-leinung-2018/index.html
@@ -1 +1 @@
-Analysis of Estradiol and Testosterone Levels with Oral Estradiol in Transfeminine People Based on Leinung et al. (2018) - Transfeminine Science
Analysis of Estradiol and Testosterone Levels with Oral Estradiol in Transfeminine People Based on Leinung et al. (2018)
By Aly | First published March 27, 2019 | Last modified February 17, 2023
Abstract / TL;DR
Analysis has been performed and information is provided on estradiol and testosterone levels with different doses of oral estradiol alone or in combination with antiandrogens including spironolactone (200 mg/day) and finasteride (5 mg/day) in transfeminine people based on published data from Leinung et al. (2018). With oral estradiol alone, mean estradiol levels ranged from 39 to 159 pg/mL across a dose range of 1 to 8 mg/day and mean testosterone levels ranged from 160 ng/dL at estradiol levels of <50 pg/mL to 44–61 ng/dL at estradiol levels of >100 pg/mL. Finasteride was associated with higher testosterone levels at all estradiol dose levels while spironolactone was associated with lower estradiol levels but only at an oral estradiol dose of 8 mg/day and not at other doses (2–6 mg/day). Mean testosterone levels in post-gonadectomy transfeminine people were 22 ng/dL. These findings suggest that oral estradiol dose-dependently suppresses testosterone levels and that estradiol and testosterone levels with oral estradiol may be modified by antiandrogens in transfeminine people.
Introduction
A retrospective chart study which quantified estradiol levels and suppression of testosterone levels with oral estradiol alone and in combination with an antiandrogen in a large sample of transfeminine people in the United States was published in May 2018 by Leinung and colleagues. The sample size for the study was 166 transfeminine people and estradiol and testosterone levels were determined with chemiluminescent immunoassay (CLIA). Here is the citation for the study:
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
This is a very useful study because it has data in transfeminine people that can provide precise estimates for answers to several open questions about transfeminine hormone therapy. These include what estradiol levels will be achieved with different doses of oral estradiol, how much testosterone levels will be suppressed with different doses/levels of estradiol, and the influence of certain antiandrogens—specifically spironolactone and finasteride—on estradiol and testosterone levels with oral estradiol.
I’ve digitized and recreated the two main graphs of interest from the paper. These new graphs are of higher image quality than the originals and I feel have an improved appearance. They aren’t perfect replicas of the originals (overlapping data points in the original graphs prevented this from being possible), but they should be quite close (e.g., ± 4 data points). In addition to the remade graphs, I’ve created two new graphs using data from the figures in the paper. These new graphs are variations of the originals that I think may be more understandable and useful. The four graphs are shown below (Figures 1–4). A supplementary spreadsheet containing the extracted data used to create these graphs can be found here. Some of the raw data in the spreadsheet is also included below in the Data Tables section.
As this study was not a randomized controlled trial (RCT) and was instead a retrospective chart review, there are limitations with these data that must be noted. For example, estradiol levels with different doses of oral estradiol may be inaccurate to a degree because transfeminine people in the study had their doses adjusted based on hormone levels (e.g., low/unsatisfactory estradiol levels or testosterone suppression resulting in increased doses and high/excessive estradiol levels resulting in dose decreases). In any case, estradiol levels with oral estradiol in this study were fairly similar to those that have been reported in other studies (e.g., Lobo & Cassidenti, 1992; Kuhl, 2005; Wiki; Graphs). It should also be noted that hormone levels vary by study and blood-testing methodology used.
Graphs
Recreated Graphs
Figure 1: Estradiol levels (pg/mL) with different doses (mg/day) of oral estradiol (E2) in transfeminine people. Estradiol levels are represented by blue circles (●) with oral estradiol alone, by red squares (■) with oral estradiol plus finasteride, and by green diamonds (◆) with oral estradiol plus spironolactone. The lines of colors corresponding to those of the points represent linear trendlines for the data points. This figure has also been uploaded to and can be found on Wikipedia (Wiki).
Figure 2: Testosterone levels (ng/dL) at different levels of estradiol (pg/mL) with oral estradiol (E2) in transfeminine people. Testosterone levels are represented by blue circles (●) with oral estradiol alone, by red squares (■) with oral estradiol plus finasteride, and by green diamonds (◆) with oral estradiol plus spironolactone. The dashed horizontal grey line is the mean testosterone level in a comparison group of post-gonadectomy transfeminine people (21.7 ± 12.4 ng/dL, with 13 determinations below 10 ng/dL, the lower limit of detection for the assay). This figure has also been uploaded to and can be found on Wikipedia (Wiki).
New Graphs
Figure 3: Estradiol levels (pg/mL) with 1 to 8 mg/day oral estradiol (E2) alone (blue line) or in combination with 200 mg/day oral spironolactone (green line) in transfeminine people. The oral estradiol alone group is actually a combination of oral estradiol alone and oral estradiol taken together with finasteride (5 mg/day); these two groups showed no significant differences in estradiol levels in the original data so they were combined for this graph. Estradiol levels with estradiol alone versus estradiol plus spironolactone seemed to be different only at the highest oral estradiol dose level (8 mg/day). The error bars represent standard deviations from the mean. This figure has also been uploaded to and can be found on Wikipedia (Wiki).
Figure 4: Testosterone levels (ng/dL) at different ranges of estradiol levels (pg/mL) with oral estradiol (E2) alone (blue line) or in combination with 5 mg/day finasteride (red line) or 200 mg/day oral spironolactone (green line) in transfeminine people. The typical oral estradiol doses (mg/day) for each range of estradiol levels are also provided. The dashed horizontal purple line is the upper limit for the normal female or castrate range (~50 ng/dL), while the dashed horizontal grey line is the mean testosterone level in a comparison group of post-gonadectomy transfeminine people (21.7 ± 12.4 ng/dL, with 13 determinations below 10 ng/dL, the lower limit of detection for the assay). The error bars represent standard deviations from the mean. This figure has also been uploaded to and can be found on Wikipedia (Wiki).
Data Tables
Estradiol Levels with Oral Estradiol
Table 1: Estradiol levels with different doses of oral estradiol alonea in transfeminine people:
Dosage
n
Estradiol level (mean ± SD)
1 mg/day
5
39 ± 25 pg/mL
2 mg/day
24
62 ± 23 pg/mL
4 mg/day
34
102 ± 59 pg/mL
6 mg/day
80
125 ± 62 pg/mL
8 mg/day
24
159 ± 76 pg/mL
a Actually pooled data for oral estradiol alone and oral estradiol combined with finasteride (5 mg/day); these two groups showed no significant differences in estradiol levels in the original data so they were pooled together for this table.
Testosterone Levels with Oral Estradiol
Table 2: Testosterone levels at different estradiol levels with oral estradiol alonea in transfeminine people:
Estradiol level range
n
Estradiol level (mean ± SD)
Testosterone level (mean ± SD)
<50 pg/mL
11
33 ± 8.4 pg/mL
160 ± 139 ng/dL
50–100 pg/mL
24
76 ± 15 pg/mL
83 ± 106 ng/dL
100–150 pg/mL
21
125 ± 14 pg/mL
50 ± 37 ng/dL
150–200 pg/mL
8
170 ± 15 pg/mL
61 ± 57 ng/dL
200–250 pg/mL
4
227 ± 14 pg/mL
44 ± 33 ng/dL
a Only oral estradiol alone; oral estradiol combined with finasteride or spironolactone not included.
Update: Jain, Kwan, & Forcier (2019)
Shortly following the publication of Leinung et al. (2018), Jain and colleagues published a similar study of sublingual estradiol in combination with spironolactone and with or without medroxyprogesterone acetate in transfeminine people (Jain, Kwan, & Forcier, 2019).
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone acetate in Gender-Affirming therapy for Transwomen: results from a retrospective study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Lobo, R. A., & Cassidenti, D. L. (1992). Pharmacokinetics of Oral 17 β-Estradiol. The Journal of Reproductive Medicine, 37(1), 77–84. [Google Scholar] [PubMed] [PDF]
\ No newline at end of file
+Analysis of Estradiol and Testosterone Levels with Oral Estradiol in Transfeminine People Based on Leinung et al. (2018) - Transfeminine Science
Analysis of Estradiol and Testosterone Levels with Oral Estradiol in Transfeminine People Based on Leinung et al. (2018)
By Aly | First published March 27, 2019 | Last modified February 17, 2023
Abstract / TL;DR
Analysis has been performed and information is provided on estradiol and testosterone levels with different doses of oral estradiol alone or in combination with antiandrogens including spironolactone (200 mg/day) and finasteride (5 mg/day) in transfeminine people based on published data from Leinung et al. (2018). With oral estradiol alone, mean estradiol levels ranged from 39 to 159 pg/mL across a dose range of 1 to 8 mg/day and mean testosterone levels ranged from 160 ng/dL at estradiol levels of <50 pg/mL to 44–61 ng/dL at estradiol levels of >100 pg/mL. Finasteride was associated with higher testosterone levels at all estradiol dose levels while spironolactone was associated with lower estradiol levels but only at an oral estradiol dose of 8 mg/day and not at other doses (2–6 mg/day). Mean testosterone levels in post-gonadectomy transfeminine people were 22 ng/dL. These findings suggest that oral estradiol dose-dependently suppresses testosterone levels and that estradiol and testosterone levels with oral estradiol may be modified by antiandrogens in transfeminine people.
Introduction
A retrospective chart study which quantified estradiol levels and suppression of testosterone levels with oral estradiol alone and in combination with an antiandrogen in a large sample of transfeminine people in the United States was published in May 2018 by Leinung and colleagues. The sample size for the study was 166 transfeminine people and estradiol and testosterone levels were determined with chemiluminescent immunoassay (CLIA). Here is the citation for the study:
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
This is a very useful study because it has data in transfeminine people that can provide precise estimates for answers to several open questions about transfeminine hormone therapy. These include what estradiol levels will be achieved with different doses of oral estradiol, how much testosterone levels will be suppressed with different doses/levels of estradiol, and the influence of certain antiandrogens—specifically spironolactone and finasteride—on estradiol and testosterone levels with oral estradiol.
I’ve digitized and recreated the two main graphs of interest from the paper. These new graphs are of higher image quality than the originals and I feel have an improved appearance. They aren’t perfect replicas of the originals (overlapping data points in the original graphs prevented this from being possible), but they should be quite close (e.g., ± 4 data points). In addition to the remade graphs, I’ve created two new graphs using data from the figures in the paper. These new graphs are variations of the originals that I think may be more understandable and useful. The four graphs are shown below (Figures 1–4). A supplementary spreadsheet containing the extracted data used to create these graphs can be found here. Some of the raw data in the spreadsheet is also included below in the Data Tables section.
As this study was not a randomized controlled trial (RCT) and was instead a retrospective chart review, there are limitations with these data that must be noted. For example, estradiol levels with different doses of oral estradiol may be inaccurate to a degree because transfeminine people in the study had their doses adjusted based on hormone levels (e.g., low/unsatisfactory estradiol levels or testosterone suppression resulting in increased doses and high/excessive estradiol levels resulting in dose decreases). In any case, estradiol levels with oral estradiol in this study were fairly similar to those that have been reported in other studies (e.g., Lobo & Cassidenti, 1992; Kuhl, 2005; Wiki; Graphs). It should also be noted that hormone levels vary by study and blood-testing methodology used.
Graphs
Recreated Graphs
Figure 1: Estradiol levels (pg/mL) with different doses (mg/day) of oral estradiol (E2) in transfeminine people. Estradiol levels are represented by blue circles (●) with oral estradiol alone, by red squares (■) with oral estradiol plus finasteride, and by green diamonds (◆) with oral estradiol plus spironolactone. The lines of colors corresponding to those of the points represent linear trendlines for the data points. This figure has also been uploaded to and can be found on Wikipedia (Wiki).
Figure 2: Testosterone levels (ng/dL) at different levels of estradiol (pg/mL) with oral estradiol (E2) in transfeminine people. Testosterone levels are represented by blue circles (●) with oral estradiol alone, by red squares (■) with oral estradiol plus finasteride, and by green diamonds (◆) with oral estradiol plus spironolactone. The dashed horizontal grey line is the mean testosterone level in a comparison group of post-gonadectomy transfeminine people (21.7 ± 12.4 ng/dL, with 13 determinations below 10 ng/dL, the lower limit of detection for the assay). This figure has also been uploaded to and can be found on Wikipedia (Wiki).
New Graphs
Figure 3: Estradiol levels (pg/mL) with 1 to 8 mg/day oral estradiol (E2) alone (blue line) or in combination with 200 mg/day oral spironolactone (green line) in transfeminine people. The oral estradiol alone group is actually a combination of oral estradiol alone and oral estradiol taken together with finasteride (5 mg/day); these two groups showed no significant differences in estradiol levels in the original data so they were combined for this graph. Estradiol levels with estradiol alone versus estradiol plus spironolactone seemed to be different only at the highest oral estradiol dose level (8 mg/day). The error bars represent standard deviations from the mean. This figure has also been uploaded to and can be found on Wikipedia (Wiki).
Figure 4: Testosterone levels (ng/dL) at different ranges of estradiol levels (pg/mL) with oral estradiol (E2) alone (blue line) or in combination with 5 mg/day finasteride (red line) or 200 mg/day oral spironolactone (green line) in transfeminine people. The typical oral estradiol doses (mg/day) for each range of estradiol levels are also provided. The dashed horizontal purple line is the upper limit for the normal female or castrate range (~50 ng/dL), while the dashed horizontal grey line is the mean testosterone level in a comparison group of post-gonadectomy transfeminine people (21.7 ± 12.4 ng/dL, with 13 determinations below 10 ng/dL, the lower limit of detection for the assay). The error bars represent standard deviations from the mean. This figure has also been uploaded to and can be found on Wikipedia (Wiki).
Data Tables
Estradiol Levels with Oral Estradiol
Table 1: Estradiol levels with different doses of oral estradiol alonea in transfeminine people:
Dosage
n
Estradiol level (mean ± SD)
1 mg/day
5
39 ± 25 pg/mL
2 mg/day
24
62 ± 23 pg/mL
4 mg/day
34
102 ± 59 pg/mL
6 mg/day
80
125 ± 62 pg/mL
8 mg/day
24
159 ± 76 pg/mL
a Actually pooled data for oral estradiol alone and oral estradiol combined with finasteride (5 mg/day); these two groups showed no significant differences in estradiol levels in the original data so they were pooled together for this table.
Testosterone Levels with Oral Estradiol
Table 2: Testosterone levels at different estradiol levels with oral estradiol alonea in transfeminine people:
Estradiol level range
n
Estradiol level (mean ± SD)
Testosterone level (mean ± SD)
<50 pg/mL
11
33 ± 8.4 pg/mL
160 ± 139 ng/dL
50–100 pg/mL
24
76 ± 15 pg/mL
83 ± 106 ng/dL
100–150 pg/mL
21
125 ± 14 pg/mL
50 ± 37 ng/dL
150–200 pg/mL
8
170 ± 15 pg/mL
61 ± 57 ng/dL
200–250 pg/mL
4
227 ± 14 pg/mL
44 ± 33 ng/dL
a Only oral estradiol alone; oral estradiol combined with finasteride or spironolactone not included.
Update: Jain, Kwan, & Forcier (2019)
Shortly following the publication of Leinung et al. (2018), Jain and colleagues published a similar study of sublingual estradiol in combination with spironolactone and with or without medroxyprogesterone acetate in transfeminine people (Jain, Kwan, & Forcier, 2019).
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone acetate in Gender-Affirming therapy for Transwomen: results from a retrospective study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Lobo, R. A., & Cassidenti, D. L. (1992). Pharmacokinetics of Oral 17 β-Estradiol. The Journal of Reproductive Medicine, 37(1), 77–84. [Google Scholar] [PubMed] [PDF]
\ No newline at end of file
diff --git a/transfemscience.org/articles/oral-p4-low-levels-literature/index.html b/transfemscience.org/articles/oral-p4-low-levels-literature/index.html
index 0c0a8f8c..83d19d93 100644
--- a/transfemscience.org/articles/oral-p4-low-levels-literature/index.html
+++ b/transfemscience.org/articles/oral-p4-low-levels-literature/index.html
@@ -1 +1 @@
-Sources/Excerpts: Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects - Transfeminine Science
Sources/Excerpts: Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects
By Aly | First published August 4, 2018 | Last modified September 25, 2022
Preface
This is a literature sources/excerpts supplement to the main article which can be found here.
Excerpts From Literature Reviews
de Ziegler et al. (1998)
de Ziegler, D., Fanchin, R., de Moustier, B., & Bulletti, C. (1998). The hormonal control of endometrial receptivity: estrogen (E2) and progesterone. Journal of Reproductive Immunology, 39(1–2), 149–166. [DOI:10.1016/S0165-0378(98)00019-9]:
4.2. The oral route paradox and the methodological flaws in progesterone measurement
High levels of progesterone have been reported after oral administration of micronized progesterone (Simon et al., 1993). Yet, despite the high levels reported, the same authors describe incomplete endometrial effects. Today, we believe that the apparent paradox between high progesterone levels and minimal endometrial effects characteristic of oral progesterone has been elucidated by Nahoul et al. (1987, 1993) revealing methodological flaw in the measurement of plasma progesterone after oral administration. Indeed, while direct RIA for progesterone is valid for the measurement of plasma levels in the luteal phase of the menstrual cycle, this is not the case after oral administration. In an elegant paper, Nahoul et al. (1987) showed that because of the high levels of progesterone metabolites encountered after oral administration direct RIA measured values 3–5 times higher than those determined when measurements are conducted after extraction and separation on celite columns using an appropriate assay. Nahoul et al. (1993) showed that plasma levels of progesterone were only minimally elevated after oral ingestion of 100 mg of progesterone (barely reaching 1 ng/ml) and, particularly, were markedly lower than after vaginal administration of the same amounts of progesterone.
de Ziegler & Fanchin (2000)
de Ziegler, D., & Fanchin, R. (2000). Progesterone and progestins: applications in gynecology. Steroids, 65(10–11), 671–679. [DOI:10.1016/S0039-128X(00)00123-9]:
3.1. Oral progesterone
Progesterone is nearly entirely absorbed after oral ingestion when prepared in micronized form. Yet, because of intense inactivation by metabolism during first liver pass, the bioavailability of oral progesterone is notoriously poor at <10% [25]. This poor bioavailability explains that despite the large amounts used (up to 600 mg/day), oral progesterone fails to trigger the full array of endometrial changes seen in the late luteal phase of the menstrual cycle. Particularly, the last step of these changes (the predecidual transformation of stroma cells) fails to be induced by oral progesterone [3].
Some confusion has existed over these findings. Early reports claimed high plasma progesterone levels after oral administration of progesterone. However, fairly low plasma levels of progesterone have been reported when the hormone is given orally, and proper assays are used. Circulating levels of progesterone and its metabolites, determined by sufficiently specific assays, after oral and vaginal administration of 100 mg of progesterone are illustrated in Fig. 4. As can be seen, when taken orally, progesterone accounts for less than 10%, whereas most of ingested progesterone is transformed to 5α-reduced metabolites. The metabolites of progesterone that bind to the GABAA receptor complex are responsible for drowsiness and other neurologic side effects. Interestingly, however, erroneous readings of circulating progesterone have been obtained after oral intake of progesterone when plasma levels are measured by direct radio-immunoassays. Direct assays have only been validated for measurements of progesterone in the luteal phase of the menstrual cycle. After oral ingestion, the unusually high levels of progesterone metabolites alter the validity of progesterone measurements by direct assays because antibody specificity is insufficient in these unusual circumstances. Conversely, when progesterone is measured after separation on celite columns, plasma progesterone levels are low after oral ingestion; these low levels are concordant with the incomplete secretory transformation of the endometrium observed [25], [26].
Similarly, repeated daily administration of nasal progesterone failed to induce predecidual changes of endometrial stroma, a finding in accordance with the low levels of plasma progesterone achieved (from 2 to 5 ng/ml).
Because transdermal administration of progesterone is impractical, the vaginal route has long been regarded as the best, if not the only, alternate option for delivering progesterone non-orally to women. With the advent of IVF and other assisted reproductive procedures, interest has refocused on vaginal progesterone. Ultimately, the data suggest that vaginal progesterone is more than merely a non-oral alternative. Early work undoubtedly showed the high efficacy of vaginal progesterone at duplicating all the endometrial changes of the luteal phase. We now know, however, that this is not solely dependent on plasma levels, which remain lower than seen in the luteal phase of the menstrual cycle [30,31].
Stanczyk (2000)
Stanczyk, F. Z. (2000). Pharmacokinetics of progesterone administered orally and parenterally. In Sitruk-Ware, R., & Mishell, D. R. (Eds.). Progestins and Antiprogestins in Clinical Practice (pp. 393–400). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books]:
Progesterone is measured by immunoassay methods that include radioimmunoassay, chemiluminescent immunoassay, fluoroimmunoassay, and enzyme immunoassay. […] Although “direct” immunoassays (i.e., without a purification step) are used most commonly for quantifying progesterone in serum or plasma, such assays should not be used when measuring progesterone in samples obtained following exogenous administration of progesterone. This is because high concentrations of both unconjugated and conjugated progesterone metabolites are found in serum following oral progesterone dosing, which results in overestimated progesterone values. To obtain absolute progesterone levels, it is essential to subject samples to purification (e.g., organic solvent extraction and chromatography) before quantification by immunoassay.
Kuhl (2005)
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]:
The oral application of progesterone is associated with an extensive metabolism in the gastrointestinal tract and the liver, which results in high, but individually variable, concentrations of circulating metabolites. Consequently, the investigation of the pharmacokinetics of progesterone by means of radioimmunoassay (RIA) may be hampered by falsely high progesterone levels due to a relative pronounced cross-reactivity of progesterone metabolites. Therefore, either the gas chromatography/mass spectrometry (GC/MS) method or RIA after chromatographic separation are suitable for the measurement of progesterone. This problem is less pronounced after vaginal administration of progesterone owing to the relative low degree of metabolism213.
Wheeler & Barnard (2010)
Wheeler, M. J., & Barnard, G. (2010). Immunoassay of Steroids. In Makin, H., & Gower, D. (Eds.). Steroid Analysis (pp. 283–327). Dordrecht: Springer. [DOI:10.1023/b135931_4]:
In addition, there is a more critical requirement for improved antibody specificity in non-extraction procedures because of the presence of relatively high concentrations of steroid conjugates (glucuronides and sulphates) in unextracted serum. This is particularly true in the case of samples from patients undergoing hormone replacement therapy, which may involve the administration of oestradiol, progesterone and/or testosterone in a variety of combinations and by a variety of routes (e.g. intramuscular, oral, nasal, intravaginal or percutaneous). The route of administration profoundly affects the metabolism of the hormone and leads to the formation of particular profiles of steroid conjugates, some of which may cross-react significantly in the immunoassay (Nisbet and Jomain, 1987; Nahoul et al., 1989). In many cases, commercial direct procedures may be totally inappropriate for use in these situations.
Kuhl (2011)
Kuhl, H. (2011). Pharmacology of progestogens. Journal für Reproduktionsmedizin und Endokrinologie [Journal of Reproductive Medicine and Endocrinology], 8(1), 157–177. [URL] [PDF]:
Progesterone is an important intermediate in the ovarian and adrenal steroid synthesis, but larger amounts are produced only in the corpus luteum and the placenta. During the luteal phase, serum concentrations of 25 ng/ml are reached which may increase during pregnancy up to 200 ng/ml. […]
[…] the half-lives [of progesterone in the circulation] are only 6 min (t1/2α) and 42 min (t1/2β). Progesterone is rapidly metabolised, predominantly by reduction of the keto groups and the Δ4- double bond, and the pattern of metabolites depends largely on the route of administration. The oral application of progesterone is associated with an extensive metabolism in the gastrointestinal tract and the liver which results in high, but individually variable concentrations of circulating metabolites. Consequently, the investigation of the pharmacokinetics of progesterone by means of RIA may be hampered by falsely high progesterone levels due to a relatively pronounced cross-reactivity of progesterone metabolites. Therefore, either the GC/MS method or radioimmunoassay (RIA) after chromatographic separation are suitable for the measurement of progesterone. This problem is less pronounced after vaginal administration of progesterone owing to the relatively low degree of metabolism [54].
After oral administration, progesterone can be metabolised to more than 30 metabolites, among which some exert specific physiological activities. The most important pathway is the formation of 5α-pregnanolone and 5β-pregnanolone that exert considerable sedative effects after binding to the GABAA receptor. Further metabolites were 20-dihydroprogesterone that has 25–50% of the progestogenic potency of progesterone, 11-deoxycorticosterone (DOC) that is a potent mineralocorticoid, 17α-hydroxyprogesterone, and the inactive end-product pregnanediol (Fig. 7).
There are large interindividual differences in the pattern of metabolites circulating after oral administration [55]. The low oral bioavailability could be increased by the use of micronized progesterone suspended in oil and packaged in a gelatine capsule.
Pharmacokinetics A single oral dose of 100 mg progesterone contained in a gelatine capsule led to a rapid rise in serum progesterone as measured by liquid chromatography–mass spectrometry to a peak level of 1.5–2.2 ng/ml after 1–2 h. Thereafter the levels decreased rapidly to baseline levels within 4–6 h [53, 55]. However, determination by means of RIA revealed a mean peak level of 19.4 ng/ml suggesting a high cross reaction of progesterone metabolites [54]. There was a pronounced rise in the serum levels of 5α- and 5β-pregnanolone up to a maximum of 14 ng/ml and 3.6 ng/ml after 2 h. The DOC levels rose from 120 pg/ml to 680 pg/ml after 2 h and decreased rapidly thereafter [56].
The results cast some doubts on the reliability of progesterone determinations by RIA if metabolites are not separated by means of chromatographically in advance.
After oral intake of 200 mg progesterone, the peak levels of progesterone as measured by RIA after 4 h were 12 ng/ml, while 5α- and 5β-pregnanolone reached serum concentrations of 30 ng/ml and 60 ng/ml [55]. Further metabolites were 20-dihydroprogesterone, DOC, 17α-hydroxyprogesterone, and pregnanediol (Fig. 7).
The results of a large prospective study indicate that oral and transdermal treatment with progesterone does not protect from estrogen-induced endometrial cancer in postmenopausal women. Compared with women treated with estrogen-only preparations who showed an elevated relative risk of 2.52 (95%-CI: 1.7–3.57), the risk of endometrial cancer did not differ significantly during therapy with estrogen plus progesterone (relative risk 2.42; 95%-CI: 1.53–3.83). Contrary to this, synthetic progestogens reduced the estrogen-dependent risk significantly [57]. The lack of endometrial protection during oral progesterone therapy may be explained by the low progesterone serum levels measured with reliable methods. The same phenomenon may also explain the results of another cohort study that, in contrast to synthetic progestins, the addition of progesterone to estrogen therapy did not increase the risk of breast cancer [58, 59].
The finding of an elevated risk of endometrial cancer in postmenopausal women during treatment with estrogens and oral progesterone are in contradiction to various trials that did not find any increase in the rate of endometrial hyperplasia in women treated with estrogens and 200 mg sequential progesterone or 100 mg continuous progesterone [60–62]. However, the effect of oral treatment with progesterone on estrogenized postmenopausal endometria is dose-dependent, and during the use of 200 mg no full secretory transformation was observed, whereas the daily dose of 300 mg seems to be appropriate as an alternative to synthetic progestogens for therapy [63].
Kuhl & Schneider (2013)
Kuhl, H., & Schneider, H. P. G. (2013). Progesterone – promoter or inhibitor of breast cancer. Climacteric, 16(Suppl 1), 54–68. [DOI:10.3109/13697137.2013.768806]:
The problem of measurement of progesterone levels by RIA
Oral treatment is associated with a rapid metabolism of micronized progesterone in the intestinal mucosa and during the first liver passage. Although the bioavailability of orally administered progesterone is improved by suspending micronized particles of progesterone in oil, the serum concentrations of progesterone are very low when measured with exact methods like liquid chromatography–mass spectrometry (Table 5) 93. Contrary to this, the metabolism of endogenous progesterone in the luteal phase and of vaginally administered progesterone is low and does not compromise measurement of serum progesterone by RIA methods.
After oral administration of progesterone, however, large amounts of certain progesterone metabolites are circulating which have a more or less pronounced binding affinity to the relatively unspecific antiserum against progesterone used in the RIA. Therefore, if not separated from progesterone, these metabolites will cause falsely high progesterone serum concentrations when using a RIA (Figure 5). Therefore, after oral treatment with micronized progesterone, the separation of progesterone from its metabolites by an appropriate chromatographic system is mandatory 93–95.
After oral administration of 100 mg and 200 mg micronized progesterone to premenopausal women during the follicular phase, average peak levels of 1.5 ± 0.2 ng/ml and 4.70 ± 1.14 ng/ml, respectively, were measured after 2–3 h using a reliable RIA method after adequate chromatographic separation 94.
A direct comparison of the progesterone levels measured by either a direct RIA or liquid chromatography–mass spectrometry revealed that, after vaginal administration of 90 mg of progesterone gel, both methods resulted in similar mean peak levels of 8 ng/ml. Contrary to this, after oral administration of 100 mg progesterone per capsule, the mean peak level of progesterone was measured as 15 ng/ml using the RIA method compared to 2 ng/ml using the gold-standard method of liquid chromatography–mass spectrometry (Figure 5, Table 5) 93.
Micronized progesterone and risk of endometrial cancer
The EPIC cohort study, carried out with 115 000 postmenopausal women, revealed that estrogen-only therapy yielded a 2.52-fold increase in the relative risk of endometrial cancer which was prevented by the addition of progesterone derivatives or nortestosterone derivatives. Contrary to this, progesterone was not able to protect from the development of estrogen-induced endometrial cancer, increasing the relative risk up to 2.42 96.
Randomized trials revealed that, after treatment of postmenopausal women with oral or percutaneous estrogens and cyclic addition of oral micronized progesterone, the rate of endometrial hyperplasia was not elevated and was similar to that in the placebo group 102,103.
The lack of endometrial hyperplasia was interpreted as evidence for a sufficient protective effect on the endometrium by oral use of micronized progesterone. However, the results of these studies refer to surrogate parameters which cannot be transferred to the clinical endpoint of endometrial cancer. The oral use of daily 200 mg micronized progesterone causes peak levels of progesterone of less than 5 ng/ml, provided that adequate methods are used for measurement of the serum levels 94. In face of the large interindividual variations in the pharmacokinetics of progesterone, there will be women with serum progesterone levels too low for a long-term protection of the endometrium 93–95.
On the other hand, the mitosis rate in invasive breast carcinomas has been observed to reach a maximum during the luteal phase. Very likely, endogenous progesterone in synergism with estradiol is responsible for an increase in mammary epithelial proliferation, vascularity, breast tenderness and mammographic density.
In the 2005 report of the French E3N cohort study, no effect of percutaneous estradiol, with or without the addition of oral micronized progesterone, was found on postmenopausal breast cancer risk, in contrast to an unfavorable effect of combined synthetic progestins. The publication in 2008 revealed that oral micronized progesterone even prevented an estrogen-induced rise in breast cancer risk. In 2009, a new report did not present risk data based on the total case numbers of the three treatment groups. Instead, the cases were divided into eight subgroups according to the gap time between menopause and initiation of HRT and the duration of HRT. Importantly, long-term treatment with estrogen and micronized progesterone was found to increase significantly the risk of breast cancer.
It is hard to believe that progesterone does not promote the estrogen-related risk of breast cancer or even has a favorable effect. […] The oral dose of 100 mg or 200 mg progesterone is too low for endometrial protection in all women, as demonstrated by an increased risk of endometrial cancer. The rapid inactivation of progesterone in the intestinal mucosa and the liver causes very low progesterone serum levels which have been measured using reliable methods (e.g. liquid chromatography–mass spectrometry) instead of a RIA which is compromised by cross-reactions of certain progesterone metabolites. Therefore, a weak proliferative effect of the low progesterone levels may need a longer time interval of oral treatment with micronized progesterone, until an increase in breast cancer can be demonstrated.
The available knowledge suggests that, similar to synthetic progestins, HRT with micronized progesterone may promote the growth of small breast carcinomas which may have developed during the fertile phase of the women. Whether or not there are differences between the various progestogens remains to be elucidated.
North American Menopause Society (2017)
North American Menopause Society. (2017). The 2017 hormone therapy position statement of the North American Menopause Society. Menopause, 24(7), 728–753. [DOI:10.1097/gme.0000000000000921]:
A higher incidence of breast cancer was seen in the WHI for CEE + MPA compared with placebo, but a reduced incidence with CEE alone (Figure 1).14 Observational studies have suggested that the risk of breast cancer may be less with the use of micronized progesterone (MP) compared with synthetic progestogens,15,16 but the bioavailability of oral and transdermal progesterone is poor.
Micronized progesterone needs to be adequately dosed for endometrial protection.17-19 Improperly formulated or dosed or delivery issues with estrogen plus MP combinations have potentially serious health consequences, including increased risk of endometrial neoplasia.20 In women using EPT, unscheduled bleeding occurring more than 6 months after initiation should be investigated.
In a meta-analysis of trials of women who began HT treatment fewer than 10 years after menopause onset or who were aged younger than 60 years, strong evidence of increased risk of VTE was found in the HT group compared with placebo (RR 1.74; 95% CI, 1.11-2.73).177 […] Micronized progesterone may be less thrombogenic than other progestins.190 […]
Some but not all observational data suggest that MP may have less effect on breast cancer risk, whereas more potent progestogens such as MPA may have a more adverse effect,15,204 but randomized trials are needed.
Davey (2018)
Davey, D. A. (2018). Menopausal hormone therapy: a better and safer future. Climacteric, 21(5), 454–461. [DOI:10.1080/13697137.2018.1439915]:
Oral and vaginal micronized progesterone
Oral micronized progesterone
In postmenopausal women with an intact uterus receiving estrogen, the addition of progesterone or progestins is regarded as essential to prevent endometrial hyperplasia and carcinoma. The combination of a progestin with estrogen increases the risk of breast carcinoma and VTE and the risk varies with the type of progestin. Oral micronized progesterone (MP) has been claimed not to increase the risk of breast cancer. When administered orally, MP is rapidly metabolized in the intestinal mucosa and the liver, and the plasma levels of progesterone are very low when measured by specific liquid chromatography and mass spectrometry (LC-MS)31.
Oral administration of MP 100 mg daily results in peak levels of less than 2.2 ± 3.06 ng/ml measured by LC-MS. With the doses of MP currently used clinically, the concentrations of MP in the plasma may be insufficient to prevent endometrial hyperplasia and carcinoma when estrogens are given in the short term but may increase the risk when given in the long term (more than 5 years)32. At the same time, the low plasma concentrations of MP following oral administration have may have a weak effect on breast tissue and may not increase the risk of breast carcinoma in the short term but may increase the risk in the long term.
The low plasma concentrations of MP may also have less effect on blood coagulation and on the risk of VTE. The claim that MP does not cause endometrial hyperplasia and carcinoma and is not associated with an increased risk of breast cancer and VTE, however, has been disputed and is discussed in the following sections.
Oral micronized progesterone, oral progestins, endometrial hyperplasia and carcinoma
After a review of 40 studies, an expert committee concluded that oral MP, if applied sequentially for 12–14 days/month at 200 mg/day, provides endometrial protection for up to 5 years33. In the European EPIC study of 115 474 postmenopausal women in Europe, the risk of endometrial carcinoma was increased both in current estrogen-only users (RR 2.52, 95% CI 1.77–3.57) and in current estrogen–progestin users (RR 1.41, 95% CI 1.08–1.83)34. In estrogen–progestin users, the risk of endometrial carcinoma depended on the type of progestin, the regimen – sequential or continuous – and duration of use. The risk of endometrial carcinoma was not increased in synthetic progestin users but was significantly increased in MP users (RR 2.42, 95% CI 1.53–3.8).
In an analysis of the 65 360 women in the French cohort of the EPIC study, the risk of endometrial cancer was increased in estrogen plus MP users (RR 1.80, 95% CI 1.38–2.34) compared with never users and increased with increased duration of use: <5 years, RR = 1.3 (95% CI 0.99–1.97), and >5 years, RR = 2.66 (95% CI 1.87–3.77). The risk of endometrial cancer with the use of estrogens and progestins other than MP was not increased35.
In a systematic review of 28 studies, continuous combined estrogen–progestin therapy had a lower risk of endometrial cancer than sequential estrogen–progestin therapy. The risk of endometrial cancer was increased with MP given either continuously or sequentially36. The claim that oral MP can prevent the increased incidence of endometrial hyperplasia and carcinoma in postmenopausal women treated with estrogens has not been substantiated.
Oral micronized progesterone, oral progestins and breast cancer
Breast carcinoma is the most common carcinoma in women and an increase in the risk of breast cancer is the most serious risk associated with MHT. The WHI trial of CEE + MPA was terminated prematurely because of the increased risk of breast cancer (RR 1.26, 95% CI 1.00–1.59)2. In the CEE-only arm of the WHI trial, in contrast, the risk of breast cancer was decreased (RR 0.77, 95% CI 0.59–1.01)3.
The Million Women Study reported that the risk of breast cancer was increased both in estrogen–progestin users (RR 2.00, 95% CI 1.88–2.12) and in estrogen-only users (RR 1.30, 95% CI 1.21–1.45) and that the increase in risk for combined estrogen–progestin users was significantly greater than in estrogen-only users37.
The UK Generations Study of 58 148 menopausal women followed for 6 years (median 5.4 years) found that the risk of breast cancer was increased in current estrogen plus progestogen users (RR 2.74, 95% CI 2.05–3.6) but was not increased in estrogen-only users (RR 1.00, 95% CI 0.66–1.54)38.
In the first report of the French cohort of the E3N-EPIC study in 2005, in 54 548 postmenopausal women with a mean duration of use of MHT of 2.8 years, the risk of breast cancer was found not to be significantly increased with MHT with MP (RR 0.9, 95% CI 0.7–1.2) but was increased with MHT containing synthetic progestins (RR 1.4, 95% CI 1.2–1.7)39. In a later report of the E3N study in 2009, the risk of breast cancer was found to be increased with MHT with MP if MHT was initiated in the 3-year period following onset of the menopause and continued for 5 or more years (RR 1.54, 95% CI 1.28–1.86) but was not increased if initiated after 3 years (RR 1.00, 95% CI 0.68–1.47)40.
A separate French CECILE case–control study of 1555 menopausal women (739 cases and 816 matched controls) found that, compared with never use, the risk of breast cancer was increased with current use of estrogen plus synthetic progestins for 4 or more years (RR 2.07, 95% CI 1.26–3.39) but was not increased with estrogen plus MP for the same period (RR 0.79, 95% CI 0.37–1.71)41.
A number of factors influence the increase in risk of breast cancer associated with MHT including the interval between the menopause and starting MHT (gap time), duration of MHT, and body weight and body mass index; the interpretation of the effect of MP and progestins on risk of breast cancer may be difficult. It has been suggested that the low plasma concentrations of MP given orally may have a weak effect on breast tissue and may not increase the risk of breast carcinoma over short periods, but may increase the risk with longer periods of 5 years or more32. MHT is usually initiated for the relief of menopausal symptoms within a year or so of the menopause and it is often necessary to continue MHT for more than 5 years. An increased risk of breast cancer with oral MP given for 5 years or more cannot be ruled out, and a possible reduction in risk of breast cancer with MP cannot be regarded as a reason for preferring MP to progestins in MHT in the light of the increased risk of endometrial hyperplasia and carcinoma with MP.
Oral micronized progesterone, progestins and venous thromboembolism
Most studies have shown an increased risk of VTE with MHT with combined estrogen and progestin compared with estrogen only, and the risk appears to vary with different progestins. In the CEE + MPA arm of the WHI trial, the risk of VTE in women age 50–59 was increased (RR 1.27, 95% CI 1.19–4.33), but in the CEE-only arm the risk was not significantly increased (RR 1.22, 95% CI 0.62–2.42)2,3.
The UK NHS record linkage study reported that the risk of VTE was significantly greater for estrogen–progestin than for oral estrogen-only therapy (RR 2.07, 95% CI 1.86–2.31 vs. RR 1.42, 95% CI 1.21–1.66). The risk of VTE with MPA was greater (RR 2.67, 95% CI 2.25–3.17) than with other progestins (RR 1.91, 95% CI 1.69-2.17), heterogeneity = 0.000723.
A Dutch study found that the risk of VTE was increased with oral CEE and MPA (RR 4.0, 95% CI 1.8–8.2) and with oral estradiol plus norethisterone acetate (RR 3.9, 95% CI 1.5–10.7) compared with oral estrogen-only MHT and that there was no significant difference between the progestins24.
In a Swedish study, the risk of VTE in combined estrogen–progestogen users was double that of estrogen-only users (RR 2.18, 95% CI 1.21–3.92, p = 0.009). The risk was increased by both medroxyprogesterone acetate (MPA) (RR 2.94, 95% CI 1.67–5.36) and norethisterone acetate (RR 2.25, 95% CI 1.50–3.40) and there was no significant difference between the progestins25.
It has been claimed that the risk of VTE is less with MHT with MP than with other progestins. In the E3N study of 80 308 postmenopausal women with 549 cases of incident VTE, the risk for VTE was not significantly increased with the use of estrogens combined with MP (RR 0.9, 95% CI 0.6–1.5), nortestosterone derivatives (RR 1.4, 95% CI 0.7–2.4) or pregnane derivatives including MPA (RR 1.3, 95% CI 0.9–2.0), compared with oral estrogens only but was increased with norpregnane derivatives combined with estrogens (RR 1.8, 95% CI 1.2–2.7)21.
In the four studies of transdermal estrogens with oral MP or progestins, the risk of VTE was not increased20,23–25. The use of transdermal rather than oral estrogens in all women receiving MHT would obviate any possible increased risk of VTE with all types of progestins as well as with MP and is another good reason for using transdermal estrogens in preference to oral estrogens in all perimenopausal and postmenopausal women.
Kuhl & Wiegratz (2021)
Kuhl, H., & Wiegratz, I. (2021). Pharmakokinetik und Pharmakodynamik der in der Assistierten Reproduktion Verwendeten Gestagene. [Pharmacokinetics and Pharmacodynamics of Progestogens Used in Assisted Reproduction.] Gynäkologische Endokrinologie [Gynecological Endocrinology], 19, 105–117. [Google Scholar] [DOI:10.1007/s10304-020-00372-5] [Translation] [Translated]:
Abstract
[…] Oral administration of progesterone causes extensive metabolism resulting in the formation of more than 30 metabolites, some of which may cause anesthetic/sedative effects. The use of a direct radioimmunoassay (RIA) without prior separation of progesterone from its metabolites may simulate falsely high serum concentrations of progesterone owing to cross-reaction of some metabolites with the specific antiserum of the RIA. This effect is not observed after parenteral treatment with progesterone due to minimal metabolism. […]
Progesterone pharmacokinetics
Problems with the determination of progesterone in serum
Because of the large number of progesterone metabolites that circulate in variable concentrations after oral administration, false-high progesterone levels must be expected when using an immunoassay. The cause is the cross-reactions of some progesterone metabolites, such as 5α- and 5β-DHP and 5α-pregnan-3β-ol-20-one [13]. For this reason, a radioimmunoassay (RIA) can only be used, for example, if the progesterone has previously been isolated by means of a suitable chromatographic separation. The determination of progesterone with exact liquid or gas chromatography-mass spectrometry (LC-MS or GC-MS; [14]; Table 1) is more reliable, albeit more complex.
The problem of cross-reactions does not play an essential role in parenteral progesterone treatment due to the low metabolism of progesterone. The specificity of the antiserum used is of particular importance for the comparability of the results of different RIAs, as it influences the cross-reactivity of the numerous progesterone metabolites [11].
The determination of the progesterone level, which was measured after oral administration of a capsule containing 100 mg of micronized progesterone, resulted in a false high peak value of 19.4 ng/mL in the RIA, which was 8 times as high as that with the exact LC-MS method which determined a true value of 2.4 ng/mL (Table 1). The cause was metabolites, the high concentrations of which simulated a false high concentration via a cross-reaction with the RIA antiserum [11]. In contrast, after intravaginal application of a gel containing 90 mg of progesterone, the metabolism of progesterone and thus the influence of cross-reactions is so low that the progesterone concentration of 10.5 ng/mL measured with the RIA agrees with the result of the exact LC-MS method (Table 1; [11]).
“Enzyme-linked immunosorbent assay”
As with the RIA, the “enzyme-linked immunosorbent assay” (ELISA) is a procedure based on an immune reaction between an antigen and a specific antibody. For this purpose, a specific antibody is produced in a separate standard process for the antigen, for example progesterone, which binds the antigen with high affinity. An enzyme is coupled to this specific antibody that cleaves an added chromogen, producing a dye, the intensity of which is measured in a photometer. The measured intensity of the color correlates with the concentration of the antigen (for example progesterone).
With regard to the cross-reaction of some progesterone metabolites, which leads to false high values of the progesterone concentration, the same restriction applies to the ELISA as to the direct RIA (Table 1).
Summary
If a progesterone determination is carried out in the serum after oral progesterone application in the RIA or in the ELISA without prior chromatographic separation, one measures incorrectly high progesterone values, since progesterone metabolites are also measured! This problem does not exist with parenteral application (intravaginal, intramuscular, or subcutaneous).
Oral application of progesterone
After separation of important progesterone metabolites by means of column chromatography, an increase in progesterone in the serum to a maximum of 4.7 ± 1.15 ng/mL was found in the RIA after oral administration of 200 mg micronized progesterone [14].
Summary
In summary, it can be stated that oral administration of 100 to 200 mg progesterone leads to an increase in serum levels to around 3–5 ng/mL, provided the laboratory diagnostics are carried out correctly, i.e. after chromatographic separation.
Conclusion for practice
After oral progesterone administration, more than 30 metabolites are formed, which in immunoassays can cause false high results of the systemic progesterone concentration. This problem can only be solved by prior chromatographic separation of the progesterone or by direct measurement methods.
Individual Studies and Papers
Comparative Immunoassay versus Immunoassay with Chromatography or Mass Spectrometry Studies
Nahoul, K., Dehennin, L., & Scholler, R. (1987). Radioimmunoassay of plasma progesterone after oral administration of micronized progesterone. Journal of Steroid Biochemistry, 26(2), 241–249. [DOI:10.1016/0022-4731(87)90078-1]
Levine, H., & Watson, N. (2000). Comparison of the pharmacokinetics of Crinone 8% administered vaginally versus Prometrium administered orally in postmenopausal women. Fertility and Sterility, 73(3), 516–521. [DOI:10.1016/S0015-0282(99)00553-1]
Sapin, R., Neamtu, D., Gasser, F., Ohl, J., Grunenberger, F., & Grucker, D. (2000). De la prudence lors de l’utilisation des dosages directs de progestérone. [Caution when using direct progesterone assays.] Immuno-analyse et Biologie Spécialisée, 3(15), 203–204. [DOI:10.1016/S0923-2532(00)80010-1] [Translation]
Non-Comparative Immunoassay with Adequate Chromatography or Mass Spectrometry Studies
Nahoul, K., Dehennin, L., Jondet, M., & Roger, M. (1993). Profiles of plasma estrogens, progesterone and their metabolites after oral or vaginal administration of estradiol or progesterone. Maturitas, 16(3), 185–202. [DOI:10.1016/0378-5122(93)90064-O]
Lobo, R. A., Liu, J., Stanczyk, F. Z., Constantine, G. D., Pickar, J. H., Shadiack, A. M., Bernick, B., & Mirkin, S. (2019). Estradiol and progesterone bioavailability for moderate to severe vasomotor symptom treatment and endometrial protection with the continuous-combined regimen of TX-001HR (oral estradiol and progesterone capsules). Menopause (New York, NY), 26(7), 720–727.[DOI:10.1097/GME.0000000000001306]
Commentaries on Studies
Nahoul, K., & de Ziegler, D. (1994). “Validity” of serum progesterone levels after oral progesterone. Fertility and Sterility, 61(4), 790–791. [DOI:10.1016/S0015-0282(16)56666-7]
\ No newline at end of file
+Sources/Excerpts: Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects - Transfeminine Science
Sources/Excerpts: Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects
By Aly | First published August 4, 2018 | Last modified September 25, 2022
Preface
This is a literature sources/excerpts supplement to the main article which can be found here.
Excerpts From Literature Reviews
de Ziegler et al. (1998)
de Ziegler, D., Fanchin, R., de Moustier, B., & Bulletti, C. (1998). The hormonal control of endometrial receptivity: estrogen (E2) and progesterone. Journal of Reproductive Immunology, 39(1–2), 149–166. [DOI:10.1016/S0165-0378(98)00019-9]:
4.2. The oral route paradox and the methodological flaws in progesterone measurement
High levels of progesterone have been reported after oral administration of micronized progesterone (Simon et al., 1993). Yet, despite the high levels reported, the same authors describe incomplete endometrial effects. Today, we believe that the apparent paradox between high progesterone levels and minimal endometrial effects characteristic of oral progesterone has been elucidated by Nahoul et al. (1987, 1993) revealing methodological flaw in the measurement of plasma progesterone after oral administration. Indeed, while direct RIA for progesterone is valid for the measurement of plasma levels in the luteal phase of the menstrual cycle, this is not the case after oral administration. In an elegant paper, Nahoul et al. (1987) showed that because of the high levels of progesterone metabolites encountered after oral administration direct RIA measured values 3–5 times higher than those determined when measurements are conducted after extraction and separation on celite columns using an appropriate assay. Nahoul et al. (1993) showed that plasma levels of progesterone were only minimally elevated after oral ingestion of 100 mg of progesterone (barely reaching 1 ng/ml) and, particularly, were markedly lower than after vaginal administration of the same amounts of progesterone.
de Ziegler & Fanchin (2000)
de Ziegler, D., & Fanchin, R. (2000). Progesterone and progestins: applications in gynecology. Steroids, 65(10–11), 671–679. [DOI:10.1016/S0039-128X(00)00123-9]:
3.1. Oral progesterone
Progesterone is nearly entirely absorbed after oral ingestion when prepared in micronized form. Yet, because of intense inactivation by metabolism during first liver pass, the bioavailability of oral progesterone is notoriously poor at <10% [25]. This poor bioavailability explains that despite the large amounts used (up to 600 mg/day), oral progesterone fails to trigger the full array of endometrial changes seen in the late luteal phase of the menstrual cycle. Particularly, the last step of these changes (the predecidual transformation of stroma cells) fails to be induced by oral progesterone [3].
Some confusion has existed over these findings. Early reports claimed high plasma progesterone levels after oral administration of progesterone. However, fairly low plasma levels of progesterone have been reported when the hormone is given orally, and proper assays are used. Circulating levels of progesterone and its metabolites, determined by sufficiently specific assays, after oral and vaginal administration of 100 mg of progesterone are illustrated in Fig. 4. As can be seen, when taken orally, progesterone accounts for less than 10%, whereas most of ingested progesterone is transformed to 5α-reduced metabolites. The metabolites of progesterone that bind to the GABAA receptor complex are responsible for drowsiness and other neurologic side effects. Interestingly, however, erroneous readings of circulating progesterone have been obtained after oral intake of progesterone when plasma levels are measured by direct radio-immunoassays. Direct assays have only been validated for measurements of progesterone in the luteal phase of the menstrual cycle. After oral ingestion, the unusually high levels of progesterone metabolites alter the validity of progesterone measurements by direct assays because antibody specificity is insufficient in these unusual circumstances. Conversely, when progesterone is measured after separation on celite columns, plasma progesterone levels are low after oral ingestion; these low levels are concordant with the incomplete secretory transformation of the endometrium observed [25], [26].
Similarly, repeated daily administration of nasal progesterone failed to induce predecidual changes of endometrial stroma, a finding in accordance with the low levels of plasma progesterone achieved (from 2 to 5 ng/ml).
Because transdermal administration of progesterone is impractical, the vaginal route has long been regarded as the best, if not the only, alternate option for delivering progesterone non-orally to women. With the advent of IVF and other assisted reproductive procedures, interest has refocused on vaginal progesterone. Ultimately, the data suggest that vaginal progesterone is more than merely a non-oral alternative. Early work undoubtedly showed the high efficacy of vaginal progesterone at duplicating all the endometrial changes of the luteal phase. We now know, however, that this is not solely dependent on plasma levels, which remain lower than seen in the luteal phase of the menstrual cycle [30,31].
Stanczyk (2000)
Stanczyk, F. Z. (2000). Pharmacokinetics of progesterone administered orally and parenterally. In Sitruk-Ware, R., & Mishell, D. R. (Eds.). Progestins and Antiprogestins in Clinical Practice (pp. 393–400). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books]:
Progesterone is measured by immunoassay methods that include radioimmunoassay, chemiluminescent immunoassay, fluoroimmunoassay, and enzyme immunoassay. […] Although “direct” immunoassays (i.e., without a purification step) are used most commonly for quantifying progesterone in serum or plasma, such assays should not be used when measuring progesterone in samples obtained following exogenous administration of progesterone. This is because high concentrations of both unconjugated and conjugated progesterone metabolites are found in serum following oral progesterone dosing, which results in overestimated progesterone values. To obtain absolute progesterone levels, it is essential to subject samples to purification (e.g., organic solvent extraction and chromatography) before quantification by immunoassay.
Kuhl (2005)
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]:
The oral application of progesterone is associated with an extensive metabolism in the gastrointestinal tract and the liver, which results in high, but individually variable, concentrations of circulating metabolites. Consequently, the investigation of the pharmacokinetics of progesterone by means of radioimmunoassay (RIA) may be hampered by falsely high progesterone levels due to a relative pronounced cross-reactivity of progesterone metabolites. Therefore, either the gas chromatography/mass spectrometry (GC/MS) method or RIA after chromatographic separation are suitable for the measurement of progesterone. This problem is less pronounced after vaginal administration of progesterone owing to the relative low degree of metabolism213.
Wheeler & Barnard (2010)
Wheeler, M. J., & Barnard, G. (2010). Immunoassay of Steroids. In Makin, H., & Gower, D. (Eds.). Steroid Analysis (pp. 283–327). Dordrecht: Springer. [DOI:10.1023/b135931_4]:
In addition, there is a more critical requirement for improved antibody specificity in non-extraction procedures because of the presence of relatively high concentrations of steroid conjugates (glucuronides and sulphates) in unextracted serum. This is particularly true in the case of samples from patients undergoing hormone replacement therapy, which may involve the administration of oestradiol, progesterone and/or testosterone in a variety of combinations and by a variety of routes (e.g. intramuscular, oral, nasal, intravaginal or percutaneous). The route of administration profoundly affects the metabolism of the hormone and leads to the formation of particular profiles of steroid conjugates, some of which may cross-react significantly in the immunoassay (Nisbet and Jomain, 1987; Nahoul et al., 1989). In many cases, commercial direct procedures may be totally inappropriate for use in these situations.
Kuhl (2011)
Kuhl, H. (2011). Pharmacology of progestogens. Journal für Reproduktionsmedizin und Endokrinologie [Journal of Reproductive Medicine and Endocrinology], 8(1), 157–177. [URL] [PDF]:
Progesterone is an important intermediate in the ovarian and adrenal steroid synthesis, but larger amounts are produced only in the corpus luteum and the placenta. During the luteal phase, serum concentrations of 25 ng/ml are reached which may increase during pregnancy up to 200 ng/ml. […]
[…] the half-lives [of progesterone in the circulation] are only 6 min (t1/2α) and 42 min (t1/2β). Progesterone is rapidly metabolised, predominantly by reduction of the keto groups and the Δ4- double bond, and the pattern of metabolites depends largely on the route of administration. The oral application of progesterone is associated with an extensive metabolism in the gastrointestinal tract and the liver which results in high, but individually variable concentrations of circulating metabolites. Consequently, the investigation of the pharmacokinetics of progesterone by means of RIA may be hampered by falsely high progesterone levels due to a relatively pronounced cross-reactivity of progesterone metabolites. Therefore, either the GC/MS method or radioimmunoassay (RIA) after chromatographic separation are suitable for the measurement of progesterone. This problem is less pronounced after vaginal administration of progesterone owing to the relatively low degree of metabolism [54].
After oral administration, progesterone can be metabolised to more than 30 metabolites, among which some exert specific physiological activities. The most important pathway is the formation of 5α-pregnanolone and 5β-pregnanolone that exert considerable sedative effects after binding to the GABAA receptor. Further metabolites were 20-dihydroprogesterone that has 25–50% of the progestogenic potency of progesterone, 11-deoxycorticosterone (DOC) that is a potent mineralocorticoid, 17α-hydroxyprogesterone, and the inactive end-product pregnanediol (Fig. 7).
There are large interindividual differences in the pattern of metabolites circulating after oral administration [55]. The low oral bioavailability could be increased by the use of micronized progesterone suspended in oil and packaged in a gelatine capsule.
Pharmacokinetics A single oral dose of 100 mg progesterone contained in a gelatine capsule led to a rapid rise in serum progesterone as measured by liquid chromatography–mass spectrometry to a peak level of 1.5–2.2 ng/ml after 1–2 h. Thereafter the levels decreased rapidly to baseline levels within 4–6 h [53, 55]. However, determination by means of RIA revealed a mean peak level of 19.4 ng/ml suggesting a high cross reaction of progesterone metabolites [54]. There was a pronounced rise in the serum levels of 5α- and 5β-pregnanolone up to a maximum of 14 ng/ml and 3.6 ng/ml after 2 h. The DOC levels rose from 120 pg/ml to 680 pg/ml after 2 h and decreased rapidly thereafter [56].
The results cast some doubts on the reliability of progesterone determinations by RIA if metabolites are not separated by means of chromatographically in advance.
After oral intake of 200 mg progesterone, the peak levels of progesterone as measured by RIA after 4 h were 12 ng/ml, while 5α- and 5β-pregnanolone reached serum concentrations of 30 ng/ml and 60 ng/ml [55]. Further metabolites were 20-dihydroprogesterone, DOC, 17α-hydroxyprogesterone, and pregnanediol (Fig. 7).
The results of a large prospective study indicate that oral and transdermal treatment with progesterone does not protect from estrogen-induced endometrial cancer in postmenopausal women. Compared with women treated with estrogen-only preparations who showed an elevated relative risk of 2.52 (95%-CI: 1.7–3.57), the risk of endometrial cancer did not differ significantly during therapy with estrogen plus progesterone (relative risk 2.42; 95%-CI: 1.53–3.83). Contrary to this, synthetic progestogens reduced the estrogen-dependent risk significantly [57]. The lack of endometrial protection during oral progesterone therapy may be explained by the low progesterone serum levels measured with reliable methods. The same phenomenon may also explain the results of another cohort study that, in contrast to synthetic progestins, the addition of progesterone to estrogen therapy did not increase the risk of breast cancer [58, 59].
The finding of an elevated risk of endometrial cancer in postmenopausal women during treatment with estrogens and oral progesterone are in contradiction to various trials that did not find any increase in the rate of endometrial hyperplasia in women treated with estrogens and 200 mg sequential progesterone or 100 mg continuous progesterone [60–62]. However, the effect of oral treatment with progesterone on estrogenized postmenopausal endometria is dose-dependent, and during the use of 200 mg no full secretory transformation was observed, whereas the daily dose of 300 mg seems to be appropriate as an alternative to synthetic progestogens for therapy [63].
Kuhl & Schneider (2013)
Kuhl, H., & Schneider, H. P. G. (2013). Progesterone – promoter or inhibitor of breast cancer. Climacteric, 16(Suppl 1), 54–68. [DOI:10.3109/13697137.2013.768806]:
The problem of measurement of progesterone levels by RIA
Oral treatment is associated with a rapid metabolism of micronized progesterone in the intestinal mucosa and during the first liver passage. Although the bioavailability of orally administered progesterone is improved by suspending micronized particles of progesterone in oil, the serum concentrations of progesterone are very low when measured with exact methods like liquid chromatography–mass spectrometry (Table 5) 93. Contrary to this, the metabolism of endogenous progesterone in the luteal phase and of vaginally administered progesterone is low and does not compromise measurement of serum progesterone by RIA methods.
After oral administration of progesterone, however, large amounts of certain progesterone metabolites are circulating which have a more or less pronounced binding affinity to the relatively unspecific antiserum against progesterone used in the RIA. Therefore, if not separated from progesterone, these metabolites will cause falsely high progesterone serum concentrations when using a RIA (Figure 5). Therefore, after oral treatment with micronized progesterone, the separation of progesterone from its metabolites by an appropriate chromatographic system is mandatory 93–95.
After oral administration of 100 mg and 200 mg micronized progesterone to premenopausal women during the follicular phase, average peak levels of 1.5 ± 0.2 ng/ml and 4.70 ± 1.14 ng/ml, respectively, were measured after 2–3 h using a reliable RIA method after adequate chromatographic separation 94.
A direct comparison of the progesterone levels measured by either a direct RIA or liquid chromatography–mass spectrometry revealed that, after vaginal administration of 90 mg of progesterone gel, both methods resulted in similar mean peak levels of 8 ng/ml. Contrary to this, after oral administration of 100 mg progesterone per capsule, the mean peak level of progesterone was measured as 15 ng/ml using the RIA method compared to 2 ng/ml using the gold-standard method of liquid chromatography–mass spectrometry (Figure 5, Table 5) 93.
Micronized progesterone and risk of endometrial cancer
The EPIC cohort study, carried out with 115 000 postmenopausal women, revealed that estrogen-only therapy yielded a 2.52-fold increase in the relative risk of endometrial cancer which was prevented by the addition of progesterone derivatives or nortestosterone derivatives. Contrary to this, progesterone was not able to protect from the development of estrogen-induced endometrial cancer, increasing the relative risk up to 2.42 96.
Randomized trials revealed that, after treatment of postmenopausal women with oral or percutaneous estrogens and cyclic addition of oral micronized progesterone, the rate of endometrial hyperplasia was not elevated and was similar to that in the placebo group 102,103.
The lack of endometrial hyperplasia was interpreted as evidence for a sufficient protective effect on the endometrium by oral use of micronized progesterone. However, the results of these studies refer to surrogate parameters which cannot be transferred to the clinical endpoint of endometrial cancer. The oral use of daily 200 mg micronized progesterone causes peak levels of progesterone of less than 5 ng/ml, provided that adequate methods are used for measurement of the serum levels 94. In face of the large interindividual variations in the pharmacokinetics of progesterone, there will be women with serum progesterone levels too low for a long-term protection of the endometrium 93–95.
On the other hand, the mitosis rate in invasive breast carcinomas has been observed to reach a maximum during the luteal phase. Very likely, endogenous progesterone in synergism with estradiol is responsible for an increase in mammary epithelial proliferation, vascularity, breast tenderness and mammographic density.
In the 2005 report of the French E3N cohort study, no effect of percutaneous estradiol, with or without the addition of oral micronized progesterone, was found on postmenopausal breast cancer risk, in contrast to an unfavorable effect of combined synthetic progestins. The publication in 2008 revealed that oral micronized progesterone even prevented an estrogen-induced rise in breast cancer risk. In 2009, a new report did not present risk data based on the total case numbers of the three treatment groups. Instead, the cases were divided into eight subgroups according to the gap time between menopause and initiation of HRT and the duration of HRT. Importantly, long-term treatment with estrogen and micronized progesterone was found to increase significantly the risk of breast cancer.
It is hard to believe that progesterone does not promote the estrogen-related risk of breast cancer or even has a favorable effect. […] The oral dose of 100 mg or 200 mg progesterone is too low for endometrial protection in all women, as demonstrated by an increased risk of endometrial cancer. The rapid inactivation of progesterone in the intestinal mucosa and the liver causes very low progesterone serum levels which have been measured using reliable methods (e.g. liquid chromatography–mass spectrometry) instead of a RIA which is compromised by cross-reactions of certain progesterone metabolites. Therefore, a weak proliferative effect of the low progesterone levels may need a longer time interval of oral treatment with micronized progesterone, until an increase in breast cancer can be demonstrated.
The available knowledge suggests that, similar to synthetic progestins, HRT with micronized progesterone may promote the growth of small breast carcinomas which may have developed during the fertile phase of the women. Whether or not there are differences between the various progestogens remains to be elucidated.
North American Menopause Society (2017)
North American Menopause Society. (2017). The 2017 hormone therapy position statement of the North American Menopause Society. Menopause, 24(7), 728–753. [DOI:10.1097/gme.0000000000000921]:
A higher incidence of breast cancer was seen in the WHI for CEE + MPA compared with placebo, but a reduced incidence with CEE alone (Figure 1).14 Observational studies have suggested that the risk of breast cancer may be less with the use of micronized progesterone (MP) compared with synthetic progestogens,15,16 but the bioavailability of oral and transdermal progesterone is poor.
Micronized progesterone needs to be adequately dosed for endometrial protection.17-19 Improperly formulated or dosed or delivery issues with estrogen plus MP combinations have potentially serious health consequences, including increased risk of endometrial neoplasia.20 In women using EPT, unscheduled bleeding occurring more than 6 months after initiation should be investigated.
In a meta-analysis of trials of women who began HT treatment fewer than 10 years after menopause onset or who were aged younger than 60 years, strong evidence of increased risk of VTE was found in the HT group compared with placebo (RR 1.74; 95% CI, 1.11-2.73).177 […] Micronized progesterone may be less thrombogenic than other progestins.190 […]
Some but not all observational data suggest that MP may have less effect on breast cancer risk, whereas more potent progestogens such as MPA may have a more adverse effect,15,204 but randomized trials are needed.
Davey (2018)
Davey, D. A. (2018). Menopausal hormone therapy: a better and safer future. Climacteric, 21(5), 454–461. [DOI:10.1080/13697137.2018.1439915]:
Oral and vaginal micronized progesterone
Oral micronized progesterone
In postmenopausal women with an intact uterus receiving estrogen, the addition of progesterone or progestins is regarded as essential to prevent endometrial hyperplasia and carcinoma. The combination of a progestin with estrogen increases the risk of breast carcinoma and VTE and the risk varies with the type of progestin. Oral micronized progesterone (MP) has been claimed not to increase the risk of breast cancer. When administered orally, MP is rapidly metabolized in the intestinal mucosa and the liver, and the plasma levels of progesterone are very low when measured by specific liquid chromatography and mass spectrometry (LC-MS)31.
Oral administration of MP 100 mg daily results in peak levels of less than 2.2 ± 3.06 ng/ml measured by LC-MS. With the doses of MP currently used clinically, the concentrations of MP in the plasma may be insufficient to prevent endometrial hyperplasia and carcinoma when estrogens are given in the short term but may increase the risk when given in the long term (more than 5 years)32. At the same time, the low plasma concentrations of MP following oral administration have may have a weak effect on breast tissue and may not increase the risk of breast carcinoma in the short term but may increase the risk in the long term.
The low plasma concentrations of MP may also have less effect on blood coagulation and on the risk of VTE. The claim that MP does not cause endometrial hyperplasia and carcinoma and is not associated with an increased risk of breast cancer and VTE, however, has been disputed and is discussed in the following sections.
Oral micronized progesterone, oral progestins, endometrial hyperplasia and carcinoma
After a review of 40 studies, an expert committee concluded that oral MP, if applied sequentially for 12–14 days/month at 200 mg/day, provides endometrial protection for up to 5 years33. In the European EPIC study of 115 474 postmenopausal women in Europe, the risk of endometrial carcinoma was increased both in current estrogen-only users (RR 2.52, 95% CI 1.77–3.57) and in current estrogen–progestin users (RR 1.41, 95% CI 1.08–1.83)34. In estrogen–progestin users, the risk of endometrial carcinoma depended on the type of progestin, the regimen – sequential or continuous – and duration of use. The risk of endometrial carcinoma was not increased in synthetic progestin users but was significantly increased in MP users (RR 2.42, 95% CI 1.53–3.8).
In an analysis of the 65 360 women in the French cohort of the EPIC study, the risk of endometrial cancer was increased in estrogen plus MP users (RR 1.80, 95% CI 1.38–2.34) compared with never users and increased with increased duration of use: <5 years, RR = 1.3 (95% CI 0.99–1.97), and >5 years, RR = 2.66 (95% CI 1.87–3.77). The risk of endometrial cancer with the use of estrogens and progestins other than MP was not increased35.
In a systematic review of 28 studies, continuous combined estrogen–progestin therapy had a lower risk of endometrial cancer than sequential estrogen–progestin therapy. The risk of endometrial cancer was increased with MP given either continuously or sequentially36. The claim that oral MP can prevent the increased incidence of endometrial hyperplasia and carcinoma in postmenopausal women treated with estrogens has not been substantiated.
Oral micronized progesterone, oral progestins and breast cancer
Breast carcinoma is the most common carcinoma in women and an increase in the risk of breast cancer is the most serious risk associated with MHT. The WHI trial of CEE + MPA was terminated prematurely because of the increased risk of breast cancer (RR 1.26, 95% CI 1.00–1.59)2. In the CEE-only arm of the WHI trial, in contrast, the risk of breast cancer was decreased (RR 0.77, 95% CI 0.59–1.01)3.
The Million Women Study reported that the risk of breast cancer was increased both in estrogen–progestin users (RR 2.00, 95% CI 1.88–2.12) and in estrogen-only users (RR 1.30, 95% CI 1.21–1.45) and that the increase in risk for combined estrogen–progestin users was significantly greater than in estrogen-only users37.
The UK Generations Study of 58 148 menopausal women followed for 6 years (median 5.4 years) found that the risk of breast cancer was increased in current estrogen plus progestogen users (RR 2.74, 95% CI 2.05–3.6) but was not increased in estrogen-only users (RR 1.00, 95% CI 0.66–1.54)38.
In the first report of the French cohort of the E3N-EPIC study in 2005, in 54 548 postmenopausal women with a mean duration of use of MHT of 2.8 years, the risk of breast cancer was found not to be significantly increased with MHT with MP (RR 0.9, 95% CI 0.7–1.2) but was increased with MHT containing synthetic progestins (RR 1.4, 95% CI 1.2–1.7)39. In a later report of the E3N study in 2009, the risk of breast cancer was found to be increased with MHT with MP if MHT was initiated in the 3-year period following onset of the menopause and continued for 5 or more years (RR 1.54, 95% CI 1.28–1.86) but was not increased if initiated after 3 years (RR 1.00, 95% CI 0.68–1.47)40.
A separate French CECILE case–control study of 1555 menopausal women (739 cases and 816 matched controls) found that, compared with never use, the risk of breast cancer was increased with current use of estrogen plus synthetic progestins for 4 or more years (RR 2.07, 95% CI 1.26–3.39) but was not increased with estrogen plus MP for the same period (RR 0.79, 95% CI 0.37–1.71)41.
A number of factors influence the increase in risk of breast cancer associated with MHT including the interval between the menopause and starting MHT (gap time), duration of MHT, and body weight and body mass index; the interpretation of the effect of MP and progestins on risk of breast cancer may be difficult. It has been suggested that the low plasma concentrations of MP given orally may have a weak effect on breast tissue and may not increase the risk of breast carcinoma over short periods, but may increase the risk with longer periods of 5 years or more32. MHT is usually initiated for the relief of menopausal symptoms within a year or so of the menopause and it is often necessary to continue MHT for more than 5 years. An increased risk of breast cancer with oral MP given for 5 years or more cannot be ruled out, and a possible reduction in risk of breast cancer with MP cannot be regarded as a reason for preferring MP to progestins in MHT in the light of the increased risk of endometrial hyperplasia and carcinoma with MP.
Oral micronized progesterone, progestins and venous thromboembolism
Most studies have shown an increased risk of VTE with MHT with combined estrogen and progestin compared with estrogen only, and the risk appears to vary with different progestins. In the CEE + MPA arm of the WHI trial, the risk of VTE in women age 50–59 was increased (RR 1.27, 95% CI 1.19–4.33), but in the CEE-only arm the risk was not significantly increased (RR 1.22, 95% CI 0.62–2.42)2,3.
The UK NHS record linkage study reported that the risk of VTE was significantly greater for estrogen–progestin than for oral estrogen-only therapy (RR 2.07, 95% CI 1.86–2.31 vs. RR 1.42, 95% CI 1.21–1.66). The risk of VTE with MPA was greater (RR 2.67, 95% CI 2.25–3.17) than with other progestins (RR 1.91, 95% CI 1.69-2.17), heterogeneity = 0.000723.
A Dutch study found that the risk of VTE was increased with oral CEE and MPA (RR 4.0, 95% CI 1.8–8.2) and with oral estradiol plus norethisterone acetate (RR 3.9, 95% CI 1.5–10.7) compared with oral estrogen-only MHT and that there was no significant difference between the progestins24.
In a Swedish study, the risk of VTE in combined estrogen–progestogen users was double that of estrogen-only users (RR 2.18, 95% CI 1.21–3.92, p = 0.009). The risk was increased by both medroxyprogesterone acetate (MPA) (RR 2.94, 95% CI 1.67–5.36) and norethisterone acetate (RR 2.25, 95% CI 1.50–3.40) and there was no significant difference between the progestins25.
It has been claimed that the risk of VTE is less with MHT with MP than with other progestins. In the E3N study of 80 308 postmenopausal women with 549 cases of incident VTE, the risk for VTE was not significantly increased with the use of estrogens combined with MP (RR 0.9, 95% CI 0.6–1.5), nortestosterone derivatives (RR 1.4, 95% CI 0.7–2.4) or pregnane derivatives including MPA (RR 1.3, 95% CI 0.9–2.0), compared with oral estrogens only but was increased with norpregnane derivatives combined with estrogens (RR 1.8, 95% CI 1.2–2.7)21.
In the four studies of transdermal estrogens with oral MP or progestins, the risk of VTE was not increased20,23–25. The use of transdermal rather than oral estrogens in all women receiving MHT would obviate any possible increased risk of VTE with all types of progestins as well as with MP and is another good reason for using transdermal estrogens in preference to oral estrogens in all perimenopausal and postmenopausal women.
Kuhl & Wiegratz (2021)
Kuhl, H., & Wiegratz, I. (2021). Pharmakokinetik und Pharmakodynamik der in der Assistierten Reproduktion Verwendeten Gestagene. [Pharmacokinetics and Pharmacodynamics of Progestogens Used in Assisted Reproduction.] Gynäkologische Endokrinologie [Gynecological Endocrinology], 19, 105–117. [Google Scholar] [DOI:10.1007/s10304-020-00372-5] [Translation] [Translated]:
Abstract
[…] Oral administration of progesterone causes extensive metabolism resulting in the formation of more than 30 metabolites, some of which may cause anesthetic/sedative effects. The use of a direct radioimmunoassay (RIA) without prior separation of progesterone from its metabolites may simulate falsely high serum concentrations of progesterone owing to cross-reaction of some metabolites with the specific antiserum of the RIA. This effect is not observed after parenteral treatment with progesterone due to minimal metabolism. […]
Progesterone pharmacokinetics
Problems with the determination of progesterone in serum
Because of the large number of progesterone metabolites that circulate in variable concentrations after oral administration, false-high progesterone levels must be expected when using an immunoassay. The cause is the cross-reactions of some progesterone metabolites, such as 5α- and 5β-DHP and 5α-pregnan-3β-ol-20-one [13]. For this reason, a radioimmunoassay (RIA) can only be used, for example, if the progesterone has previously been isolated by means of a suitable chromatographic separation. The determination of progesterone with exact liquid or gas chromatography-mass spectrometry (LC-MS or GC-MS; [14]; Table 1) is more reliable, albeit more complex.
The problem of cross-reactions does not play an essential role in parenteral progesterone treatment due to the low metabolism of progesterone. The specificity of the antiserum used is of particular importance for the comparability of the results of different RIAs, as it influences the cross-reactivity of the numerous progesterone metabolites [11].
The determination of the progesterone level, which was measured after oral administration of a capsule containing 100 mg of micronized progesterone, resulted in a false high peak value of 19.4 ng/mL in the RIA, which was 8 times as high as that with the exact LC-MS method which determined a true value of 2.4 ng/mL (Table 1). The cause was metabolites, the high concentrations of which simulated a false high concentration via a cross-reaction with the RIA antiserum [11]. In contrast, after intravaginal application of a gel containing 90 mg of progesterone, the metabolism of progesterone and thus the influence of cross-reactions is so low that the progesterone concentration of 10.5 ng/mL measured with the RIA agrees with the result of the exact LC-MS method (Table 1; [11]).
“Enzyme-linked immunosorbent assay”
As with the RIA, the “enzyme-linked immunosorbent assay” (ELISA) is a procedure based on an immune reaction between an antigen and a specific antibody. For this purpose, a specific antibody is produced in a separate standard process for the antigen, for example progesterone, which binds the antigen with high affinity. An enzyme is coupled to this specific antibody that cleaves an added chromogen, producing a dye, the intensity of which is measured in a photometer. The measured intensity of the color correlates with the concentration of the antigen (for example progesterone).
With regard to the cross-reaction of some progesterone metabolites, which leads to false high values of the progesterone concentration, the same restriction applies to the ELISA as to the direct RIA (Table 1).
Summary
If a progesterone determination is carried out in the serum after oral progesterone application in the RIA or in the ELISA without prior chromatographic separation, one measures incorrectly high progesterone values, since progesterone metabolites are also measured! This problem does not exist with parenteral application (intravaginal, intramuscular, or subcutaneous).
Oral application of progesterone
After separation of important progesterone metabolites by means of column chromatography, an increase in progesterone in the serum to a maximum of 4.7 ± 1.15 ng/mL was found in the RIA after oral administration of 200 mg micronized progesterone [14].
Summary
In summary, it can be stated that oral administration of 100 to 200 mg progesterone leads to an increase in serum levels to around 3–5 ng/mL, provided the laboratory diagnostics are carried out correctly, i.e. after chromatographic separation.
Conclusion for practice
After oral progesterone administration, more than 30 metabolites are formed, which in immunoassays can cause false high results of the systemic progesterone concentration. This problem can only be solved by prior chromatographic separation of the progesterone or by direct measurement methods.
Individual Studies and Papers
Comparative Immunoassay versus Immunoassay with Chromatography or Mass Spectrometry Studies
Nahoul, K., Dehennin, L., & Scholler, R. (1987). Radioimmunoassay of plasma progesterone after oral administration of micronized progesterone. Journal of Steroid Biochemistry, 26(2), 241–249. [DOI:10.1016/0022-4731(87)90078-1]
Levine, H., & Watson, N. (2000). Comparison of the pharmacokinetics of Crinone 8% administered vaginally versus Prometrium administered orally in postmenopausal women. Fertility and Sterility, 73(3), 516–521. [DOI:10.1016/S0015-0282(99)00553-1]
Sapin, R., Neamtu, D., Gasser, F., Ohl, J., Grunenberger, F., & Grucker, D. (2000). De la prudence lors de l’utilisation des dosages directs de progestérone. [Caution when using direct progesterone assays.] Immuno-analyse et Biologie Spécialisée, 3(15), 203–204. [DOI:10.1016/S0923-2532(00)80010-1] [Translation]
Non-Comparative Immunoassay with Adequate Chromatography or Mass Spectrometry Studies
Nahoul, K., Dehennin, L., Jondet, M., & Roger, M. (1993). Profiles of plasma estrogens, progesterone and their metabolites after oral or vaginal administration of estradiol or progesterone. Maturitas, 16(3), 185–202. [DOI:10.1016/0378-5122(93)90064-O]
Lobo, R. A., Liu, J., Stanczyk, F. Z., Constantine, G. D., Pickar, J. H., Shadiack, A. M., Bernick, B., & Mirkin, S. (2019). Estradiol and progesterone bioavailability for moderate to severe vasomotor symptom treatment and endometrial protection with the continuous-combined regimen of TX-001HR (oral estradiol and progesterone capsules). Menopause (New York, NY), 26(7), 720–727.[DOI:10.1097/GME.0000000000001306]
Commentaries on Studies
Nahoul, K., & de Ziegler, D. (1994). “Validity” of serum progesterone levels after oral progesterone. Fertility and Sterility, 61(4), 790–791. [DOI:10.1016/S0015-0282(16)56666-7]
\ No newline at end of file
diff --git a/transfemscience.org/articles/oral-p4-low-levels/index.html b/transfemscience.org/articles/oral-p4-low-levels/index.html
index d5cfede8..af2e32f8 100644
--- a/transfemscience.org/articles/oral-p4-low-levels/index.html
+++ b/transfemscience.org/articles/oral-p4-low-levels/index.html
@@ -1 +1 @@
-Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects - Transfeminine Science
Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects
By Aly | First published August 4, 2018 | Last modified August 20, 2025
Abstract / TL;DR
Oral progesterone is the most widely used form of progesterone in transfeminine hormone therapy. Because of previous studies using inaccurate blood tests (immunoassays without adequate chromatographic purification), it was thought that typical therapeutic dosages of oral progesterone produced progesterone levels that reached typical luteal-phase levels in cisgender women (which range from about 7 to 22 ng/mL). However, newer studies using more accurate blood tests (immunoassays with adequate purification and mass spectrometry-based assays) have shown that 100 mg/day progesterone—with or without food—achieves very low peak progesterone levels of only about 2 to 3 ng/mL and average progesterone levels over 24 hours of only about 0.1 to 0.6 ng/mL. In accordance, oral progesterone has often shown only weak progestogenic effects in clinical studies. Higher doses of oral progesterone that might achieve better levels are limited by persistingly low progesterone levels, pronounced neurosteroid side effects caused by the first pass of progesterone through the liver, and substantial variability between individuals. While the progesterone levels with oral progesterone are apparently sufficient for endometrial protection in cisgender women, they are unlikely to be adequate for desired effects in transfeminine people. For these reasons, transfeminine people and their clinicians may wish to avoid oral progesterone if the aim is therapeutic progestogenic effects. Instead, non-oral forms of progesterone with greater bioavailability like rectal or injectable progesterone can be used. Alternatively, progestins, which are likewise fully effective progestogens, can be employed.
Introduction
The major female sex hormones are estrogens and progestogens, and both may be used in transfeminine hormone therapy. Progestogens are useful in transfeminine people for helping to suppress testosterone levels and possibly though not certainly influencing breast development. Progestogens include progesterone as well as synthetic progestogens known as progestins. Progesterone has relatively unfavorable pharmacokinetics, which has been overcome with progestins. However, progestins have differing pharmacodynamic properties compared to progesterone, which can potentially be unfavorable. As such, there is interest in using progesterone instead of progestins in transfeminine people and other populations like cisgender women despite its poor pharmacokinetic properties.
Progesterone is available in formulations for use via a variety of different routes, including oral, sublingual, topical, vaginal, rectal, and injectable administration (Wiki; Table). Among these routes, oral administration is the easiest and most convenient, and in relation to this, oral progesterone is the most widely used form of progesterone in transfeminine people. However, the pharmacokinetic problems of progesterone limit the favorability of oral progesterone. Moreover, these limitations of oral progesterone actually appear to be much more substantial than is generally realized, a fact that has been obscured by methodological limitations of many—but not all—of the pharmacokinetic studies that have characterized oral progesterone. The purpose of this article is to explain and review these findings, as well as to explore solutions and alternatives to oral progesterone for progestogen therapy in transfeminine people.
Progesterone Levels with Oral Progesterone
Oral micronized progesterone, or simply oral progesterone, is the form of progesterone that is used by the oral route as a pharmaceutical medication. It is an oil suspension of micronized progesterone crystals contained in gelatin capsules. The formulation is marketed under brand names including Prometrium, Utrogestan, and Microgest, among many others. Oral progesterone has very low bioavailability (≤10%) due to the first pass through the intestines and liver with oral administration. As a result of the first pass, most of the delivered progesterone with oral progesterone is metabolized into neurosteroid metabolites such as allopregnanolone and pregnanolone before reaching the bloodstream (de Lignieres, Dennerstein, & Backstrom, 1995). This is why oral progesterone has alcohol-like side effects like sedation that are not shared by typical doses of non-oral progesterone such as vaginal progesterone or progesterone by injection. In spite of the low bioavailability of oral progesterone, typical clinical doses of oral progesterone, such as 100 to 300 mg/day, have been reported to produce progesterone levels measured with immunoassays that are similar to those in the normal luteal phase of the menstrual cycle in cisgender women (Simon et al., 1993). For this reason, it has been believed that oral progesterone can achieve high and physiologically adequate levels of progesterone.
Figure 1: Progesterone levels during the menstrual cycle in normal premenopausal women (Stricker et al., 2006). The dashed horizontal lines are the mean levels for each curve and the dashed vertical line demarcates mid-cycle (when ovulation occurs). Progesterone levels are normally elevated only during the luteal phase.
Figure 2: Progesterone levels measured by immunoassay after single 100 to 300 mg doses of oral micronized progesterone in postmenopausal women (Simon et al., 1993). The horizontal dashed lines are mean levels over 24 hours. Progesterone levels appeared to reach concentrations comparable to normal luteal-phase levels. However, these levels were in fact not accurate due to the use of immunoassays (Nahoul & de Ziegler, 1994).
A notable study using LC–MS found maximal progesterone levels of only about 2 ng/mL and average progesterone levels over a period of 24 hours of only about 0.14 ng/mL after a single 100 mg dose of oral progesterone (Levine & Watson, 2000; Kuhl & Schneider, 2013). Another more recent study with LC–MS found progesterone levels of around 2.5 to 3 ng/mL at peak and average progesterone levels over 24 hours of around 0.6 ng/mL after a single 100 mg dose of oral progesterone with food (Lobo et al., 2019). (It should be noted that intake with food is known to increase the bioavailability of oral progesterone by a few-fold (Wiki; Bijuva FDA Label; Simon et al., 1993; Prometrium FDA Review, 1996; Pickar et al., 2015).) These progesterone levels are well below normal luteal-phase levels of progesterone, which range from 7 to 22 ng/mL with LC–MS (Nakamoto, 2016). Studies that have directly compared quantification of progesterone with immunoassays against more reliable methods have found that immunoassays overestimate progesterone levels by 5- to 8-fold (Nahoul, Dehennin, & Scholler, 1987; Nahoul & de Ziegler, 1994; Levine & Watson, 2000; Kuhl, 2011; Kuhl & Schneider, 2013; Davey, 2018). In one small study of a few individuals, the degree of overestimation varied from 2-fold to 40-fold with several different commercial immunoassays (Sapin et al., 2000). These findings are obscure and still relatively little-known in the scientific and medical communities. In any case, it is clear that oral progesterone achieves progesterone levels that are far lower than once thought and are well below the luteal-phase levels that would be therapeutically desirable for transfeminine people.
Figure 3: Progesterone levels measured by immunoassay or LC–MS after a single dose of oral or vaginal micronized progesterone in postmenopausal women (Levine & Watson, 2000; Kuhl & Schneider, 2013). Levels of progesterone with oral progesterone measured by immunoassay were falsely high due to cross-reactivity. Conversely, progesterone levels measured by LC–MS or with vaginal progesterone can be considered accurate.
Figure 4: Progesterone levels measured by LC–MS with 100 mg/day oral micronized progesterone taken with food in postmenopausal women (Lobo et al., 2019). The horizontal dashed line is the mean level over 24 hours. Food increases progesterone levels with oral progesterone by about 2- to 3-fold (Bijuva FDA Label; Simon et al., 1993). The progesterone levels measured in this study can be considered accurate to due to the use of LC–MS.
Therapeutic Implications
Progestogenic Potency and Effects
A variety of perplexing findings on the clinical progestogenic effects of oral progesterone have been made over the decades and can now be readily explained by the newer data on oral progesterone with better analytic methods. Oral progesterone is used in clinical medicine mainly to protect the endometrium from unopposed stimulation by estrogens in menopausal cisgender women and is able to reliably prevent endometrial hyperplasia induced by estrogens even with the low progesterone levels that typical clinical doses achieve (Wiki). However, oral progesterone failed to provide adequate protection against estrogen-mediated endometrial cancer risk in a large observational study (Davey, 2018). Oral progesterone even at very high doses also is unable to produce full endometrial transformation—a normal effect of luteal-phase levels of progesterone—whereas vaginal and injectable progesterone are effective (de Ziegler et al., 2013). For this reason, oral progesterone, in contrast to parenteral progesterone, is considered to be inappropriate for use in assisted reproduction (de Ziegler et al., 2013). Oral progesterone additionally failed to suppress testosterone levels even at high doses (400 mg/day) in cisgender males (Trollan et al., 1993; Wiki). Conversely, progestins, rectal progesterone, and injectable progesterone can all produce robust testosterone suppression in cisgender males (Wiki; Aly, 2019). Similarly, oral progesterone has little or no apparent antigonadotropic effect in menopausal cisgender women, which is again in notable contrast to progestins (Holst, 1983; Holst et al., 1983; Ottosson, 1984; Maxson & Hargrove, 1985; Saarikoski, Yliskosk, & Penttilä, 1990). However, in one study, oral progesterone did seem to show a substantial antigonadotropic effect similarly to medroxyprogesterone acetate (MPA) and vaginal progesterone in premenopausal women with polycystic ovary syndrome (PCOS) (Bagis et al., 2002).
Unlike other clinically used progestogens, the addition of oral progesterone to estrogen therapy in menopausal women has not been associated with increased risk of venous thromboembolism (VTE; blood clots) (Wiki). Nor has it been associated with increased breast cancer risk in the short-term (<5 years of therapy) (Wiki). However, with long-term use (≥5 years), the combination of estrogen plus oral progesterone is associated with significantly greater risk of breast cancer relative to estrogen alone similarly to other progestogens (Aly, 2020; Sam, 2020; Wiki; Table). This has been said to be consistent with a weak proliferative effect of oral progesterone on the breasts such that a longer duration of exposure is necessary for a quantifiable increase in breast cancer risk to manifest (Kuhl & Schneider, 2013; Davey, 2018). It is also consistent with preclinical research, which clearly suggests a carcinogenic role for progesterone and progesterone receptor activation in the breast (Kuhl & Schneider, 2013; Trabert et al., 2020). The preceding clinical findings on endometrial efficacy, testosterone and gonadotropin suppression, VTE risk, and breast cancer risk with oral progesterone are in contrast to those with almost all clinically used progestins (with the exception of the oral progesterone-like dydrogesterone). These previously perplexing discrepancies can be readily explained by the very low levels of progesterone that are now known to be achieved with oral progesterone.
Bioavailability, Half-Life, and Duration
Considering the much lower levels of progesterone observed with oral progesterone in studies using reliable analytic methods, the bioavailability of oral progesterone needs to be reassessed. In studies with immunoassays, the bioavailability of oral progesterone has been reported to be around 10% (Wiki). The true oral bioavailability of progesterone is unknown at this time as studies with reliable analytic methods have not been conducted. In any case, it can be assumed that it may be closer to around 1 or 2% based on the findings that immunoassays overestimate progesterone levels by 5- to 8-fold.
The elimination half-life of progesterone with oral progesterone has been determined in studies employing immunoassays to be 16 to 18 hours (Wiki). Based on the fact that the blood half-life of progesterone administered by intravenous injection is very short at a range of only 3 minutes to 1.5 hours (Wiki), the reported half-life of progesterone with oral progesterone is much longer than one would expect. Oral estradiol has a relatively long half-life of 13 to 20 hours due to formation with the first pass of a circulating reservoir of estrogen conjugates that are slowly converted back into estradiol (Kuhl, 2005; Wiki). In contrast to estradiol however, progesterone itself has no available hydroxyl groups for conjugation and an analogous circulating reservoir of progesterone conjugates that can be converted back into progesterone is not known to be the case (Kuhl, 2005).
Studies with more reliable analytic methods like LC–MS have found a half-life of progesterone with oral progesterone of 5 to 10 hours and a duration of highly elevated progesterone levels of only about 4 to 8 hours (Wiki; Graphs). These findings indicate that oral progesterone has a much shorter duration than previously thought as well. As such, if oral progesterone is used, it may be advisable to take it in divided doses multiple times per day to allow for more sustained progestogenic exposure.
Higher Oral Progesterone Doses and Neurosteroid Side Effects
Use of higher doses of oral progesterone than typical doses is likely to achieve dose-dependently higher progesterone levels (Table). However, based on how low progesterone levels are with oral progesterone using reliable analytic techniques, even very high doses would still be expected to achieve only low progesterone levels in most cases. Moreover, high doses of oral progesterone result in very high levels of its neurosteroid metabolites and have been found to produce substantial alcohol-like side effects (i.e., central depression and effects within that umbrella) (Wiki; Wiki). These limitations are likely to preclude higher doses of oral progesterone from being practical.
Figure 5: Levels of progesterone, allopregnanolone, and pregnanolone in premenopausal women following a single dose of oral progesterone or vaginal progesterone (as a suppository) (de Lignieres, Dennerstein, & Backstrom, 1995). Allopregnanolone and pregnanolone levels were determined by MS-based assays while progesterone levels were measured by immunoassay with chromatographic separation. Hence, the levels should be reliable.
A Note on Oral Progesterone’s Metabolites
Although progesterone levels with oral progesterone are very low, the metabolites of progesterone are formed in disproportionate amounts with the first pass (Sitruk-Ware et al., 1987; de Lignieres, Dennerstein, & Backstrom, 1995; de Lignieres, 1999; de Ziegler & Fanchin, 2000; Lobo, 2000; Kuhl, 2005). Moreover, while much less potent than progesterone, some of these metabolites have been found to have progestogenic activity similarly to progesterone (e.g., Besch et al., 1965; Junkermann, Runnebaum, & Lisboa, 1977; Lobo, 2000). This activity derives either from them having intrinsic progestogenic activity of their own or from being converted back into progesterone or other progestogenic metabolites (including in an intracrine fashion within tissues, for instance in the uterus). Examples of such metabolites include 20α-dihydroprogesterone, 20β-dihydroprogesterone, 5α-dihydroprogesterone, 3β-dihydroprogesterone, allopregnanolone, and 11-deoxycorticosterone. If the metabolites of oral progesterone contribute significantly to its progestogenic activity, then the progestogenic strength of oral progesterone would be greater than that implied by the progesterone levels that occur with it alone. However, this possibility is only theoretical and there is little literature discussing it. More research would be needed to determine if the metabolites of oral progesterone do indeed play a meaningful role in its progestogenic potency. In any case, oral progesterone is still a relatively weak progestogen based on clinical studies of its progestogenic effects.
Alternative Options to Oral Progesterone
Non-Oral Forms of Progesterone
Non-oral forms of progesterone, such as vaginal progesterone, rectal progesterone, sublingual progesterone, and progesterone by injection, have been found to achieve much higher progesterone levels than oral progesterone (Wiki). They can be used instead of oral progesterone to achieve higher and more adequate progesterone levels. Unfortunately however, while more effective than oral progesterone, non-oral progesterone routes have various limitations of their own.
Vaginal progesterone is of course not possible in transfeminine people who have not undergone vaginoplasty. And in those who have undergone vaginoplasty, the lining of the neovagina is either skin (penile inversion vaginoplasty) or intestine (sigmoid colon vaginoplasty) rather than the normal vaginal mucosa. As such, the absorptive characteristics of neovaginal administration are likely not the same as vaginal administration (Aly, 2018). It is notable that transdermal progesterone achieves very low progesterone levels similarly to oral progesterone and is not a good option for progesterone therapy (Wiki; Hermann et al., 2005; Graph). Progesterone levels with neovaginal administration of progesterone in those who have undergone penile inversion vaginoplasty are likely to be low similarly.
Rectal progesterone is an excellent route that achieves high progesterone levels comparable to the levels of progesterone that occur during the normal luteal phase (Wiki; Graphs). It has a somewhat short duration and twice daily use may be warranted for more sustained levels however. A more important problem is that the availability of pharmaceutical rectal progesterone suppositories throughout the world is limited and they are not marketed in most countries (Wiki). In any case, rectal progesterone suppositories may be available from compounding pharmacies in some countries. In addition, oral micronized progesterone capsules are available ubiquitously and have been administered vaginally instead of orally with success (Miles et al., 1994; Wang et al., 2019). Administration of oral micronized progesterone capsules rectally instead of orally likewise may be effective and may achieve much higher progesterone levels than oral administration (Aly, 2018). However, rectal administration of oral progesterone capsules has not been formally studied. Although rectal progesterone is effective, it is fairly inconvenient. This may be especially true with long-term therapy. In any case, of the available non-oral forms of progesterone, rectal progesterone is probably the best overall. A significant subset of transfeminine people on progestogens take progesterone rectally (Chang et al., 2024).
Figure 6: Progesterone levels with a single suppository containing 100 mg progesterone administered rectally or vaginally in premenopausal women (Chakmakjian & Zachariah, 1987).
Sublingual progesterone appears to achieve high and more physiological progesterone levels than oral progesterone but has a short duration of highly elevated progesterone levels similarly and necessitates administration several times per day (Wiki; Graph). Moreover, although sublingual progesterone may have been more widely available in the past (Wiki), it is available today only in a couple of Eastern European countries (Wiki). It might be available from compounding pharmacies in some countries however. While never formally studied, it may be possible to use oral micronized progesterone capsules sublingually instead of orally. However, this route is complicated by the fact that this form of progesterone is suspended in oil within gelatin capsules. Hence, sublingual administration of oral micronized progesterone is likely to be difficult and potentially unpleasant.
Progesterone by intramuscular or subcutaneous injection can easily achieve very high progesterone levels (Wiki; Graphs; Wiki; Graph). However, progesterone administered by this route has a relatively short duration when compared to other injectable sex-hormone formulations and requires injection once every 1 to 3 days. This is simply too frequent for most people, especially with long-term therapy. In addition, progesterone by subcutaneous injection, which is more convenient than progesterone by intramuscular injection, has limited availability and is marketed mostly only in Europe (Wiki). In contrast to other sex hormones like estradiol and testosterone, progesterone esters that are more fat-soluble than progesterone and extend its duration when used in injectable form are not possible since progesterone has no free hydroxyl groups available for esterification. Injectable aqueous suspensions of progesterone that had much longer durations than the oil solutions and aqueous solutions that are used by injection today were previously available (Wiki; Wiki; Aly, 2019). However, they were associated with painful injection site reactions and this led to their discontinuation. In any case, injectable aqueous suspensions of progesterone do actually seem to remain available for people in a couple of Eastern European countries today (Aly, 2019).
In conclusion, oral progesterone achieves very low progesterone levels at typical clinical doses and produces only weak progestogenic effects that seem to be far from physiologically adequate. Although use of higher doses of oral progesterone is likely to achieve higher progesterone levels, such doses are likely to be impractical because progesterone levels will still be low even at higher doses and the neurosteroid side effects of oral progesterone will be much more substantial and difficult to tolerate.
Due to its limitations, transfeminine people and clinicians treating them may wish to avoid oral progesterone if the intended goal is to produce therapeutic progestogenic effects. Instead, non-oral progesterone routes, such as rectal and injected progesterone, although with various limitations such as limited availability and inconvenience, can be used. Alternatively, progestins, particularly those with more favorable profiles, can be used instead of progesterone altogether.
Oral progesterone may perhaps be most appropriately conceptualized as a potent neurosteroid prodrug with weak progestogenic effects. Conversely, non-oral progesterone, as well as progestins, can be regarded as potent progestogens with either physiological or no neurosteroid effects, respectively.
Additional Content
Literature
The sources and excerpts collected here go in-depth on much of what has been described in this article on the topic of the measurement problems and low progesterone levels with oral progesterone.
In August 2025, a conference abstract of a randomized controlled trial of oral progesterone and breast development in transfeminine people was published (Dreijerink et al., 2025). This study had been preregistered and its protocol had been previously published in 2023 (Dijkman et al., 2023). The trial found that oral micronized progesterone (Utrogestan) at doses of 200 to 400 mg/day, in conjunction with estradiol therapy and especially with higher estradiol levels in the range of 400 to 800 pmol/L (109–218 pg/mL), could significantly increase breast volume in transfeminine people. The study is discussed in greater detail elsewhere on this site (Aly, 2020). Based on the trial’s findings, although oral progesterone can only achieve low progesterone levels measured with mass spectrometry, it appears that these low levels are nonetheless able to produce significant effects on the breasts. Hence, while oral progesterone may still prove to be less efficacious than non-oral progesterone or progestins, it would appear that it is not absent of value in terms of potential therapeutic effects in transfeminine people.
References
Bagis, T., Gokcel, A., Zeyneloglu, H. B., Tarim, E., Kilicdag, E. B., & Haydardedeoglu, B. (2002). The Effects of Short-Term Medroxyprogesterone Acetate and Micronized Progesterone on Glucose Metabolism and Lipid Profiles in Patients with Polycystic Ovary Syndrome: A Prospective Randomized Study. The Journal of Clinical Endocrinology & Metabolism, 87(10), 4536–4540. [DOI:10.1210/jc.2002-020294]
Besch, P. K., Barry, R. D., Vorys, N., Stevens, V., & Ullery, J. C. (1965). A review of some aspects of the metabolism of progestational agents. Metabolism, 14(3), 432–443. [DOI:10.1016/0026-0495(65)90031-4]
Bijuva Estradiol/Progesterone Label. U.S. Food and Drug Administration. [URL] [PDF]
Chakmakjian, Z. H., & Zachariah, N. Y. (1987). Bioavailability of progesterone with different modes of administration. The Journal of Reproductive Medicine, 32(6), 443–448. [Google Scholar] [PubMed] [PDF]
Chang, J. J., Tran, N. K., Flentje, A., Lubensky, M. E., Obedin-Maliver, J., Lunn, M. R., & Ariel, D. (2024). 12330 Progestogen Experience And Perception Among Transfeminine Adults - A National Survey. Journal of the Endocrine Society, 8(Suppl 1), bvae163.1657. [DOI:10.1210/jendso/bvae163.1657]
Conklin, S. E., & Knezevic, C. E. (2020). Advancements in the gold standard: Measuring steroid sex hormones by mass spectrometry. Clinical Biochemistry, 82, 21–32. [DOI:10.1016/j.clinbiochem.2020.03.008]
Davey, D. A. (2018). Menopausal hormone therapy: a better and safer future. Climacteric, 21(5), 454–461. [DOI:10.1080/13697137.2018.1439915]
de Lignières, B., Dennerstein, L., & Backstrom, T. (1995). Influence of route of administration on progesterone metabolism. Maturitas, 21(3), 251–257. [DOI:10.1016/0378-5122(94)00882-8]
de Lignières, B. (1999). Oral micronized progesterone. Clinical Therapeutics, 21(1), 41–60. [DOI:10.1016/S0149-2918(00)88267-3]
de Ziegler, D., & Fanchin, R. (2000). Progesterone and progestins: applications in gynecology. Steroids, 65(10–11), 671–679. [DOI:10.1016/S0039-128X(00)00123-9]
de Ziegler, D., Streuli, I., Marszalek, A., Gayet, V., & Chapron, C. (2013). Preparing the Endometrium to Maximize Success: The Dynamics of Artificial Cycles. In Sauer, M. V. (Ed.). Principles of Oocyte and Embryo Donation, 2nd Edition (pp. 109–127). London: Springer. [DOI:10.1007/978-1-4471-2392-7_9]
Dijkman, B. A., Helder, D., Boogers, L. S., Gieles, N. C., van Heesewijk, J. O., Slaa, S. t., Liberton, N. P., Wiepjes, C. M., de Blok, C. J., den Heijer, M., & Dreijerink, K. M. (2023). Addition of progesterone to feminizing gender-affirming hormone therapy in transgender individuals for breast development: a randomized controlled trial. BMC Pharmacology and Toxicology, 24(1), 80. [DOI:10.1186/s40360-023-00724-4]
Dreijerink, K., den Heijer, M., Geels, R. (2025). Increased breast volume due to addition of progesterone and increasing the estradiol dose in feminizing gender-affirming hormone therapy. EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
Hermann, A. C., Nafziger, A. N., Victory, J., Kulawy, R., Rocci Jr, M. L., & Bertino Jr, J. S. (2005). Over‐the‐Counter Progesterone Cream Produces Significant Drug Exposure Compared to a Food and Drug Administration‐Approved Oral Progesterone Product. The Journal of Clinical Pharmacology, 45(6), 614–619. [DOI:10.1177/0091270005276621]
Holst, J. (1983). Percutaneous estrogen therapy: Endometrial response and metabolic effects. Acta Obstetricia et Gynecologica Scandinavica, 62(Suppl 115), 4–30. [DOI:10.3109/00016348309155363]
Holst, J., Cajander, S., Carlström, K., Damber, M. G., & von Schoultz, B. (1983). Percutaneous oestrogen therapy opposed by lynestrenol or natural progesterone-effects on circulating oestrogens, FSH, sex hormone binding globulin and pregnancy zone protein. Maturitas, 5(1), 1–8. [DOI:10.1016/0378-5122(83)90015-4]
Junkermann, H., Runnebaum, B., & Lisboa, B. P. (1977). New progesterone metabolites in human myometrium. Steroids, 30(1), 1–14. [DOI:10.1016/0039-128X(77)90131-3]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H. (2011). Pharmacology of progestogens. Journal für Reproduktionsmedizin und Endokrinologie [Journal of Reproductive Medicine and Endocrinology], 8(1), 157–177. [URL]
Kuhl, H., & Schneider, H. P. G. (2013). Progesterone–promoter or inhibitor of breast cancer. Climacteric, 16(Suppl 1), 54–68. [DOI:10.3109/13697137.2013.768806]
Lobo, R. A. (2000). Progestogens. In Lobo, R. A., Kelsey, J., & Marcus, R. (Eds.). Menopause: Biology and Pathobiology (pp. 429–444). Burlington: Academic Press. [Google Books]
Lobo, R. A., Liu, J., Stanczyk, F. Z., Constantine, G. D., Pickar, J. H., Shadiack, A. M., Bernick, B., & Mirkin, S. (2019). Estradiol and progesterone bioavailability for moderate to severe vasomotor symptom treatment and endometrial protection with the continuous-combined regimen of TX-001HR (oral estradiol and progesterone capsules). Menopause (New York, NY), 26(7), 720–727.[DOI:10.1097/GME.0000000000001306]
Levine, H., & Watson, N. (2000). Comparison of the pharmacokinetics of Crinone 8% administered vaginally versus Prometrium administered orally in postmenopausal women. Fertility and Sterility, 73(3), 516–521. [DOI:10.1016/S0015-0282(99)00553-1]
Maxson, W. S., & Hargrove, J. T. (1985). Bioavailability of oral micronized progesterone. Fertility and Sterility, 44(5), 622–626. [DOI:10.1016/S0015-0282(16)48977-6]
Miles, R. A., Paulson, R. J., Lobo, R. A., Press, M. F., Dahmoush, L., & Sauer, M. V. (1994). Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: a comparative study. Fertility and Sterility, 62(3), 485–490. [DOI:10.1016/S0015-0282(16)56935-0]
Nahoul, K., Dehennin, L., & Scholler, R. (1987). Radioimmunoassay of plasma progesterone after oral administration of micronized progesterone. Journal of Steroid Biochemistry, 26(2), 241–249. [DOI:10.1016/0022-4731(87)90078-1]
Nahoul, K., Dehennin, L., Jondet, M., & Roger, M. (1993). Profiles of plasma estrogens, progesterone and their metabolites after oral or vaginal administration of estradiol or progesterone. Maturitas, 16(3), 185–202. [DOI:10.1016/0378-5122(93)90064-O]
Nahoul, K., & de Ziegler, D. (1994). “ Validity” of serum progesterone levels after oral progesterone. Fertility and Sterility, 61(4), 790–792. [DOI:10.1016/S0015-0282(16)56666-7]
Nakamoto, J. (2016). Endocrine Testing. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition (pp. 2655–2688.e1). Philadelphia: Saunders/Elsevier. [DOI:10.1016/B978-0-323-18907-1.00154-2]
Ottosson, U. B. (1984). Oral progesterone and estrogen/progestogen therapy: effects of natural and synthetic hormones on subfractions of HDL cholesterol and liver proteins. Acta Obstetricia et Gynecologica Scandinavica, 63(Suppl 127), 1–37. [DOI:10.3109/00016348409157016]
Pickar, J. H., Bon, C., Amadio, J. M., Mirkin, S., & Bernick, B. (2015). Pharmacokinetics of the first combination 17β-estradiol/progesterone capsule in clinical development for menopausal hormone therapy. Menopause (New York, NY), 22(12), 1308–1316. [DOI:10.1097/GME.0000000000000467]
Prometrium FDA Review 1996. Application Number: NDA 19-781. Clinical Pharmacology and Biopharmaceutics Review(s) [Prometrium]. Center for Drug Evaluation and Research, U.S. Food and Drug Administration. [PDF]
Saarikoski, S., Yliskosk, M., & Penttilä, I. (1990). Sequential use of norethisterone and natural progesterone in pre-menopausal bleeding disorders. Maturitas, 12(2), 89–97. [DOI:10.1016/0378-5122(90)90086-L]
Sapin, R., Neamtu, D., Gasser, F., Ohl, J., Grunenberger, F., & Grucker, D. (2000). De la prudence lors de l’utilisation des dosages directs de progestérone. [Caution when using direct progesterone assays.] Immuno-analyse et Biologie Spécialisée, 3(15), 203–204. [DOI:10.1016/S0923-2532(00)80010-1] [Translation]
Simon, J. A., Robinson, D. E., Andrews, M. C., Hildebrand III, J. R., Rocci Jr, M. L., Blake, R. E., & Hodgen, G. D. (1993). The absorption of oral micronized progesterone: the effect of food, dose proportionality, and comparison with intramuscular progesterone. Fertility and Sterility, 60(1), 26–33. [DOI:10.1016/S0015-0282(16)56031-2]
Sitruk-Ware, R., Bricaire, C., De Lignieres, B., Yaneva, H., & Mauvais-Jarvis, P. (1987). Oral micronized progesterone: Bioavailability pharmacokinetics, pharmacological and therapeutic implications—A review. Contraception, 36(4), 373–402. [DOI:10.1016/0010-7824(87)90088-6]
Stricker, R., Eberhart, R., Chevailler, M. C., Quinn, F. A., Bischof, P., & Stricker, R. (2006). Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT® analyzer. Clinical Chemistry and Laboratory Medicine (CCLM), 44(7), 883–887. [DOI:10.1515/CCLM.2006.160]
Tollan, A., Øian, P., Kjeldsen, S. E., Eide, I., & Maltau, J. M. (1993). Progesterone reduces sympathetic tone without changing blood pressure or fluid balance in men. Gynecologic and Obstetric Investigation, 36(4), 234–238. [DOI:10.1159/000292636]
Trabert, B., Sherman, M. E., Kannan, N., & Stanczyk, F. Z. (2020). Progesterone and breast cancer. Endocrine Reviews, 41(2), 320–344. [DOI:10.1210/endrev/bnz001]
Wang, H., Liu, M., Fu, Q., & Deng, C. (2019). Pharmacokinetics of hard micronized progesterone capsules via vaginal or oral route compared with soft micronized capsules in healthy postmenopausal women: a randomized open-label clinical study. Drug Design, Development and Therapy, 13, 2475–2482. [DOI:10.2147/dddt.s204624]
Woodward, G. M., & Rumsby, G. (2019). Steroid Profiling: Analytical Perspectives. In Rumsby, G., Woodward, G. M. (Eds.). Disorders of Steroidogenesis: Guide to Steroid Profiling and Biochemical Diagnosis (pp. 27–40). Cham: Springer. [DOI:10.1007/978-3-319-96364-8_3]
\ No newline at end of file
+Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects - Transfeminine Science
Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects
By Aly | First published August 4, 2018 | Last modified August 20, 2025
Abstract / TL;DR
Oral progesterone is the most widely used form of progesterone in transfeminine hormone therapy. Because of previous studies using inaccurate blood tests (immunoassays without adequate chromatographic purification), it was thought that typical therapeutic dosages of oral progesterone produced progesterone levels that reached typical luteal-phase levels in cisgender women (which range from about 7 to 22 ng/mL). However, newer studies using more accurate blood tests (immunoassays with adequate purification and mass spectrometry-based assays) have shown that 100 mg/day progesterone—with or without food—achieves very low peak progesterone levels of only about 2 to 3 ng/mL and average progesterone levels over 24 hours of only about 0.1 to 0.6 ng/mL. In accordance, oral progesterone has often shown only weak progestogenic effects in clinical studies. Higher doses of oral progesterone that might achieve better levels are limited by persistingly low progesterone levels, pronounced neurosteroid side effects caused by the first pass of progesterone through the liver, and substantial variability between individuals. While the progesterone levels with oral progesterone are apparently sufficient for endometrial protection in cisgender women, they are unlikely to be adequate for desired effects in transfeminine people. For these reasons, transfeminine people and their clinicians may wish to avoid oral progesterone if the aim is therapeutic progestogenic effects. Instead, non-oral forms of progesterone with greater bioavailability like rectal or injectable progesterone can be used. Alternatively, progestins, which are likewise fully effective progestogens, can be employed.
Introduction
The major female sex hormones are estrogens and progestogens, and both may be used in transfeminine hormone therapy. Progestogens are useful in transfeminine people for helping to suppress testosterone levels and possibly though not certainly influencing breast development. Progestogens include progesterone as well as synthetic progestogens known as progestins. Progesterone has relatively unfavorable pharmacokinetics, which has been overcome with progestins. However, progestins have differing pharmacodynamic properties compared to progesterone, which can potentially be unfavorable. As such, there is interest in using progesterone instead of progestins in transfeminine people and other populations like cisgender women despite its poor pharmacokinetic properties.
Progesterone is available in formulations for use via a variety of different routes, including oral, sublingual, topical, vaginal, rectal, and injectable administration (Wiki; Table). Among these routes, oral administration is the easiest and most convenient, and in relation to this, oral progesterone is the most widely used form of progesterone in transfeminine people. However, the pharmacokinetic problems of progesterone limit the favorability of oral progesterone. Moreover, these limitations of oral progesterone actually appear to be much more substantial than is generally realized, a fact that has been obscured by methodological limitations of many—but not all—of the pharmacokinetic studies that have characterized oral progesterone. The purpose of this article is to explain and review these findings, as well as to explore solutions and alternatives to oral progesterone for progestogen therapy in transfeminine people.
Progesterone Levels with Oral Progesterone
Oral micronized progesterone, or simply oral progesterone, is the form of progesterone that is used by the oral route as a pharmaceutical medication. It is an oil suspension of micronized progesterone crystals contained in gelatin capsules. The formulation is marketed under brand names including Prometrium, Utrogestan, and Microgest, among many others. Oral progesterone has very low bioavailability (≤10%) due to the first pass through the intestines and liver with oral administration. As a result of the first pass, most of the delivered progesterone with oral progesterone is metabolized into neurosteroid metabolites such as allopregnanolone and pregnanolone before reaching the bloodstream (de Lignieres, Dennerstein, & Backstrom, 1995). This is why oral progesterone has alcohol-like side effects like sedation that are not shared by typical doses of non-oral progesterone such as vaginal progesterone or progesterone by injection. In spite of the low bioavailability of oral progesterone, typical clinical doses of oral progesterone, such as 100 to 300 mg/day, have been reported to produce progesterone levels measured with immunoassays that are similar to those in the normal luteal phase of the menstrual cycle in cisgender women (Simon et al., 1993). For this reason, it has been believed that oral progesterone can achieve high and physiologically adequate levels of progesterone.
Figure 1: Progesterone levels during the menstrual cycle in normal premenopausal women (Stricker et al., 2006). The dashed horizontal lines are the mean levels for each curve and the dashed vertical line demarcates mid-cycle (when ovulation occurs). Progesterone levels are normally elevated only during the luteal phase.
Figure 2: Progesterone levels measured by immunoassay after single 100 to 300 mg doses of oral micronized progesterone in postmenopausal women (Simon et al., 1993). The horizontal dashed lines are mean levels over 24 hours. Progesterone levels appeared to reach concentrations comparable to normal luteal-phase levels. However, these levels were in fact not accurate due to the use of immunoassays (Nahoul & de Ziegler, 1994).
A notable study using LC–MS found maximal progesterone levels of only about 2 ng/mL and average progesterone levels over a period of 24 hours of only about 0.14 ng/mL after a single 100 mg dose of oral progesterone (Levine & Watson, 2000; Kuhl & Schneider, 2013). Another more recent study with LC–MS found progesterone levels of around 2.5 to 3 ng/mL at peak and average progesterone levels over 24 hours of around 0.6 ng/mL after a single 100 mg dose of oral progesterone with food (Lobo et al., 2019). (It should be noted that intake with food is known to increase the bioavailability of oral progesterone by a few-fold (Wiki; Bijuva FDA Label; Simon et al., 1993; Prometrium FDA Review, 1996; Pickar et al., 2015).) These progesterone levels are well below normal luteal-phase levels of progesterone, which range from 7 to 22 ng/mL with LC–MS (Nakamoto, 2016). Studies that have directly compared quantification of progesterone with immunoassays against more reliable methods have found that immunoassays overestimate progesterone levels by 5- to 8-fold (Nahoul, Dehennin, & Scholler, 1987; Nahoul & de Ziegler, 1994; Levine & Watson, 2000; Kuhl, 2011; Kuhl & Schneider, 2013; Davey, 2018). In one small study of a few individuals, the degree of overestimation varied from 2-fold to 40-fold with several different commercial immunoassays (Sapin et al., 2000). These findings are obscure and still relatively little-known in the scientific and medical communities. In any case, it is clear that oral progesterone achieves progesterone levels that are far lower than once thought and are well below the luteal-phase levels that would be therapeutically desirable for transfeminine people.
Figure 3: Progesterone levels measured by immunoassay or LC–MS after a single dose of oral or vaginal micronized progesterone in postmenopausal women (Levine & Watson, 2000; Kuhl & Schneider, 2013). Levels of progesterone with oral progesterone measured by immunoassay were falsely high due to cross-reactivity. Conversely, progesterone levels measured by LC–MS or with vaginal progesterone can be considered accurate.
Figure 4: Progesterone levels measured by LC–MS with 100 mg/day oral micronized progesterone taken with food in postmenopausal women (Lobo et al., 2019). The horizontal dashed line is the mean level over 24 hours. Food increases progesterone levels with oral progesterone by about 2- to 3-fold (Bijuva FDA Label; Simon et al., 1993). The progesterone levels measured in this study can be considered accurate to due to the use of LC–MS.
Therapeutic Implications
Progestogenic Potency and Effects
A variety of perplexing findings on the clinical progestogenic effects of oral progesterone have been made over the decades and can now be readily explained by the newer data on oral progesterone with better analytic methods. Oral progesterone is used in clinical medicine mainly to protect the endometrium from unopposed stimulation by estrogens in menopausal cisgender women and is able to reliably prevent endometrial hyperplasia induced by estrogens even with the low progesterone levels that typical clinical doses achieve (Wiki). However, oral progesterone failed to provide adequate protection against estrogen-mediated endometrial cancer risk in a large observational study (Davey, 2018). Oral progesterone even at very high doses also is unable to produce full endometrial transformation—a normal effect of luteal-phase levels of progesterone—whereas vaginal and injectable progesterone are effective (de Ziegler et al., 2013). For this reason, oral progesterone, in contrast to parenteral progesterone, is considered to be inappropriate for use in assisted reproduction (de Ziegler et al., 2013). Oral progesterone additionally failed to suppress testosterone levels even at high doses (400 mg/day) in cisgender males (Trollan et al., 1993; Wiki). Conversely, progestins, rectal progesterone, and injectable progesterone can all produce robust testosterone suppression in cisgender males (Wiki; Aly, 2019). Similarly, oral progesterone has little or no apparent antigonadotropic effect in menopausal cisgender women, which is again in notable contrast to progestins (Holst, 1983; Holst et al., 1983; Ottosson, 1984; Maxson & Hargrove, 1985; Saarikoski, Yliskosk, & Penttilä, 1990). However, in one study, oral progesterone did seem to show a substantial antigonadotropic effect similarly to medroxyprogesterone acetate (MPA) and vaginal progesterone in premenopausal women with polycystic ovary syndrome (PCOS) (Bagis et al., 2002).
Unlike other clinically used progestogens, the addition of oral progesterone to estrogen therapy in menopausal women has not been associated with increased risk of venous thromboembolism (VTE; blood clots) (Wiki). Nor has it been associated with increased breast cancer risk in the short-term (<5 years of therapy) (Wiki). However, with long-term use (≥5 years), the combination of estrogen plus oral progesterone is associated with significantly greater risk of breast cancer relative to estrogen alone similarly to other progestogens (Aly, 2020; Sam, 2020; Wiki; Table). This has been said to be consistent with a weak proliferative effect of oral progesterone on the breasts such that a longer duration of exposure is necessary for a quantifiable increase in breast cancer risk to manifest (Kuhl & Schneider, 2013; Davey, 2018). It is also consistent with preclinical research, which clearly suggests a carcinogenic role for progesterone and progesterone receptor activation in the breast (Kuhl & Schneider, 2013; Trabert et al., 2020). The preceding clinical findings on endometrial efficacy, testosterone and gonadotropin suppression, VTE risk, and breast cancer risk with oral progesterone are in contrast to those with almost all clinically used progestins (with the exception of the oral progesterone-like dydrogesterone). These previously perplexing discrepancies can be readily explained by the very low levels of progesterone that are now known to be achieved with oral progesterone.
Bioavailability, Half-Life, and Duration
Considering the much lower levels of progesterone observed with oral progesterone in studies using reliable analytic methods, the bioavailability of oral progesterone needs to be reassessed. In studies with immunoassays, the bioavailability of oral progesterone has been reported to be around 10% (Wiki). The true oral bioavailability of progesterone is unknown at this time as studies with reliable analytic methods have not been conducted. In any case, it can be assumed that it may be closer to around 1 or 2% based on the findings that immunoassays overestimate progesterone levels by 5- to 8-fold.
The elimination half-life of progesterone with oral progesterone has been determined in studies employing immunoassays to be 16 to 18 hours (Wiki). Based on the fact that the blood half-life of progesterone administered by intravenous injection is very short at a range of only 3 minutes to 1.5 hours (Wiki), the reported half-life of progesterone with oral progesterone is much longer than one would expect. Oral estradiol has a relatively long half-life of 13 to 20 hours due to formation with the first pass of a circulating reservoir of estrogen conjugates that are slowly converted back into estradiol (Kuhl, 2005; Wiki). In contrast to estradiol however, progesterone itself has no available hydroxyl groups for conjugation and an analogous circulating reservoir of progesterone conjugates that can be converted back into progesterone is not known to be the case (Kuhl, 2005).
Studies with more reliable analytic methods like LC–MS have found a half-life of progesterone with oral progesterone of 5 to 10 hours and a duration of highly elevated progesterone levels of only about 4 to 8 hours (Wiki; Graphs). These findings indicate that oral progesterone has a much shorter duration than previously thought as well. As such, if oral progesterone is used, it may be advisable to take it in divided doses multiple times per day to allow for more sustained progestogenic exposure.
Higher Oral Progesterone Doses and Neurosteroid Side Effects
Use of higher doses of oral progesterone than typical doses is likely to achieve dose-dependently higher progesterone levels (Table). However, based on how low progesterone levels are with oral progesterone using reliable analytic techniques, even very high doses would still be expected to achieve only low progesterone levels in most cases. Moreover, high doses of oral progesterone result in very high levels of its neurosteroid metabolites and have been found to produce substantial alcohol-like side effects (i.e., central depression and effects within that umbrella) (Wiki; Wiki). These limitations are likely to preclude higher doses of oral progesterone from being practical.
Figure 5: Levels of progesterone, allopregnanolone, and pregnanolone in premenopausal women following a single dose of oral progesterone or vaginal progesterone (as a suppository) (de Lignieres, Dennerstein, & Backstrom, 1995). Allopregnanolone and pregnanolone levels were determined by MS-based assays while progesterone levels were measured by immunoassay with chromatographic separation. Hence, the levels should be reliable.
A Note on Oral Progesterone’s Metabolites
Although progesterone levels with oral progesterone are very low, the metabolites of progesterone are formed in disproportionate amounts with the first pass (Sitruk-Ware et al., 1987; de Lignieres, Dennerstein, & Backstrom, 1995; de Lignieres, 1999; de Ziegler & Fanchin, 2000; Lobo, 2000; Kuhl, 2005). Moreover, while much less potent than progesterone, some of these metabolites have been found to have progestogenic activity similarly to progesterone (e.g., Besch et al., 1965; Junkermann, Runnebaum, & Lisboa, 1977; Lobo, 2000). This activity derives either from them having intrinsic progestogenic activity of their own or from being converted back into progesterone or other progestogenic metabolites (including in an intracrine fashion within tissues, for instance in the uterus). Examples of such metabolites include 20α-dihydroprogesterone, 20β-dihydroprogesterone, 5α-dihydroprogesterone, 3β-dihydroprogesterone, allopregnanolone, and 11-deoxycorticosterone. If the metabolites of oral progesterone contribute significantly to its progestogenic activity, then the progestogenic strength of oral progesterone would be greater than that implied by the progesterone levels that occur with it alone. However, this possibility is only theoretical and there is little literature discussing it. More research would be needed to determine if the metabolites of oral progesterone do indeed play a meaningful role in its progestogenic potency. In any case, oral progesterone is still a relatively weak progestogen based on clinical studies of its progestogenic effects.
Alternative Options to Oral Progesterone
Non-Oral Forms of Progesterone
Non-oral forms of progesterone, such as vaginal progesterone, rectal progesterone, sublingual progesterone, and progesterone by injection, have been found to achieve much higher progesterone levels than oral progesterone (Wiki). They can be used instead of oral progesterone to achieve higher and more adequate progesterone levels. Unfortunately however, while more effective than oral progesterone, non-oral progesterone routes have various limitations of their own.
Vaginal progesterone is of course not possible in transfeminine people who have not undergone vaginoplasty. And in those who have undergone vaginoplasty, the lining of the neovagina is either skin (penile inversion vaginoplasty) or intestine (sigmoid colon vaginoplasty) rather than the normal vaginal mucosa. As such, the absorptive characteristics of neovaginal administration are likely not the same as vaginal administration (Aly, 2018). It is notable that transdermal progesterone achieves very low progesterone levels similarly to oral progesterone and is not a good option for progesterone therapy (Wiki; Hermann et al., 2005; Graph). Progesterone levels with neovaginal administration of progesterone in those who have undergone penile inversion vaginoplasty are likely to be low similarly.
Rectal progesterone is an excellent route that achieves high progesterone levels comparable to the levels of progesterone that occur during the normal luteal phase (Wiki; Graphs). It has a somewhat short duration and twice daily use may be warranted for more sustained levels however. A more important problem is that the availability of pharmaceutical rectal progesterone suppositories throughout the world is limited and they are not marketed in most countries (Wiki). In any case, rectal progesterone suppositories may be available from compounding pharmacies in some countries. In addition, oral micronized progesterone capsules are available ubiquitously and have been administered vaginally instead of orally with success (Miles et al., 1994; Wang et al., 2019). Administration of oral micronized progesterone capsules rectally instead of orally likewise may be effective and may achieve much higher progesterone levels than oral administration (Aly, 2018). However, rectal administration of oral progesterone capsules has not been formally studied. Although rectal progesterone is effective, it is fairly inconvenient. This may be especially true with long-term therapy. In any case, of the available non-oral forms of progesterone, rectal progesterone is probably the best overall. A significant subset of transfeminine people on progestogens take progesterone rectally (Chang et al., 2024).
Figure 6: Progesterone levels with a single suppository containing 100 mg progesterone administered rectally or vaginally in premenopausal women (Chakmakjian & Zachariah, 1987).
Sublingual progesterone appears to achieve high and more physiological progesterone levels than oral progesterone but has a short duration of highly elevated progesterone levels similarly and necessitates administration several times per day (Wiki; Graph). Moreover, although sublingual progesterone may have been more widely available in the past (Wiki), it is available today only in a couple of Eastern European countries (Wiki). It might be available from compounding pharmacies in some countries however. While never formally studied, it may be possible to use oral micronized progesterone capsules sublingually instead of orally. However, this route is complicated by the fact that this form of progesterone is suspended in oil within gelatin capsules. Hence, sublingual administration of oral micronized progesterone is likely to be difficult and potentially unpleasant.
Progesterone by intramuscular or subcutaneous injection can easily achieve very high progesterone levels (Wiki; Graphs; Wiki; Graph). However, progesterone administered by this route has a relatively short duration when compared to other injectable sex-hormone formulations and requires injection once every 1 to 3 days. This is simply too frequent for most people, especially with long-term therapy. In addition, progesterone by subcutaneous injection, which is more convenient than progesterone by intramuscular injection, has limited availability and is marketed mostly only in Europe (Wiki). In contrast to other sex hormones like estradiol and testosterone, progesterone esters that are more fat-soluble than progesterone and extend its duration when used in injectable form are not possible since progesterone has no free hydroxyl groups available for esterification. Injectable aqueous suspensions of progesterone that had much longer durations than the oil solutions and aqueous solutions that are used by injection today were previously available (Wiki; Wiki; Aly, 2019). However, they were associated with painful injection site reactions and this led to their discontinuation. In any case, injectable aqueous suspensions of progesterone do actually seem to remain available for people in a couple of Eastern European countries today (Aly, 2019).
In conclusion, oral progesterone achieves very low progesterone levels at typical clinical doses and produces only weak progestogenic effects that seem to be far from physiologically adequate. Although use of higher doses of oral progesterone is likely to achieve higher progesterone levels, such doses are likely to be impractical because progesterone levels will still be low even at higher doses and the neurosteroid side effects of oral progesterone will be much more substantial and difficult to tolerate.
Due to its limitations, transfeminine people and clinicians treating them may wish to avoid oral progesterone if the intended goal is to produce therapeutic progestogenic effects. Instead, non-oral progesterone routes, such as rectal and injected progesterone, although with various limitations such as limited availability and inconvenience, can be used. Alternatively, progestins, particularly those with more favorable profiles, can be used instead of progesterone altogether.
Oral progesterone may perhaps be most appropriately conceptualized as a potent neurosteroid prodrug with weak progestogenic effects. Conversely, non-oral progesterone, as well as progestins, can be regarded as potent progestogens with either physiological or no neurosteroid effects, respectively.
Additional Content
Literature
The sources and excerpts collected here go in-depth on much of what has been described in this article on the topic of the measurement problems and low progesterone levels with oral progesterone.
In August 2025, a conference abstract of a randomized controlled trial of oral progesterone and breast development in transfeminine people was published (Dreijerink et al., 2025). This study had been preregistered and its protocol had been previously published in 2023 (Dijkman et al., 2023). The trial found that oral micronized progesterone (Utrogestan) at doses of 200 to 400 mg/day, in conjunction with estradiol therapy and especially with higher estradiol levels in the range of 400 to 800 pmol/L (109–218 pg/mL), could significantly increase breast volume in transfeminine people. The study is discussed in greater detail elsewhere on this site (Aly, 2020). Based on the trial’s findings, although oral progesterone can only achieve low progesterone levels measured with mass spectrometry, it appears that these low levels are nonetheless able to produce significant effects on the breasts. Hence, while oral progesterone may still prove to be less efficacious than non-oral progesterone or progestins, it would appear that it is not absent of value in terms of potential therapeutic effects in transfeminine people.
References
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2018). Sources/Excerpts: Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects. Transfeminine Science. [URL]
Aly. (2019). Injectable Aqueous Suspensions of Sex Hormones and How Depot Injectables Work. Transfeminine Science. [URL]
Aly. (2019). Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Breast Cancer Risk with Hormone Therapy in Transfeminine People. Transfeminine Science. [URL]
Bagis, T., Gokcel, A., Zeyneloglu, H. B., Tarim, E., Kilicdag, E. B., & Haydardedeoglu, B. (2002). The Effects of Short-Term Medroxyprogesterone Acetate and Micronized Progesterone on Glucose Metabolism and Lipid Profiles in Patients with Polycystic Ovary Syndrome: A Prospective Randomized Study. The Journal of Clinical Endocrinology & Metabolism, 87(10), 4536–4540. [DOI:10.1210/jc.2002-020294]
Besch, P. K., Barry, R. D., Vorys, N., Stevens, V., & Ullery, J. C. (1965). A review of some aspects of the metabolism of progestational agents. Metabolism, 14(3), 432–443. [DOI:10.1016/0026-0495(65)90031-4]
Bijuva Estradiol/Progesterone Label. U.S. Food and Drug Administration. [URL] [PDF]
Chakmakjian, Z. H., & Zachariah, N. Y. (1987). Bioavailability of progesterone with different modes of administration. The Journal of Reproductive Medicine, 32(6), 443–448. [Google Scholar] [PubMed] [PDF]
Chang, J. J., Tran, N. K., Flentje, A., Lubensky, M. E., Obedin-Maliver, J., Lunn, M. R., & Ariel, D. (2024). 12330 Progestogen Experience And Perception Among Transfeminine Adults - A National Survey. Journal of the Endocrine Society, 8(Suppl 1), bvae163.1657. [DOI:10.1210/jendso/bvae163.1657]
Conklin, S. E., & Knezevic, C. E. (2020). Advancements in the gold standard: Measuring steroid sex hormones by mass spectrometry. Clinical Biochemistry, 82, 21–32. [DOI:10.1016/j.clinbiochem.2020.03.008]
Davey, D. A. (2018). Menopausal hormone therapy: a better and safer future. Climacteric, 21(5), 454–461. [DOI:10.1080/13697137.2018.1439915]
de Lignières, B., Dennerstein, L., & Backstrom, T. (1995). Influence of route of administration on progesterone metabolism. Maturitas, 21(3), 251–257. [DOI:10.1016/0378-5122(94)00882-8]
de Lignières, B. (1999). Oral micronized progesterone. Clinical Therapeutics, 21(1), 41–60. [DOI:10.1016/S0149-2918(00)88267-3]
de Ziegler, D., & Fanchin, R. (2000). Progesterone and progestins: applications in gynecology. Steroids, 65(10–11), 671–679. [DOI:10.1016/S0039-128X(00)00123-9]
de Ziegler, D., Streuli, I., Marszalek, A., Gayet, V., & Chapron, C. (2013). Preparing the Endometrium to Maximize Success: The Dynamics of Artificial Cycles. In Sauer, M. V. (Ed.). Principles of Oocyte and Embryo Donation, 2nd Edition (pp. 109–127). London: Springer. [DOI:10.1007/978-1-4471-2392-7_9]
Dijkman, B. A., Helder, D., Boogers, L. S., Gieles, N. C., van Heesewijk, J. O., Slaa, S. t., Liberton, N. P., Wiepjes, C. M., de Blok, C. J., den Heijer, M., & Dreijerink, K. M. (2023). Addition of progesterone to feminizing gender-affirming hormone therapy in transgender individuals for breast development: a randomized controlled trial. BMC Pharmacology and Toxicology, 24(1), 80. [DOI:10.1186/s40360-023-00724-4]
Dreijerink, K., den Heijer, M., Geels, R. (2025). Increased breast volume due to addition of progesterone and increasing the estradiol dose in feminizing gender-affirming hormone therapy. EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
Hermann, A. C., Nafziger, A. N., Victory, J., Kulawy, R., Rocci Jr, M. L., & Bertino Jr, J. S. (2005). Over‐the‐Counter Progesterone Cream Produces Significant Drug Exposure Compared to a Food and Drug Administration‐Approved Oral Progesterone Product. The Journal of Clinical Pharmacology, 45(6), 614–619. [DOI:10.1177/0091270005276621]
Holst, J. (1983). Percutaneous estrogen therapy: Endometrial response and metabolic effects. Acta Obstetricia et Gynecologica Scandinavica, 62(Suppl 115), 4–30. [DOI:10.3109/00016348309155363]
Holst, J., Cajander, S., Carlström, K., Damber, M. G., & von Schoultz, B. (1983). Percutaneous oestrogen therapy opposed by lynestrenol or natural progesterone-effects on circulating oestrogens, FSH, sex hormone binding globulin and pregnancy zone protein. Maturitas, 5(1), 1–8. [DOI:10.1016/0378-5122(83)90015-4]
Junkermann, H., Runnebaum, B., & Lisboa, B. P. (1977). New progesterone metabolites in human myometrium. Steroids, 30(1), 1–14. [DOI:10.1016/0039-128X(77)90131-3]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H. (2011). Pharmacology of progestogens. Journal für Reproduktionsmedizin und Endokrinologie [Journal of Reproductive Medicine and Endocrinology], 8(1), 157–177. [URL]
Kuhl, H., & Schneider, H. P. G. (2013). Progesterone–promoter or inhibitor of breast cancer. Climacteric, 16(Suppl 1), 54–68. [DOI:10.3109/13697137.2013.768806]
Lobo, R. A. (2000). Progestogens. In Lobo, R. A., Kelsey, J., & Marcus, R. (Eds.). Menopause: Biology and Pathobiology (pp. 429–444). Burlington: Academic Press. [Google Books]
Lobo, R. A., Liu, J., Stanczyk, F. Z., Constantine, G. D., Pickar, J. H., Shadiack, A. M., Bernick, B., & Mirkin, S. (2019). Estradiol and progesterone bioavailability for moderate to severe vasomotor symptom treatment and endometrial protection with the continuous-combined regimen of TX-001HR (oral estradiol and progesterone capsules). Menopause (New York, NY), 26(7), 720–727.[DOI:10.1097/GME.0000000000001306]
Levine, H., & Watson, N. (2000). Comparison of the pharmacokinetics of Crinone 8% administered vaginally versus Prometrium administered orally in postmenopausal women. Fertility and Sterility, 73(3), 516–521. [DOI:10.1016/S0015-0282(99)00553-1]
Maxson, W. S., & Hargrove, J. T. (1985). Bioavailability of oral micronized progesterone. Fertility and Sterility, 44(5), 622–626. [DOI:10.1016/S0015-0282(16)48977-6]
Miles, R. A., Paulson, R. J., Lobo, R. A., Press, M. F., Dahmoush, L., & Sauer, M. V. (1994). Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: a comparative study. Fertility and Sterility, 62(3), 485–490. [DOI:10.1016/S0015-0282(16)56935-0]
Nahoul, K., Dehennin, L., & Scholler, R. (1987). Radioimmunoassay of plasma progesterone after oral administration of micronized progesterone. Journal of Steroid Biochemistry, 26(2), 241–249. [DOI:10.1016/0022-4731(87)90078-1]
Nahoul, K., Dehennin, L., Jondet, M., & Roger, M. (1993). Profiles of plasma estrogens, progesterone and their metabolites after oral or vaginal administration of estradiol or progesterone. Maturitas, 16(3), 185–202. [DOI:10.1016/0378-5122(93)90064-O]
Nahoul, K., & de Ziegler, D. (1994). “ Validity” of serum progesterone levels after oral progesterone. Fertility and Sterility, 61(4), 790–792. [DOI:10.1016/S0015-0282(16)56666-7]
Nakamoto, J. (2016). Endocrine Testing. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition (pp. 2655–2688.e1). Philadelphia: Saunders/Elsevier. [DOI:10.1016/B978-0-323-18907-1.00154-2]
Ottosson, U. B. (1984). Oral progesterone and estrogen/progestogen therapy: effects of natural and synthetic hormones on subfractions of HDL cholesterol and liver proteins. Acta Obstetricia et Gynecologica Scandinavica, 63(Suppl 127), 1–37. [DOI:10.3109/00016348409157016]
Pickar, J. H., Bon, C., Amadio, J. M., Mirkin, S., & Bernick, B. (2015). Pharmacokinetics of the first combination 17β-estradiol/progesterone capsule in clinical development for menopausal hormone therapy. Menopause (New York, NY), 22(12), 1308–1316. [DOI:10.1097/GME.0000000000000467]
Prometrium FDA Review 1996. Application Number: NDA 19-781. Clinical Pharmacology and Biopharmaceutics Review(s) [Prometrium]. Center for Drug Evaluation and Research, U.S. Food and Drug Administration. [PDF]
Saarikoski, S., Yliskosk, M., & Penttilä, I. (1990). Sequential use of norethisterone and natural progesterone in pre-menopausal bleeding disorders. Maturitas, 12(2), 89–97. [DOI:10.1016/0378-5122(90)90086-L]
Sam. (2020). Progestogens and Breast Cancer in Transgender Women: A Review and Discussion of Risk. Transfeminine Science. [URL]
Sapin, R., Neamtu, D., Gasser, F., Ohl, J., Grunenberger, F., & Grucker, D. (2000). De la prudence lors de l’utilisation des dosages directs de progestérone. [Caution when using direct progesterone assays.] Immuno-analyse et Biologie Spécialisée, 3(15), 203–204. [DOI:10.1016/S0923-2532(00)80010-1] [Translation]
Simon, J. A., Robinson, D. E., Andrews, M. C., Hildebrand III, J. R., Rocci Jr, M. L., Blake, R. E., & Hodgen, G. D. (1993). The absorption of oral micronized progesterone: the effect of food, dose proportionality, and comparison with intramuscular progesterone. Fertility and Sterility, 60(1), 26–33. [DOI:10.1016/S0015-0282(16)56031-2]
Sitruk-Ware, R., Bricaire, C., De Lignieres, B., Yaneva, H., & Mauvais-Jarvis, P. (1987). Oral micronized progesterone: Bioavailability pharmacokinetics, pharmacological and therapeutic implications—A review. Contraception, 36(4), 373–402. [DOI:10.1016/0010-7824(87)90088-6]
Stricker, R., Eberhart, R., Chevailler, M. C., Quinn, F. A., Bischof, P., & Stricker, R. (2006). Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT® analyzer. Clinical Chemistry and Laboratory Medicine (CCLM), 44(7), 883–887. [DOI:10.1515/CCLM.2006.160]
Tollan, A., Øian, P., Kjeldsen, S. E., Eide, I., & Maltau, J. M. (1993). Progesterone reduces sympathetic tone without changing blood pressure or fluid balance in men. Gynecologic and Obstetric Investigation, 36(4), 234–238. [DOI:10.1159/000292636]
Trabert, B., Sherman, M. E., Kannan, N., & Stanczyk, F. Z. (2020). Progesterone and breast cancer. Endocrine Reviews, 41(2), 320–344. [DOI:10.1210/endrev/bnz001]
Wang, H., Liu, M., Fu, Q., & Deng, C. (2019). Pharmacokinetics of hard micronized progesterone capsules via vaginal or oral route compared with soft micronized capsules in healthy postmenopausal women: a randomized open-label clinical study. Drug Design, Development and Therapy, 13, 2475–2482. [DOI:10.2147/dddt.s204624]
Woodward, G. M., & Rumsby, G. (2019). Steroid Profiling: Analytical Perspectives. In Rumsby, G., Woodward, G. M. (Eds.). Disorders of Steroidogenesis: Guide to Steroid Profiling and Biochemical Diagnosis (pp. 27–40). Cham: Springer. [DOI:10.1007/978-3-319-96364-8_3]
\ No newline at end of file
diff --git a/transfemscience.org/articles/oral-vs-transdermal-e2-suppl/index.html b/transfemscience.org/articles/oral-vs-transdermal-e2-suppl/index.html
index 389c5cfa..41f56aa9 100644
--- a/transfemscience.org/articles/oral-vs-transdermal-e2-suppl/index.html
+++ b/transfemscience.org/articles/oral-vs-transdermal-e2-suppl/index.html
@@ -1 +1 @@
-Sources/Excerpts: Reviews, Cohort Studies and Randomised Controlled Trials Comparing the Feminising Efficacy of Estradiol Administered by Oral and Non-Oral Routes - Transfeminine Science
Sources/Excerpts: Reviews, Cohort Studies and Randomised Controlled Trials Comparing the Feminising Efficacy of Estradiol Administered by Oral and Non-Oral Routes
By Sam | First published April 10, 2020 | Last modified October 5, 2020
Preface
This is a sources and excerpts supplement to the main article which can be found here.
Shah et al. (2014)
Shah, S., Forghani, N., Durham, E., & Neely, E. K. (2014). A randomized trial of transdermal and oral estrogen therapy in adolescent girls with hypogonadism. International Journal of Pediatric Endocrinology, 2014(1), 12. [DOI:10.1186/1687-9856-2014-12]:
History and physical exams, including assessment of feminization by Tanner staging and breast diameter were obtained at baseline and every 6 months for 24 months.
The proportion of subjects in each group reaching Tanner 3 breast stage at each 6 month interval is shown in Figure 3. All subjects in the TBE and OBE group were feminized by 18 months, […]
Wierckx et al. (2014)
Wierckx, K., Van Caenegem, E., Schreiner, T., Haraldsen, I., Fisher, A. D., Toye, K., Kaufman, J. M., & T’Sjoen, G. (2014). Cross‐sex hormone therapy in trans persons is safe and effective at short‐time follow‐up: results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 11(8), 1999–2011. [DOI:10.1111/jsm.12571]:
Antiandrogen with estrogen treatment resulted in a significant increase (average 3.3 cm) in breast circumference at the nipple, with a wide interindividual range of increase. Trans women using oral estrogens experienced similar changes in physical measures as those using transdermal estrogens (Table 3).
We found no changes in night sweats, abdominal pain, anxiety, irritability, palpitations, skin dryness, joint pain, muscle soreness, headache, mood swings, fatigue, concentration difficulties, memory, or sleep-related problems (data not shown). There were no significant differences in the presence of treatment related symptoms at the 3-month time point between trans women treated with oral or transdermal estrogen.
[…] In addition, trans women using oral EV showed significantly higher E1/E2 ratio than those using transdermal therapy. A lower E1/E2 ratio has been reported in postmenopausal women receiving transdermal hormone replacement therapy [40]. No other differences were observed between these two modes of estrogen treatment, suggesting both are equally effective.
de Blok et al. (2018)
de Blok, C. J. M., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2018). Breast development in transwomen after 1 year of cross-sex hormone therapy: results of a prospective multicenter study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]:
Because most transwomen (99%) in this study received cyproterone acetate as antiandrogen treatment, no analyses on treatment regimen other than oral vs transdermal estradiol administration route could be performed.
Transwomen treated with transdermal estradiol had a faster increase in breast-chest difference until 6 months after initializing treatment. However, breast development after 1 year of treatment did not differ between oral and transdermal estradiol administration [Fig. 5(e)].
Klaver et al. (2018)
Klaver, M., de Blok, C. J. M., Wiepjes, C. M., Nota, N. M., Dekker, M. J. H. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/EJE-17-0496]:
Subgroups in transwomen were oral estradiol and transdermal estradiol. In these analyses, only transwomen and transmen using the same type of treatment during the whole year were included. Analyses on sex hormone levels were performed in quartiles of Z-scores, which is described in more detail below.
Sixty five transwomen using oral estrogens, 49 transwomen using transdermal estrogens, 41 transmen using T esters, 34 transmen using T gel and 30 transmen using T undecanoate were included. Crude analyses in transwomen showed a trend toward a larger change in some body fat outcomes in persons using oral estradiol compared with persons using transdermal estradiol, but after adjustment for BMI at the start and age, these differences disappeared (Supplementary Table 1).
Klein et al. (2018)
Klein, K. O., Rosenfield, R. L., Santen, R. J., Gawlik, A. M., Backeljauw, P. F., Gravholt, C. H., Sas, T. C. J., & Mauras, N. (2018). The Journal of Clinical Endocrinology & Metabolism, 103(5), 1790–1803. [DOI:10.1210/jc.2017-02183]:
Incremental dose increases at approximately 6-month intervals can mimic the normal pubertal tempo until adult dosing is reached over 2 to 3 years. This theoretically translates into a 25% to 100% increase in dose every 6 months for four to six dose changes between the initiation and adult doses portrayed in the Table 1. However, no studies to date have rigorously studied outcomes in relation to the rate of dose increase for the different preparations.
In general, the studies summarized in Table 3 report onset of breast buds within 6 months in most girls (17, 18, 22, 23, 27). Each of these regimens results in pubertal stage 4 breasts in an average of 2.25 years, which is similar to that in girls with TS who have spontaneous puberty (1.9 years), as well as in the general population (23).
\ No newline at end of file
+Sources/Excerpts: Reviews, Cohort Studies and Randomised Controlled Trials Comparing the Feminising Efficacy of Estradiol Administered by Oral and Non-Oral Routes - Transfeminine Science
Sources/Excerpts: Reviews, Cohort Studies and Randomised Controlled Trials Comparing the Feminising Efficacy of Estradiol Administered by Oral and Non-Oral Routes
By Sam | First published April 10, 2020 | Last modified October 5, 2020
Preface
This is a sources and excerpts supplement to the main article which can be found here.
Shah et al. (2014)
Shah, S., Forghani, N., Durham, E., & Neely, E. K. (2014). A randomized trial of transdermal and oral estrogen therapy in adolescent girls with hypogonadism. International Journal of Pediatric Endocrinology, 2014(1), 12. [DOI:10.1186/1687-9856-2014-12]:
History and physical exams, including assessment of feminization by Tanner staging and breast diameter were obtained at baseline and every 6 months for 24 months.
The proportion of subjects in each group reaching Tanner 3 breast stage at each 6 month interval is shown in Figure 3. All subjects in the TBE and OBE group were feminized by 18 months, […]
Wierckx et al. (2014)
Wierckx, K., Van Caenegem, E., Schreiner, T., Haraldsen, I., Fisher, A. D., Toye, K., Kaufman, J. M., & T’Sjoen, G. (2014). Cross‐sex hormone therapy in trans persons is safe and effective at short‐time follow‐up: results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 11(8), 1999–2011. [DOI:10.1111/jsm.12571]:
Antiandrogen with estrogen treatment resulted in a significant increase (average 3.3 cm) in breast circumference at the nipple, with a wide interindividual range of increase. Trans women using oral estrogens experienced similar changes in physical measures as those using transdermal estrogens (Table 3).
We found no changes in night sweats, abdominal pain, anxiety, irritability, palpitations, skin dryness, joint pain, muscle soreness, headache, mood swings, fatigue, concentration difficulties, memory, or sleep-related problems (data not shown). There were no significant differences in the presence of treatment related symptoms at the 3-month time point between trans women treated with oral or transdermal estrogen.
[…] In addition, trans women using oral EV showed significantly higher E1/E2 ratio than those using transdermal therapy. A lower E1/E2 ratio has been reported in postmenopausal women receiving transdermal hormone replacement therapy [40]. No other differences were observed between these two modes of estrogen treatment, suggesting both are equally effective.
de Blok et al. (2018)
de Blok, C. J. M., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2018). Breast development in transwomen after 1 year of cross-sex hormone therapy: results of a prospective multicenter study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]:
Because most transwomen (99%) in this study received cyproterone acetate as antiandrogen treatment, no analyses on treatment regimen other than oral vs transdermal estradiol administration route could be performed.
Transwomen treated with transdermal estradiol had a faster increase in breast-chest difference until 6 months after initializing treatment. However, breast development after 1 year of treatment did not differ between oral and transdermal estradiol administration [Fig. 5(e)].
Klaver et al. (2018)
Klaver, M., de Blok, C. J. M., Wiepjes, C. M., Nota, N. M., Dekker, M. J. H. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/EJE-17-0496]:
Subgroups in transwomen were oral estradiol and transdermal estradiol. In these analyses, only transwomen and transmen using the same type of treatment during the whole year were included. Analyses on sex hormone levels were performed in quartiles of Z-scores, which is described in more detail below.
Sixty five transwomen using oral estrogens, 49 transwomen using transdermal estrogens, 41 transmen using T esters, 34 transmen using T gel and 30 transmen using T undecanoate were included. Crude analyses in transwomen showed a trend toward a larger change in some body fat outcomes in persons using oral estradiol compared with persons using transdermal estradiol, but after adjustment for BMI at the start and age, these differences disappeared (Supplementary Table 1).
Klein et al. (2018)
Klein, K. O., Rosenfield, R. L., Santen, R. J., Gawlik, A. M., Backeljauw, P. F., Gravholt, C. H., Sas, T. C. J., & Mauras, N. (2018). The Journal of Clinical Endocrinology & Metabolism, 103(5), 1790–1803. [DOI:10.1210/jc.2017-02183]:
Incremental dose increases at approximately 6-month intervals can mimic the normal pubertal tempo until adult dosing is reached over 2 to 3 years. This theoretically translates into a 25% to 100% increase in dose every 6 months for four to six dose changes between the initiation and adult doses portrayed in the Table 1. However, no studies to date have rigorously studied outcomes in relation to the rate of dose increase for the different preparations.
In general, the studies summarized in Table 3 report onset of breast buds within 6 months in most girls (17, 18, 22, 23, 27). Each of these regimens results in pubertal stage 4 breasts in an average of 2.25 years, which is similar to that in girls with TS who have spontaneous puberty (1.9 years), as well as in the general population (23).
\ No newline at end of file
diff --git a/transfemscience.org/articles/oral-vs-transdermal-e2/index.html b/transfemscience.org/articles/oral-vs-transdermal-e2/index.html
index 16a34f7f..ca7dc309 100644
--- a/transfemscience.org/articles/oral-vs-transdermal-e2/index.html
+++ b/transfemscience.org/articles/oral-vs-transdermal-e2/index.html
@@ -1 +1 @@
-A Comparison of Oral and Transdermal Estradiol in Transfeminine Hormone Therapy - Transfeminine Science
A Comparison of Oral and Transdermal Estradiol in Transfeminine Hormone Therapy
By Sam | First published April 11, 2020 | Last modified July 29, 2025
Abstract / TL;DR
The most common means to administer exogenous estradiol are the oral and transdermal routes. Both are widely used as a component of gender-affirming hormone therapy. Current clinical evidence shows no difference in feminising efficacy between these formulations at equivalent doses. Although both are generally well tolerated, the oral route is unphysiological in its metabolism and is associated with a significantly greater incidence of cardiovascular and thromboembolic complications. At low adult replacement doses, transdermal forms do not have these disadvantages and may be superior to their oral counterparts in the long-term.
Introduction
Estrogen replacement is both an important and necessary intervention for many transgender people (Hembree et al., 2017; Coleman et al., 2022). In the past, feminising therapy in this group was mostly done using high dose estrogen monotherapy with parenteral esters of estradiol such as estradiol valerate or estradiol undecylate (Benjamin, 1967; Hamburger, 1969). Non-bioidentical oral estrogens such as conjugated equine estrogens and ethinylestradiol were also widely used (Meyer et al., 1986; Meyer, Walker & Suplee, 1989). However, with significant progress made in drug development, bioidentical estradiol became widely available in oral and transdermal formulations for gender-affirming hormone therapy.
Some transgender individuals prefer to use injectable formulations of estradiol (Geffen et al., 2018). However, the oral and transdermal routes of administration appear to be most commonplace (Fisher & Maggi, 2015; Hamidi & Davidge-Pitts, 2019; Seal, 2019). Many people receiving or eager to start hormone therapy may be interested to know what data exists regarding differences between oral and transdermal estradiol. As we require long-term therapy with these formulations, a discussion regarding adverse effects between these routes of administration may also be of importance. Although the focus of this review largely concerns oral estradiol as directly compared to transdermal estradiol, a good amount of the discussion specific to transdermal estradiol can likely be extrapolated to other non-oral routes of administration when the doses are known to have similar potency. For instance, estradiol administered by intramuscular or subcutaneous injection is a non-oral route of administration.
Pharmacology
Oral estradiol includes pill or tablet formulations, while transdermal estradiol is most commonly available as patches or gels (Kuhl, 2005). Oral estradiol and transdermal gel is usually administered once per day (Rohr, Volko & Schindler 2014). However, doses may be split and taken twice-daily. Theoretically, this would result in more stable estradiol levels, although this could be less convenient for the user. Estradiol pills can also be administered by the sublingual or buccal routes (Wren et al., 2003; Jain, Kwan & Forcier 2019; Doll et al., 2022). Up until only a short time ago, data pertaining to sublingual administration in transfeminine people was scarce, however in more recent years there has been considerably greater interest from the scientific community (Cortez et al., 2024). However, usage of these two routes is probably still relatively uncommon in clinical practice. In this review, the term “oral estradiol” has been used to refer exclusively to the swallowing of estradiol tablets. Estradiol patches are applied and worn continuously. Different brands exist and transdermal patches are available for twice-weekly or weekly administration. On average, a 50 μg/day dosage delivered by transdermal patch is generally considered to have approximately similar potency to a 1 to 2 mg/day dosage of oral estradiol and to a 1.5 mg/day dosage of transdermal gel (Kuhl, 2005; Järvinen, Nykänen & Paasiniemi, 1999). However, there is considerable interindividual variation in the metabolism of different estradiol formulations. Due to this variability, these doses are unlikely to correspond to one another on an individual basis.
In cisgender women, estradiol is secreted by the ovaries into systematic circulation. As a result, the liver does not receive disproportionate exposure to the hormone (Gravholt et al., 2017). Transdermal estradiol is effective in mimicking this behaviour. However, orally administered estradiol, owing to its passage through the gastrointestinal tract, is associated with disproportionate estrogenic exposure in the liver (Bińkowska, 2014). This behaviour gives rise to a number of differences between oral and transdermal estradiol. One such difference is that, on average, about 95% of oral estradiol is metabolised, as a consequence of the first pass effect, into estrone and other clinically weak/insignificant estrogens (Kuhz, Blode & Zimmermann, 1993). The ratio of estrone to estradiol is close to 1:1 in both adult women and pubertal girls and with transdermal formulations (Kuhl, 2005; Frederiksen et al., 2020). However, with a dose of oral estradiol, postmenopausal women have been found to have about 5 times the concentration of estrone as estradiol (Kuhl, 2005). In some patients, the concentration of estrone may be 20 times higher than that of estradiol (Kuhnz, Gansau & Mahler, 1993). A new retrospective study has recently confirmed these findings in transfeminine people (Kariyawasam et al., 2025) For this reason, the metabolism of oral estradiol has been described as unphysiologic (Gravholt et al., 2017; Mauras et al., 2019). It should be emphasised that these findings by themselves do not necessarily translate into clinically important differences between oral and transdermal estradiol, such as in feminisation outcomes.
Efficacy
Many transgender people taking feminising hormone therapy are concerned with which regimens might be most “effective”. In particular, satisfactory breast development is often sought after. A number of randomised controlled trials assessing the efficacy of different gender-affirming hormone regimens on objective measures of feminisation have been completed or are currently underway (Dijkman et al., 2023; Angus et al., 2025a; Angus et al., 2025b). However, there have yet to be any such studies directly comparing the influence of the different routes of administration. Moreover, there are no measures of breast development or other effects of transition that are universally agreed upon by the scientific community. For this reason, it is difficult to directly compare the findings of many studies.
In spite of the above, a number of observational studies have studied and quantified feminisation experienced with hormone therapy. Several studies of transgender women treated with cyproterone acetate plus either oral or transdermal estradiol have reported no apparent difference in physical measures such as breast circumference and breast-chest difference between oral and transdermal administration in the short-term (ie: 6 to 12 months) (Wierckx et al., 2014; de Blok et al., 2018; Tebbens et al., 2022). In the latter study, it is interesting to note that estrone concentrations were directly measured and found to not be associated with the extent of breast development. This is consistent with non-oral and oral formulations being comparable in their effect despite the differences in their pharmacology. Consequently, the authors concluded that monitoring of estrone concentrations does not appear to have a place in transfeminine hormone therapy. A longer term follow-up of another one of these study groups involving transgender women attending clinics in the Netherlands, Belgium and Italy also reported that the increase in breast volume following 3 years of therapy did not differ between those using oral and transdermal formulations (de Blok et al., 2021). As a result of unsatisfactory development, many transgender women seek breast augmentation (de Blok et al., 2020). Although breast development itself was not measured, it is interesting to note that one retrospective study found no statistical difference in the rate of augmentation requests between users of different estrogen types (Seal et al., 2012). This suggests that oral estradiol valerate might be no more or less effective than the other estrogens in the study (oral conjugated estrogens and oral ethinylestradiol) in attaining a satisfying amount of breast development. A large cohort study of transgender women found that changes in gynoid and android fat, total body fat and total lean body mass were not statistically different between the oral and transdermal estradiol groups if BMI and age were controlled for (Klaver et al., 2018).
Estrogen replacement, being a necessary therapy for the vast majority of individuals with Turner syndrome, has also been studied in adolescent girls (Gravholt et al., 2017; Klein & Phillips, 2019). Girls treated with low dose oral estradiol were described in one study as having “similar” breast development to the normal Dutch population (Bannink et al., 2009). Other studies of puberty induction therapy have found that patients using low doses of transdermal estradiol gel and low dose intramuscular estradiol cypionate also all achieved breast Tanner stage 4 or 5 at final follow-up (Piippo et al., 2004; Rosenfield et al., 2005). A small randomised controlled trial of hypogonadal girls demonstrated that the response to oral and transdermal estradiol at comparable doses was near identical (Shah et al., 2014). All the girls receiving bioidentical estrogens achieved Tanner stage 3 or greater after 18 months of treatment, irrespective of route of administration. Interestingly, a cross sectional study of breast development in women with differences of sex development (DSD), including those with Turner syndrome, reported that breast satisfaction in the sample group was much lower than in women without a DSD (van de Grift & Kreukels, 2019). Some studies have found that breast development, in addition to breast satisfaction, seems to be poorer in Turner syndrome girls than in normal cisgender girls (Guo et al., 2019). Nevertheless, a recent review concluded that all these different regimens seemed to result in similar feminising outcomes (Klein et al., 2018). In comparison to the available data of transfeminine people these findings are low quality and inconclusive on their own, since breast development itself was not measured objectively. Nevertheless, the findings fit an emerging pattern within the literature and are consistent with a recent systematic review which did not find any evidence of differences between the oral and transdermal routes in transfeminine hormone therapy (Winston-McPherson et al., 2025)
In summary, current clinical evidence appears to show no difference in objectively measured outcomes between therapy with different routes of administration when the doses have comparable potency. Rather, when taken together, these findings indicate that the extent of breast development and other feminisation is independent of what route of administration is used (excerpts).
Safety and Tolerability
In the past, estrogens in general have been associated with a greater overall incidence of adverse cardiovascular and thromboembolic events (Kuhl, 2005). These events can include deep vein thrombosis and myocardial infarction (heart attack). Such complications have been attributed to estrogenic activity in the liver which, at sufficient exposure, causes an increased synthesis of liver proteins such as as sex-hormone binding globulin (von Schoultz et al., 1989; Ockrim, Lalani & Abel, 2006). Synthesis rates of lipids and coagulation factors have also been found to change. However, the type and route of administration of estrogen has been shown to modify risk (Olié, Canonico & Scarabin, 2011; Oliver-Williams et al., 2018).
Synthetic and non-bioidentical estrogens are more resistant to enzymatic metabolism by the liver and have disproportionate estrogenic effects relative to bioidentical estrogens such as estradiol (Kuhl, 2005). Because of this behaviour, they contribute to a much greater synthesis of liver proteins and are associated with a significantly higher risk of venous thromboembolism and other cardiovascular complications (Henriksson & Edhag, 1986; Kuhl, 2005; Lycette et al., 2006). A 2015 retrospective case-control study found that venous thromboembolism was 2 to 5 times more common in young women using combined oral contraceptives containing ethinylestradiol and other synthetic progestins than in non users (Vinogradova, Coupland & Hippisley-Cox, 2015). In 2019, the same authors published another case-control study; this time investigating women receiving hormone therapy at the menopause (Vinogradova, Coupland & Hippisley-Cox, 2019). A key finding was that low doses of oral estradiol (2 mg/day or less) were associated with a slight but significant increase in the incidence of venous thromboembolism, while low transdermal doses (100 μg/day or less) were not. This has also been reported by the ESTHER case-control and E3N cohort studies (Scarabin, 2014). Therefore, a strong advantage of transdermal estradiol over oral estradiol is that the incidence of venous thromboembolism is lower (Files & Kling, 2020). As with the synthetic estrogens, this difference is thought to be attributable to the disproportionate amount of estrogenic exposure in the liver that occurs with oral administration (Olié, Canonico & Scarabin, 2011). Nevertheless, high dose polyestradiol phosphate (160 to 240 mg/month) administered by intramuscular injection has been associated with significantly increased cardiovascular and thromboembolic morbidity and mortality in at least one large study of prostate cancer patients (Mikkola et al., 2005; Mikkola et al., 2007). While the increased incidence of these adverse events is clearly much lower than with oral estradiol, it is much less clear if it may be entirely eliminated by non-oral routes of administration at higher doses (Sam, 2020).
It is difficult to accurately determine the incidence of venous thromboembolism in transgender people receiving hormone therapy because of the diverse range of regimens employed in different geographical regions; which may confer different risks (Goldstein et al., 2019). Moreover, although a number of observational and retrospective studies have reported risk as low or relatively insignificant in our community, most are not adequately powered to accurately report risk (Khan et al., 2019). Based on the available evidence, we can probably safely assume that the incidence is low overall with modern regimens (Getahun et al., 2018; Ott et al., 2010; Pyra et al., 2020). It is particularly of note that these complications are, thankfully, mostly confined to people at higher baseline risk such as elderly individuals or those with inherited mutations that predispose to such toxicity (Silverstein et al., 1998; Bezgin et al., 2016). The absolute risk is likely low for most people. Nonetheless, the association between estrogens and adverse cardiovascular and thromboembolic events is of obvious importance.
Summary and Conclusions
In conclusion, oral and transdermal estradiol is metabolised differently. Most significantly, oral estadiol is predominantly converted by the liver into estrone and other estrogen metabolites before it enters circulation. By contrast, transdermal estradiol bypasses the liver and the conversion of the medication into these weak estrogens is mostly avoided. On average, a transdermal patch that delivers a 50 μg/day dose is thought to have similar estrogenic potency to a 1 to 2 mg/day dose of oral estradiol and to a 1.5 mg/day dose of transdermal gel.
In spite of these difference, there appears to be no evidence that oral estradiol provides more effective feminisation than transdermal estradiol or vice versa if the doses are similar. Instead, the existing clinical evidence seems to show that the extent of feminising changes such as breast development and fat distribution is independent of the route that estradiol is administered by. Contrariwise, there is a large amount of epidemiological evidence that oral estradiol is associated with a higher incidence of venous thrombosis than is transdermal estradiol at a comparable dose. For this reason, transdermal estradiol at physiological doses is likely safer than oral estradiol in long term for gender-affirming hormone therapy.
References
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery, J. C., Zajac, J. D., & Cheung, A. S. (2025). Effect of spironolactone and cyproterone acetate on breast growth in transgender people: a randomized clinical trial. The Journal of Clinical Endocrinology & Metabolism, 110(6), 1874-1884. [DOI:10.1210/clinem/dgae650]
Angus, L. M., Leemaqz, S. Y., Doery, J. C., & Cheung, A. S. (2025). Effect of anti-androgens on body composition in transgender people: secondary analysis of a randomized clinical trial. International Journal of Transgender Health, 1-12. [DOI:10.1080/26895269.2025.2531421]
Bannink, E. M., Van Sassen, C., Van Buuren, S., De Jong, F. H., Lequin, M., Mulder, P. G., & De Muinck Keizer-Schrama, S. M. (2009). Puberty induction in Turner syndrome: results of oestrogen treatment on development of secondary sexual characteristics, uterine dimensions and serum hormone levels. Clinical Endocrinology, 70(2), 265–273. [DOI:10.1111/j.1365-2265.2008.03446.x]
Benjamin, H. (1967). Transvestism and Transsexualism in the male and female. Journal of Sex Research, 3(2), 107–127. [DOI:10.1080/00224496709550519]
Bezgin, T., Kaymaz, C., Akbal, Ö., Yılmaz, F., Tokgöz, H. C., & Özdemir, N. (2016). Thrombophilic Gene Mutations in Relation to Different Manifestations of Venous Thromboembolism: A Single Tertiary Center Study. Clinical and Applied Thrombosis/Hemostasis, 24(1), 100–106. [DOI:10.1177/1076029616672585]
Bińkowska, M. (2014). Featured paper Menopausal hormone therapy and venous thromboembolism. Przegląd Menopauzalny [Menopausal Review], 13(5), 267–272. [DOI:10.5114/pm.2014.46468]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., De Vries, A. L., Deutsch, M. B., … & Arcelus, J. (2022). Standards of care for the health of transgender and gender diverse people, Version 8. International Journal of Transgender Health, 23(sup1), S1-S259. [DOI:10.1080/26895269.2022.2100644]
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C. J., Fields, M. E., … & Baranski, T. (2024). Effectiveness and safety of different estradiol regimens in transgender females: a randomized controlled trial. Journal of the Endocrine Society, 8(8), bvae108. [DOI:10.1210/jendso/bvae108]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de Blok, C. J., Staphorsius, A. S., Wiepjes, C. M., Smit, J. M., Nanayakkara, P. W., & den Heijer, M. (2019). Frequency, Determinants, and Satisfaction of Breast Augmentation in Trans Women Receiving Hormone Treatment. The Journal of Sexual Medicine, 17(2), 342–348. [DOI:10.1016/j.jsxm.2019.10.021]
de Blok, C. J., Dijkman, B. A., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., … & den Heijer, M. (2021). Sustained breast development and breast anthropometric changes in 3 years of gender-affirming hormone treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), 782-790. [DOI:10.1210/clinem/dgaa841]
Dijkman, B. A., Helder, D., Boogers, L. S., Gieles, N. C., van Heesewijk, J. O., Slaa, S. T., … & Dreijerink, K. M. (2023). Addition of progesterone to feminizing gender-affirming hormone therapy in transgender individuals for breast development: a randomized controlled trial. BMC Pharmacology and Toxicology, 24(1), 80. [DOI:10.1186/s40360-023-00724-4]
Files, J., & Kling, J. M. (2019). Transdermal delivery of bioidentical estrogen in menopausal hormone therapy: a clinical review. Expert Opinion on Drug Delivery, 17(4), 543–549. [DOI:10.1080/17425247.2020.1700949]
Fisher, A. D., & Maggi, M. (2015). Endocrine Treatment of Transsexual Male-to-Female Persons. In Trombetta, C., Liguori, G., & Bertolotto, M. (Eds.). Management of Gender Dysphoria: A Multidisciplinary Approach (pp. 83–91). Milano: Springer Milan. [DOI:10.1007/978-88-470-5696-1_10]
Frederiksen, H., Johannsen, T. H., Andersen, S. E., Albrethsen, J., Landersoe, S. K., Petersen, J. H., Andersen, A. N., Vestergaard, E. T., Schorring, M. E., Linneberg, A., Main, K. M., Andersson, A., & Juul, A. (2019). Sex-specific Estrogen Levels and Reference Intervals from Infancy to Late Adulthood Determined by LC-MS/MS. The Journal of Clinical Endocrinology & Metabolism, 105(3), 754–768. [DOI:10.1210/clinem/dgz196]
Geffen, S., Horn, T., Smith, K. J., & Cahill, S. (2018). Advocacy for Gender Affirming Care: Learning from the Injectable Estrogen Shortage. Transgender Health, 3(1), 42–44. [DOI:10.1089/trgh.2017.0025]
Getahun, D., Nash, R., Flanders, W. D., Baird, T. C., Becerra-Culqui, T. A., Cromwell, L., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Silverberg, M. J., Safer, J., Slovis, J., Tangpricha, V., & Goodman, M. (2018). Cross-sex Hormones and Acute Cardiovascular Events in Transgender Persons. Annals of Internal Medicine, 169(4), 205–213. [DOI:10.7326/m17-2785]
Goldstein, Z., Khan, M., Reisman, T., & Safer, J. D. (2019). Managing the risk of venous thromboembolism in transgender adults undergoing hormone therapy. Journal of Blood Medicine, 10, 209–216. [DOI:10.2147/jbm.s166780]
Gravholt, C. H., Andersen, N. H., Conway, G. S., Dekkers, O. M., Geffner, M. E., Klein, K. O., Lin, A. E., Mauras, N., Quigley, C. A., Rubin, K., Sandberg, D. E., Sas, T. C., Silberbach, M., Söderström-Anttila, V., Stochholm, K., van Alfen-van derVelden, J. A., Woelfle, J., Backeljauw, P. F., & the International Turner Syndrome Consensus Group. (2017). Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. European Journal of Endocrinology, 177(3), G1–G70. [DOI:10.1530/eje-17-0430]
Guo, S., Zhang, J., Li, Y., Ma, H., Chen, Q., Chen, H., & Du, M. (2019). The pubertal development mode of Chinese girls with turner syndrome undergoing hormone replacement therapy. BMC Endocrine Disorders, 19(1), 72. [DOI:10.1186/s12902-019-0403-2]
Hamburger, C., & Benjamin, H. (1969). Endocrine Treatment of Male and Female Transsexualism / Appendix for the Practicing Physician: Suggestions and Guidelines for the Management of Transsexuals. In Green, R., & Money, J. (Eds.). Transsexualism and Sex Reassignment (pp. 291–307). Baltimore: John Hopkins University Press. [Google Scholar] [Google Books] [PDF]
Hamidi, O., & Davidge-Pitts, C. J. (2019). Transfeminine Hormone Therapy. Endocrinology and Metabolism Clinics of North America, 48(2), 341–355. [DOI:10.1016/j.ecl.2019.02.001]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline [2nd Version]. The Journal of Clinical Endocrinology & Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Henriksson, P., & Edhag, O. (1986). Orchidectomy versus oestrogen for prostatic cancer: cardiovascular effects. BMJ, 293(6544), 413–415. [DOI:10.1136/bmj.293.6544.413]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Järvinen, A., Nykänen, S., & Paasiniemi, L. (1999). Absorption and bioavailability of oestradiol from a gel, a patch and a tablet. Maturitas, 32(2), 103–113. [DOI:10.1016/s0378-5122(99)00021-3]
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2025). Comparison of estrone/estradiol ratio and levels in transfeminine individuals on different routes of estradiol. Transgender Health, 10(3), 261-268. [DOI:10.1089/trgh.2023.0138]
Khan, J., Schmidt, R. L., Spittal, M. J., Goldstein, Z., Smock, K. J., & Greene, D. N. (2019). Venous Thrombotic Risk in Transgender Women Undergoing Estrogen Therapy: A Systematic Review and Metaanalysis. Clinical Chemistry, 65(1), 57–66. [DOI:10.1373/clinchem.2018.288316]
Klaver, M., de Blok, C. J., Wiepjes, C. M., Nota, N. M., Dekker, M. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/eje-17-0496]
Klein, K. O., Rosenfield, R. L., Santen, R. J., Gawlik, A. M., Backeljauw, P. F., Gravholt, C. H., Sas, T. C., & Mauras, N. (2018). Estrogen Replacement in Turner Syndrome: Literature Review and Practical Considerations. The Journal of Clinical Endocrinology & Metabolism, 103(5), 1790–1803. [DOI:10.1210/jc.2017-02183]
Klein, K. O., & Phillips, S. A. (2019). Review of Hormone Replacement Therapy in Girls and Adolescents with Hypogonadism. Journal of Pediatric and Adolescent Gynecology, 32(5), 460–468. [DOI:10.1016/j.jpag.2019.04.010]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhnz, W., Blode, H., & Zimmermann, H. (1993). Pharmacokinetics of exogenous natural and synthetic estrogens and antiestrogens. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 261–322). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_15]
Kuhnz, W., Gansau, C., & Mahler, M. (1993). Pharmacokinetics of estradiol, free and total estrone, in young women following single intravenous and oral administration of 17β-estradiol. Arzneimittelforschung, 43(9), 966–973. [Google Scholar] [PubMed] [PDF]
Mauras, N., Torres-Santiago, L., Santen, R., Mericq, V., Ross, J., Colon-Otero, G., Damaso, L., Hossain, J., Wang, Q., Mesaros, C., & Blair, I. A. (2018). Impact of route of administration on genotoxic oestrogens concentrations using oral vs transdermal oestradiol in girls with Turner syndrome. Clinical Endocrinology, 90(1), 155–161. [DOI:10.1111/cen.13869]
Meyer, W. J., Walker, P. A., & Suplee, Z. R. (1981). A survey of transsexual hormonal treatment in twenty gender‐treatment centers. The Journal of Sex Research, 17(4), 344–349. [DOI:10.1080/00224498109551125]
Meyer, W. J., Webb, A., Stuart, C. A., Finkelstein, J. W., Lawrence, B., & Walker, P. A. (1986). Physical and hormonal evaluation of transsexual patients: A longitudinal study. Archives of Sexual Behavior, 15(2), 121–138. [DOI:10.1007/bf01542220]
Mikkola, A., Aro, J., Rannikko, S., Oksanen, H., Ruutu, M., & (2005). Cardiovascular complications in patients with advanced prostatic cancer treated by means of orchiectomy or polyestradiol phosphate. Scandinavian Journal of Urology and Nephrology, 39(4), 294–300. [DOI:10.1080/00365590510031228]
Mikkola, A., Aro, J., Rannikko, S., Ruutu, M., & (2007). Ten-year survival and cardiovascular mortality in patients with advanced prostate cancer primarily treated by intramuscular polyestradiol phosphate or orchiectomy. The Prostate, 67(4), 447–455. [DOI:10.1002/pros.20547]
Ockrim, J., Lalani, E., & Abel, P. (2006). Therapy Insight: parenteral estrogen treatment for prostate cancer—a new dawn for an old therapy. Nature Clinical Practice Oncology, 3(10), 552–563. [DOI:10.1038/ncponc0602]
Olié, V., Canonico, M., & Scarabin, P. (2011). Postmenopausal hormone therapy and venous thromboembolism. Thrombosis Research, 127, S26–S29. [DOI:10.1016/s0049-3848(11)70008-1]
Oliver-Williams, C., Glisic, M., Shahzad, S., Brown, E., Pellegrino Baena, C., Chadni, M., Chowdhury, R., Franco, O. H., & Muka, T. (2018). The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Human Reproduction Update, 25(2), 257–271. [DOI:10.1093/humupd/dmy039]
Ott, J., Kaufmann, U., Bentz, E., Huber, J. C., & Tempfer, C. B. (2010). Incidence of thrombophilia and venous thrombosis in transsexuals under cross-sex hormone therapy. Fertility and Sterility, 93(4), 1267–1272. [DOI:10.1016/j.fertnstert.2008.12.017]
Piippo, S., Lenko, H., Kainulainen, P., & Sipilä, I. (2004). Use of Percutaneous Estrogen Gel for Induction of Puberty in Girls with Turner Syndrome. The Journal of Clinical Endocrinology & Metabolism, 89(7), 3241–3247. [DOI:10.1210/jc.2003-032069]
Pyra, M., Casimiro, I., Rusie, L., Ross, N., Blum, C., Keglovitz Baker, K., Baker, A., & Schneider, J. (2020). An Observational Study of Hypertension and Thromboembolism Among Transgender Patients Using Gender-Affirming Hormone Therapy. Transgender Health, 5(1), 1–9. [DOI:10.1089/trgh.2019.0061]
Rohr, U. D., Volko, C. D., & Schindler, A. E. (2014). Comparison of steady state development and reduction of menopausal symptoms after oral or transdermal delivery of 17-β-estradiol in young healthy symptomatic menopausal women. Hormone Molecular Biology and Clinical Investigation, 18(3), 123–126. [DOI:10.1515/hmbci-2013-0047]
Rosenfield, R. L., Devine, N., Hunold, J. J., Mauras, N., Moshang, T., & Root, A. W. (2005). Salutary Effects of Combining Early Very Low-Dose Systemic Estradiol with Growth Hormone Therapy in Girls with Turner Syndrome. The Journal of Clinical Endocrinology & Metabolism, 90(12), 6424–6430. [DOI:10.1210/jc.2005-1081]
Scarabin, P. (2014). Hormone Therapy and Venous Thromboembolism among Postmenopausal Women. Frontiers of Hormone Research, 43, 21–32. / Granata, R., & Isgaard, J. (Eds.). Cardiovascular Issues in Endocrinology (Frontiers of Hormone Research, Volume 43) (pp. 21–32). Basel: Karger. [DOI:10.1159/000360554] [Google Books]
Seal, L. J., Franklin, S., Richards, C., Shishkareva, A., Sinclaire, C., & Barrett, J. (2012). Predictive Markers for Mammoplasty and a Comparison of Side Effect Profiles in Transwomen Taking Various Hormonal Regimens. The Journal of Clinical Endocrinology & Metabolism, 97(12), 4422–4428. [DOI:10.1210/jc.2012-2030]
Seal, L. J. (2019). Cardiovascular disease in transgendered people: A review of the literature and discussion of risk. JRSM Cardiovascular Disease, 8, 204800401988074. [DOI:10.1177/2048004019880745]
Shah, S., Forghani, N., Durham, E., & Neely, E. K. (2014). A randomized trial of transdermal and oral estrogen therapy in adolescent girls with hypogonadism. International Journal of Pediatric Endocrinology, 2014(1), 12. [DOI:10.1186/1687-9856-2014-12]
Silverstein, M. D., Heit, J. A., Mohr, D. N., Petterson, T. M., O’Fallon, W. M., & Melton, L. J. (1998). Trends in the Incidence of Deep Vein Thrombosis and Pulmonary Embolism. Archives of Internal Medicine, 158(6), 585–593. [DOI:10.1001/archinte.158.6.585]
Tebbens, M., Heijboer, A. C., T’Sjoen, G., Bisschop, P. H., & den Heijer, M. (2022). The role of estrone in feminizing hormone treatment. The Journal of Clinical Endocrinology & Metabolism, 107(2), 458-466. [DOI:10.1210/clinem/dgab741]
van de Grift, T. C., Kreukels, B. P., & (2019). Breast development and satisfaction in women with disorders/differences of sex development. Human Reproduction, 34(12), 2410–2417. [DOI:10.1093/humrep/dez230]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2015). Use of combined oral contraceptives and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 350, h2135. [DOI:10.1136/bmj.h2135]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2019). Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 364, k4810. [DOI:10.1136/bmj.k4810]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
Wierckx, K., Van Caenegem, E., Schreiner, T., Haraldsen, I., Fisher, A., Toye, K., Kaufman, J. M., & T’Sjoen, G. (2014). Cross‐Sex Hormone Therapy in Trans Persons Is Safe and Effective at Short‐Time Follow‐Up: Results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 11(8), 1999–2011. [DOI:10.1111/jsm.12571]
Winston-McPherson, G. N., Thomas, T. A., Krasowski, M. D., Ahmed, S. B., Cirrincione, L. R., Katzman, B. M., … & Greene, D. N. (2025). Estradiol Concentrations for Adequate Gender-Affirming Feminizing Therapy: A Systematic Review. LGBT Health. [DOI:10.1089/lgbt.2024.0407]
Wren, B. G., Day, R. O., McLachlan, A. J., & Williams, K. M. (2003). Pharmacokinetics of estradiol, progesterone, testosterone and dehydroepiandrosterone after transbuccal administration to postmenopausal women. Climacteric, 6(2), 104–111. [DOI:10.1080/cmt.6.2.104.111]
\ No newline at end of file
+A Comparison of Oral and Transdermal Estradiol in Transfeminine Hormone Therapy - Transfeminine Science
A Comparison of Oral and Transdermal Estradiol in Transfeminine Hormone Therapy
By Sam | First published April 11, 2020 | Last modified July 29, 2025
Abstract / TL;DR
The most common means to administer exogenous estradiol are the oral and transdermal routes. Both are widely used as a component of gender-affirming hormone therapy. Current clinical evidence shows no difference in feminising efficacy between these formulations at equivalent doses. Although both are generally well tolerated, the oral route is unphysiological in its metabolism and is associated with a significantly greater incidence of cardiovascular and thromboembolic complications. At low adult replacement doses, transdermal forms do not have these disadvantages and may be superior to their oral counterparts in the long-term.
Introduction
Estrogen replacement is both an important and necessary intervention for many transgender people (Hembree et al., 2017; Coleman et al., 2022). In the past, feminising therapy in this group was mostly done using high dose estrogen monotherapy with parenteral esters of estradiol such as estradiol valerate or estradiol undecylate (Benjamin, 1967; Hamburger, 1969). Non-bioidentical oral estrogens such as conjugated equine estrogens and ethinylestradiol were also widely used (Meyer et al., 1986; Meyer, Walker, & Suplee, 1989). However, with significant progress made in drug development, bioidentical estradiol became widely available in oral and transdermal formulations for gender-affirming hormone therapy.
Some transgender individuals prefer to use injectable formulations of estradiol (Geffen et al., 2018). However, the oral and transdermal routes of administration appear to be most commonplace (Fisher & Maggi, 2015; Hamidi & Davidge-Pitts, 2019; Seal, 2019). Many people receiving or eager to start hormone therapy may be interested to know what data exists regarding differences between oral and transdermal estradiol. As we require long-term therapy with these formulations, a discussion regarding adverse effects between these routes of administration may also be of importance. Although the focus of this review largely concerns oral estradiol as directly compared to transdermal estradiol, a good amount of the discussion specific to transdermal estradiol can likely be extrapolated to other non-oral routes of administration when the doses are known to have similar potency. For instance, estradiol administered by intramuscular or subcutaneous injection is a non-oral route of administration.
Pharmacology
Oral estradiol includes pill or tablet formulations, while transdermal estradiol is most commonly available as patches or gels (Kuhl, 2005). Oral estradiol and transdermal gel is usually administered once per day (Rohr, Volko, & Schindler 2014). However, doses may be split and taken twice-daily. Theoretically, this would result in more stable estradiol levels, although this could be less convenient for the user. Estradiol pills can also be administered by the sublingual or buccal routes (Wren et al., 2003; Jain, Kwan, & Forcier 2019; Doll et al., 2022). Up until only a short time ago, data pertaining to sublingual administration in transfeminine people was scarce, however in more recent years there has been considerably greater interest from the scientific community (Cortez et al., 2024). However, usage of these two routes is probably still relatively uncommon in clinical practice. In this review, the term “oral estradiol” has been used to refer exclusively to the swallowing of estradiol tablets. Estradiol patches are applied and worn continuously. Different brands exist and transdermal patches are available for twice-weekly or weekly administration. On average, a 50 μg/day dosage delivered by transdermal patch is generally considered to have approximately similar potency to a 1 to 2 mg/day dosage of oral estradiol and to a 1.5 mg/day dosage of transdermal gel (Kuhl, 2005; Järvinen, Nykänen, & Paasiniemi, 1999). However, there is considerable interindividual variation in the metabolism of different estradiol formulations. Due to this variability, these doses are unlikely to correspond to one another on an individual basis.
In cisgender women, estradiol is secreted by the ovaries into systematic circulation. As a result, the liver does not receive disproportionate exposure to the hormone (Gravholt et al., 2017). Transdermal estradiol is effective in mimicking this behaviour. However, orally administered estradiol, owing to its passage through the gastrointestinal tract, is associated with disproportionate estrogenic exposure in the liver (Bińkowska, 2014). This behaviour gives rise to a number of differences between oral and transdermal estradiol. One such difference is that, on average, about 95% of oral estradiol is metabolised, as a consequence of the first pass effect, into estrone and other clinically weak/insignificant estrogens (Kuhz, Blode & Zimmermann, 1993). The ratio of estrone to estradiol is close to 1:1 in both adult women and pubertal girls and with transdermal formulations (Kuhl, 2005; Frederiksen et al., 2020). However, with a dose of oral estradiol, postmenopausal women have been found to have about 5 times the concentration of estrone as estradiol (Kuhl, 2005). In some patients, the concentration of estrone may be 20 times higher than that of estradiol (Kuhnz, Gansau, & Mahler, 1993). A new retrospective study has recently confirmed these findings in transfeminine people (Kariyawasam et al., 2025) For this reason, the metabolism of oral estradiol has been described as unphysiologic (Gravholt et al., 2017; Mauras et al., 2019). It should be emphasised that these findings by themselves do not necessarily translate into clinically important differences between oral and transdermal estradiol, such as in feminisation outcomes.
Efficacy
Many transgender people taking feminising hormone therapy are concerned with which regimens might be most “effective”. In particular, satisfactory breast development is often sought after. A number of randomised controlled trials assessing the efficacy of different gender-affirming hormone regimens on objective measures of feminisation have been completed or are currently underway (Dijkman et al., 2023; Angus et al., 2025a; Angus et al., 2025b). However, there have yet to be any such studies directly comparing the influence of the different routes of administration. Moreover, there are no measures of breast development or other effects of transition that are universally agreed upon by the scientific community. For this reason, it is difficult to directly compare the findings of many studies.
In spite of the above, a number of observational studies have studied and quantified feminisation experienced with hormone therapy. Several studies of transgender women treated with cyproterone acetate plus either oral or transdermal estradiol have reported no apparent difference in physical measures such as breast circumference and breast-chest difference between oral and transdermal administration in the short-term (ie: 6 to 12 months) (Wierckx et al., 2014; de Blok et al., 2018; Tebbens et al., 2022). In the latter study, it is interesting to note that estrone concentrations were directly measured and found to not be associated with the extent of breast development. This is consistent with non-oral and oral formulations being comparable in their effect despite the differences in their pharmacology. Consequently, the authors concluded that monitoring of estrone concentrations does not appear to have a place in transfeminine hormone therapy. A longer term follow-up of another one of these study groups involving transgender women attending clinics in the Netherlands, Belgium and Italy also reported that the increase in breast volume following 3 years of therapy did not differ between those using oral and transdermal formulations (de Blok et al., 2021). As a result of unsatisfactory development, many transgender women seek breast augmentation (de Blok et al., 2020). Although breast development itself was not measured, it is interesting to note that one retrospective study found no statistical difference in the rate of augmentation requests between users of different estrogen types (Seal et al., 2012). This suggests that oral estradiol valerate might be no more or less effective than the other estrogens in the study (oral conjugated estrogens and oral ethinylestradiol) in attaining a satisfying amount of breast development. A large cohort study of transgender women found that changes in gynoid and android fat, total body fat and total lean body mass were not statistically different between the oral and transdermal estradiol groups if BMI and age were controlled for (Klaver et al., 2018).
Estrogen replacement, being a necessary therapy for the vast majority of individuals with Turner syndrome, has also been studied in adolescent girls (Gravholt et al., 2017; Klein & Phillips, 2019). Girls treated with low dose oral estradiol were described in one study as having “similar” breast development to the normal Dutch population (Bannink et al., 2009). Other studies of puberty induction therapy have found that patients using low doses of transdermal estradiol gel and low dose intramuscular estradiol cypionate also all achieved breast Tanner stage 4 or 5 at final follow-up (Piippo et al., 2004; Rosenfield et al., 2005). A small randomised controlled trial of hypogonadal girls demonstrated that the response to oral and transdermal estradiol at comparable doses was near identical (Shah et al., 2014). All the girls receiving bioidentical estrogens achieved Tanner stage 3 or greater after 18 months of treatment, irrespective of route of administration. Interestingly, a cross sectional study of breast development in women with differences of sex development (DSD), including those with Turner syndrome, reported that breast satisfaction in the sample group was much lower than in women without a DSD (van de Grift & Kreukels, 2019). Some studies have found that breast development, in addition to breast satisfaction, seems to be poorer in Turner syndrome girls than in normal cisgender girls (Guo et al., 2019). Nevertheless, a recent review concluded that all these different regimens seemed to result in similar feminising outcomes (Klein et al., 2018). In comparison to the available data of transfeminine people these findings are low quality and inconclusive on their own, since breast development itself was not measured objectively. Nevertheless, the findings fit an emerging pattern within the literature and are consistent with a recent systematic review which did not find any evidence of differences between the oral and transdermal routes in transfeminine hormone therapy (Winston-McPherson et al., 2025)
In summary, current clinical evidence appears to show no difference in objectively measured outcomes between therapy with different routes of administration when the doses have comparable potency. Rather, when taken together, these findings indicate that the extent of breast development and other feminisation is independent of what route of administration is used (excerpts).
Safety and Tolerability
In the past, estrogens in general have been associated with a greater overall incidence of adverse cardiovascular and thromboembolic events (Kuhl, 2005). These events can include deep vein thrombosis and myocardial infarction (heart attack). Such complications have been attributed to estrogenic activity in the liver which, at sufficient exposure, causes an increased synthesis of liver proteins such as as sex-hormone binding globulin (von Schoultz et al., 1989; Ockrim, Lalani, & Abel, 2006). Synthesis rates of lipids and coagulation factors have also been found to change. However, the type and route of administration of estrogen has been shown to modify risk (Olié, Canonico, & Scarabin, 2011; Oliver-Williams et al., 2018).
Synthetic and non-bioidentical estrogens are more resistant to enzymatic metabolism by the liver and have disproportionate estrogenic effects relative to bioidentical estrogens such as estradiol (Kuhl, 2005). Because of this behaviour, they contribute to a much greater synthesis of liver proteins and are associated with a significantly higher risk of venous thromboembolism and other cardiovascular complications (Henriksson & Edhag, 1986; Kuhl, 2005; Lycette et al., 2006). A 2015 retrospective case-control study found that venous thromboembolism was 2 to 5 times more common in young women using combined oral contraceptives containing ethinylestradiol and other synthetic progestins than in non users (Vinogradova, Coupland, & Hippisley-Cox, 2015). In 2019, the same authors published another case-control study; this time investigating women receiving hormone therapy at the menopause (Vinogradova, Coupland, & Hippisley-Cox, 2019). A key finding was that low doses of oral estradiol (2 mg/day or less) were associated with a slight but significant increase in the incidence of venous thromboembolism, while low transdermal doses (100 μg/day or less) were not. This has also been reported by the ESTHER case-control and E3N cohort studies (Scarabin, 2014). Therefore, a strong advantage of transdermal estradiol over oral estradiol is that the incidence of venous thromboembolism is lower (Files & Kling, 2020). As with the synthetic estrogens, this difference is thought to be attributable to the disproportionate amount of estrogenic exposure in the liver that occurs with oral administration (Olié, Canonico, & Scarabin, 2011). Nevertheless, high dose polyestradiol phosphate (160 to 240 mg/month) administered by intramuscular injection has been associated with significantly increased cardiovascular and thromboembolic morbidity and mortality in at least one large study of prostate cancer patients (Mikkola et al., 2005; Mikkola et al., 2007). While the increased incidence of these adverse events is clearly much lower than with oral estradiol, it is much less clear if it may be entirely eliminated by non-oral routes of administration at higher doses (Sam, 2020).
It is difficult to accurately determine the incidence of venous thromboembolism in transgender people receiving hormone therapy because of the diverse range of regimens employed in different geographical regions; which may confer different risks (Goldstein et al., 2019). Moreover, although a number of observational and retrospective studies have reported risk as low or relatively insignificant in our community, most are not adequately powered to accurately report risk (Khan et al., 2019). Based on the available evidence, we can probably safely assume that the incidence is low overall with modern regimens (Getahun et al., 2018; Ott et al., 2010; Pyra et al., 2020). It is particularly of note that these complications are, thankfully, mostly confined to people at higher baseline risk such as elderly individuals or those with inherited mutations that predispose to such toxicity (Silverstein et al., 1998; Bezgin et al., 2016). The absolute risk is likely low for most people. Nonetheless, the association between estrogens and adverse cardiovascular and thromboembolic events is of obvious importance.
Summary and Conclusions
In conclusion, oral and transdermal estradiol is metabolised differently. Most significantly, oral estadiol is predominantly converted by the liver into estrone and other estrogen metabolites before it enters circulation. By contrast, transdermal estradiol bypasses the liver and the conversion of the medication into these weak estrogens is mostly avoided. On average, a transdermal patch that delivers a 50 μg/day dose is thought to have similar estrogenic potency to a 1 to 2 mg/day dose of oral estradiol and to a 1.5 mg/day dose of transdermal gel.
In spite of these difference, there appears to be no evidence that oral estradiol provides more effective feminisation than transdermal estradiol or vice versa if the doses are similar. Instead, the existing clinical evidence seems to show that the extent of feminising changes such as breast development and fat distribution is independent of the route that estradiol is administered by. Contrariwise, there is a large amount of epidemiological evidence that oral estradiol is associated with a higher incidence of venous thrombosis than is transdermal estradiol at a comparable dose. For this reason, transdermal estradiol at physiological doses is likely safer than oral estradiol in long term for gender-affirming hormone therapy.
References
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery, J. C., Zajac, J. D., & Cheung, A. S. (2025). Effect of spironolactone and cyproterone acetate on breast growth in transgender people: a randomized clinical trial. The Journal of Clinical Endocrinology & Metabolism, 110(6), 1874–1884. [DOI:10.1210/clinem/dgae650]
Angus, L. M., Leemaqz, S. Y., Doery, J. C., & Cheung, A. S. (2025). Effect of anti-androgens on body composition in transgender people: secondary analysis of a randomized clinical trial. International Journal of Transgender Health, online ahead of print. [DOI:10.1080/26895269.2025.2531421]
Bannink, E. M., Van Sassen, C., Van Buuren, S., De Jong, F. H., Lequin, M., Mulder, P. G., & De Muinck Keizer-Schrama, S. M. (2009). Puberty induction in Turner syndrome: results of oestrogen treatment on development of secondary sexual characteristics, uterine dimensions and serum hormone levels. Clinical Endocrinology, 70(2), 265–273. [DOI:10.1111/j.1365-2265.2008.03446.x]
Benjamin, H. (1967). Transvestism and Transsexualism in the male and female. Journal of Sex Research, 3(2), 107–127. [DOI:10.1080/00224496709550519]
Bezgin, T., Kaymaz, C., Akbal, Ö., Yılmaz, F., Tokgöz, H. C., & Özdemir, N. (2016). Thrombophilic Gene Mutations in Relation to Different Manifestations of Venous Thromboembolism: A Single Tertiary Center Study. Clinical and Applied Thrombosis/Hemostasis, 24(1), 100–106. [DOI:10.1177/1076029616672585]
Bińkowska, M. (2014). Featured paper Menopausal hormone therapy and venous thromboembolism. Przegląd Menopauzalny [Menopausal Review], 13(5), 267–272. [DOI:10.5114/pm.2014.46468]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C. J., Fields, M. E., Gray, T., Guo, Z., Nicol, G., & Baranski, T. (2024). Effectiveness and Safety of Different Estradiol Regimens in Transgender Females: A Randomized Controlled Trial. Journal of the Endocrine Society, 8(8), bvae108. [DOI:10.1210/jendso/bvae108]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de Blok, C. J., Staphorsius, A. S., Wiepjes, C. M., Smit, J. M., Nanayakkara, P. W., & den Heijer, M. (2019). Frequency, Determinants, and Satisfaction of Breast Augmentation in Trans Women Receiving Hormone Treatment. The Journal of Sexual Medicine, 17(2), 342–348. [DOI:10.1016/j.jsxm.2019.10.021]
de Blok, C. J. M., Dijkman, B. A. M., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K. M. A., & den Heijer, M. (2021). Sustained breast development and breast anthropometric changes in 3 years of gender-affirming hormone treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), 782–790. [DOI:10.1210/clinem/dgaa841]
Dijkman, B. A. M., Helder, D., Boogers, L. S., Gieles, N. C., van Heesewijk, J. O., Slaa, S. T., Liberton, N. P. T. J., Wiepjes, C. M., de Blok, C. J. M., den Heijer, M., & Dreijerink, K. M. A. (2023). Addition of progesterone to feminizing gender-affirming hormone therapy in transgender individuals for breast development: a randomized controlled trial. BMC Pharmacology and Toxicology, 24(1), 80. [DOI:10.1186/s40360-023-00724-4]
Doll, E., Gunsolus, I., Thorgerson, A., Tangpricha, V., Lamberton, N., & Sarvaideo, J. L. (2022). Pharmacokinetics of Sublingual Versus Oral Estradiol in Transgender Women. Endocrine Practice, 28(3), 237–242. [DOI:10.1016/j.eprac.2021.11.081]
Files, J., & Kling, J. M. (2019). Transdermal delivery of bioidentical estrogen in menopausal hormone therapy: a clinical review. Expert Opinion on Drug Delivery, 17(4), 543–549. [DOI:10.1080/17425247.2020.1700949]
Fisher, A. D., & Maggi, M. (2015). Endocrine Treatment of Transsexual Male-to-Female Persons. In Trombetta, C., Liguori, G., & Bertolotto, M. (Eds.). Management of Gender Dysphoria: A Multidisciplinary Approach (pp. 83–91). Milano: Springer Milan. [DOI:10.1007/978-88-470-5696-1_10]
Frederiksen, H., Johannsen, T. H., Andersen, S. E., Albrethsen, J., Landersoe, S. K., Petersen, J. H., Andersen, A. N., Vestergaard, E. T., Schorring, M. E., Linneberg, A., Main, K. M., Andersson, A., & Juul, A. (2019). Sex-specific Estrogen Levels and Reference Intervals from Infancy to Late Adulthood Determined by LC-MS/MS. The Journal of Clinical Endocrinology & Metabolism, 105(3), 754–768. [DOI:10.1210/clinem/dgz196]
Geffen, S., Horn, T., Smith, K. J., & Cahill, S. (2018). Advocacy for Gender Affirming Care: Learning from the Injectable Estrogen Shortage. Transgender Health, 3(1), 42–44. [DOI:10.1089/trgh.2017.0025]
Getahun, D., Nash, R., Flanders, W. D., Baird, T. C., Becerra-Culqui, T. A., Cromwell, L., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Silverberg, M. J., Safer, J., Slovis, J., Tangpricha, V., & Goodman, M. (2018). Cross-sex Hormones and Acute Cardiovascular Events in Transgender Persons. Annals of Internal Medicine, 169(4), 205–213. [DOI:10.7326/m17-2785]
Goldstein, Z., Khan, M., Reisman, T., & Safer, J. D. (2019). Managing the risk of venous thromboembolism in transgender adults undergoing hormone therapy. Journal of Blood Medicine, 10, 209–216. [DOI:10.2147/jbm.s166780]
Gravholt, C. H., Andersen, N. H., Conway, G. S., Dekkers, O. M., Geffner, M. E., Klein, K. O., Lin, A. E., Mauras, N., Quigley, C. A., Rubin, K., Sandberg, D. E., Sas, T. C., Silberbach, M., Söderström-Anttila, V., Stochholm, K., van Alfen-van derVelden, J. A., Woelfle, J., Backeljauw, P. F., & the International Turner Syndrome Consensus Group. (2017). Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. European Journal of Endocrinology, 177(3), G1–G70. [DOI:10.1530/eje-17-0430]
Guo, S., Zhang, J., Li, Y., Ma, H., Chen, Q., Chen, H., & Du, M. (2019). The pubertal development mode of Chinese girls with turner syndrome undergoing hormone replacement therapy. BMC Endocrine Disorders, 19(1), 72. [DOI:10.1186/s12902-019-0403-2]
Hamburger, C., & Benjamin, H. (1969). Endocrine Treatment of Male and Female Transsexualism / Appendix for the Practicing Physician: Suggestions and Guidelines for the Management of Transsexuals. In Green, R., & Money, J. (Eds.). Transsexualism and Sex Reassignment (pp. 291–307). Baltimore: John Hopkins University Press. [Google Scholar] [Google Books] [PDF]
Hamidi, O., & Davidge-Pitts, C. J. (2019). Transfeminine Hormone Therapy. Endocrinology and Metabolism Clinics of North America, 48(2), 341–355. [DOI:10.1016/j.ecl.2019.02.001]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline [2nd Version]. The Journal of Clinical Endocrinology & Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Henriksson, P., & Edhag, O. (1986). Orchidectomy versus oestrogen for prostatic cancer: cardiovascular effects. BMJ, 293(6544), 413–415. [DOI:10.1136/bmj.293.6544.413]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Järvinen, A., Nykänen, S., & Paasiniemi, L. (1999). Absorption and bioavailability of oestradiol from a gel, a patch and a tablet. Maturitas, 32(2), 103–113. [DOI:10.1016/s0378-5122(99)00021-3]
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2025). Comparison of estrone/estradiol ratio and levels in transfeminine individuals on different routes of estradiol. Transgender Health, 10(3), 261–268. [DOI:10.1089/trgh.2023.0138]
Khan, J., Schmidt, R. L., Spittal, M. J., Goldstein, Z., Smock, K. J., & Greene, D. N. (2019). Venous Thrombotic Risk in Transgender Women Undergoing Estrogen Therapy: A Systematic Review and Metaanalysis. Clinical Chemistry, 65(1), 57–66. [DOI:10.1373/clinchem.2018.288316]
Klaver, M., de Blok, C. J., Wiepjes, C. M., Nota, N. M., Dekker, M. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/eje-17-0496]
Klein, K. O., Rosenfield, R. L., Santen, R. J., Gawlik, A. M., Backeljauw, P. F., Gravholt, C. H., Sas, T. C., & Mauras, N. (2018). Estrogen Replacement in Turner Syndrome: Literature Review and Practical Considerations. The Journal of Clinical Endocrinology & Metabolism, 103(5), 1790–1803. [DOI:10.1210/jc.2017-02183]
Klein, K. O., & Phillips, S. A. (2019). Review of Hormone Replacement Therapy in Girls and Adolescents with Hypogonadism. Journal of Pediatric and Adolescent Gynecology, 32(5), 460–468. [DOI:10.1016/j.jpag.2019.04.010]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhnz, W., Blode, H., & Zimmermann, H. (1993). Pharmacokinetics of exogenous natural and synthetic estrogens and antiestrogens. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 261–322). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_15]
Kuhnz, W., Gansau, C., & Mahler, M. (1993). Pharmacokinetics of estradiol, free and total estrone, in young women following single intravenous and oral administration of 17β-estradiol. Arzneimittelforschung, 43(9), 966–973. [Google Scholar] [PubMed] [PDF]
Mauras, N., Torres-Santiago, L., Santen, R., Mericq, V., Ross, J., Colon-Otero, G., Damaso, L., Hossain, J., Wang, Q., Mesaros, C., & Blair, I. A. (2018). Impact of route of administration on genotoxic oestrogens concentrations using oral vs transdermal oestradiol in girls with Turner syndrome. Clinical Endocrinology, 90(1), 155–161. [DOI:10.1111/cen.13869]
Meyer, W. J., Walker, P. A., & Suplee, Z. R. (1981). A survey of transsexual hormonal treatment in twenty gender‐treatment centers. The Journal of Sex Research, 17(4), 344–349. [DOI:10.1080/00224498109551125]
Meyer, W. J., Webb, A., Stuart, C. A., Finkelstein, J. W., Lawrence, B., & Walker, P. A. (1986). Physical and hormonal evaluation of transsexual patients: A longitudinal study. Archives of Sexual Behavior, 15(2), 121–138. [DOI:10.1007/bf01542220]
Mikkola, A., Aro, J., Rannikko, S., Oksanen, H., Ruutu, M., & (2005). Cardiovascular complications in patients with advanced prostatic cancer treated by means of orchiectomy or polyestradiol phosphate. Scandinavian Journal of Urology and Nephrology, 39(4), 294–300. [DOI:10.1080/00365590510031228]
Mikkola, A., Aro, J., Rannikko, S., Ruutu, M., & (2007). Ten-year survival and cardiovascular mortality in patients with advanced prostate cancer primarily treated by intramuscular polyestradiol phosphate or orchiectomy. The Prostate, 67(4), 447–455. [DOI:10.1002/pros.20547]
Ockrim, J., Lalani, E., & Abel, P. (2006). Therapy Insight: parenteral estrogen treatment for prostate cancer—a new dawn for an old therapy. Nature Clinical Practice Oncology, 3(10), 552–563. [DOI:10.1038/ncponc0602]
Olié, V., Canonico, M., & Scarabin, P. (2011). Postmenopausal hormone therapy and venous thromboembolism. Thrombosis Research, 127, S26–S29. [DOI:10.1016/s0049-3848(11)70008-1]
Oliver-Williams, C., Glisic, M., Shahzad, S., Brown, E., Pellegrino Baena, C., Chadni, M., Chowdhury, R., Franco, O. H., & Muka, T. (2018). The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Human Reproduction Update, 25(2), 257–271. [DOI:10.1093/humupd/dmy039]
Ott, J., Kaufmann, U., Bentz, E., Huber, J. C., & Tempfer, C. B. (2010). Incidence of thrombophilia and venous thrombosis in transsexuals under cross-sex hormone therapy. Fertility and Sterility, 93(4), 1267–1272. [DOI:10.1016/j.fertnstert.2008.12.017]
Piippo, S., Lenko, H., Kainulainen, P., & Sipilä, I. (2004). Use of Percutaneous Estrogen Gel for Induction of Puberty in Girls with Turner Syndrome. The Journal of Clinical Endocrinology & Metabolism, 89(7), 3241–3247. [DOI:10.1210/jc.2003-032069]
Pyra, M., Casimiro, I., Rusie, L., Ross, N., Blum, C., Keglovitz Baker, K., Baker, A., & Schneider, J. (2020). An Observational Study of Hypertension and Thromboembolism Among Transgender Patients Using Gender-Affirming Hormone Therapy. Transgender Health, 5(1), 1–9. [DOI:10.1089/trgh.2019.0061]
Rohr, U. D., Volko, C. D., & Schindler, A. E. (2014). Comparison of steady state development and reduction of menopausal symptoms after oral or transdermal delivery of 17-β-estradiol in young healthy symptomatic menopausal women. Hormone Molecular Biology and Clinical Investigation, 18(3), 123–126. [DOI:10.1515/hmbci-2013-0047]
Rosenfield, R. L., Devine, N., Hunold, J. J., Mauras, N., Moshang, T., & Root, A. W. (2005). Salutary Effects of Combining Early Very Low-Dose Systemic Estradiol with Growth Hormone Therapy in Girls with Turner Syndrome. The Journal of Clinical Endocrinology & Metabolism, 90(12), 6424–6430. [DOI:10.1210/jc.2005-1081]
Sam. (2020). Analysis of Cardiovascular and Thromboembolic Toxicity with High Dose Parenteral Polyestradiol Phosphate in the Treatment of Prostate Cancer. Transfeminine Science. [URL]
Sam. (2020). Sources/Excerpts: Reviews, Cohort Studies and Randomised Controlled Trials Comparing the Feminising Efficacy of Estradiol Administered by Oral and Non-Oral Routes. Transfeminine Science. [URL]
Scarabin, P. (2014). Hormone Therapy and Venous Thromboembolism among Postmenopausal Women. Frontiers of Hormone Research, 43, 21–32. / Granata, R., & Isgaard, J. (Eds.). Cardiovascular Issues in Endocrinology (Frontiers of Hormone Research, Volume 43) (pp. 21–32). Basel: Karger. [DOI:10.1159/000360554] [Google Books]
Seal, L. J., Franklin, S., Richards, C., Shishkareva, A., Sinclaire, C., & Barrett, J. (2012). Predictive Markers for Mammoplasty and a Comparison of Side Effect Profiles in Transwomen Taking Various Hormonal Regimens. The Journal of Clinical Endocrinology & Metabolism, 97(12), 4422–4428. [DOI:10.1210/jc.2012-2030]
Seal, L. J. (2019). Cardiovascular disease in transgendered people: A review of the literature and discussion of risk. JRSM Cardiovascular Disease, 8, 204800401988074. [DOI:10.1177/2048004019880745]
Shah, S., Forghani, N., Durham, E., & Neely, E. K. (2014). A randomized trial of transdermal and oral estrogen therapy in adolescent girls with hypogonadism. International Journal of Pediatric Endocrinology, 2014(1), 12. [DOI:10.1186/1687-9856-2014-12]
Silverstein, M. D., Heit, J. A., Mohr, D. N., Petterson, T. M., O’Fallon, W. M., & Melton, L. J. (1998). Trends in the Incidence of Deep Vein Thrombosis and Pulmonary Embolism. Archives of Internal Medicine, 158(6), 585–593. [DOI:10.1001/archinte.158.6.585]
Tebbens, M., Heijboer, A. C., T’Sjoen, G., Bisschop, P. H., & den Heijer, M. (2022). The role of estrone in feminizing hormone treatment. The Journal of Clinical Endocrinology & Metabolism, 107(2), 458–466. [DOI:10.1210/clinem/dgab741]
van de Grift, T. C., Kreukels, B. P., & (2019). Breast development and satisfaction in women with disorders/differences of sex development. Human Reproduction, 34(12), 2410–2417. [DOI:10.1093/humrep/dez230]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2015). Use of combined oral contraceptives and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 350, h2135. [DOI:10.1136/bmj.h2135]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2019). Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 364, k4810. [DOI:10.1136/bmj.k4810]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
Wierckx, K., Van Caenegem, E., Schreiner, T., Haraldsen, I., Fisher, A., Toye, K., Kaufman, J. M., & T’Sjoen, G. (2014). Cross‐Sex Hormone Therapy in Trans Persons Is Safe and Effective at Short‐Time Follow‐Up: Results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 11(8), 1999–2011. [DOI:10.1111/jsm.12571]
Winston-McPherson, G. N., Thomas, T. A., Krasowski, M. D., Ahmed, S. B., Cirrincione, L. R., Katzman, B. M., Pierre, C. C., Rytz, C. L., Turino Miranda, K., Goldstein, Z., & Greene, D. N. (2025). Estradiol Concentrations for Adequate Gender-Affirming Feminizing Therapy: A Systematic Review. LGBT Health. [DOI:10.1089/lgbt.2024.0407]
Wren, B. G., Day, R. O., McLachlan, A. J., & Williams, K. M. (2003). Pharmacokinetics of estradiol, progesterone, testosterone and dehydroepiandrosterone after transbuccal administration to postmenopausal women. Climacteric, 6(2), 104–111. [DOI:10.1080/cmt.6.2.104.111]
\ No newline at end of file
diff --git a/transfemscience.org/articles/pep-cardiovascular-analysis/index.html b/transfemscience.org/articles/pep-cardiovascular-analysis/index.html
index 5457a2ba..d0fc76a0 100644
--- a/transfemscience.org/articles/pep-cardiovascular-analysis/index.html
+++ b/transfemscience.org/articles/pep-cardiovascular-analysis/index.html
@@ -1 +1 @@
-Analysis of Cardiovascular and Thromboembolic Toxicity with High Dose Parenteral Polyestradiol Phosphate in the Treatment of Prostate Cancer - Transfeminine Science
In the late 1980s, there was a renewed interest in the Nordic countries for estrogen therapy as a treatment for prostate cancer. The reason for this was that, in small pilot studies, high doses of polyestradiol phosphate (PEP) administered parenterally did not seem to result in a greater incidence of cardiovascular complications (Henriksson et al., 1988; Stege et al., 1989; Carlström et al., 1989; Henriksson et al., 1999). This finding was in direct contrast to earlier studies that had reported significantly greater incidences of cardiovascular morbidity and mortality in elderly male prostate cancer patients treated with oral ethinylestradiol (EE) and diethylstilbestrol (DES) (Bailar & Byar, 1972; Henriksson & Edhag, 1986).
At sufficient exposure, systemic estrogenic activity has been associated with a greater synthesis of liver proteins, lipids and coagulation factors such as sex-hormone binding globulin (SHBG) and triglycerides (Ottosson et al., 1986). In turn, many of these unphysiologic homeostatic changes are correlated with increased cardiovascular and thromboembolic toxicity (Scarabin, 2014; Seal, 2019; Khan et al., 2019). In addition to pure estrogens, raloxifene usage also has been shown to confer increased risk of venous thromboembolism (Vinogradova, Coupland & Hippisley-Cox, 2019). This is of significance as it was also found in the pilot studies that, while high dose EE therapy greatly increased the synthesis of SHBG and coagulation factor VIII, the effect was significantly less marked with parenteral PEP (Stege et al., 1988; Henriksson et al., 1990). However, both have been shown to induce a significant decrease in antithrombin III; a liver protein that prevents the formation of abnormal blood clots (Aro et al., 1990). By contrast, LHRH agonists do not appear to alter the production of this protein (Varenhorst et al., 1986). These differences were attributed to the disproportionate estrogenic exposure that occurs within the liver relative to other tissues via the oral route (von Schoultz et al., 1989; Ockrim, Lalani & Abel, 2006). Moreover, synthetic estrogens such as EE are resistant to enzymatic liver metabolism due to their augmented or non steroidal chemical structures and cause further disproportionate hepatic receptor activation (Kuhl, 2005). For this reason, intramuscular therapy with PEP; a prodrug for estradiol, was predicted to have much lesser cardiovascular and thromboembolic toxicity than with conventional estrogen therapy (Hedlund, 1999).
During the 1990s and 2000s, several multicenter clinical trials were conducted to establish the efficacy and safety of high dose parenteral estradiol therapy; as compared to bilateral orchiectomy or LHRH agonists (Lycette et al., 2006). The results of these clinical trials are of obvious importance to gender affirming hormone therapy as transgender women usually require long term use of estrogen formulations and a good number choose parenteral forms. It may not be possible to extrapolate these findings to all doses and types of estrogen esters. Nonetheless, these clinical trials give us some idea of the cardiovascular and thromboembolic toxicity of parenteral estradiol. For reference, a 160 to 240 mg/month dose of PEP has been found to produce estradiol levels of between approximately 300 to 500 pg/mL at steady state (Graph). It is obvious from findings that parenteral PEP is associated with a much lesser incidence of adverse cardiovascular and thromboembolic events than was oral EE (Mikkola et al., 1998; Hedlund & Henriksson, 2000; Hedlund et al., 2002). However, it is much less clear if toxicity is eliminated completely. In most of these studies there was inadequate sample power to show if the incidence was changed significantly (Bishop et al., 1989; Lukkarinen & Kontturi, 1994). Nevertheless, one of the largest studies did conclude that parenteral PEP ought not to be used as primary therapy for prostate cancer because of a significant increase in the incidence of such complications (Mikkola et al., 2005; Mikkola et al., 2007). The results of this clinical trial are probably one factor in why the use of high dose estrogen therapy in the treatment of prostate cancer diminished thereafter in many countries.
I thought it would be interesting to bring together all of the data in a pooled meta-analysis. In total, there were six different clinical trials evaluating polyestradiol phosphate. I noted the number of cardiovascular and thromboembolic events in each study for the treatment group exposed to high dose parenteral estradiol and for the comparison group (which had no exposure to estrogens). Unfortunately, one of these clinical trials (FinnProstate-4) did not differentiate between which complications were cardiovascular and which were thromboembolic and so it was not possible to include. There was patient follow-up for a total of 1,890 patients. From this information, I was able to calculate the odds of cardiovascular and thromboembolic complications in patients exposed to therapy for each individual study and for the overall sample. In addition, I calculated a 95% confidence interval. I may turn these findings into part of a more comprehensive article in the future. For now, I have presented them here in the form of some (hopefully useful) infographics (Sources for reference values in Fig. 4 were Vinogradova, Coupland & Hippisley-Cox, 2019 and Langley et al., 2013).
Pooling together the findings of six randomised controlled trials reveals that parenteral estrogen therapy is associated with an increased incidence of cardiovascular and thromboembolic complications; both separately and combined. Nevertheless, due to poor study precision, the exact risk with high doses of parenteral estrogens remains to be accurately quantified.
Figure 1: Odds of CVD with parenteral PEP vs. orchiectomy or LHRH agonist (p = 0.0017; significant at p<0.01).
Figure 2: Odds of VTE with parenteral PEP vs. orchiectomy or LHRH agonist (p = 0.052; significant at p<0.10).
Figure 3: Combined odds of VTE and CVD with parenteral PEP vs. orchiectomy or LHRH agonist (p = 0.0003).
Figure 4: Odds of VTE with parenteral PEP compared to other formulations and doses.
References
Aro, J. L., Haapiainen, R. K., Rannikko, S. A., & Alfthan, O. S. (1989). High Dose Polyoestradiol Phosphate with and without Acetosalicylic Acid versus Orchiectomy in the Treatment of Prostatic Cancer. British Journal of Urology, 63(5), 512–514. [DOI:10.1111/j.1464-410x.1989.tb05946.x]
Aro, J., Haapiainen, R., Rasi, V., Rannikko, S., & Alfthan, O. (1990). The Effect of Parenteral Estrogen versus Orchiectomy on Blood Coagulation and Fibrinolysis in Prostatic Cancer Patients. European Urology, 17(2), 161–165. [DOI:10.1159/000464026]
Aro, J., Ruutu, M., Juusela, H., Hansson, E., & Permi, J. (1993). Polyestradiol phosphate (160 mg/month) or LHRH analog (buserelin depot) in the treatment of locally advanced or metastasized prostatic cancer. The Finnprostate Group. Annales Chirurgiae et Gynaecologiae, 82(Supplementum 206), 5–8. [Google Scholar] [PubMed] [PDF]
Bishop, M. C., Lemberger, R. J., Selby, C., & Lawrence, W. T. (1989). Oestrogen Dosage in Prostatic Cancer: the Threshold Effect? British Journal of Urology, 64(3), 290–296. [DOI:10.1111/j.1464-410x.1989.tb06016.x]
Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., Stege, R., & von Schoultz, B. (1989). A comparison of androgen status in patients with prostatic cancer treated with oral and/or parenteral estrogens or by orchidectomy. The Prostate, 14(2), 177–182. [DOI:10.1002/pros.2990140210]
Haapiainen, R., Rannikko, S., & Alfthan, O. (1990). Comparison of Primary Orchiectomy and Polyoestradiol Phosphate in the Treatment of Advanced Prostatic Cancer. British Journal of Urology, 66(1), 94–97. [DOI:10.1111/j.1464-410x.1990.tb14872.x]
Hedlund, P. O. (1999). The Scandinavian Prostatic Cancer Group: A Short Review of its History and Work. Scandinavian Journal of Urology and Nephrology, 33(203), 53–56. [DOI:10.1080/003655999750016285-2]
Hedlund, P. O., & Henriksson, P. (2000). Parenteral estrogen versus total androgen ablation in the treatment of advanced prostate carcinoma: effects on overall survival and cardiovascular mortality. Urology, 55(3), 328–332. [DOI:10.1016/s0090-4295(99)00580-4]
Hedlund, P. O., Ala-Opas, M., Brekkan, E., Damber, J. E., Damber, L., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Pousette, Å., Rasmussen, F., Salo, J., Vaage, S., & Varenhorst, E. (2002). Parenteral Estrogen versus Combined Androgen Deprivation in the Treatment of Metastatic Prostatic Cancer - Scandinavian Prostatic Cancer Group (SPCG) Study No. 5. Scandinavian Journal of Urology and Nephrology, 36(6), 405–413. [DOI:10.1080/003655902762467549]
Hedlund, P. O., Damber, J., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Johansson, R., Klarskov, P., Lundbeck, F., Rasmussen, F., Varenhorst, E., Viitanen, J., Olov Hedlund, P., Damber, J., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Johansson, R., Klarskov, P., Lundbeck, F., Rasmussen, F., Varenhorst, E., Viitanen, J., , & (2008). Parenteral estrogen versus combined androgen deprivation in the treatment of metastatic prostatic cancer: Part 2. Final evaluation of the Scandinavian Prostatic Cancer Group (SPCG) Study No. 5. Scandinavian Journal of Urology and Nephrology, 42(3), 220–229. [DOI:10.1080/00365590801943274]
Hedlund, P. O., Johansson, R., Damber, J. E., Hagerman, I., Henriksson, P., Iversen, P., Klarskov, P., Mogensen, P., Rasmussen, F., & Varenhorst, E. (2011). Significance of pretreatment cardiovascular morbidity as a risk factor during treatment with parenteral oestrogen or combined androgen deprivation of 915 patients with metastasized prostate cancer: Evaluation of cardiovascular events in a randomized trial. Scandinavian Journal of Urology and Nephrology, 45(5), 346–353. [DOI:10.3109/00365599.2011.585820]
Henriksson, P., & Edhag, O. (1986). Orchidectomy versus oestrogen for prostatic cancer: cardiovascular effects. BMJ, 293(6544), 413–415. [DOI:10.1136/bmj.293.6544.413]
Henriksson, P., Eriksson, A., Stege, R., Collste, L., Pousette, Å., Von Schoultz, B., & Carlström, K. (1988). Cardiovascular follow-up of patients with prostatic cancer treated with single-drug polyestradiol phosphate. The Prostate, 13(3), 257–261. [DOI:10.1002/pros.2990130308]
Henriksson, P., Blombäck, M., Eriksson, A., Stege, R., & Carlström, K. (1990). Effect of Parenteral Oestrogen on the Coagulation System in Patients with Prostatic Carcinoma. British Journal of Urology, 65(3), 282–285. [DOI:10.1111/j.1464-410x.1990.tb14728.x]
Henriksson, P., Carlström, K., Pousette, A., Gunnarsson, P. O., Johansson, C. J., Eriksson, B., Altersgård-Brorsson, A. K., Nordle, O., & Stege, R. (1999). Time for revival of estrogens in the treatment of advanced prostatic carcinoma? Pharmacokinetics, and endocrine and clinical effects, of a parenteral estrogen regimen. The Prostate, 40(2), 76–82. [DOI:10.1002/(SICI)1097-0045(19990701)40:2<76::AID-PROS2>3.0.CO;2-Q]
Jacobi, G. H., Altwein, J. E., Kurth, K. H., Basting, R., & Hohenfellner, R. (1980). Treatment of Advanced Prostatic Cancer with Parenteral Cyproterone Acetate: A Phase III Randomised Trial. British Journal of Urology, 52(3), 208–215. [DOI:10.1111/j.1464-410x.1980.tb02961.x]
Jacobi, G. H., Tunn, U., & Senge, T. (1982). Clinical experience with cyproterone acetate for palliation of inoperable prostate cancer. In Jacobi, G. H., & Hohenfellner, R. (Eds.). Prostate Cancer (International Perspectives in Urology, Volume 3) (pp. 305–319). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Khan, J., Schmidt, R. L., Spittal, M. J., Goldstein, Z., Smock, K. J., & Greene, D. N. (2019). Venous Thrombotic Risk in Transgender Women Undergoing Estrogen Therapy: A Systematic Review and Metaanalysis. Clinical Chemistry, 65(1), 57–66. [DOI:10.1373/clinchem.2018.288316]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Langley, R. E., Cafferty, F. H., Alhasso, A. A., Rosen, S. D., Sundaram, S. K., Freeman, S. C., Pollock, P., Jinks, R. C., Godsland, I. F., Kockelbergh, R., Clarke, N. W., Kynaston, H. G., Parmar, M. K., & Abel, P. D. (2013). Cardiovascular outcomes in patients with locally advanced and metastatic prostate cancer treated with luteinising-hormone-releasing-hormone agonists or transdermal oestrogen: the randomised, phase 2 MRC PATCH trial (PR09). The Lancet Oncology, 14(4), 306–316. [DOI:10.1016/s1470-2045(13)70025-1]
Lukkarinen, O., Kontturi, M., & (1994). Comparison of a Long-Acting LHRH Agonist and Polyoestradiol Phosphate in the Treatment of Advanced Prostatic Carcinoma. Scandinavian Journal of Urology and Nephrology, 28(2), 171–178. [DOI:10.3109/00365599409180495]
Lycette, J. L., Bland, L. B., Garzotto, M., & Beer, T. M. (2006). Parenteral Estrogens for Prostate Cancer: Can a New Route of Administration Overcome Old Toxicities? Clinical Genitourinary Cancer, 5(3), 198–205. [DOI:10.3816/cgc.2006.n.037]
Mikkola, A. K., Ruutu, M. L., Aro, J. L., Rannikko, S. A., Salo, J. O., & the Finnprostate Group. (1998). Parenteral polyoestradiol phosphate vs orchidectomy in the treatment of advanced prostatic cancer. Efficacy and cardiovascular complications: a 2-year follow-up report of a national, prospective prostatic cancer study. BJU International, 82(1), 63–68. [DOI:10.1046/j.1464-410x.1998.00688.x]
Mikkola, A., Aro, J., Rannikko, S., Oksanen, H., Ruutu, M., & (2005). Cardiovascular complications in patients with advanced prostatic cancer treated by means of orchiectomy or polyestradiol phosphate. Scandinavian Journal of Urology and Nephrology, 39(4), 294–300. [DOI:10.1080/00365590510031228]
Mikkola, A., Aro, J., Rannikko, S., Ruutu, M., & (2007). Ten-year survival and cardiovascular mortality in patients with advanced prostate cancer primarily treated by intramuscular polyestradiol phosphate or orchiectomy. The Prostate, 67(4), 447–455. [DOI:10.1002/pros.20547]
Namer, M. (1988). Clinical applications of antiandrogens. Journal of Steroid Biochemistry, 31(4), 719–729. [DOI:10.1016/0022-4731(88)90023-4]
Ockrim, J., Lalani, E., & Abel, P. (2006). Therapy Insight: parenteral estrogen treatment for prostate cancer—a new dawn for an old therapy. Nature Clinical Practice Oncology, 3(10), 552–563. [DOI:10.1038/ncponc0602]
Ottosson, U., Carlström, K., Johansson, B., & von Schoultz, B. (1986). Estrogen Induction of Liver Proteins and High-Density Lipoprotein Cholesterol: Comparison between Estradiol Valerate and Ethinyl Estradiol. Gynecologic and Obstetric Investigation, 22(4), 198–205. [DOI:10.1159/000298914]
Scarabin, P. (2014). Hormone Therapy and Venous Thromboembolism among Postmenopausal Women. Frontiers of Hormone Research, 43, 21–32. / Granata, R., & Isgaard, J. (Eds.). Cardiovascular Issues in Endocrinology (Frontiers of Hormone Research, Volume 43) (pp. 21–32). Basel: Karger. [DOI:10.1159/000360554] [Google Books]
Schröder, F. H., & Radlmaier, A. (2002). Steroidal Antiandrogens. In Jordan, C. V., & Furr, B. J. A. (Eds.). Hormone Therapy in Breast and Prostate Cancer (pp. 325–346). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-59259-152-7_15]
Seal, L. J. (2019). Cardiovascular disease in transgendered people: A review of the literature and discussion of risk. JRSM Cardiovascular Disease, 8, 204800401988074. [DOI:10.1177/2048004019880745]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & von Schoultz, B. (1989). Single-drug parenteral estrogen treatment in prostatic cancer: A study of two maintenance-dose regimens. The Prostate, 14(2), 183–188. [DOI:10.1002/pros.2990140211]
Tunn, U. W., Radlmaier, A., & Neumann, F. (1988). Antiandrogens in Cancer Treatment. In Stoll, B. A. (Ed.). Endocrine Management of Cancer: Contemporary Therapy (Contemporary Therapy, Volume 2) (pp. 43–56). Basel: Karger. [DOI:10.1159/000415355]
Varenhorst, E., Svensson, M., Hjertberg, H., & Malmqvist, E. (1986). Antithrombin III concentration, thrombosis, and treatment with luteinising hormone releasing hormone agonist in prostatic carcinoma. BMJ, 292(6525), 935–936. [DOI:10.1136/bmj.292.6525.935]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2019). Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 364, k4810. [DOI:10.1136/bmj.k4810]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
Wenderoth, U. K., & Jacobi, G. H. (1983). Gonadotropin-releasing hormone analogues for palliation of carcinoma of the prostate. World Journal of Urology, 1(1), 40–48. [DOI:10.1007/bf00326861]
\ No newline at end of file
+Analysis of Cardiovascular and Thromboembolic Toxicity with High Dose Parenteral Polyestradiol Phosphate in the Treatment of Prostate Cancer - Transfeminine Science
In the late 1980s, there was a renewed interest in the Nordic countries for estrogen therapy as a treatment for prostate cancer. The reason for this was that, in small pilot studies, high doses of polyestradiol phosphate (PEP) administered parenterally did not seem to result in a greater incidence of cardiovascular complications (Henriksson et al., 1988; Stege et al., 1989; Carlström et al., 1989; Henriksson et al., 1999). This finding was in direct contrast to earlier studies that had reported significantly greater incidences of cardiovascular morbidity and mortality in elderly male prostate cancer patients treated with oral ethinylestradiol (EE) and diethylstilbestrol (DES) (Bailar & Byar, 1972; Henriksson & Edhag, 1986).
At sufficient exposure, systemic estrogenic activity has been associated with a greater synthesis of liver proteins, lipids and coagulation factors such as sex-hormone binding globulin (SHBG) and triglycerides (Ottosson et al., 1986). In turn, many of these unphysiologic homeostatic changes are correlated with increased cardiovascular and thromboembolic toxicity (Scarabin, 2014; Seal, 2019; Khan et al., 2019). In addition to pure estrogens, raloxifene usage also has been shown to confer increased risk of venous thromboembolism (Vinogradova, Coupland & Hippisley-Cox, 2019). This is of significance as it was also found in the pilot studies that, while high dose EE therapy greatly increased the synthesis of SHBG and coagulation factor VIII, the effect was significantly less marked with parenteral PEP (Stege et al., 1988; Henriksson et al., 1990). However, both have been shown to induce a significant decrease in antithrombin III; a liver protein that prevents the formation of abnormal blood clots (Aro et al., 1990). By contrast, LHRH agonists do not appear to alter the production of this protein (Varenhorst et al., 1986). These differences were attributed to the disproportionate estrogenic exposure that occurs within the liver relative to other tissues via the oral route (von Schoultz et al., 1989; Ockrim, Lalani & Abel, 2006). Moreover, synthetic estrogens such as EE are resistant to enzymatic liver metabolism due to their augmented or non steroidal chemical structures and cause further disproportionate hepatic receptor activation (Kuhl, 2005). For this reason, intramuscular therapy with PEP; a prodrug for estradiol, was predicted to have much lesser cardiovascular and thromboembolic toxicity than with conventional estrogen therapy (Hedlund, 1999).
During the 1990s and 2000s, several multicenter clinical trials were conducted to establish the efficacy and safety of high dose parenteral estradiol therapy; as compared to bilateral orchiectomy or LHRH agonists (Lycette et al., 2006). The results of these clinical trials are of obvious importance to gender affirming hormone therapy as transgender women usually require long term use of estrogen formulations and a good number choose parenteral forms. It may not be possible to extrapolate these findings to all doses and types of estrogen esters. Nonetheless, these clinical trials give us some idea of the cardiovascular and thromboembolic toxicity of parenteral estradiol. For reference, a 160 to 240 mg/month dose of PEP has been found to produce estradiol levels of between approximately 300 to 500 pg/mL at steady state (Graph). It is obvious from findings that parenteral PEP is associated with a much lesser incidence of adverse cardiovascular and thromboembolic events than was oral EE (Mikkola et al., 1998; Hedlund & Henriksson, 2000; Hedlund et al., 2002). However, it is much less clear if toxicity is eliminated completely. In most of these studies there was inadequate sample power to show if the incidence was changed significantly (Bishop et al., 1989; Lukkarinen & Kontturi, 1994). Nevertheless, one of the largest studies did conclude that parenteral PEP ought not to be used as primary therapy for prostate cancer because of a significant increase in the incidence of such complications (Mikkola et al., 2005; Mikkola et al., 2007). The results of this clinical trial are probably one factor in why the use of high dose estrogen therapy in the treatment of prostate cancer diminished thereafter in many countries.
I thought it would be interesting to bring together all of the data in a pooled meta-analysis. In total, there were six different clinical trials evaluating polyestradiol phosphate. I noted the number of cardiovascular and thromboembolic events in each study for the treatment group exposed to high dose parenteral estradiol and for the comparison group (which had no exposure to estrogens). Unfortunately, one of these clinical trials (FinnProstate-4) did not differentiate between which complications were cardiovascular and which were thromboembolic and so it was not possible to include. There was patient follow-up for a total of 1,890 patients. From this information, I was able to calculate the odds of cardiovascular and thromboembolic complications in patients exposed to therapy for each individual study and for the overall sample. In addition, I calculated a 95% confidence interval. I may turn these findings into part of a more comprehensive article in the future. For now, I have presented them here in the form of some (hopefully useful) infographics (Sources for reference values in Fig. 4 were Vinogradova, Coupland & Hippisley-Cox, 2019 and Langley et al., 2013).
Pooling together the findings of six randomised controlled trials reveals that parenteral estrogen therapy is associated with an increased incidence of cardiovascular and thromboembolic complications; both separately and combined. Nevertheless, due to poor study precision, the exact risk with high doses of parenteral estrogens remains to be accurately quantified.
Figure 1: Odds of CVD with parenteral PEP vs. orchiectomy or LHRH agonist (p = 0.0017; significant at p<0.01).
Figure 2: Odds of VTE with parenteral PEP vs. orchiectomy or LHRH agonist (p = 0.052; significant at p<0.10).
Figure 3: Combined odds of VTE and CVD with parenteral PEP vs. orchiectomy or LHRH agonist (p = 0.0003).
Figure 4: Odds of VTE with parenteral PEP compared to other formulations and doses.
References
Aro, J. L., Haapiainen, R. K., Rannikko, S. A., & Alfthan, O. S. (1989). High Dose Polyoestradiol Phosphate with and without Acetosalicylic Acid versus Orchiectomy in the Treatment of Prostatic Cancer. British Journal of Urology, 63(5), 512–514. [DOI:10.1111/j.1464-410x.1989.tb05946.x]
Aro, J., Haapiainen, R., Rasi, V., Rannikko, S., & Alfthan, O. (1990). The Effect of Parenteral Estrogen versus Orchiectomy on Blood Coagulation and Fibrinolysis in Prostatic Cancer Patients. European Urology, 17(2), 161–165. [DOI:10.1159/000464026]
Aro, J., Ruutu, M., Juusela, H., Hansson, E., & Permi, J. (1993). Polyestradiol phosphate (160 mg/month) or LHRH analog (buserelin depot) in the treatment of locally advanced or metastasized prostatic cancer. The Finnprostate Group. Annales Chirurgiae et Gynaecologiae, 82(Supplementum 206), 5–8. [Google Scholar] [PubMed] [PDF]
Bishop, M. C., Lemberger, R. J., Selby, C., & Lawrence, W. T. (1989). Oestrogen Dosage in Prostatic Cancer: the Threshold Effect? British Journal of Urology, 64(3), 290–296. [DOI:10.1111/j.1464-410x.1989.tb06016.x]
Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., Stege, R., & von Schoultz, B. (1989). A comparison of androgen status in patients with prostatic cancer treated with oral and/or parenteral estrogens or by orchidectomy. The Prostate, 14(2), 177–182. [DOI:10.1002/pros.2990140210]
Haapiainen, R., Rannikko, S., & Alfthan, O. (1990). Comparison of Primary Orchiectomy and Polyoestradiol Phosphate in the Treatment of Advanced Prostatic Cancer. British Journal of Urology, 66(1), 94–97. [DOI:10.1111/j.1464-410x.1990.tb14872.x]
Hedlund, P. O. (1999). The Scandinavian Prostatic Cancer Group: A Short Review of its History and Work. Scandinavian Journal of Urology and Nephrology, 33(203), 53–56. [DOI:10.1080/003655999750016285-2]
Hedlund, P. O., & Henriksson, P. (2000). Parenteral estrogen versus total androgen ablation in the treatment of advanced prostate carcinoma: effects on overall survival and cardiovascular mortality. Urology, 55(3), 328–332. [DOI:10.1016/s0090-4295(99)00580-4]
Hedlund, P. O., Ala-Opas, M., Brekkan, E., Damber, J. E., Damber, L., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Pousette, Å., Rasmussen, F., Salo, J., Vaage, S., & Varenhorst, E. (2002). Parenteral Estrogen versus Combined Androgen Deprivation in the Treatment of Metastatic Prostatic Cancer - Scandinavian Prostatic Cancer Group (SPCG) Study No. 5. Scandinavian Journal of Urology and Nephrology, 36(6), 405–413. [DOI:10.1080/003655902762467549]
Hedlund, P. O., Damber, J., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Johansson, R., Klarskov, P., Lundbeck, F., Rasmussen, F., Varenhorst, E., Viitanen, J., Olov Hedlund, P., Damber, J., Hagerman, I., Haukaas, S., Henriksson, P., Iversen, P., Johansson, R., Klarskov, P., Lundbeck, F., Rasmussen, F., Varenhorst, E., Viitanen, J., , & (2008). Parenteral estrogen versus combined androgen deprivation in the treatment of metastatic prostatic cancer: Part 2. Final evaluation of the Scandinavian Prostatic Cancer Group (SPCG) Study No. 5. Scandinavian Journal of Urology and Nephrology, 42(3), 220–229. [DOI:10.1080/00365590801943274]
Hedlund, P. O., Johansson, R., Damber, J. E., Hagerman, I., Henriksson, P., Iversen, P., Klarskov, P., Mogensen, P., Rasmussen, F., & Varenhorst, E. (2011). Significance of pretreatment cardiovascular morbidity as a risk factor during treatment with parenteral oestrogen or combined androgen deprivation of 915 patients with metastasized prostate cancer: Evaluation of cardiovascular events in a randomized trial. Scandinavian Journal of Urology and Nephrology, 45(5), 346–353. [DOI:10.3109/00365599.2011.585820]
Henriksson, P., & Edhag, O. (1986). Orchidectomy versus oestrogen for prostatic cancer: cardiovascular effects. BMJ, 293(6544), 413–415. [DOI:10.1136/bmj.293.6544.413]
Henriksson, P., Eriksson, A., Stege, R., Collste, L., Pousette, Å., Von Schoultz, B., & Carlström, K. (1988). Cardiovascular follow-up of patients with prostatic cancer treated with single-drug polyestradiol phosphate. The Prostate, 13(3), 257–261. [DOI:10.1002/pros.2990130308]
Henriksson, P., Blombäck, M., Eriksson, A., Stege, R., & Carlström, K. (1990). Effect of Parenteral Oestrogen on the Coagulation System in Patients with Prostatic Carcinoma. British Journal of Urology, 65(3), 282–285. [DOI:10.1111/j.1464-410x.1990.tb14728.x]
Henriksson, P., Carlström, K., Pousette, A., Gunnarsson, P. O., Johansson, C. J., Eriksson, B., Altersgård-Brorsson, A. K., Nordle, O., & Stege, R. (1999). Time for revival of estrogens in the treatment of advanced prostatic carcinoma? Pharmacokinetics, and endocrine and clinical effects, of a parenteral estrogen regimen. The Prostate, 40(2), 76–82. [DOI:10.1002/(SICI)1097-0045(19990701)40:2<76::AID-PROS2>3.0.CO;2-Q]
Jacobi, G. H., Altwein, J. E., Kurth, K. H., Basting, R., & Hohenfellner, R. (1980). Treatment of Advanced Prostatic Cancer with Parenteral Cyproterone Acetate: A Phase III Randomised Trial. British Journal of Urology, 52(3), 208–215. [DOI:10.1111/j.1464-410x.1980.tb02961.x]
Jacobi, G. H., Tunn, U., & Senge, T. (1982). Clinical experience with cyproterone acetate for palliation of inoperable prostate cancer. In Jacobi, G. H., & Hohenfellner, R. (Eds.). Prostate Cancer (International Perspectives in Urology, Volume 3) (pp. 305–319). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Khan, J., Schmidt, R. L., Spittal, M. J., Goldstein, Z., Smock, K. J., & Greene, D. N. (2019). Venous Thrombotic Risk in Transgender Women Undergoing Estrogen Therapy: A Systematic Review and Metaanalysis. Clinical Chemistry, 65(1), 57–66. [DOI:10.1373/clinchem.2018.288316]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Langley, R. E., Cafferty, F. H., Alhasso, A. A., Rosen, S. D., Sundaram, S. K., Freeman, S. C., Pollock, P., Jinks, R. C., Godsland, I. F., Kockelbergh, R., Clarke, N. W., Kynaston, H. G., Parmar, M. K., & Abel, P. D. (2013). Cardiovascular outcomes in patients with locally advanced and metastatic prostate cancer treated with luteinising-hormone-releasing-hormone agonists or transdermal oestrogen: the randomised, phase 2 MRC PATCH trial (PR09). The Lancet Oncology, 14(4), 306–316. [DOI:10.1016/s1470-2045(13)70025-1]
Lukkarinen, O., Kontturi, M., & (1994). Comparison of a Long-Acting LHRH Agonist and Polyoestradiol Phosphate in the Treatment of Advanced Prostatic Carcinoma. Scandinavian Journal of Urology and Nephrology, 28(2), 171–178. [DOI:10.3109/00365599409180495]
Lycette, J. L., Bland, L. B., Garzotto, M., & Beer, T. M. (2006). Parenteral Estrogens for Prostate Cancer: Can a New Route of Administration Overcome Old Toxicities? Clinical Genitourinary Cancer, 5(3), 198–205. [DOI:10.3816/cgc.2006.n.037]
Mikkola, A. K., Ruutu, M. L., Aro, J. L., Rannikko, S. A., Salo, J. O., & the Finnprostate Group. (1998). Parenteral polyoestradiol phosphate vs orchidectomy in the treatment of advanced prostatic cancer. Efficacy and cardiovascular complications: a 2-year follow-up report of a national, prospective prostatic cancer study. BJU International, 82(1), 63–68. [DOI:10.1046/j.1464-410x.1998.00688.x]
Mikkola, A., Aro, J., Rannikko, S., Oksanen, H., Ruutu, M., & (2005). Cardiovascular complications in patients with advanced prostatic cancer treated by means of orchiectomy or polyestradiol phosphate. Scandinavian Journal of Urology and Nephrology, 39(4), 294–300. [DOI:10.1080/00365590510031228]
Mikkola, A., Aro, J., Rannikko, S., Ruutu, M., & (2007). Ten-year survival and cardiovascular mortality in patients with advanced prostate cancer primarily treated by intramuscular polyestradiol phosphate or orchiectomy. The Prostate, 67(4), 447–455. [DOI:10.1002/pros.20547]
Namer, M. (1988). Clinical applications of antiandrogens. Journal of Steroid Biochemistry, 31(4), 719–729. [DOI:10.1016/0022-4731(88)90023-4]
Ockrim, J., Lalani, E., & Abel, P. (2006). Therapy Insight: parenteral estrogen treatment for prostate cancer—a new dawn for an old therapy. Nature Clinical Practice Oncology, 3(10), 552–563. [DOI:10.1038/ncponc0602]
Ottosson, U., Carlström, K., Johansson, B., & von Schoultz, B. (1986). Estrogen Induction of Liver Proteins and High-Density Lipoprotein Cholesterol: Comparison between Estradiol Valerate and Ethinyl Estradiol. Gynecologic and Obstetric Investigation, 22(4), 198–205. [DOI:10.1159/000298914]
Scarabin, P. (2014). Hormone Therapy and Venous Thromboembolism among Postmenopausal Women. Frontiers of Hormone Research, 43, 21–32. / Granata, R., & Isgaard, J. (Eds.). Cardiovascular Issues in Endocrinology (Frontiers of Hormone Research, Volume 43) (pp. 21–32). Basel: Karger. [DOI:10.1159/000360554] [Google Books]
Schröder, F. H., & Radlmaier, A. (2002). Steroidal Antiandrogens. In Jordan, C. V., & Furr, B. J. A. (Eds.). Hormone Therapy in Breast and Prostate Cancer (pp. 325–346). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-59259-152-7_15]
Seal, L. J. (2019). Cardiovascular disease in transgendered people: A review of the literature and discussion of risk. JRSM Cardiovascular Disease, 8, 204800401988074. [DOI:10.1177/2048004019880745]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & von Schoultz, B. (1989). Single-drug parenteral estrogen treatment in prostatic cancer: A study of two maintenance-dose regimens. The Prostate, 14(2), 183–188. [DOI:10.1002/pros.2990140211]
Tunn, U. W., Radlmaier, A., & Neumann, F. (1988). Antiandrogens in Cancer Treatment. In Stoll, B. A. (Ed.). Endocrine Management of Cancer: Contemporary Therapy (Contemporary Therapy, Volume 2) (pp. 43–56). Basel: Karger. [DOI:10.1159/000415355]
Varenhorst, E., Svensson, M., Hjertberg, H., & Malmqvist, E. (1986). Antithrombin III concentration, thrombosis, and treatment with luteinising hormone releasing hormone agonist in prostatic carcinoma. BMJ, 292(6525), 935–936. [DOI:10.1136/bmj.292.6525.935]
Vinogradova, Y., Coupland, C., & Hippisley-Cox, J. (2019). Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases. BMJ, 364, k4810. [DOI:10.1136/bmj.k4810]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
Wenderoth, U. K., & Jacobi, G. H. (1983). Gonadotropin-releasing hormone analogues for palliation of carcinoma of the prostate. World Journal of Urology, 1(1), 40–48. [DOI:10.1007/bf00326861]
\ No newline at end of file
diff --git a/transfemscience.org/articles/progestogens-breast-cancer/index.html b/transfemscience.org/articles/progestogens-breast-cancer/index.html
index 6ff04131..83d53b29 100644
--- a/transfemscience.org/articles/progestogens-breast-cancer/index.html
+++ b/transfemscience.org/articles/progestogens-breast-cancer/index.html
@@ -1 +1 @@
-Progestogens and Breast Cancer in Transgender Women: A Review and Discussion of Risk - Transfeminine Science
Progestogens and Breast Cancer in Transgender Women: A Review and Discussion of Risk
By Sam | First published February 14, 2020 | Last modified October 5, 2020
Introduction
Breast cancer is the most common invasive cancer in women. The worldwide incidence in this group is about 1 in 8 (Balasubramanian et al., 2019; McGuire et al., 2015). There are likely many genetic, hormonal and lifestyle factors that influence risk. For instance, breast cancer is strongly associated with age. Indeed, only 7% of breast cancer cases have been found to be in women under 40 years old (Anders et al., 2009).
Sex steroids have profound influences on the breast. Mammary gland development begins during embryogenesis and marked differentiation takes place in the adult female at puberty and during pregnancy as a consequence of such influences (Javed et al., 2013; Sun et al., 2018; Leitch et al., 2018). Estrogens such as estradiol, in tandem with growth factors, promote morphological breast development at puberty while androgens inhibit (Hynes et al., 2010; Dimitrakakis et al., 2009). Moreover, estradiol and progesterone act synergistically to induce lobuloalveolar development in preparation for lactation. Owing to the effect of sex steroids, there is a positive relationship between breast development and breast cancer (Folkerd et al., 2013; Rezvanpour et al., 2016). In accordance, it is well established in large epidemiological studies that use of estrogen medications are strongly associated with an increased risk of breast cancer relative to non-use (Kotsopoulos, 2019). Women with larger breasts and hence more breast tissue are also apparently at a greater risk for breast cancer (Jansen et al., 2014).
Some patients and clinicians believe that progesterone may have benefits to breast development or otherwise. However, the use of progesterone in transfeminine hormone therapy for these hypothetical benefits is controversial and not evidence-based (Hembree et al., 2017; Wierckx et al., 2014; Randolph, 2018). It is sometimes argued that because these benefits are unsupported and usage is associated with an increased risk of cancer, progestogens (and specifically progesterone) ought not to be indicated for therapy in transgender women (Coxon et al., 2018). I decided to evaluate the strength of this claim in a short literature review.
Predictive Endocrine Markers of Breast Cancer
Oophorectomy (removal of the ovaries) can be used both as effective active treatment and prophylaxis against breast cancer (Singh, 2012; Moscucci et al., 2007). A large meta-analysis of over 100 epidemiological studies determined that both younger age at menarche and older age at menopause were associated with a greater risk of breast cancer (CGHFBC, 2012). While it is not clear how much this risk is attributable to estrogens vs progestogens, these findings are definitive evidence that breast cancer risk is linked to and enhanced by exposure to ovarian steroids.
High mammographic density is thought to be an important risk factor for breast cancer (Boyd et al., 2011; McCormack et al., 2006). Owing to this, it is interesting to note that use of hormone replacement therapy at the menopause is associated with both higher breast density and a higher incidence of breast cancer (Azam et al., 2018; CGHFBC, 2019). However, evidence suggesting association between sex hormones and breast cancer is conflicting. One study found no association between levels of free or total estradiol and breast density, but a positive association between levels of progesterone. (Sprague et al., 2010). Contrariwise, other studies reported no association between estradiol and progesterone and a negative association between estradiol and breast density, respectively (Jung et al., 2015; Tamimi et al., 2005). A review reported that in therapy with estrogens plus oral progesterone, breast density was increased significantly in three studies but unchanged in two other studies (Mirkin, 2018). Even though mammographic density is an important marker of breast cancer, the effects of sex steroids on this variable appear to be highly inconsistent between different studies. There may not be much to be learned from these findings.
Epidemiological Data
The risk of breast cancer with different estrogen and progestogen medications in menopausal hormone therapy has also been directly assessed by high-quality meta-analysis and with large sample sizes in the E3N/E3N-EPIC studies (CGHFBC, 2019; Fournier et al., 2014). In these trials, women using estradiol in combination with the synthetic progestins such as medroxyprogesterone acetate and chlormadinone acetate, have been found to have significantly greater rates of breast cancer relative to estradiol alone (Fournier et al., 2008; Lambrinoudaki, 2014). Conversely, in these large observational studies, the use of estradiol in combination with oral micronised progesterone has not been associated with a higher risk of breast cancer in the short term.
On the basis of these findings, it has been claimed that micronized progesterone differs from synthetic progestins in that it is not associated with a higher risk of developing breast cancer (Regidor, 2014; Yang et al., 2016; Rymer et al., 2019). But this does not appear to be the case. In the E3N-EPIC cohort, the relative risks (RR) [95% confidence interval (CI)] of breast cancer with transdermal estrogens and oral micronised progesterone were 0.9 [0.6–1.4], 0.7 [0.4–1.2] and 1.2 [0.7–2.0] at less than 2 years, between 2 to 4 years and at over 4 years of use, respectively (Mirkin, 2018). However, in contrast to these short-term findings, long-term observational studies have consistently found the use of micronized oral progesterone to be associated with higher breast cancer risk. In the same cohort, the relative risks of estrogens plus oral progesterone were reported between 4 to 6 years and beyond 6 years as 1.26 [0.87–1.82] and 1.22 [0.89–1.67], respectively (Fournier et al., 2008). At the conclusion of this study, the relative risks for estrogen plus oral progesterone were determined to be 1.13 [0.99–1.29] and 1.31 [1.15–1.48] below 5 years and at and beyond 5 years (Fournier et al., 2014). A recent systematic review concluded, on the basis of these findings, that breast cancer risk is not increased in the short-term by oral progesterone in combination with estrogens, but that long-term exposure has greater risk (Stute et al., 2018). The more recent CGHFBC meta-analysis determined that relative risk for estrogens plus oral progesterone was 0.91 [0.47–1.78] at less 5 years of use (CGHFBC, 2019; Table). Yet, in the long term, after 5 to 14 years of exposure, the risk was increased to 2.05 [1.38–3.06] and hence as high as medroxyprogesterone acetate and other oral progestins (2.07 [1.96–2.19]). There is no adequately powered evidence to demonstrate that estrogens plus micronized progesterone is associated with a lower incidence of breast cancer.
The notion that micronized progesterone does not increase the risk of breast cancer had been criticised before the long-term follow up of the E3N cohort and CGHFBC meta-analysis (Kuhl et al., 2013). It is important to consider the pharmacology of oral progesterone. Unlike with the synthetic progestins, this route of administration is associated with rapid metabolism and marked inactivation of the active medication into metabolites of progesterone by the gastrointestinal tract (Lobo et al., 2019; Levine et al., 2001). As a result, bioavailability is very low and, in accordance, oral progesterone is considerably less potent than the oral progestins (Davey et al., 2018; Kuhl, 2005). Taking the fact that it is far less potent into consideration, it would seem logical to assume as a default null hypothesis that breast cancer takes longer to appear and may be less common with oral progesterone (Kuhl et al., 2013). It is difficult to rationalise why all these other progestins, in spite of their different structures and biochemistry, would increase the incidence of breast cancer while micronized progesterone would not or have even an opposing effect. Conversely, other parenteral routes of progesterone such as vaginal or rectal administration have considerably higher bioavailability and more potent systematic effects (Kuhl, 2005). These non-oral routes likely have a similar incidence of breast cancer to those of the synthetic progestins.
In the CGHFBC meta-analysis, it was found that bioidentical estradiol has a similar, if not greater, incidence to conjugated (and therefore non-bioidentical) estrogens (1.78 (1.58–1.99) vs 1.68 [1.57–1.80]) (CGHFBC, 2019). Therefore, the rate of breast cancer with bioidentical estrogen usage is not significantly lower than with non-bioidentical estrogen usage. This is of note because, unlike oral progesterone and its synthetic derivatives, oral estradiol can have similar potency to these conjugated estrogens at practical clinical doses (Kuhl, 2005). One might expect the same to be true of bioidentical and non-identical progestogens at comparable potencies. The ELITE trial reported that there were more cases of breast cancer in a group of women using estradiol and relatively low-dose vaginal progesterone (45 mg/day) versus controls (Hodis et al., 2016). However, as the sample sizes were not large enough, it is not possible to attribute the higher rate of breast cancer to parenteral progesterone (Stute et al., 2018). Higher quality evidence is necessary to confirm, beyond a shadow of a doubt, that parenteral progesterone does in fact have comparable breast cancer risk to oral progestins.
Breast Cancer in Transgender Women
There is a positive association between breast cancer risk and a greater volume of breast tissue (Jansen et al., 2014; Eriksson et al., 2012). As transgender women develop breast tissue from hormone therapy, one might expect to find that their breast cancer rate would also increase. Surprisingly, however, the findings of epidemiological studies investigating the rate of breast cancer in transgender women are conflicting. Two large cohort studies (n = 2,307 and n = 1,259, respectively) reported no greater risk relative to cisgender men (Gooren et al., 2013a; Brown et al., 2014). However, another more recent study (n = 2,260) found a rate of breast cancer in transgender women that was between that of cisgender men and cisgender women (de Blok et al., 2019). The reason for this enormous difference in findings is unclear. A recent review found that there were just 20 cases of breast cancer in transgender women reported in the literature and hence almost as many as in transgender men (Hartley et al., 2018). It is of note that another review found only 7 reported cases of prostate cancer in transgender women (Gooren et al., 2013b). In accordance, prostate cancer seems to be an exceptionally rare diagnosis. One could argue on this basis, that the occurrence of breast cancer in transgender women is possibly also very rare.
Nonetheless, we should probably take the available evidence with some scepticism. It is well-known breast cancer takes many years to appear; after many decades of hormone exposure. Data from SEER in 2004 found that the incidences of breast cancer in women at 30, 40 and 50 years of age were only 1 in 1,523, 1 in 173 and 1 in 45, respectively (Anders et al., 2009). Given the secretion of ovarian steroid hormones begins at about 10-years-old in cisgender females, by the age of 50 these women have had about 40 years of exposure. But the follow-up times in these epidemiological studies of transgender women are very short in comparison (typically less than 20 years) and, following a similar timeframe of hormone exposure, breast cancer risk may be higher (Gooren et al., 2013a; Brown et al., 2014). This is particularly relevant because there appears to have been a recent surge in the number of transgender people commencing hormone therapy at a young age (MacGregor et al., 2019; Cartaya et al., 2018). Klinefelter syndrome affects about 1 in 600 men and, usually being accompanied by a state of mild hypoandrogenism and mild hyperestrogenism, these men develop gynecomastia (Høst et al., 2014). It is of note that the breast cancer risk for Klinefelter syndrome males is significantly higher (about 3% absolute risk) that the rest of the male population (Niewoehner et al., 2008). As transgender women effectively achieve a induced state of marked “hypoandrogenism” and “hyperestrogenism” with hormone therapy, their breast cancer risk may theoretically be at least as high, or higher, than these males with Klinefelter syndrome. A recent case report concluded that the risk of breast cancer in transgender women is “acceptably safe in the short term” but that “the safety and potential risks in the longer term are unknown at present” (Sattari, 2015). As the incidence appears to be low, breast cancer may not be an overwhelming concern for transgender women. It is not, at present, possible to be sure.
Summary and Conclusions
Based on all available evidence, as with estrogens, progestogen usage appears to be associated with a higher incidence of breast cancer. Longer and more intense exposure may exemplify risk. This is likely the case for both synthetic progestins and bioidentical progesterone (particularly with some parenteral routes). However, more rigorous and adequately powered studies on breast cancer risk with these routes of progesterone administration are necessary to confirm this.
Because breast cancer in transgender women may not be nearly as common as breast cancer in cisgender women, it could be argued that the increased risk associated with progestogen exposure is of far lesser significance. Contrariwise, it cannot be argued that progesterone has no effect on or reduces the incidence of breast cancer. Given the limited and conflicting evidence for the notion that breast cancer remains very low in transgender women with long-term therapy, it ought not to be dismissed that an increased risk of breast cancer may form the basis of contraindication for progesterone therapy. This may be a particularly important consideration for those with a family history of the disease. Nonetheless, based on the extremely low number of case reports of breast cancer in transgender women, progestogens and progesterone might be well tolerated in this respect; even over long periods of time. There are probably more convincing reasons for us not to add micronised progesterone to our regimens.
References
Anders, C. K., Johnson, R., Litton, J., Phillips, M., & Bleyer, A. (2009). Breast Cancer Before Age 40 Years. Seminars in Oncology, 36(3), 237–249. [DOI:10.1053/j.seminoncol.2009.03.001]
Azam, S., Lange, T., Huynh, S., Aro, A. R., von Euler-Chelpin, M., Vejborg, I., Tjønneland, A., Lynge, E., & Andersen, Z. J. (2018). Hormone replacement therapy, mammographic density, and breast cancer risk: a cohort study. Cancer Causes & Control, 29(6), 495–505. [DOI:10.1007/s10552-018-1033-0]
Balasubramanian, R., Rolph, R., Morgan, C., & Hamed, H. (2019). Genetics of breast cancer: management strategies and risk-reducing surgery. British Journal of Hospital Medicine, 80(12), 720–725. [DOI:10.12968/hmed.2019.80.12.720]
Boyd, N. F., Martin, L. J., Yaffe, M. J., & Minkin, S. (2011). Mammographic density and breast cancer risk: current understanding and future prospects. Breast Cancer Research, 13(6), 223. [DOI:10.1186/bcr2942]
Brown, G. R., & Jones, K. T. (2014). Incidence of breast cancer in a cohort of 5,135 transgender veterans. Breast Cancer Research and Treatment, 149(1), 191–198. [DOI:10.1007/s10549-014-3213-2]
Cartaya, J., & Lopez, X. (2018). Gender dysphoria in youth. Current Opinion in Endocrinology & Diabetes and Obesity, 25(1), 44–48. [DOI:10.1097/med.0000000000000378]
Collaborative Group on Hormonal Factors in Breast Cancer. (2012). Menarche, menopause, and breast cancer risk: individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. The Lancet Oncology, 13(11), 1141–1151. [DOI:10.1016/s1470-2045(12)70425-4]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/s0140-6736(19)31709-x]
Coxon, J., & Seal, L. (2018). Hormone management of trans women. Trends in Urology & Men’s Health, 9(6), 10–14. [DOI:10.1002/tre.663]
Davey, D. A. (2018). Menopausal hormone therapy: a better and safer future. Climacteric, 21(5), 454–461. [DOI:10.1080/13697137.2018.1439915]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, I. R., & den Heijer, M. (2019). Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in the Netherlands. BMJ, 365, l1652. [DOI:10.1136/bmj.l1652]
Dimitrakakis, C., & Bondy, C. (2009). Androgens and the breast. Breast Cancer Research, 11(5), 212. [DOI:10.1186/bcr2413]
Eriksson, N., Benton, G. M., Do, C. B., Kiefer, A. K., Mountain, J. L., Hinds, D. A., Francke, U., & Tung, J. Y. (2012). Genetic variants associated with breast size also influence breast cancer risk. BMC Medical Genetics, 13(1), 53. [DOI:10.1186/1471-2350-13-53]
Folkerd, E., & Dowsett, M. (2013). Sex hormones and breast cancer risk and prognosis. The Breast, 22, S38–S43. [DOI:10.1016/j.breast.2013.07.007]
Fournier, A., Berrino, F., & Clavel-Chapelon, F. (2007). Unequal risks for breast cancer associated with different hormone replacement therapies: results from the E3N cohort study. Breast Cancer Research and Treatment, 107(1), 103–111. [DOI:10.1007/s10549-007-9523-x]
Fournier, A., Mesrine, S., Dossus, L., Boutron-Ruault, M., Clavel-Chapelon, F., & Chabbert-Buffet, N. (2014). Risk of breast cancer after stopping menopausal hormone therapy in the E3N cohort. Breast Cancer Research and Treatment, 145(2), 535–543. [DOI:10.1007/s10549-014-2934-6]
Gooren, L., & Morgentaler, A. (2013). Prostate cancer incidence in orchidectomised male-to-female transsexual persons treated with oestrogens. Andrologia, 46(10), 1156–1160. [DOI:10.1111/and.12208]
Gooren, L. J., van Trotsenburg, M. A., Giltay, E. J., & van Diest, P. J. (2013). Breast Cancer Development in Transsexual Subjects Receiving Cross-Sex Hormone Treatment. The Journal of Sexual Medicine, 10(12), 3129–3134. [DOI:10.1111/jsm.12319]
Hartley, R. L., Stone, J. P., & Temple-Oberle, C. (2018). Breast cancer in transgender patients: A systematic review. Part 1: Male to female. European Journal of Surgical Oncology, 44(10), 1455–1462. [DOI:10.1016/j.ejso.2018.06.035]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Hodis, H. N., Mack, W. J., Henderson, V. W., Shoupe, D., Budoff, M. J., Hwang-Levine, J., Li, Y., Feng, M., Dustin, L., Kono, N., Stanczyk, F. Z., Selzer, R. H., & Azen, S. P. (2016). Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. New England Journal of Medicine, 374(13), 1221–1231. [DOI:10.1056/nejmoa1505241]
Høst, C., Skakkebæk, A., Groth, K. A., & Bojesen, A. (2014). The role of hypogonadism in Klinefelter syndrome. Asian Journal of Andrology, 16(2), 185–191. [DOI:10.4103/1008-682X.122201]
Hynes, N. E., & Watson, C. J. (2010). Mammary Gland Growth Factors: Roles in Normal Development and in Cancer. Cold Spring Harbor Perspectives in Biology, 2(8), a003186. [DOI:10.1101/cshperspect.a003186]
Jansen, L., Backstein, R., & Brown, M. (2014). Breast size and breast cancer: A systematic review. Journal of Plastic, Reconstructive & Aesthetic Surgery, 67(12), 1615–1623. [DOI:10.1016/j.bjps.2014.10.001]
Jung, S., Stanczyk, F. Z., Egleston, B. L., Snetselaar, L. G., Stevens, V. J., Shepherd, J. A., Van Horn, L., LeBlanc, E. S., Paris, K., Klifa, C., & Dorgan, J. F. (2015). Endogenous Sex Hormones and Breast Density in Young Women. Cancer Epidemiology, Biomarkers & Prevention, 24(2), 369–378. [DOI:10.1158/1055-9965.epi-14-0939]
Kotsopoulos, J. (2019). Menopausal hormones: definitive evidence for breast cancer. The Lancet, 394(10204), 1116–1118. [DOI:10.1016/s0140-6736(19)31901-4]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H., & Schneider, H. P. (2013). Progesterone – promoter or inhibitor of breast cancer. Climacteric, 16(Suppl 1), 54–68. [DOI:10.3109/13697137.2013.768806]
Lambrinoudaki, I. (2014). Progestogens in postmenopausal hormone therapy and the risk of breast cancer. Maturitas, 77(4), 311–317. [DOI:10.1016/j.maturitas.2014.01.001]
Leitch, A. M., & Ashfaq, R. (2018). Discharges and Secretions of the Nipple. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 57–78.e3). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00004-0]
Levine, H., & Watson, N. (2000). Comparison of the pharmacokinetics of Crinone 8% administered vaginally versus Prometrium administered orally in postmenopausal women. Fertility and Sterility, 73(3), 516–521. [DOI:10.1016/s0015-0282(99)00553-1]
Lobo, R. A., Liu, J., Stanczyk, F. Z., Constantine, G. D., Pickar, J. H., Shadiack, A. M., Bernick, B., & Mirkin, S. (2019). Estradiol and progesterone bioavailability for moderate to severe vasomotor symptom treatment and endometrial protection with the continuous-combined regimen of TX-001HR (oral estradiol and progesterone capsules). Menopause, 26(7), 720–727. [DOI:10.1097/gme.0000000000001306]
Lteif, A., & Javed, A. (2013). Development of the Human Breast. Seminars in Plastic Surgery, 27(1), 5–12. [DOI:10.1055/s-0033-1343989]
MacGregor, E. A., & van den Brink, A. M. (2019). Transgender and Migraine. In van den Brink, A. M., & MacGregor, E. A. (Eds.). Gender and Migraine (pp. 113–127). Cham: Springer International Publishing. [DOI:10.1007/978-3-030-02988-3_9]
McCormack, V. A., & dos Santos Silva, I. (2006). Breast Density and Parenchymal Patterns as Markers of Breast Cancer Risk: A Meta-analysis. Cancer Epidemiology, Biomarkers & Prevention, 15(6), 1159–1169. [DOI:10.1158/1055-9965.epi-06-0034]
McGuire, A., Brown, J. A., Malone, C., McLaughlin, R., & Kerin, M. J. (2015). Effects of age on the detection and management of breast cancer. Cancers, 7(2), 908–929. [DOI:10.3390/cancers7020815]
Mirkin, S. (2018). Evidence on the use of progesterone in menopausal hormone therapy. Climacteric, 21(4), 346–354. [DOI:10.1080/13697137.2018.1455657]
Moscucci, O., & Clarke, A. (2007). Prophylactic oophorectomy: a historical perspective. Journal of Epidemiology & Community Health, 61(3), 182–184. [DOI:10.1136/jech.2006.046474]
Niewoehner, C. B., & Schorer, A. E. (2008). Gynaecomastia and breast cancer in men. BMJ, 336(7646), 709–713. [DOI:10.1136/bmj.39511.493391.be]
Randolph, J. F. (2018). Gender-Affirming Hormone Therapy for Transgender Females. Clinical Obstetrics & Gynecology, 61(4), 705–721. [DOI:10.1097/grf.0000000000000396]
Regidor, P. (2014). Progesterone in Peri- and Postmenopause: A Review. Geburtshilfe und Frauenheilkunde, 74(11), 995–1002. [DOI:10.1055/s-0034-1383297]
Rezvanpour, A., & Don-Wauchope, A. C. (2016). Clinical implications of estrone sulfate measurement in laboratory medicine. Critical Reviews in Clinical Laboratory Sciences, 54(2), 73–86. [DOI:10.1080/10408363.2016.1252310]
Rymer, J., Brian, K., & Regan, L. (2019). HRT and breast cancer risk. BMJ, 367, l5928. [DOI:10.1136/bmj.l5928]
Sattari, M. (2015). Breast Cancer in Male-to-Female Transgender Patients: A Case for Caution. Clinical Breast Cancer, 15(1), e67–e69. [DOI:10.1016/j.clbc.2014.08.004]
Singh, G. (2012). Oophorectomy in Breast Cancer—Controversies and Current Status. Indian Journal of Surgery, 74(3), 210–212. [DOI:10.1007/s12262-012-0584-7]
Sprague, B. L., Trentham-Dietz, A., Gangnon, R. E., Buist, D. S., Burnside, E. S., Bowles, E. J., Stanczyk, F. Z., & Sisney, G. S. (2010). Circulating Sex Hormones and Mammographic Breast Density among Postmenopausal Women. Hormones and Cancer, 2(1), 62–72. [DOI:10.1007/s12672-010-0056-0]
Stute, P., Wildt, L., & Neulen, J. (2018). The impact of micronized progesterone on breast cancer risk: a systematic review. Climacteric, 21(2), 111–122. [DOI:10.1080/13697137.2017.1421925]
Sun, S. X., Bostanci, Z., Kass, R. B., Mancino, A. T., Rosenbloom, A. L., Klimberg, V. S., & Bland, K. I. (2018). Breast Physiology. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 37–56.e6). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00003-9]
Tamimi, R. M., Hankinson, S. E., Colditz, G. A., & Byrne, C. (2005). Endogenous Sex Hormone Levels and Mammographic Density among Postmenopausal Women. Cancer Epidemiology, Biomarkers & Prevention, 14(11), 2641–2647. [DOI:10.1158/1055-9965.epi-05-0558]
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical Review: Breast Development in Trans Women Receiving Cross-Sex Hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]
Yang, Z., Hu, Y., Zhang, J., Xu, L., Zeng, R., & Kang, D. (2016). Estradiol therapy and breast cancer risk in perimenopausal and postmenopausal women: a systematic review and meta-analysis. Gynecological Endocrinology, 33(2), 87–92. [DOI:10.1080/09513590.2016.1248932]
\ No newline at end of file
+Progestogens and Breast Cancer in Transgender Women: A Review and Discussion of Risk - Transfeminine Science
Progestogens and Breast Cancer in Transgender Women: A Review and Discussion of Risk
By Sam | First published February 14, 2020 | Last modified October 5, 2020
Introduction
Breast cancer is the most common invasive cancer in women. The worldwide incidence in this group is about 1 in 8 (Balasubramanian et al., 2019; McGuire et al., 2015). There are likely many genetic, hormonal and lifestyle factors that influence risk. For instance, breast cancer is strongly associated with age. Indeed, only 7% of breast cancer cases have been found to be in women under 40 years old (Anders et al., 2009).
Sex steroids have profound influences on the breast. Mammary gland development begins during embryogenesis and marked differentiation takes place in the adult female at puberty and during pregnancy as a consequence of such influences (Javed et al., 2013; Sun et al., 2018; Leitch et al., 2018). Estrogens such as estradiol, in tandem with growth factors, promote morphological breast development at puberty while androgens inhibit (Hynes et al., 2010; Dimitrakakis et al., 2009). Moreover, estradiol and progesterone act synergistically to induce lobuloalveolar development in preparation for lactation. Owing to the effect of sex steroids, there is a positive relationship between breast development and breast cancer (Folkerd et al., 2013; Rezvanpour et al., 2016). In accordance, it is well established in large epidemiological studies that use of estrogen medications are strongly associated with an increased risk of breast cancer relative to non-use (Kotsopoulos, 2019). Women with larger breasts and hence more breast tissue are also apparently at a greater risk for breast cancer (Jansen et al., 2014).
Some patients and clinicians believe that progesterone may have benefits to breast development or otherwise. However, the use of progesterone in transfeminine hormone therapy for these hypothetical benefits is controversial and not evidence-based (Hembree et al., 2017; Wierckx et al., 2014; Randolph, 2018). It is sometimes argued that because these benefits are unsupported and usage is associated with an increased risk of cancer, progestogens (and specifically progesterone) ought not to be indicated for therapy in transgender women (Coxon et al., 2018). I decided to evaluate the strength of this claim in a short literature review.
Predictive Endocrine Markers of Breast Cancer
Oophorectomy (removal of the ovaries) can be used both as effective active treatment and prophylaxis against breast cancer (Singh, 2012; Moscucci et al., 2007). A large meta-analysis of over 100 epidemiological studies determined that both younger age at menarche and older age at menopause were associated with a greater risk of breast cancer (CGHFBC, 2012). While it is not clear how much this risk is attributable to estrogens vs progestogens, these findings are definitive evidence that breast cancer risk is linked to and enhanced by exposure to ovarian steroids.
High mammographic density is thought to be an important risk factor for breast cancer (Boyd et al., 2011; McCormack et al., 2006). Owing to this, it is interesting to note that use of hormone replacement therapy at the menopause is associated with both higher breast density and a higher incidence of breast cancer (Azam et al., 2018; CGHFBC, 2019). However, evidence suggesting association between sex hormones and breast cancer is conflicting. One study found no association between levels of free or total estradiol and breast density, but a positive association between levels of progesterone. (Sprague et al., 2010). Contrariwise, other studies reported no association between estradiol and progesterone and a negative association between estradiol and breast density, respectively (Jung et al., 2015; Tamimi et al., 2005). A review reported that in therapy with estrogens plus oral progesterone, breast density was increased significantly in three studies but unchanged in two other studies (Mirkin, 2018). Even though mammographic density is an important marker of breast cancer, the effects of sex steroids on this variable appear to be highly inconsistent between different studies. There may not be much to be learned from these findings.
Epidemiological Data
The risk of breast cancer with different estrogen and progestogen medications in menopausal hormone therapy has also been directly assessed by high-quality meta-analysis and with large sample sizes in the E3N/E3N-EPIC studies (CGHFBC, 2019; Fournier et al., 2014). In these trials, women using estradiol in combination with the synthetic progestins such as medroxyprogesterone acetate and chlormadinone acetate, have been found to have significantly greater rates of breast cancer relative to estradiol alone (Fournier et al., 2008; Lambrinoudaki, 2014). Conversely, in these large observational studies, the use of estradiol in combination with oral micronised progesterone has not been associated with a higher risk of breast cancer in the short term.
On the basis of these findings, it has been claimed that micronized progesterone differs from synthetic progestins in that it is not associated with a higher risk of developing breast cancer (Regidor, 2014; Yang et al., 2016; Rymer et al., 2019). But this does not appear to be the case. In the E3N-EPIC cohort, the relative risks (RR) [95% confidence interval (CI)] of breast cancer with transdermal estrogens and oral micronised progesterone were 0.9 [0.6–1.4], 0.7 [0.4–1.2] and 1.2 [0.7–2.0] at less than 2 years, between 2 to 4 years and at over 4 years of use, respectively (Mirkin, 2018). However, in contrast to these short-term findings, long-term observational studies have consistently found the use of micronized oral progesterone to be associated with higher breast cancer risk. In the same cohort, the relative risks of estrogens plus oral progesterone were reported between 4 to 6 years and beyond 6 years as 1.26 [0.87–1.82] and 1.22 [0.89–1.67], respectively (Fournier et al., 2008). At the conclusion of this study, the relative risks for estrogen plus oral progesterone were determined to be 1.13 [0.99–1.29] and 1.31 [1.15–1.48] below 5 years and at and beyond 5 years (Fournier et al., 2014). A recent systematic review concluded, on the basis of these findings, that breast cancer risk is not increased in the short-term by oral progesterone in combination with estrogens, but that long-term exposure has greater risk (Stute et al., 2018). The more recent CGHFBC meta-analysis determined that relative risk for estrogens plus oral progesterone was 0.91 [0.47–1.78] at less 5 years of use (CGHFBC, 2019; Table). Yet, in the long term, after 5 to 14 years of exposure, the risk was increased to 2.05 [1.38–3.06] and hence as high as medroxyprogesterone acetate and other oral progestins (2.07 [1.96–2.19]). There is no adequately powered evidence to demonstrate that estrogens plus micronized progesterone is associated with a lower incidence of breast cancer.
The notion that micronized progesterone does not increase the risk of breast cancer had been criticised before the long-term follow up of the E3N cohort and CGHFBC meta-analysis (Kuhl et al., 2013). It is important to consider the pharmacology of oral progesterone. Unlike with the synthetic progestins, this route of administration is associated with rapid metabolism and marked inactivation of the active medication into metabolites of progesterone by the gastrointestinal tract (Lobo et al., 2019; Levine et al., 2001). As a result, bioavailability is very low and, in accordance, oral progesterone is considerably less potent than the oral progestins (Davey et al., 2018; Kuhl, 2005). Taking the fact that it is far less potent into consideration, it would seem logical to assume as a default null hypothesis that breast cancer takes longer to appear and may be less common with oral progesterone (Kuhl et al., 2013). It is difficult to rationalise why all these other progestins, in spite of their different structures and biochemistry, would increase the incidence of breast cancer while micronized progesterone would not or have even an opposing effect. Conversely, other parenteral routes of progesterone such as vaginal or rectal administration have considerably higher bioavailability and more potent systematic effects (Kuhl, 2005). These non-oral routes likely have a similar incidence of breast cancer to those of the synthetic progestins.
In the CGHFBC meta-analysis, it was found that bioidentical estradiol has a similar, if not greater, incidence to conjugated (and therefore non-bioidentical) estrogens (1.78 (1.58–1.99) vs 1.68 [1.57–1.80]) (CGHFBC, 2019). Therefore, the rate of breast cancer with bioidentical estrogen usage is not significantly lower than with non-bioidentical estrogen usage. This is of note because, unlike oral progesterone and its synthetic derivatives, oral estradiol can have similar potency to these conjugated estrogens at practical clinical doses (Kuhl, 2005). One might expect the same to be true of bioidentical and non-identical progestogens at comparable potencies. The ELITE trial reported that there were more cases of breast cancer in a group of women using estradiol and relatively low-dose vaginal progesterone (45 mg/day) versus controls (Hodis et al., 2016). However, as the sample sizes were not large enough, it is not possible to attribute the higher rate of breast cancer to parenteral progesterone (Stute et al., 2018). Higher quality evidence is necessary to confirm, beyond a shadow of a doubt, that parenteral progesterone does in fact have comparable breast cancer risk to oral progestins.
Breast Cancer in Transgender Women
There is a positive association between breast cancer risk and a greater volume of breast tissue (Jansen et al., 2014; Eriksson et al., 2012). As transgender women develop breast tissue from hormone therapy, one might expect to find that their breast cancer rate would also increase. Surprisingly, however, the findings of epidemiological studies investigating the rate of breast cancer in transgender women are conflicting. Two large cohort studies (n = 2,307 and n = 1,259, respectively) reported no greater risk relative to cisgender men (Gooren et al., 2013a; Brown et al., 2014). However, another more recent study (n = 2,260) found a rate of breast cancer in transgender women that was between that of cisgender men and cisgender women (de Blok et al., 2019). The reason for this enormous difference in findings is unclear. A recent review found that there were just 20 cases of breast cancer in transgender women reported in the literature and hence almost as many as in transgender men (Hartley et al., 2018). It is of note that another review found only 7 reported cases of prostate cancer in transgender women (Gooren et al., 2013b). In accordance, prostate cancer seems to be an exceptionally rare diagnosis. One could argue on this basis, that the occurrence of breast cancer in transgender women is possibly also very rare.
Nonetheless, we should probably take the available evidence with some scepticism. It is well-known breast cancer takes many years to appear; after many decades of hormone exposure. Data from SEER in 2004 found that the incidences of breast cancer in women at 30, 40 and 50 years of age were only 1 in 1,523, 1 in 173 and 1 in 45, respectively (Anders et al., 2009). Given the secretion of ovarian steroid hormones begins at about 10-years-old in cisgender females, by the age of 50 these women have had about 40 years of exposure. But the follow-up times in these epidemiological studies of transgender women are very short in comparison (typically less than 20 years) and, following a similar timeframe of hormone exposure, breast cancer risk may be higher (Gooren et al., 2013a; Brown et al., 2014). This is particularly relevant because there appears to have been a recent surge in the number of transgender people commencing hormone therapy at a young age (MacGregor et al., 2019; Cartaya et al., 2018). Klinefelter syndrome affects about 1 in 600 men and, usually being accompanied by a state of mild hypoandrogenism and mild hyperestrogenism, these men develop gynecomastia (Høst et al., 2014). It is of note that the breast cancer risk for Klinefelter syndrome males is significantly higher (about 3% absolute risk) that the rest of the male population (Niewoehner et al., 2008). As transgender women effectively achieve a induced state of marked “hypoandrogenism” and “hyperestrogenism” with hormone therapy, their breast cancer risk may theoretically be at least as high, or higher, than these males with Klinefelter syndrome. A recent case report concluded that the risk of breast cancer in transgender women is “acceptably safe in the short term” but that “the safety and potential risks in the longer term are unknown at present” (Sattari, 2015). As the incidence appears to be low, breast cancer may not be an overwhelming concern for transgender women. It is not, at present, possible to be sure.
Summary and Conclusions
Based on all available evidence, as with estrogens, progestogen usage appears to be associated with a higher incidence of breast cancer. Longer and more intense exposure may exemplify risk. This is likely the case for both synthetic progestins and bioidentical progesterone (particularly with some parenteral routes). However, more rigorous and adequately powered studies on breast cancer risk with these routes of progesterone administration are necessary to confirm this.
Because breast cancer in transgender women may not be nearly as common as breast cancer in cisgender women, it could be argued that the increased risk associated with progestogen exposure is of far lesser significance. Contrariwise, it cannot be argued that progesterone has no effect on or reduces the incidence of breast cancer. Given the limited and conflicting evidence for the notion that breast cancer remains very low in transgender women with long-term therapy, it ought not to be dismissed that an increased risk of breast cancer may form the basis of contraindication for progesterone therapy. This may be a particularly important consideration for those with a family history of the disease. Nonetheless, based on the extremely low number of case reports of breast cancer in transgender women, progestogens and progesterone might be well tolerated in this respect; even over long periods of time. There are probably more convincing reasons for us not to add micronised progesterone to our regimens.
References
Anders, C. K., Johnson, R., Litton, J., Phillips, M., & Bleyer, A. (2009). Breast Cancer Before Age 40 Years. Seminars in Oncology, 36(3), 237–249. [DOI:10.1053/j.seminoncol.2009.03.001]
Azam, S., Lange, T., Huynh, S., Aro, A. R., von Euler-Chelpin, M., Vejborg, I., Tjønneland, A., Lynge, E., & Andersen, Z. J. (2018). Hormone replacement therapy, mammographic density, and breast cancer risk: a cohort study. Cancer Causes & Control, 29(6), 495–505. [DOI:10.1007/s10552-018-1033-0]
Balasubramanian, R., Rolph, R., Morgan, C., & Hamed, H. (2019). Genetics of breast cancer: management strategies and risk-reducing surgery. British Journal of Hospital Medicine, 80(12), 720–725. [DOI:10.12968/hmed.2019.80.12.720]
Boyd, N. F., Martin, L. J., Yaffe, M. J., & Minkin, S. (2011). Mammographic density and breast cancer risk: current understanding and future prospects. Breast Cancer Research, 13(6), 223. [DOI:10.1186/bcr2942]
Brown, G. R., & Jones, K. T. (2014). Incidence of breast cancer in a cohort of 5,135 transgender veterans. Breast Cancer Research and Treatment, 149(1), 191–198. [DOI:10.1007/s10549-014-3213-2]
Cartaya, J., & Lopez, X. (2018). Gender dysphoria in youth. Current Opinion in Endocrinology & Diabetes and Obesity, 25(1), 44–48. [DOI:10.1097/med.0000000000000378]
Collaborative Group on Hormonal Factors in Breast Cancer. (2012). Menarche, menopause, and breast cancer risk: individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. The Lancet Oncology, 13(11), 1141–1151. [DOI:10.1016/s1470-2045(12)70425-4]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/s0140-6736(19)31709-x]
Coxon, J., & Seal, L. (2018). Hormone management of trans women. Trends in Urology & Men’s Health, 9(6), 10–14. [DOI:10.1002/tre.663]
Davey, D. A. (2018). Menopausal hormone therapy: a better and safer future. Climacteric, 21(5), 454–461. [DOI:10.1080/13697137.2018.1439915]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, I. R., & den Heijer, M. (2019). Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in the Netherlands. BMJ, 365, l1652. [DOI:10.1136/bmj.l1652]
Dimitrakakis, C., & Bondy, C. (2009). Androgens and the breast. Breast Cancer Research, 11(5), 212. [DOI:10.1186/bcr2413]
Eriksson, N., Benton, G. M., Do, C. B., Kiefer, A. K., Mountain, J. L., Hinds, D. A., Francke, U., & Tung, J. Y. (2012). Genetic variants associated with breast size also influence breast cancer risk. BMC Medical Genetics, 13(1), 53. [DOI:10.1186/1471-2350-13-53]
Folkerd, E., & Dowsett, M. (2013). Sex hormones and breast cancer risk and prognosis. The Breast, 22, S38–S43. [DOI:10.1016/j.breast.2013.07.007]
Fournier, A., Berrino, F., & Clavel-Chapelon, F. (2007). Unequal risks for breast cancer associated with different hormone replacement therapies: results from the E3N cohort study. Breast Cancer Research and Treatment, 107(1), 103–111. [DOI:10.1007/s10549-007-9523-x]
Fournier, A., Mesrine, S., Dossus, L., Boutron-Ruault, M., Clavel-Chapelon, F., & Chabbert-Buffet, N. (2014). Risk of breast cancer after stopping menopausal hormone therapy in the E3N cohort. Breast Cancer Research and Treatment, 145(2), 535–543. [DOI:10.1007/s10549-014-2934-6]
Gooren, L., & Morgentaler, A. (2013). Prostate cancer incidence in orchidectomised male-to-female transsexual persons treated with oestrogens. Andrologia, 46(10), 1156–1160. [DOI:10.1111/and.12208]
Gooren, L. J., van Trotsenburg, M. A., Giltay, E. J., & van Diest, P. J. (2013). Breast Cancer Development in Transsexual Subjects Receiving Cross-Sex Hormone Treatment. The Journal of Sexual Medicine, 10(12), 3129–3134. [DOI:10.1111/jsm.12319]
Hartley, R. L., Stone, J. P., & Temple-Oberle, C. (2018). Breast cancer in transgender patients: A systematic review. Part 1: Male to female. European Journal of Surgical Oncology, 44(10), 1455–1462. [DOI:10.1016/j.ejso.2018.06.035]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Hodis, H. N., Mack, W. J., Henderson, V. W., Shoupe, D., Budoff, M. J., Hwang-Levine, J., Li, Y., Feng, M., Dustin, L., Kono, N., Stanczyk, F. Z., Selzer, R. H., & Azen, S. P. (2016). Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. New England Journal of Medicine, 374(13), 1221–1231. [DOI:10.1056/nejmoa1505241]
Høst, C., Skakkebæk, A., Groth, K. A., & Bojesen, A. (2014). The role of hypogonadism in Klinefelter syndrome. Asian Journal of Andrology, 16(2), 185–191. [DOI:10.4103/1008-682X.122201]
Hynes, N. E., & Watson, C. J. (2010). Mammary Gland Growth Factors: Roles in Normal Development and in Cancer. Cold Spring Harbor Perspectives in Biology, 2(8), a003186. [DOI:10.1101/cshperspect.a003186]
Jansen, L., Backstein, R., & Brown, M. (2014). Breast size and breast cancer: A systematic review. Journal of Plastic, Reconstructive & Aesthetic Surgery, 67(12), 1615–1623. [DOI:10.1016/j.bjps.2014.10.001]
Jung, S., Stanczyk, F. Z., Egleston, B. L., Snetselaar, L. G., Stevens, V. J., Shepherd, J. A., Van Horn, L., LeBlanc, E. S., Paris, K., Klifa, C., & Dorgan, J. F. (2015). Endogenous Sex Hormones and Breast Density in Young Women. Cancer Epidemiology, Biomarkers & Prevention, 24(2), 369–378. [DOI:10.1158/1055-9965.epi-14-0939]
Kotsopoulos, J. (2019). Menopausal hormones: definitive evidence for breast cancer. The Lancet, 394(10204), 1116–1118. [DOI:10.1016/s0140-6736(19)31901-4]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H., & Schneider, H. P. (2013). Progesterone – promoter or inhibitor of breast cancer. Climacteric, 16(Suppl 1), 54–68. [DOI:10.3109/13697137.2013.768806]
Lambrinoudaki, I. (2014). Progestogens in postmenopausal hormone therapy and the risk of breast cancer. Maturitas, 77(4), 311–317. [DOI:10.1016/j.maturitas.2014.01.001]
Leitch, A. M., & Ashfaq, R. (2018). Discharges and Secretions of the Nipple. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 57–78.e3). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00004-0]
Levine, H., & Watson, N. (2000). Comparison of the pharmacokinetics of Crinone 8% administered vaginally versus Prometrium administered orally in postmenopausal women. Fertility and Sterility, 73(3), 516–521. [DOI:10.1016/s0015-0282(99)00553-1]
Lobo, R. A., Liu, J., Stanczyk, F. Z., Constantine, G. D., Pickar, J. H., Shadiack, A. M., Bernick, B., & Mirkin, S. (2019). Estradiol and progesterone bioavailability for moderate to severe vasomotor symptom treatment and endometrial protection with the continuous-combined regimen of TX-001HR (oral estradiol and progesterone capsules). Menopause, 26(7), 720–727. [DOI:10.1097/gme.0000000000001306]
Lteif, A., & Javed, A. (2013). Development of the Human Breast. Seminars in Plastic Surgery, 27(1), 5–12. [DOI:10.1055/s-0033-1343989]
MacGregor, E. A., & van den Brink, A. M. (2019). Transgender and Migraine. In van den Brink, A. M., & MacGregor, E. A. (Eds.). Gender and Migraine (pp. 113–127). Cham: Springer International Publishing. [DOI:10.1007/978-3-030-02988-3_9]
McCormack, V. A., & dos Santos Silva, I. (2006). Breast Density and Parenchymal Patterns as Markers of Breast Cancer Risk: A Meta-analysis. Cancer Epidemiology, Biomarkers & Prevention, 15(6), 1159–1169. [DOI:10.1158/1055-9965.epi-06-0034]
McGuire, A., Brown, J. A., Malone, C., McLaughlin, R., & Kerin, M. J. (2015). Effects of age on the detection and management of breast cancer. Cancers, 7(2), 908–929. [DOI:10.3390/cancers7020815]
Mirkin, S. (2018). Evidence on the use of progesterone in menopausal hormone therapy. Climacteric, 21(4), 346–354. [DOI:10.1080/13697137.2018.1455657]
Moscucci, O., & Clarke, A. (2007). Prophylactic oophorectomy: a historical perspective. Journal of Epidemiology & Community Health, 61(3), 182–184. [DOI:10.1136/jech.2006.046474]
Niewoehner, C. B., & Schorer, A. E. (2008). Gynaecomastia and breast cancer in men. BMJ, 336(7646), 709–713. [DOI:10.1136/bmj.39511.493391.be]
Randolph, J. F. (2018). Gender-Affirming Hormone Therapy for Transgender Females. Clinical Obstetrics & Gynecology, 61(4), 705–721. [DOI:10.1097/grf.0000000000000396]
Regidor, P. (2014). Progesterone in Peri- and Postmenopause: A Review. Geburtshilfe und Frauenheilkunde, 74(11), 995–1002. [DOI:10.1055/s-0034-1383297]
Rezvanpour, A., & Don-Wauchope, A. C. (2016). Clinical implications of estrone sulfate measurement in laboratory medicine. Critical Reviews in Clinical Laboratory Sciences, 54(2), 73–86. [DOI:10.1080/10408363.2016.1252310]
Rymer, J., Brian, K., & Regan, L. (2019). HRT and breast cancer risk. BMJ, 367, l5928. [DOI:10.1136/bmj.l5928]
Sattari, M. (2015). Breast Cancer in Male-to-Female Transgender Patients: A Case for Caution. Clinical Breast Cancer, 15(1), e67–e69. [DOI:10.1016/j.clbc.2014.08.004]
Singh, G. (2012). Oophorectomy in Breast Cancer—Controversies and Current Status. Indian Journal of Surgery, 74(3), 210–212. [DOI:10.1007/s12262-012-0584-7]
Sprague, B. L., Trentham-Dietz, A., Gangnon, R. E., Buist, D. S., Burnside, E. S., Bowles, E. J., Stanczyk, F. Z., & Sisney, G. S. (2010). Circulating Sex Hormones and Mammographic Breast Density among Postmenopausal Women. Hormones and Cancer, 2(1), 62–72. [DOI:10.1007/s12672-010-0056-0]
Stute, P., Wildt, L., & Neulen, J. (2018). The impact of micronized progesterone on breast cancer risk: a systematic review. Climacteric, 21(2), 111–122. [DOI:10.1080/13697137.2017.1421925]
Sun, S. X., Bostanci, Z., Kass, R. B., Mancino, A. T., Rosenbloom, A. L., Klimberg, V. S., & Bland, K. I. (2018). Breast Physiology. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 37–56.e6). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00003-9]
Tamimi, R. M., Hankinson, S. E., Colditz, G. A., & Byrne, C. (2005). Endogenous Sex Hormone Levels and Mammographic Density among Postmenopausal Women. Cancer Epidemiology, Biomarkers & Prevention, 14(11), 2641–2647. [DOI:10.1158/1055-9965.epi-05-0558]
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical Review: Breast Development in Trans Women Receiving Cross-Sex Hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]
Yang, Z., Hu, Y., Zhang, J., Xu, L., Zeng, R., & Kang, D. (2016). Estradiol therapy and breast cancer risk in perimenopausal and postmenopausal women: a systematic review and meta-analysis. Gynecological Endocrinology, 33(2), 87–92. [DOI:10.1080/09513590.2016.1248932]
\ No newline at end of file
diff --git a/transfemscience.org/articles/progestogens-breast-dev/index.html b/transfemscience.org/articles/progestogens-breast-dev/index.html
index ef6aab61..d893f852 100644
--- a/transfemscience.org/articles/progestogens-breast-dev/index.html
+++ b/transfemscience.org/articles/progestogens-breast-dev/index.html
@@ -1 +1 @@
-A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People - Transfeminine Science
A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People
By Aly | First published February 14, 2020 | Last modified August 23, 2025
Abstract / TL;DR
The major female sex hormones are estrogen and progesterone. Both of these hormones are known to be importantly involved in the development of the breasts at different stages of life. Speculation, use, and anecdotes of progestogens for enhancing breast development in transfeminine people date back to at least the 1960s. A limited number of clinical studies have assessed breast development with progestogens in transfeminine people, but current evidence on progestogens for improving breast development is of very low quality and is inconclusive. Studies of progestogens and breast development in cisgender girls and women are similarly limited. In any case, more studies evaluating progestogens and breast development are currently underway. The possible role of progestogens in enhancing breast development can also be informed by indirect and circumstantial evidence, including notably findings on progesterone and breast changes during normal puberty, the menstrual cycle, and pregnancy in humans and animals. Available evidence overall is not suggestive of an essential role for progesterone in breast growth during puberty, but progesterone does have a clear and key role in lobuloalveolar development of the breasts during pregnancy. However, breast changes in pregnancy revert following cessation of lactation and breastfeeding. Progesterone may additionally contribute to reversible breast enlargement during the luteal phase of the menstrual cycle. There are some findings to suggest that progestogens may have antiestrogenic effects in the breasts and may have a stunting influence on breast development if introduced too early following initiation of hormone therapy. However, more research is needed to assess this possibility. In any case, if progestogens are used, it may be advisable to delay their introduction until most or all estrogen-mediated breast development is complete. Options for progestogen therapy in transfeminine people include bioidentical progesterone and progestins. However, oral progesterone has major bioavailability problems and does not achieve satisfactory progesterone levels. Progestogens, including progesterone, have been variously linked to significant health risks, which is an important consideration in terms of their use in transfeminine people. Overall, based on current knowledge, progestogens do not seem to be promising for lastingly improving breast development in transfeminine people, but more research and data are still needed for clear conclusions.
Introduction
Breast development in terms of size and shape is often less than desired in transfeminine people, and there is a need for therapeutic approaches that improve breast growth in this population. There are two major types of female hormones, estrogens and progestogens. Estrogens are almost universally employed in transfeminine hormone therapy, while progestogens are used in a subset of transfeminine people. Progestogens that have been commonly employed in transfeminine people include bioidenticalprogesterone, the progestin (synthetic progestogen) medroxyprogesterone acetate (MPA), and the strongly progestogenic antiandrogen cyproterone acetate (CPA). Estrogens are the major mediators of feminization and breast development in females. However, progestogens also have physiological effects on the breasts, and in relation to this, may or may not provide benefits to breast development as well.
The topic of progestogens and breast development has been discussed for many years in the transgender community and is a controversial subject (Coleman et al., 2012). Use of progestogens to improve breast development in transfeminine people goes back at least as far as Harry Benjamin and Christian Hamburger in the 1960s (Benjamin, 1966; Benjamin, 1967; Hamburger & Benjamin, 1969; Wiki). Arguments have been made both for (e.g., Bevan, 2012; Bellwether, 2019; Bevan, 2019) and against (e.g., Curtis, 2009) a possible role of progestogens in terms of breast development. It is often claimed that progestogens can enhance breast development or are even required for full breast development in cisgender females and transfeminine people. With respect to the latter, it is sometimes said that progestogens are necessary for people to move from Tanner stage 4 to Tanner stage 5 pubertal breast development and that progestogens help to fill and round out the breasts (e.g., Vorherr, 1974a; Basson & Prior, 1998; Kaiser & Ho, 2015; Prior, 2011; Prior, 2019a; Prior, 2020). It has even been claimed by some that without progestogens, the breasts will remain conical and “pointy” like they are in the earlier Tanner stages. On the other extreme, certain critics have claimed that there are “no biologically significant progesterone receptor sites for biological males” and that progesterone is not produced during normal female puberty until after breast development has been fully completed (Barrett, 2009; Seal, 2017; Coxon & Seal, 2018; Price, McManus, & Barrett, 2019; Richards & Barrett, 2020). In turn, these particular authors have argued against the use of progestogens in transfeminine people in various of their publications (Google Scholar). In general, the use of progestogens in transfeminine people has longstandingly been controversial, with positions both for and against (Sam, 2020).
The purpose of this article is to review the available direct and circumstantial evidence on the topic of progestogens and breast development in order to help inform whether progestogen therapy may be able to enhance breast development in transfeminine people. Aside from an involvement in breast development, progestogens are not otherwise currently thought to be or known to be involved in physical feminization (e.g., Coleman et al., 2012; Gooren, 2016). In relation to this, the present article will limit its discussion to breast development with progestogens, and will not explore feminization in general.
Progestogen Therapy and Breast Development in Humans
Progestogens and Breast Development in Transfeminine People
Orentreich & Durr (1974) was one of the earliest studies on breast development in transfeminine people. They employed combinations of estrogens and progestogens as well as gonadectomy to produce feminization and breast development in a case series of 5 transfeminine people. The employed estrogens were estradiol valerate 30 mg/2 weeks by intramuscular injection and oral conjugated estrogens 1.25–5.0 mg/day and the used progestogens were “60 mg medroxyprogesterone caproate” every 2 weeks by intramuscular injection and oral medroxyprogesterone acetate 0–10 mg/day. Medroxyprogesterone caproate (MPC) has never been used pharmaceutically, so this was likely a typo and the actual progestogen employed was likely either MPA or hydroxyprogesterone caproate (OHPC). The authors reported that estrogen and progestogen therapy produced modest to significant breast development in the transfeminine people that was not strictly dose-related and included clinical photographs of the breasts. They concluded that the breast development was comparable to that of adult cisgender women. Orentreich and colleagues also discussed the topic of lobuloalveolar maturation of the breasts, which was known to be progestogen-dependent, but noted that they had not done histological assessment and that the degree of lobuloalveolar development of the breasts does not necessarily correlate with clinical breast size per findings in cisgender women. The findings of Orentreich and colleagues are limited by methodological problems like lack of objective measurements, lack of estrogen-only controls, and the small sample size of only 5 transfeminine people, and hence the study is of limited value in terms of assessing the involvement of progestogens in breast development.
Meyer et al. (1986) assessed the effects of progestogens added to estrogen therapy on breast development and other clinical parameters in transfeminine people. Of the 60 transfeminine people in the study, 15 (25%) received an oral progestogen, usually MPA at a dosage of 10 mg/day, for “at least for a short time”, and with only 8 (13.3%) receiving progestogen therapy for the full treatment period. In an earlier report of the study, it was noted that in 90% of observation periods the dose was 10 mg/day and for the remainder it was 20 mg/day (Meyer et al., 1981). A dosage of 10 mg/day MPA is roughly comparable to luteal-phase progesterone exposure in terms of progestogenic potency (Wiki). Breast development was measured in the study via breast hemicircumference (Diagram). Progestogen therapy was reported to not modify estrogen-induced changes, including laboratory measurements, hormone levels, and physical parameters like weight and breast growth. The lack of apparent changes in hormone levels is unexpected, as MPA in higher-quality studies has shown clear testosterone suppression (e.g., Jain, Kwan, & Forcier, 2019; Wiki). Meyer and colleagues concluded that adding progestogens to estrogen does not seem to enhance breast development in transfeminine people. However, they noted that the number of individuals who received progestogens was small and further studies were needed.
Prior et al. (1986) and Prior, Vigna, & Watson (1989) studied estrogen, high-dose spironolactone (100–600 mg/day), and MPA (10–20 mg/day cylically or continuously) in transfeminine people who were either pre-hormone therapy or had previously been on higher doses of estrogens (and/or progestogens) without spironolactone prior to the study. The researchers reported that following 12 months of treatment with the study’s hormone therapy regimen, there was increased breast size and increased nipple development. Most individuals reached an A cup size, or approximately 8 to 14 cm in diameter of breast tissue, by the end of the study. Breast development was measured in part with photographic documentation. Although breast development reportedly improved, the researchers themselves noted that it was difficult to determine whether the enhanced breast development could be attributed to spironolactone or to MPA. Moreover, testosterone suppression was inadequate before the study and improved with the study’s hormone therapy regimen, which may have helped to improve breast development regardless of any potential direct progestogenic action of MPA on the breasts. Finally, it is possible that breast development with estrogen may not yet have been complete, and that the improved breast development may have simply been continued progression due to estrogen alone. In other publications, Jerilynn Prior, the lead study author, has claimed that progesterone enhances breast development, and has cited the preceding studies by her in support of this claim (Prior, 2011; Prior, 2019a; Prior, 2019b; Prior, 2020). However, her claim is not well-supported due to the study limitations discussed.
Dittrich et al. (2005) reported that the combination of oral estradiol valerate and a gonadotropin-releasing hormone (GnRH) agonist for 2 years in transfeminine people resulted in self-reported breast cup sizes of C cup or greater in 5%, B cup in 30%, A cup in 35%, and less than A cup in 30%. They noted however that 70% of the individuals were unsatisfied with their breast development and wished to undergo breast augmentation surgery. The researchers claimed that the regimen had similar effectiveness in terms of feminization, including increases in breast size, compared to prior reported treatment regimens of ethinylestradiol and CPA. No other details or specifics were given. The claim about similar breast development to regimens containing CPA is relevant as CPA is a very strong progestogen at the doses used historically in transfeminine people (Aly, 2019). It should be cautioned however that this study did not actually employ or study progestogen therapy itself. In addition, self-reported breast cup size is a subjective and low-quality means of measuring breast development and size. As such, the findings of this study are of questionable value in terms of understanding progestogens and breast development.
Estrogen is primarily involved in ductal development of the breasts, whereas progesterone is mainly involved in lobuloalveolar development. Kanhai et al. (2000) compared internal histological breast tissue changes with estrogen and CPA 100 mg/day (i.e. very-high-dose progestogen) therapy in 14 transfeminine people versus nonsteroidal antiandrogen monotherapy with flutamide or bicalutamide in 2 cisgender men with prostate cancer. Both types of treatments block androgens, increase estrogen levels, and are known to induce breast development or gynecomastia at similarly high rates. However, nonsteroidal antiandrogen monotherapy differs from combined estrogen and progestogen therapy in that it lacks any progestogenic effects. In the transfeminine people, full lobuloalveolar formation was apparent in the biopsied breast tissue, whereas in the men with prostate cancer, only “moderate” and incomplete lobuloalveolar maturation was found. It was also noted that lobuloalveolar formation tended to regress upon discontinuation of CPA following gonadectomy in transfeminine people. The researchers concluded that progestogenic exposure is needed for the breasts to fully develop on a histological level and for the breast tissue of transfeminine people to completely mimic the histology of the mature female breast. In accordance with these findings, estrogen plus high doses of CPA, as well as certain other regimens, have been associated with galactorrhea (lactation) as a side effect in transfeminine people (Dewhurst & Underhill, 1979; Futterweit, 1980; Gooren, Harmsen-Louman, & van Kessel, 1985; Schlatterer et al., 1998; Levy, Crown, & Reid, 2003; Bazarra-Castro, 2009). While the findings of Kanhai and colleagues’ study are interesting, they only concern tissue characteristics and do not actually provide any information about breast development in terms of physical form or appearance. With regard to this, tissue-level differences may or may not translate to relevant differences in for instance breast size or shape. As such, the study is of limited value in understanding whether progestogens improve breast development in transfeminine people in the ways that are actually valued.
Seal and colleagues conducted a retrospective chart review assessing clinical predictors for surgical breast augmentation in transfeminine people (Seal et al., 2012). In the transfeminine people who underwent breast augmentation, significantly more of them were taking spironolactone than were those who did not undergo breast augmentation. Conversely, the differential rates of use of specific antiandrogens were not significantly discordant between those who did and did not undergo breast augmentation in the case of the other prescribed antiandrogens, including CPA, the 5α-reductase inhibitors, and GnRH analogues. However, this study had many methodological limitations, including the use of almost three dozen t-tests with no adjustment for multiple comparisons (and hence risk of false positives and concerns about p-hacking), small sample sizes for individual antiandrogens, use of undergoing breast augmentation as a surrogate for breast development with no actual physical measurement of the breasts or breast sizes, and a correlational design with lack of control for potential confounding variables. As such, the study does not show that different antiandrogens result in differences in breast development, and its findings must be considered with due caution.
Jain, Kwan, & Forcier (2019) studied sublingual estradiol and spironolactone with and without MPA in 92 transfeminine people. MPA was given at a dose of 5 to 10 mg/day sublingually or at a dose of 150 mg once every 3 months by intramuscular injection. Of 39 transfeminine people who received MPA, 26 (67%) self-reported improved breast development. No further details were provided, but measurement of breast development was presumably subjective and anecdotal. Igo & Visram (2021) studied addition of progesterone to hormone therapy in transfeminine people. Progesterone was provided as 100 mg micronized progesterone (probably oral) and was prescribed when progesterone was specifically requested by the patient or when the patient expressed dissatisfaction with feminization and/or breast development. Of 190 individuals, 51 (26.8%) received progesterone therapy. Treatment with progesterone on average began after 12.7 months of estradiol therapy, and the mean total follow-up time was 14.3 months of hormone therapy. Of those who received progesterone, only 6 (11.8%) reported benefit to breast development. No further details were provided, but as with other studies, breast development was likely quantified anecdotally via self-report. As breast development does not appear to have been objectively measured or compared to a control group in either Jain, Kwan, & Forcier (2019) or Igo & Visram (2021), the findings of these studies are limitedly informative.
Nolan and colleagues assessed the short-term effects of low-dose oral micronized progesterone on breast development in transfeminine people on stable hormone therapy in a prospective controlled study (Nolan et al., 2022a; Nolan et al., 2022b). Progesterone was given at a dose of 100 mg/day for 3 months to 23 transfeminine people and findings were compared to those of a control group of 19 transfeminine people. Breast development was measured using self-reported Tanner stage, with participants provided photographs of different Tanner stages to self-select from. At the end of the 3 months, Tanner stage was not significantly different between groups (mean 3.5, 95% CI 3.2–3.7 for progesterone vs. mean 3.6, 95% CI 3.3–3.9 for controls; p = 0.42). A limitation of this study is that oral progesterone has very low bioavailability and 100 mg/day oral progesterone achieves very low progesterone levels that are well below normal luteal-phase progesterone levels (Aly, 2018a; Wiki). As such, progestogenic exposure in this study, and notably also in Igo & Visram (2021) and other studies, is likely to have been inadequate. Besides the issue of progestogenic strength, the very short duration of the study (3 months) and the reliance on self-reported subjective Tanner stages (as opposed to more objective physical breast measurements) are also major limitations. In any case, this study is of higher quality than previous studies, and is notably likely to continue and report further follow-up at later points in the future.
Bahr et al. (2024) conducted a retrospective chart review at their clinic and compared 29 transfeminine people who had received progestogens versus 59 transfeminine people who had not. The form of progestogen used was oral or rectal progesterone in 93% of cases and MPA by intramuscular injection in the remaining 7% of cases. Of those who took progesterone, 25 (93%) used it orally and 2 (7%) used oral progesterone capsules rectally. Progestogen doses were not reported, except that 100 mg progesterone capsules were employed. Most of those in the progestogen-treated group (59%) had started it 1 to 6 months following initiation of standard hormone therapy. The researchers found that progestogen-treated group had significantly better self-reported breast development satisfaction (rated as satisfied, neutral, or unsatisfied) compared to the group that did not receive progestogens at 6 months (satisfied: 53.8% vs. 19.6%; p = 0.004) and 9 months (satisfied: 71.4% vs. 20.8%; p = 0.003) of hormone therapy. Limitations of this study include the lack of objective measurement of breast development, the restrospective nature of the study, and the lack of randomization of treatment, among others.
Aside from the above studies, a variety of other studies have also reported breast development with estrogen and CPA in transfeminine people. These studies have often employed objective physical measurements of breast development (e.g., breast volume, breast–chest difference, breast cup size, breast hemicircumference). However, they have lacked comparison groups, thereby precluding comparison of progestogenic versus non-progestogenic hormone therapy. In addition, CPA is unusual among progestogens in that it is employed at very high doses in transfeminine people (Aly, 2019), which may result in different and potentially stunted outcomes in terms of breast development than more physiological progestogenic exposure. As such, most studies of breast development with estrogen and CPA in transfeminine people have not been discussed in the present section and are instead discussed elsewhere in this article (see the section below). In any case, to briefly summarize the findings, breast development in transfeminine people with estrogen and CPA has generally been poor in these studies. The outcomes have included incomplete maturation in terms of Tanner staging (stage 2–4), small cup sizes, small breast volumes, and breasts much smaller in size than those in cisgender women.
The findings from the preceding studies in transfeminine people are of very low-quality due to methodological limitations, including lack of control groups, lack of randomization, reliance on poor measures of breast development (e.g., subjective and self-report) rather than objective physical measurements (Wiki), short treatment durations, and small sample sizes, among others. This may explain the conflicting results of the studies. More research is still needed to assess the influence of progestogens on breast development in transfeminine people. There is specifically a need for randomized controlled trials (RCTs) of feminizing hormone therapy with versus without progestogen therapy that employ objective measures of breast development, have adequate sample sizes, and have sufficient follow-up durations. Additional variables like progestogen type, route, dose, and timing of introduction would also be of value to explore. A 2014 review on hormone therapy in transfeminine people summarizes the state of research on progestogens and breast development in transfeminine people, with their conclusions still holding true today (Wierckx, Gooren, & T’Sjoen, 2014):
Our knowledge concerning the natural history and effects of different cross-sex hormone therapies on breast development in trans women is extremely sparse and based on low quality of evidence. Current evidence does not provide evidence that progestogens enhance breast development in trans women. Neither do they prove the absence of such an effect. This prevents us from drawing any firm conclusion at this moment and demonstrates the need for further research to clarify these important clinical questions.
Several studies of progesterone and other progestogens in transfeminine people are currently underway. These studies include (1) an RCT of oral progesterone added to hormone therapy by Sandeep Dhindsa and colleagues in St. Louis, Missouri in the United States (ClinicalTrials.gov; MediFind; ICH GCP); (2) a prospectiveobservational study and/or RCT of addition of oral progesterone to hormone therapy by Ada Cheung and colleagues in Melbourne, Australia (University of Melbourne; University of Melbourne); (3) an RCT of estradiol plus spironolactone versus estradiol plus CPA also by Ada Cheung and colleagues (ANZCTR; WHO ICTRP; Trans Health Research [Flyer] [Poster]; University of Melbourne) (update: see below); and (4) a large RCT of oral progesterone at different doses added to hormone therapy by Martin den Heijer and colleagues at the Vrije Universiteit University Medical Center (VUMC) in Amsterdam, the Netherlands (Dijkman et al., 2023a; General Info/Links; Info Sheet Dutch; Info Sheet English Translated) (update: see below). Unfortunately however, all of the studies using progesterone employ oral progesterone, which has major bioavailability and potency problems (Aly, 2018a; Wiki). In any case, it was said that the VUMC researchers may follow their trial up with studies of other progesterone routes (General Info/Links). The preceding studies may provide more insight on the question of whether progestogen therapy is of therapeutic benefit to breast development in transfeminine people.
Progestogens and Breast Development in Cisgender Females
To date, there appear to be no useful studies on breast development with progesterone or other progestogens in cisgender females. There seem to mostly only be a few brief and conflicting anecdotal clinical statements in this area that are scattered throughout the literature. These include the following literature excerpts, which are specifically in the context of progestogens as part of puberty induction in cisgender girls and women with delayed or absent puberty due to hypogonadism:
I […] performed studies on three women lacking mammary development and exhibiting signs of marked hypogonadism. […] Corpus luteum extract, 5 international units daily for a period of thirty days, when given alone produced no detectable change in the breasts. This is in accord with the experimental observations on animals of Turner,2 Corner 3 and others. When, however, patients were given alternate daily injections of 1 international unit of progesterone and from 20,000 to 50,000 international units of estrone or of estradiol benzoate, breast growth was more rapid than that produced by the estrogenic hormones alone. The simultaneous use of the corpus luteum and estrogenic therapy definitely produced a much firmer breast growth, which was distinctly lobular to palpation, whereas the growth produced by the estrogenic hormones alone was smooth and the borders of the glandular tissue were difficult to define. Rapid regression in the size of the breasts followed the omission of the hormone injections, but the regression was less rapid when the combined therapy had been used. [MacBryde (1939)]
There are authorities who consider that breast growth is better if a progestogen is combined with oestrogen for the latter part of the cycle of treatment (Capraro, 1971). Shearman (1971) employs sequential therapy in his cases. Huffman (1971) however, does not believe that there is any improvement with the addition of progestogens. [Dewhurst (1971a)]
The effects of progesterone on the human breast remain obscure. Although widely stated to cause glandular development, the evidence for this is slender (Benson et al 1959). [Shearman (1972a)]
Many people use oestrogens alone, but the addition of a progestin for 6 or 10 days each month gives much better cycle control and appears to cause better breast development. [Shearman (1972b)]
Some authorities consider that breast growth is better if a progestogen is given for the latter part of each course of treatment. [Capraro & Dewhurst (1975)]
It has been suggested that progestins be added during the last week of each cycle of estrogen therapy in order to develop more rounded breasts rather than the conical breasts many of these patients develop, but we have been unable to detect any difference in breast contour with or without progestins. [Davajan & Kletzky (1979)]
I have been satisfied that the addition of a progestogen was necessary to get a good breast response to hormone treatment although the progestogen, as I have said, is required after the first year if the uterus is present. [Dewhurst (1982)]
In addition to the preceding instances, Werner (1935) and Geschickter (1945) assessed the effects of progesterone on the breasts in cisgender women. Werner (1935) attempted to induce lactation in 8 surgically gonadectomized cisgender women with combinations of estrogen, progesterone, and prolactin, all in the form of crude extracts by injection. In two women who were given progesterone, he claimed that a marked increase in the size of the breasts beyond that with estrogen alone was observed. Additionally, he claimed that the breasts were more firm, the glandular tissue “more tortuous and nodular”, and the nipples more prominent. He was not successful in inducing lactation in the women in this study. The doses of hormones used were unclear as they were in the form of extracts, and were likely supraphysiological, potentially pregnancy-like due to the nature of the experiment. Werner’s study was also briefly discussed by Nelson (1936), among other citations. Geschickter (1945) observed lobuloalveolar growth on histological examination with administration of progesterone for 6 weeks to 2 months in one woman but not in another woman. However, the exterior physical changes of the breasts were not assessed or reported by this author and hence his findings are limitedly informative.
Surprisingly, there have been few analogous studies of the effects of progestogens on the breasts in cisgender girls and women following the preceding reports and anecdotes. Although there are very little data on progestogens and breast growth in cisgender females, clinical studies are finally starting to look more closely at the specifics of hormonal medications, including progestogens, in terms of breast development in girls undergoing puberty induction (e.g., Rodari et al., 2023). As such, future studies may provide more insight on the subject of progestogens and breast development in cisgender females.
Progesterone and its Physiological Role in Breast Development in Humans
Progesterone and Breast Development in Puberty
The role of progesterone in breast development and its possible usefulness for helping with breast development in transfeminine hormone therapy can be informed by the normal biological circumstances of puberty in cisgender females. Puberty in cisgender girls usually starts around age 11 (range 8–13 years) and completes around age 15 years (range 12–19 years), taking on average 3 to 4 years (but with a range of about 1.5–6 years in most cases) (Schauffler, 1942; Marshall & Tanner, 1969; Marshall, 1978; Begley, Firth, & Hoult, 1980; Drife, 1986). Progesterone essentially does not appear during puberty until ovulatorymenstrual cycles begin. Menarche, the onset of menstruation and hence of menstrual cycling, occurs on average at Tanner breast stage 4 or about 13 years of age, although it occurs at Tanner breast stage 3 or Tanner breast stage 5 in significant subsets of girls (26% for Tanner stage 3, 62% for Tanner stage 4, and 10% for Tanner stage 5) (Marshall & Tanner, 1969; Marshall, 1978; Drife, 1986; Hillard, 2007). Hence, the appearance of progesterone in normal female puberty is a relatively late event (Scott et al., 1950; Marshall, 1978; Begley, Firth, & Hoult, 1980; Drife, 1986), and most breast development appears to be complete by menarche and thus by the time that progesterone is first produced (Huffman, Dewhurst, & Capraro, 1981; Drife, 1982). Moreover, a small but significant subset of girls reaches Tanner breast stage 5 and hence fully developed breasts before menarche (Edmonds, 1989), which suggests that progesterone may not be essential for complete pubertal breast development.
Only a handful of studies and sources have reported progesterone levels during puberty across Tanner stages or by age in cisgender girls (e.g., Sizonenko, 1978 [Graph]; Kühnel, 2000; Lee, 2001 [Table]; Aly, 2020a). They corroborate the above findings with regard to limited progesterone exposure during puberty. The “A Girl’s First Period Study” is an ambitious research project announced in 2022 that aims to better characterize reproductive hormone levels in pubertal and adolescent girls and may shed more light on the physiological role of progesterone during puberty (Lucien et al., 2022). The researchers have specifically highlighted the possible role of progesterone in breast development as part of their interests:
Does exposure to low levels of [progesterone (P4)], as occurs before menarche, during anovulatory cycles with some degree of follicle luteinization, and during early, immature ovulatory cycles play an important role in normal breast development during puberty? This question has important clinical implications as hormone replacement during puberty does not typically include low-dose P4; rather, it is conducted using a staggered approach of estrogen-only therapy followed by the addition of full adult doses of exogenous P4 only after 2 years or when breakthrough bleeding occurs.27 This is done to avoid development of tubular breasts, although there are limited data linking early P4 exposure to suboptimal breast development.28
Taken together, production of progesterone is a late event in normal female puberty, and even once it does begin, exposure to progesterone is low and sporadic until well after puberty has completed. Moreover, a subset of girls complete breast development before progesterone production starts. These facts call into some question the role of progesterone in breast development in female puberty, as most breast development appears to be complete prior to the appearance of progesterone. However, more research is still needed on the role of progesterone in breast development during normal puberty.
On the basis of normal female puberty, it seems it may be advisable that if progestogens are introduced in an attempt to enhance breast development in transfeminine people, their introduction be delayed until after 2 or 3 years of hormone therapy, so as to mimic the normal progestogenic exposure of puberty.
Progesterone and Breast Development in Pregnancy
During pregnancy, under the influence of ovarian hyperstimulation and placental formation, there are profound changes in hormonal profiles, including of hormones like estrogen, progesterone, and prolactin, among many others (Table 1). Comparing hormone levels during the menstrual cycle to those during the third trimester of pregnancy, estradiol levels increase on the order of 100-fold, progesterone levels increase on the order of 10- to 20-fold, and prolactin levels increase by around 10-fold (Table 1). Levels of numerous other hormones also change considerably during pregnancy, for instance other estrogens besides estradiol, androgens, gonadotropins (e.g., human choronic gonadotropin or hCG), human placental lactogen (hPL), relaxin, adrenocorticotropic hormone (ACTH), cortisol, aldosterone, growth hormone (GH), and insulin-like growth factor 1 (IGF-1), among others (Goodman, 2009 [Figure]; Mesiano, 2019). These hormones are variously produced by the ovaries, the placenta, and the pituitary gland, among other glands. In response to the myriad hormonal changes during pregnancy, there are dramatic changes to the breasts, which prepare the mother for postpartumlactation and breastfeeding.
Table 1: Changes in hormone levels (estradiol, progesterone, and prolactin) during normal pregnancy:
There are large and dramatic changes in levels of numerous hormones during pregnancy, and the exact hormones responsible for the breast changes during pregnancy are not known (Hytten & Leitch, 1971a; Hytten, 1976). However, it is considered likely, on the basis of animal studies, that a variety of hormones, including estrogen, progesterone, prolactin, placental lactogen, glucocorticoids, and growth hormone, are all importantly involved in different aspects of the maturation (Hytten & Leitch, 1971a; Hytten, 1976; Cox et al., 1999). Moreover, in a quantitative clinical study of breast changes during pregnancy, increases in breast volume and areola size were positively correlated with levels of hPL, while increases in nipple size were positively correlated with levels of prolactin (Cox et al., 1999). Progesterone and prolactin have specifically been implicated in the lobuloalveolar development of the breasts during pregnancy (Bässler, 1970; Lee & Ormandy, 2012; Obr & Edwards, 2012). Both hormones appear to be independently essential in normal lobuloalveolar growth per animal studies (Obr & Edwards, 2012; McNally & Stein, 2017; Hannan et al., 2023). Prolactin likewise appears to be essential in humans, based on case reports of lactation failure in women with isolated prolactin deficiency (Buhimschi, 2004). Conversely, hPL may not be essential for lactation based on case reports of normal lactation in women with very low levels of hPL during pregnancy (Gaede, Trolle, & Pedersen, 1978; Hannan et al., 2023).
On the basis of the preceding, in spite of rather extreme hormonal stimulation, the breast changes of pregnancy, although quite dramatic, are essentially temporary and fully reversible, remaining only as long as continuous hormonal exposure is maintained. This hormonal stimulation includes exposure to extremely high levels of progesterone. It would seem, based on pregnancy, that once pubertal breast development is completed, the breasts are rather unamenable to permanent further growth, whether that involves exposure to progestogens or to a variety of other hormones known to act on the breasts.
Breast Composition and Lobuloalveolar Tissue Proportion
The breasts are made up of two main types of tissue: (1) epithelial tissue, the actual functional internal mammary glandular tissue, including ducts and alveoli or lobules; and (2) stromal tissue, a mixture of connective tissue and adipose (fat) tissue. Lobuloalveolar development refers to growth and maturation of the alveoli and lobules, and hence is a form of epithelial or glandular development. Progestogens are involved primarily in lobuloalveolar development of the breasts, which is the type of breast development that is necessary for lactation and breastfeeding and that occurs mainly during pregnancy.
During pregnancy and lactation in humans, the breasts undergo dramatic changes, and epithelial tissue comes to make up a much greater proportion of the breasts (Ramsay et al., 2005; Bland, Copeland, & Klimberg, 2018). In fact, sources state that glandular tissue comprises a majority of the breast during pregnancy and lactation, with one study of lactating women finding that the breasts were composed 63% (range 46–83%) of glandular tissue (Ramsay et al., 2005). This is not merely due to lobuloalveolar development and glandular growth, but is also due to a marked reversible reduction in mammary adipose tissue (Wang & Scherer, 2019; Alex, Bhandary, & McGuire, 2020). In any case, under more normal physiological circumstances and progesterone exposure, the contribution of lobuloalveolar tissue to the size of the breasts would appear to be quite small. In relation to this, outside of pregnancy levels of progesterone, the significance of progestogen-mediated breast lobuloalveolar growth in terms of breast size is unclear but seemingly questionable (Orentreich & Durr, 1974; Wierkcx, Gooren, & T’Sjoen, 2014).
Breast Development in Cisgender Women with Complete Androgen Insensitivity Syndrome and Consequent Absence of Progesterone
It has been claimed that progesterone helps to move transfeminine people and cisgender females from Tanner stage 4 to 5 breast development and that it helps to round out the breasts (e.g., Vorherr, 1974a; Prior, 2011; Prior, 2019a; Prior, 2020). It has also sometimes been claimed in the online transgender community that cisgender women with complete androgen insensitivity syndrome (CAIS), an experiment of nature of women who lack progesterone, are stuck at Tanner stage 4 breast growth and have “cone-shaped” breasts due to their absence of progesterone. In actuality however, there is no good evidence at this time that progesterone is required for normal pubertal breast development, that progesterone is needed to reach Tanner stage 5, or that it helps to round out the breasts. Such claims are contradicted by extensive available literature and evidence, including notably the literature on CAIS women themselves.
Women with CAIS are individuals who have a 46,XY karyotype (i.e., are genetically “male”), testes, and who would otherwise have physically developed as males, but did not because they have a mutation in the gene encoding the androgen receptor that makes them completely insensitive to the effects of androgens. There are also incomplete forms of the syndrome, like partial androgen insensitivity syndrome (PAIS) and mild androgen insensitivity syndrome (MAIS). CAIS women have a male-typical hormonal profile, generated by their testes, including high male-range levels of testosterone, low female-range estradiol levels, and negligible progesterone levels (Wiki; Table). Instead of developing physically as males however, CAIS women are perfectly phenotypically female, with a normal female body, vagina, and breasts (Wiki; Photo). Their testosterone has been unable to masculinize them, while their estradiol, unopposed by androgens, is able to fully feminize them. The internal reproductive system in CAIS women is essentially that of a highly underdeveloped male, with testes instead of ovaries, no uterus, fallopian tubes, or cervix, and no prostate gland or seminal vesicles. The testes are internally located, either intra-abdominally, inguinally, or labially. They are usually surgically removed by early adulthood, as they otherwise have a high risk of developing testicular cancer because of their location. The vagina in CAIS women is often short and is blind-ending, which is related to their lack of a uterus. In terms of behavior, gender, and sexuality, CAIS women are described as feminine.
Despite claims that CAIS women have generous breast sizes however, in actuality, some CAIS women have large breasts, while some have small breasts. One study found a wide range of breast size measurements of 16×14 cm to 41×31 cm, which equates to an almost 6-fold variation in breast size as quantified by area (Wisniewski et al., 2000). Moreover, the breasts of CAIS women have never been directly compared to those of normal women. Hence, there are no clear data at this time that the breasts of CAIS women are actually larger than average for women. The variation in breast growth in CAIS women parallels the same large variation in breast size between individuals that is seen in cisgender women in general. Here is a collection of photos of CAIS women and their breast development from published case reports and reviews throughout the literature. As can be seen from these photos, breast development in CAIS women is normal and often excellent, although subject to considerable variation between individuals in terms of breast size and shape as in women generally.
If CAIS women truly do have enhanced breast development and breast sizes compared to normal women, it may be that their androgen insensitivity, and hence lack of inhibition of estrogen-mediated breast development by androgens, is responsible for this (Wilson, 1968; Sobrinho, Kase, & Grunt, 1971; Andler & Zachmann, 1979; Zachmann et al., 1986; Patterson, McPhaul, & Hughes, 1994; Barbieri, 2019). Another theoretical possibility is that the high testosterone levels may be aromatized into greater amounts of estradiol locally within the breasts and other tissues in CAIS women and that this may somehow allow for enhanced breast development (Ladjouze & Donaldson, 2019). Interestingly, it has been claimed anecdotally by some researchers that breast development is much better in CAIS women who are allowed to naturally undergo puberty with their own endogenous hormones compared to CAIS women who undergo gonadectomy before puberty and have pubertal maturation induced with exogenous estrogen therapy (Dewhurst, 1972; Glenn, 1976; Dewhurst, 1981; Reindollar & McDonough, 1985; Shearman, 1985; Laufer, Goldstein, & Hendren, 2005). This is to the extent that some CAIS women who have had induced puberty have needed to undergo surgical breast augmentation due to poorly developed breasts (Dewhurst, 1981; Shearman, 1985). In relation to the preceding, it is usually standard clinical practice to delay gonadectomy in CAIS women until puberty has fully completed (Laufer, Goldstein, & Hendren, 2005). However, one clinical study reported good breast development rated as Tanner stage 5 in all cases in CAIS women who experienced either spontaneous or therapeutic puberty (Cheikhelard et al., 2008). It may be important to mimic normal pubertal estrogen exposure with puberty induction in CAIS females by employing low physiological estradiol levels that are slowly and gradually increased over a few years (Dewhurst, 1981; Cheikhelard et al., 2008; Bertelloni et al., 2011).
Baron evaluated a total of 41 people with androgen insensitivity syndrome (AIS) and found that 97% of CAIS women had normal breast development while 63% of individuals with “incomplete AIS” (likely PAIS) had normal breast development (Baron, 1993; Baron, 1994a; Baron, 1994b). In another earlier published study of 50 CAIS females, by Sir Christopher John Dewhurst, 76% were rated as having full breast development, 14% as having moderate breast development, 10% as having “mild” breast development, and 0% as having absent breast development (Dewhurst, 1971b). Hence, based on findings in large samples of CAIS females, most to almost all have normal or full breast development. That a minority of CAIS females have had less breast growth may be due to factors like low and inadequate estradiol levels in some individuals, young age at time of assessment by which point breast development has not fully completed, and/or a small subset of women in general having underdeveloped or small breasts.
CAIS women have never been described in the literature as having “cone-shaped”, “pointy”, or otherwise abnormal breasts. The only exception is that they are often said to have nipples and areolas that are described as “juvenile”, “infantile”, “small”, “pale”, and “non-pigmented” (e.g., Photo) (e.g., Morris, 1953; Morris & Mahesh, 1963; Simmer, Pion, & Dignam, 1965; Dewhurst, 1967; Khoo & Mackay, 1972; Perez-Palacios & Jaffe, 1972; Dewhurst & Spence, 1977). This has been said to be the case regardless of breast size or maturation (Khoo & Mackay, 1972). A possible reason for this phenomenon is that estradiol levels in CAIS women are relatively low, only about 35 pg/mL (130 pmol/L) on average (Wiki; Table). This is relevant as estrogens are known to concentration-dependently produce nipple and areolar pigmentation and enlargement (e.g., Davis et al., 1945 [Figure]; Kennedy & Nathanson, 1953). In contrast to estrogens, progestogens have not been implicated in nipple or areolar pigmentation. Hence, it seems that higher estrogen levels may be necessary for full adult-like nipple and areolar maturation.
Despite their often large breasts, CAIS women are said to have relatively little breast glandular tissue, as opposed to fat and connective tissue, and to have minimal breast lobuloalveolar development (Morris, 1953; Morris & Mahesh, 1963; Simmer, Pion, & Dignam, 1965; McMillan, 1966; Perez-Palacios & Jaffe, 1972; Dewhurst & Spence, 1977; Shapiro, 1982). This is in accordance with the lack of progesterone in CAIS women, since progesterone is important in mediating lobuloalveolar growth. The retained breast sizes of CAIS women despite reduced glandular and lobuloalveolar structures is consistent with the fact that the breasts are composed mostly of stromal adipose and connective tissue. Hence, as touched on previously in this article, greater glandular or lobuloalveolar formation in the breasts may not necessarily translate to greater breast size, which seems readily apparent in CAIS women.
The normal and excellent breast development of CAIS women is notable because these individuals, owing to their testes and hence absence of significant gonadal progesterone production, have very low and negligible levels of progesterone (Wiki; Table; Barbieri, 2019). CAIS womens’ normal breast development, often large breasts, and ability to reach complete breast maturation, as measured by the Tanner scale, are collectively suggestive that progesterone is not required for normal or complete pubertal breast development (Barbieri, 2019). In any case, it must be noted and cautioned again that the breasts of CAIS women have never been directly compared to those in normal women. In addition, quantitative studies of the breasts of CAIS women are very scarce, and much of our knowledge in this area is based on anecdotal clinical experience and subjective breast evaluation. This is in large part due to the rarity of CAIS women and the difficulty in obtaining decent samples of them for study. Furthermore, CAIS women also have other differences from regular women besides their lack of progesterone, for instance their relatively low circulating estradiol levels, high testosterone levels (which can be aromatized into estradiol within tissues like the breasts), androgen insensitivity, and XY karyotype, among others. Hence, the insights into breast development provided by CAIS women come with a variety of caveats.
Interestingly, in spite of their well-developed breasts, breast cancer has never been reported in CAIS women, and would appear to be very rare in these individuals (Aly, 2020b; Aly, 2020c). This may be related to factors like the lack of progesterone and lobuloalveolar maturation in CAIS women and/or their absence of a second X chromosome (Aly, 2020b; Aly, 2020c). CAIS women suggest that breast cancer is not an inherent eventual consequence of excellent breast development.
Menstrual Cycles and Temporary Cyclic Breast Enlargement
The enlargement of the breasts during the luteal phase of the menstrual cycle is believed to be due to temporary glandular and stromal tissue growth, luminal dilation of the ducts and alveoli, fluid retention in the glandular and stromal structures, and increased vascularization and blood flow (Scott et al., 1950; Drife, 1989; Fowler et al., 1990; Hussain et al., 1999; Alekseev, 2021; Biswas et al., 2022). However, studies suggest that most of the changes are merely due to water fluctuations and that change in breast glandular volume is relatively small (Rix et al., 2023). The breast changes during the menstrual cycle, such as breast enlargement, have been positively correlated with increased levels of estradiol and progesterone during the luteal phase (Jemström & Olsson, 1997; Jasieńska et al., 2004; Clendenen et al., 2013; Rix et al., 2023). Correspondingly, combined estrogen and progestogen therapy has been found to reversibly increase breast size (e.g., Hartmann et al., 1998). Estradiol levels are also positively associated with breast tenderness during estrogen therapy, whereas progestogens may actually reduce breast tenderness (e.g., de Lignières & Mauvais-Jarvis, 1981 [Figures]; Sitruk-Ware et al., 1984; Wiki; Wiki). Both estradiol and progesterone can promote water retention via distinct hormonal mechanisms as well as mediate breast glandular growth and changes (Rix et al., 2023). As such, the breast changes during the menstrual cycle are assumed to be due to changing levels of estradiol and progesterone, though it is noteworthy that progesterone has been particularly implicated owing to the breast volume increase occurring during the luteal phase (Lawrence & Lawrence, 2015; Rix et al., 2023). There is a delay in breast volume increases following the peaks of estradiol and progesterone levels during the menstrual cycle and hence the changes are not instantaneous (Rix et al., 2023).
Combined oral contraceptives, which are estrogen–progestogen preparations, as well as menopausal estrogen–progestogen hormone therapy, may produce temporary breast enlargement and feelings of breast fullness analogous to those that occur during the luteal phase of the menstrual cycle (Milligan, Drife, & Short, 1975; Dennerstein et al., 1980 [Figure]; Malini, Smith, & Goldzieher, 1985; Jemström & Olsson, 1997; Jernström et al., 2005). In one study, breast volume was around 100 mL greater (~30% higher) in women who were currently taking oral contraceptives relative to those who had not taken or had previously taken oral contraceptives (Jemström & Olsson, 1997). In some women, the increase in breast size with oral contraceptives was subjectively reported to be up to a single bra cup size in volume (Jemström & Olsson, 1997). However, in another study by the same group of researchers that had a much larger sample size (n=258 vs. n=65), breast volumes were not significantly different between current hormonal contraceptive users and non-users (Jernström et al., 2005). Additionally, another study found no significant differences in breast volume in women between different estrogen–progestogen oral contraceptives that had about 6-fold variation in dose of the same progestin (0.4 to 2.5 mg/day norethisterone) as well as non-users (Malini, Smith, & Goldzieher, 1985). However, this study was underpowered due to small sample sizes (n=5 to n=15 per group) (Malini, Smith, & Goldzieher, 1985).
Engman et al. (2008) conducted an RCT of treatment with mifepristone, a selective progesterone receptor modulator (SPRM) with predominantly antiprogestogenic effects, versus placebo for 3 months in normally cycling premenopausal cisgender women, and evaluated the effects of this progesterone receptor blockade on the breasts. They found that mifepristone significantly reduced Ki-67 index, a measure of cellular proliferation in the breasts, and reduced subjectively rated symptom scores on the Breast Symptom Index (BSI). More specifically, breast soreness, breast swelling, sense of increased breast volume, and the total breast symptoms score were all significantly reduced on the BSI. However, breast volume was not objectively measured in this study. A major limitation of this study is that mifepristone inhibits ovulation and modifies levels of estradiol and other hormones (Spitz et al., 1989; Spitz et al., 1994; Engman et al., 2008, Spitz, 2010). As such, it is unclear whether the effects observed by Engman and colleagues were specifically due to progesterone receptor antagonism in the breasts or due to disruption of the hypothalamic–pituitary–gonadal (HPG) axis, for instance lowered estradiol levels.
An interesting case report of an adult woman with CAIS documented a significant increase in breast volume with combined estrogen–progestogen therapy relative to estrogen monotherapy (Dijkman et al., 2023b). The woman was started on cyclic oral estradiol 2 mg/day and dydrogesterone 10 mg/day and subjectively experienced breast pain and fluctuations in breast volume of about one cup size while on this regimen. Subsequently, she was switched to oral estradiol valerate 3 mg/day monotherapy and the fluctuations in breast volume ceased. However, her overall breast volume was reduced as well, and the woman decided to resume combined estradiol and dydrogesterone therapy. Her clinicians proceeded to measure her breast volume using 3D body scanning. Her left breast was 758 mL and right breast was 673 mL with estrogen monotherapy, and her breasts increased to respective volumes of 875 mL and 784 mL during combined estrogen–progestogen therapy, giving net volume increases of 117 mL (+16%) and 111 mL (+17%). These differences in volume corresponded to an almost one bra cup difference in size. The researchers noted that estradiol and progesterone are associated with cyclical breast changes, and hypothesized that the changes in their patient were due to increased fluid retention in the breasts. Taken together, the case report demonstrates that progestogens can cause rapid and considerable reversible breast enlargement in some women analogous to that during the normal menstrual cycle.
Progesterone and Mammary Development in Animals
Progesterone and Pubertal Mammary Development in Animals
Although progesterone does not seem to be essential in normal pubertal mammary development in mice, studies have interestingly found that it is able to substitute for estrogen in mediating pubertal ductal mammary development in this species. Ruan, Monaco, & Kleinberg (2005) studied the effects of various combinations of exogenous estradiol, progesterone, and IGF-1 on mammary development in oophorectomized female IGF-1-knockout mice. In terms of stimulation of ductal development to occupy the mammary gland fat pad, the combination of progesterone and IGF-1 produced 92% occupation, estradiol and IGF-1 resulted in 92% occupation, estradiol, progesterone, and IGF-1 achieved 96% occupation, and IGF-1 alone resulted in only 28% occupation (Ruan, Monaco, & Kleinberg, 2005; Kleinberg & Ruan, 2008). In terms of gross anatomical appearance, the ductal tree with progesterone and IGF-1 was said to resemble that of a normal fully developed pubertal mammary gland (Ruan, Monaco, & Kleinberg, 2005). However, differences in mammary development between the combination of estradiol and IGF-1 and the combination of progesterone and IGF-1 were apparent, with estradiol and IGF-1 having greater effect on terminal end bud formation, ductal decorations, and slight alveolar maturation, and progesterone and IGF-1 having more effect on ductal formation, extension, and branching (Ruan, Monaco, & Kleinberg, 2005; Kleinberg & Ruan, 2008). The effects of progesterone on mammary development were reversed by the progesterone receptor antagonist mifepristone (Ruan, Monaco, & Kleinberg, 2005). Only the combination of estradiol, progesterone, and IGF-1 produced mammary development that resembled that during mid-pregnancy, with full maturation of secretory alveolar structures (Ruan, Monaco, & Kleinberg, 2005; Kleinberg & Ruan, 2008).
A limitation of studies that have used exogenous progesterone to stimulate pubertal ductal mammary development in mice is that the doses of progesterone employed, in conjunction with other hormones like estradiol, have been sufficient to mediate mammary growth to a level typical of pregnancy, with robust maturation of mammary lobuloalveolar structures (e.g., Škarda, Fremrová, & Bezecný, 1989; Ruan, Monaco, & Kleinberg, 2005). Pregnancy is a time when hormone levels are much higher than usual. Hence, the progesterone exposure in these studies may have been supraphysiological relative to normal puberty, and may have produced effects on mammary growth that would not otherwise occur during this time. Accordingly, Škarda, Fremrová, & Bezecný (1989) found that whereas untreated normal female mice naturally grew to a mammary gland area of 26.4 mm2 and normal female mice treated with exogenous estradiol grew to a mammary gland area of 25.3 mm2, normal female mice treated with exogenous estradiol and progesterone grew to a mammary gland area of 43.5 mm2 and with exogenous progesterone alone to a mammary gland area of 64.6 mm2. The untreated control mice did not show alveolar buds, whereas the progesterone-treated groups did have alveolar maturation, indicating supraphysiological and pregnancy-like development compared to non-pregnant mice (Škarda, Fremrová, & Bezecný, 1989). In any case, one study employed low doses of progesterone (0.1 mg/day), one-tenth of that used in most other studies (1 mg/day), and found that progesterone still stimulated significant ductal development in mice at these doses (Aupperlee et al., 2013; Berryhill, Trott, & Hovey, 2016). Hence, progesterone is still able to stimulate some level of ductal growth in mice even at lower levels.
Although progestogens by themselves can apparently stimulate normal pubertal mammary development in lieu of estrogen exposure in mice, it is not clear that they do so similarly in humans. It is well-known that progestogens alone, without concomitant estrogenic activity, do not generally produce breast development in humans. As an example, progestogens, for instance MPA and CPA, have been used as puberty blockers in boys and girls at very high doses, and do not produce breast development in this context, instead causing arrest and regression of breast development via gonadal suppression (Lyon, De Bruyn, & Grant, 1985; Fuqua & Eugster, 2022). Cases of gynecomastia in boys have occurred with CPA, but only in a minority and with this easily attributable to other causes than progestogenic activity, for instance the antiandrogenic activity of CPA and disruption of the HPG axis (Kauli et al., 1984; Laron & Kauli, 2000). Similarly, progestogens like MPA and CPA have been used at very high doses in men to treat prostate conditions and sexual disorders, and likewise do not usually produce gynecomastia under these circumstances. Rates of gynecomastia with CPA used in the treatment of prostate cancer are low and are not noticeably different from the rates with surgical or medical castration (~10%) (Fourcade & McLeod, 2004; Di Lorenzo et al., 2005). This is in major contrast to the high rates of gynecomastia with estrogens and nonsteroidal antiandrogens (up to 70–80%) (Fourcade & McLeod, 2004; Di Lorenzo et al., 2005; Deepinder & Braunstein, 2012). Species differences may be present such that progestogens can produce robust pubertal mammary development in mice but do not do so in humans.
Progesterone and Gestational Mammary Development in Animals
Therapeutic or pharmacological pseudopregnancy is a type of hormone therapy that attempts to replicate the hormonal mileu of pregnancy for certain medical indications in cisgender females by administering exogenous hormones. In practice, this has involved the administration of very high doses of estrogens and progestogens, with most other pregnancy hormones not included. Therapeutic pseudopregnancy was first developed in the 1950s and is largely no longer used in medicine today (Kaiser, 1993).
The effects of therapeutic pseudopregnancy on the breasts are of interest due to the breast changes that occur during pregnancy, for instance lobuloalveolar development and substantial reversible breast enlargement. In the 1980s, Lauritzen and colleagues conducted a study of therapeutic pseudopregnancy for treatment of breast hypoplasia (small/underdeveloped breasts) in cisgender women (Lauritzen, 1980; Lauritzen, 1982; Lauritzen, 1989; Göretzlehner & Lauritzen, 1992). They employed the estrogen estradiol valerate 40 mg/week and the progestogen hydroxyprogesterone caproate (OHPC) 250 to 500 mg/week both by intramuscular injection for 4 to 5 months. The estradiol valerate dosage employed was very high, with other studies by the same authors reporting that this dosage of estradiol valerate resulted in first-trimester pregnancy levels of estradiol in women (~3,000 pg/mL [~11,000 pmol/L]) (Ulrich, Pfeifer, & Lauritzen, 1994; Ulrich et al., 1995). These estradiol levels are roughly 30 times the normal concentrations outside of pregnancy (Aly, 2018b). Similarly, the OHPC doses were very high, with 250 to 500 mg per month being similar in strength to luteal-phase progestogenic exposure (Wiki). Hence, as the same OHPC doses were used weekly in the study, the doses were roughly around 4.5 times luteal-phase exposure and thus were analogously similar to first- or second-trimester progesterone levels in terms of strength (Aly, 2020d). The authors noted that they had initially tried lower hormone doses, similar to those originally used in the 1950s, but did not achieve significant breast growth with these doses, and so increased the dosage. Breast changes were measured in the study with a tape measure (applied horizontally and vertically to the breast area), photographs, breast imaging using mammography and sonography, and, later in the study, plasticine impressions/molds with determination of the filling volume.
Lauritzen and colleagues reported the study findings in four different publications with different follow-up times and growing sample sizes. In the final follow-up, a total of 221 women had been treated. In the second follow-up, when 78 women had been treated, it was noted that 29 of the cases (37%) were less than 18 years old. However, in the final follow-up of 221 women, the age range was listed as 18 to 42 years. The researchers found that breast volume increased by 10 to 30% above baseline in 65% of the women. This was also accompanied by breast tenderness in almost all of the women, though the breast tenderness progressively declined during the treatment period. Other breast-related side effects like pigmentation and stretch marks were rarely observed. Prolactin levels slightly increased to 14 to 28 pg/mL by the end of treatment. Breast imaging showed an increase in the density of breast glandular tissue. The researchers claimed that the increase in breast size in their study was due to increased adipose tissue, water retention, and moderate hypertrophy of the glandular tissue.
Following treatment discontinuation, the increases in breast volume gradually and partially regressed in 40% of the women, to an increase of 10 to 20% above baseline. However, the authors claimed that the regression in breast volume could be reduced with adequate-dose combined estrogen–progestogen birth control pills or with topical estrogen and progestogen therapy applied to the breasts. In addition, they noted that therapeutic pseudopregnancy could be repeated to increase breast volume again. This was performed in a subset of the women, with treatment repeated 1 to 2 times after 6 months. In the second follow-up, which had 78 women, it was noted that 12 women (15%) had undergone multiple treatments. Aside from Lauritzen and colleagues, many other researchers have also reported substantial or full regression in breast size following estrogen and/or progestogen therapy to increase breast size in cisgender women (e.g., Cernea, 1944; Müller, 1953; Anderson, 1962; Bruck & Müller, 1967; Keller, 1984; Kaiser & Leidenberger, 1991; Keller, 1995; Hartmann et al., 1998).
The findings of Lauritzen and colleagues were reported very informally, in the form of non-peer-reviewed book chapters, conference papers, and medical magazines, and were never published in a peer-reviewed journal article. In relation to this, the methodology and results of the study were only briefly and imprecisely described. There are also additional concerns related to study design, such as lack of controls, randomization, and the quality of the breast measurement methods. As a result of the preceding issues, it is difficult to fully interpret the results of the study and to have complete confidence in its findings. In any case, Lauritzen and colleages’ results suggest that treatment with high-dose combined estrogen–progestogen therapy, achieving earlier-pregnancy estrogenic and progestogenic exposure, may be able to produce a significant temporary increase in breast size and a smaller long-term increase. The findings of a permanent increase in breast size conflict with those of other researchers who have reported complete regression in breast changes following treatment discontinuation. Moreover, the results are contradicted by findings in pregnant women, who, as described previously, show complete reversion to pre-pregnancy breast size or to even slightly smaller breasts following cessation of lactation.
It is difficult to evaluate the relative roles of the estrogen and the progestogen in the findings of Lauritzen and colleagues, as there were no comparison groups employing estrogen or progestogen therapy alone in the study. Both estrogens and progestogens have been implicated in causing breast enlargement and plausibly could have contributed to the breast changes. As such, it is unclear to what extent the breast changes were specifically due to progestogenic exposure rather than to estrogenic exposure.
The breast size increases observed by Lauritzen and colleagues were seemingly more modest relative to those that occur normally during pregnancy. They also lacked certain characteristics of pregnancy-related breast changes, like nipple and areolar pigmentation. The reasons for this are not fully clear. The subject populations between these studies were different, for instance in terms of factors like initial breast size and age, which may be contributing reasons. Another possible contributing factor is that only estrogen and progestogen levels increased in the study, whereas levels of other pregnancy hormones, besides the slight increase in prolactin levels, did not increase. These other pregnancy hormones, for instance hPL and IGF-1, may also be involved in breast development during pregnancy. Finally, the treatment duration was only 4 to 5 months, and the estrogen and progestogen exposure was only similar to that during early-to-mid pregnancy, whereas normal pregnancy lasts 9 months and involves continued dramatic increases in estrogen and progesterone levels through to childbirth.
It should be noted that, owing to the highly supraphysiological estrogen and progestogen levels required, which can cause serious health complications like blood clots and cardiovascular problems (Aly, 2020e), as well as the small to negligible lasting increase in breast volume, therapeutic pseudopregnancy is inadvisable for transfeminine people and should not be pursued or employed. Nonetheless, the historical findings of therapeutic pseudopregnancy for increasing breast size in cisgender females are of significant theoretical interest in exploring the roles of estrogens and progestogens in breast growth.
Early Progestogen Exposure and the Possibility of Suboptimal Breast Development
While progestogens are typically sought after by transfeminine people for their potential in improving breast development, there have also been various suggestions in the literature that early or premature exposure to progestogens may result in suboptimal breast development and that progestogens may suppress or reduce estrogen-mediated breast development. These suggestions include progestogens having known antiestrogenic effects in the breasts, animal studies finding stunted mammary development with high doses of progestogens, clinical publications cautioning against premature introduction of progestogens in female puberty induction due to concerns about possibly stunted breast growth, clinical use of progestogens to treat macromastia in cisgender females, poor breast development with estrogen therapy in cisgender girls with a disorder of sexual development that results in high progesterone exposure, and breast development with estrogen and CPA (a very strong progestogen) typically being poor in transfeminine people. As with the question of whether progestogens can enhance breast development, it is currently unknown whether progestogens could worsen breast development. It is also unknown what dosage level and timing of introduction would be required for such an effect. In any case, for informational purposes, the preceding topics will each be discussed in the subsequent sections.
Antiestrogenic Effects of Progestogens in the Breasts
Stunted Mammary Growth with Progestogens in Animal Studies
Animal studies using progestogens including bioidentical progesterone and chlormadinone acetate (CMA), a progestin closely related to CPA, have found that high doses of these progestogens substantially stunt mammary gland development in rabbits, whereas lower doses do not do so (Lyons & McGinty, 1941; Beyer, Cruz, & Martinez-Manautou, 1970). See here for relevant literature excerpts as well as figures from these studies. Lyons & McGinty (1941) [Figure] found that estrogen alone induced ductal mammary development and estrogen plus progesterone 0.25 to 1 mg/day produced ductal development and slight to “fair” lobuloalveolar development. Conversely, estrogen plus progesterone 4 to 8 mg/day, which were 4- to 8-fold higher doses of progesterone than the most optimal dose, produced stunted mammary development with inhibited ductal development, only slight lobuloalveolar development, and, at the highest dosage, resulted in a much smaller mammary gland in terms of size than in the ≤1 mg/day groups. They concluded that high doses of progesterone are inhibitory and result in relatively poor mammary development. In the paper, doses of progesterone in international units (IU) were reported, but a citing review, Pfeiffer (1943), indicated that 1 IU progesterone is equal to 1 mg progesterone. As such, the milligram doses are listed above instead. Beyer, Cruz, & Martinez-Manautou (1970) [Figure] found that estrogen alone produced good ductal development without lobuloalveolar growth (mean mammary area = 376 mm2) and both estrogen plus CMA 0.5 mg/day and estrogen plus progesterone 2.5 mg/day produced optimal ductal and lobuloalveolar development (mean mammary area = 765 mm2 and mean mammary area = 688 mm2, respectively). Conversely, estrogen plus CMA 2.5 mg/day, a 5-fold higher dose of CMA than the optimal dose, resulted in dramatically reduced ductal development and mammary gland size albeit with significant lobuloalveolar growth (mean mammary area = 284 mm2). The authors concluded that moderate doses of progestogens stimulate mammary gland growth whereas large doses inhibit mammary gland development.
While these animal studies are suggestive that high doses of progestogens may be able to stunt breast development in humans, this is far from a certainty. There are species differences in hormone-mediated mammary development such that findings in one species, such as rabbits, may not translate to another species, like humans, or sometimes even to closely related species, like rats or guinea pigs (Bässler, 1970). As far as the present author is aware, stunted mammary development with high doses of progestogens has not been studied or reported in other animal species, for instance other rodent species or monkeys. It is also unclear that the doses employed in these animal studies are necessarily relevant to progestogen therapy in humans. This is because pregnancy levels of progesterone, which are much higher than luteal-phase progesterone levels, are necessary for substantial mammary lobuloalveolar development, and the doses of progestogens used in these studies were above that magnitude of progestogenic exposure. Hence, the doses may have corresponded to what in humans would be extremely high doses. However, such doses could still be relevant in the case of CPA used as an antiandrogen in humans, as CPA is used in this context at very high doses (see section below). The present author is unaware of any animal studies finding that physiological non-pregnancy levels of progesterone have any stunting or other adverse influence on mammary development, suggesting that only high doses of progestogens may have such effects. Finally, it seems notable that the estrogen and progestogen were initiated simultaneously in these animal studies and yet produced optimal pregnancy-like mammary development at the right doses. This suggests that early or immediate progestogen exposure might not be unfavorable in terms of breast development in humans. However, once again species differences may be present and confirmatory clinical studies are needed in humans.
Clinical Publications Cautioning Against Premature Introduction of Progestogens Due to Possibly Stunted Breast Development
A large number of clinical publications largely in the pediatric endocrinology literature have warned that premature exposure to progestogens during for instance puberty induction may result in suboptimal breast development in cisgender girls and/or transfeminine people (Zacharin, 2000; Bondy et al., 2007; Colvin, Devineni, & Ashraf, 2014; Wierckx, Gooren, & T’Sjoen, 2014; Kaiser & Ho, 2015; Bauman, Novello, & Kreitzer, 2016; Gawlik et al., 2016; Randolph, 2018; Donaldson et al., 2019; Heath & Wynne, 2019a; Heath & Wynne, 2019b; Iwamoto et al., 2019; Crowley & Pitteloud, 2020; Naseem, Lokman, & Fitzgerald, 2021; Federici et al., 2022; Lucien et al., 2022; Rothman & Iwamoto, 2022). The full relevant excerpts from these sources can be found here. In relation to these claims, and in order to mimic normal female puberty, a progestogen is not typically added to estrogen therapy during puberty induction in cisgender girls with delayed puberty until after about 2 to 3 years of treatment, by which point breast growth is generally considered complete. Additionally, progestogens are generally never added as part of puberty induction in transfeminine adolescents. Despite the preceding widespread literature statements and accepted clinical practices in the field of puberty induction however, it is important to note that the claims that premature introduction of progestogens might stunt breast development in this context are currently not based on any actual reliable clinical evidence and hence remain unsubstantiated. It is not even clear that these statements are based on anecdotal clinical experience as opposed to simple conjecture. The absence of data in this area may finally change in the future as more clinical studies of progestogens in puberty induction in cisgender girls are conducted (e.g., Rodari et al., 2023).
Rodari and colleagues studied optimization of puberty induction with estrogen therapy followed by eventual introduction of progestogen therapy in 49 cisgender girls with hypogonadism (e.g., Rodari et al., 2022; Rodari, 2022; Rodari et al., 2023). The researchers employed incrementally titrated low-dose transdermal estradiol to mimic the low and gradually increasing estradiol levels during normal puberty and added a progestogen only once menstrual bleeding began. The total duration of treatment was mean 2.65 ± 1 years, the time of first menstrual bleeding occurrence was 2.3 ± 1 years, and the time of progestogen introduction was median 2.22 years (IQR 1.56–2.87 years). Of the girls, 90% reached Tanner breast stage 4, but only 41% reached Tanner breast stage 5. Reaching the final Tanner breast stage was significantly associated with the number of estradiol dose increases (i.e., gradual estradiol dose titration) and the estradiol dose at progestogen introduction. The researchers interpreted the latter finding as progestogen exposure potentially hampering breast development. They questioned introducing progestogen therapy in the presence of incompletely developed breasts and suggested that instead of adding a progestogen upon onset of menstrual bleeding, clinicians should consider slightly reducing the estradiol dosage to delay progestogen introduction until the breasts complete maturation. While interesting, it must be noted that the findings of Rodari and colleagues are merely correlational, are open to multiple interpretations, and do not causally show that progestogens impair breast maturation.
Progestogens in the Treatment of Breast Hypertrophy
More recently, a couple of studies, both by the same group of researchers, assessed the impact of different types of hormonal contraception on macromastia in adolescent cisgender females with macromastia (Nuzzi et al., 2021; Nuzzi et al., 2022). They found that use of progestin-only contraceptives was associated with significantly more breast tissue removed upon surgical breast reduction (959.9 g/m2 vs. 735.9 g/m2 [+30%]; p = 0.04) and worse clinical symptoms (e.g., breast pain—odds ratio, 4.94, p = 0.005) relative to non-users of hormonal contraception (Nuzzi et al., 2021). Conversely, use of combined oral contraceptives, which are estrogen–progestogen preparations, was associated with significantly less breast tissue removed with breast reduction (639.5 g/m2 vs. 735.9 g/m2 [−13%]; p = 0.003), though not with any differences in clinical symptoms, relative to those naive to hormonal contraception (Nuzzi et al., 2022). It should be noted that progestin-only contraceptives suppress the HPG axis and result in low estradiol levels, whereas combined oral contraceptives suppress the HPG axis and lower estradiol production but simultaneously supplement estrogen signaling by delivering exogenous estrogen. This difference may somehow be responsible for the opposite influence of estrogen–progestogen therapy versus progestogen-alone therapy on macromastia severity. While the findings of Nuzzi and colleagues are interesting, it is noteworthy that the methodology and findings of their research were criticized on various grounds in a letter to the editor concerning one of the articles (Karp, 2022).
Santen et al. (2024), in a case series of cisgender girls with juvenile gigantomastia, noted that breast growth continues for only a number of years following onset and hence there must be some form of stop signal that is activated and that prevents further breast growth. They speculated that this signal may be related to apoptosis (programmed cell death). Santen and colleagues noted that in adult cisgender women, proliferation of breast cells is increased during the follicular phase of the menstrual cycle, whereas apoptosis in breast cells is increased during the luteal phase of the cycle. They hypothesized that the apoptosis during the luteal phase may block further breast development. Since progesterone is produced during the luteal phase and may mediate said apoptosis, this would substantiate the use of progestogens in the treatment of breast hypertrophy. However, the researchers noted that no data exist on apoptosis in the breasts of girls with juvenile gigantomastia. Moreover, an important point against the authors’ hypothesis is that estrogen-induced breast growth gradually slows and ceases in people who do not have menstrual cycles and luteal phases or progestogenic exposure just as it does in normal cisgender girls. Prominent examples of such individuals include CAIS women, transfeminine people, and cisgender men with prostate cancer treated with estrogen therapy.
Poor Breast Development in 17α-Hydroxylase/17,20-Lyase Deficiency
Non-Comparative Clinical Studies of Breast Development with Estrogen and Cyproterone Acetate in Transfeminine People
The possibility of suboptimal breast development with premature exposure to progestogens is of particular relevance in the case of CPA used as an antiandrogen in transfeminine people. This is because CPA is a potent progestogen in addition to antiandrogen, starts to be taken at the initiation of hormone therapy, and happens to be used in transfeminine people at doses that result in very strong to profound progestogenic exposure (Aly, 2019). In terms of progestogenic strength, CPA at a dosage of 2 mg/day is comparable to the progesterone exposure during the luteal phase of the menstrual cycle (Aly, 2019; Wiki). For comparison, CPA has been used in transfeminine people at doses ranging from 10 to 100 mg/day (Aly, 2019). This would mean that CPA provides roughly 6.25 times the progestogenic impact of luteal-phase progesterone exposure at a dosage of 12.5 mg/day, 12.5 times the impact at 25 mg/day, 25 times the impact at 50 mg/day, and 50 times the impact at 100 mg/day. Moreover, this does not consider the fact that progesterone is only produced during the luteal phase, or half of the menstrual cycle, whereas CPA is taken continuously every day of the month. The preceding magnitudes of progestogenic exposure with CPA are on par with and even beyond those during pregnancy. Only recently have lower doses of CPA (e.g., ≤12.5 mg/day) started to be used in transfeminine hormone therapy.
Studies in pubertal and adolescent transfeminine people given GnRH agonists to block puberty plus estrogen therapy have reported good breast development in these individuals as assessed by subjective clinical impression or Tanner staging (de Vries et al., 2010; Hannema et al., 2017). However, quality objective measures of breast development were not employed in these studies. Conversely, non-comparative studies using estrogen plus CPA in adult transfeminine people have commonly reported modest breast development, including incomplete breast development only to Tanner stage 2 to 4, small breast cup sizes, and small breast volumes (Kanhai et al., 1999; Sosa et al., 2003; Sosa et al., 2004; Wierckx et al., 2014; Fisher et al., 2016; Tack et al., 2017; de Blok et al., 2018; Reisman, Goldstein, & Safer, 2019; Meyer et al., 2020; de Blok et al., 2021). Additionally, breast sizes smaller than those in cisgender women have been reported (Asscheman & Gooren, 1992; Kanhai et al., 1999). In one study, breast development with estrogen plus CPA was also poor in late-adolescent transfeminine people (Tack et al., 2017). However, in this particular study, the estrogen dose used was likely too low and resulted in inadequate estradiol levels, as noted by the authors themselves, and this is a potential confounding factor in their findings (Tack et al., 2017). In any case, breast growth with estrogen plus CPA in transfeminine people would seem to consistently be poor. In contrast to the regimen of estrogen and CPA, breast development with other hormone therapy regimens, for instance estrogen with non-progestogenic antiandrogens like spironolactone, bicalutamide, and GnRH modulators, has not been nearly as well-studied in comparison, and hence comparisons of outcomes between regimens is difficult.
In one of the highest quality studies of estrogen and CPA and breast development in adult transfeminine people, breast volume measured with 3D body scanning (Vectra XT) was approximately mean 100 mL (95% CI ~75–125 mL; range up to ~750 mL), equating to less than an A cup size on average, after 3 years of hormone therapy with estrogen and CPA in 69 transfeminine people (de Blok et al., 2021 [Figure]). In this study, breast changes over time had clearly plateaued, suggesting that breast development was either complete or was nearly so (de Blok et al., 2021 [Figure]). Although most of the transfeminine people in this study had less than an A cup breast size (71%), a minority had cup sizes ranging from an A cup (9%), B cup (16%), C cup (3%), to E cup (1%) (de Blok et al., 2021 [Figure]). For comparison, a study of normative data on breast volumes in cisgender women, using a different 3D body scanning device (Artec Eva 3D), found breast volumes of median ~515 mL and mean ~650 mL (IQR ~310–850 mL; range ~50–3,100 mL) in 378 cisgender women (Coltman, Steele, & McGhee, 2017). As such, adult transfeminine people treated with estrogen and CPA would appear to have substantially smaller breasts than cisgender women. However, it must be emphasized that the preceding data come from separate clinical studies and hence are not directly comparative. It is noteworthy in this regard that breast volumes can vary considerably between different studies even using similar measurement methods (e.g., magnetic resonance imaging) (Sindi et al., 2019 [Table]). Hence, there is a need for studies directly comparing breast volumes in transfeminine people to those in cisgender women using the same measurement method in order to comparatively evaluate breast development.
Regardless of the preceding, transfeminine people could simply have poor breast development in general without this necessarily being related to CPA or progestogenic exposure. Indeed, a more recent study in transfeminine people who underwent pubertal suppression in adolescence, presumably with GnRH agonists and then estrogen therapy, found similarly poor breast development as has been reported in adults (Boogers et al., 2022; c.f. de Blok et al., 2021). This study used breast volume via 3D body scanning to measure breast development and found a mean breast volume of 114 mL (IQR 58–203 mL), equating to less than an A cup size, after 4.2 years of hormone therapy (Boogers et al., 2022). It was notably conducted by the same group of researchers who did the earlier higher-quality study in adult transfeminine people, and hence likely used the same 3D scanning method (de Blok et al., 2021).
No directly comparative studies of breast development with CPA versus other antiandrogens in transfeminine people are currently available. Hence, it’s not fully known whether the findings are specific to CPA or also generalize to other antiandrogens that are not also strongly progestogenic. The RCT of estradiol and spironolactone versus estradiol and CPA in transfeminine people by Ada Cheung and colleagues underway in Australia may provide more insight on this issue, as spironolactone is only a weakly or clinically non-progestogenic antiandrogen (Aly, 2018b; Wiki; update: see below).
Additional Considerations for Progestogen Therapy and Breast Development in Transfeminine People
Anecdotes About Progestogens and Breast Development
Many transfeminine people who have taken progestogens as part of hormone therapy have anedotally reported that the progestogens improved their breast development. At the same time, many other transfeminine people have anecdotally reported no benefit of progestogens to breast development. It must be cautioned in general that anecdotal reports are unreliable and represent a very low form of medical evidence. This is because subjective observations and attributions are often erroneous. Perceptions can be faulty and inaccurate, especially with slowly developing physical changes, and true physical changes can be due to coincidence and unrelated confounding factors rather than due to a person’s causal attributions. A couple notable examples of potential confounding factors with regard to progestogens and breast development include: (1) continued breast development from estrogen acting on its own; and (2) temporary breast enlargement due to local fluid retention, increased blood flow, and reversible lobuloalveolar growth caused by progestogens. Such factors have the potential to mislead, and may contribute significantly to anecdotal reports of enhanced breast development with progestogens in transfeminine people. Clinical studies that are well-designed, controlled, and employ reliable objective measures, with long-term follow-up and eventual discontinuation of the progestogen to control for reversible effects, are needed to properly evaluate the effects of progestogens on breast development.
Therapeutic Limitations of Oral Progesterone
Oral progesterone produces very low progesterone levels and has only weak progestogenic effects even at high doses (Aly, 2018a; Wiki). These low progesterone levels are likely to be inadequate in terms of desired physiological progestogenic effects, for instance in the breasts. Oral progesterone also uniquely has potent neurosteroid actions via active metabolites like allopregnanolone, which can result in prominent side effects such as alcohol-like central nervous system inhibition as well as mood swings (Aly, 2018b; Wiki; Wiki). These neurosteroid effects are dose-dependent and are more severe at high doses. Non-oral progesterone forms like rectal or injectable progesterone or progestins, which do not have the preceding problems, can be used instead to avoid such concerns (Aly, 2018a; Aly, 2018b).
Tolerability and Safety Considerations for Progestogens
Progestogens have a variety of tolerability issues and safety risks (Aly, 2018b). Examples of such risks variously include adverse mood changes, breast cancer, blood clots, cardiovascular complications, benignbrain tumors including prolactinomas and meningiomas, and off-target actions with undesirable effects (e.g., androgenic or glucocorticoid activity), among others (Aly, 2018b). CPA at high doses also uniquely has a significant risk of serious liver toxicity (Aly, 2018b). The risks of progestogens vary depending on the specific progestogen and dosage, but all progestogens, including even bioidentical progesterone, have significant known risks. The risks of progestogens, along with lack of evidence of beneficial effects in terms of feminization, well-being, and health, are why there are concerns about and hesitations on their use in transfeminine people (Aly, 2018b). However, cisgender women naturally have progesterone in their bodies, and the absolute risks of progestogens are low (Aly, 2018b). The risks of progestogens can be minimized by use for a limited duration of time (e.g., a few years), by using the lowest dosages expected to be effective in terms of desired effects, and by selection of progestogens with more favorable pharmacological profiles (Aly, 2018a; Aly, 2018b).
Updates
Update 1: Angus et al. (2023–2024)
It was previously reported in this article that an RCT assessing breast development with estradiol plus spironolactone versus estradiol plus CPA in transfeminine people was being conducted by Ada Cheung and colleagues. This study could provide more insight into breast development with progestogens, as CPA is a very potent progestogen whereas spironolactone is not meaningfully progestogenic. Cheung and colleagues’ study, led by Lachlan Angus, has now been published in the form of the following two conference abstracts, with a journal article also currently in the process of being published:
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (November 2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (November 2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
The study assessed estradiol plus spironolactone 100 mg/day versus estradiol plus CPA 12.5 mg/day in 55 transfeminine people, with 27 in the spironolactone group and 28 in the CPA group. Hormone therapy duration, at least at this follow-up point in the study, was 6 months. The measures of breast development included breast–chest difference (primary) and estimated breast volume (secondary).
Breast development, measured by breast–chest difference (mean ± SD), was 8.3 ± 2.7 cm with spironolactone and 9.2 ± 3.0 cm with CPA, with the differences between groups not statistically significant (p = 0.27). In addition, breast development, measured by estimated breast volume (mean ± SD), was 158 ± 112 mL with spironolactone and 190 ± 159 mL with CPA, with the differences between groups not statistically significant (p = 0.39). There was variability between individuals in estimated breast volume, with breast volume measurements ranging from 20 to 788 mL. Besides breast growth, the researchers found that CPA also resulted in a greater increase in body fat percentage and gynoid fat compared to spironolactone. Estradiol levels were comparable between antiandrogen groups, whereas total testosterone levels were (mean ± SD) 4.29 ± 5.44 nmol/L (124 ± 157 ng/dL) with spironolactone and 1.48 ± 3.45 nmol/L (43 ± 99 ng/dL) with CPA, a difference that was statistically significant (p = 0.04).
The researchers concluded that there was no difference in breast development with spironolactone versus CPA in their study and that antiandrogen choice should be individualized based on patient and clinician preference as well as consideration of associated side effects. Moreover, they concluded that further research is needed to optimize breast development in transfeminine people.
Angus, L., Mikolajczyk, M., Cheung, A., Zajac, J., Antoszewski, B., & Kasielska-Trojan, A. (2022). Estimation of breast volume in transgender women using 2D photography: validation of the BreastIdea Volume Estimator in men and transgender women. ESA/SRB/APEG/NZSE ASM 2022, November 13-16, Christchurch, Abstracts and Programme, 127–127 (abstract no. 279). [URL] [PDF] [Full Abstract Book]
In studies by the developers of the BreastIdea Volume Estimator, they reported breast volumes measured with the tool in cisgender women. These estimated breast volumes can provide comparison to the breast-volume findings in transfeminine people by Cheung and Angus and colleagues. The developers of the BreastIdea Volume Estimator reported that breast volume (mean ± SD) in cisgender women with normal breasts (n=30) was 283 ± 144 mL and in cisgender women with macromastia or gigantomastia (n=35) was 888 ± 277 mL (Kasielska-Trojan, Zawadzki, & Antoszewski, 2022). In another study, they reported that breast volume (mean ± SD) in cisgender women was 272 ± 150 mL, with a range of 99 to 694 mL (Kasielska-Trojan, Mikołajczyk, & Antoszewski, 2020).
Although the BreastIdea Volume Estimator is an interesting and promising tool for quantifying breast development, it has notable limitations, such as its resolution and accuracy being much less than that with 3D scanners like the Artec Eva and Vectra XT (Mikołajczyk, Kasielska-Trojan, & Antoszewski, 2019). Vectra and Artec 3D scanners have been and are being employed to measure breast development with hormone therapy in other studies in transfeminine people (de Blok et al., 2021; Boogers et al., 2022; Dijkman et al., 2023a; Dijkman et al., 2023b; Lopez et al., 2023). The accuracy limitations of the BreastIdea Volume Estimator may explain why the breast volume findings with it in transfeminine people and cisgender women were different from those seen in other studies that employed more advanced 3D scanning methods. Aside from the breast volume measurement, breast–chest difference also has limitations as a measure of breast development in transfeminine people, for instance failing to identify continued breast growth that can be detected with breast volume measurement (de Blok et al., 2021).
Besides the employed measurement methods for breast development, limitations of Lachlan Angus and colleagues’ RCT of breast development with spironolactone and CPA in transfeminine people include its limited duration of follow-up of only 6 months, the fact that testosterone levels were non-equivalent between the spironolactone and CPA groups, and its limited sample size. The incompletely suppressed testosterone levels with spironolactone are notable as androgens oppose estrogen-mediated breast development and could have reduced breast development in the spironolactone group. The limited sample size of the study was responsible for the numeric difference in breast measurements between antiandrogen groups not being statistically significant. In any case, Angus and colleagues’ findings are suggestive that CPA, which is highly progestogenic, neither enhances nor stunts breast development, at least relative to non-progestogenic spironolactone for up to 6 months of hormone therapy. It seems likely that the RCT will continue to longer follow-up times and durations of hormone therapy in the future.
In January 2025, the full paper for the study was published:
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery JCG, Zajac, J. D., & Cheung, A. S. (2025). Effect of Spironolactone and Cyproterone Acetate on Breast Growth in Transgender People: A Randomized Clinical Trial. The Journal of Clinical Endocrinology and Metabolism, 110(6), e1874–e1884. [DOI:10.1210/clinem/dgae650]
Update 2: Flamant, Vervalcke, & T’Sjoen (2023) and Yang et al. (2024)
The following two recent studies provide additional information on the topic of breast development with progestogen exposure—specifically with CPA—in transfeminine people:
Flamant, T., Vervalcke, J., & T’Sjoen, G. (November 2023). Dose Reduction of Cyproterone Acetate in Trans Women and the Effect on Patient-reported Outcomes: Results from the ENIGI Study. Endocrine Abstracts, 97 [Belgian Endocrine Society 2023], 5–5 (abstract no. 007). [URL] [PDF]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
In the first study, Flamant, Vervalcke, & T’Sjoen (2023), clinical outcomes in transfeminine people at the University of Ghent, Belgium clinic were compared in 72 people taking CPA at low doses (10–12.5 mg/day) or high doses (25–50 mg/day). Testosterone suppression was equivalent between the two dose groups. Breast development satisfaction, measured with the Body Image Scale, was not significantly different with low-dose CPA versus high-dose CPA following 1 year of hormone therapy (p = 0.078). However, the p-value indicates that there was almost a statistically significant difference between groups, though it was not stated which group was numerically higher in terms of satisfaction. In any case, the researchers stated that breast development satisfaction was “non-inferior” with low-dose CPA compared to high-dose CPA, which seems suggestive that satisfaction may have been higher in the high-dose CPA group. These findings suggest that higher doses of CPA may not stunt breast development relative to doses of CPA that are lower, although still quite high in terms of progestogenic activity.
In the second study, Yang et al. (2024), clinical outcomes in transfeminine people at the Peking University Third Hospital in China with estradiol plus spironolactone (n=43) versus estradiol plus CPA (n=53) were retrospectively compared. Testosterone levels were much higher in the spironolactone group relative to the CPA group (374 ng/dL [13.0 nmol/L] vs. 20 ng/dL [0.7 nmol/L]; p < 0.001) and duration of hormone therapy was shorter in the spironolactone group than in the CPA group (median 12 months vs. 18 months). Breast development satisfaction, measured with a visual analogue scale (VAS), was median 6.0 (IQR 4.0–7.0) with spironolactone and 6.0 (IQR 4.0–7.0) with CPA, and was not statistically different. On the other hand, the CPA group outperformed the spironolactone group in terms of several other VAS-based clinical-outcome measures, including figure feminization, testicular atrophy, decreased penile erections, and in terms of a composite overall satifaction score. These findings suggest, as with the RCT by Lachlan Angus and colleagues, that spironolactone and CPA result in similar breast development in transfeminine people despite differences in testosterone levels and other clinical outcomes.
In 2023, a study protocol for a randomized controlled trial of oral progesterone and breast development in transfeminine people was published (Dijkman et al., 2023). The protocol was published by Benthe Dijkman and colleagues at the Vrije Universiteit University Medical Center (VUMC) in Amsterdam, the Netherlands. The trial would be the first prospective randomized controlled trial of progesterone and breast development in transfeminine people.
In this non-blinded non-placebo-controlled randomized trial, 90 transfeminine people would be randomized into 6 study arms with 15 people each. The transfeminine people would be individuals who had been on hormone therapy for at least one year and had undergone vaginoplasty or orchiectomy. Those who were currently or previously taking a progestogen, including CPA, would be excluded. The study’s treatment arms or groups would include the following:
Standard-dose estradiol alone (control group)
Double-dose estradiol alone
Standard-dose estradiol plus progesterone 200 mg/day
Double-dose estradiol plus progesterone 200 mg/day
Standard-dose estradiol plus progesterone 400 mg/day
Double-dose estradiol plus progesterone 400 mg/day
The estradiol therapy was specifically oral estradol valerate, oral estradiol hemihydrate, transdermal estradiol patches, transdermal estradiol gel, or transdermal estradiol spray, at doses resulting in estradiol levels of 200 to 400 pmol/L (54–109 pg/mL) in the standard-dose group and 400 to 800 pmol/L (109–218 pg/mL) in the double-dose group. The progesterone therapy was specifically oral micronized progesterone (Utrogestan). It was noted that in order to maximize adherence, progesterone would be prescribed for limited 1 to 3 month intervals, but no further details on this were provided.
The duration of the study would be 3 years and initial phase would be 12 months, with breast development and/or hormone levels measured at baseline, 3 months, 6 months, and 12 months of treatment. Estradiol levels would be measured with mass spectrometry, whereas progesterone levels would be measured with immunoassays. Breast development would be measured with 3D scanning (Artec Leo 3D) and breast–chest difference. Bra cup size would additionally be calculated from these measures. In the protocol, it was stated that an average breast volume increase of 30%, which was said to correspond to one bra cup size increase, would be considered a clinically relevant outcome. There would also be a number of secondary outcomes, including side effects/safety, satisfaction, mood, sleep, and sexual pleasure. It was noted that the study may serve as a pilot project for a larger future study of progesterone and breast development initiated at the start of hormone therapy prior to surgery.
In August 2025, an EPATH conference abstract with briefly described results of the study was published online in advance of the 6th EPATH conference to be held in September 2025 (Dreijerink et al., 2025):
Dreijerink, K., den Heijer, M., Geels, R. (2025). Increased breast volume due to addition of progesterone and increasing the estradiol dose in feminizing gender-affirming hormone therapy. EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
It was reported that mean breast volume, relative to standard-dose estradiol alone, changed as follows:
Treatment group
n
Breast volume change
E2 double-dose alone
15
+6% (95% CI, –13 to 24)
E2 standard-dose plus P4 200 mg/day
15
+13% (95% CI, –7 to 33)
E2 double-dose plus P4 200 mg/day
15
+37% (95% CI, 18 to 57)
E2 standard-dose plus P4 400 mg/day
15
+20% (95% CI, 0 to 40)
E2 double-dose plus P4 400 mg/day
15
+27% (95% CI, 8 to 47)
The authors concluded that progesterone and higher estradiol dose increased breast volume in transfeminine people. The results of significance tests for breast volume between individual treatment groups or relative to controls were not provided in the abstract. Subjective satisfaction with breast growth and size was said to be improved in all treatment groups relative to the control group (p < 0.05). Aside from breast size changes, side effects with oral progesterone included tiredness (44%), breast/nipple tenderness (27%), and mood changes (22%). There were no treatment-related serious adverse events. No other results or data were provided in the abstract. The full results of the this trial by Dreijerink and colleagues will be published in a journal article at some point in the future. It was concluded that oral progesterone was safe but did cause some side effects. Moreover, the study concluded that their results supported a future role of progesterone in transfeminine hormone therapy. However, it was noted that the long-term effects of progesterone in transfeminine people still need to be studied.
The findings of Dreijerink and colleagues are the highest-quality data on progesterone and breast changes in transfeminine people that are currently available. Their findings suggest that addition of oral progesterone to estradiol increases breast volume and that higher-dose estradiol levels synergize with progesterone to increase breast volume. There was a 13 to 37% increase in volume with oral progesterone depending on the estradiol and progesterone doses. It is important to note however that, as extensively reviewed in the present article, higher estradiol levels and progesterone are associated with increased breast volume due to effects like increased local fluid retention, increased blood flow, and/or temporary growth, but these effects are reversible and regress following withdrawal of the hormonal exposure. Unfortunately, Dreijerink and colleagues do not appear to have included a discontinuation phase to assess whether the breast volume increases observed in the trial were reversible or not. As such, while higher-dose estradiol and oral progesterone can significantly increase breast volume during treatment in transfeminine people, it is still not possible to draw conclusions about whether these interventions actually improve breast development—that is, lasting/permanent breast growth. Only future research that includes discontinuation phases will be able to answer this question.
Other limitations of Dreijerink and colleagues’ study include the use of oral progesterone, the employment of immunoassays to measure progesterone levels, the relatively small sample sizes of the individual treatment subgroups in the study and consequent risk of statistical error, and the patient population being transfeminine people who were post-vaginoplasty or -orchiectomy and hence had already been on hormone therapy for a long period of time (at least 1 year but likely longer on average, such as 2 or 3 years). Oral progesterone is known to achieve relatively low progesterone levels and may be inferior in general effectiveness to non-oral progesterone and progestins (Aly, 2019). Immunoassays are known to substantially overestimate and hence provide a misleading idea of progesterone levels, whereas mass spectrometry-based assays provide accurate progesterone levels (Aly, 2019). Individuals who have been on hormone therapy for many years may have near- or fully-complete breast development and hence less potential for enhancement of true breast development. In any case, caveats aside, Dreijerink and colleagues’ findings are relatively high-quality data, and demonstrate with decent confidence that oral progesterone can, at least exposure-dependently and in conjunction with sufficiently high estradiol levels, provide an increase in breast size in transfeminine people.
References
Abduljalil, K., Furness, P., Johnson, T. N., Rostami-Hodjegan, A., & Soltani, H. (2012). Anatomical, Physiological and Metabolic Changes with Gestational Age during Normal Pregnancy. Clinical Pharmacokinetics, 51(6), 365–396. [DOI:10.2165/11597440-000000000-00000]
Adams, R. D., Kliman, B., Federman, D. D., Ulfelder, H. S., & Holmes, L. B. (1970). Syndromes of Testicular Feminization. Clinical Pediatrics, 9(3), 165–178. [DOI:10.1177/000992287000900312]
Alekseev, N. P. (2021). Origin and Development of the Mammary Glands. In Alekseev, N. P. Physiology of Human Female Lactation (pp. 11–66). Cham: Springer. [DOI:10.1007/978-3-030-66364-3_2]
Alex, A., Bhandary, E., & McGuire, K. P. (2020). Anatomy and Physiology of the Breast during Pregnancy and Lactation. In Alipour, S., & Omranipour, R. (Eds.). Diseases of the Breast during Pregnancy and Lactation (Advances in Experimental Medicine and Biology, Volume 1252) (pp. 3–7). Cham: Springer. [DOI:10.1007/978-3-030-41596-9]
Anderson, W. A. (1962). Experimental stimulation of breast development in the teen-age female. The Journal of the Medical Society of New Jersey, 59(10), 541–543. [Google Scholar] [PubMed] [PDF]
Andler, W., & Zachmann, M. (1979). Spontaneous breast development in an adolescent girl with testicular feminization after castration in early childhood. The Journal of Pediatrics, 94(2), 304–305. [DOI:10.1016/s0022-3476(79)80852-5]
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Angus, L., Mikolajczyk, M., Cheung, A., Zajac, J., Antoszewski, B., & Kasielska-Trojan, A. (2022). Estimation of breast volume in transgender women using 2D photography: validation of the BreastIdea Volume Estimator in men and transgender women. ESA/SRB/APEG/NZSE ASM 2022, November 13-16, Christchurch, Abstracts and Programme, 127–127 (abstract no. 279). [URL] [PDF] [Full Abstract Book]
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery JCG, Zajac, J. D., & Cheung, A. S. (2025). Effect of Spironolactone and Cyproterone Acetate on Breast Growth in Transgender People: A Randomized Clinical Trial. The Journal of Clinical Endocrinology and Metabolism, 110(6), e1874–e1884. [DOI:10.1210/clinem/dgae650]
Apter, D. (1980). Serum steroids and pituitary hormones in female puberty: a partly longitudinal study. Clinical Endocrinology, 12(2), 107–120. [DOI:10.1111/j.1365-2265.1980.tb02125.x]
Apter, D., Räisänen, I., Ylöstalo, P., & Vihko, R. (1987). Follicular growth in relation to serum hormonal patterns in adolescent compared with adult menstrual cycles. Fertility and Sterility, 47(1), 82–88. [DOI:10.1016/s0015-0282(16)49940-1]
Aritaki, S., Miyazawa, H., Ogihara, M., Ushio, M., & Izumizawa, A. (1992). An Endocrinological Study of Persistent Pubertal Macromastia. The Tohoku Journal of Experimental Medicine, 167(3), 189–196. [DOI:10.1620/tjem.167.189]
Arya, S., Barnabas, R., Lila, A. R., Sarathi, V., Memon, S. S., Bhandare, V. V., Thakkar, K., Patil, V., Shah, N. S., Kunwar, A., & Bandgar, T. (2021). Clinical, Hormonal, Genetic, and Molecular Characteristics in Androgen Insensitivity Syndrome in an Asian Indian Cohort from a Single Centre in Western India. Sexual Development, 15(4), 253–261. [DOI:10.1159/000517763]
Asscheman, H., & Gooren, L. J. (1992). Hormone Treatment in Transsexuals. In Bocking, W. O., Coleman, E. (Eds). Gender Dysphoria: Interdisciplinary Approaches in Clinical Management (pp. 39–54). Binghamton: Haworth Press. / Journal of Psychology & Human Sexuality, 5(4), 39–54. [Google Scholar] [Google Books] [DOI:10.1300/J056v05n04_03]
Athanasoulia, A., Auer, M., Riepe, F., & Stalla, G. (2013). Rare Missense P450c17 (CYP17A1) Mutation in Exon 1 as a Cause of 46,XY Disorder of Sexual Development: Implications of Breast Tissue ‘Unresponsiveness’ despite Adequate Estradiol Substitution. Sexual Development, 7(4), 212–215. [DOI:10.1159/000348301]
Atwood, C., Hovey, R., Glover, J., Chepko, G., Ginsburg, E., Robison, W., & Vonderhaar, B. (2000). Progesterone induces side-branching of the ductal epithelium in the mammary glands of peripubertal mice. Journal of Endocrinology, 167(1), 39–52. [DOI:10.1677/joe.0.1670039]
Aupperlee, M. D., Leipprandt, J. R., Bennett, J. M., Schwartz, R. C., & Haslam, S. Z. (2013). Amphiregulin mediates progesterone-induced mammary ductal development during puberty. Breast Cancer Research, 15(3), R44. [DOI:10.1186/bcr3431]
Bahr, C., Ewald, J., Dragovich, R., & Gothard, M. D. (2024). Effects of progesterone on gender affirmation outcomes as part of feminizing hormone therapy. Journal of the American Pharmacists Association, 64(1), 268–272. [DOI:10.1016/j.japh.2023.08.001]
Baird, D., Hytten, F. E., & Thomson, A. M. (1958). Age and Human Reproduction. BJOG, 65(6), 865–876. [DOI:10.1111/j.1471-0528.1958.tb08582.x]
Baker, S. B., Burkey, B. A., Thornton, P., & LaRossa, D. (2001). Juvenile Gigantomastia: Presentation of Four Cases and Review of the Literature. Annals of Plastic Surgery, 46(5), 517–526. [DOI:10.1097/00000637-200105000-00011]
Bames, H. O. (1948). Reduction of massive breast hypertrophy. Plastic and Reconstructive Surgery, 3(5), 560–569. [DOI:10.1097/00006534-194809000-00006]
Barbieri, R. L. (2019). Breast. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 248–255.e3). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/B978-0-323-47912-7.00010-X]
Baron, J. (1993). Zespół braku wrazliwości na androgeny. Studium kliniczne i endokrynologiczne 40 osobników [Androgen insensitivity syndrome. Clinical and endocrinologic study of 40 cases]. Endokrynologia Polska, 44(2), 175–186. [Google Scholar] [PubMed]
Baron, J. (1994). Klasyczne i niepełne zespoły braku wrazliwości na androgeny [Classical and incomplete androgen insensitivity syndromes]. Ginekologia Polska, 65(7), 377–386. [Google Scholar] [PubMed]
Baron, J. (1994). Niepełny lub cześciowy zespół braku wrazliwości na androgeny [Partial androgen insensitivity syndrome]. Ginekologia Polska, 65(6), 319–325. [Google Scholar] [PubMed]
Barrat, J., de Lignières, B., Marpeau, L., Larue, L., Fournier, S., Nahoul, K., Linares, G., Giorgi, H., & Contesso, G. (1990). Effet in vivo de l’administration locale de progestérone sur l’activité mitotique des galactophores humains. Résultat d’une étude pilote. [The in vivo effect of the local administration of progesterone on the mitotic activity of human ductal breast tissue. Results of a pilot study.] Journal de Gynecologie, Obstetrique et Biologie de la Reproduction, 19(3), 269–274. [Google Scholar 1] [Google Scholar 2] [PubMed]
Barrett, J. (2009). The clinical risks associated with the diagnosis and management of disorders of gender identity. Clinical Risk, 15(4), 131–134. [DOI:10.1258/cr.2008.080069]
Bässler, R. (1970). The Morphology of Hormone Induced Structural Changes in the Female Breast. In Altmann, H.-W., et al. (Eds.). Current Topics in Pathology: Ergebnisse der Pathology, Volume 53 (pp. 1–89). Heidelberg: Springer Berlin. [DOI:10.1007/978-3-662-30514-0_1]
Basson, R., & Prior, J. C. (1998). Hormonal Therapy of Gender Dysphoria: The Male-to-Female Transsexual. In Denny, D. (Ed.). Concepts in Transgender Identity (Garland Gay and Lesbian Studies, Volume 11) (pp. 277–296). New York: Garland Publishing Inc. [Google Scholar] [Google Books] [DOI:10.4324/9780203775134-19] [PDF]
Bauman, A., Novello, L., & Kreitzer, P. (2016). Endocrine Disorders and Delayed Puberty. In Appelbaum, H. (Ed.). Abnormal Female Puberty: A Clinical Casebook (pp. 87–107). Cham: Springer. [DOI:10.1007/978-3-319-27225-2_5]
Bayer, C. M., Bani, M. R., Schneider, M., Dammer, U., Raabe, E., Haeberle, L., Faschingbauer, F., Schneeberger, S., Renner, S. P., Fischer, D., Schulz-Wendtland, R., Fasching, P. A., Beckmann, M. W., & Jud, S. M. (2014). Assessment of breast volume changes during human pregnancy using a three-dimensional surface assessment technique in the prospective CGATE study. European Journal of Cancer Prevention, 23(3), 151–157. [DOI:10.1097/cej.0b013e3283651ccb]
Bazarra-Castro, M. A. (2009). Etiological aspects, therapy regimes, side effects and treatment satisfaction of transsexual patients. (Doctoral dissertation, Ludwig Maximilian University of Munich.) [DOI:10.5282/edoc.9984] [URN:urn:nbn:de:bvb:19-99840] [PDF]
Beck, P. (1972). Lactogenic Activity of Human Chorionic Somatomammotropin in Rhesus Monkeys. Experimental Biology and Medicine, 140(1), 183–187. [DOI:10.3181/00379727-140-36422]
Begley, D. J., Firth, J. A., & Hoult, J. R. (1980). The Breast and Lactation. In Begley, D. J., Firth, J. A., & Hoult, J. R. Human Reproduction and Developmental Biology (pp. 204–219). London: Macmillan Education UK. [DOI:10.1007/978-1-349-16260-4_14]
Bellwether, C. J. (2020). Why Should a Transfeminine Person Consider Progesterone? Google Docs. [URL]
Benjamin, H. (1966). Nonsurgical Management of Transsexualism. In Benjamin, H. The Transsexual Phenomenon (pp. 86–99). New York: Julian Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Benjamin, H. (1967). Transvestism and Transsexualism in the male and female. Journal of Sex Research, 3(2), 107–127. [DOI:10.1080/00224496709550519]
Berliere, M., Coche, M., Lacroix, C., Riggi, J., Coyette, M., Coulie, J., Galant, C., Fellah, L., Leconte, I., Maiter, D., Duhoux, F. P., & François, A. (2022). Effects of Hormones on Breast Development and Breast Cancer Risk in Transgender Women. Cancers, 15(1), 245. [DOI:10.3390/cancers15010245]
Berryhill, G. E., Trott, J. F., & Hovey, R. C. (2016). Mammary gland development—It’s not just about estrogen. Journal of Dairy Science, 99(1), 875–883. [DOI:10.3168/jds.2015-10105]
Bertelloni, S., Dati, E., Baroncelli, G. I., & Hiort, O. (2011). Hormonal Management of Complete Androgen Insensitivity Syndrome from Adolescence Onward. Hormone Research in Paediatrics, 76(6), 428–433. [DOI:10.1159/000334162]
Bethea, C. L., Kohama, S. G., & Pecins-Thompson, M. (1997). Pituitary and Brain Actions of Estrogen and Progesterone in the Regulation of Primate Prolactin Secretion. In Pavlik, E. J. (Ed.). Estrogens, Progestins, and Their Antagonists: Functions and Mechanisms of Action (pp. 3–46). Boston: Birkhäuser. [DOI:10.1007/978-1-4612-2004-6_1]
Bevan, D. J. (2012). Progesterone for Breast Development? / Should Male-to-Female Transsexuals Take Progesterone as part of Hormone Therapy (HT) for Better Breast Development? Biopsychology of TSTG / Transgender Forum. [URL 1] [URL 2]
Bevan, D. J. (2019). Grow Your Own : Breast Development Update. Transgender Forum. [URL]
Beyer, C., Cruz, M. L., & Martinez-Manautou, J. (1970). Effect of Chlormadinone Acetate on Mammary Development and Lactation in the Rabbit. Endocrinology, 86(5), 1172–1174. [DOI:10.1210/endo-86-5-1172]
Biswas, S. K., Banerjee, S., Baker, G. W., Kuo, C., & Chowdhury, I. (2022). The Mammary Gland: Basic Structure and Molecular Signaling during Development. International Journal of Molecular Sciences, 23(7), 3883. [DOI:10.3390/ijms23073883]
Bland, K. I., Copeland, E. M., & Klimberg, V. S. (2018). Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 20–36.e2). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00002-7]
Bland, K. I., Harrison Howard, J., & Romrell, L. J. (2009). Congenital and Acquired Disturbances of Breast Development and Growth. In Bland, K. I., & Copeland, E. M. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 4th Edition (pp. 189–207). Philadelphia: Saunders/Elsevier. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Bondy, C. A., & Turner Syndrome Consensus Study Group. (2007). Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. The Journal of Clinical Endocrinology & Metabolism, 92(1), 10–25. [DOI:10.1210/jc.2006-1374]
Boogers, L., Infirri, S. S., Bouchareb, A., de Blok, C., Liberton, N., van Trotsenburg, P., Dreijerink, K., den Heijer, M., Wiepjes, C., & Hannema, S. (2022). The effect of timing of puberty suppression on breast development in trans girls; a cross-sectional study. Hormone Research in Paediatrics, 95(Suppl 2) [60th Annual Meeting of the European Society for Paediatric Endocrinology (ESPE), Rome, Italy, September 15–17, 2022], 390–391 (abstract no. P1-379). [Google Scholar] [DOI:10.1159/000525606] [URL] [PDF 1] [PDF 2]
Boyce, S. W., Hoffman, P. G., & Mathes, S. J. (1984). Recurrent Macromastia after Subcutaneous Mastectomy. Annals of Plastic Surgery, 13(6), 511–518. [DOI:10.1097/00000637-198412000-00008]
Bruck, H. G., & Müller, G. (1967). Zur Hormontherapie der Hypoplastischen Weiblichen Brust. [On Hormonal Therapy of the Hypoplastic Female Breast.] Ästhetische Medizin [Ästhetische Medizin : Kongreß-Organ der Deutschen Gesellschaft für die Ästhetische Medizin und ihre Grenzgebiete], 16(12), 365–366. [ISSN:0400-6755] [Google Scholar] [PubMed] [WorldCat] [PDF] [Translation]
Bryant, R., Underwood, A., Robinson, A., Stephenson, T., & Underwood, J. (1998). Determination of breast tissue composition for improved accuracy in estimating radiation doses and risks in mammographic screening. The Breast, 7(2), 95–98. [DOI:10.1016/s0960-9776(98)90064-9]
Buhimschi, C. S. (2004). Endocrinology of lactation. Obstetrics and Gynecology Clinics of North America, 31(4), 963–979. [DOI:10.1016/j.ogc.2004.08.002]
Camilletti, M. A., Abeledo-Machado, A., Faraoni, E. Y., Thomas, P., & Díaz-Torga, G. (2019). New insights into progesterone actions on prolactin secretion and prolactinoma development. Steroids, 152, 108496. [DOI:10.1016/j.steroids.2019.108496]
Çamtosun, E., Şıklar, Z., Ceylaner, S., Kocaay, P., & Berberoğlu, M. (2017). Delayed Diagnosis of a 17-Hydroxylase/17,20-Lyase Deficient Patient Presenting as a 46,XY Female: A Low Normal Potassium Level Can Be an Alerting Diagnostic Sign. Journal of Clinical Research in Pediatric Endocrinology, 9(2), 163–167. [DOI:10.4274/jcrpe.3839]
Capraro, V. J., & Dewhurst, C. J. (1975). Breast Disorders in Childhood and Adolescence. Clinical Obstetrics and Gynecology, 18(2), 25–50. [DOI:10.1097/00003081-197506000-00003]
Carlson, L. J., & Shaw, N. D. (2019). Development of Ovulatory Menstrual Cycles in Adolescent Girls. Journal of Pediatric and Adolescent Gynecology, 32(3), 249–253. [DOI:10.1016/j.jpag.2019.02.119]
Caro, T. M. (1987). Human breasts: Unsupported hypotheses reviewed. Human Evolution, 2(3), 271–282. [DOI:10.1007/bf03016112]
Ceriani, R. L. (1974). Hormones and Other Factors Controlling Growth in the Mammary Gland: A Review. Journal of Investigative Dermatology, 63(1), 93–108. [DOI:10.1111/1523-1747.ep12678104]
Cernea, R. (1944). Lokale Hormonbehandlung bei Mammaatrophie und Unterentwicklung. [Local Hormone Treatment in Mammary Atrophy and Underdevelopment.] Medizinische Klinik, 40(11/12), 169–170. [Google Scholar] [PDF] [Translation]
Chang, K., Lee, T. T., Linares-Cruz, G., Fournier, S., & de Ligniéres, B. (1995). Influences of percutaneous administration of estradiol and progesterone on human breast epithelial cell cycle in vivo. Fertility and Sterility, 63(4), 785–791. [DOI:10.1016/s0015-0282(16)57482-2]
Cheikhelard, A., Morel, Y., Thibaud, E., Lortat-Jacob, S., Jaubert, F., Polak, M., & Nihoul-Fekete, C. (2008). Long-Term Followup and Comparison Between Genotype and Phenotype in 29 Cases of Complete Androgen Insensitivity Syndrome. Journal of Urology, 180(4), 1496–1501. [DOI:10.1016/j.juro.2008.06.045]
Ciarloni, L., Mallepell, S., & Brisken, C. (2007). Amphiregulin is an essential mediator of estrogen receptor α function in mammary gland development. Proceedings of the National Academy of Sciences, 104(13), 5455–5460. [DOI:10.1073/pnas.0611647104]
Clendenen, T. V., Kim, S., Moy, L., Wan, L., Rusinek, H., Stanczyk, F. Z., Pike, M. C., & Zeleniuch-Jacquotte, A. (2013). Magnetic Resonance Imaging (MRI) of hormone-induced breast changes in young premenopausal women. Magnetic Resonance Imaging, 31(1), 1–9. [DOI:10.1016/j.mri.2012.06.022]
Cline, J. M., & Wood, C. E. (2006). Hormonal Effects on the Mammary Gland of Postmenopausal Nonhuman Primates. Breast Disease, 24(1), 59–70. [DOI:10.3233/bd-2006-24105]
Cline, J. M., & Wood, C. E. (2008). The Mammary Glands of Macaques. Toxicologic Pathology, 36(7 Suppl), 130S–141S. [DOI:10.1177/0192623308327411]
Cole, R. D., & Hopkins, T. R. (1962). A Biochemical Test of Artificial Mammogenesis and Lactogenesis As Models of the Natural Processes. Endocrinology, 70(3), 375–380. [DOI:10.1210/endo-70-3-375]
Coleman, E., Bockting, W., Botzer, M., Cohen-Kettenis, P., DeCuypere, G., Feldman, J., Fraser, L., Green, J., Knudson, G., Meyer, W. J., Monstrey, S., Adler, R. K., Brown, G. R., Devor, A. H., Ehrbar, R., Ettner, R., Eyler, E., Garofalo, R., Karasic, D. H., Lev, A. I., Mayer, G., Meyer-Bahlburg, H., Hall, B. P., Pfaefflin, F., Rachlin, K., Robinson, B., Schechter, L. S., Tangpricha, V., van Trotsenburg, M., Vitale, A., Winter, S., Whittle, S., Wylie, K. R., & Zucker, K. (2012). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, Version 7. International Journal of Transgenderism, 13(4), 165–232. [DOI:10.1080/15532739.2011.700873] [URL] [PDF]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Coltman, C. E., Steele, J. R., & McGhee, D. E. (2017). Breast volume is affected by body mass index but not age. Ergonomics, 60(11), 1576–1585. [DOI:10.1080/00140139.2017.1330968]
Colvin, C., Devineni, G., & Ashraf, A. P. (2014). Delayed Puberty. In Bandeira, F., Gharib, H., Golbert, A., Griz, L., & Faria, M. (Eds.). Endocrinology and Diabetes (pp. 203–217). New York: Springer. [DOI:10.1007/978-1-4614-8684-8_17]
Cowie, A. T., & Folley, S. J. (1961). The Mammary Gland and Lactation. In Young, W. C. (Ed.). Sex and Internal Secretions, 3rd Edition, Volume I (pp. 590–642). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Cowie, A. T., Forsyth, I. A., & Hart, I. C. (1980). Growth and Development of the Mammary Gland. In Cowie, A. T., Forsyth, I. A., & Hart, I. C. Hormonal Control of Lactation (Monographs on Endocrinology, Volume 15) (pp. 58–145). Berlin/Heidelberg: Springer Berlin Heidelberg. [DOI:10.1007/978-3-642-81389-4_3]
Cox, D. B., Kent, J. C., Casey, T. M., Owens, R. A., & Hartmann, P. E. (1999). Breast Growth and the Urinary Excretion of Lactose During Human Pregnancy and Early Lactation: Endocrine Relationships. Experimental Physiology, 84(2), 421–434. [DOI:10.1111/j.1469-445x.1999.01807.x]
Cox, C. B., Kent, J. C., Owens, R., & Hartmann, P. E. (1994). Mammary morphological and functional changes during pregnancy in women. In Thompson, J. (Ed.). Proceedings of the Twenty-Sixth Annual Conference, Hilton Hotel, Brisbane, 26–28 September, 1994 [The Australian Society for Reproductive Biology Inc., Twenty Sixth Annual Conference, The Hilton Hotel, Brisbane, September 26 - 28 1994, Programme and Miniposters of Papers] (pp. 47–47). Australia: Australian Society for Reproductive Biology. [Google Scholar] [WorldCat] [URL] [PDF]
Coxon, J., & Seal, L. (2018). Hormone management of trans women. Trends in Urology & Men’s Health, 9(6), 10–14. [DOI:10.1002/tre.663]
Cregan, M. D., & Hartmann, P. E. (1999). Computerized Breast Measurement from Conception to Weaning: Clinical Implications. Journal of Human Lactation, 15(2), 89–96. [DOI:10.1177/089033449901500202]
Crowley, W. F., & Pitteloud, N. (2020). Approach to the patient with delayed puberty. UpToDate. [Google Scholar] [URL]
Cruz-Korchin, N., Korchin, L., González-Keelan, C., Climent, C., & Morales, I. (2002). Macromastia. Plastic and Reconstructive Surgery, 109(1), 64–68. [DOI:10.1097/00006534-200201000-00011]
Curtis, R. J. (2009 July 10). The Lowdown on Progesterone. London: The London Gender Clinic. [Google Scholar] [URL] [PDF]
Dallmann, A., Ince, I., Meyer, M., Willmann, S., Eissing, T., & Hempel, G. (2017). Gestation-Specific Changes in the Anatomy and Physiology of Healthy Pregnant Women: An Extended Repository of Model Parameters for Physiologically Based Pharmacokinetic Modeling in Pregnancy. Clinical Pharmacokinetics, 56(11), 1303–1330. [DOI:10.1007/s40262-017-0539-z]
Dancey, A., Khan, M., Dawson, J., & Peart, F. (2008). Gigantomastia – a classification and review of the literature. Journal of Plastic, Reconstructive & Aesthetic Surgery, 61(5), 493–502. [DOI:10.1016/j.bjps.2007.10.041]
Davajan, V., & Kletzky, O. A. (1979). Amenorrhea without Galactorrhea or Hirsutism. In Mishell, D. R., & Davajan, V. (Eds.). Reproductive Endocrinology, Infertility, and Contraception (pp. 219–248). Philadelphia: F. A. Davis Co. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Davis, M. E., Boynton, M. W., Ferguson, J. H., & Rothman, S. (1945). Studies on Pigmentation of Endocrine Origin. The Journal of Clinical Endocrinology & Metabolism, 5(3), 138–146. [DOI:10.1210/jcem-5-3-138]
de Blok, C. J., Dijkman, B. A., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K. M., & den Heijer, M. (2021). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de Lignières, B. (2002). Effects of progestogens on the postmenopausal breast. Climacteric, 5(3), 229–235. [DOI:10.1080/cmt.5.3.229.235]
de Lignières, B., & Mauvais-Jarvis, P. (1981). Hormonal Dependence of Benign Breast Disease, Gynecomastia and Breast Cancer. In Hollman, K. H., Brux, J., & Verley, J. M. (Eds.). New Frontiers in Mammary Pathology (pp. 287–308). Boston: Springer US. [DOI:10.1007/978-1-4757-0019-0_17]
de Vries, A. L., Steensma, T. D., Wagemaar, E. C. F., Doreleijers, T. A., & Cohen-Kettenis, P. T. (2010). Puberty suppression followed by cross-sex hormones and gender reassignment surgery: A prospective follow-up of gender dysphoric adolescents into adulthood. In de Vries, A. L. (Ed.). Gender Dysphoria in Adolescents: Mental Health and Treatment Evaluation (pp. 91–106). (Doctoral thesis, Vrije Universiteit Amsterdam.) [Google Scholar] [URL] [PDF]
Deeb, A., Al Suwaidi, H., Attia, S., & Al Ameri, A. (2015). 17-hydroxylase/17,20-lyase deficiency due to a R96Q mutation causing hypertension and poor breast development. Endocrinology, Diabetes & Metabolism Case Reports, 2015(1), 15-0069. [DOI:10.1530/EDM-15-0069]
Deepinder, F., & Braunstein, G. D. (2012). Drug-induced gynecomastia: an evidence-based review. Expert Opinion on Drug Safety, 11(5), 779–795. [DOI:10.1517/14740338.2012.712109]
Dennerstein, L., Burrows, G. D., Hyman, G. J., & Sharpe, K. (1980). Some clinical effects of oestrogen-progestogen therapy in surgically castrated women. Maturitas, 2(1), 19–28. [DOI:10.1016/0378-5122(80)90056-0]
Dewhurst, C. J. (1971). Sex Chromosome Abnormalities and the Gynaecologist. BJOG, 78(12), 1058–1076. [DOI:10.1111/j.1471-0528.1971.tb00227.x]
Dewhurst, C. J. (1971). The XY female. American Journal of Obstetrics and Gynecology, 109(5) [Transactions of the Eighty-First Annual Meeting of the American Association of Obstetricians and Gynecologists], 675–688. [DOI:10.1016/0002-9378(71)90753-8]
Dewhurst, C. (1972). Amenorrhoea and the Paediatrician. Pediatric Clinics of North America, 19(3), 605–618. [DOI:10.1016/s0031-3955(16)32741-9]
Dewhurst, C. J., & Spence, J. E. (1977). The XY female. British Journal of Hospital Medicine, 17(5), 498, 501–506. [Google Scholar] [PubMed] [PDF]
Dewhurst, J., & Underhill, R. (1979). The Suppression of Facial Hair Growth in Transsexuals Using Cyproterone Acetate. British Journal of Sexual Medicine, 6, 19–23. [Google Scholar] [PDF]
Dewhurst, J. (1981). Breast Disorders in Children and Adolescents. Pediatric Clinics of North America, 28(2), 287–308. [DOI:10.1016/s0031-3955(16)33997-9]
Dewhurst, J. (1982). Breast Hypoplasia. In Bruni, V., Gasparri, F., Dewhurst, J., & Rey-Stocker, I. (Eds.). Pediatric and Adolescent Gynaecology [Proceedings of the IVth International Symposium, Florence, October 5-7, 1978] (pp. 205–212). Rome, Italy: Serono Symposia. [Google Scholar] [Google Books] [WorldCat] [PDF]
Di Lorenzo, G., Autorino, R., Perdonà, S., & De Placido, S. (2005). Management of gynaecomastia in patients with prostate cancer: a systematic review. The Lancet Oncology, 6(12), 972–979. [DOI:10.1016/s1470-2045(05)70464-2]
Dickson, L. M., & Hewer, E. E. (1950). The structure of the breast. In Saner, F. D. (Ed.). The Breast: Structure, Function, Disease (pp. 1–52). Baltimore: William & Wilins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL] [PDF]
Dijkman, B. A., Blok, C. J., Dreijerink, K. M., & den Heijer, M. (2023). Progestin-related breast volume changes in a woman with complete androgen insensitivity syndrome (CAIS). Endocrinology, Diabetes & Metabolism Case Reports, 2023(2), 22-0346. [DOI:10.1530/edm-22-0346]
Dijkman, B. A., Helder, D., Boogers, L. S., Gieles, N. C., van Heesewijk, J. O., Slaa, S. t., Liberton, N. P., Wiepjes, C. M., de Blok, C. J., den Heijer, M., & Dreijerink, K. M. (2023). Addition of progesterone to feminizing gender-affirming hormone therapy in transgender individuals for breast development: a randomized controlled trial. BMC Pharmacology and Toxicology, 24(1), 80. [DOI:10.1186/s40360-023-00724-4]
Dittrich, R., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Mueller, A. (2005). Endocrine Treatment of Male-to-Female Transsexuals Using Gonadotropin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 113(10), 586–592. [DOI:10.1055/s-2005-865900]
Donaldson, M., Kriström, B., Ankarberg-Lindgren, C., Verlinde, S., van Alfen-van der Velden, J., Gawlik, A., van Gelder, M., Sas, T., & (2019). Optimal Pubertal Induction in Girls with Turner Syndrome Using Either Oral or Transdermal Estradiol: A Proposed Modern Strategy. Hormone Research in Paediatrics, 91(3), 153–163. [DOI:10.1159/000500050]
Dorgan, J. F., Klifa, C., Deshmukh, S., Egleston, B. L., Shepherd, J. A., Kwiterovich, P. O., Van Horn, L., Snetselaar, L. G., Stevens, V. J., Robson, A. M., Lasser, N. L., & Hylton, N. M. (2013). Menstrual and reproductive characteristics and breast density in young women. Cancer Causes & Control, 24(11), 1973–1983. [DOI:10.1007/s10552-013-0273-2]
Döring, G. K. (1963). Über die relative Häufigkeit des anovulatorischen Cyclus im Leben der Frau. [On the relative frequency of the anovulatory cycle in women’s lives.] Archiv für Gynäkologie, 199(2), 115–123. [DOI:10.1007/bf00668062]
Drąsutis, J. (2017). Changes in breast morphological parameters, body size and shape, blood serum prolactin and lipids during pregnancy, multiple relationships of these indicators and morphological markers for health risk. (Doctoral dissertation, Vilniaus Universitetas.) [Google Scholar] [URL]
Dreijerink, K., den Heijer, M., Geels, R. (2025). Increased breast volume due to addition of progesterone and increasing the estradiol dose in feminizing gender-affirming hormone therapy. EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
Drife, J. O. (1982). The effects of parity and the menstrual cycle on the normal mammary gland and their possible relationship to malignant change. (Doctoral dissertation, University of Edinburgh). [Google Scholar] [Google Books] [WorldCat] [URL] [PDF]
Drife, J. O. (1984). The pill and the breast. IPPF Medical Bulletin, 18(6), 1–2. [Google Scholar] [PubMed]
Drife, J. O. (1986). Breast Development in Puberty. Annals of the New York Academy of Sciences, 464(1) [Endocrinology of the Breast: Basic and Clinical Aspects], 58–65. [DOI:10.1111/j.1749-6632.1986.tb15993.x]
Drife, J. O. (1989). Breast modifications during the menstrual cycle. Supplement to International Journal of Gynecology and Obstetrics, 1, 19–24. / International Journal of Gynecology and Obstetrics, 1989(Suppl 1), 19–24. [Google Scholar] [PubMed] [Archive.org] [PDF]
Drife, J. O. (1990). Premenopausal Hormone Therapy. In Drife, J. O., & Studd, J. W. W. (Eds.). HRT and Osteoporosis (pp. 351–362). London: Springer London. [DOI:10.1007/978-1-4471-1799-5_25]
Duncan, M. (2010). Sexual Selection and Human Breast Morphology. (Doctoral dissertation, Te Herenga Waka-Victoria University of Wellington.) [Google Scholar] [URL]
Edmonds, D. K. (1989). Normal Puberty. In Edmonds, D. K. Dewhurst’s Practical Paediatric and Adolescent Gynaecology, 2nd Edition (pp. 56–62). London: Butterworths. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL] [Archive.org]
Elling, S. V., & Powell, F. C. (1997). Physiological changes in the skin during pregnancy. Clinics in Dermatology, 15(1), 35–43. [DOI:10.1016/s0738-081x(96)00108-3]
Engman, M., Skoog, L., Soderqvist, G., & Gemzell-Danielsson, K. (2008). The effect of mifepristone on breast cell proliferation in premenopausal women evaluated through fine needle aspiration cytology. Human Reproduction, 23(9), 2072–2079. [DOI:10.1093/humrep/den228]
Federici, S., Goggi, G., Quinton, R., Giovanelli, L., Persani, L., Cangiano, B., & Bonomi, M. (2021). New and Consolidated Therapeutic Options for Pubertal Induction in Hypogonadism: In-depth Review of the Literature. Endocrine Reviews, 43(5), 824–851. [DOI:10.1210/endrev/bnab043]
Fernández-Cancio, M., García-García, E., González-Cejudo, C., Martínez-Maestre, M., Mangas-Cruz, M., Guerra-Junior, G., Pandi de Mello, M., Arnhold, I. J., Nishi, M. Y., Bilharinho Mendonça, B., García-Arumí, E., Audí, L., Tizzano, E., & Carrascosa, A. (2017). Discordant Genotypic Sex and Phenotype Variations in Two Spanish Siblings with 17α-Hydroxylase/17,20-Lyase Deficiency Carrying the Most Prevalent Mutated CYP17A1 Alleles of Brazilian Patients. Sexual Development, 11(2), 70–77. [DOI:10.1159/000468160]
Fernandez-Valdivia, R., Mukherjee, A., Mulac-Jericevic, B., Conneely, O. M., DeMayo, F. J., Amato, P., & Lydon, J. P. (2005). Revealing Progesterone’s Role in Uterine and Mammary Gland Biology: Insights from the Mouse. Seminars in Reproductive Medicine, 23(1), 22–37. [DOI:10.1055/s-2005-864031]
Finkenzeller, D. A., & Loveless, M. B. (2007). Pediatric Gynecology. In Fortner, K. B., Szymanski, L. M., Fox, H. E., & Wallach, E. E. (Eds.). The Johns Hopkins Manual of Gynecology and Obstetrics, 3rd Edition (Spiral Manual Series) (pp. 363–379). Philadelphia: Lippincott Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Fisher, A. D., Castellini, G., Ristori, J., Casale, H., Cassioli, E., Sensi, C., Fanni, E., Amato, A. M., Bettini, E., Mosconi, M., Dèttore, D., Ricca, V., & Maggi, M. (2016). Cross-Sex Hormone Treatment and Psychobiological Changes in Transsexual Persons: Two-Year Follow-Up Data. The Journal of Clinical Endocrinology & Metabolism, 101(11), 4260–4269. [DOI:10.1210/jc.2016-1276]
Foidart, J. M., Colin, C., Denoo, X., Desreux, J., Fournier, S., & de Linières, B. (1996). Influence of percutaneous administration of estradiol and progesterone on the proliferation of human breast epithelial cells. In Calvo, F., Crépin, M., & Magdelenat, H. (Eds.). Breast Cancer: Advances in Biology and Therapeutics [21st Meeting of the International Association for Breast Cancer Research, July 3-4-5, 1996, Paris] (pp. 329–334). Montrouge/Paris: John Libbey Eurotext. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Foidart, J., Colin, C., Denoo, X., Desreux, J., Béliard, A., Fournier, S., & de Lignières, B. (1998). Estradiol and Progesterone Regulate the Proliferation of Human Breast Epithelial Cells. Fertility and Sterility, 69(5), 963–969. [DOI:10.1016/s0015-0282(98)00042-9]
Folley, S. J. (1947). Endocrine Control of the Mammary Gland. British Medical Bulletin, 5(2–3), 130–134. [DOI:10.1093/oxfordjournals.bmb.a073121]
Folley, S. J. (1950). Lactational Physiology. In Bowes, K. (Ed.). Modern Trends in Obstetrics and Gynaecology (pp. 441–453). London: Butterworth. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Folley, S. J. (1952). Lactation. In Parkes, A. S. (Ed.). Marshall’s Physiology of Reproduction, 3rd Edition, Volume II (pp. 525–647). Longmans, Green & Co.: London. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Folley, S. J. (1956). Recent Studies on the Development of the Mammary Gland. In Folley, S. J. The Physiology and Biochemistry of Lactation, 1st Edition (pp. 1–22). Edinburgh: Oliver & Boyd. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Folley, S. J., & Malpress, F. H. (1948). Hormonal Control of Mammary Growth. In Pincus, G., & Thimann, K. V. (Eds.). The Hormones: Physiology, Chemistry and Applications, Volume I (pp. 695–743). New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Fourcade, R., & McLeod, D. (2004). Tolerability of Antiandrogens in the Treatment of Prostate Cancer. UroOncology, 4(1), 5–13. [DOI:10.1080/1561095042000191655]
Fowler, P. A., Casey, C. E., Cameron, G. G., Foster, M. A., & Knight, C. H. (1990). Cyclic changes in composition and volume of the breast during the menstrual cycle, measured by magnetic resonance imaging. BJOG, 97(7), 595–602. [DOI:10.1111/j.1471-0528.1990.tb02546.x]
Fridriksdottir, A. J., Petersen, O. W., & Rnnov-Jessen, L. (2011). Mammary gland stem cells: current status and future challenges. The International Journal of Developmental Biology, 55(7–8–9), 719–729. [DOI:10.1387/ijdb.113373af]
Fuqua, J. S., & Eugster, E. A. (2022). History of Puberty: Normal and Precocious. Hormone Research in Paediatrics, 95(6), 568–578. [DOI:10.1159/000526464]
Futterweit, W. (1980). Endocrine management of transsexual. Hormonal profiles of serum prolactin, testosterone, and estradiol. New York State Journal of Medicine, 80(8), 1260–1264. [Google Scholar] [PubMed] [Archive.org] [PDF]
Gaede, P., Trolle, D., & Pedersen, H. (1978). Extremely low placental lactogen hormone (hpl) values in an otherwise uneventful pregnancy preceding delivery of a normal baby. Acta Obstetricia et Gynecologica Scandinavica, 57(3), 203–209. [DOI:10.3109/00016347809154883]
Galani, A., Kitsiou-Tzeli, S., Sofokleous, C., Kanavakis, E., & Kalpini-Mavrou, A. (2008). Androgen insensitivity syndrome: clinical features and molecular defects. Hormones, 7(3), 217–229. [DOI:10.14310/horm.2002.1201]
Galbarczyk, A. (2011). Unexpected changes in maternal breast size during pregnancy in relation to infant sex: An evolutionary interpretation. American Journal of Human Biology, 23(4), 560–562. [DOI:10.1002/ajhb.21177]
Gawlik, A., Hankus, M., Such, K., Drosdzol-Cop, A., Madej, P., Borkowska, M., Zachurzok, A., & Malecka-Tendera, E. (2016). Hypogonadism and Sex Steroid Replacement Therapy in Girls with Turner Syndrome. Journal of Pediatric and Adolescent Gynecology, 29(6), 542–550. [DOI:10.1016/j.jpag.2016.03.005]
Gershon-Cohen, J. (1970). The Normal Breast. In Gershon-Cohen, J. Atlas of Mammography (pp. 23–38). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-85678-5_4] [Google Books]
Gertig, D. M., Stillman, I. E., Byrne, C., Spiegelman, D., Schnitt, S. J., Connolly, J. L., Colditz, G. A., & Hunter, D. J. (1999). Association of age and reproductive factors with benign breast tissue composition. Cancer Epidemiology, Biomarkers & Prevention, 8(10), 873–879. [Google Scholar] [PubMed] [URL]
Geschickter, C. F. (1945). Endocrine Physiology of the Breast. In Geschickter, C. F. Diseases of the Breast: Diagnosis, Pathology, Treatment, 2nd Edition (pp. 42–81). Philadelphia: J.B. Lippincott. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Glenn, J. F. (1976). Testicular feminization syndrome current clinical considerations. Urology, 7(6), 569–577. [DOI:10.1016/0090-4295(76)90079-0]
Gliosci, A., & Presutti, F. (1993). Virginal gigantomastia: Validity of combined surgical and hormonal treatments. Aesthetic Plastic Surgery, 17(1), 61–65. [DOI:10.1007/bf00455051]
Gompel, A., & Plu-Bureau, G. (2018). Progesterone, progestins and the breast in menopause treatment. Climacteric, 21(4), 326–332. [DOI:10.1080/13697137.2018.1476483]
Goodman, H. M. (2009). Hormonal Control of Pregnancy and Lactation. In Goodman, H. M. Basic Medical Endocrinology, 4th Edition (pp. 277–301). Amsterdam: Academic Press/Elsevier. [DOI:10.1016/b978-0-12-373975-9.00014-8]
Gooren, L. J., Harmsen-Louman, W., & Kessel, H. (1985). Follow-up of prolactin levels in long-term oestrogen-treated male-to-female transsexuals with regard to prolactinoma induction. Clinical Endocrinology, 22(2), 201–207. [DOI:10.1111/j.1365-2265.1985.tb01081.x]
Gooren, L. J. (2016). Hormone Treatment of Adult Transgender People. In Ettner, R., Monstrey, S., & Coleman, E. (Eds.). Principles of Transgender Medicine and Surgery, 2nd Edition (pp. 167–179). New York: Routledge. [Google Scholar] [Google Books] [DOI:10.4324/9781315718972-11]
Gordon, C. M., & Laufer, M. R. (2005) The Physiology of Puberty. In Emans, S. J., Laufer, M. R., & Goldstein, D. P. (Eds.). Pediatric and Adolescent Gynecology, 5th Edition (pp. 120–155). Philadelphia: Lippincott Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Göretzlehner, G. & Lauritzen, C. (1992). Hormontherapie bei Gynäkologischen Erkrankungen [Hormone Therapy for Gynecological Diseases]. In Göretzlehner, G. & Lauritzen, C. Praktische Hormontherapie in der Gynäkologie, 1. Auflage [Practical Hormone Therapy in Gynecology, 1st Edition] (pp. 245–299). Berlin/New York: de Gruyter. [DOI:10.1515/9783112417706-007] [Google Books] [PDF] [Translation]
Graham, J. D., & Clarke, C. L. (1997). Physiological Action of Progesterone in Target Tissues. Endocrine Reviews, 18(4), 502–519. [DOI:10.1210/edrv.18.4.0308]
Graham, S. J., Stanchev, P. L., Lloyd‐Smith, J. O., Bronskill, M. J., & Plewes, D. B. (1995). Changes in Fibroglandular Volume and Water Content of Breast Tissue During the Menstrual Cycle Observed by MR Imaging at 1.5 T. Journal of Magnetic Resonance Imaging, 5(6), 695–701. [DOI:10.1002/jmri.1880050613]
Greydanus, D. E., Omar, H. A., Matytsina, L. A., & Tsitsika, A. (2010). Breast Disorders in Children and Adolescents. In Omar, H. A., Greydanus, D. E., Tsitsika, A. K., Patel, D. R., & Merrick, J. (Eds.). Pediatric and Adolescent Sexuality and Gynecology: Principles for the Primary Care Clinician (pp. 245–316). Hauppauge: Nova Science Publishers. [Google Scholar] [Google Books] [URL] [OpenLibrary] [WorldCat] [PDF]
Guaragna-Filho, G., Guerra-Junior, G., Tadokoro-Cuccaro, R., Hughes, I. A., Barros, B. A., Hiort, O., Balsamo, A., Guran, T., Holterhus, P. M., Hannema, S., Poyrazoglu, S., Darendeliler, F., Bryce, J., Ahmed, S. F., & Quigley, C. A. (2023). Pubertal and Gonadal Outcomes in 46,XY Individuals with Partial Androgen Insensitivity Syndrome Raised as Girls. Sexual Development, 17(1), 16–25. [DOI:10.1159/000526997]
Gunn, H. M., Tsai, M., McRae, A., & Steinbeck, K. S. (2018). Menstrual Patterns in the First Gynecological Year: A Systematic Review. Journal of Pediatric and Adolescent Gynecology, 31(6), 557–565.e6. [DOI:10.1016/j.jpag.2018.07.009]
Günzel, P., Hasan, S. H., Düsterberg, B., Hümpel, M., Putz, B., & Lehmann, M. (1987). Zur toxikologischen Prüfung von Steroidhormonen. [For the Toxicological Testing of Steroid Hormones.] In Burger, O. K., Grosdanoff, P., Henschler, D., Kraupp, O., & Schnieders, B. (Eds.). Aktuelle Probleme der Biomedizin (pp. 93–112). Berlin/New York: De Gruyter. [Google Scholar] [Google Books] [DOI:10.1515/9783110898231-014]
Hagisawa, S., Shimura, N., & Arisaka, O. (2012). Effect of Excess Estrogen on Breast and External Genitalia Development in Growth Hormone Deficiency. Journal of Pediatric and Adolescent Gynecology, 25(3), e61–e63. [DOI:10.1016/j.jpag.2011.11.005]
Hamburger, C., & Benjamin, H. (1969). Endocrine Treatment of Male and Female Transsexualism / Appendix for the Practicing Physician: Suggestions and Guidelines for the Management of Transsexuals. In Green, R., & Money, J. (Eds.). Transsexualism and Sex Reassignment (pp. 291–307). Baltimore: John Hopkins University Press. [Google Scholar] [Google Books] [PDF]
Hannan, F. M., Elajnaf, T., Vandenberg, L. N., Kennedy, S. H., & Thakker, R. V. (2022). Hormonal regulation of mammary gland development and lactation. Nature Reviews Endocrinology, 19(1), 46–61. [DOI:10.1038/s41574-022-00742-y]
Hannema, S. E., Schagen, S. E., Cohen-Kettenis, P. T., & Delemarre-van de Waal, H. A. (2017). Efficacy and Safety of Pubertal Induction Using 17β-Estradiol in Transgirls. The Journal of Clinical Endocrinology & Metabolism, 102(7), 2356–2363. [DOI:10.1210/jc.2017-00373]
Hartmann, B. W., Laml, T., Albrecht, A. E., Huber, J. C., & Kirchengast, S. (1998). Hormonal Breast Augmentation: Prognostic Relevance of Insulin-Like Growth Factor-I. Gynecological Endocrinology, 12(2), 123–127. [DOI:10.3109/09513599809024960]
Hartmann, P. E., Owens, R. A., Cox, D. B., & Kent, J. C. (1996). Breast Development and Control of Milk Synthesis. Food and Nutrition Bulletin, 17(4), 1–12. [DOI:10.1177/156482659601700404]
Hasan, S. (1974). Steroid Hormone Levels During Pregnancy in Various Species. In Bernhard, S., & Raspé, G. (Ed.). Hormones and Embryonic Development (Advances in the Biosciences, Volume 13) (pp. 181–197). Oxford/New York: Pergamon Press. [Google Scholar] [Google Books] [DOI:10.1016/b978-0-08-018239-1.50014-3] [OpenLibrary] [Archive.org]
Hassiotou, F., & Geddes, D. (2012). Anatomy of the human mammary gland: Current status of knowledge. Clinical Anatomy, 26(1), 29–48. [DOI:10.1002/ca.22165]
Heath, R. A., & Wynne, K. (2019). Children and Adolescents. In Heath, R. A., & Wynne, K. A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (pp. 87–106). Santa Barbara: Praeger/ABC-CLIO. [Google Books]
Heath, R. A., & Wynne, K. (2019). Hormone and Surgical Therapies for Adults. In Heath, R. A., & Wynne, K. A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (pp. 107–146). Santa Barbara: Praeger/ABC-CLIO. [Google Books]
Hertz, R., Odell, W. D., & Ross, G. T. (1966). Diagnostic Implications of Primary Amenorrhea: Combined Clinical Staff Conference at the National Institutes of Health. Annals of Internal Medicine, 65(4), 800–820. [DOI:10.7326/0003-4819-65-4-800]
Hillard, P. J. A. (2007). Benign Diseases of the Female Reproductive Tract. In Berek, J. S., & Novak, E. (Eds.). Berek & Novak’s Gynecology, 14th Edition (pp. 431–496). Philadelphia: Lippincott Williams and Wilkins. [Google Scholar] [OpenLibrary] [WorldCat] [Archive.org]
Hoppe, I. C., Patel, P. P., Singer-Granick, C. J., & Granick, M. S. (2011). Virginal Mammary Hypertrophy: A Meta-Analysis and Treatment Algorithm. Plastic and Reconstructive Surgery, 127(6), 2224–2231. [DOI:10.1097/prs.0b013e3182131bd1]
Hovey, R. C., Trott, J. F., Ginsburg, E., Goldhar, A., Sasaki, M. M., Fountain, S. J., Sundararajan, K., & Vonderhaar, B. K. (2001). Transcriptional and spatiotemporal regulation of prolactin receptor mRNA and cooperativity with progesterone receptor function during ductal branch growth in the mammary gland. Developmental Dynamics, 222(2), 192–205. [DOI:10.1002/dvdy.1179]
Howard, B. A., & Gusterson, B. A. (2000). Human Breast Development. Journal of Mammary Gland Biology and Neoplasia, 5(2), 119–137. [DOI:10.1023/a:1026487120779]
Huffman, J., Dewhurst, C. J., & Capraro, V. J. (1981). The Breast and its Disorders in Childhood and Adolescence. In Huffman, J., Dewhurst, J., & Capraro, V. The Gynecology of Childhood and Adolescence, 2nd Edition (pp. 542–559). Philadelphia: Saunders. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Hussain, Z., Brooks, J., & Percy, D. (2008). Menstrual variation of breast volume and T2 relaxation times in cyclical mastalgia. Radiography, 14(1), 8–16. [DOI:10.1016/j.radi.2006.07.003]
Hussain, Z., Roberts, N., Whitehouse, G. H., García-Fiñana, M., & Percy, D. (1999). Estimation of breast volume and its variation during the menstrual cycle using MRI and stereology. The British Journal of Radiology, 72(855), 236–245. [DOI:10.1259/bjr.72.855.10396212]
Hutson, S. W., Cowen, P. N., & Bird, C. C. (1985). Morphometric studies of age related changes in normal human breast and their significance for evolution of mammary cancer. Journal of Clinical Pathology, 38(3), 281–287. [DOI:10.1136/jcp.38.3.281]
Hytten, F. E. (1954). Clinical and Chemical Studies in Human Lactation–VI. BMJ, 1(4867), 912–915. [DOI:10.1136/bmj.1.4867.912]
Hytten, F. E. (1976). The physiology of lactation. International Journal of Food Sciences and Nutrition, 30(4), 225–232. [DOI:10.3109/09637487609142745]
Hytten, F. E., & Baird, D. (1958). The Development of the Nipple in Pregnancy. The Lancet, 271(7032), 1201–1204. [DOI:10.1016/s0140-6736(58)91908-1]
Hytten, F. E., & Leitch, I. (1971). Preparations for Breast Feeding. In Hytten, F. E., & Leitch, I. The Physiology of Human Pregnancy, 2nd Edition (pp. 234–241). Oxford: Blackwell Scientific Publications. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Hytten, F. E., & Leitch, I. (1971). Preparations for Breast Feeding. In Hytten, F. E., & Leitch, I. The Physiology of Human Pregnancy, 2nd Edition (pp. 234–241). Oxford: Blackwell Scientific Publications. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Hytten, F. E., & Thomson, A. M. (1965). Pregnancy, childbirth and lactation. In Edholm, O. G., & Bacharach, A. L. (Eds.). The Physiology of Human Survival (pp. 327–350). London/New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Hytten, F. E., & Thomson, A. M. (1968). Maternal physiological adjustments. In Assali, N. S. (Ed.). The Maternal Organism, Volume I (Biology of Gestation) (pp. 449–479). New York: Academic Press. [Google Scholar] [Google Books] [WorldCat]
Igo, J., & Visram, H. (2021). Progesterone Therapy Use and Safety in Male to Female Transgender Patients. Canadian Journal of Diabetes, 45(7 Suppl), S39–S39 (abstract no. 109). [DOI:10.1016/j.jcjd.2021.09.119] [URL]
Ingleby, H. (1949). Changes in breast volume in a group of normal young women. Bulletin of the International Association of Medical Museums, 29, 87–92. [Google Scholar] [Google Books] [HathiTrust]
Ingleby, H., Moore, L., & Gershon-Cohen, J. (1957). Gestational breast changes: x-ray studies of the human breast. Obstetrics & Gynecology, 10(2), 149–157. [Google Scholar] [PubMed] [URL]
Ismail, P. M., Amato, P., Soyal, S. M., DeMayo, F. J., Conneely, O. M., O’Malley, B. W., & Lydon, J. P. (2003). Progesterone involvement in breast development and tumorigenesis—as revealed by progesterone receptor “knockout” and “knockin” mouse models. Steroids, 68(10–13), 779–787. [DOI:10.1016/s0039-128x(03)00133-8]
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 204201881987116. [DOI:10.1177/2042018819871166]
Jacobsohn, D. (1961). Hormonal Regulation of Mammary Gland Growth. In Kon, S. K., & Cowie, A. T. (Eds.). Milk: The Mammary Gland and Its Secretion, Volume 1 (pp. 127–160). New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Jamal, N., Ng, K., McLean, D., Looi, L., & Moosa, F. (2004). Mammographic Breast Glandularity in Malaysian Women: Data Derived from Radiography. American Journal of Roentgenology, 182(3), 713–717. [DOI:10.2214/ajr.182.3.1820713]
Jasieńska, G., Ziomkiewicz, A., Ellison, P. T., Lipson, S. F., & Thune, I. (2004). Large breasts and narrow waists indicate high reproductive potential in women. Proceedings of the Royal Society of London. Series B: Biological Sciences, 271(1545), 1213–1217. [DOI:10.1098/rspb.2004.2712]
Jemstrom, H., & Olsson, H. (1997). Breast Size in Relation to Endogenous Hormone Levels, Body Constitution, and Oral Contraceptive Use in Healthy Nulligravid Women Aged 19-25 Years. American Journal of Epidemiology, 145(7), 571–580. [DOI:10.1093/oxfordjournals.aje.a009153]
Jernström, H., Sandberg, T., Bågeman, E., Borg, Å., & Olsson, H. (2005). Insulin-like growth factor-1 (IGF1) genotype predicts breast volume after pregnancy and hormonal contraception and is associated with circulating IGF-1 levels: implications for risk of early-onset breast cancer in young women from hereditary breast cancer families. British Journal of Cancer, 92(5), 857–866. [DOI:10.1038/sj.bjc.6602389]
Jeruss, J. S. (2006). Molecular Basis of Breast Cancer. In Winchester, D. J., Winchester, D. P., Hudis, C. A., & Norton, L. (Eds.). Breast Cancer, 2nd Edition (pp. 83–95). Hamilton: B.C. Decker. [Google Scholar] [Google Books] [WorldCat]
Johnson, M. C., & Cutler, M. L. (2016). Anatomy and Physiology of the Breast. In Jatoi, I., & Rody, A. (Eds.). Management of Breast Diseases (pp. 1–39). Cham: Springer. [DOI:10.1007/978-3-319-46356-8_1]
Kaiser, R. (1993). Gestagen-Östrogen-Kombinationen in der Gynäkologie. Zur Geschichte, Dosierung und Anwendung eines Hormonprinzips. [Gestagen-Oestrogen Combinations in Gynaecology. History, Dosage and Use of a Hormonal Principle.] Geburtshilfe und Frauenheilkunde, 53(7), 503–513. [Google Scholar] [PubMed] [DOI:10.1055/s-2007-1022924]
Kaiser, R., & Leidenberger, F. (1991). Hormonbehandlung bei Benign Mammaerkrankungen: Mammahypoplasie. [Hormonal Treatment in Benign Breast Diseases: Breast Hypoplasia.] In Kaiser, R., & Leidenberger, F. Hormonbehandlung in der Gynäkologischen Praxis, 7. Auflage [Hormone Treatment in Gynecological Practice, 7th Edition] (pp. 138–138). Stuttgart: Georg Thieme Verlag. [Google Books] [OpenLibrary] [WorldCat] [PDF] [Translation]
Kaiser, U., & Ho, K. K. (2015). Pituitary Physiology and Diagnostic Evaluation. In Melmed, S., Polonsky, K. S., Larsen, P. R., Kronenberg, & H. M. (Eds.). Williams Textbook of Endocrinology, 13th Edition (pp. 176–231). Philadelphia: Elsevier. [DOI:10.1016/B978-0-323-29738-7.00008-3] [Google Books]
Kanhai, R. C., Hage, J. J., Asscheman, H., Mulder, W. J., & Hage, J. J. (1999). Augmentation Mammaplasty in Male-to-Female Transsexuals. Plastic and Reconstructive Surgery, 104(2), 542–549. [DOI:10.1097/00006534-199908000-00040]
Kanhai, R. C., Hage, J. J., van Diest, P. J., Bloemena, E., & Mulder, J. W. (2000). Short-Term and Long-Term Histologic Effects of Castration and Estrogen Treatment on Breast Tissue of 14 Male-to-Female Transsexuals in Comparison With Two Chemically Castrated Men. The American Journal of Surgical Pathology, 24(1), 74–80. [DOI:10.1097/00000478-200001000-00009]
Kardelen, A. D., Toksoy, G., Baş, F., Yavaş Abalı, Z., Gençay, G., Poyrazoğlu, Ş., Bundak, R., Altunoğlu, U., Avcı, Ş., Najaflı, A., Uyguner, O., Karaman, B., Başaran, S., & Darendeliler, F. (2018). A Rare Cause of Congenital Adrenal Hyperplasia: Clinical and Genetic Findings and Follow-up Characteristics of Six Patients with 17-Hydroxylase Deficiency Including Two Novel Mutations. Journal of Clinical Research in Pediatric Endocrinology, 10(3), 206–215. [DOI:10.4274/jcrpe.0032]
Kariagina, A., Xie, J., Leipprandt, J. R., & Haslam, S. Z. (2010). Amphiregulin Mediates Estrogen, Progesterone, and EGFR Signaling in the Normal Rat Mammary Gland and in Hormone-Dependent Rat Mammary Cancers. Hormones and Cancer, 1(5), 229–244. [DOI:10.1007/s12672-010-0048-0]
Karp, N. S. (2022). Discussion: The Impact of Combined Oral Contraceptives on Adolescents with Macromastia. Plastic & Reconstructive Surgery, 150(4), 739–740. [DOI:10.1097/prs.0000000000009514]
Kasielska-Trojan, A., Mikołajczyk, M., & Antoszewski, B. (2020). BreastIdea Volume Estimator: A New Tool for Breast Volume Estimation—Presentation and Validation for Women. Plastic & Reconstructive Surgery, 146(6), 744e–748e. [DOI:10.1097/prs.0000000000007373]
Kasielska-Trojan, A., Zawadzki, T., & Antoszewski, B. (2022). Breast Fluctuating Asymmetry in Women with Macromastia/Gigantomastia. International Journal of Environmental Research and Public Health, 19(24), 16895. [DOI:10.3390/ijerph192416895]
Kauli, R., Pertzelan, A., Ben‐Zeev, Z., Lewin, R. P., Kaufman, H., Schally, A. C., Schally, A. V., & Laron, Z. (1984). Treatment of precocious puberty with LHRH analogue in combination with cyproterone acetate—further experience. Clinical Endocrinology, 20(4), 377–387. [DOI:10.1111/j.1365-2265.1984.tb03433.x]
Keller, P. J. (1984). Diagnostik und Therapie wichtiger hormonaler Störungen: Mammahypoplasie. [Diagnosis and Treatment of Important Hormonal Disorders: Mammary Hypoplasia.] In Keller, P. J. Hormonale Störungen in der Gynäkologie: Diagnostik und Behandlung, 3. Auflage [Hormonal Disorders in Gynecology: Diagnosis and Treatment, 3rd Edition] (Kliniktaschenbücher) (pp. 133–134). Berlin/Heidelberg: Springer. [Google Books] [DOI:10.1007/978-3-662-00442-5_3]
Keller, P. J. (1995). Diagnostik und Therapie wichtiger Störungen: Mammahypoplasie. [Diagnosis and Treatment of Important Disorders: Mammary Hypoplasia.] In Keller, P. J. Hormon- und Fertilitätsstörungen in der Gynäkologie, 4. Auflage [Hormonal and Fertility Disorders in Gynecology, 4th Edition] (pp. 145–146). Berlin/Heidelberg: Springer. [Google Books] [DOI:10.1007/978-3-662-12026-2_3]
Kennedy, B. J. (1953). Effects of intensive sex steroid hormone therapy in advanced breast cancer. JAMA, 152(12), 1135–1141. [DOI:10.1001/jama.1953.63690120004013]
Kent, J. C., Mitoulas, L., Cox, D. B., Owens, R. A., & Hartmann, P. E. (1999). Breast Volume and Milk Production During Extended Lactation in Women. Experimental Physiology, 84(2), 435–447. [DOI:10.1111/j.1469-445x.1999.01808.x]
Khoo, S. K., & Mackay, E. V. (1972). Testicular Feminization: The Clinical Features, Endocrine Function and Gonadal Pathology in Six Patients. Australian and New Zealand Journal of Obstetrics and Gynaecology, 12(1), 1–13. [DOI:10.1111/j.1479-828x.1972.tb00721.x]
Klein, R., Aichinger, H., Dierker, J., Jansen, J. T., Joite-Barfuß, S., Säbel, M., Schulz-Wendtland, R., & Zoetelief, J. (1997). Determination of average glandular dose with modern mammography units for two large groups of patients. Physics in Medicine and Biology, 42(4), 651–671. [DOI:10.1088/0031-9155/42/4/004]
Kleinberg, D. L. (2006). Endocrinology of lactation. In DeGroot, L. J., & Jameson, J. L. (Eds). Endocrinology, 5th Edition (pp. 3461–3473). Philadelphia: Elsevier Saunders. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Kleinberg, D. L., & Barcellos-Hoff, M. H. (2011). The Pivotal Role of Insulin-Like Growth Factor I in Normal Mammary Development. Endocrinology and Metabolism Clinics of North America, 40(3), 461–471. [DOI:10.1016/j.ecl.2011.06.001]
Kleinberg, D. L., & Ruan, W. (2008). IGF-I, GH, and Sex Steroid Effects in Normal Mammary Gland Development. Journal of Mammary Gland Biology and Neoplasia, 13(4), 353–360. [DOI:10.1007/s10911-008-9103-7]
Kleinberg, D. L., Wood, T. L., Furth, P. A., & Lee, A. V. (2008). Growth Hormone and Insulin-Like Growth Factor-I in the Transition from Normal Mammary Development to Preneoplastic Mammary Lesions. Endocrine Reviews, 30(1), 51–74. [DOI:10.1210/er.2008-0022]
Kühnel, W. (2000). IMMULITE® and IMMULITE® 2000 Reference Range Compendium, First English Edition. Los Angeles, California: Diagnostic Products Corporation. [Google Scholar] [URL] [PDF 1] [PDF 2]
Kustin, J., & Rebar, R. W. (1987). Menstrual Disorders in the Adolescent Age Group. Primary Care: Clinics in Office Practice, 14(1), 139–166. [DOI:10.1016/s0095-4543(21)01004-6]
Kutten, F., Malet, C., & Leygue, E. (1994). Antiestrogen action of progestogens in human breast cells. In Berg, G., & Hammar, M. (Eds.). The Modern Management of the Menopause: A Perspective for the 21st Century [The Proceedings of the VII International Congress on the Menopause, Stockholm, Sweden 1993] (pp. 419–433). Canforth: Parthenon. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Ladjouze, A., & Donaldson, M. (2019). Primary gonadal failure. Best Practice & Research Clinical Endocrinology & Metabolism, 33(3), 101295. [DOI:10.1016/j.beem.2019.101295]
Laessle, R. G., Tuschi, R. J., Schweiger, U., & Pirke, K. M. (1990). Mood changes and physical complaints during the normal menstrual cycle in healthy young women. Psychoneuroendocrinology, 15(2), 131–138. [DOI:10.1016/0306-4530(90)90021-z]
LaMarca, H. L., & Rosen, J. M. (2007). Estrogen regulation of mammary gland development and breast cancer: amphiregulin takes center stage. Breast Cancer Research, 9(4), 304. [DOI:10.1186/bcr1740]
Laron, Z., & Kauli, R. (2000). Experience with Cyproterone Acetate in the Treatment of Precocious Puberty. Journal of Pediatric Endocrinology and Metabolism, 13(Suppl 1), 805–810. [DOI:10.1515/jpem.2000.13.s1.805]
Laufer, M. R., Goldstein, D. P., & Hendren, W. H. (2005). Structural Abnormalities of the Female Reproductive Tract. In Emans, J. E., Laufer, M. R., & Goldstein, D. P. (Eds.). Pediatric and Adolescent Gynecology, 5th Edition (pp. 334–416). Philadelphia: Lippincott Williams and Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Lauritzen, C. (1980 October 27). Hormonkur Kann Hypoplastischer Mamma Aufhelfen. [Hormone Therapy Can Help Hypoplastic Breasts.] Selecta: Medizin aktuell; das Magazin für ärztliche Fortbildung [Selecta: Medicine Currently; the Magazine for Medical Training], 22(43), 3798–3801. Planegg: Selecta-Verlag. [ISSN:0582-4877] [WorldCat] [PDF] [Translation]
Lauritzen, C. (1982). Treatment of mammary- and uterus hypoplasia by estrogen-progestagen pseudopregnancy. In Richter, K., Huber, A., Terruhn, V. (Eds.) 1. Europäisches Symposium für Kinder- und Jugendgynäkologie. München 19.-21.3.1981, Band 2 [First European Symposium on Pediatric and Adolescent Gynaecology. München, March 19–21, 1981, Volume 2] (pp. 487–489). Friedrichsdorf/Taunus: Milupa-Aktiengesellschaft, Wissenschaftliche Abteilung. [Google Scholar] [PDF]
Lauritzen, C. (1989). Hormonelle Substitutionstherapie zur Brustvergrößerung. [Hormonal Substitution Therapy for Breast Enlargement.] In Beller, F. K. & Seitzer, D. (Eds.). 7. Siebte Wissenschaftliche Tagung der Deutschen Gesellschaft für Senologie, Münster, 25. - 27. September 1987: Ausgewählte Referate [7th Scientific Conference of the German Society for Senology, Münster, [Germany,] September 25–27, 1987: Selected Papers] (pp. 39–41). Mülheim, Germany: H.U.F. Verlag. [Google Scholar] [WorldCat 1] [WorldCat 2] [WorldCat 3] [Cited By 1] [Cited By 2] [Cited By 3] [PDF] [Translation]
Lawrence, R. A., & Lawrence, R. M. (2015). Physiology of Lactation. In Lawrence, R. A., & Lawrence, R. M. Breastfeeding: A Guide for the Medical Profession, 8th Edition (pp. 56–90). Philadelphia: Elsevier. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Lee, H. J., & Ormandy, C. J. (2012). Interplay between progesterone and prolactin in mammary development and implications for breast cancer. Molecular and Cellular Endocrinology, 357(1–2), 101–107. [DOI:10.1016/j.mce.2011.09.020]
Lee, P. A. (2001). Physiology of Puberty. In Becker, K. L., Bilezikian, J. P., Bremner, W. J., Hung, W., Kahn, C. R., Loriaux, D. L., Nylén, E. S., Rebar, R. W., Robertson, G. L., Snider, R. H., Wartofsky, L. (Eds.). Principles and Practice of Endocrinology and Metabolism, 3rd Edition (pp. 885–893). Philadelphia: Lippincott Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Lee, S. W., Kwak, D. S., Jung, I. S., Kwak, J. H., Park, J. H., Hong, S. M., Lee, C. B., Park, Y. S., Kim, D. S., Choi, W. H., & Ahn, Y. H. (2015). Partial Androgen Insensitivity Syndrome Presenting with Gynecomastia. Endocrinology and Metabolism, 30(2), 226–230. [DOI:10.3803/enm.2015.30.2.226]
Lejour, M. (1994). Vertical Mammaplasty and Liposuction of the Breast. Plastic and Reconstructive Surgery, 94(1), 100–114. [DOI:10.1097/00006534-199407000-00010]
Lejour, M. (1997). Evaluation of Fat in Breast Tissue Removed by Vertical Mammaplasty. Plastic and Reconstructive Surgery, 99(2), 386–393. [DOI:10.1097/00006534-199702000-00012]
Lemarchand-Béraud, T., Zufferey, M., Reymond, M., & Rey, I. (1982). Maturation of the Hypothalamo-Pituitary-Ovarian Axis in Adolescent Girls. The Journal of Clinical Endocrinology & Metabolism, 54(2), 241–246. [DOI:10.1210/jcem-54-2-241]
Levy, A., Crown, A., & Reid, R. (2003). Endocrine intervention for transsexuals. Clinical Endocrinology, 59(4), 409–418. [DOI:10.1046/j.1365-2265.2003.01821.x]
Lewin, R. (2016). Breast Hypertrophy and Outcome of Breast Reduction Surgery. (Doctoral thesis, University of Gothenburg. Sahlgrenska Academy.) [Google Scholar] [URL]
Lim, L. Y., Ho, P. J., Liu, J., Chay, W. Y., Tan, M., Hartman, M., & Li, J. (2018). Determinants of breast size in Asian women. Scientific Reports, 8(1), 1201. [DOI:10.1038/s41598-018-19437-4]
Lloyd, C. W., & Leathem, J. H. (1964). Growth and development of the breast and lactation. In Lloyd, C. W. (Ed.). Human Reproduction and Sexual Behavior (pp. 117–134). Philadelphia: Lea & Febiger. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Lopez, X., Panton, J., Nagarkar, P., Preston, S., Abramowitz, J., & Amirlak, B. (2023). Initial Assessment of VECTRA Three-Dimensional Imaging to Accurately Simulate Breast Volume Changes in Transfeminine Patients: A Mannequin Study. Aesthetic Surgery Journal Open Forum, 5, online ahead of print. [DOI:10.1093/asjof/ojad015]
Lorincz, A. M., & Sukumar, S. (2006). Molecular links between obesity and breast cancer. Endocrine-Related Cancer, 13(2), 279–292. [DOI:10.1677/erc.1.00729]
Lucien, J. N., Ortega, M. T., Calvert, M. E., Smith, C., White, X., Rogers, H., Mosley, B., Agrawal, R., Drude, A., McGee, C., George, M., Brown, A., Downey, K., Wild, C., Njunge, A., Kuzmiak, C. M., Zava, D., Zava, T., Pollard, J., Francis, J., Beery, B. L., Harlin, M., Gonzalez, G. R., & Shaw, N. D. (2022). The Launch of A Girl’s First Period Study: Demystifying Reproductive Hormone Profiles in Adolescent Girls. Journal of Pediatric and Adolescent Gynecology, 35(4), 420–425. [DOI:10.1016/j.jpag.2021.12.018]
Lydon, J. P., DeMayo, F. J., Funk, C. R., Mani, S. K., Hughes, A. R., Montgomery, C. A., Shyamala, G., Conneely, O. M., & O’Malley, B. W. (1995). Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes & Development, 9(18), 2266–2278. [DOI:10.1101/gad.9.18.2266]
Lyon, A. J., De Bruyn, R., & Grant, D. B. (1985). Isosexual Precocious Puberty in Girls. Acta Paediatrica, 74(6), 950–955. [DOI:10.1111/j.1651-2227.1985.tb10063.x]
Lyons, W. R. (1958). Hormonal synergism in mammary growth. Proceedings of the Royal Society of London. Series B - Biological Sciences, 149(936), 303–325. [DOI:10.1098/rspb.1958.0071]
Lyons, W. R., & McGinty, D. A. (1941). Effects of estrone and progesterone on male rabbit mammary glands. I. Varying doses of progesterone. Proceedings of the Society for Experimental Biology and Medicine, 48(1), 83–86. [DOI:10.3181/00379727-48-13227]
Lyons, W. R., Li, C. H., & Johnson, R. E. (1958). The Hormonal Control of Mammary Growth and Lactation. In Pincus, G. (Ed.). Recent Progress in Hormone Research, Volume 14 [Proceedings of the Laurentian Hormone Conference 1957, Held at Mont Tremblant, Quebec] (pp. 219–254). New York/London: Academic Press. [Google Scholar] [Google Books] [PubMed] [OpenLibrary] [WorldCat] [PDF]
MacBryde, C. M. (1939). The Production of Breast Growth in the Human Female. Journal of the American Medical Association, 112(11), 1045–1049. [DOI:10.1001/jama.1939.02800110025006]
Malet, C., Gompel, A., Yaneva, H., Cren, H., Fidji, N., Mowszowicz, I., Kuttenn, F., & Mauvais-Jarvis, P. (1991). Estradiol and Progesterone Receptors in Cultured Normal Human Breast Epithelial Cells and Fibroblasts: Immunocytochemical Studies. The Journal of Clinical Endocrinology & Metabolism, 73(1), 8–17. [DOI:10.1210/jcem-73-1-8]
Malini, S., Smith, E. O., & Goldzieher, J. W. (1985). Measurement of breast volume by ultrasound during normal menstrual cycles and with oral contraceptive use. Obstetrics and Gynecology, 66(4), 538–541. [Google Scholar] [PubMed] [URL] [PDF]
Marshall, W. A. (1978). Puberty. In Falkner, F., & Tanner, J. M. (Eds.). Human Growth: Postnatal Growth (pp. 141–181). Boston: Springer US. [DOI:10.1007/978-1-4684-2622-9_6]
Marshall, W. A., & Tanner, J. M. (1969). Variations in pattern of pubertal changes in girls. Archives of Disease in Childhood, 44(235), 291–303. [DOI:10.1136/adc.44.235.291]
Mauvais-Jarvis, P., Kuttenn, F., & Gompel, A. (1986). Antiestrogen action of progesterone in breast tissue. Breast Cancer Research and Treatment, 8(3), 179–188. [DOI:10.1007/bf01807330]
Mauvais-Jarvis, P., Kuttenn, F., & Gompel, A. (1986). Estradiol/Progesterone Interaction in Normal and Pathologic Breast Cells. Annals of the New York Academy of Sciences, 464(1), 152–167. [DOI:10.1111/j.1749-6632.1986.tb16002.x]
Mauvais-Jarvis, P., Kuttenn, F., & Gompel, A. (1987). Antiestrogen Action of Progesterone in Breast Tissue. Hormone Research, 28(2–4), 212–218. [DOI:10.1159/000180946]
Mauvais-Jarvis, P., Kuttenn, F., Gompel, A., & Benotmane, A. (1987). Action anti-estrogène de la progestérone dans le sein. [Antiestrogen action of progesterone in the breast]. Pathologie-Biologie, 35(7), 1081–1086. [Google Scholar 1] [Google Scholar 2] [PubMed]
Mauvais-Jarvis, P., Sitruk-Ware, R., & Kuttenn, F. (1981). Benign Breast Disease. In McGuire, W. L. (Ed.). Breast Cancer 4: Advances in Research and Treatment (pp. 51–94). Boston: Springer US. [DOI:10.1007/978-1-4615-6571-0_3]
McArthur, J. W. (1966). The Reproductive Endocrinology of Adolescence. In Heald, F. P. (Ed.). Adolescent Gynecology (pp. 9–20). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
McBryan, J., Howlin, J., Napoletano, S., & Martin, F. (2008). Amphiregulin: Role in Mammary Gland Development and Breast Cancer. Journal of Mammary Gland Biology and Neoplasia, 13(2), 159–169. [DOI:10.1007/s10911-008-9075-7]
McDonough, P. G., Spicer, D. V., Ursin, G., & Pike, M. C. (1996). Progesterone Concentrations—Physiologic or Pharmacologic? Fertility and Sterility, 65(5), 1077–1078. [DOI:10.1016/s0015-0282(16)58295-8]
McMillan, M. (1966). Five cases of testicular feminisation including one with a teratoma of the testis. The Journal of Pathology and Bacteriology, 91(2), 417–427. [DOI:10.1002/path.1700910216]
McNally, S., & Stein, T. (2016). Overview of Mammary Gland Development: A Comparison of Mouse and Human. In Martin, F., Stein, T., & Howlin, J. (Eds.). Mammary Gland Development (Methods in Molecular Biology, Volume 1501) (pp. 1–17). New York: Springer New York. [DOI:10.1007/978-1-4939-6475-8_1]
McPhaul, M. J. (2002). Androgen receptor mutations and androgen insensitivity. Molecular and Cellular Endocrinology, 198(1–2), 61–67. [DOI:10.1016/s0303-7207(02)00369-6]
Mesiano, S. (2019). Endocrinology of Human Pregnancy and Fetal-Placental Neuroendocrine Development. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 256–284.e9). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/b978-0-323-47912-7.00011-1]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463]
Meyer, W. J., Finkelstein, J. W., Stuart, C. A., Webb, A., Smith, E. R., Payer, A. F., & Walker, P. A. (1981). Physical and hormonal evaluation of transsexual patients during hormonal therapy. Archives of Sexual Behavior, 10(4), 347–356. [DOI:10.1007/bf01565538]
Meyer, W. J., Webb, A., Stuart, C. A., Finkelstein, J. W., Lawrence, B., & Walker, P. A. (1986). Physical and hormonal evaluation of transsexual patients: A longitudinal study. Archives of Sexual Behavior, 15(2), 121–138. [DOI:10.1007/bf01542220]
Mikołajczyk, M., Kasielska-Trojan, A., & Antoszewski, B. (2019). A New Tool for Breast Anthropometric Measurements: Presentation and Validation for Women and Men. Aesthetic Plastic Surgery, 43(5), 1160–1170. [DOI:10.1007/s00266-019-01467-6]
Milionis, C., Ilias, I., & Koukkou, E. (2022). Progesterone in gender-affirming therapy of trans women. World Journal of Biological Chemistry, 13(3), 66–71. [DOI:10.4331/wjbc.v13.i3.66]
Milligan, D., Drife, J. O., & Short, R. V. (1975). Changes in breast volume during normal menstrual cycle and after oral contraceptives. BMJ, 4(5995), 494–496. [DOI:10.1136/bmj.4.5995.494]
Morris, J. M. (1953). The syndrome of testicular feminization in male pseudohermaphrodites. American Journal of Obstetrics and Gynecology, 65(6), 1192–1211. [DOI:10.1016/0002-9378(53)90359-7]
Morris, J. M., & Mahesh, V. B. (1963). Further observations on the syndrome, “testicular feminization”. American Journal of Obstetrics and Gynecology, 87(6), 731–748. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF]
Muallem, M. M., & Rubeiz, N. G. (2006). Physiological and biological skin changes in pregnancy. Clinics in Dermatology, 24(2), 80–83. [DOI:10.1016/j.clindermatol.2005.10.002]
Müller, C. (1953). Zur Hormonalen Kosmetik der Weiblichen Brust: Wirkung und Nebenwirkung Östrogenhaltiger Kosmetischer Präparate. [Hormonal Cosmetics of the Female Breast: Effects and Side Effects of Estrogen-Containing Cosmetic Preparations.] Schweizerische Medizinische Wochenschrift [Swiss Medical Weekly], 83(4), 81–84. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF] [Translation]
Muzaffar, F., Hussain, I., & Haroon, T. S. (1998). Physiologic skin changes during pregnancy: a study of 140 cases. International Journal of Dermatology, 37(6), 429–431. [DOI:10.1046/j.1365-4362.1998.00281.x]
Naik, M., Diwakar, R. K., Patre, S., & Singh, S. (2015). Gigantomastia: A Rare Complication In Pregnancy. Journal of Medical Science and Clinical Research, 3(7), 6873–6876. [Google Scholar] [URL] [PDF]
Naseem, H., Lokman, M., & Fitzgerald, C. (2021). Management of congenital hypogonadotropic hypogonadism in females. Human Fertility, 26(3), 622–631. [DOI:10.1080/14647273.2021.1998929]
Nelson, W. O. (1936). Endocrine Control of the Mammary Gland. Physiological Reviews, 16(3), 488–526. [DOI:10.1152/physrev.1936.16.3.488]
Nolan, B. J., Frydman, A. S., Leemaqz, S. Y., Carroll, M., Grossmann, M., Zajac, J. D., & Cheung, A. S. (2022). Effects of low-dose oral micronised progesterone on sleep, psychological distress, and breast development in transgender individuals undergoing feminising hormone therapy: a prospective controlled study. Endocrine Connections, 11(5), e220170. [DOI:10.1530/EC-22-0170]
Nolan, B. J., Frydman, A. S., Leemaqz, S. Y., Carroll, M., Grossmann, M., Zajac, J. D., & Cheung, A. S. (2022). Effects Of Low-dose Oral Micronised Progesterone On Sleep, Psychological Distress And Breast Development In Transgender Individuals Undergoing Feminising Hormone Therapy: A Prospective Controlled Study. Journal of the Endocrine Society, 6(Suppl 1), A653–A654 (abstract no. LBODP089). [DOI:10.1210/jendso/bvac150.1351]
Nussbaum, R., & Benedetto, A. V. (2006). Cosmetic aspects of pregnancy. Clinics in Dermatology, 24(2), 133–141. [DOI:10.1016/j.clindermatol.2005.10.007]
Nuzzi, L. C., Pramanick, T., Massey, G. G., Walsh, L. R., McNamara, C. T., Firriolo, J. M., DiVasta, A. D., & Labow, B. I. (2021). The Impact of Progestin-only Contraception on Adolescents with Macromastia. Plastic and Reconstructive Surgery - Global Open, 9(2), e3421. [DOI:10.1097/gox.0000000000003421]
Nuzzi, L. C., Pramanick, T., Massey, G. G., Walsh, L. R., McNamara, C. T., Firriolo, J. M., DiVasta, A. D., & Labow, B. I. (2022). The Impact of Combined Oral Contraceptives on Adolescents with Macromastia. Plastic and Reconstructive Surgery, 150(4), 731–738. [DOI:10.1097/prs.0000000000009513]
Oakes, M. B., Eyvazzadeh, A. D., Quint, E., & Smith, Y. R. (2008). Complete Androgen Insensitivity Syndrome—A Review. Journal of Pediatric and Adolescent Gynecology, 21(6), 305–310. [DOI:10.1016/j.jpag.2007.09.006]
Obr, A. E., & Edwards, D. P. (2012). The biology of progesterone receptor in the normal mammary gland and in breast cancer. Molecular and Cellular Endocrinology, 357(1–2), 4–17. [DOI:10.1016/j.mce.2011.10.030]
Olanrewaju, F., Onayemi, O., Olasode, O., Adeyemi, A., Oninla, A., Oripelaye, M., Ezejiofor, O., & Oke, O. (2017). Prevalence and Pattern of Pigmentary Changes among Primigravidae Attending a Tertiary Health Facility in South-Western Nigeria. British Journal of Medicine and Medical Research, 21(5), 1–9. [DOI:10.9734/bjmmr/2017/33382]
Orentreich, N., & Durr, N. P. (1974). Mammogenesis in Transsexuals. Journal of Investigative Dermatology, 63(1), 142–146. [DOI:10.1111/1523-1747.ep12678272]
Pandya, S., & Moore, R. G. (2011). Breast Development and Anatomy. Clinical Obstetrics & Gynecology, 54(1), 91–95. [DOI:10.1097/grf.0b013e318207ffe9]
Park, I. Y., Kim, M. R., Jo, H. H., Lee, M. K., & Kim, M. J. (2014). Association of the Nipple-Areola Complexes with Age, Parity, and Breastfeeding in Korean Premenopausal Women. Journal of Human Lactation, 30(4), 474–479. [DOI:10.1177/0890334414549049]
Pasqualini, J. R. (2007). Progestins and breast cancer. Gynecological Endocrinology, 23(Suppl 1), 32–41. [DOI:10.1080/09513590701585003]
Pasqualini, J. R. (2009). Breast cancer and steroid metabolizing enzymes: The role of progestogens. Maturitas, 65(Suppl 1), S17–S21. [DOI:10.1016/j.maturitas.2009.11.006]
Pasqualini, J. R., & Kincl, F. A. (1985). Hormone Production and Concentrations During Pregnancy in Humans and in Other Mammalian Species. In Pasqualini, J. R., & Kincl, F. A. Hormones and the Fetus, Volume I: Production, Concentration and Metabolism During Pregnancy (pp. 173–334). Oxford: Pergamon Press. [DOI:10.1016/b978-0-08-019708-1.50007-6]
Patel, H., Arruarana, V., Yao, L., Cui, X., & Ray, E. (2020). Effects of hormones and hormone therapy on breast tissue in transgender patients: a concise review. Endocrine, 68(1), 6–15. [DOI:10.1007/s12020-020-02197-5]
Patel, K. T., Adeel, S., Rodrigues Miragaya, J., & Tangpricha, V. (2022). Progestogen Use in Gender-Affirming Hormone Therapy: A Systematic Review. Endocrine Practice, 28(12), 1244–1252. [DOI:10.1016/j.eprac.2022.08.012]
Patterson, M. N., McPhaul, M. J., & Hughes, I. A. (1994). Androgen insensitivity syndrome. Baillière’s Clinical Endocrinology and Metabolism, 8(2), 379–404. [DOI:10.1016/s0950-351x(05)80258-7]
Pawłowski, B., & Żelaźniewicz, A. (2021). The evolution of perennially enlarged breasts in women: a critical review and a novel hypothesis. Biological Reviews, 96(6), 2794–2809. [DOI:10.1111/brv.12778]
Perez-Palacios, G., & Jaffe, R. B. (1972). The Syndrome of Testicular Feminization. Pediatric Clinics of North America, 19(3), 653–667. [DOI:10.1016/s0031-3955(16)32744-4]
Petrakis, N. (1978). Recurrent extreme breast involution following pregnancy and susceptibility to breast cancer: A hypothesis. Medical Hypotheses, 4(3), 268–272. [DOI:10.1016/0306-9877(78)90006-3]
Pfeiffer, C. A. (1943). Endocrinology of Reproduction. Annual Review of Physiology, 5(1), 413–452. [DOI:10.1146/annurev.ph.05.030143.002213]
Pipkin, F. B. (2019). Physiological Changes in Pregnancy. In Symonds, I. M., & Arulkumaran, S. (Eds.). Essential Obstetrics and Gynaecology, 6th Edition (pp. 22–40). Edinburgh: Elsevier. [Google Books] [OpenLibrary] [WorldCat]
Plu-Bureau, G., Touraine, P., & Mauvais-Jarvis, P. (1999). Interactions Between Estradiol and Progesterone in Normal Breast: Implications for Mammary Carcinogenesis. In Manni, A. (Ed.). Endocrinology of Breast Cancer (Contemporary Endocrinology, Volume 11) (pp. 21–37). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-59259-699-7_2] [OpenLibrary] [Archive.org]
Pocock, G., Richards, C. D., & Richards, D. A. (2013). Fertilization, Pregnancy, and Lactation. In Pocock, G., Richards, C. D., & Richards, D. A. Human Physiology, 4th Edition (pp. 655–752). Oxford: Oxford University Press. [Google Scholar] [Google Books]
Polani, P. E. (1970). Hormonal and clinical aspects of hermaphroditism and the testicular feminizing syndrome in man. Philosophical Transactions of the Royal Society of London. B, Biological Sciences, 259(828), 187–206. [DOI:10.1098/rstb.1970.0058]
Price, S., McManus, J., & Barrett, J. (2019). The transgender population: improving awareness for gynaecologists and their role in the provision of care. The Obstetrician & Gynaecologist, 21(1), 11–20. [DOI:10.1111/tog.12521]
Prior, J. C. (2011). Progesterone for Symptomatic Perimenopause Treatment - Progesterone politics, physiology and potential for perimenopause. Facts, Views & Vision in ObGyn, 3(2), 109–120. [PubMed] [PubMed Central] [PDF]
Prior, J. C. (2019). Progesterone Is Important for Transgender Women’s Therapy—Applying Evidence for the Benefits of Progesterone in Ciswomen. The Journal of Clinical Endocrinology & Metabolism, 104(4), 1181–1186. [DOI:10.1210/jc.2018-01777]
Prior, J. C. (2019). Response to Letter to the Editor: “Progesterone Is Important for Transgender Women’s Therapy—Applying Evidence for the Benefits of Progesterone in Ciswomen”. The Journal of Clinical Endocrinology & Metabolism, 104(8), 3129–3130. [DOI:10.1210/jc.2019-00524]
Prior, J. C. (2020). Women’s Reproductive System as Balanced Estradiol and Progesterone Actions—A revolutionary, paradigm-shifting concept in women’s health. Drug Discovery Today: Disease Models, 32(Part B), 31–40. [DOI:10.1016/j.ddmod.2020.11.005]
Prior, J. C., Vigna, Y. M., & Watson, D. (1989). Spironolactone with physiological female steroids for presurgical therapy of male-to-female transsexualism. Archives of Sexual Behavior, 18(1), 49–57. [DOI:10.1007/bf01579291]
Prior, J. C., Vigna, Y. M., Watson, D., Diewold, P., & Robinow, O. (1986). Spironolactone in the presurgical therapy of male to female transsexuals: Philosophy and experience of the Vancouver Gender Dysphoria Clinic. Journal of Sex Information & Education Council of Canada, 1(1), 1–7. [Google Scholar] [PDF]
Quigley, C. A. (1998). The androgen receptor: Physiology and pathophysiology. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 33–106). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_2]
Quigley, C. A., Bellis, A. D., Marschke, K. B., El-Awady, M. K., Wilson, E. M., & French, F. S. (1995). Androgen Receptor Defects: Historical, Clinical, and Molecular Perspectives. Endocrine Reviews, 16(3), 271–321. [DOI:10.1210/edrv-16-3-271]
Radisky, D. C., & Hartmann, L. C. (2009). Mammary Involution and Breast Cancer Risk: Transgenic Models and Clinical Studies. Journal of Mammary Gland Biology and Neoplasia, 14(2), 181–191. [DOI:10.1007/s10911-009-9123-y]
Ramos, L., Chávez, B., Mares, L., Valdés, E., & Vilchis, F. (2018). Mutational analysis of the androgen receptor (NR3C4) gene in patients with 46,XY DSD. Gene, 641, 86–93. [DOI:10.1016/j.gene.2017.10.038]
Ramsay, D. T., Kent, J. C., Hartmann, R. A., & Hartmann, P. E. (2005). Anatomy of the lactating human breast redefined with ultrasound imaging. Journal of Anatomy, 206(6), 525–534. [DOI:10.1111/j.1469-7580.2005.00417.x]
Randolph, J. F. (2018). Gender-Affirming Hormone Therapy for Transgender Females. Clinical Obstetrics & Gynecology, 61(4), 705–721. [DOI:10.1097/grf.0000000000000396]
Rauh, C., Faschingbauer, F., Haeberle, L., Jud, S. M., Heusinger, K., Fasching, P. A., Goecke, T. W., Rajakaruna, N., Voigt, F., Bani, M. R., Lux, M. P., Renner, S. P., Loehberg, C. R., Hartmann, A., Schulz-Wendtland, R., Beckmann, M. W., & Bayer, C. M. (2013). Factors influencing breast changes after pregnancy. European Journal of Cancer Prevention, 22(3), 259–261. [DOI:10.1097/cej.0b013e328359cb81]
Rebar, R. W. (1988). The Ovaries. In Wyngaarden, J. B., & Smith, L. H. (Eds.). Cecil Textbook of Medicine, 18th Edition (pp. 1425–1446). Philadelphia: W. B. Saunders. [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Rebar, R. W. (1990). Disorders of Menstruation, Ovulation, and Sexual Response. In Becker, K. L., Bilezikian, J. P., Bremner, W. J., Hung, W., Kahn, C. R., Loriaux, D. L., Rebar, R. W., Robertson, G. L., & Wartofsky, L. (Eds.). Principles and Practice of Endocrinology and Metabolism, 1st Edition (pp. 798–814). Philadelphia: Lippincott. [Google Scholar—3rd/2001 Edition] [Google Books—3rd/2001 Edition] [OpenLibrary] [WorldCat] [Archive.org]
Rebar, R. W. (1993). Normal and Abnormal Sexual Differentiation and Pubertal Development. In Moore, T. R., Reiter, R. C., Rebar, R. W., & Baker, W. (Eds.). Gynecology & Obstetrics: A Longitudinal Approach (pp. 97–133). New York: Churchill Livingstone. [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Rebar, R. W. (1996). Puberty. In Berek, J. S., Adashi, E. Y., & Hillard, P. A. (Eds.). Novak’s Gynecology, 12th Edition (pp. 771–807). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Reindollar, R. H., & McDonough, P. G. (1985). The Child with Ambiguous Genitalia. In Lavery, J. P., & Sanfilippo, J. S. (Eds.). Pediatric and Adolescent Obstetrics and Gynecology, 1st Edition (Clinical Perspective in Obstetrics and Gynecology) (pp. 38–60). New York: Springer. [DOI:10.1007/978-1-4612-5064-7_5]
Reisman, T., Goldstein, Z., & Safer, J. D. (2019). A Review of Breast Development in Cisgender Women and Implications for Transgender Women. Endocrine Practice, 25(12), 1338–1345. [DOI:10.4158/ep-2019-0183]
Richards, C., & Barrett, J. (2020). Physical Treatments for Trans People and Their Interactions with Psychiatric Treatments. In Richards, C., & Barrett, J. Trans and Non-binary Gender Healthcare for Psychiatrists, Psychologists, and Other Health Professionals (pp. 31–44). Cambridge: Cambridge University Press. [DOI:10.1017/9781108628419.003]
Richert, M. M., Schwertfeger, K. L., Ryder, J. W., & Anderson, S. M. (2000). An Atlas of Mouse Mammary Gland Development. Journal of Mammary Gland Biology and Neoplasia, 5(2), 227–241. [DOI:10.1023/a:1026499523505]
Rix, J., Mills, C., Ross, E., Allen, S., Lai, A., & Wakefield-Scurr, J. (2023). Breast volume fluctuations are associated with oestradiol and progesterone changes across the menstrual cycle. Research Square, preprint. [Google Scholar] [DOI:10.21203/rs.3.rs-3753080/v1] [PDF]
Rodari, G. (2022). Pubertal induction in girls with hypogonadism: estrogen replacement therapy outcomes and optimization of progesterone introduction. (Doctoral thesis, Università degli Studi di Milano.) [URL]
Rodari, G., Federici, S., Cattoni, A., Todisco, T., Ubertini, G., Giacchetti, F., Profka, E., Dall’Antonia, A., Cangiano, B., Arosio, M., Bonomi, M., Cappa, M., & Giavoli, C. (2022). Pubertal induction in girls with hypogonadism: insight into estrogen replacement therapy outcomes and optimization of progesterone introduction. Endocrine Abstracts, 81 [24th European Congress of Endocrinology 2022 21–24 May 2022, Milan, Italy], 107–107 (abstract no. RC12.7). [DOI:10.1530/endoabs.81.rc12.7] [PDF]
Rodari, G., Federici, S., Todisco, T., Ubertini, G., Cattoni, A., Pagano, M., Giacchetti, F., Profka, E., Citterio, V., Messetti, D., Collini, V., Soranna, D., Carbone, E., Arosio, M., Mantovani, G., Persani, L., Cappa, M., Bonomi, M., & Giavoli, C. (2023). Towards an individualized management of pubertal induction in girls with hypogonadism: insight into the best replacement outcomes from a large multicentre registry. European Journal of Endocrinology, 188(6), 467–476. [DOI:10.1093/ejendo/lvad056]
Rohn, R. D. (1989). Nipple (papilla) development in girls: III. Journal of Adolescent Health Care, 10(1), 39–40. [DOI:10.1016/0197-0070(89)90045-4]
Rosenfield, R. L. (2013). Adolescent Anovulation: Maturational Mechanisms and Implications. The Journal of Clinical Endocrinology & Metabolism, 98(9), 3572–3583. [DOI:10.1210/jc.2013-1770]
Rosenfield, R. L., Cooke, D. W., & Radovick, S. (2021). Puberty in the Female and Its Disorders. In Sperling, M. A., Majzoub, J. A., Menon, R. K., & Stratakis, C. A. (Eds.). Sperling Pediatric Endocrinology, 5th Edition (pp. 528–626). Philadelphia: Elsevier. [DOI:10.1016/B978-0-323-62520-3.00016-6]
Rothman, M. S., & Iwamoto, S. J. (2022). Feminizing Gender-Affirming Hormone Therapy: Special Considerations for Older Adults. In Davis, T. F. (Ed.). A Case-Based Guide to Clinical Endocrinology (pp. 513–523). Cham: Springer. [DOI:10.1007/978-3-030-84367-0_58]
Ruan, W., Monaco, M. E., & Kleinberg, D. L. (2005). Progesterone Stimulates Mammary Gland Ductal Morphogenesis by Synergizing with and Enhancing Insulin-Like Growth Factor-I Action. Endocrinology, 146(3), 1170–1178. [DOI:10.1210/en.2004-1360]
Rutgers, J. L., & Scully, R. E. (1991). The Androgen Insensitivity Syndrome (Testicular Feminization). International Journal of Gynecological Pathology, 10(2), 126–144. [DOI:10.1097/00004347-199104000-00002]
Ryan, R. F., & Pernoll, M. L. (1985). Virginal Hypertrophy. Plastic and Reconstructive Surgery, 75(5), 737–742. [DOI:10.1097/00006534-198505000-00024]
Saito, R., Yamamoto, Y., Goto, M., Araki, S., Kubo, K., Kawagoe, R., Kawada, Y., Kusuhara, K., Igarashi, M., & Fukami, M. (2014). Tamoxifen Treatment for Pubertal Gynecomastia in Two Siblings with Partial Androgen Insensitivity Syndrome. Hormone Research in Paediatrics, 81(3), 211–216. [DOI:10.1159/000356923]
Sandhu, R., Chollet-Hinton, L., Kirk, E. L., Midkiff, B., & Troester, M. A. (2016). Digital histologic analysis reveals morphometric patterns of age-related involution in breast epithelium and stroma. Human Pathology, 48, 60–68. [DOI:10.1016/j.humpath.2015.09.031]
Santen, R. J., Karaguzel, G., Livaoglu, M., Yue, W., Cline, J. M., Ratan, A., & Sasano, H. (2024). Role of ERα and aromatase in juvenile gigantomastia. The Journal of Clinical Endocrinology and Metabolism, online ahead of print. [DOI:10.1210/clinem/dgae019]
Sanuki, J., Fukuma, E., & Uchida, Y. (2008). Morphologic Study of Nipple-Areola Complex in 600 Breasts. Aesthetic Plastic Surgery, 33(3), 295–297. [DOI:10.1007/s00266-008-9194-y]
Satoh, K., Hovey, R. C., Malewski, T., Warri, A., Goldhar, A. S., Ginsburg, E., Saito, K., Lydon, J. P., & Vonderhaar, B. K. (2007). Progesterone enhances branching morphogenesis in the mouse mammary gland by increased expression of Msx2. Oncogene, 26(54), 7526–7534. [DOI:10.1038/sj.onc.1210555]
Schauffler, G. C. (1942). Disorders During Adolescence—The Onset of Menstruation. In Schauffler, G. C. Pediatric Gynecology; with Sections on Urology and Proctology, 1st Edition (pp. 169–211). Chicago: Year Book. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL]
Schindler, A. E. (2010). Dydrogesterone and other progestins in benign breast disease: an overview. Archives of Gynecology and Obstetrics, 283(2), 369–371. [DOI:10.1007/s00404-010-1456-7]
Schlatterer, K., Yassouridis, A., Werder, K. V., Poland, D., Kemper, J., & Stalla, G. K. (1998). Archives of Sexual Behavior, 27(5), 475–492. [DOI:10.1023/a:1018704630036]
Scott, P. P., Greene, R., Lane-Roberts, C. S., & Swain, V. (1950). The Function of the Breast. In Saner, F. D. (Ed.). The Breast: Structure, Function, Disease (pp. 53–86). Baltimore: Williams and Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL] [PDF]
Seal, L. (2017). Adult Endocrinology. In Richards, C., Bouman, W. P., & Barker, M.-J. (Eds.). Genderqueer and Non-Binary Genders (Critical and Applied Approaches in Sexuality, Gender and Identity) (pp. 183–223). London: Palgrave Macmillan UK. [DOI:10.1057/978-1-137-51053-2_10]
Seal, L. J., Franklin, S., Richards, C., Shishkareva, A., Sinclaire, C., & Barrett, J. (2012). Predictive Markers for Mammoplasty and a Comparison of Side Effect Profiles in Transwomen Taking Various Hormonal Regimens. The Journal of Clinical Endocrinology & Metabolism, 97(12), 4422–4428. [DOI:10.1210/jc.2012-2030]
Seibert, B., & Günzel, P. (1994). Animal toxicity studies performed for risk assessment of the once-a-month injectable contraceptive Mesigyna®. Contraception, 49(4), 303–333. [DOI:10.1016/0010-7824(94)90030-2]
Shapiro, L. R. (1982). Disorders of Female Sex Differentiation. In Blaustein, A. (Ed.). Pathology of the Female Genital Tract, 2nd Edition (pp. 479–510). New York: Springer New York. [DOI:10.1007/978-1-4757-1767-9_20]
Shearman, R. P. (1972). Ovarian Function and its Control. In Shearman, R. P. (Ed.). Human Reproductive Physiology, 1st Edition (pp. 45–90). Oxford: Blackwell Scientific Publications. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Shearman, R. P. (1972). Primary Ammenorhoea. In Dewhurst, C. J. (Ed.). Integrated Obstetrics and Gynaecology for Postgraduates, 1st Edition (pp. 55–62). Oxford: Blackwell Scientific Publications. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Shi, H. Y., Lydon, J. P., & Zhang, M. (2004). Hormonal Defect in Maspin Heterozygous Mice Reveals a Role of Progesterone in Pubertal Ductal Development. Molecular Endocrinology, 18(9), 2196–2207. [DOI:10.1210/me.2004-0052]
Shuttleworth, F. K. (1938). The Adolescent Period: A Graphic and Pictorial Atlas (Monographs of the Society for Research in Child Development, Volume 3, Number 3 / Serial No. 16). Washington, D. C.: Society for Research in Child Development. [Google Scholar] [Google Books] [DOI:10.2307/1165482]
Simmer, H. H., Pion, R. J., & Dignam, W. J. (1965). Testicular Feminization: Endocrine Function of Feminizing Testes, Comparison with Normal Testes. Springfield, Illinois: Thomas. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Simpson, J. L., & Rebar, R. W. (1990). Normal and Abnormal Sexual Differentiation and Development. In Becker, K. L., Bilezikian, J. P., Bremner, W. J., Hung, W., Kahn, C. R., Loriaux, D. L., Rebar, R. W., Robertson, G. L., & Wartofsky, L. (Eds.). Principles and Practice of Endocrinology and Metabolism, 1st Edition (pp. 710–739). Philadelphia: Lippincott. [Google Scholar] [Google Books—3rd/2001 Edition] [OpenLibrary] [WorldCat] [Archive.org]
Sindi, R., Sá Dos Reis, C., Bennett, C., Stevenson, G., & Sun, Z. (2019). Quantitative Measurements of Breast Density Using Magnetic Resonance Imaging: A Systematic Review and Meta-Analysis. Journal of Clinical Medicine, 8(5), 745. [DOI:10.3390/jcm8050745]
Sitruk-Ware, R., Basdevant, A., de Lignières, B., & Mauvais-Jarvis, P. (1984). Percutaneous oestrogen therapy. In van Herendael, H., van Herendael, B., Riphagen, F. E., Goessens, L., & van der Plas, H. (Eds.). The Climacteric: An Update: Proceedings of the fourth Jan Palfijn Symposium, European Conference on the Menopause, held in Antwerp, Belgium, on September 1–2, 1983, under the auspices of ‘De Vereniging voor Nederlandstalige gynecologen van België’ and ‘The International Menopause Society’ (pp. 127–139). Lancaster: MTP Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [DOI:10.1007/978-94-009-5608-7_14]
Sizonenko, P. C. (1978). Endocrinology in Preadolescents and Adolescents. American Journal of Diseases of Children, 132(7), 704–712. [DOI:10.1001/archpedi.1978.02120320064015]
Škarda, J., Fremrová, V., & Bezecný, I. (1989). Progesterone alone is responsible for stimulation of the growth of ducts and of mammary alveolar structures in mice. Endocrinologia Experimentalis, 23(1), 17–28. [Google Scholar] [PubMed] [PDF]
Sobrinho, L. G., Kase, N., & Grunt, J. A. (1971). Spontaneous pubertal breast growth in a castrated patient with the syndrome of testicular feminization. The Yale Journal of Biology and Medicine, 44(2), 225–9. [Google Scholar] [PubMed] [PubMed Central] [PDF]
Sosa, M., Jódar, E., Arbelo, E., Domínguez, C., Saavedra, P., Torres, A., Salido, E., de Tejada, M. J., & Hernández, D. (2003). Bone Mass, Bone Turnover, Vitamin D, and Estrogen Receptor Gene Polymorphisms in Male to Female Transsexuals. Journal of Clinical Densitometry, 6(3), 297–304. [DOI:10.1385/jcd:6:3:297]
Sosa, M., Jódar, E., Arbelo, E., Domı́nguez, C., Saavedra, P., Torres, A., Salido, E., Limiñana, J., Gómez de Tejada, M. J., & Hernández, D. (2004). Serum lipids and estrogen receptor gene polymorphisms in male-to-female transsexuals: effects of estrogen treatment. European Journal of Internal Medicine, 15(4), 231–237. [DOI:10.1016/j.ejim.2004.04.009]
Soyal, S., Ismail, P. M., Li, J., Mulac-Jericevic, B., Conneely, O. M., & Lydon, J. P. (2002). Progesterone receptors - animal models and cell signaling in breast cancer: Progesterone’s role in mammary gland development and tumorigenesis as disclosed by experimental mouse genetics. Breast Cancer Research, 4(5), 191. [DOI:10.1186/bcr451]
Sperling, R. L., & Gold, J. J. (1973). Use of an anti-estrogen after a reduction mammaplasty to prevent recurrence of virginal hypertrophy of breasts. Plastic and Reconstructive Surgery, 52(4), 439–442. [DOI:10.1097/00006534-197352040-00030]
Spitz, I. M., Shoupe, D., Sitruk-Ware, R., & Mishell, D. R. (1989). Response to the antiprogestagen RU 486 (mifepristone) during early pregnancy and the menstrual cycle in women. Journal of Reproduction and Fertility Supplement, 37, 253–260. [Google Scholar] [PubMed]
Spitz, I., Croxatto, H., Lahteenmaki, P., Heikinheimo, O., & Bardin, C. (1994). Effect of mifepristone on inhibition of ovulation and induction of luteolysis. Human Reproduction, 9(Suppl 1), 69–76. [DOI:10.1093/humrep/9.suppl_1.69]
Spitz, I. M. (2010). Mifepristone: where do we come from and where are we going? Contraception, 82(5), 442–452. [DOI:10.1016/j.contraception.2009.12.012]
Sridhar, G. R., & Sinha, M. J. (1995). Macromastia in adolescent girls. Indian Pediatrics, 32(4), 496–499. [Google Scholar] [PubMed] [PDF]
Sun, B. Z., Kangarloo, T., Adams, J. M., Sluss, P. M., Welt, C. K., Chandler, D. W., Zava, D. T., McGrath, J. A., Umbach, D. M., Hall, J. E., & Shaw, N. D. (2018). Healthy Post-Menarchal Adolescent Girls Demonstrate Multi-Level Reproductive Axis Immaturity. The Journal of Clinical Endocrinology & Metabolism, 104(2), 613–623. [DOI:10.1210/jc.2018-00595]
Sun, S. X., Bostanci, Z., Kass, R. B., Mancino, A. T., Rosenbloom, A. L., Klimberg, V. S., & Bland, K. I. (2018). Breast Physiology: Normal and Abnormal Development and Function. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 37–56.e6). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00003-9]
Swelstad, M. R., Swelstad, B. B., Rao, V. K., & Gutowski, K. A. (2006). Management of Gestational Gigantomastia. Plastic and Reconstructive Surgery, 118(4), 840–848. [DOI:10.1097/01.prs.0000232364.40958.47]
Tack, L. J., Heyse, R., Craen, M., Dhondt, K., Bossche, H. V., Laridaen, J., & Cools, M. (2017). Consecutive Cyproterone Acetate and Estradiol Treatment in Late-Pubertal Transgender Female Adolescents. The Journal of Sexual Medicine, 14(5), 747–757. [DOI:10.1016/j.jsxm.2017.03.251]
Talbert, L. M., Hammond, M. G., Groff, T., & Udry, J. R. (1985). Relationship of age and pubertal development to ovulation in adolescent girls. Obstetrics & Gynecology, 66(4), 542–544. [Google Scholar] [PubMed] [URL]
Tanos, T., & Brisken, C. (2008). What Signals Operate in the Mammary Niche? Breast Disease, 29(1), 69–82. [DOI:10.3233/bd-2008-29108]
Thanaboonyawat, I., Chanprapaph, P., Lattalapkul, J., & Rongluen, S. (2013). Pilot Study of Normal Development of Nipples during Pregnancy. Journal of Human Lactation, 29(4), 480–483. [DOI:10.1177/0890334413493350]
Thody, A. J., & Smith, A. G. (1977). Hormones and Skin Pigmentation in the Mammal. International Journal of Dermatology, 16(8), 657–664. [DOI:10.1111/j.1365-4362.1977.tb01876.x]
Thoresen, M., & Wesche, J. (1988). Doppler measurements of changes in human mammary and uterine blood flow during pregnancy and lactation. Acta Obstetricia et Gynecologica Scandinavica, 67(8), 741–745. [DOI:10.3109/00016349809004301]
Tiefenbacher, K., & Daxenbichler, G. (2008). The Role of Androgens in Normal and Malignant Breast Tissue. Breast Care, 3(5), 325–331. [DOI:10.1159/000158055]
Tim, F., Jeroen, V., & T’Sjoen, G. (2023). Dose Reduction of Cyproterone Acetate in Trans Women and the Effect on Patient-reported Outcomes: Results from the ENIGI Study. Endocrine Abstracts, 97 [Belgian Endocrine Society 2023], 5–5 (abstract no. 007). [URL] [PDF]
Trabert, B., Sherman, M. E., Kannan, N., & Stanczyk, F. Z. (2019). Progesterone and Breast Cancer. Endocrine Reviews, 41(2), 320–344. [DOI:10.1210/endrev/bnz001]
Tucker, H. (2000). Hormones, Mammary Growth, and Lactation: a 41-Year Perspective. Journal of Dairy Science, 83(4), 874–884. [DOI:10.3168/jds.s0022-0302(00)74951-4]
Turan, S., Bereket, A., Guran, T., Akcay, T., Papari-Zareei, M., & Auchus, R. J. (2009). Puberty in a case with novel 17-hydroxylase mutation and the putative role of estrogen in development of pubic hair. European Journal of Endocrinology, 160(2), 325–330. [DOI:10.1530/eje-08-0632]
Turner, C. W. (1939). The Mammary Glands. In Allen, E., Danforth, C. H., & Doisy, E. A. (Eds.). Sex and Internal Secretions: A Survey of Recent Research, 2nd Edition (pp. 740–803). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Ulrich, U., Pfeifer, T., & Lauritzen, C. (1994). Rapid Increase in Lumbar Spine Bone Density in Osteopenic Women by High-Dose Intramuscular Estrogen-Progestogen Injections. Hormone and Metabolic Research, 26(9), 428–431. [DOI:10.1055/s-2007-1001723]
Ulrich, U., Pfeifer, T., Buck, G., Keckstein, J., & Lauritzen, C. (1995). High-dose estrogen-progestogen injections in gonadal dysgenesis, ovarian hypoplasia, and androgen insensitivity syndrome: Impact on bone density. Adolescent and Pediatric Gynecology, 8(1), 20–23. [DOI:10.1016/s0932-8610(12)80156-3]
Valentine, G. H. (1969). Chromosome Anomalies in the Female. In Valentine, G. H. The Chromosome Disorders: An Introduction for Clinicians, 2nd Edition (pp. 126–143). London: W. Heinemann Medical Books. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
van der Meulen, A. J. (1974). An unusual case of massive hypertrophy of the breasts. South African Medical Journal=Suid-Afrikaanse Tydskrif vir Geneeskunde, 48(34), 1465–1466. [Google Scholar] [PubMed] [URL 1] [URL 2]
Vandeweyer, E., & Hertens, D. (2002). Quantification of glands and fat in breast tissue: An experimental determination. Annals of Anatomy - Anatomischer Anzeiger, 184(2), 181–184. [DOI:10.1016/s0940-9602(02)80016-4]
Venturoli, S., Fabbri, R., Porcu, E., Paradisi, R., Orsini, L. F., Brondelli, L., Ruggeri, S., & Flamigni, C. (1989). Endocrine and ovarian parameters at various frequencies of ovulation in adolescents. Archives of Gynecology and Obstetrics, 246(2), 107–114. [DOI:10.1007/bf00934127]
Venturoli, S., Porcu, E., Fabbri, R., Magrini, O., Paradisi, R., Pallotti, G., Gammi, L., & Flamigni, C. (1987). Postmenarchal evolution of endocrine pattern and ovarian aspects in adolescents with menstrual irregularities. Fertility and Sterility, 48(1), 78–85. [DOI:10.1016/s0015-0282(16)59294-2]
Vorherr, H. (1974). Development of the Female Breast. In Vorherr, H. The Breast: Morphology, Physiology, and Lactation (pp. 1–19). New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Vorherr, H. (1974). Morphology of the Mature Female Breast. In Vorherr, H. The Breast: Morphology, Physiology, and Lactation (pp. 20–70). New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Wade, T. R., Wade, S. L., & Jones, H. E. (1978). Skin changes and diseases associated with pregnancy. Obstetrics and Gynecology, 52(2), 233–242. [Google Scholar] [PubMed] [URL]
Walker, M., Baker, G., & Lamb, M. (2013). Physiology of the Breast During Pregnancy and Lactation. In Mannel, R., Martens, P. J., & Walker, M. (Eds.). Core Curriculum for Lactation Consultant Practice, 3rd Edition (pp. 287–300). Burlington: Jones & Bartlett Learning. [Google Scholar] [Google Books—2013/3rd Edition] [Google Books—2008/2nd Edition] [OpenLibrary] [WorldCat]
Wang, C., Luan, J., Cheng, H., Chen, L., Li, Z., Panayi, A. C., & Liu, C. (2018). Menstrual Cycle-Related Fluctuations in Breast Volume Measured Using Three-Dimensional Imaging: Implications for Volumetric Evaluation in Breast Augmentation. Aesthetic Plastic Surgery, 43(1), 1–6. [DOI:10.1007/s00266-018-1243-6]
Wang, Q. A., & Scherer, P. E. (2019). Remodeling of Murine Mammary Adipose Tissue during Pregnancy, Lactation, and Involution. Journal of Mammary Gland Biology and Neoplasia, 24(3), 207–212. [DOI:10.1007/s10911-019-09434-2]
Weisberg, M. G., Malkasian, G. D., & Pratt, J. H. (1970). Testicular feminization syndrome. American Journal of Obstetrics and Gynecology, 107(8), 1181–1187. [DOI:10.1016/s0002-9378(15)30367-7]
Wellons, M. F., & Rebar, R. W. (2013). Amenorrhea. In Falcone, T., & Hurd, W. W. (Eds.). Clinical Reproductive Medicine and Surgery: A Practical Guide, 2nd Edition (pp. 105–112). New York: Springer New York. [DOI:10.1007/978-1-4614-6837-0_7]
Wellons, M. F., Weeber, K. M., & Rebar, R. W. (2017). Amenorrhea. In Falcone, T., & Hurd, W. W. (Eds.). Clinical Reproductive Medicine and Surgery, 3rd Edition (pp. 109–122). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-52210-4_6]
Werner, A. A. (1935). Experiment to produce lactation in castrate women. Endocrinology, 19(2), 144–150. [DOI:10.1210/endo-19-2-144]
Whiteley, S. (1994). Predictors of Milk Production in Lactating Women. (Master’s thesis, The Open University.) [Google Scholar] [DOI:10.21954/ou.ro.0000fe02] [URL]
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical Review: Breast Development in Trans Women Receiving Cross-Sex Hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]
Wierckx, K., Van Caenegem, E., Schreiner, T., Haraldsen, I., Fisher, A., Toye, K., Kaufman, J. M., & T’Sjoen, G. (2014). Cross‐Sex Hormone Therapy in Trans Persons Is Safe and Effective at Short‐Time Follow‐Up: Results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 11(8), 1999–2011. [DOI:10.1111/jsm.12571]
Wilson, J. D. (1968 March 28). Medical Grand Rounds: Testicular Feminization. Parkland Memorial Hospital, UT Southwestern Medical Center. [Google Scholar] [URL] [PDF]
Wilson, C. L., Sims, A. H., Howell, A., Miller, C. J., & Clarke, R. B. (2006). Effects of oestrogen on gene expression in epithelium and stroma of normal human breast tissue. Endocrine-Related Cancer, 13(2), 617–628. [DOI:10.1677/erc.1.01165]
Winkler, U. H., Schindler, A. E., Brinkmann, U. S., Ebert, C., & Oberhoff, C. (2001). Cyclic progestin therapy for the management of mastopathy and mastodynia. Gynecological Endocrinology, 15(Suppl 6), 37–43. [DOI:10.1080/gye.15.s6.37.43]
Wisniewski, A. B., Migeon, C. J., Meyer-Bahlburg, H. F., Gearhart, J. P., Berkovitz, G. D., Brown, T. R., & Money, J. (2000). Complete Androgen Insensitivity Syndrome: Long-Term Medical, Surgical, and Psychosexual Outcome. The Journal of Clinical Endocrinology & Metabolism, 85(8), 2664–2669. [DOI:10.1210/jcem.85.8.6742]
Wong, R. C., & Ellis, C. N. (1984). Physiologic skin changes in pregnancy. Journal of the American Academy of Dermatology, 10(6), 929–940. [DOI:10.1016/s0190-9622(84)80305-9]
Wren, B. G., & Eden, J. A. (1996). Do Progestogens Reduce The Risk of Breast Cancer? A Review of the Evidence. Menopause, 3(1), 4–12. [DOI:10.1097/00042192-199603010-00003]
Wright, E. M. (2015). Breastfeeding and the Mother–Newborn Dyad (pp. 1157–1182). In King, T. L., Brucker, M. C., Kriebs, J. M., Fahey, J. O., Gegor, C. L., & Varney, H. (Eds.). Varney’s Midwifery, 5th Edition. Burlington: Jones & Bartlett Learning. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Xu, P., Ye, W., Zhong, S., Li, H., Feng, E., Lin, S. H., Kuo, C. T., Liu, J. Y., & Lin, Y. C. (2010). Leptin and zeranol up-regulate cyclin D1 expression in primary cultured normal human breast pre-adipocytes. Molecular Medicine Reports, 3(6), 983–990. [DOI:10.3892/mmr.2010.370]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Zacharin, M. (2000). Use of androgens and oestrogens in adolescents - A review of hormone replacement treatment. Journal of Pediatric Endocrinology and Metabolism, 13(1), 3–12. [DOI:10.1515/JPEM.2000.13.1.3]
Zachmann, M., Prader, A., Sobel, E. H., Crigler, J. F., Ritzén, E. M., Atarés, M., & Ferrandez, A. (1986). Pubertal growth in patients with androgen insensitivity: Indirect evidence for the importance of estrogens in pubertal growth of girls. The Journal of Pediatrics, 108(5), 694–697. [DOI:10.1016/s0022-3476(86)81043-5]
Żelaźniewicz, A., & Pawłowski, B. (2015). Breast size and asymmetry during pregnancy in dependence of a fetus’s sex. American Journal of Human Biology, 27(5), 690–696. [DOI:10.1002/ajhb.22716]
Żelaźniewicz, A., & Pawłowski, B. (2018). Maternal breast volume in pregnancy and lactation capacity. American Journal of Physical Anthropology, 168(1), 180–189. [DOI:10.1002/ajpa.23734]
Zhang, D., Yao, F., Tian, T., Deng, S., Luo, M., & Tian, Q. (2021). Clinical characteristics and molecular genetics of complete androgen insensitivity syndrome patients: a series study of 30 cases from a Chinese tertiary medical center. Fertility and Sterility, 115(5), 1270–1279. [DOI:10.1016/j.fertnstert.2020.12.008]
\ No newline at end of file
+A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People - Transfeminine Science
A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People
By Aly | First published February 14, 2020 | Last modified August 23, 2025
Abstract / TL;DR
The major female sex hormones are estrogen and progesterone. Both of these hormones are known to be importantly involved in the development of the breasts at different stages of life. Speculation, use, and anecdotes of progestogens for enhancing breast development in transfeminine people date back to at least the 1960s. A limited number of clinical studies have assessed breast development with progestogens in transfeminine people, but current evidence on progestogens for improving breast development is of very low quality and is inconclusive. Studies of progestogens and breast development in cisgender girls and women are similarly limited. In any case, more studies evaluating progestogens and breast development are currently underway. The possible role of progestogens in enhancing breast development can also be informed by indirect and circumstantial evidence, including notably findings on progesterone and breast changes during normal puberty, the menstrual cycle, and pregnancy in humans and animals. Available evidence overall is not suggestive of an essential role for progesterone in breast growth during puberty, but progesterone does have a clear and key role in lobuloalveolar development of the breasts during pregnancy. However, breast changes in pregnancy revert following cessation of lactation and breastfeeding. Progesterone may additionally contribute to reversible breast enlargement during the luteal phase of the menstrual cycle. There are some findings to suggest that progestogens may have antiestrogenic effects in the breasts and may have a stunting influence on breast development if introduced too early following initiation of hormone therapy. However, more research is needed to assess this possibility. In any case, if progestogens are used, it may be advisable to delay their introduction until most or all estrogen-mediated breast development is complete. Options for progestogen therapy in transfeminine people include bioidentical progesterone and progestins. However, oral progesterone has major bioavailability problems and does not achieve satisfactory progesterone levels. Progestogens, including progesterone, have been variously linked to significant health risks, which is an important consideration in terms of their use in transfeminine people. Overall, based on current knowledge, progestogens do not seem to be promising for lastingly improving breast development in transfeminine people, but more research and data are still needed for clear conclusions.
Introduction
Breast development in terms of size and shape is often less than desired in transfeminine people, and there is a need for therapeutic approaches that improve breast growth in this population. There are two major types of female hormones, estrogens and progestogens. Estrogens are almost universally employed in transfeminine hormone therapy, while progestogens are used in a subset of transfeminine people. Progestogens that have been commonly employed in transfeminine people include bioidenticalprogesterone, the progestin (synthetic progestogen) medroxyprogesterone acetate (MPA), and the strongly progestogenic antiandrogen cyproterone acetate (CPA). Estrogens are the major mediators of feminization and breast development in females. However, progestogens also have physiological effects on the breasts, and in relation to this, may or may not provide benefits to breast development as well.
The topic of progestogens and breast development has been discussed for many years in the transgender community and is a controversial subject (Coleman et al., 2012). Use of progestogens to improve breast development in transfeminine people goes back at least as far as Harry Benjamin and Christian Hamburger in the 1960s (Benjamin, 1966; Benjamin, 1967; Hamburger & Benjamin, 1969; Wiki). Arguments have been made both for (e.g., Bevan, 2012; Bellwether, 2019; Bevan, 2019) and against (e.g., Curtis, 2009) a possible role of progestogens in terms of breast development. It is often claimed that progestogens can enhance breast development or are even required for full breast development in cisgender females and transfeminine people. With respect to the latter, it is sometimes said that progestogens are necessary for people to move from Tanner stage 4 to Tanner stage 5 pubertal breast development and that progestogens help to fill and round out the breasts (e.g., Vorherr, 1974a; Basson & Prior, 1998; Kaiser & Ho, 2015; Prior, 2011; Prior, 2019a; Prior, 2020). It has even been claimed by some that without progestogens, the breasts will remain conical and “pointy” like they are in the earlier Tanner stages. On the other extreme, certain critics have claimed that there are “no biologically significant progesterone receptor sites for biological males” and that progesterone is not produced during normal female puberty until after breast development has been fully completed (Barrett, 2009; Seal, 2017; Coxon & Seal, 2018; Price, McManus, & Barrett, 2019; Richards & Barrett, 2020). In turn, these particular authors have argued against the use of progestogens in transfeminine people in various of their publications (Google Scholar). In general, the use of progestogens in transfeminine people has longstandingly been controversial, with positions both for and against (Sam, 2020).
The purpose of this article is to review the available direct and circumstantial evidence on the topic of progestogens and breast development in order to help inform whether progestogen therapy may be able to enhance breast development in transfeminine people. Aside from an involvement in breast development, progestogens are not otherwise currently thought to be or known to be involved in physical feminization (e.g., Coleman et al., 2012; Gooren, 2016). In relation to this, the present article will limit its discussion to breast development with progestogens, and will not explore feminization in general.
Progestogen Therapy and Breast Development in Humans
Progestogens and Breast Development in Transfeminine People
Orentreich & Durr (1974) was one of the earliest studies on breast development in transfeminine people. They employed combinations of estrogens and progestogens as well as gonadectomy to produce feminization and breast development in a case series of 5 transfeminine people. The employed estrogens were estradiol valerate 30 mg/2 weeks by intramuscular injection and oral conjugated estrogens 1.25–5.0 mg/day and the used progestogens were “60 mg medroxyprogesterone caproate” every 2 weeks by intramuscular injection and oral medroxyprogesterone acetate 0–10 mg/day. Medroxyprogesterone caproate (MPC) has never been used pharmaceutically, so this was likely a typo and the actual progestogen employed was likely either MPA or hydroxyprogesterone caproate (OHPC). The authors reported that estrogen and progestogen therapy produced modest to significant breast development in the transfeminine people that was not strictly dose-related and included clinical photographs of the breasts. They concluded that the breast development was comparable to that of adult cisgender women. Orentreich and colleagues also discussed the topic of lobuloalveolar maturation of the breasts, which was known to be progestogen-dependent, but noted that they had not done histological assessment and that the degree of lobuloalveolar development of the breasts does not necessarily correlate with clinical breast size per findings in cisgender women. The findings of Orentreich and colleagues are limited by methodological problems like lack of objective measurements, lack of estrogen-only controls, and the small sample size of only 5 transfeminine people, and hence the study is of limited value in terms of assessing the involvement of progestogens in breast development.
Meyer et al. (1986) assessed the effects of progestogens added to estrogen therapy on breast development and other clinical parameters in transfeminine people. Of the 60 transfeminine people in the study, 15 (25%) received an oral progestogen, usually MPA at a dosage of 10 mg/day, for “at least for a short time”, and with only 8 (13.3%) receiving progestogen therapy for the full treatment period. In an earlier report of the study, it was noted that in 90% of observation periods the dose was 10 mg/day and for the remainder it was 20 mg/day (Meyer et al., 1981). A dosage of 10 mg/day MPA is roughly comparable to luteal-phase progesterone exposure in terms of progestogenic potency (Wiki). Breast development was measured in the study via breast hemicircumference (Diagram). Progestogen therapy was reported to not modify estrogen-induced changes, including laboratory measurements, hormone levels, and physical parameters like weight and breast growth. The lack of apparent changes in hormone levels is unexpected, as MPA in higher-quality studies has shown clear testosterone suppression (e.g., Jain, Kwan, & Forcier, 2019; Wiki). Meyer and colleagues concluded that adding progestogens to estrogen does not seem to enhance breast development in transfeminine people. However, they noted that the number of individuals who received progestogens was small and further studies were needed.
Prior et al. (1986) and Prior, Vigna, & Watson (1989) studied estrogen, high-dose spironolactone (100–600 mg/day), and MPA (10–20 mg/day cylically or continuously) in transfeminine people who were either pre-hormone therapy or had previously been on higher doses of estrogens (and/or progestogens) without spironolactone prior to the study. The researchers reported that following 12 months of treatment with the study’s hormone therapy regimen, there was increased breast size and increased nipple development. Most individuals reached an A cup size, or approximately 8 to 14 cm in diameter of breast tissue, by the end of the study. Breast development was measured in part with photographic documentation. Although breast development reportedly improved, the researchers themselves noted that it was difficult to determine whether the enhanced breast development could be attributed to spironolactone or to MPA. Moreover, testosterone suppression was inadequate before the study and improved with the study’s hormone therapy regimen, which may have helped to improve breast development regardless of any potential direct progestogenic action of MPA on the breasts. Finally, it is possible that breast development with estrogen may not yet have been complete, and that the improved breast development may have simply been continued progression due to estrogen alone. In other publications, Jerilynn Prior, the lead study author, has claimed that progesterone enhances breast development, and has cited the preceding studies by her in support of this claim (Prior, 2011; Prior, 2019a; Prior, 2019b; Prior, 2020). However, her claim is not well-supported due to the study limitations discussed.
Dittrich et al. (2005) reported that the combination of oral estradiol valerate and a gonadotropin-releasing hormone (GnRH) agonist for 2 years in transfeminine people resulted in self-reported breast cup sizes of C cup or greater in 5%, B cup in 30%, A cup in 35%, and less than A cup in 30%. They noted however that 70% of the individuals were unsatisfied with their breast development and wished to undergo breast augmentation surgery. The researchers claimed that the regimen had similar effectiveness in terms of feminization, including increases in breast size, compared to prior reported treatment regimens of ethinylestradiol and CPA. No other details or specifics were given. The claim about similar breast development to regimens containing CPA is relevant as CPA is a very strong progestogen at the doses used historically in transfeminine people (Aly, 2019). It should be cautioned however that this study did not actually employ or study progestogen therapy itself. In addition, self-reported breast cup size is a subjective and low-quality means of measuring breast development and size. As such, the findings of this study are of questionable value in terms of understanding progestogens and breast development.
Estrogen is primarily involved in ductal development of the breasts, whereas progesterone is mainly involved in lobuloalveolar development. Kanhai et al. (2000) compared internal histological breast tissue changes with estrogen and CPA 100 mg/day (i.e. very-high-dose progestogen) therapy in 14 transfeminine people versus nonsteroidal antiandrogen monotherapy with flutamide or bicalutamide in 2 cisgender men with prostate cancer. Both types of treatments block androgens, increase estrogen levels, and are known to induce breast development or gynecomastia at similarly high rates. However, nonsteroidal antiandrogen monotherapy differs from combined estrogen and progestogen therapy in that it lacks any progestogenic effects. In the transfeminine people, full lobuloalveolar formation was apparent in the biopsied breast tissue, whereas in the men with prostate cancer, only “moderate” and incomplete lobuloalveolar maturation was found. It was also noted that lobuloalveolar formation tended to regress upon discontinuation of CPA following gonadectomy in transfeminine people. The researchers concluded that progestogenic exposure is needed for the breasts to fully develop on a histological level and for the breast tissue of transfeminine people to completely mimic the histology of the mature female breast. In accordance with these findings, estrogen plus high doses of CPA, as well as certain other regimens, have been associated with galactorrhea (lactation) as a side effect in transfeminine people (Dewhurst & Underhill, 1979; Futterweit, 1980; Gooren, Harmsen-Louman, & van Kessel, 1985; Schlatterer et al., 1998; Levy, Crown, & Reid, 2003; Bazarra-Castro, 2009). While the findings of Kanhai and colleagues’ study are interesting, they only concern tissue characteristics and do not actually provide any information about breast development in terms of physical form or appearance. With regard to this, tissue-level differences may or may not translate to relevant differences in for instance breast size or shape. As such, the study is of limited value in understanding whether progestogens improve breast development in transfeminine people in the ways that are actually valued.
Seal and colleagues conducted a retrospective chart review assessing clinical predictors for surgical breast augmentation in transfeminine people (Seal et al., 2012). In the transfeminine people who underwent breast augmentation, significantly more of them were taking spironolactone than were those who did not undergo breast augmentation. Conversely, the differential rates of use of specific antiandrogens were not significantly discordant between those who did and did not undergo breast augmentation in the case of the other prescribed antiandrogens, including CPA, the 5α-reductase inhibitors, and GnRH analogues. However, this study had many methodological limitations, including the use of almost three dozen t-tests with no adjustment for multiple comparisons (and hence risk of false positives and concerns about p-hacking), small sample sizes for individual antiandrogens, use of undergoing breast augmentation as a surrogate for breast development with no actual physical measurement of the breasts or breast sizes, and a correlational design with lack of control for potential confounding variables. As such, the study does not show that different antiandrogens result in differences in breast development, and its findings must be considered with due caution.
Jain, Kwan, & Forcier (2019) studied sublingual estradiol and spironolactone with and without MPA in 92 transfeminine people. MPA was given at a dose of 5 to 10 mg/day sublingually or at a dose of 150 mg once every 3 months by intramuscular injection. Of 39 transfeminine people who received MPA, 26 (67%) self-reported improved breast development. No further details were provided, but measurement of breast development was presumably subjective and anecdotal. Igo & Visram (2021) studied addition of progesterone to hormone therapy in transfeminine people. Progesterone was provided as 100 mg micronized progesterone (probably oral) and was prescribed when progesterone was specifically requested by the patient or when the patient expressed dissatisfaction with feminization and/or breast development. Of 190 individuals, 51 (26.8%) received progesterone therapy. Treatment with progesterone on average began after 12.7 months of estradiol therapy, and the mean total follow-up time was 14.3 months of hormone therapy. Of those who received progesterone, only 6 (11.8%) reported benefit to breast development. No further details were provided, but as with other studies, breast development was likely quantified anecdotally via self-report. As breast development does not appear to have been objectively measured or compared to a control group in either Jain, Kwan, & Forcier (2019) or Igo & Visram (2021), the findings of these studies are limitedly informative.
Nolan and colleagues assessed the short-term effects of low-dose oral micronized progesterone on breast development in transfeminine people on stable hormone therapy in a prospective controlled study (Nolan et al., 2022a; Nolan et al., 2022b). Progesterone was given at a dose of 100 mg/day for 3 months to 23 transfeminine people and findings were compared to those of a control group of 19 transfeminine people. Breast development was measured using self-reported Tanner stage, with participants provided photographs of different Tanner stages to self-select from. At the end of the 3 months, Tanner stage was not significantly different between groups (mean 3.5, 95% CI 3.2–3.7 for progesterone vs. mean 3.6, 95% CI 3.3–3.9 for controls; p = 0.42). A limitation of this study is that oral progesterone has very low bioavailability and 100 mg/day oral progesterone achieves very low progesterone levels that are well below normal luteal-phase progesterone levels (Aly, 2018a; Wiki). As such, progestogenic exposure in this study, and notably also in Igo & Visram (2021) and other studies, is likely to have been inadequate. Besides the issue of progestogenic strength, the very short duration of the study (3 months) and the reliance on self-reported subjective Tanner stages (as opposed to more objective physical breast measurements) are also major limitations. In any case, this study is of higher quality than previous studies, and is notably likely to continue and report further follow-up at later points in the future.
Bahr et al. (2024) conducted a retrospective chart review at their clinic and compared 29 transfeminine people who had received progestogens versus 59 transfeminine people who had not. The form of progestogen used was oral or rectal progesterone in 93% of cases and MPA by intramuscular injection in the remaining 7% of cases. Of those who took progesterone, 25 (93%) used it orally and 2 (7%) used oral progesterone capsules rectally. Progestogen doses were not reported, except that 100 mg progesterone capsules were employed. Most of those in the progestogen-treated group (59%) had started it 1 to 6 months following initiation of standard hormone therapy. The researchers found that progestogen-treated group had significantly better self-reported breast development satisfaction (rated as satisfied, neutral, or unsatisfied) compared to the group that did not receive progestogens at 6 months (satisfied: 53.8% vs. 19.6%; p = 0.004) and 9 months (satisfied: 71.4% vs. 20.8%; p = 0.003) of hormone therapy. Limitations of this study include the lack of objective measurement of breast development, the restrospective nature of the study, and the lack of randomization of treatment, among others.
Aside from the above studies, a variety of other studies have also reported breast development with estrogen and CPA in transfeminine people. These studies have often employed objective physical measurements of breast development (e.g., breast volume, breast–chest difference, breast cup size, breast hemicircumference). However, they have lacked comparison groups, thereby precluding comparison of progestogenic versus non-progestogenic hormone therapy. In addition, CPA is unusual among progestogens in that it is employed at very high doses in transfeminine people (Aly, 2019), which may result in different and potentially stunted outcomes in terms of breast development than more physiological progestogenic exposure. As such, most studies of breast development with estrogen and CPA in transfeminine people have not been discussed in the present section and are instead discussed elsewhere in this article (see the section below). In any case, to briefly summarize the findings, breast development in transfeminine people with estrogen and CPA has generally been poor in these studies. The outcomes have included incomplete maturation in terms of Tanner staging (stage 2–4), small cup sizes, small breast volumes, and breasts much smaller in size than those in cisgender women.
The findings from the preceding studies in transfeminine people are of very low-quality due to methodological limitations, including lack of control groups, lack of randomization, reliance on poor measures of breast development (e.g., subjective and self-report) rather than objective physical measurements (Wiki), short treatment durations, and small sample sizes, among others. This may explain the conflicting results of the studies. More research is still needed to assess the influence of progestogens on breast development in transfeminine people. There is specifically a need for randomized controlled trials (RCTs) of feminizing hormone therapy with versus without progestogen therapy that employ objective measures of breast development, have adequate sample sizes, and have sufficient follow-up durations. Additional variables like progestogen type, route, dose, and timing of introduction would also be of value to explore. A 2014 review on hormone therapy in transfeminine people summarizes the state of research on progestogens and breast development in transfeminine people, with their conclusions still holding true today (Wierckx, Gooren, & T’Sjoen, 2014):
Our knowledge concerning the natural history and effects of different cross-sex hormone therapies on breast development in trans women is extremely sparse and based on low quality of evidence. Current evidence does not provide evidence that progestogens enhance breast development in trans women. Neither do they prove the absence of such an effect. This prevents us from drawing any firm conclusion at this moment and demonstrates the need for further research to clarify these important clinical questions.
Several studies of progesterone and other progestogens in transfeminine people are currently underway. These studies include (1) an RCT of oral progesterone added to hormone therapy by Sandeep Dhindsa and colleagues in St. Louis, Missouri in the United States (ClinicalTrials.gov; MediFind; ICH GCP); (2) a prospectiveobservational study and/or RCT of addition of oral progesterone to hormone therapy by Ada Cheung and colleagues in Melbourne, Australia (University of Melbourne; University of Melbourne); (3) an RCT of estradiol plus spironolactone versus estradiol plus CPA also by Ada Cheung and colleagues (ANZCTR; WHO ICTRP; Trans Health Research [Flyer] [Poster]; University of Melbourne) (update: see below); and (4) a large RCT of oral progesterone at different doses added to hormone therapy by Martin den Heijer and colleagues at the Vrije Universiteit University Medical Center (VUMC) in Amsterdam, the Netherlands (Dijkman et al., 2023a; General Info/Links; Info Sheet Dutch; Info Sheet English Translated) (update: see below). Unfortunately however, all of the studies using progesterone employ oral progesterone, which has major bioavailability and potency problems (Aly, 2018a; Wiki). In any case, it was said that the VUMC researchers may follow their trial up with studies of other progesterone routes (General Info/Links). The preceding studies may provide more insight on the question of whether progestogen therapy is of therapeutic benefit to breast development in transfeminine people.
Progestogens and Breast Development in Cisgender Females
To date, there appear to be no useful studies on breast development with progesterone or other progestogens in cisgender females. There seem to mostly only be a few brief and conflicting anecdotal clinical statements in this area that are scattered throughout the literature. These include the following literature excerpts, which are specifically in the context of progestogens as part of puberty induction in cisgender girls and women with delayed or absent puberty due to hypogonadism:
I […] performed studies on three women lacking mammary development and exhibiting signs of marked hypogonadism. […] Corpus luteum extract, 5 international units daily for a period of thirty days, when given alone produced no detectable change in the breasts. This is in accord with the experimental observations on animals of Turner,2 Corner 3 and others. When, however, patients were given alternate daily injections of 1 international unit of progesterone and from 20,000 to 50,000 international units of estrone or of estradiol benzoate, breast growth was more rapid than that produced by the estrogenic hormones alone. The simultaneous use of the corpus luteum and estrogenic therapy definitely produced a much firmer breast growth, which was distinctly lobular to palpation, whereas the growth produced by the estrogenic hormones alone was smooth and the borders of the glandular tissue were difficult to define. Rapid regression in the size of the breasts followed the omission of the hormone injections, but the regression was less rapid when the combined therapy had been used. [MacBryde (1939)]
There are authorities who consider that breast growth is better if a progestogen is combined with oestrogen for the latter part of the cycle of treatment (Capraro, 1971). Shearman (1971) employs sequential therapy in his cases. Huffman (1971) however, does not believe that there is any improvement with the addition of progestogens. [Dewhurst (1971a)]
The effects of progesterone on the human breast remain obscure. Although widely stated to cause glandular development, the evidence for this is slender (Benson et al 1959). [Shearman (1972a)]
Many people use oestrogens alone, but the addition of a progestin for 6 or 10 days each month gives much better cycle control and appears to cause better breast development. [Shearman (1972b)]
Some authorities consider that breast growth is better if a progestogen is given for the latter part of each course of treatment. [Capraro & Dewhurst (1975)]
It has been suggested that progestins be added during the last week of each cycle of estrogen therapy in order to develop more rounded breasts rather than the conical breasts many of these patients develop, but we have been unable to detect any difference in breast contour with or without progestins. [Davajan & Kletzky (1979)]
I have been satisfied that the addition of a progestogen was necessary to get a good breast response to hormone treatment although the progestogen, as I have said, is required after the first year if the uterus is present. [Dewhurst (1982)]
In addition to the preceding instances, Werner (1935) and Geschickter (1945) assessed the effects of progesterone on the breasts in cisgender women. Werner (1935) attempted to induce lactation in 8 surgically gonadectomized cisgender women with combinations of estrogen, progesterone, and prolactin, all in the form of crude extracts by injection. In two women who were given progesterone, he claimed that a marked increase in the size of the breasts beyond that with estrogen alone was observed. Additionally, he claimed that the breasts were more firm, the glandular tissue “more tortuous and nodular”, and the nipples more prominent. He was not successful in inducing lactation in the women in this study. The doses of hormones used were unclear as they were in the form of extracts, and were likely supraphysiological, potentially pregnancy-like due to the nature of the experiment. Werner’s study was also briefly discussed by Nelson (1936), among other citations. Geschickter (1945) observed lobuloalveolar growth on histological examination with administration of progesterone for 6 weeks to 2 months in one woman but not in another woman. However, the exterior physical changes of the breasts were not assessed or reported by this author and hence his findings are limitedly informative.
Surprisingly, there have been few analogous studies of the effects of progestogens on the breasts in cisgender girls and women following the preceding reports and anecdotes. Although there are very little data on progestogens and breast growth in cisgender females, clinical studies are finally starting to look more closely at the specifics of hormonal medications, including progestogens, in terms of breast development in girls undergoing puberty induction (e.g., Rodari et al., 2023). As such, future studies may provide more insight on the subject of progestogens and breast development in cisgender females.
Progesterone and its Physiological Role in Breast Development in Humans
Progesterone and Breast Development in Puberty
The role of progesterone in breast development and its possible usefulness for helping with breast development in transfeminine hormone therapy can be informed by the normal biological circumstances of puberty in cisgender females. Puberty in cisgender girls usually starts around age 11 (range 8–13 years) and completes around age 15 years (range 12–19 years), taking on average 3 to 4 years (but with a range of about 1.5–6 years in most cases) (Schauffler, 1942; Marshall & Tanner, 1969; Marshall, 1978; Begley, Firth, & Hoult, 1980; Drife, 1986). Progesterone essentially does not appear during puberty until ovulatorymenstrual cycles begin. Menarche, the onset of menstruation and hence of menstrual cycling, occurs on average at Tanner breast stage 4 or about 13 years of age, although it occurs at Tanner breast stage 3 or Tanner breast stage 5 in significant subsets of girls (26% for Tanner stage 3, 62% for Tanner stage 4, and 10% for Tanner stage 5) (Marshall & Tanner, 1969; Marshall, 1978; Drife, 1986; Hillard, 2007). Hence, the appearance of progesterone in normal female puberty is a relatively late event (Scott et al., 1950; Marshall, 1978; Begley, Firth, & Hoult, 1980; Drife, 1986), and most breast development appears to be complete by menarche and thus by the time that progesterone is first produced (Huffman, Dewhurst, & Capraro, 1981; Drife, 1982). Moreover, a small but significant subset of girls reaches Tanner breast stage 5 and hence fully developed breasts before menarche (Edmonds, 1989), which suggests that progesterone may not be essential for complete pubertal breast development.
Only a handful of studies and sources have reported progesterone levels during puberty across Tanner stages or by age in cisgender girls (e.g., Sizonenko, 1978 [Graph]; Kühnel, 2000; Lee, 2001 [Table]; Aly, 2020a). They corroborate the above findings with regard to limited progesterone exposure during puberty. The “A Girl’s First Period Study” is an ambitious research project announced in 2022 that aims to better characterize reproductive hormone levels in pubertal and adolescent girls and may shed more light on the physiological role of progesterone during puberty (Lucien et al., 2022). The researchers have specifically highlighted the possible role of progesterone in breast development as part of their interests:
Does exposure to low levels of [progesterone (P4)], as occurs before menarche, during anovulatory cycles with some degree of follicle luteinization, and during early, immature ovulatory cycles play an important role in normal breast development during puberty? This question has important clinical implications as hormone replacement during puberty does not typically include low-dose P4; rather, it is conducted using a staggered approach of estrogen-only therapy followed by the addition of full adult doses of exogenous P4 only after 2 years or when breakthrough bleeding occurs.27 This is done to avoid development of tubular breasts, although there are limited data linking early P4 exposure to suboptimal breast development.28
Taken together, production of progesterone is a late event in normal female puberty, and even once it does begin, exposure to progesterone is low and sporadic until well after puberty has completed. Moreover, a subset of girls complete breast development before progesterone production starts. These facts call into some question the role of progesterone in breast development in female puberty, as most breast development appears to be complete prior to the appearance of progesterone. However, more research is still needed on the role of progesterone in breast development during normal puberty.
On the basis of normal female puberty, it seems it may be advisable that if progestogens are introduced in an attempt to enhance breast development in transfeminine people, their introduction be delayed until after 2 or 3 years of hormone therapy, so as to mimic the normal progestogenic exposure of puberty.
Progesterone and Breast Development in Pregnancy
During pregnancy, under the influence of ovarian hyperstimulation and placental formation, there are profound changes in hormonal profiles, including of hormones like estrogen, progesterone, and prolactin, among many others (Table 1). Comparing hormone levels during the menstrual cycle to those during the third trimester of pregnancy, estradiol levels increase on the order of 100-fold, progesterone levels increase on the order of 10- to 20-fold, and prolactin levels increase by around 10-fold (Table 1). Levels of numerous other hormones also change considerably during pregnancy, for instance other estrogens besides estradiol, androgens, gonadotropins (e.g., human choronic gonadotropin or hCG), human placental lactogen (hPL), relaxin, adrenocorticotropic hormone (ACTH), cortisol, aldosterone, growth hormone (GH), and insulin-like growth factor 1 (IGF-1), among others (Goodman, 2009 [Figure]; Mesiano, 2019). These hormones are variously produced by the ovaries, the placenta, and the pituitary gland, among other glands. In response to the myriad hormonal changes during pregnancy, there are dramatic changes to the breasts, which prepare the mother for postpartumlactation and breastfeeding.
Table 1: Changes in hormone levels (estradiol, progesterone, and prolactin) during normal pregnancy:
There are large and dramatic changes in levels of numerous hormones during pregnancy, and the exact hormones responsible for the breast changes during pregnancy are not known (Hytten & Leitch, 1971a; Hytten, 1976). However, it is considered likely, on the basis of animal studies, that a variety of hormones, including estrogen, progesterone, prolactin, placental lactogen, glucocorticoids, and growth hormone, are all importantly involved in different aspects of the maturation (Hytten & Leitch, 1971a; Hytten, 1976; Cox et al., 1999). Moreover, in a quantitative clinical study of breast changes during pregnancy, increases in breast volume and areola size were positively correlated with levels of hPL, while increases in nipple size were positively correlated with levels of prolactin (Cox et al., 1999). Progesterone and prolactin have specifically been implicated in the lobuloalveolar development of the breasts during pregnancy (Bässler, 1970; Lee & Ormandy, 2012; Obr & Edwards, 2012). Both hormones appear to be independently essential in normal lobuloalveolar growth per animal studies (Obr & Edwards, 2012; McNally & Stein, 2017; Hannan et al., 2023). Prolactin likewise appears to be essential in humans, based on case reports of lactation failure in women with isolated prolactin deficiency (Buhimschi, 2004). Conversely, hPL may not be essential for lactation based on case reports of normal lactation in women with very low levels of hPL during pregnancy (Gaede, Trolle, & Pedersen, 1978; Hannan et al., 2023).
On the basis of the preceding, in spite of rather extreme hormonal stimulation, the breast changes of pregnancy, although quite dramatic, are essentially temporary and fully reversible, remaining only as long as continuous hormonal exposure is maintained. This hormonal stimulation includes exposure to extremely high levels of progesterone. It would seem, based on pregnancy, that once pubertal breast development is completed, the breasts are rather unamenable to permanent further growth, whether that involves exposure to progestogens or to a variety of other hormones known to act on the breasts.
Breast Composition and Lobuloalveolar Tissue Proportion
The breasts are made up of two main types of tissue: (1) epithelial tissue, the actual functional internal mammary glandular tissue, including ducts and alveoli or lobules; and (2) stromal tissue, a mixture of connective tissue and adipose (fat) tissue. Lobuloalveolar development refers to growth and maturation of the alveoli and lobules, and hence is a form of epithelial or glandular development. Progestogens are involved primarily in lobuloalveolar development of the breasts, which is the type of breast development that is necessary for lactation and breastfeeding and that occurs mainly during pregnancy.
During pregnancy and lactation in humans, the breasts undergo dramatic changes, and epithelial tissue comes to make up a much greater proportion of the breasts (Ramsay et al., 2005; Bland, Copeland, & Klimberg, 2018). In fact, sources state that glandular tissue comprises a majority of the breast during pregnancy and lactation, with one study of lactating women finding that the breasts were composed 63% (range 46–83%) of glandular tissue (Ramsay et al., 2005). This is not merely due to lobuloalveolar development and glandular growth, but is also due to a marked reversible reduction in mammary adipose tissue (Wang & Scherer, 2019; Alex, Bhandary, & McGuire, 2020). In any case, under more normal physiological circumstances and progesterone exposure, the contribution of lobuloalveolar tissue to the size of the breasts would appear to be quite small. In relation to this, outside of pregnancy levels of progesterone, the significance of progestogen-mediated breast lobuloalveolar growth in terms of breast size is unclear but seemingly questionable (Orentreich & Durr, 1974; Wierkcx, Gooren, & T’Sjoen, 2014).
Breast Development in Cisgender Women with Complete Androgen Insensitivity Syndrome and Consequent Absence of Progesterone
It has been claimed that progesterone helps to move transfeminine people and cisgender females from Tanner stage 4 to 5 breast development and that it helps to round out the breasts (e.g., Vorherr, 1974a; Prior, 2011; Prior, 2019a; Prior, 2020). It has also sometimes been claimed in the online transgender community that cisgender women with complete androgen insensitivity syndrome (CAIS), an experiment of nature of women who lack progesterone, are stuck at Tanner stage 4 breast growth and have “cone-shaped” breasts due to their absence of progesterone. In actuality however, there is no good evidence at this time that progesterone is required for normal pubertal breast development, that progesterone is needed to reach Tanner stage 5, or that it helps to round out the breasts. Such claims are contradicted by extensive available literature and evidence, including notably the literature on CAIS women themselves.
Women with CAIS are individuals who have a 46,XY karyotype (i.e., are genetically “male”), testes, and who would otherwise have physically developed as males, but did not because they have a mutation in the gene encoding the androgen receptor that makes them completely insensitive to the effects of androgens. There are also incomplete forms of the syndrome, like partial androgen insensitivity syndrome (PAIS) and mild androgen insensitivity syndrome (MAIS). CAIS women have a male-typical hormonal profile, generated by their testes, including high male-range levels of testosterone, low female-range estradiol levels, and negligible progesterone levels (Wiki; Table). Instead of developing physically as males however, CAIS women are perfectly phenotypically female, with a normal female body, vagina, and breasts (Wiki; Photo). Their testosterone has been unable to masculinize them, while their estradiol, unopposed by androgens, is able to fully feminize them. The internal reproductive system in CAIS women is essentially that of a highly underdeveloped male, with testes instead of ovaries, no uterus, fallopian tubes, or cervix, and no prostate gland or seminal vesicles. The testes are internally located, either intra-abdominally, inguinally, or labially. They are usually surgically removed by early adulthood, as they otherwise have a high risk of developing testicular cancer because of their location. The vagina in CAIS women is often short and is blind-ending, which is related to their lack of a uterus. In terms of behavior, gender, and sexuality, CAIS women are described as feminine.
Despite claims that CAIS women have generous breast sizes however, in actuality, some CAIS women have large breasts, while some have small breasts. One study found a wide range of breast size measurements of 16×14 cm to 41×31 cm, which equates to an almost 6-fold variation in breast size as quantified by area (Wisniewski et al., 2000). Moreover, the breasts of CAIS women have never been directly compared to those of normal women. Hence, there are no clear data at this time that the breasts of CAIS women are actually larger than average for women. The variation in breast growth in CAIS women parallels the same large variation in breast size between individuals that is seen in cisgender women in general. Here is a collection of photos of CAIS women and their breast development from published case reports and reviews throughout the literature. As can be seen from these photos, breast development in CAIS women is normal and often excellent, although subject to considerable variation between individuals in terms of breast size and shape as in women generally.
If CAIS women truly do have enhanced breast development and breast sizes compared to normal women, it may be that their androgen insensitivity, and hence lack of inhibition of estrogen-mediated breast development by androgens, is responsible for this (Wilson, 1968; Sobrinho, Kase, & Grunt, 1971; Andler & Zachmann, 1979; Zachmann et al., 1986; Patterson, McPhaul, & Hughes, 1994; Barbieri, 2019). Another theoretical possibility is that the high testosterone levels may be aromatized into greater amounts of estradiol locally within the breasts and other tissues in CAIS women and that this may somehow allow for enhanced breast development (Ladjouze & Donaldson, 2019). Interestingly, it has been claimed anecdotally by some researchers that breast development is much better in CAIS women who are allowed to naturally undergo puberty with their own endogenous hormones compared to CAIS women who undergo gonadectomy before puberty and have pubertal maturation induced with exogenous estrogen therapy (Dewhurst, 1972; Glenn, 1976; Dewhurst, 1981; Reindollar & McDonough, 1985; Shearman, 1985; Laufer, Goldstein, & Hendren, 2005). This is to the extent that some CAIS women who have had induced puberty have needed to undergo surgical breast augmentation due to poorly developed breasts (Dewhurst, 1981; Shearman, 1985). In relation to the preceding, it is usually standard clinical practice to delay gonadectomy in CAIS women until puberty has fully completed (Laufer, Goldstein, & Hendren, 2005). However, one clinical study reported good breast development rated as Tanner stage 5 in all cases in CAIS women who experienced either spontaneous or therapeutic puberty (Cheikhelard et al., 2008). It may be important to mimic normal pubertal estrogen exposure with puberty induction in CAIS females by employing low physiological estradiol levels that are slowly and gradually increased over a few years (Dewhurst, 1981; Cheikhelard et al., 2008; Bertelloni et al., 2011).
Baron evaluated a total of 41 people with androgen insensitivity syndrome (AIS) and found that 97% of CAIS women had normal breast development while 63% of individuals with “incomplete AIS” (likely PAIS) had normal breast development (Baron, 1993; Baron, 1994a; Baron, 1994b). In another earlier published study of 50 CAIS females, by Sir Christopher John Dewhurst, 76% were rated as having full breast development, 14% as having moderate breast development, 10% as having “mild” breast development, and 0% as having absent breast development (Dewhurst, 1971b). Hence, based on findings in large samples of CAIS females, most to almost all have normal or full breast development. That a minority of CAIS females have had less breast growth may be due to factors like low and inadequate estradiol levels in some individuals, young age at time of assessment by which point breast development has not fully completed, and/or a small subset of women in general having underdeveloped or small breasts.
CAIS women have never been described in the literature as having “cone-shaped”, “pointy”, or otherwise abnormal breasts. The only exception is that they are often said to have nipples and areolas that are described as “juvenile”, “infantile”, “small”, “pale”, and “non-pigmented” (e.g., Photo) (e.g., Morris, 1953; Morris & Mahesh, 1963; Simmer, Pion, & Dignam, 1965; Dewhurst, 1967; Khoo & Mackay, 1972; Perez-Palacios & Jaffe, 1972; Dewhurst & Spence, 1977). This has been said to be the case regardless of breast size or maturation (Khoo & Mackay, 1972). A possible reason for this phenomenon is that estradiol levels in CAIS women are relatively low, only about 35 pg/mL (130 pmol/L) on average (Wiki; Table). This is relevant as estrogens are known to concentration-dependently produce nipple and areolar pigmentation and enlargement (e.g., Davis et al., 1945 [Figure]; Kennedy & Nathanson, 1953). In contrast to estrogens, progestogens have not been implicated in nipple or areolar pigmentation. Hence, it seems that higher estrogen levels may be necessary for full adult-like nipple and areolar maturation.
Despite their often large breasts, CAIS women are said to have relatively little breast glandular tissue, as opposed to fat and connective tissue, and to have minimal breast lobuloalveolar development (Morris, 1953; Morris & Mahesh, 1963; Simmer, Pion, & Dignam, 1965; McMillan, 1966; Perez-Palacios & Jaffe, 1972; Dewhurst & Spence, 1977; Shapiro, 1982). This is in accordance with the lack of progesterone in CAIS women, since progesterone is important in mediating lobuloalveolar growth. The retained breast sizes of CAIS women despite reduced glandular and lobuloalveolar structures is consistent with the fact that the breasts are composed mostly of stromal adipose and connective tissue. Hence, as touched on previously in this article, greater glandular or lobuloalveolar formation in the breasts may not necessarily translate to greater breast size, which seems readily apparent in CAIS women.
The normal and excellent breast development of CAIS women is notable because these individuals, owing to their testes and hence absence of significant gonadal progesterone production, have very low and negligible levels of progesterone (Wiki; Table; Barbieri, 2019). CAIS womens’ normal breast development, often large breasts, and ability to reach complete breast maturation, as measured by the Tanner scale, are collectively suggestive that progesterone is not required for normal or complete pubertal breast development (Barbieri, 2019). In any case, it must be noted and cautioned again that the breasts of CAIS women have never been directly compared to those in normal women. In addition, quantitative studies of the breasts of CAIS women are very scarce, and much of our knowledge in this area is based on anecdotal clinical experience and subjective breast evaluation. This is in large part due to the rarity of CAIS women and the difficulty in obtaining decent samples of them for study. Furthermore, CAIS women also have other differences from regular women besides their lack of progesterone, for instance their relatively low circulating estradiol levels, high testosterone levels (which can be aromatized into estradiol within tissues like the breasts), androgen insensitivity, and XY karyotype, among others. Hence, the insights into breast development provided by CAIS women come with a variety of caveats.
Interestingly, in spite of their well-developed breasts, breast cancer has never been reported in CAIS women, and would appear to be very rare in these individuals (Aly, 2020b; Aly, 2020c). This may be related to factors like the lack of progesterone and lobuloalveolar maturation in CAIS women and/or their absence of a second X chromosome (Aly, 2020b; Aly, 2020c). CAIS women suggest that breast cancer is not an inherent eventual consequence of excellent breast development.
Menstrual Cycles and Temporary Cyclic Breast Enlargement
The enlargement of the breasts during the luteal phase of the menstrual cycle is believed to be due to temporary glandular and stromal tissue growth, luminal dilation of the ducts and alveoli, fluid retention in the glandular and stromal structures, and increased vascularization and blood flow (Scott et al., 1950; Drife, 1989; Fowler et al., 1990; Hussain et al., 1999; Alekseev, 2021; Biswas et al., 2022). However, studies suggest that most of the changes are merely due to water fluctuations and that change in breast glandular volume is relatively small (Rix et al., 2023). The breast changes during the menstrual cycle, such as breast enlargement, have been positively correlated with increased levels of estradiol and progesterone during the luteal phase (Jemström & Olsson, 1997; Jasieńska et al., 2004; Clendenen et al., 2013; Rix et al., 2023). Correspondingly, combined estrogen and progestogen therapy has been found to reversibly increase breast size (e.g., Hartmann et al., 1998). Estradiol levels are also positively associated with breast tenderness during estrogen therapy, whereas progestogens may actually reduce breast tenderness (e.g., de Lignières & Mauvais-Jarvis, 1981 [Figures]; Sitruk-Ware et al., 1984; Wiki; Wiki). Both estradiol and progesterone can promote water retention via distinct hormonal mechanisms as well as mediate breast glandular growth and changes (Rix et al., 2023). As such, the breast changes during the menstrual cycle are assumed to be due to changing levels of estradiol and progesterone, though it is noteworthy that progesterone has been particularly implicated owing to the breast volume increase occurring during the luteal phase (Lawrence & Lawrence, 2015; Rix et al., 2023). There is a delay in breast volume increases following the peaks of estradiol and progesterone levels during the menstrual cycle and hence the changes are not instantaneous (Rix et al., 2023).
Combined oral contraceptives, which are estrogen–progestogen preparations, as well as menopausal estrogen–progestogen hormone therapy, may produce temporary breast enlargement and feelings of breast fullness analogous to those that occur during the luteal phase of the menstrual cycle (Milligan, Drife, & Short, 1975; Dennerstein et al., 1980 [Figure]; Malini, Smith, & Goldzieher, 1985; Jemström & Olsson, 1997; Jernström et al., 2005). In one study, breast volume was around 100 mL greater (~30% higher) in women who were currently taking oral contraceptives relative to those who had not taken or had previously taken oral contraceptives (Jemström & Olsson, 1997). In some women, the increase in breast size with oral contraceptives was subjectively reported to be up to a single bra cup size in volume (Jemström & Olsson, 1997). However, in another study by the same group of researchers that had a much larger sample size (n=258 vs. n=65), breast volumes were not significantly different between current hormonal contraceptive users and non-users (Jernström et al., 2005). Additionally, another study found no significant differences in breast volume in women between different estrogen–progestogen oral contraceptives that had about 6-fold variation in dose of the same progestin (0.4 to 2.5 mg/day norethisterone) as well as non-users (Malini, Smith, & Goldzieher, 1985). However, this study was underpowered due to small sample sizes (n=5 to n=15 per group) (Malini, Smith, & Goldzieher, 1985).
Engman et al. (2008) conducted an RCT of treatment with mifepristone, a selective progesterone receptor modulator (SPRM) with predominantly antiprogestogenic effects, versus placebo for 3 months in normally cycling premenopausal cisgender women, and evaluated the effects of this progesterone receptor blockade on the breasts. They found that mifepristone significantly reduced Ki-67 index, a measure of cellular proliferation in the breasts, and reduced subjectively rated symptom scores on the Breast Symptom Index (BSI). More specifically, breast soreness, breast swelling, sense of increased breast volume, and the total breast symptoms score were all significantly reduced on the BSI. However, breast volume was not objectively measured in this study. A major limitation of this study is that mifepristone inhibits ovulation and modifies levels of estradiol and other hormones (Spitz et al., 1989; Spitz et al., 1994; Engman et al., 2008, Spitz, 2010). As such, it is unclear whether the effects observed by Engman and colleagues were specifically due to progesterone receptor antagonism in the breasts or due to disruption of the hypothalamic–pituitary–gonadal (HPG) axis, for instance lowered estradiol levels.
An interesting case report of an adult woman with CAIS documented a significant increase in breast volume with combined estrogen–progestogen therapy relative to estrogen monotherapy (Dijkman et al., 2023b). The woman was started on cyclic oral estradiol 2 mg/day and dydrogesterone 10 mg/day and subjectively experienced breast pain and fluctuations in breast volume of about one cup size while on this regimen. Subsequently, she was switched to oral estradiol valerate 3 mg/day monotherapy and the fluctuations in breast volume ceased. However, her overall breast volume was reduced as well, and the woman decided to resume combined estradiol and dydrogesterone therapy. Her clinicians proceeded to measure her breast volume using 3D body scanning. Her left breast was 758 mL and right breast was 673 mL with estrogen monotherapy, and her breasts increased to respective volumes of 875 mL and 784 mL during combined estrogen–progestogen therapy, giving net volume increases of 117 mL (+16%) and 111 mL (+17%). These differences in volume corresponded to an almost one bra cup difference in size. The researchers noted that estradiol and progesterone are associated with cyclical breast changes, and hypothesized that the changes in their patient were due to increased fluid retention in the breasts. Taken together, the case report demonstrates that progestogens can cause rapid and considerable reversible breast enlargement in some women analogous to that during the normal menstrual cycle.
Progesterone and Mammary Development in Animals
Progesterone and Pubertal Mammary Development in Animals
Although progesterone does not seem to be essential in normal pubertal mammary development in mice, studies have interestingly found that it is able to substitute for estrogen in mediating pubertal ductal mammary development in this species. Ruan, Monaco, & Kleinberg (2005) studied the effects of various combinations of exogenous estradiol, progesterone, and IGF-1 on mammary development in oophorectomized female IGF-1-knockout mice. In terms of stimulation of ductal development to occupy the mammary gland fat pad, the combination of progesterone and IGF-1 produced 92% occupation, estradiol and IGF-1 resulted in 92% occupation, estradiol, progesterone, and IGF-1 achieved 96% occupation, and IGF-1 alone resulted in only 28% occupation (Ruan, Monaco, & Kleinberg, 2005; Kleinberg & Ruan, 2008). In terms of gross anatomical appearance, the ductal tree with progesterone and IGF-1 was said to resemble that of a normal fully developed pubertal mammary gland (Ruan, Monaco, & Kleinberg, 2005). However, differences in mammary development between the combination of estradiol and IGF-1 and the combination of progesterone and IGF-1 were apparent, with estradiol and IGF-1 having greater effect on terminal end bud formation, ductal decorations, and slight alveolar maturation, and progesterone and IGF-1 having more effect on ductal formation, extension, and branching (Ruan, Monaco, & Kleinberg, 2005; Kleinberg & Ruan, 2008). The effects of progesterone on mammary development were reversed by the progesterone receptor antagonist mifepristone (Ruan, Monaco, & Kleinberg, 2005). Only the combination of estradiol, progesterone, and IGF-1 produced mammary development that resembled that during mid-pregnancy, with full maturation of secretory alveolar structures (Ruan, Monaco, & Kleinberg, 2005; Kleinberg & Ruan, 2008).
A limitation of studies that have used exogenous progesterone to stimulate pubertal ductal mammary development in mice is that the doses of progesterone employed, in conjunction with other hormones like estradiol, have been sufficient to mediate mammary growth to a level typical of pregnancy, with robust maturation of mammary lobuloalveolar structures (e.g., Škarda, Fremrová, & Bezecný, 1989; Ruan, Monaco, & Kleinberg, 2005). Pregnancy is a time when hormone levels are much higher than usual. Hence, the progesterone exposure in these studies may have been supraphysiological relative to normal puberty, and may have produced effects on mammary growth that would not otherwise occur during this time. Accordingly, Škarda, Fremrová, & Bezecný (1989) found that whereas untreated normal female mice naturally grew to a mammary gland area of 26.4 mm2 and normal female mice treated with exogenous estradiol grew to a mammary gland area of 25.3 mm2, normal female mice treated with exogenous estradiol and progesterone grew to a mammary gland area of 43.5 mm2 and with exogenous progesterone alone to a mammary gland area of 64.6 mm2. The untreated control mice did not show alveolar buds, whereas the progesterone-treated groups did have alveolar maturation, indicating supraphysiological and pregnancy-like development compared to non-pregnant mice (Škarda, Fremrová, & Bezecný, 1989). In any case, one study employed low doses of progesterone (0.1 mg/day), one-tenth of that used in most other studies (1 mg/day), and found that progesterone still stimulated significant ductal development in mice at these doses (Aupperlee et al., 2013; Berryhill, Trott, & Hovey, 2016). Hence, progesterone is still able to stimulate some level of ductal growth in mice even at lower levels.
Although progestogens by themselves can apparently stimulate normal pubertal mammary development in lieu of estrogen exposure in mice, it is not clear that they do so similarly in humans. It is well-known that progestogens alone, without concomitant estrogenic activity, do not generally produce breast development in humans. As an example, progestogens, for instance MPA and CPA, have been used as puberty blockers in boys and girls at very high doses, and do not produce breast development in this context, instead causing arrest and regression of breast development via gonadal suppression (Lyon, De Bruyn, & Grant, 1985; Fuqua & Eugster, 2022). Cases of gynecomastia in boys have occurred with CPA, but only in a minority and with this easily attributable to other causes than progestogenic activity, for instance the antiandrogenic activity of CPA and disruption of the HPG axis (Kauli et al., 1984; Laron & Kauli, 2000). Similarly, progestogens like MPA and CPA have been used at very high doses in men to treat prostate conditions and sexual disorders, and likewise do not usually produce gynecomastia under these circumstances. Rates of gynecomastia with CPA used in the treatment of prostate cancer are low and are not noticeably different from the rates with surgical or medical castration (~10%) (Fourcade & McLeod, 2004; Di Lorenzo et al., 2005). This is in major contrast to the high rates of gynecomastia with estrogens and nonsteroidal antiandrogens (up to 70–80%) (Fourcade & McLeod, 2004; Di Lorenzo et al., 2005; Deepinder & Braunstein, 2012). Species differences may be present such that progestogens can produce robust pubertal mammary development in mice but do not do so in humans.
Progesterone and Gestational Mammary Development in Animals
Therapeutic or pharmacological pseudopregnancy is a type of hormone therapy that attempts to replicate the hormonal mileu of pregnancy for certain medical indications in cisgender females by administering exogenous hormones. In practice, this has involved the administration of very high doses of estrogens and progestogens, with most other pregnancy hormones not included. Therapeutic pseudopregnancy was first developed in the 1950s and is largely no longer used in medicine today (Kaiser, 1993).
The effects of therapeutic pseudopregnancy on the breasts are of interest due to the breast changes that occur during pregnancy, for instance lobuloalveolar development and substantial reversible breast enlargement. In the 1980s, Lauritzen and colleagues conducted a study of therapeutic pseudopregnancy for treatment of breast hypoplasia (small/underdeveloped breasts) in cisgender women (Lauritzen, 1980; Lauritzen, 1982; Lauritzen, 1989; Göretzlehner & Lauritzen, 1992). They employed the estrogen estradiol valerate 40 mg/week and the progestogen hydroxyprogesterone caproate (OHPC) 250 to 500 mg/week both by intramuscular injection for 4 to 5 months. The estradiol valerate dosage employed was very high, with other studies by the same authors reporting that this dosage of estradiol valerate resulted in first-trimester pregnancy levels of estradiol in women (~3,000 pg/mL [~11,000 pmol/L]) (Ulrich, Pfeifer, & Lauritzen, 1994; Ulrich et al., 1995). These estradiol levels are roughly 30 times the normal concentrations outside of pregnancy (Aly, 2018b). Similarly, the OHPC doses were very high, with 250 to 500 mg per month being similar in strength to luteal-phase progestogenic exposure (Wiki). Hence, as the same OHPC doses were used weekly in the study, the doses were roughly around 4.5 times luteal-phase exposure and thus were analogously similar to first- or second-trimester progesterone levels in terms of strength (Aly, 2020d). The authors noted that they had initially tried lower hormone doses, similar to those originally used in the 1950s, but did not achieve significant breast growth with these doses, and so increased the dosage. Breast changes were measured in the study with a tape measure (applied horizontally and vertically to the breast area), photographs, breast imaging using mammography and sonography, and, later in the study, plasticine impressions/molds with determination of the filling volume.
Lauritzen and colleagues reported the study findings in four different publications with different follow-up times and growing sample sizes. In the final follow-up, a total of 221 women had been treated. In the second follow-up, when 78 women had been treated, it was noted that 29 of the cases (37%) were less than 18 years old. However, in the final follow-up of 221 women, the age range was listed as 18 to 42 years. The researchers found that breast volume increased by 10 to 30% above baseline in 65% of the women. This was also accompanied by breast tenderness in almost all of the women, though the breast tenderness progressively declined during the treatment period. Other breast-related side effects like pigmentation and stretch marks were rarely observed. Prolactin levels slightly increased to 14 to 28 pg/mL by the end of treatment. Breast imaging showed an increase in the density of breast glandular tissue. The researchers claimed that the increase in breast size in their study was due to increased adipose tissue, water retention, and moderate hypertrophy of the glandular tissue.
Following treatment discontinuation, the increases in breast volume gradually and partially regressed in 40% of the women, to an increase of 10 to 20% above baseline. However, the authors claimed that the regression in breast volume could be reduced with adequate-dose combined estrogen–progestogen birth control pills or with topical estrogen and progestogen therapy applied to the breasts. In addition, they noted that therapeutic pseudopregnancy could be repeated to increase breast volume again. This was performed in a subset of the women, with treatment repeated 1 to 2 times after 6 months. In the second follow-up, which had 78 women, it was noted that 12 women (15%) had undergone multiple treatments. Aside from Lauritzen and colleagues, many other researchers have also reported substantial or full regression in breast size following estrogen and/or progestogen therapy to increase breast size in cisgender women (e.g., Cernea, 1944; Müller, 1953; Anderson, 1962; Bruck & Müller, 1967; Keller, 1984; Kaiser & Leidenberger, 1991; Keller, 1995; Hartmann et al., 1998).
The findings of Lauritzen and colleagues were reported very informally, in the form of non-peer-reviewed book chapters, conference papers, and medical magazines, and were never published in a peer-reviewed journal article. In relation to this, the methodology and results of the study were only briefly and imprecisely described. There are also additional concerns related to study design, such as lack of controls, randomization, and the quality of the breast measurement methods. As a result of the preceding issues, it is difficult to fully interpret the results of the study and to have complete confidence in its findings. In any case, Lauritzen and colleages’ results suggest that treatment with high-dose combined estrogen–progestogen therapy, achieving earlier-pregnancy estrogenic and progestogenic exposure, may be able to produce a significant temporary increase in breast size and a smaller long-term increase. The findings of a permanent increase in breast size conflict with those of other researchers who have reported complete regression in breast changes following treatment discontinuation. Moreover, the results are contradicted by findings in pregnant women, who, as described previously, show complete reversion to pre-pregnancy breast size or to even slightly smaller breasts following cessation of lactation.
It is difficult to evaluate the relative roles of the estrogen and the progestogen in the findings of Lauritzen and colleagues, as there were no comparison groups employing estrogen or progestogen therapy alone in the study. Both estrogens and progestogens have been implicated in causing breast enlargement and plausibly could have contributed to the breast changes. As such, it is unclear to what extent the breast changes were specifically due to progestogenic exposure rather than to estrogenic exposure.
The breast size increases observed by Lauritzen and colleagues were seemingly more modest relative to those that occur normally during pregnancy. They also lacked certain characteristics of pregnancy-related breast changes, like nipple and areolar pigmentation. The reasons for this are not fully clear. The subject populations between these studies were different, for instance in terms of factors like initial breast size and age, which may be contributing reasons. Another possible contributing factor is that only estrogen and progestogen levels increased in the study, whereas levels of other pregnancy hormones, besides the slight increase in prolactin levels, did not increase. These other pregnancy hormones, for instance hPL and IGF-1, may also be involved in breast development during pregnancy. Finally, the treatment duration was only 4 to 5 months, and the estrogen and progestogen exposure was only similar to that during early-to-mid pregnancy, whereas normal pregnancy lasts 9 months and involves continued dramatic increases in estrogen and progesterone levels through to childbirth.
It should be noted that, owing to the highly supraphysiological estrogen and progestogen levels required, which can cause serious health complications like blood clots and cardiovascular problems (Aly, 2020e), as well as the small to negligible lasting increase in breast volume, therapeutic pseudopregnancy is inadvisable for transfeminine people and should not be pursued or employed. Nonetheless, the historical findings of therapeutic pseudopregnancy for increasing breast size in cisgender females are of significant theoretical interest in exploring the roles of estrogens and progestogens in breast growth.
Early Progestogen Exposure and the Possibility of Suboptimal Breast Development
While progestogens are typically sought after by transfeminine people for their potential in improving breast development, there have also been various suggestions in the literature that early or premature exposure to progestogens may result in suboptimal breast development and that progestogens may suppress or reduce estrogen-mediated breast development. These suggestions include progestogens having known antiestrogenic effects in the breasts, animal studies finding stunted mammary development with high doses of progestogens, clinical publications cautioning against premature introduction of progestogens in female puberty induction due to concerns about possibly stunted breast growth, clinical use of progestogens to treat macromastia in cisgender females, poor breast development with estrogen therapy in cisgender girls with a disorder of sexual development that results in high progesterone exposure, and breast development with estrogen and CPA (a very strong progestogen) typically being poor in transfeminine people. As with the question of whether progestogens can enhance breast development, it is currently unknown whether progestogens could worsen breast development. It is also unknown what dosage level and timing of introduction would be required for such an effect. In any case, for informational purposes, the preceding topics will each be discussed in the subsequent sections.
Antiestrogenic Effects of Progestogens in the Breasts
Stunted Mammary Growth with Progestogens in Animal Studies
Animal studies using progestogens including bioidentical progesterone and chlormadinone acetate (CMA), a progestin closely related to CPA, have found that high doses of these progestogens substantially stunt mammary gland development in rabbits, whereas lower doses do not do so (Lyons & McGinty, 1941; Beyer, Cruz, & Martinez-Manautou, 1970). See here for relevant literature excerpts as well as figures from these studies. Lyons & McGinty (1941) [Figure] found that estrogen alone induced ductal mammary development and estrogen plus progesterone 0.25 to 1 mg/day produced ductal development and slight to “fair” lobuloalveolar development. Conversely, estrogen plus progesterone 4 to 8 mg/day, which were 4- to 8-fold higher doses of progesterone than the most optimal dose, produced stunted mammary development with inhibited ductal development, only slight lobuloalveolar development, and, at the highest dosage, resulted in a much smaller mammary gland in terms of size than in the ≤1 mg/day groups. They concluded that high doses of progesterone are inhibitory and result in relatively poor mammary development. In the paper, doses of progesterone in international units (IU) were reported, but a citing review, Pfeiffer (1943), indicated that 1 IU progesterone is equal to 1 mg progesterone. As such, the milligram doses are listed above instead. Beyer, Cruz, & Martinez-Manautou (1970) [Figure] found that estrogen alone produced good ductal development without lobuloalveolar growth (mean mammary area = 376 mm2) and both estrogen plus CMA 0.5 mg/day and estrogen plus progesterone 2.5 mg/day produced optimal ductal and lobuloalveolar development (mean mammary area = 765 mm2 and mean mammary area = 688 mm2, respectively). Conversely, estrogen plus CMA 2.5 mg/day, a 5-fold higher dose of CMA than the optimal dose, resulted in dramatically reduced ductal development and mammary gland size albeit with significant lobuloalveolar growth (mean mammary area = 284 mm2). The authors concluded that moderate doses of progestogens stimulate mammary gland growth whereas large doses inhibit mammary gland development.
While these animal studies are suggestive that high doses of progestogens may be able to stunt breast development in humans, this is far from a certainty. There are species differences in hormone-mediated mammary development such that findings in one species, such as rabbits, may not translate to another species, like humans, or sometimes even to closely related species, like rats or guinea pigs (Bässler, 1970). As far as the present author is aware, stunted mammary development with high doses of progestogens has not been studied or reported in other animal species, for instance other rodent species or monkeys. It is also unclear that the doses employed in these animal studies are necessarily relevant to progestogen therapy in humans. This is because pregnancy levels of progesterone, which are much higher than luteal-phase progesterone levels, are necessary for substantial mammary lobuloalveolar development, and the doses of progestogens used in these studies were above that magnitude of progestogenic exposure. Hence, the doses may have corresponded to what in humans would be extremely high doses. However, such doses could still be relevant in the case of CPA used as an antiandrogen in humans, as CPA is used in this context at very high doses (see section below). The present author is unaware of any animal studies finding that physiological non-pregnancy levels of progesterone have any stunting or other adverse influence on mammary development, suggesting that only high doses of progestogens may have such effects. Finally, it seems notable that the estrogen and progestogen were initiated simultaneously in these animal studies and yet produced optimal pregnancy-like mammary development at the right doses. This suggests that early or immediate progestogen exposure might not be unfavorable in terms of breast development in humans. However, once again species differences may be present and confirmatory clinical studies are needed in humans.
Clinical Publications Cautioning Against Premature Introduction of Progestogens Due to Possibly Stunted Breast Development
A large number of clinical publications largely in the pediatric endocrinology literature have warned that premature exposure to progestogens during for instance puberty induction may result in suboptimal breast development in cisgender girls and/or transfeminine people (Zacharin, 2000; Bondy et al., 2007; Colvin, Devineni, & Ashraf, 2014; Wierckx, Gooren, & T’Sjoen, 2014; Kaiser & Ho, 2015; Bauman, Novello, & Kreitzer, 2016; Gawlik et al., 2016; Randolph, 2018; Donaldson et al., 2019; Heath & Wynne, 2019a; Heath & Wynne, 2019b; Iwamoto et al., 2019; Crowley & Pitteloud, 2020; Naseem, Lokman, & Fitzgerald, 2021; Federici et al., 2022; Lucien et al., 2022; Rothman & Iwamoto, 2022). The full relevant excerpts from these sources can be found here. In relation to these claims, and in order to mimic normal female puberty, a progestogen is not typically added to estrogen therapy during puberty induction in cisgender girls with delayed puberty until after about 2 to 3 years of treatment, by which point breast growth is generally considered complete. Additionally, progestogens are generally never added as part of puberty induction in transfeminine adolescents. Despite the preceding widespread literature statements and accepted clinical practices in the field of puberty induction however, it is important to note that the claims that premature introduction of progestogens might stunt breast development in this context are currently not based on any actual reliable clinical evidence and hence remain unsubstantiated. It is not even clear that these statements are based on anecdotal clinical experience as opposed to simple conjecture. The absence of data in this area may finally change in the future as more clinical studies of progestogens in puberty induction in cisgender girls are conducted (e.g., Rodari et al., 2023).
Rodari and colleagues studied optimization of puberty induction with estrogen therapy followed by eventual introduction of progestogen therapy in 49 cisgender girls with hypogonadism (e.g., Rodari et al., 2022; Rodari, 2022; Rodari et al., 2023). The researchers employed incrementally titrated low-dose transdermal estradiol to mimic the low and gradually increasing estradiol levels during normal puberty and added a progestogen only once menstrual bleeding began. The total duration of treatment was mean 2.65 ± 1 years, the time of first menstrual bleeding occurrence was 2.3 ± 1 years, and the time of progestogen introduction was median 2.22 years (IQR 1.56–2.87 years). Of the girls, 90% reached Tanner breast stage 4, but only 41% reached Tanner breast stage 5. Reaching the final Tanner breast stage was significantly associated with the number of estradiol dose increases (i.e., gradual estradiol dose titration) and the estradiol dose at progestogen introduction. The researchers interpreted the latter finding as progestogen exposure potentially hampering breast development. They questioned introducing progestogen therapy in the presence of incompletely developed breasts and suggested that instead of adding a progestogen upon onset of menstrual bleeding, clinicians should consider slightly reducing the estradiol dosage to delay progestogen introduction until the breasts complete maturation. While interesting, it must be noted that the findings of Rodari and colleagues are merely correlational, are open to multiple interpretations, and do not causally show that progestogens impair breast maturation.
Progestogens in the Treatment of Breast Hypertrophy
More recently, a couple of studies, both by the same group of researchers, assessed the impact of different types of hormonal contraception on macromastia in adolescent cisgender females with macromastia (Nuzzi et al., 2021; Nuzzi et al., 2022). They found that use of progestin-only contraceptives was associated with significantly more breast tissue removed upon surgical breast reduction (959.9 g/m2 vs. 735.9 g/m2 [+30%]; p = 0.04) and worse clinical symptoms (e.g., breast pain—odds ratio, 4.94, p = 0.005) relative to non-users of hormonal contraception (Nuzzi et al., 2021). Conversely, use of combined oral contraceptives, which are estrogen–progestogen preparations, was associated with significantly less breast tissue removed with breast reduction (639.5 g/m2 vs. 735.9 g/m2 [−13%]; p = 0.003), though not with any differences in clinical symptoms, relative to those naive to hormonal contraception (Nuzzi et al., 2022). It should be noted that progestin-only contraceptives suppress the HPG axis and result in low estradiol levels, whereas combined oral contraceptives suppress the HPG axis and lower estradiol production but simultaneously supplement estrogen signaling by delivering exogenous estrogen. This difference may somehow be responsible for the opposite influence of estrogen–progestogen therapy versus progestogen-alone therapy on macromastia severity. While the findings of Nuzzi and colleagues are interesting, it is noteworthy that the methodology and findings of their research were criticized on various grounds in a letter to the editor concerning one of the articles (Karp, 2022).
Santen et al. (2024), in a case series of cisgender girls with juvenile gigantomastia, noted that breast growth continues for only a number of years following onset and hence there must be some form of stop signal that is activated and that prevents further breast growth. They speculated that this signal may be related to apoptosis (programmed cell death). Santen and colleagues noted that in adult cisgender women, proliferation of breast cells is increased during the follicular phase of the menstrual cycle, whereas apoptosis in breast cells is increased during the luteal phase of the cycle. They hypothesized that the apoptosis during the luteal phase may block further breast development. Since progesterone is produced during the luteal phase and may mediate said apoptosis, this would substantiate the use of progestogens in the treatment of breast hypertrophy. However, the researchers noted that no data exist on apoptosis in the breasts of girls with juvenile gigantomastia. Moreover, an important point against the authors’ hypothesis is that estrogen-induced breast growth gradually slows and ceases in people who do not have menstrual cycles and luteal phases or progestogenic exposure just as it does in normal cisgender girls. Prominent examples of such individuals include CAIS women, transfeminine people, and cisgender men with prostate cancer treated with estrogen therapy.
Poor Breast Development in 17α-Hydroxylase/17,20-Lyase Deficiency
Non-Comparative Clinical Studies of Breast Development with Estrogen and Cyproterone Acetate in Transfeminine People
The possibility of suboptimal breast development with premature exposure to progestogens is of particular relevance in the case of CPA used as an antiandrogen in transfeminine people. This is because CPA is a potent progestogen in addition to antiandrogen, starts to be taken at the initiation of hormone therapy, and happens to be used in transfeminine people at doses that result in very strong to profound progestogenic exposure (Aly, 2019). In terms of progestogenic strength, CPA at a dosage of 2 mg/day is comparable to the progesterone exposure during the luteal phase of the menstrual cycle (Aly, 2019; Wiki). For comparison, CPA has been used in transfeminine people at doses ranging from 10 to 100 mg/day (Aly, 2019). This would mean that CPA provides roughly 6.25 times the progestogenic impact of luteal-phase progesterone exposure at a dosage of 12.5 mg/day, 12.5 times the impact at 25 mg/day, 25 times the impact at 50 mg/day, and 50 times the impact at 100 mg/day. Moreover, this does not consider the fact that progesterone is only produced during the luteal phase, or half of the menstrual cycle, whereas CPA is taken continuously every day of the month. The preceding magnitudes of progestogenic exposure with CPA are on par with and even beyond those during pregnancy. Only recently have lower doses of CPA (e.g., ≤12.5 mg/day) started to be used in transfeminine hormone therapy.
Studies in pubertal and adolescent transfeminine people given GnRH agonists to block puberty plus estrogen therapy have reported good breast development in these individuals as assessed by subjective clinical impression or Tanner staging (de Vries et al., 2010; Hannema et al., 2017). However, quality objective measures of breast development were not employed in these studies. Conversely, non-comparative studies using estrogen plus CPA in adult transfeminine people have commonly reported modest breast development, including incomplete breast development only to Tanner stage 2 to 4, small breast cup sizes, and small breast volumes (Kanhai et al., 1999; Sosa et al., 2003; Sosa et al., 2004; Wierckx et al., 2014; Fisher et al., 2016; Tack et al., 2017; de Blok et al., 2018; Reisman, Goldstein, & Safer, 2019; Meyer et al., 2020; de Blok et al., 2021). Additionally, breast sizes smaller than those in cisgender women have been reported (Asscheman & Gooren, 1992; Kanhai et al., 1999). In one study, breast development with estrogen plus CPA was also poor in late-adolescent transfeminine people (Tack et al., 2017). However, in this particular study, the estrogen dose used was likely too low and resulted in inadequate estradiol levels, as noted by the authors themselves, and this is a potential confounding factor in their findings (Tack et al., 2017). In any case, breast growth with estrogen plus CPA in transfeminine people would seem to consistently be poor. In contrast to the regimen of estrogen and CPA, breast development with other hormone therapy regimens, for instance estrogen with non-progestogenic antiandrogens like spironolactone, bicalutamide, and GnRH modulators, has not been nearly as well-studied in comparison, and hence comparisons of outcomes between regimens is difficult.
In one of the highest quality studies of estrogen and CPA and breast development in adult transfeminine people, breast volume measured with 3D body scanning (Vectra XT) was approximately mean 100 mL (95% CI ~75–125 mL; range up to ~750 mL), equating to less than an A cup size on average, after 3 years of hormone therapy with estrogen and CPA in 69 transfeminine people (de Blok et al., 2021 [Figure]). In this study, breast changes over time had clearly plateaued, suggesting that breast development was either complete or was nearly so (de Blok et al., 2021 [Figure]). Although most of the transfeminine people in this study had less than an A cup breast size (71%), a minority had cup sizes ranging from an A cup (9%), B cup (16%), C cup (3%), to E cup (1%) (de Blok et al., 2021 [Figure]). For comparison, a study of normative data on breast volumes in cisgender women, using a different 3D body scanning device (Artec Eva 3D), found breast volumes of median ~515 mL and mean ~650 mL (IQR ~310–850 mL; range ~50–3,100 mL) in 378 cisgender women (Coltman, Steele, & McGhee, 2017). As such, adult transfeminine people treated with estrogen and CPA would appear to have substantially smaller breasts than cisgender women. However, it must be emphasized that the preceding data come from separate clinical studies and hence are not directly comparative. It is noteworthy in this regard that breast volumes can vary considerably between different studies even using similar measurement methods (e.g., magnetic resonance imaging) (Sindi et al., 2019 [Table]). Hence, there is a need for studies directly comparing breast volumes in transfeminine people to those in cisgender women using the same measurement method in order to comparatively evaluate breast development.
Regardless of the preceding, transfeminine people could simply have poor breast development in general without this necessarily being related to CPA or progestogenic exposure. Indeed, a more recent study in transfeminine people who underwent pubertal suppression in adolescence, presumably with GnRH agonists and then estrogen therapy, found similarly poor breast development as has been reported in adults (Boogers et al., 2022; c.f. de Blok et al., 2021). This study used breast volume via 3D body scanning to measure breast development and found a mean breast volume of 114 mL (IQR 58–203 mL), equating to less than an A cup size, after 4.2 years of hormone therapy (Boogers et al., 2022). It was notably conducted by the same group of researchers who did the earlier higher-quality study in adult transfeminine people, and hence likely used the same 3D scanning method (de Blok et al., 2021).
No directly comparative studies of breast development with CPA versus other antiandrogens in transfeminine people are currently available. Hence, it’s not fully known whether the findings are specific to CPA or also generalize to other antiandrogens that are not also strongly progestogenic. The RCT of estradiol and spironolactone versus estradiol and CPA in transfeminine people by Ada Cheung and colleagues underway in Australia may provide more insight on this issue, as spironolactone is only a weakly or clinically non-progestogenic antiandrogen (Aly, 2018b; Wiki; update: see below).
Additional Considerations for Progestogen Therapy and Breast Development in Transfeminine People
Anecdotes About Progestogens and Breast Development
Many transfeminine people who have taken progestogens as part of hormone therapy have anedotally reported that the progestogens improved their breast development. At the same time, many other transfeminine people have anecdotally reported no benefit of progestogens to breast development. It must be cautioned in general that anecdotal reports are unreliable and represent a very low form of medical evidence. This is because subjective observations and attributions are often erroneous. Perceptions can be faulty and inaccurate, especially with slowly developing physical changes, and true physical changes can be due to coincidence and unrelated confounding factors rather than due to a person’s causal attributions. A couple notable examples of potential confounding factors with regard to progestogens and breast development include: (1) continued breast development from estrogen acting on its own; and (2) temporary breast enlargement due to local fluid retention, increased blood flow, and reversible lobuloalveolar growth caused by progestogens. Such factors have the potential to mislead, and may contribute significantly to anecdotal reports of enhanced breast development with progestogens in transfeminine people. Clinical studies that are well-designed, controlled, and employ reliable objective measures, with long-term follow-up and eventual discontinuation of the progestogen to control for reversible effects, are needed to properly evaluate the effects of progestogens on breast development.
Therapeutic Limitations of Oral Progesterone
Oral progesterone produces very low progesterone levels and has only weak progestogenic effects even at high doses (Aly, 2018a; Wiki). These low progesterone levels are likely to be inadequate in terms of desired physiological progestogenic effects, for instance in the breasts. Oral progesterone also uniquely has potent neurosteroid actions via active metabolites like allopregnanolone, which can result in prominent side effects such as alcohol-like central nervous system inhibition as well as mood swings (Aly, 2018b; Wiki; Wiki). These neurosteroid effects are dose-dependent and are more severe at high doses. Non-oral progesterone forms like rectal or injectable progesterone or progestins, which do not have the preceding problems, can be used instead to avoid such concerns (Aly, 2018a; Aly, 2018b).
Tolerability and Safety Considerations for Progestogens
Progestogens have a variety of tolerability issues and safety risks (Aly, 2018b). Examples of such risks variously include adverse mood changes, breast cancer, blood clots, cardiovascular complications, benignbrain tumors including prolactinomas and meningiomas, and off-target actions with undesirable effects (e.g., androgenic or glucocorticoid activity), among others (Aly, 2018b). CPA at high doses also uniquely has a significant risk of serious liver toxicity (Aly, 2018b). The risks of progestogens vary depending on the specific progestogen and dosage, but all progestogens, including even bioidentical progesterone, have significant known risks. The risks of progestogens, along with lack of evidence of beneficial effects in terms of feminization, well-being, and health, are why there are concerns about and hesitations on their use in transfeminine people (Aly, 2018b). However, cisgender women naturally have progesterone in their bodies, and the absolute risks of progestogens are low (Aly, 2018b). The risks of progestogens can be minimized by use for a limited duration of time (e.g., a few years), by using the lowest dosages expected to be effective in terms of desired effects, and by selection of progestogens with more favorable pharmacological profiles (Aly, 2018a; Aly, 2018b).
Updates
Update 1: Angus et al. (2023–2024)
It was previously reported in this article that an RCT assessing breast development with estradiol plus spironolactone versus estradiol plus CPA in transfeminine people was being conducted by Ada Cheung and colleagues. This study could provide more insight into breast development with progestogens, as CPA is a very potent progestogen whereas spironolactone is not meaningfully progestogenic. Cheung and colleagues’ study, led by Lachlan Angus, has now been published in the form of the following two conference abstracts, with a journal article also currently in the process of being published:
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (November 2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (November 2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
The study assessed estradiol plus spironolactone 100 mg/day versus estradiol plus CPA 12.5 mg/day in 55 transfeminine people, with 27 in the spironolactone group and 28 in the CPA group. Hormone therapy duration, at least at this follow-up point in the study, was 6 months. The measures of breast development included breast–chest difference (primary) and estimated breast volume (secondary).
Breast development, measured by breast–chest difference (mean ± SD), was 8.3 ± 2.7 cm with spironolactone and 9.2 ± 3.0 cm with CPA, with the differences between groups not statistically significant (p = 0.27). In addition, breast development, measured by estimated breast volume (mean ± SD), was 158 ± 112 mL with spironolactone and 190 ± 159 mL with CPA, with the differences between groups not statistically significant (p = 0.39). There was variability between individuals in estimated breast volume, with breast volume measurements ranging from 20 to 788 mL. Besides breast growth, the researchers found that CPA also resulted in a greater increase in body fat percentage and gynoid fat compared to spironolactone. Estradiol levels were comparable between antiandrogen groups, whereas total testosterone levels were (mean ± SD) 4.29 ± 5.44 nmol/L (124 ± 157 ng/dL) with spironolactone and 1.48 ± 3.45 nmol/L (43 ± 99 ng/dL) with CPA, a difference that was statistically significant (p = 0.04).
The researchers concluded that there was no difference in breast development with spironolactone versus CPA in their study and that antiandrogen choice should be individualized based on patient and clinician preference as well as consideration of associated side effects. Moreover, they concluded that further research is needed to optimize breast development in transfeminine people.
Angus, L., Mikolajczyk, M., Cheung, A., Zajac, J., Antoszewski, B., & Kasielska-Trojan, A. (2022). Estimation of breast volume in transgender women using 2D photography: validation of the BreastIdea Volume Estimator in men and transgender women. ESA/SRB/APEG/NZSE ASM 2022, November 13-16, Christchurch, Abstracts and Programme, 127–127 (abstract no. 279). [URL] [PDF] [Full Abstract Book]
In studies by the developers of the BreastIdea Volume Estimator, they reported breast volumes measured with the tool in cisgender women. These estimated breast volumes can provide comparison to the breast-volume findings in transfeminine people by Cheung and Angus and colleagues. The developers of the BreastIdea Volume Estimator reported that breast volume (mean ± SD) in cisgender women with normal breasts (n=30) was 283 ± 144 mL and in cisgender women with macromastia or gigantomastia (n=35) was 888 ± 277 mL (Kasielska-Trojan, Zawadzki, & Antoszewski, 2022). In another study, they reported that breast volume (mean ± SD) in cisgender women was 272 ± 150 mL, with a range of 99 to 694 mL (Kasielska-Trojan, Mikołajczyk, & Antoszewski, 2020).
Although the BreastIdea Volume Estimator is an interesting and promising tool for quantifying breast development, it has notable limitations, such as its resolution and accuracy being much less than that with 3D scanners like the Artec Eva and Vectra XT (Mikołajczyk, Kasielska-Trojan, & Antoszewski, 2019). Vectra and Artec 3D scanners have been and are being employed to measure breast development with hormone therapy in other studies in transfeminine people (de Blok et al., 2021; Boogers et al., 2022; Dijkman et al., 2023a; Dijkman et al., 2023b; Lopez et al., 2023). The accuracy limitations of the BreastIdea Volume Estimator may explain why the breast volume findings with it in transfeminine people and cisgender women were different from those seen in other studies that employed more advanced 3D scanning methods. Aside from the breast volume measurement, breast–chest difference also has limitations as a measure of breast development in transfeminine people, for instance failing to identify continued breast growth that can be detected with breast volume measurement (de Blok et al., 2021).
Besides the employed measurement methods for breast development, limitations of Lachlan Angus and colleagues’ RCT of breast development with spironolactone and CPA in transfeminine people include its limited duration of follow-up of only 6 months, the fact that testosterone levels were non-equivalent between the spironolactone and CPA groups, and its limited sample size. The incompletely suppressed testosterone levels with spironolactone are notable as androgens oppose estrogen-mediated breast development and could have reduced breast development in the spironolactone group. The limited sample size of the study was responsible for the numeric difference in breast measurements between antiandrogen groups not being statistically significant. In any case, Angus and colleagues’ findings are suggestive that CPA, which is highly progestogenic, neither enhances nor stunts breast development, at least relative to non-progestogenic spironolactone for up to 6 months of hormone therapy. It seems likely that the RCT will continue to longer follow-up times and durations of hormone therapy in the future.
In January 2025, the full paper for the study was published:
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery JCG, Zajac, J. D., & Cheung, A. S. (2025). Effect of Spironolactone and Cyproterone Acetate on Breast Growth in Transgender People: A Randomized Clinical Trial. The Journal of Clinical Endocrinology and Metabolism, 110(6), e1874–e1884. [DOI:10.1210/clinem/dgae650]
Update 2: Flamant, Vervalcke, & T’Sjoen (2023) and Yang et al. (2024)
The following two recent studies provide additional information on the topic of breast development with progestogen exposure—specifically with CPA—in transfeminine people:
Flamant, T., Vervalcke, J., & T’Sjoen, G. (November 2023). Dose Reduction of Cyproterone Acetate in Trans Women and the Effect on Patient-reported Outcomes: Results from the ENIGI Study. Endocrine Abstracts, 97 [Belgian Endocrine Society 2023], 5–5 (abstract no. 007). [URL] [PDF]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
In the first study, Flamant, Vervalcke, & T’Sjoen (2023), clinical outcomes in transfeminine people at the University of Ghent, Belgium clinic were compared in 72 people taking CPA at low doses (10–12.5 mg/day) or high doses (25–50 mg/day). Testosterone suppression was equivalent between the two dose groups. Breast development satisfaction, measured with the Body Image Scale, was not significantly different with low-dose CPA versus high-dose CPA following 1 year of hormone therapy (p = 0.078). However, the p-value indicates that there was almost a statistically significant difference between groups, though it was not stated which group was numerically higher in terms of satisfaction. In any case, the researchers stated that breast development satisfaction was “non-inferior” with low-dose CPA compared to high-dose CPA, which seems suggestive that satisfaction may have been higher in the high-dose CPA group. These findings suggest that higher doses of CPA may not stunt breast development relative to doses of CPA that are lower, although still quite high in terms of progestogenic activity.
In the second study, Yang et al. (2024), clinical outcomes in transfeminine people at the Peking University Third Hospital in China with estradiol plus spironolactone (n=43) versus estradiol plus CPA (n=53) were retrospectively compared. Testosterone levels were much higher in the spironolactone group relative to the CPA group (374 ng/dL [13.0 nmol/L] vs. 20 ng/dL [0.7 nmol/L]; p < 0.001) and duration of hormone therapy was shorter in the spironolactone group than in the CPA group (median 12 months vs. 18 months). Breast development satisfaction, measured with a visual analogue scale (VAS), was median 6.0 (IQR 4.0–7.0) with spironolactone and 6.0 (IQR 4.0–7.0) with CPA, and was not statistically different. On the other hand, the CPA group outperformed the spironolactone group in terms of several other VAS-based clinical-outcome measures, including figure feminization, testicular atrophy, decreased penile erections, and in terms of a composite overall satifaction score. These findings suggest, as with the RCT by Lachlan Angus and colleagues, that spironolactone and CPA result in similar breast development in transfeminine people despite differences in testosterone levels and other clinical outcomes.
In 2023, a study protocol for a randomized controlled trial of oral progesterone and breast development in transfeminine people was published (Dijkman et al., 2023). The protocol was published by Benthe Dijkman and colleagues at the Vrije Universiteit University Medical Center (VUMC) in Amsterdam, the Netherlands. The trial would be the first prospective randomized controlled trial of progesterone and breast development in transfeminine people.
In this non-blinded non-placebo-controlled randomized trial, 90 transfeminine people would be randomized into 6 study arms with 15 people each. The transfeminine people would be individuals who had been on hormone therapy for at least one year and had undergone vaginoplasty or orchiectomy. Those who were currently or previously taking a progestogen, including CPA, would be excluded. The study’s treatment arms or groups would include the following:
Standard-dose estradiol alone (control group)
Double-dose estradiol alone
Standard-dose estradiol plus progesterone 200 mg/day
Double-dose estradiol plus progesterone 200 mg/day
Standard-dose estradiol plus progesterone 400 mg/day
Double-dose estradiol plus progesterone 400 mg/day
The estradiol therapy was specifically oral estradol valerate, oral estradiol hemihydrate, transdermal estradiol patches, transdermal estradiol gel, or transdermal estradiol spray, at doses resulting in estradiol levels of 200 to 400 pmol/L (54–109 pg/mL) in the standard-dose group and 400 to 800 pmol/L (109–218 pg/mL) in the double-dose group. The progesterone therapy was specifically oral micronized progesterone (Utrogestan). It was noted that in order to maximize adherence, progesterone would be prescribed for limited 1 to 3 month intervals, but no further details on this were provided.
The duration of the study would be 3 years and initial phase would be 12 months, with breast development and/or hormone levels measured at baseline, 3 months, 6 months, and 12 months of treatment. Estradiol levels would be measured with mass spectrometry, whereas progesterone levels would be measured with immunoassays. Breast development would be measured with 3D scanning (Artec Leo 3D) and breast–chest difference. Bra cup size would additionally be calculated from these measures. In the protocol, it was stated that an average breast volume increase of 30%, which was said to correspond to one bra cup size increase, would be considered a clinically relevant outcome. There would also be a number of secondary outcomes, including side effects/safety, satisfaction, mood, sleep, and sexual pleasure. It was noted that the study may serve as a pilot project for a larger future study of progesterone and breast development initiated at the start of hormone therapy prior to surgery.
In August 2025, an EPATH conference abstract with briefly described results of the study was published online in advance of the 6th EPATH conference to be held in September 2025 (Dreijerink et al., 2025):
Dreijerink, K., den Heijer, M., Geels, R. (2025). Increased breast volume due to addition of progesterone and increasing the estradiol dose in feminizing gender-affirming hormone therapy. EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
It was reported that mean breast volume, relative to standard-dose estradiol alone, changed as follows:
Treatment group
n
Breast volume change
E2 double-dose alone
15
+6% (95% CI, –13 to 24)
E2 standard-dose plus P4 200 mg/day
15
+13% (95% CI, –7 to 33)
E2 double-dose plus P4 200 mg/day
15
+37% (95% CI, 18 to 57)
E2 standard-dose plus P4 400 mg/day
15
+20% (95% CI, 0 to 40)
E2 double-dose plus P4 400 mg/day
15
+27% (95% CI, 8 to 47)
The authors concluded that progesterone and higher estradiol dose increased breast volume in transfeminine people. The results of significance tests for breast volume between individual treatment groups or relative to controls were not provided in the abstract. Subjective satisfaction with breast growth and size was said to be improved in all treatment groups relative to the control group (p < 0.05). Aside from breast size changes, side effects with oral progesterone included tiredness (44%), breast/nipple tenderness (27%), and mood changes (22%). There were no treatment-related serious adverse events. No other results or data were provided in the abstract. The full results of the this trial by Dreijerink and colleagues will be published in a journal article at some point in the future. It was concluded that oral progesterone was safe but did cause some side effects. Moreover, the study concluded that their results supported a future role of progesterone in transfeminine hormone therapy. However, it was noted that the long-term effects of progesterone in transfeminine people still need to be studied.
The findings of Dreijerink and colleagues are the highest-quality data on progesterone and breast changes in transfeminine people that are currently available. Their findings suggest that addition of oral progesterone to estradiol increases breast volume and that higher-dose estradiol levels synergize with progesterone to increase breast volume. There was a 13 to 37% increase in volume with oral progesterone depending on the estradiol and progesterone doses. It is important to note however that, as extensively reviewed in the present article, higher estradiol levels and progesterone are associated with increased breast volume due to effects like increased local fluid retention, increased blood flow, and/or temporary growth, but these effects are reversible and regress following withdrawal of the hormonal exposure. Unfortunately, Dreijerink and colleagues do not appear to have included a discontinuation phase to assess whether the breast volume increases observed in the trial were reversible or not. As such, while higher-dose estradiol and oral progesterone can significantly increase breast volume during treatment in transfeminine people, it is still not possible to draw conclusions about whether these interventions actually improve breast development—that is, lasting/permanent breast growth. Only future research that includes discontinuation phases will be able to answer this question.
Other limitations of Dreijerink and colleagues’ study include the use of oral progesterone, the employment of immunoassays to measure progesterone levels, the relatively small sample sizes of the individual treatment subgroups in the study and consequent risk of statistical error, and the patient population being transfeminine people who were post-vaginoplasty or -orchiectomy and hence had already been on hormone therapy for a long period of time (at least 1 year but likely longer on average, such as 2 or 3 years). Oral progesterone is known to achieve relatively low progesterone levels and may be inferior in general effectiveness to non-oral progesterone and progestins (Aly, 2019). Immunoassays are known to substantially overestimate and hence provide a misleading idea of progesterone levels, whereas mass spectrometry-based assays provide accurate progesterone levels (Aly, 2019). Individuals who have been on hormone therapy for many years may have near- or fully-complete breast development and hence less potential for enhancement of true breast development. In any case, caveats aside, Dreijerink and colleagues’ findings are relatively high-quality data, and demonstrate with decent confidence that oral progesterone can, at least exposure-dependently and in conjunction with sufficiently high estradiol levels, provide an increase in breast size in transfeminine people.
References
Abduljalil, K., Furness, P., Johnson, T. N., Rostami-Hodjegan, A., & Soltani, H. (2012). Anatomical, Physiological and Metabolic Changes with Gestational Age during Normal Pregnancy. Clinical Pharmacokinetics, 51(6), 365–396. [DOI:10.2165/11597440-000000000-00000]
Adams, R. D., Kliman, B., Federman, D. D., Ulfelder, H. S., & Holmes, L. B. (1970). Syndromes of Testicular Feminization. Clinical Pediatrics, 9(3), 165–178. [DOI:10.1177/000992287000900312]
Alekseev, N. P. (2021). Origin and Development of the Mammary Glands. In Alekseev, N. P. Physiology of Human Female Lactation (pp. 11–66). Cham: Springer. [DOI:10.1007/978-3-030-66364-3_2]
Alex, A., Bhandary, E., & McGuire, K. P. (2020). Anatomy and Physiology of the Breast during Pregnancy and Lactation. In Alipour, S., & Omranipour, R. (Eds.). Diseases of the Breast during Pregnancy and Lactation (Advances in Experimental Medicine and Biology, Volume 1252) (pp. 3–7). Cham: Springer. [DOI:10.1007/978-3-030-41596-9]
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2018). Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects. Transfeminine Science. [URL]
Aly. (2019). Literature on Early Progestogen Exposure and Breast Development. Transfeminine Science. [URL]
Aly. (2019). Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Breast Cancer Risk with Hormone Therapy in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
Aly. (2020). Hormone Levels During Normal Puberty in Cisgender Girls. Transfeminine Science. [URL]
Aly. (2020). The Possible Role of a Second X Chromosome in Breast Cancer Risk. Transfeminine Science. [URL]
Anderson, W. A. (1962). Experimental stimulation of breast development in the teen-age female. The Journal of the Medical Society of New Jersey, 59(10), 541–543. [Google Scholar] [PubMed] [PDF]
Andler, W., & Zachmann, M. (1979). Spontaneous breast development in an adolescent girl with testicular feminization after castration in early childhood. The Journal of Pediatrics, 94(2), 304–305. [DOI:10.1016/s0022-3476(79)80852-5]
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Angus, L., Mikolajczyk, M., Cheung, A., Zajac, J., Antoszewski, B., & Kasielska-Trojan, A. (2022). Estimation of breast volume in transgender women using 2D photography: validation of the BreastIdea Volume Estimator in men and transgender women. ESA/SRB/APEG/NZSE ASM 2022, November 13-16, Christchurch, Abstracts and Programme, 127–127 (abstract no. 279). [URL] [PDF] [Full Abstract Book]
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery JCG, Zajac, J. D., & Cheung, A. S. (2025). Effect of Spironolactone and Cyproterone Acetate on Breast Growth in Transgender People: A Randomized Clinical Trial. The Journal of Clinical Endocrinology and Metabolism, 110(6), e1874–e1884. [DOI:10.1210/clinem/dgae650]
Apter, D. (1980). Serum steroids and pituitary hormones in female puberty: a partly longitudinal study. Clinical Endocrinology, 12(2), 107–120. [DOI:10.1111/j.1365-2265.1980.tb02125.x]
Apter, D., Räisänen, I., Ylöstalo, P., & Vihko, R. (1987). Follicular growth in relation to serum hormonal patterns in adolescent compared with adult menstrual cycles. Fertility and Sterility, 47(1), 82–88. [DOI:10.1016/s0015-0282(16)49940-1]
Aritaki, S., Miyazawa, H., Ogihara, M., Ushio, M., & Izumizawa, A. (1992). An Endocrinological Study of Persistent Pubertal Macromastia. The Tohoku Journal of Experimental Medicine, 167(3), 189–196. [DOI:10.1620/tjem.167.189]
Arya, S., Barnabas, R., Lila, A. R., Sarathi, V., Memon, S. S., Bhandare, V. V., Thakkar, K., Patil, V., Shah, N. S., Kunwar, A., & Bandgar, T. (2021). Clinical, Hormonal, Genetic, and Molecular Characteristics in Androgen Insensitivity Syndrome in an Asian Indian Cohort from a Single Centre in Western India. Sexual Development, 15(4), 253–261. [DOI:10.1159/000517763]
Asscheman, H., & Gooren, L. J. (1992). Hormone Treatment in Transsexuals. In Bocking, W. O., Coleman, E. (Eds). Gender Dysphoria: Interdisciplinary Approaches in Clinical Management (pp. 39–54). Binghamton: Haworth Press. / Journal of Psychology & Human Sexuality, 5(4), 39–54. [Google Scholar] [Google Books] [DOI:10.1300/J056v05n04_03]
Athanasoulia, A., Auer, M., Riepe, F., & Stalla, G. (2013). Rare Missense P450c17 (CYP17A1) Mutation in Exon 1 as a Cause of 46,XY Disorder of Sexual Development: Implications of Breast Tissue ‘Unresponsiveness’ despite Adequate Estradiol Substitution. Sexual Development, 7(4), 212–215. [DOI:10.1159/000348301]
Atwood, C., Hovey, R., Glover, J., Chepko, G., Ginsburg, E., Robison, W., & Vonderhaar, B. (2000). Progesterone induces side-branching of the ductal epithelium in the mammary glands of peripubertal mice. Journal of Endocrinology, 167(1), 39–52. [DOI:10.1677/joe.0.1670039]
Aupperlee, M. D., Leipprandt, J. R., Bennett, J. M., Schwartz, R. C., & Haslam, S. Z. (2013). Amphiregulin mediates progesterone-induced mammary ductal development during puberty. Breast Cancer Research, 15(3), R44. [DOI:10.1186/bcr3431]
Bahr, C., Ewald, J., Dragovich, R., & Gothard, M. D. (2024). Effects of progesterone on gender affirmation outcomes as part of feminizing hormone therapy. Journal of the American Pharmacists Association, 64(1), 268–272. [DOI:10.1016/j.japh.2023.08.001]
Baird, D., Hytten, F. E., & Thomson, A. M. (1958). Age and Human Reproduction. BJOG, 65(6), 865–876. [DOI:10.1111/j.1471-0528.1958.tb08582.x]
Baker, S. B., Burkey, B. A., Thornton, P., & LaRossa, D. (2001). Juvenile Gigantomastia: Presentation of Four Cases and Review of the Literature. Annals of Plastic Surgery, 46(5), 517–526. [DOI:10.1097/00000637-200105000-00011]
Bames, H. O. (1948). Reduction of massive breast hypertrophy. Plastic and Reconstructive Surgery, 3(5), 560–569. [DOI:10.1097/00006534-194809000-00006]
Barbieri, R. L. (2019). Breast. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 248–255.e3). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/B978-0-323-47912-7.00010-X]
Baron, J. (1993). Zespół braku wrazliwości na androgeny. Studium kliniczne i endokrynologiczne 40 osobników [Androgen insensitivity syndrome. Clinical and endocrinologic study of 40 cases]. Endokrynologia Polska, 44(2), 175–186. [Google Scholar] [PubMed]
Baron, J. (1994). Klasyczne i niepełne zespoły braku wrazliwości na androgeny [Classical and incomplete androgen insensitivity syndromes]. Ginekologia Polska, 65(7), 377–386. [Google Scholar] [PubMed]
Baron, J. (1994). Niepełny lub cześciowy zespół braku wrazliwości na androgeny [Partial androgen insensitivity syndrome]. Ginekologia Polska, 65(6), 319–325. [Google Scholar] [PubMed]
Barrat, J., de Lignières, B., Marpeau, L., Larue, L., Fournier, S., Nahoul, K., Linares, G., Giorgi, H., & Contesso, G. (1990). Effet in vivo de l’administration locale de progestérone sur l’activité mitotique des galactophores humains. Résultat d’une étude pilote. [The in vivo effect of the local administration of progesterone on the mitotic activity of human ductal breast tissue. Results of a pilot study.] Journal de Gynecologie, Obstetrique et Biologie de la Reproduction, 19(3), 269–274. [Google Scholar 1] [Google Scholar 2] [PubMed]
Barrett, J. (2009). The clinical risks associated with the diagnosis and management of disorders of gender identity. Clinical Risk, 15(4), 131–134. [DOI:10.1258/cr.2008.080069]
Bässler, R. (1970). The Morphology of Hormone Induced Structural Changes in the Female Breast. In Altmann, H.-W., et al. (Eds.). Current Topics in Pathology: Ergebnisse der Pathology, Volume 53 (pp. 1–89). Heidelberg: Springer Berlin. [DOI:10.1007/978-3-662-30514-0_1]
Basson, R., & Prior, J. C. (1998). Hormonal Therapy of Gender Dysphoria: The Male-to-Female Transsexual. In Denny, D. (Ed.). Concepts in Transgender Identity (Garland Gay and Lesbian Studies, Volume 11) (pp. 277–296). New York: Garland Publishing Inc. [Google Scholar] [Google Books] [DOI:10.4324/9780203775134-19] [PDF]
Bauman, A., Novello, L., & Kreitzer, P. (2016). Endocrine Disorders and Delayed Puberty. In Appelbaum, H. (Ed.). Abnormal Female Puberty: A Clinical Casebook (pp. 87–107). Cham: Springer. [DOI:10.1007/978-3-319-27225-2_5]
Bayer, C. M., Bani, M. R., Schneider, M., Dammer, U., Raabe, E., Haeberle, L., Faschingbauer, F., Schneeberger, S., Renner, S. P., Fischer, D., Schulz-Wendtland, R., Fasching, P. A., Beckmann, M. W., & Jud, S. M. (2014). Assessment of breast volume changes during human pregnancy using a three-dimensional surface assessment technique in the prospective CGATE study. European Journal of Cancer Prevention, 23(3), 151–157. [DOI:10.1097/cej.0b013e3283651ccb]
Bazarra-Castro, M. A. (2009). Etiological aspects, therapy regimes, side effects and treatment satisfaction of transsexual patients. (Doctoral dissertation, Ludwig Maximilian University of Munich.) [DOI:10.5282/edoc.9984] [URN:urn:nbn:de:bvb:19-99840] [PDF]
Beck, P. (1972). Lactogenic Activity of Human Chorionic Somatomammotropin in Rhesus Monkeys. Experimental Biology and Medicine, 140(1), 183–187. [DOI:10.3181/00379727-140-36422]
Begley, D. J., Firth, J. A., & Hoult, J. R. (1980). The Breast and Lactation. In Begley, D. J., Firth, J. A., & Hoult, J. R. Human Reproduction and Developmental Biology (pp. 204–219). London: Macmillan Education UK. [DOI:10.1007/978-1-349-16260-4_14]
Bellwether, C. J. (2020). Why Should a Transfeminine Person Consider Progesterone? Google Docs. [URL]
Benjamin, H. (1966). Nonsurgical Management of Transsexualism. In Benjamin, H. The Transsexual Phenomenon (pp. 86–99). New York: Julian Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Benjamin, H. (1967). Transvestism and Transsexualism in the male and female. Journal of Sex Research, 3(2), 107–127. [DOI:10.1080/00224496709550519]
Berliere, M., Coche, M., Lacroix, C., Riggi, J., Coyette, M., Coulie, J., Galant, C., Fellah, L., Leconte, I., Maiter, D., Duhoux, F. P., & François, A. (2022). Effects of Hormones on Breast Development and Breast Cancer Risk in Transgender Women. Cancers, 15(1), 245. [DOI:10.3390/cancers15010245]
Berryhill, G. E., Trott, J. F., & Hovey, R. C. (2016). Mammary gland development—It’s not just about estrogen. Journal of Dairy Science, 99(1), 875–883. [DOI:10.3168/jds.2015-10105]
Bertelloni, S., Dati, E., Baroncelli, G. I., & Hiort, O. (2011). Hormonal Management of Complete Androgen Insensitivity Syndrome from Adolescence Onward. Hormone Research in Paediatrics, 76(6), 428–433. [DOI:10.1159/000334162]
Bethea, C. L., Kohama, S. G., & Pecins-Thompson, M. (1997). Pituitary and Brain Actions of Estrogen and Progesterone in the Regulation of Primate Prolactin Secretion. In Pavlik, E. J. (Ed.). Estrogens, Progestins, and Their Antagonists: Functions and Mechanisms of Action (pp. 3–46). Boston: Birkhäuser. [DOI:10.1007/978-1-4612-2004-6_1]
Bevan, D. J. (2012). Progesterone for Breast Development? / Should Male-to-Female Transsexuals Take Progesterone as part of Hormone Therapy (HT) for Better Breast Development? Biopsychology of TSTG / Transgender Forum. [URL 1] [URL 2]
Bevan, D. J. (2019). Grow Your Own : Breast Development Update. Transgender Forum. [URL]
Beyer, C., Cruz, M. L., & Martinez-Manautou, J. (1970). Effect of Chlormadinone Acetate on Mammary Development and Lactation in the Rabbit. Endocrinology, 86(5), 1172–1174. [DOI:10.1210/endo-86-5-1172]
Biswas, S. K., Banerjee, S., Baker, G. W., Kuo, C., & Chowdhury, I. (2022). The Mammary Gland: Basic Structure and Molecular Signaling during Development. International Journal of Molecular Sciences, 23(7), 3883. [DOI:10.3390/ijms23073883]
Bland, K. I., Copeland, E. M., & Klimberg, V. S. (2018). Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 20–36.e2). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00002-7]
Bland, K. I., Harrison Howard, J., & Romrell, L. J. (2009). Congenital and Acquired Disturbances of Breast Development and Growth. In Bland, K. I., & Copeland, E. M. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 4th Edition (pp. 189–207). Philadelphia: Saunders/Elsevier. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Bondy, C. A., & Turner Syndrome Consensus Study Group. (2007). Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. The Journal of Clinical Endocrinology & Metabolism, 92(1), 10–25. [DOI:10.1210/jc.2006-1374]
Boogers, L., Infirri, S. S., Bouchareb, A., de Blok, C., Liberton, N., van Trotsenburg, P., Dreijerink, K., den Heijer, M., Wiepjes, C., & Hannema, S. (2022). The effect of timing of puberty suppression on breast development in trans girls; a cross-sectional study. Hormone Research in Paediatrics, 95(Suppl 2) [60th Annual Meeting of the European Society for Paediatric Endocrinology (ESPE), Rome, Italy, September 15–17, 2022], 390–391 (abstract no. P1-379). [Google Scholar] [DOI:10.1159/000525606] [URL] [PDF 1] [PDF 2]
Boyce, S. W., Hoffman, P. G., & Mathes, S. J. (1984). Recurrent Macromastia after Subcutaneous Mastectomy. Annals of Plastic Surgery, 13(6), 511–518. [DOI:10.1097/00000637-198412000-00008]
Bruck, H. G., & Müller, G. (1967). Zur Hormontherapie der Hypoplastischen Weiblichen Brust. [On Hormonal Therapy of the Hypoplastic Female Breast.] Ästhetische Medizin [Ästhetische Medizin : Kongreß-Organ der Deutschen Gesellschaft für die Ästhetische Medizin und ihre Grenzgebiete], 16(12), 365–366. [ISSN:0400-6755] [Google Scholar] [PubMed] [WorldCat] [PDF] [Translation]
Bryant, R., Underwood, A., Robinson, A., Stephenson, T., & Underwood, J. (1998). Determination of breast tissue composition for improved accuracy in estimating radiation doses and risks in mammographic screening. The Breast, 7(2), 95–98. [DOI:10.1016/s0960-9776(98)90064-9]
Buhimschi, C. S. (2004). Endocrinology of lactation. Obstetrics and Gynecology Clinics of North America, 31(4), 963–979. [DOI:10.1016/j.ogc.2004.08.002]
Camilletti, M. A., Abeledo-Machado, A., Faraoni, E. Y., Thomas, P., & Díaz-Torga, G. (2019). New insights into progesterone actions on prolactin secretion and prolactinoma development. Steroids, 152, 108496. [DOI:10.1016/j.steroids.2019.108496]
Çamtosun, E., Şıklar, Z., Ceylaner, S., Kocaay, P., & Berberoğlu, M. (2017). Delayed Diagnosis of a 17-Hydroxylase/17,20-Lyase Deficient Patient Presenting as a 46,XY Female: A Low Normal Potassium Level Can Be an Alerting Diagnostic Sign. Journal of Clinical Research in Pediatric Endocrinology, 9(2), 163–167. [DOI:10.4274/jcrpe.3839]
Capraro, V. J., & Dewhurst, C. J. (1975). Breast Disorders in Childhood and Adolescence. Clinical Obstetrics and Gynecology, 18(2), 25–50. [DOI:10.1097/00003081-197506000-00003]
Carlson, L. J., & Shaw, N. D. (2019). Development of Ovulatory Menstrual Cycles in Adolescent Girls. Journal of Pediatric and Adolescent Gynecology, 32(3), 249–253. [DOI:10.1016/j.jpag.2019.02.119]
Caro, T. M. (1987). Human breasts: Unsupported hypotheses reviewed. Human Evolution, 2(3), 271–282. [DOI:10.1007/bf03016112]
Ceriani, R. L. (1974). Hormones and Other Factors Controlling Growth in the Mammary Gland: A Review. Journal of Investigative Dermatology, 63(1), 93–108. [DOI:10.1111/1523-1747.ep12678104]
Cernea, R. (1944). Lokale Hormonbehandlung bei Mammaatrophie und Unterentwicklung. [Local Hormone Treatment in Mammary Atrophy and Underdevelopment.] Medizinische Klinik, 40(11/12), 169–170. [Google Scholar] [PDF] [Translation]
Chang, K., Lee, T. T., Linares-Cruz, G., Fournier, S., & de Ligniéres, B. (1995). Influences of percutaneous administration of estradiol and progesterone on human breast epithelial cell cycle in vivo. Fertility and Sterility, 63(4), 785–791. [DOI:10.1016/s0015-0282(16)57482-2]
Cheikhelard, A., Morel, Y., Thibaud, E., Lortat-Jacob, S., Jaubert, F., Polak, M., & Nihoul-Fekete, C. (2008). Long-Term Followup and Comparison Between Genotype and Phenotype in 29 Cases of Complete Androgen Insensitivity Syndrome. Journal of Urology, 180(4), 1496–1501. [DOI:10.1016/j.juro.2008.06.045]
Ciarloni, L., Mallepell, S., & Brisken, C. (2007). Amphiregulin is an essential mediator of estrogen receptor α function in mammary gland development. Proceedings of the National Academy of Sciences, 104(13), 5455–5460. [DOI:10.1073/pnas.0611647104]
Clendenen, T. V., Kim, S., Moy, L., Wan, L., Rusinek, H., Stanczyk, F. Z., Pike, M. C., & Zeleniuch-Jacquotte, A. (2013). Magnetic Resonance Imaging (MRI) of hormone-induced breast changes in young premenopausal women. Magnetic Resonance Imaging, 31(1), 1–9. [DOI:10.1016/j.mri.2012.06.022]
Cline, J. M., & Wood, C. E. (2006). Hormonal Effects on the Mammary Gland of Postmenopausal Nonhuman Primates. Breast Disease, 24(1), 59–70. [DOI:10.3233/bd-2006-24105]
Cline, J. M., & Wood, C. E. (2008). The Mammary Glands of Macaques. Toxicologic Pathology, 36(7 Suppl), 130S–141S. [DOI:10.1177/0192623308327411]
Cole, R. D., & Hopkins, T. R. (1962). A Biochemical Test of Artificial Mammogenesis and Lactogenesis As Models of the Natural Processes. Endocrinology, 70(3), 375–380. [DOI:10.1210/endo-70-3-375]
Coleman, E., Bockting, W., Botzer, M., Cohen-Kettenis, P., DeCuypere, G., Feldman, J., Fraser, L., Green, J., Knudson, G., Meyer, W. J., Monstrey, S., Adler, R. K., Brown, G. R., Devor, A. H., Ehrbar, R., Ettner, R., Eyler, E., Garofalo, R., Karasic, D. H., Lev, A. I., Mayer, G., Meyer-Bahlburg, H., Hall, B. P., Pfaefflin, F., Rachlin, K., Robinson, B., Schechter, L. S., Tangpricha, V., van Trotsenburg, M., Vitale, A., Winter, S., Whittle, S., Wylie, K. R., & Zucker, K. (2012). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, Version 7. International Journal of Transgenderism, 13(4), 165–232. [DOI:10.1080/15532739.2011.700873] [URL] [PDF]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Coltman, C. E., Steele, J. R., & McGhee, D. E. (2017). Breast volume is affected by body mass index but not age. Ergonomics, 60(11), 1576–1585. [DOI:10.1080/00140139.2017.1330968]
Colvin, C., Devineni, G., & Ashraf, A. P. (2014). Delayed Puberty. In Bandeira, F., Gharib, H., Golbert, A., Griz, L., & Faria, M. (Eds.). Endocrinology and Diabetes (pp. 203–217). New York: Springer. [DOI:10.1007/978-1-4614-8684-8_17]
Cowie, A. T., & Folley, S. J. (1961). The Mammary Gland and Lactation. In Young, W. C. (Ed.). Sex and Internal Secretions, 3rd Edition, Volume I (pp. 590–642). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Cowie, A. T., Forsyth, I. A., & Hart, I. C. (1980). Growth and Development of the Mammary Gland. In Cowie, A. T., Forsyth, I. A., & Hart, I. C. Hormonal Control of Lactation (Monographs on Endocrinology, Volume 15) (pp. 58–145). Berlin/Heidelberg: Springer Berlin Heidelberg. [DOI:10.1007/978-3-642-81389-4_3]
Cox, D. B., Kent, J. C., Casey, T. M., Owens, R. A., & Hartmann, P. E. (1999). Breast Growth and the Urinary Excretion of Lactose During Human Pregnancy and Early Lactation: Endocrine Relationships. Experimental Physiology, 84(2), 421–434. [DOI:10.1111/j.1469-445x.1999.01807.x]
Cox, C. B., Kent, J. C., Owens, R., & Hartmann, P. E. (1994). Mammary morphological and functional changes during pregnancy in women. In Thompson, J. (Ed.). Proceedings of the Twenty-Sixth Annual Conference, Hilton Hotel, Brisbane, 26–28 September, 1994 [The Australian Society for Reproductive Biology Inc., Twenty Sixth Annual Conference, The Hilton Hotel, Brisbane, September 26 - 28 1994, Programme and Miniposters of Papers] (pp. 47–47). Australia: Australian Society for Reproductive Biology. [Google Scholar] [WorldCat] [URL] [PDF]
Coxon, J., & Seal, L. (2018). Hormone management of trans women. Trends in Urology & Men’s Health, 9(6), 10–14. [DOI:10.1002/tre.663]
Cregan, M. D., & Hartmann, P. E. (1999). Computerized Breast Measurement from Conception to Weaning: Clinical Implications. Journal of Human Lactation, 15(2), 89–96. [DOI:10.1177/089033449901500202]
Crowley, W. F., & Pitteloud, N. (2020). Approach to the patient with delayed puberty. UpToDate. [Google Scholar] [URL]
Cruz-Korchin, N., Korchin, L., González-Keelan, C., Climent, C., & Morales, I. (2002). Macromastia. Plastic and Reconstructive Surgery, 109(1), 64–68. [DOI:10.1097/00006534-200201000-00011]
Curtis, R. J. (2009 July 10). The Lowdown on Progesterone. London: The London Gender Clinic. [Google Scholar] [URL] [PDF]
Dallmann, A., Ince, I., Meyer, M., Willmann, S., Eissing, T., & Hempel, G. (2017). Gestation-Specific Changes in the Anatomy and Physiology of Healthy Pregnant Women: An Extended Repository of Model Parameters for Physiologically Based Pharmacokinetic Modeling in Pregnancy. Clinical Pharmacokinetics, 56(11), 1303–1330. [DOI:10.1007/s40262-017-0539-z]
Dancey, A., Khan, M., Dawson, J., & Peart, F. (2008). Gigantomastia – a classification and review of the literature. Journal of Plastic, Reconstructive & Aesthetic Surgery, 61(5), 493–502. [DOI:10.1016/j.bjps.2007.10.041]
Davajan, V., & Kletzky, O. A. (1979). Amenorrhea without Galactorrhea or Hirsutism. In Mishell, D. R., & Davajan, V. (Eds.). Reproductive Endocrinology, Infertility, and Contraception (pp. 219–248). Philadelphia: F. A. Davis Co. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Davis, M. E., Boynton, M. W., Ferguson, J. H., & Rothman, S. (1945). Studies on Pigmentation of Endocrine Origin. The Journal of Clinical Endocrinology & Metabolism, 5(3), 138–146. [DOI:10.1210/jcem-5-3-138]
de Blok, C. J., Dijkman, B. A., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K. M., & den Heijer, M. (2021). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de Lignières, B. (2002). Effects of progestogens on the postmenopausal breast. Climacteric, 5(3), 229–235. [DOI:10.1080/cmt.5.3.229.235]
de Lignières, B., & Mauvais-Jarvis, P. (1981). Hormonal Dependence of Benign Breast Disease, Gynecomastia and Breast Cancer. In Hollman, K. H., Brux, J., & Verley, J. M. (Eds.). New Frontiers in Mammary Pathology (pp. 287–308). Boston: Springer US. [DOI:10.1007/978-1-4757-0019-0_17]
de Vries, A. L., Steensma, T. D., Wagemaar, E. C. F., Doreleijers, T. A., & Cohen-Kettenis, P. T. (2010). Puberty suppression followed by cross-sex hormones and gender reassignment surgery: A prospective follow-up of gender dysphoric adolescents into adulthood. In de Vries, A. L. (Ed.). Gender Dysphoria in Adolescents: Mental Health and Treatment Evaluation (pp. 91–106). (Doctoral thesis, Vrije Universiteit Amsterdam.) [Google Scholar] [URL] [PDF]
Deeb, A., Al Suwaidi, H., Attia, S., & Al Ameri, A. (2015). 17-hydroxylase/17,20-lyase deficiency due to a R96Q mutation causing hypertension and poor breast development. Endocrinology, Diabetes & Metabolism Case Reports, 2015(1), 15-0069. [DOI:10.1530/EDM-15-0069]
Deepinder, F., & Braunstein, G. D. (2012). Drug-induced gynecomastia: an evidence-based review. Expert Opinion on Drug Safety, 11(5), 779–795. [DOI:10.1517/14740338.2012.712109]
Dennerstein, L., Burrows, G. D., Hyman, G. J., & Sharpe, K. (1980). Some clinical effects of oestrogen-progestogen therapy in surgically castrated women. Maturitas, 2(1), 19–28. [DOI:10.1016/0378-5122(80)90056-0]
Dewhurst, C. J. (1971). Sex Chromosome Abnormalities and the Gynaecologist. BJOG, 78(12), 1058–1076. [DOI:10.1111/j.1471-0528.1971.tb00227.x]
Dewhurst, C. J. (1971). The XY female. American Journal of Obstetrics and Gynecology, 109(5) [Transactions of the Eighty-First Annual Meeting of the American Association of Obstetricians and Gynecologists], 675–688. [DOI:10.1016/0002-9378(71)90753-8]
Dewhurst, C. (1972). Amenorrhoea and the Paediatrician. Pediatric Clinics of North America, 19(3), 605–618. [DOI:10.1016/s0031-3955(16)32741-9]
Dewhurst, C. J., & Spence, J. E. (1977). The XY female. British Journal of Hospital Medicine, 17(5), 498, 501–506. [Google Scholar] [PubMed] [PDF]
Dewhurst, J., & Underhill, R. (1979). The Suppression of Facial Hair Growth in Transsexuals Using Cyproterone Acetate. British Journal of Sexual Medicine, 6, 19–23. [Google Scholar] [PDF]
Dewhurst, J. (1981). Breast Disorders in Children and Adolescents. Pediatric Clinics of North America, 28(2), 287–308. [DOI:10.1016/s0031-3955(16)33997-9]
Dewhurst, J. (1982). Breast Hypoplasia. In Bruni, V., Gasparri, F., Dewhurst, J., & Rey-Stocker, I. (Eds.). Pediatric and Adolescent Gynaecology [Proceedings of the IVth International Symposium, Florence, October 5-7, 1978] (pp. 205–212). Rome, Italy: Serono Symposia. [Google Scholar] [Google Books] [WorldCat] [PDF]
Di Lorenzo, G., Autorino, R., Perdonà, S., & De Placido, S. (2005). Management of gynaecomastia in patients with prostate cancer: a systematic review. The Lancet Oncology, 6(12), 972–979. [DOI:10.1016/s1470-2045(05)70464-2]
Dickson, L. M., & Hewer, E. E. (1950). The structure of the breast. In Saner, F. D. (Ed.). The Breast: Structure, Function, Disease (pp. 1–52). Baltimore: William & Wilins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL] [PDF]
Dijkman, B. A., Blok, C. J., Dreijerink, K. M., & den Heijer, M. (2023). Progestin-related breast volume changes in a woman with complete androgen insensitivity syndrome (CAIS). Endocrinology, Diabetes & Metabolism Case Reports, 2023(2), 22-0346. [DOI:10.1530/edm-22-0346]
Dijkman, B. A., Helder, D., Boogers, L. S., Gieles, N. C., van Heesewijk, J. O., Slaa, S. t., Liberton, N. P., Wiepjes, C. M., de Blok, C. J., den Heijer, M., & Dreijerink, K. M. (2023). Addition of progesterone to feminizing gender-affirming hormone therapy in transgender individuals for breast development: a randomized controlled trial. BMC Pharmacology and Toxicology, 24(1), 80. [DOI:10.1186/s40360-023-00724-4]
Dittrich, R., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Mueller, A. (2005). Endocrine Treatment of Male-to-Female Transsexuals Using Gonadotropin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 113(10), 586–592. [DOI:10.1055/s-2005-865900]
Donaldson, M., Kriström, B., Ankarberg-Lindgren, C., Verlinde, S., van Alfen-van der Velden, J., Gawlik, A., van Gelder, M., Sas, T., & (2019). Optimal Pubertal Induction in Girls with Turner Syndrome Using Either Oral or Transdermal Estradiol: A Proposed Modern Strategy. Hormone Research in Paediatrics, 91(3), 153–163. [DOI:10.1159/000500050]
Dorgan, J. F., Klifa, C., Deshmukh, S., Egleston, B. L., Shepherd, J. A., Kwiterovich, P. O., Van Horn, L., Snetselaar, L. G., Stevens, V. J., Robson, A. M., Lasser, N. L., & Hylton, N. M. (2013). Menstrual and reproductive characteristics and breast density in young women. Cancer Causes & Control, 24(11), 1973–1983. [DOI:10.1007/s10552-013-0273-2]
Döring, G. K. (1963). Über die relative Häufigkeit des anovulatorischen Cyclus im Leben der Frau. [On the relative frequency of the anovulatory cycle in women’s lives.] Archiv für Gynäkologie, 199(2), 115–123. [DOI:10.1007/bf00668062]
Drąsutis, J. (2017). Changes in breast morphological parameters, body size and shape, blood serum prolactin and lipids during pregnancy, multiple relationships of these indicators and morphological markers for health risk. (Doctoral dissertation, Vilniaus Universitetas.) [Google Scholar] [URL]
Dreijerink, K., den Heijer, M., Geels, R. (2025). Increased breast volume due to addition of progesterone and increasing the estradiol dose in feminizing gender-affirming hormone therapy. EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
Drife, J. O. (1982). The effects of parity and the menstrual cycle on the normal mammary gland and their possible relationship to malignant change. (Doctoral dissertation, University of Edinburgh). [Google Scholar] [Google Books] [WorldCat] [URL] [PDF]
Drife, J. O. (1984). The pill and the breast. IPPF Medical Bulletin, 18(6), 1–2. [Google Scholar] [PubMed]
Drife, J. O. (1986). Breast Development in Puberty. Annals of the New York Academy of Sciences, 464(1) [Endocrinology of the Breast: Basic and Clinical Aspects], 58–65. [DOI:10.1111/j.1749-6632.1986.tb15993.x]
Drife, J. O. (1989). Breast modifications during the menstrual cycle. Supplement to International Journal of Gynecology and Obstetrics, 1, 19–24. / International Journal of Gynecology and Obstetrics, 1989(Suppl 1), 19–24. [Google Scholar] [PubMed] [Archive.org] [PDF]
Drife, J. O. (1990). Premenopausal Hormone Therapy. In Drife, J. O., & Studd, J. W. W. (Eds.). HRT and Osteoporosis (pp. 351–362). London: Springer London. [DOI:10.1007/978-1-4471-1799-5_25]
Duncan, M. (2010). Sexual Selection and Human Breast Morphology. (Doctoral dissertation, Te Herenga Waka-Victoria University of Wellington.) [Google Scholar] [URL]
Edmonds, D. K. (1989). Normal Puberty. In Edmonds, D. K. Dewhurst’s Practical Paediatric and Adolescent Gynaecology, 2nd Edition (pp. 56–62). London: Butterworths. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL] [Archive.org]
Elling, S. V., & Powell, F. C. (1997). Physiological changes in the skin during pregnancy. Clinics in Dermatology, 15(1), 35–43. [DOI:10.1016/s0738-081x(96)00108-3]
Engman, M., Skoog, L., Soderqvist, G., & Gemzell-Danielsson, K. (2008). The effect of mifepristone on breast cell proliferation in premenopausal women evaluated through fine needle aspiration cytology. Human Reproduction, 23(9), 2072–2079. [DOI:10.1093/humrep/den228]
Federici, S., Goggi, G., Quinton, R., Giovanelli, L., Persani, L., Cangiano, B., & Bonomi, M. (2021). New and Consolidated Therapeutic Options for Pubertal Induction in Hypogonadism: In-depth Review of the Literature. Endocrine Reviews, 43(5), 824–851. [DOI:10.1210/endrev/bnab043]
Fernández-Cancio, M., García-García, E., González-Cejudo, C., Martínez-Maestre, M., Mangas-Cruz, M., Guerra-Junior, G., Pandi de Mello, M., Arnhold, I. J., Nishi, M. Y., Bilharinho Mendonça, B., García-Arumí, E., Audí, L., Tizzano, E., & Carrascosa, A. (2017). Discordant Genotypic Sex and Phenotype Variations in Two Spanish Siblings with 17α-Hydroxylase/17,20-Lyase Deficiency Carrying the Most Prevalent Mutated CYP17A1 Alleles of Brazilian Patients. Sexual Development, 11(2), 70–77. [DOI:10.1159/000468160]
Fernandez-Valdivia, R., Mukherjee, A., Mulac-Jericevic, B., Conneely, O. M., DeMayo, F. J., Amato, P., & Lydon, J. P. (2005). Revealing Progesterone’s Role in Uterine and Mammary Gland Biology: Insights from the Mouse. Seminars in Reproductive Medicine, 23(1), 22–37. [DOI:10.1055/s-2005-864031]
Finkenzeller, D. A., & Loveless, M. B. (2007). Pediatric Gynecology. In Fortner, K. B., Szymanski, L. M., Fox, H. E., & Wallach, E. E. (Eds.). The Johns Hopkins Manual of Gynecology and Obstetrics, 3rd Edition (Spiral Manual Series) (pp. 363–379). Philadelphia: Lippincott Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Fisher, A. D., Castellini, G., Ristori, J., Casale, H., Cassioli, E., Sensi, C., Fanni, E., Amato, A. M., Bettini, E., Mosconi, M., Dèttore, D., Ricca, V., & Maggi, M. (2016). Cross-Sex Hormone Treatment and Psychobiological Changes in Transsexual Persons: Two-Year Follow-Up Data. The Journal of Clinical Endocrinology & Metabolism, 101(11), 4260–4269. [DOI:10.1210/jc.2016-1276]
Foidart, J. M., Colin, C., Denoo, X., Desreux, J., Fournier, S., & de Linières, B. (1996). Influence of percutaneous administration of estradiol and progesterone on the proliferation of human breast epithelial cells. In Calvo, F., Crépin, M., & Magdelenat, H. (Eds.). Breast Cancer: Advances in Biology and Therapeutics [21st Meeting of the International Association for Breast Cancer Research, July 3-4-5, 1996, Paris] (pp. 329–334). Montrouge/Paris: John Libbey Eurotext. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Foidart, J., Colin, C., Denoo, X., Desreux, J., Béliard, A., Fournier, S., & de Lignières, B. (1998). Estradiol and Progesterone Regulate the Proliferation of Human Breast Epithelial Cells. Fertility and Sterility, 69(5), 963–969. [DOI:10.1016/s0015-0282(98)00042-9]
Folley, S. J. (1947). Endocrine Control of the Mammary Gland. British Medical Bulletin, 5(2–3), 130–134. [DOI:10.1093/oxfordjournals.bmb.a073121]
Folley, S. J. (1950). Lactational Physiology. In Bowes, K. (Ed.). Modern Trends in Obstetrics and Gynaecology (pp. 441–453). London: Butterworth. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Folley, S. J. (1952). Lactation. In Parkes, A. S. (Ed.). Marshall’s Physiology of Reproduction, 3rd Edition, Volume II (pp. 525–647). Longmans, Green & Co.: London. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Folley, S. J. (1956). Recent Studies on the Development of the Mammary Gland. In Folley, S. J. The Physiology and Biochemistry of Lactation, 1st Edition (pp. 1–22). Edinburgh: Oliver & Boyd. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Folley, S. J., & Malpress, F. H. (1948). Hormonal Control of Mammary Growth. In Pincus, G., & Thimann, K. V. (Eds.). The Hormones: Physiology, Chemistry and Applications, Volume I (pp. 695–743). New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Fourcade, R., & McLeod, D. (2004). Tolerability of Antiandrogens in the Treatment of Prostate Cancer. UroOncology, 4(1), 5–13. [DOI:10.1080/1561095042000191655]
Fowler, P. A., Casey, C. E., Cameron, G. G., Foster, M. A., & Knight, C. H. (1990). Cyclic changes in composition and volume of the breast during the menstrual cycle, measured by magnetic resonance imaging. BJOG, 97(7), 595–602. [DOI:10.1111/j.1471-0528.1990.tb02546.x]
Fridriksdottir, A. J., Petersen, O. W., & Rnnov-Jessen, L. (2011). Mammary gland stem cells: current status and future challenges. The International Journal of Developmental Biology, 55(7–8–9), 719–729. [DOI:10.1387/ijdb.113373af]
Fuqua, J. S., & Eugster, E. A. (2022). History of Puberty: Normal and Precocious. Hormone Research in Paediatrics, 95(6), 568–578. [DOI:10.1159/000526464]
Futterweit, W. (1980). Endocrine management of transsexual. Hormonal profiles of serum prolactin, testosterone, and estradiol. New York State Journal of Medicine, 80(8), 1260–1264. [Google Scholar] [PubMed] [Archive.org] [PDF]
Gaede, P., Trolle, D., & Pedersen, H. (1978). Extremely low placental lactogen hormone (hpl) values in an otherwise uneventful pregnancy preceding delivery of a normal baby. Acta Obstetricia et Gynecologica Scandinavica, 57(3), 203–209. [DOI:10.3109/00016347809154883]
Galani, A., Kitsiou-Tzeli, S., Sofokleous, C., Kanavakis, E., & Kalpini-Mavrou, A. (2008). Androgen insensitivity syndrome: clinical features and molecular defects. Hormones, 7(3), 217–229. [DOI:10.14310/horm.2002.1201]
Galbarczyk, A. (2011). Unexpected changes in maternal breast size during pregnancy in relation to infant sex: An evolutionary interpretation. American Journal of Human Biology, 23(4), 560–562. [DOI:10.1002/ajhb.21177]
Gawlik, A., Hankus, M., Such, K., Drosdzol-Cop, A., Madej, P., Borkowska, M., Zachurzok, A., & Malecka-Tendera, E. (2016). Hypogonadism and Sex Steroid Replacement Therapy in Girls with Turner Syndrome. Journal of Pediatric and Adolescent Gynecology, 29(6), 542–550. [DOI:10.1016/j.jpag.2016.03.005]
Gershon-Cohen, J. (1970). The Normal Breast. In Gershon-Cohen, J. Atlas of Mammography (pp. 23–38). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-85678-5_4] [Google Books]
Gertig, D. M., Stillman, I. E., Byrne, C., Spiegelman, D., Schnitt, S. J., Connolly, J. L., Colditz, G. A., & Hunter, D. J. (1999). Association of age and reproductive factors with benign breast tissue composition. Cancer Epidemiology, Biomarkers & Prevention, 8(10), 873–879. [Google Scholar] [PubMed] [URL]
Geschickter, C. F. (1945). Endocrine Physiology of the Breast. In Geschickter, C. F. Diseases of the Breast: Diagnosis, Pathology, Treatment, 2nd Edition (pp. 42–81). Philadelphia: J.B. Lippincott. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Glenn, J. F. (1976). Testicular feminization syndrome current clinical considerations. Urology, 7(6), 569–577. [DOI:10.1016/0090-4295(76)90079-0]
Gliosci, A., & Presutti, F. (1993). Virginal gigantomastia: Validity of combined surgical and hormonal treatments. Aesthetic Plastic Surgery, 17(1), 61–65. [DOI:10.1007/bf00455051]
Gompel, A., & Plu-Bureau, G. (2018). Progesterone, progestins and the breast in menopause treatment. Climacteric, 21(4), 326–332. [DOI:10.1080/13697137.2018.1476483]
Goodman, H. M. (2009). Hormonal Control of Pregnancy and Lactation. In Goodman, H. M. Basic Medical Endocrinology, 4th Edition (pp. 277–301). Amsterdam: Academic Press/Elsevier. [DOI:10.1016/b978-0-12-373975-9.00014-8]
Gooren, L. J., Harmsen-Louman, W., & Kessel, H. (1985). Follow-up of prolactin levels in long-term oestrogen-treated male-to-female transsexuals with regard to prolactinoma induction. Clinical Endocrinology, 22(2), 201–207. [DOI:10.1111/j.1365-2265.1985.tb01081.x]
Gooren, L. J. (2016). Hormone Treatment of Adult Transgender People. In Ettner, R., Monstrey, S., & Coleman, E. (Eds.). Principles of Transgender Medicine and Surgery, 2nd Edition (pp. 167–179). New York: Routledge. [Google Scholar] [Google Books] [DOI:10.4324/9781315718972-11]
Gordon, C. M., & Laufer, M. R. (2005) The Physiology of Puberty. In Emans, S. J., Laufer, M. R., & Goldstein, D. P. (Eds.). Pediatric and Adolescent Gynecology, 5th Edition (pp. 120–155). Philadelphia: Lippincott Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Göretzlehner, G. & Lauritzen, C. (1992). Hormontherapie bei Gynäkologischen Erkrankungen [Hormone Therapy for Gynecological Diseases]. In Göretzlehner, G. & Lauritzen, C. Praktische Hormontherapie in der Gynäkologie, 1. Auflage [Practical Hormone Therapy in Gynecology, 1st Edition] (pp. 245–299). Berlin/New York: de Gruyter. [DOI:10.1515/9783112417706-007] [Google Books] [PDF] [Translation]
Graham, J. D., & Clarke, C. L. (1997). Physiological Action of Progesterone in Target Tissues. Endocrine Reviews, 18(4), 502–519. [DOI:10.1210/edrv.18.4.0308]
Graham, S. J., Stanchev, P. L., Lloyd‐Smith, J. O., Bronskill, M. J., & Plewes, D. B. (1995). Changes in Fibroglandular Volume and Water Content of Breast Tissue During the Menstrual Cycle Observed by MR Imaging at 1.5 T. Journal of Magnetic Resonance Imaging, 5(6), 695–701. [DOI:10.1002/jmri.1880050613]
Greydanus, D. E., Omar, H. A., Matytsina, L. A., & Tsitsika, A. (2010). Breast Disorders in Children and Adolescents. In Omar, H. A., Greydanus, D. E., Tsitsika, A. K., Patel, D. R., & Merrick, J. (Eds.). Pediatric and Adolescent Sexuality and Gynecology: Principles for the Primary Care Clinician (pp. 245–316). Hauppauge: Nova Science Publishers. [Google Scholar] [Google Books] [URL] [OpenLibrary] [WorldCat] [PDF]
Guaragna-Filho, G., Guerra-Junior, G., Tadokoro-Cuccaro, R., Hughes, I. A., Barros, B. A., Hiort, O., Balsamo, A., Guran, T., Holterhus, P. M., Hannema, S., Poyrazoglu, S., Darendeliler, F., Bryce, J., Ahmed, S. F., & Quigley, C. A. (2023). Pubertal and Gonadal Outcomes in 46,XY Individuals with Partial Androgen Insensitivity Syndrome Raised as Girls. Sexual Development, 17(1), 16–25. [DOI:10.1159/000526997]
Gunn, H. M., Tsai, M., McRae, A., & Steinbeck, K. S. (2018). Menstrual Patterns in the First Gynecological Year: A Systematic Review. Journal of Pediatric and Adolescent Gynecology, 31(6), 557–565.e6. [DOI:10.1016/j.jpag.2018.07.009]
Günzel, P., Hasan, S. H., Düsterberg, B., Hümpel, M., Putz, B., & Lehmann, M. (1987). Zur toxikologischen Prüfung von Steroidhormonen. [For the Toxicological Testing of Steroid Hormones.] In Burger, O. K., Grosdanoff, P., Henschler, D., Kraupp, O., & Schnieders, B. (Eds.). Aktuelle Probleme der Biomedizin (pp. 93–112). Berlin/New York: De Gruyter. [Google Scholar] [Google Books] [DOI:10.1515/9783110898231-014]
Hagisawa, S., Shimura, N., & Arisaka, O. (2012). Effect of Excess Estrogen on Breast and External Genitalia Development in Growth Hormone Deficiency. Journal of Pediatric and Adolescent Gynecology, 25(3), e61–e63. [DOI:10.1016/j.jpag.2011.11.005]
Hamburger, C., & Benjamin, H. (1969). Endocrine Treatment of Male and Female Transsexualism / Appendix for the Practicing Physician: Suggestions and Guidelines for the Management of Transsexuals. In Green, R., & Money, J. (Eds.). Transsexualism and Sex Reassignment (pp. 291–307). Baltimore: John Hopkins University Press. [Google Scholar] [Google Books] [PDF]
Hannan, F. M., Elajnaf, T., Vandenberg, L. N., Kennedy, S. H., & Thakker, R. V. (2022). Hormonal regulation of mammary gland development and lactation. Nature Reviews Endocrinology, 19(1), 46–61. [DOI:10.1038/s41574-022-00742-y]
Hannema, S. E., Schagen, S. E., Cohen-Kettenis, P. T., & Delemarre-van de Waal, H. A. (2017). Efficacy and Safety of Pubertal Induction Using 17β-Estradiol in Transgirls. The Journal of Clinical Endocrinology & Metabolism, 102(7), 2356–2363. [DOI:10.1210/jc.2017-00373]
Hartmann, B. W., Laml, T., Albrecht, A. E., Huber, J. C., & Kirchengast, S. (1998). Hormonal Breast Augmentation: Prognostic Relevance of Insulin-Like Growth Factor-I. Gynecological Endocrinology, 12(2), 123–127. [DOI:10.3109/09513599809024960]
Hartmann, P. E., Owens, R. A., Cox, D. B., & Kent, J. C. (1996). Breast Development and Control of Milk Synthesis. Food and Nutrition Bulletin, 17(4), 1–12. [DOI:10.1177/156482659601700404]
Hasan, S. (1974). Steroid Hormone Levels During Pregnancy in Various Species. In Bernhard, S., & Raspé, G. (Ed.). Hormones and Embryonic Development (Advances in the Biosciences, Volume 13) (pp. 181–197). Oxford/New York: Pergamon Press. [Google Scholar] [Google Books] [DOI:10.1016/b978-0-08-018239-1.50014-3] [OpenLibrary] [Archive.org]
Hassiotou, F., & Geddes, D. (2012). Anatomy of the human mammary gland: Current status of knowledge. Clinical Anatomy, 26(1), 29–48. [DOI:10.1002/ca.22165]
Heath, R. A., & Wynne, K. (2019). Children and Adolescents. In Heath, R. A., & Wynne, K. A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (pp. 87–106). Santa Barbara: Praeger/ABC-CLIO. [Google Books]
Heath, R. A., & Wynne, K. (2019). Hormone and Surgical Therapies for Adults. In Heath, R. A., & Wynne, K. A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (pp. 107–146). Santa Barbara: Praeger/ABC-CLIO. [Google Books]
Hertz, R., Odell, W. D., & Ross, G. T. (1966). Diagnostic Implications of Primary Amenorrhea: Combined Clinical Staff Conference at the National Institutes of Health. Annals of Internal Medicine, 65(4), 800–820. [DOI:10.7326/0003-4819-65-4-800]
Hillard, P. J. A. (2007). Benign Diseases of the Female Reproductive Tract. In Berek, J. S., & Novak, E. (Eds.). Berek & Novak’s Gynecology, 14th Edition (pp. 431–496). Philadelphia: Lippincott Williams and Wilkins. [Google Scholar] [OpenLibrary] [WorldCat] [Archive.org]
Hoppe, I. C., Patel, P. P., Singer-Granick, C. J., & Granick, M. S. (2011). Virginal Mammary Hypertrophy: A Meta-Analysis and Treatment Algorithm. Plastic and Reconstructive Surgery, 127(6), 2224–2231. [DOI:10.1097/prs.0b013e3182131bd1]
Hovey, R. C., Trott, J. F., Ginsburg, E., Goldhar, A., Sasaki, M. M., Fountain, S. J., Sundararajan, K., & Vonderhaar, B. K. (2001). Transcriptional and spatiotemporal regulation of prolactin receptor mRNA and cooperativity with progesterone receptor function during ductal branch growth in the mammary gland. Developmental Dynamics, 222(2), 192–205. [DOI:10.1002/dvdy.1179]
Howard, B. A., & Gusterson, B. A. (2000). Human Breast Development. Journal of Mammary Gland Biology and Neoplasia, 5(2), 119–137. [DOI:10.1023/a:1026487120779]
Huffman, J., Dewhurst, C. J., & Capraro, V. J. (1981). The Breast and its Disorders in Childhood and Adolescence. In Huffman, J., Dewhurst, J., & Capraro, V. The Gynecology of Childhood and Adolescence, 2nd Edition (pp. 542–559). Philadelphia: Saunders. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Hussain, Z., Brooks, J., & Percy, D. (2008). Menstrual variation of breast volume and T2 relaxation times in cyclical mastalgia. Radiography, 14(1), 8–16. [DOI:10.1016/j.radi.2006.07.003]
Hussain, Z., Roberts, N., Whitehouse, G. H., García-Fiñana, M., & Percy, D. (1999). Estimation of breast volume and its variation during the menstrual cycle using MRI and stereology. The British Journal of Radiology, 72(855), 236–245. [DOI:10.1259/bjr.72.855.10396212]
Hutson, S. W., Cowen, P. N., & Bird, C. C. (1985). Morphometric studies of age related changes in normal human breast and their significance for evolution of mammary cancer. Journal of Clinical Pathology, 38(3), 281–287. [DOI:10.1136/jcp.38.3.281]
Hytten, F. E. (1954). Clinical and Chemical Studies in Human Lactation–VI. BMJ, 1(4867), 912–915. [DOI:10.1136/bmj.1.4867.912]
Hytten, F. E. (1976). The physiology of lactation. International Journal of Food Sciences and Nutrition, 30(4), 225–232. [DOI:10.3109/09637487609142745]
Hytten, F. E., & Baird, D. (1958). The Development of the Nipple in Pregnancy. The Lancet, 271(7032), 1201–1204. [DOI:10.1016/s0140-6736(58)91908-1]
Hytten, F. E., & Leitch, I. (1971). Preparations for Breast Feeding. In Hytten, F. E., & Leitch, I. The Physiology of Human Pregnancy, 2nd Edition (pp. 234–241). Oxford: Blackwell Scientific Publications. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Hytten, F. E., & Leitch, I. (1971). Preparations for Breast Feeding. In Hytten, F. E., & Leitch, I. The Physiology of Human Pregnancy, 2nd Edition (pp. 234–241). Oxford: Blackwell Scientific Publications. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Hytten, F. E., & Thomson, A. M. (1965). Pregnancy, childbirth and lactation. In Edholm, O. G., & Bacharach, A. L. (Eds.). The Physiology of Human Survival (pp. 327–350). London/New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Hytten, F. E., & Thomson, A. M. (1968). Maternal physiological adjustments. In Assali, N. S. (Ed.). The Maternal Organism, Volume I (Biology of Gestation) (pp. 449–479). New York: Academic Press. [Google Scholar] [Google Books] [WorldCat]
Igo, J., & Visram, H. (2021). Progesterone Therapy Use and Safety in Male to Female Transgender Patients. Canadian Journal of Diabetes, 45(7 Suppl), S39–S39 (abstract no. 109). [DOI:10.1016/j.jcjd.2021.09.119] [URL]
Ingleby, H. (1949). Changes in breast volume in a group of normal young women. Bulletin of the International Association of Medical Museums, 29, 87–92. [Google Scholar] [Google Books] [HathiTrust]
Ingleby, H., Moore, L., & Gershon-Cohen, J. (1957). Gestational breast changes: x-ray studies of the human breast. Obstetrics & Gynecology, 10(2), 149–157. [Google Scholar] [PubMed] [URL]
Ismail, P. M., Amato, P., Soyal, S. M., DeMayo, F. J., Conneely, O. M., O’Malley, B. W., & Lydon, J. P. (2003). Progesterone involvement in breast development and tumorigenesis—as revealed by progesterone receptor “knockout” and “knockin” mouse models. Steroids, 68(10–13), 779–787. [DOI:10.1016/s0039-128x(03)00133-8]
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 204201881987116. [DOI:10.1177/2042018819871166]
Jacobsohn, D. (1961). Hormonal Regulation of Mammary Gland Growth. In Kon, S. K., & Cowie, A. T. (Eds.). Milk: The Mammary Gland and Its Secretion, Volume 1 (pp. 127–160). New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Jamal, N., Ng, K., McLean, D., Looi, L., & Moosa, F. (2004). Mammographic Breast Glandularity in Malaysian Women: Data Derived from Radiography. American Journal of Roentgenology, 182(3), 713–717. [DOI:10.2214/ajr.182.3.1820713]
Jasieńska, G., Ziomkiewicz, A., Ellison, P. T., Lipson, S. F., & Thune, I. (2004). Large breasts and narrow waists indicate high reproductive potential in women. Proceedings of the Royal Society of London. Series B: Biological Sciences, 271(1545), 1213–1217. [DOI:10.1098/rspb.2004.2712]
Jemstrom, H., & Olsson, H. (1997). Breast Size in Relation to Endogenous Hormone Levels, Body Constitution, and Oral Contraceptive Use in Healthy Nulligravid Women Aged 19-25 Years. American Journal of Epidemiology, 145(7), 571–580. [DOI:10.1093/oxfordjournals.aje.a009153]
Jernström, H., Sandberg, T., Bågeman, E., Borg, Å., & Olsson, H. (2005). Insulin-like growth factor-1 (IGF1) genotype predicts breast volume after pregnancy and hormonal contraception and is associated with circulating IGF-1 levels: implications for risk of early-onset breast cancer in young women from hereditary breast cancer families. British Journal of Cancer, 92(5), 857–866. [DOI:10.1038/sj.bjc.6602389]
Jeruss, J. S. (2006). Molecular Basis of Breast Cancer. In Winchester, D. J., Winchester, D. P., Hudis, C. A., & Norton, L. (Eds.). Breast Cancer, 2nd Edition (pp. 83–95). Hamilton: B.C. Decker. [Google Scholar] [Google Books] [WorldCat]
Johnson, M. C., & Cutler, M. L. (2016). Anatomy and Physiology of the Breast. In Jatoi, I., & Rody, A. (Eds.). Management of Breast Diseases (pp. 1–39). Cham: Springer. [DOI:10.1007/978-3-319-46356-8_1]
Kaiser, R. (1993). Gestagen-Östrogen-Kombinationen in der Gynäkologie. Zur Geschichte, Dosierung und Anwendung eines Hormonprinzips. [Gestagen-Oestrogen Combinations in Gynaecology. History, Dosage and Use of a Hormonal Principle.] Geburtshilfe und Frauenheilkunde, 53(7), 503–513. [Google Scholar] [PubMed] [DOI:10.1055/s-2007-1022924]
Kaiser, R., & Leidenberger, F. (1991). Hormonbehandlung bei Benign Mammaerkrankungen: Mammahypoplasie. [Hormonal Treatment in Benign Breast Diseases: Breast Hypoplasia.] In Kaiser, R., & Leidenberger, F. Hormonbehandlung in der Gynäkologischen Praxis, 7. Auflage [Hormone Treatment in Gynecological Practice, 7th Edition] (pp. 138–138). Stuttgart: Georg Thieme Verlag. [Google Books] [OpenLibrary] [WorldCat] [PDF] [Translation]
Kaiser, U., & Ho, K. K. (2015). Pituitary Physiology and Diagnostic Evaluation. In Melmed, S., Polonsky, K. S., Larsen, P. R., Kronenberg, & H. M. (Eds.). Williams Textbook of Endocrinology, 13th Edition (pp. 176–231). Philadelphia: Elsevier. [DOI:10.1016/B978-0-323-29738-7.00008-3] [Google Books]
Kanhai, R. C., Hage, J. J., Asscheman, H., Mulder, W. J., & Hage, J. J. (1999). Augmentation Mammaplasty in Male-to-Female Transsexuals. Plastic and Reconstructive Surgery, 104(2), 542–549. [DOI:10.1097/00006534-199908000-00040]
Kanhai, R. C., Hage, J. J., van Diest, P. J., Bloemena, E., & Mulder, J. W. (2000). Short-Term and Long-Term Histologic Effects of Castration and Estrogen Treatment on Breast Tissue of 14 Male-to-Female Transsexuals in Comparison With Two Chemically Castrated Men. The American Journal of Surgical Pathology, 24(1), 74–80. [DOI:10.1097/00000478-200001000-00009]
Kardelen, A. D., Toksoy, G., Baş, F., Yavaş Abalı, Z., Gençay, G., Poyrazoğlu, Ş., Bundak, R., Altunoğlu, U., Avcı, Ş., Najaflı, A., Uyguner, O., Karaman, B., Başaran, S., & Darendeliler, F. (2018). A Rare Cause of Congenital Adrenal Hyperplasia: Clinical and Genetic Findings and Follow-up Characteristics of Six Patients with 17-Hydroxylase Deficiency Including Two Novel Mutations. Journal of Clinical Research in Pediatric Endocrinology, 10(3), 206–215. [DOI:10.4274/jcrpe.0032]
Kariagina, A., Xie, J., Leipprandt, J. R., & Haslam, S. Z. (2010). Amphiregulin Mediates Estrogen, Progesterone, and EGFR Signaling in the Normal Rat Mammary Gland and in Hormone-Dependent Rat Mammary Cancers. Hormones and Cancer, 1(5), 229–244. [DOI:10.1007/s12672-010-0048-0]
Karp, N. S. (2022). Discussion: The Impact of Combined Oral Contraceptives on Adolescents with Macromastia. Plastic & Reconstructive Surgery, 150(4), 739–740. [DOI:10.1097/prs.0000000000009514]
Kasielska-Trojan, A., Mikołajczyk, M., & Antoszewski, B. (2020). BreastIdea Volume Estimator: A New Tool for Breast Volume Estimation—Presentation and Validation for Women. Plastic & Reconstructive Surgery, 146(6), 744e–748e. [DOI:10.1097/prs.0000000000007373]
Kasielska-Trojan, A., Zawadzki, T., & Antoszewski, B. (2022). Breast Fluctuating Asymmetry in Women with Macromastia/Gigantomastia. International Journal of Environmental Research and Public Health, 19(24), 16895. [DOI:10.3390/ijerph192416895]
Kauli, R., Pertzelan, A., Ben‐Zeev, Z., Lewin, R. P., Kaufman, H., Schally, A. C., Schally, A. V., & Laron, Z. (1984). Treatment of precocious puberty with LHRH analogue in combination with cyproterone acetate—further experience. Clinical Endocrinology, 20(4), 377–387. [DOI:10.1111/j.1365-2265.1984.tb03433.x]
Keller, P. J. (1984). Diagnostik und Therapie wichtiger hormonaler Störungen: Mammahypoplasie. [Diagnosis and Treatment of Important Hormonal Disorders: Mammary Hypoplasia.] In Keller, P. J. Hormonale Störungen in der Gynäkologie: Diagnostik und Behandlung, 3. Auflage [Hormonal Disorders in Gynecology: Diagnosis and Treatment, 3rd Edition] (Kliniktaschenbücher) (pp. 133–134). Berlin/Heidelberg: Springer. [Google Books] [DOI:10.1007/978-3-662-00442-5_3]
Keller, P. J. (1995). Diagnostik und Therapie wichtiger Störungen: Mammahypoplasie. [Diagnosis and Treatment of Important Disorders: Mammary Hypoplasia.] In Keller, P. J. Hormon- und Fertilitätsstörungen in der Gynäkologie, 4. Auflage [Hormonal and Fertility Disorders in Gynecology, 4th Edition] (pp. 145–146). Berlin/Heidelberg: Springer. [Google Books] [DOI:10.1007/978-3-662-12026-2_3]
Kennedy, B. J. (1953). Effects of intensive sex steroid hormone therapy in advanced breast cancer. JAMA, 152(12), 1135–1141. [DOI:10.1001/jama.1953.63690120004013]
Kent, J. C., Mitoulas, L., Cox, D. B., Owens, R. A., & Hartmann, P. E. (1999). Breast Volume and Milk Production During Extended Lactation in Women. Experimental Physiology, 84(2), 435–447. [DOI:10.1111/j.1469-445x.1999.01808.x]
Khoo, S. K., & Mackay, E. V. (1972). Testicular Feminization: The Clinical Features, Endocrine Function and Gonadal Pathology in Six Patients. Australian and New Zealand Journal of Obstetrics and Gynaecology, 12(1), 1–13. [DOI:10.1111/j.1479-828x.1972.tb00721.x]
Klein, R., Aichinger, H., Dierker, J., Jansen, J. T., Joite-Barfuß, S., Säbel, M., Schulz-Wendtland, R., & Zoetelief, J. (1997). Determination of average glandular dose with modern mammography units for two large groups of patients. Physics in Medicine and Biology, 42(4), 651–671. [DOI:10.1088/0031-9155/42/4/004]
Kleinberg, D. L. (2006). Endocrinology of lactation. In DeGroot, L. J., & Jameson, J. L. (Eds). Endocrinology, 5th Edition (pp. 3461–3473). Philadelphia: Elsevier Saunders. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Kleinberg, D. L., & Barcellos-Hoff, M. H. (2011). The Pivotal Role of Insulin-Like Growth Factor I in Normal Mammary Development. Endocrinology and Metabolism Clinics of North America, 40(3), 461–471. [DOI:10.1016/j.ecl.2011.06.001]
Kleinberg, D. L., & Ruan, W. (2008). IGF-I, GH, and Sex Steroid Effects in Normal Mammary Gland Development. Journal of Mammary Gland Biology and Neoplasia, 13(4), 353–360. [DOI:10.1007/s10911-008-9103-7]
Kleinberg, D. L., Wood, T. L., Furth, P. A., & Lee, A. V. (2008). Growth Hormone and Insulin-Like Growth Factor-I in the Transition from Normal Mammary Development to Preneoplastic Mammary Lesions. Endocrine Reviews, 30(1), 51–74. [DOI:10.1210/er.2008-0022]
Kühnel, W. (2000). IMMULITE® and IMMULITE® 2000 Reference Range Compendium, First English Edition. Los Angeles, California: Diagnostic Products Corporation. [Google Scholar] [URL] [PDF 1] [PDF 2]
Kustin, J., & Rebar, R. W. (1987). Menstrual Disorders in the Adolescent Age Group. Primary Care: Clinics in Office Practice, 14(1), 139–166. [DOI:10.1016/s0095-4543(21)01004-6]
Kutten, F., Malet, C., & Leygue, E. (1994). Antiestrogen action of progestogens in human breast cells. In Berg, G., & Hammar, M. (Eds.). The Modern Management of the Menopause: A Perspective for the 21st Century [The Proceedings of the VII International Congress on the Menopause, Stockholm, Sweden 1993] (pp. 419–433). Canforth: Parthenon. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Ladjouze, A., & Donaldson, M. (2019). Primary gonadal failure. Best Practice & Research Clinical Endocrinology & Metabolism, 33(3), 101295. [DOI:10.1016/j.beem.2019.101295]
Laessle, R. G., Tuschi, R. J., Schweiger, U., & Pirke, K. M. (1990). Mood changes and physical complaints during the normal menstrual cycle in healthy young women. Psychoneuroendocrinology, 15(2), 131–138. [DOI:10.1016/0306-4530(90)90021-z]
LaMarca, H. L., & Rosen, J. M. (2007). Estrogen regulation of mammary gland development and breast cancer: amphiregulin takes center stage. Breast Cancer Research, 9(4), 304. [DOI:10.1186/bcr1740]
Laron, Z., & Kauli, R. (2000). Experience with Cyproterone Acetate in the Treatment of Precocious Puberty. Journal of Pediatric Endocrinology and Metabolism, 13(Suppl 1), 805–810. [DOI:10.1515/jpem.2000.13.s1.805]
Laufer, M. R., Goldstein, D. P., & Hendren, W. H. (2005). Structural Abnormalities of the Female Reproductive Tract. In Emans, J. E., Laufer, M. R., & Goldstein, D. P. (Eds.). Pediatric and Adolescent Gynecology, 5th Edition (pp. 334–416). Philadelphia: Lippincott Williams and Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Lauritzen, C. (1980 October 27). Hormonkur Kann Hypoplastischer Mamma Aufhelfen. [Hormone Therapy Can Help Hypoplastic Breasts.] Selecta: Medizin aktuell; das Magazin für ärztliche Fortbildung [Selecta: Medicine Currently; the Magazine for Medical Training], 22(43), 3798–3801. Planegg: Selecta-Verlag. [ISSN:0582-4877] [WorldCat] [PDF] [Translation]
Lauritzen, C. (1982). Treatment of mammary- and uterus hypoplasia by estrogen-progestagen pseudopregnancy. In Richter, K., Huber, A., Terruhn, V. (Eds.) 1. Europäisches Symposium für Kinder- und Jugendgynäkologie. München 19.-21.3.1981, Band 2 [First European Symposium on Pediatric and Adolescent Gynaecology. München, March 19–21, 1981, Volume 2] (pp. 487–489). Friedrichsdorf/Taunus: Milupa-Aktiengesellschaft, Wissenschaftliche Abteilung. [Google Scholar] [PDF]
Lauritzen, C. (1989). Hormonelle Substitutionstherapie zur Brustvergrößerung. [Hormonal Substitution Therapy for Breast Enlargement.] In Beller, F. K. & Seitzer, D. (Eds.). 7. Siebte Wissenschaftliche Tagung der Deutschen Gesellschaft für Senologie, Münster, 25. - 27. September 1987: Ausgewählte Referate [7th Scientific Conference of the German Society for Senology, Münster, [Germany,] September 25–27, 1987: Selected Papers] (pp. 39–41). Mülheim, Germany: H.U.F. Verlag. [Google Scholar] [WorldCat 1] [WorldCat 2] [WorldCat 3] [Cited By 1] [Cited By 2] [Cited By 3] [PDF] [Translation]
Lawrence, R. A., & Lawrence, R. M. (2015). Physiology of Lactation. In Lawrence, R. A., & Lawrence, R. M. Breastfeeding: A Guide for the Medical Profession, 8th Edition (pp. 56–90). Philadelphia: Elsevier. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Lee, H. J., & Ormandy, C. J. (2012). Interplay between progesterone and prolactin in mammary development and implications for breast cancer. Molecular and Cellular Endocrinology, 357(1–2), 101–107. [DOI:10.1016/j.mce.2011.09.020]
Lee, P. A. (2001). Physiology of Puberty. In Becker, K. L., Bilezikian, J. P., Bremner, W. J., Hung, W., Kahn, C. R., Loriaux, D. L., Nylén, E. S., Rebar, R. W., Robertson, G. L., Snider, R. H., Wartofsky, L. (Eds.). Principles and Practice of Endocrinology and Metabolism, 3rd Edition (pp. 885–893). Philadelphia: Lippincott Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Lee, S. W., Kwak, D. S., Jung, I. S., Kwak, J. H., Park, J. H., Hong, S. M., Lee, C. B., Park, Y. S., Kim, D. S., Choi, W. H., & Ahn, Y. H. (2015). Partial Androgen Insensitivity Syndrome Presenting with Gynecomastia. Endocrinology and Metabolism, 30(2), 226–230. [DOI:10.3803/enm.2015.30.2.226]
Lejour, M. (1994). Vertical Mammaplasty and Liposuction of the Breast. Plastic and Reconstructive Surgery, 94(1), 100–114. [DOI:10.1097/00006534-199407000-00010]
Lejour, M. (1997). Evaluation of Fat in Breast Tissue Removed by Vertical Mammaplasty. Plastic and Reconstructive Surgery, 99(2), 386–393. [DOI:10.1097/00006534-199702000-00012]
Lemarchand-Béraud, T., Zufferey, M., Reymond, M., & Rey, I. (1982). Maturation of the Hypothalamo-Pituitary-Ovarian Axis in Adolescent Girls. The Journal of Clinical Endocrinology & Metabolism, 54(2), 241–246. [DOI:10.1210/jcem-54-2-241]
Levy, A., Crown, A., & Reid, R. (2003). Endocrine intervention for transsexuals. Clinical Endocrinology, 59(4), 409–418. [DOI:10.1046/j.1365-2265.2003.01821.x]
Lewin, R. (2016). Breast Hypertrophy and Outcome of Breast Reduction Surgery. (Doctoral thesis, University of Gothenburg. Sahlgrenska Academy.) [Google Scholar] [URL]
Lim, L. Y., Ho, P. J., Liu, J., Chay, W. Y., Tan, M., Hartman, M., & Li, J. (2018). Determinants of breast size in Asian women. Scientific Reports, 8(1), 1201. [DOI:10.1038/s41598-018-19437-4]
Lloyd, C. W., & Leathem, J. H. (1964). Growth and development of the breast and lactation. In Lloyd, C. W. (Ed.). Human Reproduction and Sexual Behavior (pp. 117–134). Philadelphia: Lea & Febiger. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Lopez, X., Panton, J., Nagarkar, P., Preston, S., Abramowitz, J., & Amirlak, B. (2023). Initial Assessment of VECTRA Three-Dimensional Imaging to Accurately Simulate Breast Volume Changes in Transfeminine Patients: A Mannequin Study. Aesthetic Surgery Journal Open Forum, 5, online ahead of print. [DOI:10.1093/asjof/ojad015]
Lorincz, A. M., & Sukumar, S. (2006). Molecular links between obesity and breast cancer. Endocrine-Related Cancer, 13(2), 279–292. [DOI:10.1677/erc.1.00729]
Lucien, J. N., Ortega, M. T., Calvert, M. E., Smith, C., White, X., Rogers, H., Mosley, B., Agrawal, R., Drude, A., McGee, C., George, M., Brown, A., Downey, K., Wild, C., Njunge, A., Kuzmiak, C. M., Zava, D., Zava, T., Pollard, J., Francis, J., Beery, B. L., Harlin, M., Gonzalez, G. R., & Shaw, N. D. (2022). The Launch of A Girl’s First Period Study: Demystifying Reproductive Hormone Profiles in Adolescent Girls. Journal of Pediatric and Adolescent Gynecology, 35(4), 420–425. [DOI:10.1016/j.jpag.2021.12.018]
Lydon, J. P., DeMayo, F. J., Funk, C. R., Mani, S. K., Hughes, A. R., Montgomery, C. A., Shyamala, G., Conneely, O. M., & O’Malley, B. W. (1995). Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes & Development, 9(18), 2266–2278. [DOI:10.1101/gad.9.18.2266]
Lyon, A. J., De Bruyn, R., & Grant, D. B. (1985). Isosexual Precocious Puberty in Girls. Acta Paediatrica, 74(6), 950–955. [DOI:10.1111/j.1651-2227.1985.tb10063.x]
Lyons, W. R. (1958). Hormonal synergism in mammary growth. Proceedings of the Royal Society of London. Series B - Biological Sciences, 149(936), 303–325. [DOI:10.1098/rspb.1958.0071]
Lyons, W. R., & McGinty, D. A. (1941). Effects of estrone and progesterone on male rabbit mammary glands. I. Varying doses of progesterone. Proceedings of the Society for Experimental Biology and Medicine, 48(1), 83–86. [DOI:10.3181/00379727-48-13227]
Lyons, W. R., Li, C. H., & Johnson, R. E. (1958). The Hormonal Control of Mammary Growth and Lactation. In Pincus, G. (Ed.). Recent Progress in Hormone Research, Volume 14 [Proceedings of the Laurentian Hormone Conference 1957, Held at Mont Tremblant, Quebec] (pp. 219–254). New York/London: Academic Press. [Google Scholar] [Google Books] [PubMed] [OpenLibrary] [WorldCat] [PDF]
MacBryde, C. M. (1939). The Production of Breast Growth in the Human Female. Journal of the American Medical Association, 112(11), 1045–1049. [DOI:10.1001/jama.1939.02800110025006]
Malet, C., Gompel, A., Yaneva, H., Cren, H., Fidji, N., Mowszowicz, I., Kuttenn, F., & Mauvais-Jarvis, P. (1991). Estradiol and Progesterone Receptors in Cultured Normal Human Breast Epithelial Cells and Fibroblasts: Immunocytochemical Studies. The Journal of Clinical Endocrinology & Metabolism, 73(1), 8–17. [DOI:10.1210/jcem-73-1-8]
Malini, S., Smith, E. O., & Goldzieher, J. W. (1985). Measurement of breast volume by ultrasound during normal menstrual cycles and with oral contraceptive use. Obstetrics and Gynecology, 66(4), 538–541. [Google Scholar] [PubMed] [URL] [PDF]
Marshall, W. A. (1978). Puberty. In Falkner, F., & Tanner, J. M. (Eds.). Human Growth: Postnatal Growth (pp. 141–181). Boston: Springer US. [DOI:10.1007/978-1-4684-2622-9_6]
Marshall, W. A., & Tanner, J. M. (1969). Variations in pattern of pubertal changes in girls. Archives of Disease in Childhood, 44(235), 291–303. [DOI:10.1136/adc.44.235.291]
Mauvais-Jarvis, P., Kuttenn, F., & Gompel, A. (1986). Antiestrogen action of progesterone in breast tissue. Breast Cancer Research and Treatment, 8(3), 179–188. [DOI:10.1007/bf01807330]
Mauvais-Jarvis, P., Kuttenn, F., & Gompel, A. (1986). Estradiol/Progesterone Interaction in Normal and Pathologic Breast Cells. Annals of the New York Academy of Sciences, 464(1), 152–167. [DOI:10.1111/j.1749-6632.1986.tb16002.x]
Mauvais-Jarvis, P., Kuttenn, F., & Gompel, A. (1987). Antiestrogen Action of Progesterone in Breast Tissue. Hormone Research, 28(2–4), 212–218. [DOI:10.1159/000180946]
Mauvais-Jarvis, P., Kuttenn, F., Gompel, A., & Benotmane, A. (1987). Action anti-estrogène de la progestérone dans le sein. [Antiestrogen action of progesterone in the breast]. Pathologie-Biologie, 35(7), 1081–1086. [Google Scholar 1] [Google Scholar 2] [PubMed]
Mauvais-Jarvis, P., Sitruk-Ware, R., & Kuttenn, F. (1981). Benign Breast Disease. In McGuire, W. L. (Ed.). Breast Cancer 4: Advances in Research and Treatment (pp. 51–94). Boston: Springer US. [DOI:10.1007/978-1-4615-6571-0_3]
McArthur, J. W. (1966). The Reproductive Endocrinology of Adolescence. In Heald, F. P. (Ed.). Adolescent Gynecology (pp. 9–20). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
McBryan, J., Howlin, J., Napoletano, S., & Martin, F. (2008). Amphiregulin: Role in Mammary Gland Development and Breast Cancer. Journal of Mammary Gland Biology and Neoplasia, 13(2), 159–169. [DOI:10.1007/s10911-008-9075-7]
McDonough, P. G., Spicer, D. V., Ursin, G., & Pike, M. C. (1996). Progesterone Concentrations—Physiologic or Pharmacologic? Fertility and Sterility, 65(5), 1077–1078. [DOI:10.1016/s0015-0282(16)58295-8]
McMillan, M. (1966). Five cases of testicular feminisation including one with a teratoma of the testis. The Journal of Pathology and Bacteriology, 91(2), 417–427. [DOI:10.1002/path.1700910216]
McNally, S., & Stein, T. (2016). Overview of Mammary Gland Development: A Comparison of Mouse and Human. In Martin, F., Stein, T., & Howlin, J. (Eds.). Mammary Gland Development (Methods in Molecular Biology, Volume 1501) (pp. 1–17). New York: Springer New York. [DOI:10.1007/978-1-4939-6475-8_1]
McPhaul, M. J. (2002). Androgen receptor mutations and androgen insensitivity. Molecular and Cellular Endocrinology, 198(1–2), 61–67. [DOI:10.1016/s0303-7207(02)00369-6]
Mesiano, S. (2019). Endocrinology of Human Pregnancy and Fetal-Placental Neuroendocrine Development. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 256–284.e9). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/b978-0-323-47912-7.00011-1]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463]
Meyer, W. J., Finkelstein, J. W., Stuart, C. A., Webb, A., Smith, E. R., Payer, A. F., & Walker, P. A. (1981). Physical and hormonal evaluation of transsexual patients during hormonal therapy. Archives of Sexual Behavior, 10(4), 347–356. [DOI:10.1007/bf01565538]
Meyer, W. J., Webb, A., Stuart, C. A., Finkelstein, J. W., Lawrence, B., & Walker, P. A. (1986). Physical and hormonal evaluation of transsexual patients: A longitudinal study. Archives of Sexual Behavior, 15(2), 121–138. [DOI:10.1007/bf01542220]
Mikołajczyk, M., Kasielska-Trojan, A., & Antoszewski, B. (2019). A New Tool for Breast Anthropometric Measurements: Presentation and Validation for Women and Men. Aesthetic Plastic Surgery, 43(5), 1160–1170. [DOI:10.1007/s00266-019-01467-6]
Milionis, C., Ilias, I., & Koukkou, E. (2022). Progesterone in gender-affirming therapy of trans women. World Journal of Biological Chemistry, 13(3), 66–71. [DOI:10.4331/wjbc.v13.i3.66]
Milligan, D., Drife, J. O., & Short, R. V. (1975). Changes in breast volume during normal menstrual cycle and after oral contraceptives. BMJ, 4(5995), 494–496. [DOI:10.1136/bmj.4.5995.494]
Morris, J. M. (1953). The syndrome of testicular feminization in male pseudohermaphrodites. American Journal of Obstetrics and Gynecology, 65(6), 1192–1211. [DOI:10.1016/0002-9378(53)90359-7]
Morris, J. M., & Mahesh, V. B. (1963). Further observations on the syndrome, “testicular feminization”. American Journal of Obstetrics and Gynecology, 87(6), 731–748. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF]
Muallem, M. M., & Rubeiz, N. G. (2006). Physiological and biological skin changes in pregnancy. Clinics in Dermatology, 24(2), 80–83. [DOI:10.1016/j.clindermatol.2005.10.002]
Müller, C. (1953). Zur Hormonalen Kosmetik der Weiblichen Brust: Wirkung und Nebenwirkung Östrogenhaltiger Kosmetischer Präparate. [Hormonal Cosmetics of the Female Breast: Effects and Side Effects of Estrogen-Containing Cosmetic Preparations.] Schweizerische Medizinische Wochenschrift [Swiss Medical Weekly], 83(4), 81–84. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF] [Translation]
Muzaffar, F., Hussain, I., & Haroon, T. S. (1998). Physiologic skin changes during pregnancy: a study of 140 cases. International Journal of Dermatology, 37(6), 429–431. [DOI:10.1046/j.1365-4362.1998.00281.x]
Naik, M., Diwakar, R. K., Patre, S., & Singh, S. (2015). Gigantomastia: A Rare Complication In Pregnancy. Journal of Medical Science and Clinical Research, 3(7), 6873–6876. [Google Scholar] [URL] [PDF]
Naseem, H., Lokman, M., & Fitzgerald, C. (2021). Management of congenital hypogonadotropic hypogonadism in females. Human Fertility, 26(3), 622–631. [DOI:10.1080/14647273.2021.1998929]
Nelson, W. O. (1936). Endocrine Control of the Mammary Gland. Physiological Reviews, 16(3), 488–526. [DOI:10.1152/physrev.1936.16.3.488]
Nolan, B. J., Frydman, A. S., Leemaqz, S. Y., Carroll, M., Grossmann, M., Zajac, J. D., & Cheung, A. S. (2022). Effects of low-dose oral micronised progesterone on sleep, psychological distress, and breast development in transgender individuals undergoing feminising hormone therapy: a prospective controlled study. Endocrine Connections, 11(5), e220170. [DOI:10.1530/EC-22-0170]
Nolan, B. J., Frydman, A. S., Leemaqz, S. Y., Carroll, M., Grossmann, M., Zajac, J. D., & Cheung, A. S. (2022). Effects Of Low-dose Oral Micronised Progesterone On Sleep, Psychological Distress And Breast Development In Transgender Individuals Undergoing Feminising Hormone Therapy: A Prospective Controlled Study. Journal of the Endocrine Society, 6(Suppl 1), A653–A654 (abstract no. LBODP089). [DOI:10.1210/jendso/bvac150.1351]
Nussbaum, R., & Benedetto, A. V. (2006). Cosmetic aspects of pregnancy. Clinics in Dermatology, 24(2), 133–141. [DOI:10.1016/j.clindermatol.2005.10.007]
Nuzzi, L. C., Pramanick, T., Massey, G. G., Walsh, L. R., McNamara, C. T., Firriolo, J. M., DiVasta, A. D., & Labow, B. I. (2021). The Impact of Progestin-only Contraception on Adolescents with Macromastia. Plastic and Reconstructive Surgery - Global Open, 9(2), e3421. [DOI:10.1097/gox.0000000000003421]
Nuzzi, L. C., Pramanick, T., Massey, G. G., Walsh, L. R., McNamara, C. T., Firriolo, J. M., DiVasta, A. D., & Labow, B. I. (2022). The Impact of Combined Oral Contraceptives on Adolescents with Macromastia. Plastic and Reconstructive Surgery, 150(4), 731–738. [DOI:10.1097/prs.0000000000009513]
Oakes, M. B., Eyvazzadeh, A. D., Quint, E., & Smith, Y. R. (2008). Complete Androgen Insensitivity Syndrome—A Review. Journal of Pediatric and Adolescent Gynecology, 21(6), 305–310. [DOI:10.1016/j.jpag.2007.09.006]
Obr, A. E., & Edwards, D. P. (2012). The biology of progesterone receptor in the normal mammary gland and in breast cancer. Molecular and Cellular Endocrinology, 357(1–2), 4–17. [DOI:10.1016/j.mce.2011.10.030]
Olanrewaju, F., Onayemi, O., Olasode, O., Adeyemi, A., Oninla, A., Oripelaye, M., Ezejiofor, O., & Oke, O. (2017). Prevalence and Pattern of Pigmentary Changes among Primigravidae Attending a Tertiary Health Facility in South-Western Nigeria. British Journal of Medicine and Medical Research, 21(5), 1–9. [DOI:10.9734/bjmmr/2017/33382]
Orentreich, N., & Durr, N. P. (1974). Mammogenesis in Transsexuals. Journal of Investigative Dermatology, 63(1), 142–146. [DOI:10.1111/1523-1747.ep12678272]
Pandya, S., & Moore, R. G. (2011). Breast Development and Anatomy. Clinical Obstetrics & Gynecology, 54(1), 91–95. [DOI:10.1097/grf.0b013e318207ffe9]
Park, I. Y., Kim, M. R., Jo, H. H., Lee, M. K., & Kim, M. J. (2014). Association of the Nipple-Areola Complexes with Age, Parity, and Breastfeeding in Korean Premenopausal Women. Journal of Human Lactation, 30(4), 474–479. [DOI:10.1177/0890334414549049]
Pasqualini, J. R. (2007). Progestins and breast cancer. Gynecological Endocrinology, 23(Suppl 1), 32–41. [DOI:10.1080/09513590701585003]
Pasqualini, J. R. (2009). Breast cancer and steroid metabolizing enzymes: The role of progestogens. Maturitas, 65(Suppl 1), S17–S21. [DOI:10.1016/j.maturitas.2009.11.006]
Pasqualini, J. R., & Kincl, F. A. (1985). Hormone Production and Concentrations During Pregnancy in Humans and in Other Mammalian Species. In Pasqualini, J. R., & Kincl, F. A. Hormones and the Fetus, Volume I: Production, Concentration and Metabolism During Pregnancy (pp. 173–334). Oxford: Pergamon Press. [DOI:10.1016/b978-0-08-019708-1.50007-6]
Patel, H., Arruarana, V., Yao, L., Cui, X., & Ray, E. (2020). Effects of hormones and hormone therapy on breast tissue in transgender patients: a concise review. Endocrine, 68(1), 6–15. [DOI:10.1007/s12020-020-02197-5]
Patel, K. T., Adeel, S., Rodrigues Miragaya, J., & Tangpricha, V. (2022). Progestogen Use in Gender-Affirming Hormone Therapy: A Systematic Review. Endocrine Practice, 28(12), 1244–1252. [DOI:10.1016/j.eprac.2022.08.012]
Patterson, M. N., McPhaul, M. J., & Hughes, I. A. (1994). Androgen insensitivity syndrome. Baillière’s Clinical Endocrinology and Metabolism, 8(2), 379–404. [DOI:10.1016/s0950-351x(05)80258-7]
Pawłowski, B., & Żelaźniewicz, A. (2021). The evolution of perennially enlarged breasts in women: a critical review and a novel hypothesis. Biological Reviews, 96(6), 2794–2809. [DOI:10.1111/brv.12778]
Perez-Palacios, G., & Jaffe, R. B. (1972). The Syndrome of Testicular Feminization. Pediatric Clinics of North America, 19(3), 653–667. [DOI:10.1016/s0031-3955(16)32744-4]
Petrakis, N. (1978). Recurrent extreme breast involution following pregnancy and susceptibility to breast cancer: A hypothesis. Medical Hypotheses, 4(3), 268–272. [DOI:10.1016/0306-9877(78)90006-3]
Pfeiffer, C. A. (1943). Endocrinology of Reproduction. Annual Review of Physiology, 5(1), 413–452. [DOI:10.1146/annurev.ph.05.030143.002213]
Pipkin, F. B. (2019). Physiological Changes in Pregnancy. In Symonds, I. M., & Arulkumaran, S. (Eds.). Essential Obstetrics and Gynaecology, 6th Edition (pp. 22–40). Edinburgh: Elsevier. [Google Books] [OpenLibrary] [WorldCat]
Plu-Bureau, G., Touraine, P., & Mauvais-Jarvis, P. (1999). Interactions Between Estradiol and Progesterone in Normal Breast: Implications for Mammary Carcinogenesis. In Manni, A. (Ed.). Endocrinology of Breast Cancer (Contemporary Endocrinology, Volume 11) (pp. 21–37). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-59259-699-7_2] [OpenLibrary] [Archive.org]
Pocock, G., Richards, C. D., & Richards, D. A. (2013). Fertilization, Pregnancy, and Lactation. In Pocock, G., Richards, C. D., & Richards, D. A. Human Physiology, 4th Edition (pp. 655–752). Oxford: Oxford University Press. [Google Scholar] [Google Books]
Polani, P. E. (1970). Hormonal and clinical aspects of hermaphroditism and the testicular feminizing syndrome in man. Philosophical Transactions of the Royal Society of London. B, Biological Sciences, 259(828), 187–206. [DOI:10.1098/rstb.1970.0058]
Price, S., McManus, J., & Barrett, J. (2019). The transgender population: improving awareness for gynaecologists and their role in the provision of care. The Obstetrician & Gynaecologist, 21(1), 11–20. [DOI:10.1111/tog.12521]
Prior, J. C. (2011). Progesterone for Symptomatic Perimenopause Treatment - Progesterone politics, physiology and potential for perimenopause. Facts, Views & Vision in ObGyn, 3(2), 109–120. [PubMed] [PubMed Central] [PDF]
Prior, J. C. (2019). Progesterone Is Important for Transgender Women’s Therapy—Applying Evidence for the Benefits of Progesterone in Ciswomen. The Journal of Clinical Endocrinology & Metabolism, 104(4), 1181–1186. [DOI:10.1210/jc.2018-01777]
Prior, J. C. (2019). Response to Letter to the Editor: “Progesterone Is Important for Transgender Women’s Therapy—Applying Evidence for the Benefits of Progesterone in Ciswomen”. The Journal of Clinical Endocrinology & Metabolism, 104(8), 3129–3130. [DOI:10.1210/jc.2019-00524]
Prior, J. C. (2020). Women’s Reproductive System as Balanced Estradiol and Progesterone Actions—A revolutionary, paradigm-shifting concept in women’s health. Drug Discovery Today: Disease Models, 32(Part B), 31–40. [DOI:10.1016/j.ddmod.2020.11.005]
Prior, J. C., Vigna, Y. M., & Watson, D. (1989). Spironolactone with physiological female steroids for presurgical therapy of male-to-female transsexualism. Archives of Sexual Behavior, 18(1), 49–57. [DOI:10.1007/bf01579291]
Prior, J. C., Vigna, Y. M., Watson, D., Diewold, P., & Robinow, O. (1986). Spironolactone in the presurgical therapy of male to female transsexuals: Philosophy and experience of the Vancouver Gender Dysphoria Clinic. Journal of Sex Information & Education Council of Canada, 1(1), 1–7. [Google Scholar] [PDF]
Quigley, C. A. (1998). The androgen receptor: Physiology and pathophysiology. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 33–106). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_2]
Quigley, C. A., Bellis, A. D., Marschke, K. B., El-Awady, M. K., Wilson, E. M., & French, F. S. (1995). Androgen Receptor Defects: Historical, Clinical, and Molecular Perspectives. Endocrine Reviews, 16(3), 271–321. [DOI:10.1210/edrv-16-3-271]
Radisky, D. C., & Hartmann, L. C. (2009). Mammary Involution and Breast Cancer Risk: Transgenic Models and Clinical Studies. Journal of Mammary Gland Biology and Neoplasia, 14(2), 181–191. [DOI:10.1007/s10911-009-9123-y]
Ramos, L., Chávez, B., Mares, L., Valdés, E., & Vilchis, F. (2018). Mutational analysis of the androgen receptor (NR3C4) gene in patients with 46,XY DSD. Gene, 641, 86–93. [DOI:10.1016/j.gene.2017.10.038]
Ramsay, D. T., Kent, J. C., Hartmann, R. A., & Hartmann, P. E. (2005). Anatomy of the lactating human breast redefined with ultrasound imaging. Journal of Anatomy, 206(6), 525–534. [DOI:10.1111/j.1469-7580.2005.00417.x]
Randolph, J. F. (2018). Gender-Affirming Hormone Therapy for Transgender Females. Clinical Obstetrics & Gynecology, 61(4), 705–721. [DOI:10.1097/grf.0000000000000396]
Rauh, C., Faschingbauer, F., Haeberle, L., Jud, S. M., Heusinger, K., Fasching, P. A., Goecke, T. W., Rajakaruna, N., Voigt, F., Bani, M. R., Lux, M. P., Renner, S. P., Loehberg, C. R., Hartmann, A., Schulz-Wendtland, R., Beckmann, M. W., & Bayer, C. M. (2013). Factors influencing breast changes after pregnancy. European Journal of Cancer Prevention, 22(3), 259–261. [DOI:10.1097/cej.0b013e328359cb81]
Rebar, R. W. (1988). The Ovaries. In Wyngaarden, J. B., & Smith, L. H. (Eds.). Cecil Textbook of Medicine, 18th Edition (pp. 1425–1446). Philadelphia: W. B. Saunders. [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Rebar, R. W. (1990). Disorders of Menstruation, Ovulation, and Sexual Response. In Becker, K. L., Bilezikian, J. P., Bremner, W. J., Hung, W., Kahn, C. R., Loriaux, D. L., Rebar, R. W., Robertson, G. L., & Wartofsky, L. (Eds.). Principles and Practice of Endocrinology and Metabolism, 1st Edition (pp. 798–814). Philadelphia: Lippincott. [Google Scholar—3rd/2001 Edition] [Google Books—3rd/2001 Edition] [OpenLibrary] [WorldCat] [Archive.org]
Rebar, R. W. (1993). Normal and Abnormal Sexual Differentiation and Pubertal Development. In Moore, T. R., Reiter, R. C., Rebar, R. W., & Baker, W. (Eds.). Gynecology & Obstetrics: A Longitudinal Approach (pp. 97–133). New York: Churchill Livingstone. [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Rebar, R. W. (1996). Puberty. In Berek, J. S., Adashi, E. Y., & Hillard, P. A. (Eds.). Novak’s Gynecology, 12th Edition (pp. 771–807). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Reindollar, R. H., & McDonough, P. G. (1985). The Child with Ambiguous Genitalia. In Lavery, J. P., & Sanfilippo, J. S. (Eds.). Pediatric and Adolescent Obstetrics and Gynecology, 1st Edition (Clinical Perspective in Obstetrics and Gynecology) (pp. 38–60). New York: Springer. [DOI:10.1007/978-1-4612-5064-7_5]
Reisman, T., Goldstein, Z., & Safer, J. D. (2019). A Review of Breast Development in Cisgender Women and Implications for Transgender Women. Endocrine Practice, 25(12), 1338–1345. [DOI:10.4158/ep-2019-0183]
Richards, C., & Barrett, J. (2020). Physical Treatments for Trans People and Their Interactions with Psychiatric Treatments. In Richards, C., & Barrett, J. Trans and Non-binary Gender Healthcare for Psychiatrists, Psychologists, and Other Health Professionals (pp. 31–44). Cambridge: Cambridge University Press. [DOI:10.1017/9781108628419.003]
Richert, M. M., Schwertfeger, K. L., Ryder, J. W., & Anderson, S. M. (2000). An Atlas of Mouse Mammary Gland Development. Journal of Mammary Gland Biology and Neoplasia, 5(2), 227–241. [DOI:10.1023/a:1026499523505]
Rix, J., Mills, C., Ross, E., Allen, S., Lai, A., & Wakefield-Scurr, J. (2023). Breast volume fluctuations are associated with oestradiol and progesterone changes across the menstrual cycle. Research Square, preprint. [Google Scholar] [DOI:10.21203/rs.3.rs-3753080/v1] [PDF]
Rodari, G. (2022). Pubertal induction in girls with hypogonadism: estrogen replacement therapy outcomes and optimization of progesterone introduction. (Doctoral thesis, Università degli Studi di Milano.) [URL]
Rodari, G., Federici, S., Cattoni, A., Todisco, T., Ubertini, G., Giacchetti, F., Profka, E., Dall’Antonia, A., Cangiano, B., Arosio, M., Bonomi, M., Cappa, M., & Giavoli, C. (2022). Pubertal induction in girls with hypogonadism: insight into estrogen replacement therapy outcomes and optimization of progesterone introduction. Endocrine Abstracts, 81 [24th European Congress of Endocrinology 2022 21–24 May 2022, Milan, Italy], 107–107 (abstract no. RC12.7). [DOI:10.1530/endoabs.81.rc12.7] [PDF]
Rodari, G., Federici, S., Todisco, T., Ubertini, G., Cattoni, A., Pagano, M., Giacchetti, F., Profka, E., Citterio, V., Messetti, D., Collini, V., Soranna, D., Carbone, E., Arosio, M., Mantovani, G., Persani, L., Cappa, M., Bonomi, M., & Giavoli, C. (2023). Towards an individualized management of pubertal induction in girls with hypogonadism: insight into the best replacement outcomes from a large multicentre registry. European Journal of Endocrinology, 188(6), 467–476. [DOI:10.1093/ejendo/lvad056]
Rohn, R. D. (1989). Nipple (papilla) development in girls: III. Journal of Adolescent Health Care, 10(1), 39–40. [DOI:10.1016/0197-0070(89)90045-4]
Rosenfield, R. L. (2013). Adolescent Anovulation: Maturational Mechanisms and Implications. The Journal of Clinical Endocrinology & Metabolism, 98(9), 3572–3583. [DOI:10.1210/jc.2013-1770]
Rosenfield, R. L., Cooke, D. W., & Radovick, S. (2021). Puberty in the Female and Its Disorders. In Sperling, M. A., Majzoub, J. A., Menon, R. K., & Stratakis, C. A. (Eds.). Sperling Pediatric Endocrinology, 5th Edition (pp. 528–626). Philadelphia: Elsevier. [DOI:10.1016/B978-0-323-62520-3.00016-6]
Rothman, M. S., & Iwamoto, S. J. (2022). Feminizing Gender-Affirming Hormone Therapy: Special Considerations for Older Adults. In Davis, T. F. (Ed.). A Case-Based Guide to Clinical Endocrinology (pp. 513–523). Cham: Springer. [DOI:10.1007/978-3-030-84367-0_58]
Ruan, W., Monaco, M. E., & Kleinberg, D. L. (2005). Progesterone Stimulates Mammary Gland Ductal Morphogenesis by Synergizing with and Enhancing Insulin-Like Growth Factor-I Action. Endocrinology, 146(3), 1170–1178. [DOI:10.1210/en.2004-1360]
Rutgers, J. L., & Scully, R. E. (1991). The Androgen Insensitivity Syndrome (Testicular Feminization). International Journal of Gynecological Pathology, 10(2), 126–144. [DOI:10.1097/00004347-199104000-00002]
Ryan, R. F., & Pernoll, M. L. (1985). Virginal Hypertrophy. Plastic and Reconstructive Surgery, 75(5), 737–742. [DOI:10.1097/00006534-198505000-00024]
Saito, R., Yamamoto, Y., Goto, M., Araki, S., Kubo, K., Kawagoe, R., Kawada, Y., Kusuhara, K., Igarashi, M., & Fukami, M. (2014). Tamoxifen Treatment for Pubertal Gynecomastia in Two Siblings with Partial Androgen Insensitivity Syndrome. Hormone Research in Paediatrics, 81(3), 211–216. [DOI:10.1159/000356923]
Sam. (2020). Sources/Excerpts: Perspectives and Opinions of Clinicians and Researchers Concerning the Use of Progestogens in Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Sandhu, R., Chollet-Hinton, L., Kirk, E. L., Midkiff, B., & Troester, M. A. (2016). Digital histologic analysis reveals morphometric patterns of age-related involution in breast epithelium and stroma. Human Pathology, 48, 60–68. [DOI:10.1016/j.humpath.2015.09.031]
Santen, R. J., Karaguzel, G., Livaoglu, M., Yue, W., Cline, J. M., Ratan, A., & Sasano, H. (2024). Role of ERα and aromatase in juvenile gigantomastia. The Journal of Clinical Endocrinology and Metabolism, online ahead of print. [DOI:10.1210/clinem/dgae019]
Sanuki, J., Fukuma, E., & Uchida, Y. (2008). Morphologic Study of Nipple-Areola Complex in 600 Breasts. Aesthetic Plastic Surgery, 33(3), 295–297. [DOI:10.1007/s00266-008-9194-y]
Satoh, K., Hovey, R. C., Malewski, T., Warri, A., Goldhar, A. S., Ginsburg, E., Saito, K., Lydon, J. P., & Vonderhaar, B. K. (2007). Progesterone enhances branching morphogenesis in the mouse mammary gland by increased expression of Msx2. Oncogene, 26(54), 7526–7534. [DOI:10.1038/sj.onc.1210555]
Schauffler, G. C. (1942). Disorders During Adolescence—The Onset of Menstruation. In Schauffler, G. C. Pediatric Gynecology; with Sections on Urology and Proctology, 1st Edition (pp. 169–211). Chicago: Year Book. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL]
Schindler, A. E. (2010). Dydrogesterone and other progestins in benign breast disease: an overview. Archives of Gynecology and Obstetrics, 283(2), 369–371. [DOI:10.1007/s00404-010-1456-7]
Schlatterer, K., Yassouridis, A., Werder, K. V., Poland, D., Kemper, J., & Stalla, G. K. (1998). Archives of Sexual Behavior, 27(5), 475–492. [DOI:10.1023/a:1018704630036]
Scott, P. P., Greene, R., Lane-Roberts, C. S., & Swain, V. (1950). The Function of the Breast. In Saner, F. D. (Ed.). The Breast: Structure, Function, Disease (pp. 53–86). Baltimore: Williams and Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL] [PDF]
Seal, L. (2017). Adult Endocrinology. In Richards, C., Bouman, W. P., & Barker, M.-J. (Eds.). Genderqueer and Non-Binary Genders (Critical and Applied Approaches in Sexuality, Gender and Identity) (pp. 183–223). London: Palgrave Macmillan UK. [DOI:10.1057/978-1-137-51053-2_10]
Seal, L. J., Franklin, S., Richards, C., Shishkareva, A., Sinclaire, C., & Barrett, J. (2012). Predictive Markers for Mammoplasty and a Comparison of Side Effect Profiles in Transwomen Taking Various Hormonal Regimens. The Journal of Clinical Endocrinology & Metabolism, 97(12), 4422–4428. [DOI:10.1210/jc.2012-2030]
Seibert, B., & Günzel, P. (1994). Animal toxicity studies performed for risk assessment of the once-a-month injectable contraceptive Mesigyna®. Contraception, 49(4), 303–333. [DOI:10.1016/0010-7824(94)90030-2]
Shapiro, L. R. (1982). Disorders of Female Sex Differentiation. In Blaustein, A. (Ed.). Pathology of the Female Genital Tract, 2nd Edition (pp. 479–510). New York: Springer New York. [DOI:10.1007/978-1-4757-1767-9_20]
Shearman, R. P. (1972). Ovarian Function and its Control. In Shearman, R. P. (Ed.). Human Reproductive Physiology, 1st Edition (pp. 45–90). Oxford: Blackwell Scientific Publications. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Shearman, R. P. (1972). Primary Ammenorhoea. In Dewhurst, C. J. (Ed.). Integrated Obstetrics and Gynaecology for Postgraduates, 1st Edition (pp. 55–62). Oxford: Blackwell Scientific Publications. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
Shi, H. Y., Lydon, J. P., & Zhang, M. (2004). Hormonal Defect in Maspin Heterozygous Mice Reveals a Role of Progesterone in Pubertal Ductal Development. Molecular Endocrinology, 18(9), 2196–2207. [DOI:10.1210/me.2004-0052]
Shuttleworth, F. K. (1938). The Adolescent Period: A Graphic and Pictorial Atlas (Monographs of the Society for Research in Child Development, Volume 3, Number 3 / Serial No. 16). Washington, D. C.: Society for Research in Child Development. [Google Scholar] [Google Books] [DOI:10.2307/1165482]
Simmer, H. H., Pion, R. J., & Dignam, W. J. (1965). Testicular Feminization: Endocrine Function of Feminizing Testes, Comparison with Normal Testes. Springfield, Illinois: Thomas. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Simpson, J. L., & Rebar, R. W. (1990). Normal and Abnormal Sexual Differentiation and Development. In Becker, K. L., Bilezikian, J. P., Bremner, W. J., Hung, W., Kahn, C. R., Loriaux, D. L., Rebar, R. W., Robertson, G. L., & Wartofsky, L. (Eds.). Principles and Practice of Endocrinology and Metabolism, 1st Edition (pp. 710–739). Philadelphia: Lippincott. [Google Scholar] [Google Books—3rd/2001 Edition] [OpenLibrary] [WorldCat] [Archive.org]
Sindi, R., Sá Dos Reis, C., Bennett, C., Stevenson, G., & Sun, Z. (2019). Quantitative Measurements of Breast Density Using Magnetic Resonance Imaging: A Systematic Review and Meta-Analysis. Journal of Clinical Medicine, 8(5), 745. [DOI:10.3390/jcm8050745]
Sitruk-Ware, R., Basdevant, A., de Lignières, B., & Mauvais-Jarvis, P. (1984). Percutaneous oestrogen therapy. In van Herendael, H., van Herendael, B., Riphagen, F. E., Goessens, L., & van der Plas, H. (Eds.). The Climacteric: An Update: Proceedings of the fourth Jan Palfijn Symposium, European Conference on the Menopause, held in Antwerp, Belgium, on September 1–2, 1983, under the auspices of ‘De Vereniging voor Nederlandstalige gynecologen van België’ and ‘The International Menopause Society’ (pp. 127–139). Lancaster: MTP Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [DOI:10.1007/978-94-009-5608-7_14]
Sizonenko, P. C. (1978). Endocrinology in Preadolescents and Adolescents. American Journal of Diseases of Children, 132(7), 704–712. [DOI:10.1001/archpedi.1978.02120320064015]
Škarda, J., Fremrová, V., & Bezecný, I. (1989). Progesterone alone is responsible for stimulation of the growth of ducts and of mammary alveolar structures in mice. Endocrinologia Experimentalis, 23(1), 17–28. [Google Scholar] [PubMed] [PDF]
Sobrinho, L. G., Kase, N., & Grunt, J. A. (1971). Spontaneous pubertal breast growth in a castrated patient with the syndrome of testicular feminization. The Yale Journal of Biology and Medicine, 44(2), 225–9. [Google Scholar] [PubMed] [PubMed Central] [PDF]
Sosa, M., Jódar, E., Arbelo, E., Domínguez, C., Saavedra, P., Torres, A., Salido, E., de Tejada, M. J., & Hernández, D. (2003). Bone Mass, Bone Turnover, Vitamin D, and Estrogen Receptor Gene Polymorphisms in Male to Female Transsexuals. Journal of Clinical Densitometry, 6(3), 297–304. [DOI:10.1385/jcd:6:3:297]
Sosa, M., Jódar, E., Arbelo, E., Domı́nguez, C., Saavedra, P., Torres, A., Salido, E., Limiñana, J., Gómez de Tejada, M. J., & Hernández, D. (2004). Serum lipids and estrogen receptor gene polymorphisms in male-to-female transsexuals: effects of estrogen treatment. European Journal of Internal Medicine, 15(4), 231–237. [DOI:10.1016/j.ejim.2004.04.009]
Soyal, S., Ismail, P. M., Li, J., Mulac-Jericevic, B., Conneely, O. M., & Lydon, J. P. (2002). Progesterone receptors - animal models and cell signaling in breast cancer: Progesterone’s role in mammary gland development and tumorigenesis as disclosed by experimental mouse genetics. Breast Cancer Research, 4(5), 191. [DOI:10.1186/bcr451]
Sperling, R. L., & Gold, J. J. (1973). Use of an anti-estrogen after a reduction mammaplasty to prevent recurrence of virginal hypertrophy of breasts. Plastic and Reconstructive Surgery, 52(4), 439–442. [DOI:10.1097/00006534-197352040-00030]
Spitz, I. M., Shoupe, D., Sitruk-Ware, R., & Mishell, D. R. (1989). Response to the antiprogestagen RU 486 (mifepristone) during early pregnancy and the menstrual cycle in women. Journal of Reproduction and Fertility Supplement, 37, 253–260. [Google Scholar] [PubMed]
Spitz, I., Croxatto, H., Lahteenmaki, P., Heikinheimo, O., & Bardin, C. (1994). Effect of mifepristone on inhibition of ovulation and induction of luteolysis. Human Reproduction, 9(Suppl 1), 69–76. [DOI:10.1093/humrep/9.suppl_1.69]
Spitz, I. M. (2010). Mifepristone: where do we come from and where are we going? Contraception, 82(5), 442–452. [DOI:10.1016/j.contraception.2009.12.012]
Sridhar, G. R., & Sinha, M. J. (1995). Macromastia in adolescent girls. Indian Pediatrics, 32(4), 496–499. [Google Scholar] [PubMed] [PDF]
Sun, B. Z., Kangarloo, T., Adams, J. M., Sluss, P. M., Welt, C. K., Chandler, D. W., Zava, D. T., McGrath, J. A., Umbach, D. M., Hall, J. E., & Shaw, N. D. (2018). Healthy Post-Menarchal Adolescent Girls Demonstrate Multi-Level Reproductive Axis Immaturity. The Journal of Clinical Endocrinology & Metabolism, 104(2), 613–623. [DOI:10.1210/jc.2018-00595]
Sun, S. X., Bostanci, Z., Kass, R. B., Mancino, A. T., Rosenbloom, A. L., Klimberg, V. S., & Bland, K. I. (2018). Breast Physiology: Normal and Abnormal Development and Function. In Bland, K. I., Copeland, E. M., Klimberg, V. S., Gradishar, W. J., White, J., & Korourian, S. (Eds.). The Breast: Comprehensive Management of Benign and Malignant Diseases, 5th Edition (pp. 37–56.e6). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-35955-9.00003-9]
Swelstad, M. R., Swelstad, B. B., Rao, V. K., & Gutowski, K. A. (2006). Management of Gestational Gigantomastia. Plastic and Reconstructive Surgery, 118(4), 840–848. [DOI:10.1097/01.prs.0000232364.40958.47]
Tack, L. J., Heyse, R., Craen, M., Dhondt, K., Bossche, H. V., Laridaen, J., & Cools, M. (2017). Consecutive Cyproterone Acetate and Estradiol Treatment in Late-Pubertal Transgender Female Adolescents. The Journal of Sexual Medicine, 14(5), 747–757. [DOI:10.1016/j.jsxm.2017.03.251]
Talbert, L. M., Hammond, M. G., Groff, T., & Udry, J. R. (1985). Relationship of age and pubertal development to ovulation in adolescent girls. Obstetrics & Gynecology, 66(4), 542–544. [Google Scholar] [PubMed] [URL]
Tanos, T., & Brisken, C. (2008). What Signals Operate in the Mammary Niche? Breast Disease, 29(1), 69–82. [DOI:10.3233/bd-2008-29108]
Thanaboonyawat, I., Chanprapaph, P., Lattalapkul, J., & Rongluen, S. (2013). Pilot Study of Normal Development of Nipples during Pregnancy. Journal of Human Lactation, 29(4), 480–483. [DOI:10.1177/0890334413493350]
Thody, A. J., & Smith, A. G. (1977). Hormones and Skin Pigmentation in the Mammal. International Journal of Dermatology, 16(8), 657–664. [DOI:10.1111/j.1365-4362.1977.tb01876.x]
Thoresen, M., & Wesche, J. (1988). Doppler measurements of changes in human mammary and uterine blood flow during pregnancy and lactation. Acta Obstetricia et Gynecologica Scandinavica, 67(8), 741–745. [DOI:10.3109/00016349809004301]
Tiefenbacher, K., & Daxenbichler, G. (2008). The Role of Androgens in Normal and Malignant Breast Tissue. Breast Care, 3(5), 325–331. [DOI:10.1159/000158055]
Tim, F., Jeroen, V., & T’Sjoen, G. (2023). Dose Reduction of Cyproterone Acetate in Trans Women and the Effect on Patient-reported Outcomes: Results from the ENIGI Study. Endocrine Abstracts, 97 [Belgian Endocrine Society 2023], 5–5 (abstract no. 007). [URL] [PDF]
Trabert, B., Sherman, M. E., Kannan, N., & Stanczyk, F. Z. (2019). Progesterone and Breast Cancer. Endocrine Reviews, 41(2), 320–344. [DOI:10.1210/endrev/bnz001]
Tucker, H. (2000). Hormones, Mammary Growth, and Lactation: a 41-Year Perspective. Journal of Dairy Science, 83(4), 874–884. [DOI:10.3168/jds.s0022-0302(00)74951-4]
Turan, S., Bereket, A., Guran, T., Akcay, T., Papari-Zareei, M., & Auchus, R. J. (2009). Puberty in a case with novel 17-hydroxylase mutation and the putative role of estrogen in development of pubic hair. European Journal of Endocrinology, 160(2), 325–330. [DOI:10.1530/eje-08-0632]
Turner, C. W. (1939). The Mammary Glands. In Allen, E., Danforth, C. H., & Doisy, E. A. (Eds.). Sex and Internal Secretions: A Survey of Recent Research, 2nd Edition (pp. 740–803). Baltimore: Williams & Wilkins. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Ulrich, U., Pfeifer, T., & Lauritzen, C. (1994). Rapid Increase in Lumbar Spine Bone Density in Osteopenic Women by High-Dose Intramuscular Estrogen-Progestogen Injections. Hormone and Metabolic Research, 26(9), 428–431. [DOI:10.1055/s-2007-1001723]
Ulrich, U., Pfeifer, T., Buck, G., Keckstein, J., & Lauritzen, C. (1995). High-dose estrogen-progestogen injections in gonadal dysgenesis, ovarian hypoplasia, and androgen insensitivity syndrome: Impact on bone density. Adolescent and Pediatric Gynecology, 8(1), 20–23. [DOI:10.1016/s0932-8610(12)80156-3]
Valentine, G. H. (1969). Chromosome Anomalies in the Female. In Valentine, G. H. The Chromosome Disorders: An Introduction for Clinicians, 2nd Edition (pp. 126–143). London: W. Heinemann Medical Books. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]
van der Meulen, A. J. (1974). An unusual case of massive hypertrophy of the breasts. South African Medical Journal=Suid-Afrikaanse Tydskrif vir Geneeskunde, 48(34), 1465–1466. [Google Scholar] [PubMed] [URL 1] [URL 2]
Vandeweyer, E., & Hertens, D. (2002). Quantification of glands and fat in breast tissue: An experimental determination. Annals of Anatomy - Anatomischer Anzeiger, 184(2), 181–184. [DOI:10.1016/s0940-9602(02)80016-4]
Venturoli, S., Fabbri, R., Porcu, E., Paradisi, R., Orsini, L. F., Brondelli, L., Ruggeri, S., & Flamigni, C. (1989). Endocrine and ovarian parameters at various frequencies of ovulation in adolescents. Archives of Gynecology and Obstetrics, 246(2), 107–114. [DOI:10.1007/bf00934127]
Venturoli, S., Porcu, E., Fabbri, R., Magrini, O., Paradisi, R., Pallotti, G., Gammi, L., & Flamigni, C. (1987). Postmenarchal evolution of endocrine pattern and ovarian aspects in adolescents with menstrual irregularities. Fertility and Sterility, 48(1), 78–85. [DOI:10.1016/s0015-0282(16)59294-2]
Vorherr, H. (1974). Development of the Female Breast. In Vorherr, H. The Breast: Morphology, Physiology, and Lactation (pp. 1–19). New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Vorherr, H. (1974). Morphology of the Mature Female Breast. In Vorherr, H. The Breast: Morphology, Physiology, and Lactation (pp. 20–70). New York: Academic Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Wade, T. R., Wade, S. L., & Jones, H. E. (1978). Skin changes and diseases associated with pregnancy. Obstetrics and Gynecology, 52(2), 233–242. [Google Scholar] [PubMed] [URL]
Walker, M., Baker, G., & Lamb, M. (2013). Physiology of the Breast During Pregnancy and Lactation. In Mannel, R., Martens, P. J., & Walker, M. (Eds.). Core Curriculum for Lactation Consultant Practice, 3rd Edition (pp. 287–300). Burlington: Jones & Bartlett Learning. [Google Scholar] [Google Books—2013/3rd Edition] [Google Books—2008/2nd Edition] [OpenLibrary] [WorldCat]
Wang, C., Luan, J., Cheng, H., Chen, L., Li, Z., Panayi, A. C., & Liu, C. (2018). Menstrual Cycle-Related Fluctuations in Breast Volume Measured Using Three-Dimensional Imaging: Implications for Volumetric Evaluation in Breast Augmentation. Aesthetic Plastic Surgery, 43(1), 1–6. [DOI:10.1007/s00266-018-1243-6]
Wang, Q. A., & Scherer, P. E. (2019). Remodeling of Murine Mammary Adipose Tissue during Pregnancy, Lactation, and Involution. Journal of Mammary Gland Biology and Neoplasia, 24(3), 207–212. [DOI:10.1007/s10911-019-09434-2]
Weisberg, M. G., Malkasian, G. D., & Pratt, J. H. (1970). Testicular feminization syndrome. American Journal of Obstetrics and Gynecology, 107(8), 1181–1187. [DOI:10.1016/s0002-9378(15)30367-7]
Wellons, M. F., & Rebar, R. W. (2013). Amenorrhea. In Falcone, T., & Hurd, W. W. (Eds.). Clinical Reproductive Medicine and Surgery: A Practical Guide, 2nd Edition (pp. 105–112). New York: Springer New York. [DOI:10.1007/978-1-4614-6837-0_7]
Wellons, M. F., Weeber, K. M., & Rebar, R. W. (2017). Amenorrhea. In Falcone, T., & Hurd, W. W. (Eds.). Clinical Reproductive Medicine and Surgery, 3rd Edition (pp. 109–122). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-52210-4_6]
Werner, A. A. (1935). Experiment to produce lactation in castrate women. Endocrinology, 19(2), 144–150. [DOI:10.1210/endo-19-2-144]
Whiteley, S. (1994). Predictors of Milk Production in Lactating Women. (Master’s thesis, The Open University.) [Google Scholar] [DOI:10.21954/ou.ro.0000fe02] [URL]
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical Review: Breast Development in Trans Women Receiving Cross-Sex Hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]
Wierckx, K., Van Caenegem, E., Schreiner, T., Haraldsen, I., Fisher, A., Toye, K., Kaufman, J. M., & T’Sjoen, G. (2014). Cross‐Sex Hormone Therapy in Trans Persons Is Safe and Effective at Short‐Time Follow‐Up: Results from the European Network for the Investigation of Gender Incongruence. The Journal of Sexual Medicine, 11(8), 1999–2011. [DOI:10.1111/jsm.12571]
Wilson, J. D. (1968 March 28). Medical Grand Rounds: Testicular Feminization. Parkland Memorial Hospital, UT Southwestern Medical Center. [Google Scholar] [URL] [PDF]
Wilson, C. L., Sims, A. H., Howell, A., Miller, C. J., & Clarke, R. B. (2006). Effects of oestrogen on gene expression in epithelium and stroma of normal human breast tissue. Endocrine-Related Cancer, 13(2), 617–628. [DOI:10.1677/erc.1.01165]
Winkler, U. H., Schindler, A. E., Brinkmann, U. S., Ebert, C., & Oberhoff, C. (2001). Cyclic progestin therapy for the management of mastopathy and mastodynia. Gynecological Endocrinology, 15(Suppl 6), 37–43. [DOI:10.1080/gye.15.s6.37.43]
Wisniewski, A. B., Migeon, C. J., Meyer-Bahlburg, H. F., Gearhart, J. P., Berkovitz, G. D., Brown, T. R., & Money, J. (2000). Complete Androgen Insensitivity Syndrome: Long-Term Medical, Surgical, and Psychosexual Outcome. The Journal of Clinical Endocrinology & Metabolism, 85(8), 2664–2669. [DOI:10.1210/jcem.85.8.6742]
Wong, R. C., & Ellis, C. N. (1984). Physiologic skin changes in pregnancy. Journal of the American Academy of Dermatology, 10(6), 929–940. [DOI:10.1016/s0190-9622(84)80305-9]
Wren, B. G., & Eden, J. A. (1996). Do Progestogens Reduce The Risk of Breast Cancer? A Review of the Evidence. Menopause, 3(1), 4–12. [DOI:10.1097/00042192-199603010-00003]
Wright, E. M. (2015). Breastfeeding and the Mother–Newborn Dyad (pp. 1157–1182). In King, T. L., Brucker, M. C., Kriebs, J. M., Fahey, J. O., Gegor, C. L., & Varney, H. (Eds.). Varney’s Midwifery, 5th Edition. Burlington: Jones & Bartlett Learning. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Xu, P., Ye, W., Zhong, S., Li, H., Feng, E., Lin, S. H., Kuo, C. T., Liu, J. Y., & Lin, Y. C. (2010). Leptin and zeranol up-regulate cyclin D1 expression in primary cultured normal human breast pre-adipocytes. Molecular Medicine Reports, 3(6), 983–990. [DOI:10.3892/mmr.2010.370]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Zacharin, M. (2000). Use of androgens and oestrogens in adolescents - A review of hormone replacement treatment. Journal of Pediatric Endocrinology and Metabolism, 13(1), 3–12. [DOI:10.1515/JPEM.2000.13.1.3]
Zachmann, M., Prader, A., Sobel, E. H., Crigler, J. F., Ritzén, E. M., Atarés, M., & Ferrandez, A. (1986). Pubertal growth in patients with androgen insensitivity: Indirect evidence for the importance of estrogens in pubertal growth of girls. The Journal of Pediatrics, 108(5), 694–697. [DOI:10.1016/s0022-3476(86)81043-5]
Żelaźniewicz, A., & Pawłowski, B. (2015). Breast size and asymmetry during pregnancy in dependence of a fetus’s sex. American Journal of Human Biology, 27(5), 690–696. [DOI:10.1002/ajhb.22716]
Żelaźniewicz, A., & Pawłowski, B. (2018). Maternal breast volume in pregnancy and lactation capacity. American Journal of Physical Anthropology, 168(1), 180–189. [DOI:10.1002/ajpa.23734]
Zhang, D., Yao, F., Tian, T., Deng, S., Luo, M., & Tian, Q. (2021). Clinical characteristics and molecular genetics of complete androgen insensitivity syndrome patients: a series study of 30 cases from a Chinese tertiary medical center. Fertility and Sterility, 115(5), 1270–1279. [DOI:10.1016/j.fertnstert.2020.12.008]
\ No newline at end of file
diff --git a/transfemscience.org/articles/progestogens-early-exposure-breast-dev/index.html b/transfemscience.org/articles/progestogens-early-exposure-breast-dev/index.html
index dd017153..6653055f 100644
--- a/transfemscience.org/articles/progestogens-early-exposure-breast-dev/index.html
+++ b/transfemscience.org/articles/progestogens-early-exposure-breast-dev/index.html
@@ -1 +1 @@
-Literature on Early Progestogen Exposure and Breast Development - Transfeminine Science
Literature on Early Progestogen Exposure and Breast Development
By Aly | First published July 21, 2019 | Last modified March 31, 2024
Preface
This is a sources and excerpts supplement for the main article section which can be found here.
Animal Studies
Lyons & McGinty (1941)
Lyons, W. R., & McGinty, D. A. (1941). Effects of estrone and progesterone on male rabbit mammary glands. I. Varying doses of progesterone. Proceedings of the Society for Experimental Biology and Medicine, 48(1), 83–86. [DOI:10.3181/00379727-48-13227]:
Summary. Eighteen doses of 0.25, 1.0, 4.0 and 8.0 I.U. of crystalline progesterone were given, simultaneously with 120 I.U. of estrone, to immature male rabbits during a period of 4 weeks. Of these 4 levels of progesterone, the 1.0 I.U. dose synergized best although the prolactational proliferation induced was not maximal. The 4.0 and 8.0 I.U. doses of progesterone were apparently inhibiting as shown by the relatively poor mammary growth obtained.
[Introduction.] Turner and Frank1 showed that whereas estrogen caused growth of the duct system and slight lobule proliferation in the male rabbit mammary gland, the combination of estrogen and progestogen caused lobule-alveolar growth simulating that seen in pregnancy. The hormones used were impure, but were assayed for rat units of estrogen and rabbit units of progestogen and judged by the results, were uncontaminated, one with the other. Until recently few investigators have had at their disposal sufficient progesterone with which to investigate this problem further, and, as, far as we are aware, the doses of estrone and progesterone that will synergize best to cause optimal mammary growth have not as yet been satisfactorily determined in any animal form. In the preliminary investigation reported herein, an attempt was made to determine the approximate dose of progesterone that would function optimally with a given daily dose (120 I.U.) of estrone in causing prolactational* growth of the male rabbit mammary gland.
Experimental. Immature, New Zealand White male rabbits weighing approximately 1.5 kg at the beginning of the experiment were treated in groups of 3 with 18 daily doses (Monday through Friday, from 2/28/39 until 3/23/39) of the following:
Group 1, 120 I.U. of estrone (theelin); Group 2, 1 I.U. of progesterone; Group 3, 120 I.U. of E and 0.25 I.U. of P; Group 4, 120 I. U. of E and 1 I.U. of P; Group 5, 1201 I.U. of E and 4 I.U. of P; Group 6, 120 I.U. of E and 8 I.U. of P. The 2 hormones were given separately, subcutaneously, in peanut oil. On 3/28/39, 5 days after the last injection, a biopsy specimen of the second left (thoracic) mammary gland was taken from each animal, fixed in formol, stained in toto with alum-carmine and cleared in methyl salicylate.
Results. The maniniary spreads from Group 1, showed that the dose of 120 I.U. of E produced good duct growth with almost negligible alveolar formation (Fig. 2 [E Alone]). Those from Group 2 (1 I.U. of P) showed but little more development than that seen in normal immature rabbit glands (Fig. 1 [Untreated]). The duct system of the glands from animals in Group 3 (120 I.U. of E plus 0.25 I.U. of P) was as extensive as that seen in animals treated with 120 I.U. of E alone, but the main ducts were narrower and, as though in compensation. more alveolar buds were present. Thus, just as progesterone in some experimental animals prevents the estrogen-induced uterine dilatation and stimulates a proliferation of luminal and glandular epithelium, so also in the rabbit it counteracts an estrogen-induced mammary duct dilatation and permits instead an extensive alveolar proliferation. The amount of incomplete lobule formation shown in Fig. 3 [E + P4 0.25 mg] was found typical of all rabbits in this group and probably may be interpreted as barely minimal prolactational proliferation in response to a low dose of progesterone. The glands from animals in Group 4 (120 I.U. of E and 1.0 I.U. of P) showed the best evidence of synergism obtained in this preliminary experiment (Fig. 4 [E + P4 1 mg]), although judged on the basis of a set of glands taken at different stages of pregnancy, they could not be said to show maximal prolactational proliferation. Interesting results were obtained in Groups 5 and 6, where 2 of the animals in Group 5 and all in Group 6 showed only scanty alveolar development and an inhibition of the duct growth (Fig. 5 [E + P4 8 mg]). The third animal in Group 5 showed good prolactational development with no inhibition of duct growth, indicating that 4 I.U. of progesterone approximated the border-line inhibiting dose when given with 120 I.U. of estrone by this particular routine.
All figures represent approximately one-half of a male rabbit mammary spread fixed in formol and stained in alum-carmine. × 1.5.
Treatment was as follows:
FIG. 1. None. Control gland. [Untreated]
FIG. 2. 18 subcutaneous doses of 120 I.U. estrone in oil over a 28-day period. Duct growth with almost no alveolar development. [E Alone]
FIG. 3. 18 subcutaneous doses of 120 I.U. estrone and 0.25 I.U. progesterone over same period. Very little prolactational proliferation. [E + P4 0.25 mg]
FIG. 4. 18 subcutaneous doses of 120 I.U. estrone and 1 I.U. progesterone over same period. Fair prolactational proliferation. [E + P4 1 mg]
FIG. 5. 18 subcutaneous doses of 120 I.U. estrone and 8 I.U. progesterone over same period. Inhibited duct growth and only slight alveolar development. [E + P4 8 mg]
Note: A citing review indicated that 1 IU progesterone is equal to 1 mg progesterone (Pfeiffer, 1943).
Beyer, Cruz, & Martinez-Manautou (1970)
Beyer, C., Cruz, M. L., & Martinez-Manautou, J. (1970). Effect of Chlormadinone Acetate on Mammary Development and Lactation in the Rabbit. Endocrinology, 86(5), 1172–1174. [DOI:10.1210/endo-86-5-1172]:
ABSTRACT. The effect of chlormadinone acetate (CA) on mammary growth and lactation was studied in the New Zealand white rabbit. Mammary development studies were performed in 5-month males castrated prepuberally. All rabbits were treated for 28 days. Ten μg estrone (E) produced good ductal development without alveolar growth (mean mammary area = 376 mm2). CA alone did not stimulate mammary growth. Administration of 0.5 mg CA plus 10 μg E produced optimal ductal and alveolar development (mean mammary area = 765 mm2). By contrast, a larger dose of CA (2.5 mg) combined with 10 μg E resulted in a much smaller mammary response (mean mammary area = 284 mm2). Administration of 100 μg or 2.5 mg CA from day 10 to 25 of lactation neither inhibited nor facilitated milk production in lactating rabbits. It is concluded that prolonged administration of CA when combined with estrogen stimulates mammary growth at moderate doses (0.5 mg) and inhibits it at large doses (2.5 mg). CA has no clear effect on milk production and/or removal in lactating rabbits. (Endocrinology86: 1172, 1970)
[Introduction]
CHLORMADINONE acetate (CA) is a potent progestational steroid widely employed in controlling human fertility (1). It has been reported that prolonged treatment with some progestational compounds can alter mammary structure and lactation in some mammalian species, including women (2). Yet no detailed study has been done on the action of CA on the mammary gland. Therefore, we considered it interesting to analyze the effect of prolonged administration of CA upon mammary growth and lactation in New Zealand white rabbits.
Results
The mammary glands of the control castrated males were not visible. When studied microscopically, they were found to consist of short primary ducts that hardly extended beyond the nipple area. Injections of 500 μg CA did not stimulate mammary growth. Administration of 10 μg E induced clear growth and ramification of the ductal system. The mean area (376 ± 41 mm2 SE) occupied by these glands was significantly larger than in the control or CA treated rabbits. These glands were composed exclusively of ductal epithelium without alveoli. Combined administration of 10 μg E and 2.5 mg progesterone (P) resulted in a good ductoalveolar development. As can be seen in Fig. 1A [E + P4 2.5 mg] the ducts were almost covered with alveoli. The area occupied by these glands was significantly larger than that of the E treated rabbits (688 ± 77 mm2 SE, group 3 vs. 4, p = .004, U test). No milk secretion was noted. Administration of both E and CA at the low dose level (0.5 mg) produced marked ducto-alveolar development (Fig. 1B [E + CMA 0.5 mg]). The mammary area in these animals was more extended than in all other treated groups (765 ± 358 mm2 SE) but also no mammary secretion was observed. Injections of E plus 2.5 mg CA resulted in a dramatic reduction in mammary extension (284 ± 38 mm2 SE), when compared with the effects obtained with the low dose of CA (group 5 vs. group 6, p = .004, U test). Limited ductal extension and ramification was noted, though alveolar development existed (Fig. 1C [E + CMA 2.5 mg]).
FIG. 1. Whole mounts of mammary glands showing development after diverse treatments: A) estrone (10 μg) plus 2 mg progesterone [E + P4 2.5 mg]; B) estrone (10 μg) plus 0.5 mg chlormadinone acetate [E + CMA 0.5 mg]; C) estrone (10 μg) plus 2.5 mg chlormadinone acetate [E + CMA 2.5 mg]. Note that the large dosage of chlormadinone acetate resulted in poor development of the ductal system.
Discussion
Our observations indicate that CA alone does not exert any clear effect on mammary gland growth. Similarly, P per se lacks stimulatory effects on mammary development in the gonadectomized rabbit (3). On the other hand, when low doses of CA (0.5 mg) were combined with E, maximal ducto-alveolar development occurred. The development of these glands was comparable to that observed in late pregnancy in the rabbit. The fact that the mammary growth obtained with 0.5 mg CA plus E was greater than that found after combination of P and E agrees with the observation that CA is a more potent progestational agent than P (4).
[…] It is interesting that CA in large doses inhibited mammary growth. Similarly, Lyons and McGinty (7) reported an inhibitory effect of large dosages of P (8 mg) in the intact male rabbit treated with estrogen. This inhibitory effect is probably due to the antiestrogenic action of these compounds (8).
In summary, prolonged administration of CA, when acting on an estrogenic background, might influence mammary development in the rabbit, stimulating it at moderate and inhibiting it at high doses. By contrast, CA has no significant effect on milk production or removal.
Clinical Publications
Zacharin (2000)
Zacharin, M. (2000). Use of androgens and oestrogens in adolescents - A review of hormone replacement treatment. Journal of Pediatric Endocrinology and Metabolism, 13(1), 3–12. [DOI:10.1515/JPEM.2000.13.1.3]:
Progestogen is not required for induction of puberty. Cyclical progestogen should be added when the oestradiol dosage reaches the equivalent of 15 μg/day of ethinyl oestradiol, at which time breakthrough bleeding is almost inevitable, or earlier if vaginal bleeding has already occurred.
Emans (2005)
Emans, S. J. (2005). Delayed Puberty. In Emans, S. J., Laufer, M. R., Goldstein, D. P. (Eds.). Pediatric & Adolescent Gynecology, 5th Edition (pp. 181–213). Philadelphia: Lippincott Williams & Wilkins. [Google Scholar] [Google Books] [WorldCat] [Archive.org]:
Oral contraceptives are not recommended for initial therapy because they contain progestin throughout the cycle, which is not a physiologic approach to the induction of normal breast development. During normal puberty, there is a long period of unopposed low levels of estrogen until ovulatory cycles begin (see Chapter 4). There has been speculation that particular doses of progestin or estrogen may be more likely to lead to tubular breasts, but a randomized study has not been done.
[…] The timing of the introduction of progestin varies among centers. Although in the past some girls received unopposed estrogen daily until breakthrough bleeding occurred, we believe that this method should be discouraged because adolescents benefit from a predictable onset of menses, and many girls experience significant dysfunctional bleeding that may require intervention. We suggest the addition of a short course of progestin (5 or 10 mg of medroxyprogesterone) to the continuous estrogen within 2 to 3 months of the Phase 2 increase in dose: e.g., daily estrogen plus medroxyprogesterone 10 mg each day for the first 5 days of each month. This dose of progestin is used only until breast development is completed over the next 6 months and then the dose of progestin is increased to 10 days and ultimately to 12 to 14 days for optimal protection of the endometrium if long-term hormone replacement is planned.
Bondy et al. (2007)
Bondy, C. A., & Turner Syndrome Consensus Study Group. (2007). Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. The Journal of Clinical Endocrinology & Metabolism, 92(1), 10–25. [DOI:10.1210/jc.2006-1374]:
To allow for normal breast and uterine development, it seems advisable to delay the addition of progestin at least 2 yr after starting estrogen or until breakthrough bleeding occurs. The use of oral contraceptive pills to achieve pubertal development is best avoided, because the synthetic estrogen doses in most formulations are too high and the typical synthetic progestin may interfere with optimal breast and uterine development.
Colvin, Devineni, & Ashraf (2014)
Colvin, C., Devineni, G., & Ashraf, A. P. (2014). Delayed Puberty. In Bandeira, F., Gharib, H., Golbert, A., Griz, L., & Faria, M. (Eds.). Endocrinology and Diabetes (pp. 203–217). New York: Springer. [DOI:10.1007/978-1-4614-8684-8_17]:
Initial therapy is with estrogen alone to maximize breast growth and to induce uterine and endometrial proliferation. Adding a progestin prematurely or administering combinations of estrogens and progestins early on may reduce ultimate breast size. Progestin is added to mimic the normal menstrual cycle after breast growth ceases (when full contour breast growth plateaus) or menses occur.
Wierckx, Gooren, & T’Sjoen (2014)
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical review: breast development in trans women receiving cross-sex hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]:
The available evidence does not provide support for better effects on breast size of adding progestogens to cross-sex hormone administration in trans women as suggested by some authors [14,18,48–51]. However, it should be said that the quality and amount of available evidence are extremely poor and hamper any firm conclusion at this moment. Also, many centers use antiandrogens with some progestational action and complicate the available evidence. In addition, some occasionally use progestins to lower testosterone levels after maximum estrogen levels when a patient cannot tolerate an estrogen-based regimen, abnormal psychological irritability, and mammary tenderness [52,53]. Furthermore, all progestogens by definition have some progestational activity, but they differ in chemical structure, metabolism, pharmacokinetics, affinity, potency, and efficacy via steroid receptors and intracellular action. All these differences can translate into very different biological and clinical effects and advocate the absence of a class effect of progestogens [54].
Nevertheless, breast development in trans women might be similar as in cisgender women indicating a major role for estrogen rather than progesterone in the early stages of breast development. The central role of estradiol in initiating breast growth at puberty is revealed by the poor-developed breast of estrogen receptor-alpha knockout mice [55], whereas progesterone knockout mice showed to have a morphologically indistinguishable ductal architecture from wild-type virgin mice [56]. Moreover, during pubertal induction in girls, early administration of progesterone is not recommended as premature initiation of progestin therapy can compromise ultimate breast growth [57]. It is however of note that progesterone is known to be an important determinant of the histology of the breast in cis women. When the mammary epithelial of the progesterone knockout mouse is transplanted into a wild-type parous mouse, the obligatory role of progesterone in acinar and lobular development is demonstrated [58,59]. Additionally, other theoretical advantages of progesterone administration might be the fact that breast epithelium exhibits maximal proliferation in the luteal phase of menstruation, when progesterone levels are at their highest [60] and increased mammographic breast density is observed when progestogens are administered [61]. However, importantly, there is no evidence that these histological and mammographic differences result in clinically significant breast size differences. Another consideration is that the increased breast density by progestogens rapidly decreases after hormone withdrawal [62], which raises the question how long progestogens then should be prescribed.
Kaiser & Ho (2015)
Kaiser, U., & Ho, K. K. (2015). Pituitary Physiology and Diagnostic Evaluation. In Melmed, S., Polonsky, K. S., Larsen, P. R., Kronenberg, & H. M. (Eds.). Williams Textbook of Endocrinology, 13th Edition (pp. 176–231). Philadelphia: Elsevier. [DOI:10.1016/B978-0-323-29738-7.00008-3] [Google Books]:
Sex Steroid Replacement Therapy. Estrogen or testosterone replacement is required for inducing and maintaining primary and secondary sexual characteristics [in patients with delayed puberty due to hypogonadism], […] Initial therapy should consist of estrogen alone to maximize breast growth and to induce uterine and endometrial proliferation. […] A progestin eventually needs to be added to prevent endometrial hyperplasia but should be avoided before completion of breast development, because it is likely to reduce ultimate breast size.
Bauman, Novello, & Kreitzer (2016)
Bauman, A., Novello, L., & Kreitzer, P. (2016). Endocrine Disorders and Delayed Puberty. In Appelbaum, H. (Ed.). Abnormal Female Puberty: A Clinical Casebook (pp. 87–107). Cham: Springer. [DOI:10.1007/978-3-319-27225-2_5]:
Depending upon when ovarian failure occurs, it may also be necessary to assist in the completion of puberty if pubertal progression was interrupted. If the breasts are not fully developed, lower doses of estrogen replacement must be initiated to allow for pubertal progression. Estrogen is gradually increased to adult or maintenance replacement dosing and then progesterone is added to protect the endometrium from long-term complications associated with unopposed estrogen. Pubertal induction is initiated with estrogen treatment alone followed by the sequential addition of progesterone. Progesterone is added sequentially, rather than concomitantly, in order to allow for estrogen to independently stimulate normal breast development because early progesterone exposure to the breast tissue can result in tubular breast formation [12].
To induce puberty, the patient will first receive low-dose estrogen, which can be given orally or transdermally. The dose is increased slowly over 2 years. This allows the patient to undergo the physical changes of puberty, including breast development. The slow increase of estradiol will also provide the opportunity for continued vertical growth. After about 12–24 months of unopposed estrogen, or if a patient experiences vaginal bleeding, she is given progesterone in cycles [47]. If the progesterone is given too early, it may result in breast deformity, but it must be started once the patient develops a uterine lining to prevent endometrial hyperplasia [40].
Gawlik et al. (2016)
Gawlik, A., Hankus, M., Such, K., Drosdzol-Cop, A., Madej, P., Borkowska, M., Zachurzok, A., & Malecka-Tendera, E. (2016). Hypogonadism and sex steroid replacement therapy in girls with Turner syndrome. Journal of Pediatric and Adolescent Gynecology, 29(6), 542–550. [DOI:10.1016/j.jpag.2016.03.005]:
It is advisable to delay the addition of progestin [in hypogonadal cisgender girls] by at least 2 years or until breakthrough bleeding occurs, so as to enable normal breast and uterine development.
Randolph (2018)
Randolph, J. F. (2018). Gender-affirming hormone therapy for transgender females. Clinical Obstetrics and Gynecology, 61(4), 705–721. [DOI:10.1097/GRF.0000000000000396]:
The use of progesterone, or progestins, to enhance breast development is controversial and not based on any reliable evidence. Although there are many anecdotal reports of breast growth with the addition of such agents to estrogen therapy in transwomen, no objective clinical trials are available to provide guidance on choice of medication, dose, duration, or response rate. Extrapolation from the experience in inducing breast growth in adolescent girls with absent or delayed pubertal development suggests that simultaneous initial administration of progestins with estrogen may result in abnormal and limited growth due to the simultaneous induction of ductal proliferation and terminal lobular differentiation. It is therefore recommended to initiate breast growth with estrogen alone until stability is reached with a consideration for trial of progesterone/progestin at that time. The risks of long-term progesterone/progestin therapy are unknown in transwomen. […] In view of the known course of development in normal puberty, and a description of abnormal breast growth with the early addition of progestins, it seems prudent to hold off on adding progesterone/progestin therapy until initial estrogen-induced ductal growth is complete.
Donaldson et al. (2019)
Donaldson, M., Kriström, B., Ankarberg-Lindgren, C., Verlinde, S., van Alfen-van der Velden, J., Gawlik, A., van Gelder, M. M. H. J., & Sas, T. (2019). Optimal pubertal induction in girls with Turner syndrome using either oral or transdermal estradiol: a proposed modern strategy. Hormone Research in Paediatrics, 91(3), 153–163. [DOI:10.1159/000500050]:
In contrast to the Cincinnati guidelines [12], advice from gynaecology and reproductive endocrine colleagues indicated that oral progesterone should not be given pre-emptively after 2 years, or automatically at the time of the first breakthrough bleed. Instead, to allow maximum time for uterine and breast development with unopposed estrogen, it was recommended that pubertal staging and where possible pelvic ultrasound examination should be carried out at the time of bleeding so that uterine size and endometrial thickness could be determined. In cases where the endometrium is still thin and the uterus relatively small, progesterone treatment should be deferred, but introduced if ultrasound shows a mature uterus with thick endometrium.
Heath & Wynne (2019)
Heath, R. A., & Wynne, K. (2019). [Chapter 6:] Children and Adolescents. In Heath, R. A., & Wynne, K. A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (pp. 87–106). Santa Barbara: Praeger/ABC-CLIO. [Google Books]:
In young people assigned female at birth, the first sign of estrogen exposure is usually the development of breast buds that slowly progress toward a mature breast size and shape. The beginning of breast development coincides with a growth spurt and an increase in body fat. Hair develops gradually in the armpits and pubic region. The menstrual cycle indicated by monthly periods usually starts around two years later, after breast growth is well underway. During puberty, progesterone is not made until the ovaries have started to produce eggs, at a point when breast development has finished.
Heath, R. A., & Wynne, K. (2019). [Chapter 7:] Hormone and Surgical Therapies for Adults. In Heath, R. A., & Wynne, K. A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (pp. 107–146). Santa Barbara: Praeger/ABC-CLIO. [Google Books]:
Progesterone is not involved in breast development in cisgender females. The levels of progesterone only increase late in puberty when breast development is complete.28 Progesterone is therefore not included in the initiating hormonal regimens for young cisgender women who have not spontaneously entered puberty.29 Neither does the current available evidence suggest that progestins enhance breast development for transgender women. However, there is insufficient high-quality evidence to form an absolute conclusion regarding the usefulness of progestins, and some Internet resources and clinical services still recommend their use.
These excerpts are somewhat inaccurate though—menarche occurs on average during Tanner breast stage 4, and in the first year after menarche a minority of cycles are ovulatory, resulting in significant although intermittent progesterone exposure (Aly, 2020).
Iwamoto et al. (2019)
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 2042018819871166. [DOI:10.1177/2042018819871166]:
Pubertal data in people assigned female at birth (AFAB) (e.g. girls with Turner syndrome) argue for delaying progesterone as it causes ductal differentiation and may interfere with optimal breast development.68
Crowley & Pitteloud (2020)
Crowley, W. F., & Pitteloud, N. (2020). Approach to the patient with delayed puberty. UpToDate. [Google Scholar] [URL]:
The initial estradiol doses used are below those required to induce menstruation. We add cyclic progestin therapy after two years of estradiol or when breakthrough bleeding occurs on unopposed estradiol. Our first choice for progestin therapy is oral micronized progesterone 200 mg days 1 to 12 of the calendar month. The progestin should not be added until there is substantial breast development that is not solely confined to the areolae and full contour breast growth has plateaued, because premature initiation of progestin therapy can compromise ultimate breast growth.
Naseem, Lokman, & Fitzgerald (2021)
Naseem, H., Lokman, M., & Fitzgerald, C. (2021). Management of congenital hypogonadotropic hypogonadism in females. Human Fertility, advance online publcation. [DOI:10.1080/14647273.2021.1998929]:
The aims of pubertal induction in CHH, are to achieve timely secondary sex characteristics which include breast and uterine development, to attain projected final height, acquire peak bone mass, optimise reproductive potential and maintain psychological well-being. It is important to recognise that randomised controlled trials on hormonal treatment in CHH are scarce, and data on clinical observational studies are also limited. Primary variables to determine the dose of hormone replacement therapy are age, patient satisfaction and clinical response.
Most breast development occurs in the two years of unopposed oestrogen prior to menarche. It has been stated that progesterone should be deferred until after adequate development of breast and uterus are achieved or with first breakthrough bleeding (Shifren, Gass, & NAMS Recommendations for Clinical Care of Midlife Women Working Group, 2014). However, if at first breakthrough bleed, B4 breast development or mature sonographic uterine dimensions have not been achieved, a rational approach is to slightly reduce oestradiol dose whilst continuing to defer progesterone treatment. Cyclical progestogens found in pre-packed preparations for post-menopausal women can be used or prescribed separately for 12 to 14 days in each cycle.
The combined oral contraceptive pill’s (COCP) relatively higher dose of oestrogen and progestogen in early puberty would disrupt breast development causing breast hypoplasia, leading to early epiphyseal closure and reduced bone mass (Matthews et al., 2017). Although COCP is commonly used as HRT after puberty, its use is not endorsed by the European Consensus Guidelines for CHH, which instead recommends oestradiol-based HRT (Boehm et al., 2015). Evidence suggests that COCP does not provide optimal oestrogen replacement in young women postpuberty and the use of COCP represents inadequate treatment in these women (O’Donnell et al., 2012; Swee et al., 2019).
Federici et al. (2022)
Federici, S., Goggi, G., Quinton, R., Giovanelli, L., Persani, L., Cangiano, B., & Bonomi, M. (2022). New and Consolidated Therapeutic Options for Pubertal Induction in Hypogonadism: In-depth Review of the Literature. Endocrine Reviews, 43(5), 824–851. [DOI:10.1210/endrev/bnab043]:
Induction of puberty in females
In females, adequate maturation of secondary sex characteristics is achieved with estrogen alone, while the main role of progesterone is to prevent endometrial hyperplasia. Indeed, clinical experience suggests that premature treatment with a progestogen may be deleterious to both final breast and probably also final uterine maturation.
In summary, treatment should be initiated with a starting dose of approximately 10% of the adult replacement dose and increased (by increments of of 1.5 to 2-fold) every 6 months over a 2 to 3-year period (or less in older adolescents or young women). After at least 2 years of unopposed oestrogen, or if more than one episode of significant breakthrough bleeding occurs, it is necessary to consider a progestin to induce withdrawal bleeding, but only if adult breast and uterine conformation has been achieved. We recommend that, if symptoms of endometrial hyperplasia develop when breasts or uterus are not yet fully developed, then a slight reduction in 17β-E2 dose should be considered instead of introducing the progestin, although we acknowledge the lack of direct evidence. However, in recognition that physiologically progesterone levels only rise substantially in the late stages of puberty (174), when ovulatory cycling becomes effective, and that it plays a role in both the breast and the uterus (175), it seems to us that following this approach that aims to recapitulate normal physiology represents the path of lowest risk.
Lucien et al. (2022)
Lucien, J. N., Ortega, M. T., Calvert, M. E., Smith, C., White, X., Rogers, H., Mosley, B., Agrawal, R., Drude, A., McGee, C., George, M., Brown, A., Downey, K., Wild, C., Njunge, A., Kuzmiak, C. M., Zava, D., Zava, T., Pollard, J., Francis, J., Beery, B. L., Harlin, M., Gonzalez, G. R., & Shaw, N. D. (2022). The Launch of A Girl’s First Period Study: Demystifying Reproductive Hormone Profiles in Adolescent Girls. Journal of Pediatric and Adolescent Gynecology, 35(4), 420–425. [DOI:10.1016/j.jpag.2021.12.018]:
Does exposure to low levels of [progesterone (P4)], as occurs before menarche, during anovulatory cycles with some degree of follicle luteinization, and during early, immature ovulatory cycles play an important role in normal breast development during puberty? This question has important clinical implications as hormone replacement during puberty does not typically include low-dose P4; rather, it is conducted using a staggered approach of estrogen-only therapy followed by the addition of full adult doses of exogenous P4 only after 2 years or when breakthrough bleeding occurs.27 This is done to avoid development of tubular breasts, although there are limited data linking early P4 exposure to suboptimal breast development.28
Rothman & Iwamoto (2022)
Rothman, M. S., & Iwamoto, S. J. (2022). Feminizing Gender-Affirming Hormone Therapy: Special Considerations for Older Adults. In Davis, T. F. (Ed.). A Case-Based Guide to Clinical Endocrinology (pp. 513–523). Cham: Springer International Publishing. [DOI:10.1007/978-3-030-84367-0_58]:
What Role Does Progesterone Have in Breast Development and Feminizing GAHT?
Although progesterone does not appear in feminizing GAHT recommendations in WPATH or the Endocrine Society clinical practice guidelines, many patients have questions surrounding its role in breast growth. We lack evidence for beneft from progesterone [6] in transfeminine patients although its use has been advocated for by some [7]. Much of what we know about progesterone’s risks and benefts come from data in cisgender female populations. In cisgender girls, early use of progesterone may lead to premature ductal differentiation and may be suboptimal for breast growth (e.g., Turner syndrome) [8]. […] Studies exploring progesterone’s impact on breast development have not found a difference in breast growth or referrals for breast augmentation surgery when progestins are added to estrogen treatment relative to estrogen alone [6]. In JD’s case, we would encourage her to start with estrogen and spironolactone and then reassess goals for breast growth after 1–2 years on this regimen.
\ No newline at end of file
+Literature on Early Progestogen Exposure and Breast Development - Transfeminine Science
Literature on Early Progestogen Exposure and Breast Development
By Aly | First published July 21, 2019 | Last modified March 31, 2024
Preface
This is a sources and excerpts supplement for the main article section which can be found here.
Animal Studies
Lyons & McGinty (1941)
Lyons, W. R., & McGinty, D. A. (1941). Effects of estrone and progesterone on male rabbit mammary glands. I. Varying doses of progesterone. Proceedings of the Society for Experimental Biology and Medicine, 48(1), 83–86. [DOI:10.3181/00379727-48-13227]:
Summary. Eighteen doses of 0.25, 1.0, 4.0 and 8.0 I.U. of crystalline progesterone were given, simultaneously with 120 I.U. of estrone, to immature male rabbits during a period of 4 weeks. Of these 4 levels of progesterone, the 1.0 I.U. dose synergized best although the prolactational proliferation induced was not maximal. The 4.0 and 8.0 I.U. doses of progesterone were apparently inhibiting as shown by the relatively poor mammary growth obtained.
[Introduction.] Turner and Frank1 showed that whereas estrogen caused growth of the duct system and slight lobule proliferation in the male rabbit mammary gland, the combination of estrogen and progestogen caused lobule-alveolar growth simulating that seen in pregnancy. The hormones used were impure, but were assayed for rat units of estrogen and rabbit units of progestogen and judged by the results, were uncontaminated, one with the other. Until recently few investigators have had at their disposal sufficient progesterone with which to investigate this problem further, and, as, far as we are aware, the doses of estrone and progesterone that will synergize best to cause optimal mammary growth have not as yet been satisfactorily determined in any animal form. In the preliminary investigation reported herein, an attempt was made to determine the approximate dose of progesterone that would function optimally with a given daily dose (120 I.U.) of estrone in causing prolactational* growth of the male rabbit mammary gland.
Experimental. Immature, New Zealand White male rabbits weighing approximately 1.5 kg at the beginning of the experiment were treated in groups of 3 with 18 daily doses (Monday through Friday, from 2/28/39 until 3/23/39) of the following:
Group 1, 120 I.U. of estrone (theelin); Group 2, 1 I.U. of progesterone; Group 3, 120 I.U. of E and 0.25 I.U. of P; Group 4, 120 I. U. of E and 1 I.U. of P; Group 5, 1201 I.U. of E and 4 I.U. of P; Group 6, 120 I.U. of E and 8 I.U. of P. The 2 hormones were given separately, subcutaneously, in peanut oil. On 3/28/39, 5 days after the last injection, a biopsy specimen of the second left (thoracic) mammary gland was taken from each animal, fixed in formol, stained in toto with alum-carmine and cleared in methyl salicylate.
Results. The maniniary spreads from Group 1, showed that the dose of 120 I.U. of E produced good duct growth with almost negligible alveolar formation (Fig. 2 [E Alone]). Those from Group 2 (1 I.U. of P) showed but little more development than that seen in normal immature rabbit glands (Fig. 1 [Untreated]). The duct system of the glands from animals in Group 3 (120 I.U. of E plus 0.25 I.U. of P) was as extensive as that seen in animals treated with 120 I.U. of E alone, but the main ducts were narrower and, as though in compensation. more alveolar buds were present. Thus, just as progesterone in some experimental animals prevents the estrogen-induced uterine dilatation and stimulates a proliferation of luminal and glandular epithelium, so also in the rabbit it counteracts an estrogen-induced mammary duct dilatation and permits instead an extensive alveolar proliferation. The amount of incomplete lobule formation shown in Fig. 3 [E + P4 0.25 mg] was found typical of all rabbits in this group and probably may be interpreted as barely minimal prolactational proliferation in response to a low dose of progesterone. The glands from animals in Group 4 (120 I.U. of E and 1.0 I.U. of P) showed the best evidence of synergism obtained in this preliminary experiment (Fig. 4 [E + P4 1 mg]), although judged on the basis of a set of glands taken at different stages of pregnancy, they could not be said to show maximal prolactational proliferation. Interesting results were obtained in Groups 5 and 6, where 2 of the animals in Group 5 and all in Group 6 showed only scanty alveolar development and an inhibition of the duct growth (Fig. 5 [E + P4 8 mg]). The third animal in Group 5 showed good prolactational development with no inhibition of duct growth, indicating that 4 I.U. of progesterone approximated the border-line inhibiting dose when given with 120 I.U. of estrone by this particular routine.
All figures represent approximately one-half of a male rabbit mammary spread fixed in formol and stained in alum-carmine. × 1.5.
Treatment was as follows:
FIG. 1. None. Control gland. [Untreated]
FIG. 2. 18 subcutaneous doses of 120 I.U. estrone in oil over a 28-day period. Duct growth with almost no alveolar development. [E Alone]
FIG. 3. 18 subcutaneous doses of 120 I.U. estrone and 0.25 I.U. progesterone over same period. Very little prolactational proliferation. [E + P4 0.25 mg]
FIG. 4. 18 subcutaneous doses of 120 I.U. estrone and 1 I.U. progesterone over same period. Fair prolactational proliferation. [E + P4 1 mg]
FIG. 5. 18 subcutaneous doses of 120 I.U. estrone and 8 I.U. progesterone over same period. Inhibited duct growth and only slight alveolar development. [E + P4 8 mg]
Note: A citing review indicated that 1 IU progesterone is equal to 1 mg progesterone (Pfeiffer, 1943).
Beyer, Cruz, & Martinez-Manautou (1970)
Beyer, C., Cruz, M. L., & Martinez-Manautou, J. (1970). Effect of Chlormadinone Acetate on Mammary Development and Lactation in the Rabbit. Endocrinology, 86(5), 1172–1174. [DOI:10.1210/endo-86-5-1172]:
ABSTRACT. The effect of chlormadinone acetate (CA) on mammary growth and lactation was studied in the New Zealand white rabbit. Mammary development studies were performed in 5-month males castrated prepuberally. All rabbits were treated for 28 days. Ten μg estrone (E) produced good ductal development without alveolar growth (mean mammary area = 376 mm2). CA alone did not stimulate mammary growth. Administration of 0.5 mg CA plus 10 μg E produced optimal ductal and alveolar development (mean mammary area = 765 mm2). By contrast, a larger dose of CA (2.5 mg) combined with 10 μg E resulted in a much smaller mammary response (mean mammary area = 284 mm2). Administration of 100 μg or 2.5 mg CA from day 10 to 25 of lactation neither inhibited nor facilitated milk production in lactating rabbits. It is concluded that prolonged administration of CA when combined with estrogen stimulates mammary growth at moderate doses (0.5 mg) and inhibits it at large doses (2.5 mg). CA has no clear effect on milk production and/or removal in lactating rabbits. (Endocrinology86: 1172, 1970)
[Introduction]
CHLORMADINONE acetate (CA) is a potent progestational steroid widely employed in controlling human fertility (1). It has been reported that prolonged treatment with some progestational compounds can alter mammary structure and lactation in some mammalian species, including women (2). Yet no detailed study has been done on the action of CA on the mammary gland. Therefore, we considered it interesting to analyze the effect of prolonged administration of CA upon mammary growth and lactation in New Zealand white rabbits.
Results
The mammary glands of the control castrated males were not visible. When studied microscopically, they were found to consist of short primary ducts that hardly extended beyond the nipple area. Injections of 500 μg CA did not stimulate mammary growth. Administration of 10 μg E induced clear growth and ramification of the ductal system. The mean area (376 ± 41 mm2 SE) occupied by these glands was significantly larger than in the control or CA treated rabbits. These glands were composed exclusively of ductal epithelium without alveoli. Combined administration of 10 μg E and 2.5 mg progesterone (P) resulted in a good ductoalveolar development. As can be seen in Fig. 1A [E + P4 2.5 mg] the ducts were almost covered with alveoli. The area occupied by these glands was significantly larger than that of the E treated rabbits (688 ± 77 mm2 SE, group 3 vs. 4, p = .004, U test). No milk secretion was noted. Administration of both E and CA at the low dose level (0.5 mg) produced marked ducto-alveolar development (Fig. 1B [E + CMA 0.5 mg]). The mammary area in these animals was more extended than in all other treated groups (765 ± 358 mm2 SE) but also no mammary secretion was observed. Injections of E plus 2.5 mg CA resulted in a dramatic reduction in mammary extension (284 ± 38 mm2 SE), when compared with the effects obtained with the low dose of CA (group 5 vs. group 6, p = .004, U test). Limited ductal extension and ramification was noted, though alveolar development existed (Fig. 1C [E + CMA 2.5 mg]).
FIG. 1. Whole mounts of mammary glands showing development after diverse treatments: A) estrone (10 μg) plus 2 mg progesterone [E + P4 2.5 mg]; B) estrone (10 μg) plus 0.5 mg chlormadinone acetate [E + CMA 0.5 mg]; C) estrone (10 μg) plus 2.5 mg chlormadinone acetate [E + CMA 2.5 mg]. Note that the large dosage of chlormadinone acetate resulted in poor development of the ductal system.
Discussion
Our observations indicate that CA alone does not exert any clear effect on mammary gland growth. Similarly, P per se lacks stimulatory effects on mammary development in the gonadectomized rabbit (3). On the other hand, when low doses of CA (0.5 mg) were combined with E, maximal ducto-alveolar development occurred. The development of these glands was comparable to that observed in late pregnancy in the rabbit. The fact that the mammary growth obtained with 0.5 mg CA plus E was greater than that found after combination of P and E agrees with the observation that CA is a more potent progestational agent than P (4).
[…] It is interesting that CA in large doses inhibited mammary growth. Similarly, Lyons and McGinty (7) reported an inhibitory effect of large dosages of P (8 mg) in the intact male rabbit treated with estrogen. This inhibitory effect is probably due to the antiestrogenic action of these compounds (8).
In summary, prolonged administration of CA, when acting on an estrogenic background, might influence mammary development in the rabbit, stimulating it at moderate and inhibiting it at high doses. By contrast, CA has no significant effect on milk production or removal.
Clinical Publications
Zacharin (2000)
Zacharin, M. (2000). Use of androgens and oestrogens in adolescents - A review of hormone replacement treatment. Journal of Pediatric Endocrinology and Metabolism, 13(1), 3–12. [DOI:10.1515/JPEM.2000.13.1.3]:
Progestogen is not required for induction of puberty. Cyclical progestogen should be added when the oestradiol dosage reaches the equivalent of 15 μg/day of ethinyl oestradiol, at which time breakthrough bleeding is almost inevitable, or earlier if vaginal bleeding has already occurred.
Emans (2005)
Emans, S. J. (2005). Delayed Puberty. In Emans, S. J., Laufer, M. R., Goldstein, D. P. (Eds.). Pediatric & Adolescent Gynecology, 5th Edition (pp. 181–213). Philadelphia: Lippincott Williams & Wilkins. [Google Scholar] [Google Books] [WorldCat] [Archive.org]:
Oral contraceptives are not recommended for initial therapy because they contain progestin throughout the cycle, which is not a physiologic approach to the induction of normal breast development. During normal puberty, there is a long period of unopposed low levels of estrogen until ovulatory cycles begin (see Chapter 4). There has been speculation that particular doses of progestin or estrogen may be more likely to lead to tubular breasts, but a randomized study has not been done.
[…] The timing of the introduction of progestin varies among centers. Although in the past some girls received unopposed estrogen daily until breakthrough bleeding occurred, we believe that this method should be discouraged because adolescents benefit from a predictable onset of menses, and many girls experience significant dysfunctional bleeding that may require intervention. We suggest the addition of a short course of progestin (5 or 10 mg of medroxyprogesterone) to the continuous estrogen within 2 to 3 months of the Phase 2 increase in dose: e.g., daily estrogen plus medroxyprogesterone 10 mg each day for the first 5 days of each month. This dose of progestin is used only until breast development is completed over the next 6 months and then the dose of progestin is increased to 10 days and ultimately to 12 to 14 days for optimal protection of the endometrium if long-term hormone replacement is planned.
Bondy et al. (2007)
Bondy, C. A., & Turner Syndrome Consensus Study Group. (2007). Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. The Journal of Clinical Endocrinology & Metabolism, 92(1), 10–25. [DOI:10.1210/jc.2006-1374]:
To allow for normal breast and uterine development, it seems advisable to delay the addition of progestin at least 2 yr after starting estrogen or until breakthrough bleeding occurs. The use of oral contraceptive pills to achieve pubertal development is best avoided, because the synthetic estrogen doses in most formulations are too high and the typical synthetic progestin may interfere with optimal breast and uterine development.
Colvin, Devineni, & Ashraf (2014)
Colvin, C., Devineni, G., & Ashraf, A. P. (2014). Delayed Puberty. In Bandeira, F., Gharib, H., Golbert, A., Griz, L., & Faria, M. (Eds.). Endocrinology and Diabetes (pp. 203–217). New York: Springer. [DOI:10.1007/978-1-4614-8684-8_17]:
Initial therapy is with estrogen alone to maximize breast growth and to induce uterine and endometrial proliferation. Adding a progestin prematurely or administering combinations of estrogens and progestins early on may reduce ultimate breast size. Progestin is added to mimic the normal menstrual cycle after breast growth ceases (when full contour breast growth plateaus) or menses occur.
Wierckx, Gooren, & T’Sjoen (2014)
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical review: breast development in trans women receiving cross-sex hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]:
The available evidence does not provide support for better effects on breast size of adding progestogens to cross-sex hormone administration in trans women as suggested by some authors [14,18,48–51]. However, it should be said that the quality and amount of available evidence are extremely poor and hamper any firm conclusion at this moment. Also, many centers use antiandrogens with some progestational action and complicate the available evidence. In addition, some occasionally use progestins to lower testosterone levels after maximum estrogen levels when a patient cannot tolerate an estrogen-based regimen, abnormal psychological irritability, and mammary tenderness [52,53]. Furthermore, all progestogens by definition have some progestational activity, but they differ in chemical structure, metabolism, pharmacokinetics, affinity, potency, and efficacy via steroid receptors and intracellular action. All these differences can translate into very different biological and clinical effects and advocate the absence of a class effect of progestogens [54].
Nevertheless, breast development in trans women might be similar as in cisgender women indicating a major role for estrogen rather than progesterone in the early stages of breast development. The central role of estradiol in initiating breast growth at puberty is revealed by the poor-developed breast of estrogen receptor-alpha knockout mice [55], whereas progesterone knockout mice showed to have a morphologically indistinguishable ductal architecture from wild-type virgin mice [56]. Moreover, during pubertal induction in girls, early administration of progesterone is not recommended as premature initiation of progestin therapy can compromise ultimate breast growth [57]. It is however of note that progesterone is known to be an important determinant of the histology of the breast in cis women. When the mammary epithelial of the progesterone knockout mouse is transplanted into a wild-type parous mouse, the obligatory role of progesterone in acinar and lobular development is demonstrated [58,59]. Additionally, other theoretical advantages of progesterone administration might be the fact that breast epithelium exhibits maximal proliferation in the luteal phase of menstruation, when progesterone levels are at their highest [60] and increased mammographic breast density is observed when progestogens are administered [61]. However, importantly, there is no evidence that these histological and mammographic differences result in clinically significant breast size differences. Another consideration is that the increased breast density by progestogens rapidly decreases after hormone withdrawal [62], which raises the question how long progestogens then should be prescribed.
Kaiser & Ho (2015)
Kaiser, U., & Ho, K. K. (2015). Pituitary Physiology and Diagnostic Evaluation. In Melmed, S., Polonsky, K. S., Larsen, P. R., Kronenberg, & H. M. (Eds.). Williams Textbook of Endocrinology, 13th Edition (pp. 176–231). Philadelphia: Elsevier. [DOI:10.1016/B978-0-323-29738-7.00008-3] [Google Books]:
Sex Steroid Replacement Therapy. Estrogen or testosterone replacement is required for inducing and maintaining primary and secondary sexual characteristics [in patients with delayed puberty due to hypogonadism], […] Initial therapy should consist of estrogen alone to maximize breast growth and to induce uterine and endometrial proliferation. […] A progestin eventually needs to be added to prevent endometrial hyperplasia but should be avoided before completion of breast development, because it is likely to reduce ultimate breast size.
Bauman, Novello, & Kreitzer (2016)
Bauman, A., Novello, L., & Kreitzer, P. (2016). Endocrine Disorders and Delayed Puberty. In Appelbaum, H. (Ed.). Abnormal Female Puberty: A Clinical Casebook (pp. 87–107). Cham: Springer. [DOI:10.1007/978-3-319-27225-2_5]:
Depending upon when ovarian failure occurs, it may also be necessary to assist in the completion of puberty if pubertal progression was interrupted. If the breasts are not fully developed, lower doses of estrogen replacement must be initiated to allow for pubertal progression. Estrogen is gradually increased to adult or maintenance replacement dosing and then progesterone is added to protect the endometrium from long-term complications associated with unopposed estrogen. Pubertal induction is initiated with estrogen treatment alone followed by the sequential addition of progesterone. Progesterone is added sequentially, rather than concomitantly, in order to allow for estrogen to independently stimulate normal breast development because early progesterone exposure to the breast tissue can result in tubular breast formation [12].
To induce puberty, the patient will first receive low-dose estrogen, which can be given orally or transdermally. The dose is increased slowly over 2 years. This allows the patient to undergo the physical changes of puberty, including breast development. The slow increase of estradiol will also provide the opportunity for continued vertical growth. After about 12–24 months of unopposed estrogen, or if a patient experiences vaginal bleeding, she is given progesterone in cycles [47]. If the progesterone is given too early, it may result in breast deformity, but it must be started once the patient develops a uterine lining to prevent endometrial hyperplasia [40].
Gawlik et al. (2016)
Gawlik, A., Hankus, M., Such, K., Drosdzol-Cop, A., Madej, P., Borkowska, M., Zachurzok, A., & Malecka-Tendera, E. (2016). Hypogonadism and sex steroid replacement therapy in girls with Turner syndrome. Journal of Pediatric and Adolescent Gynecology, 29(6), 542–550. [DOI:10.1016/j.jpag.2016.03.005]:
It is advisable to delay the addition of progestin [in hypogonadal cisgender girls] by at least 2 years or until breakthrough bleeding occurs, so as to enable normal breast and uterine development.
Randolph (2018)
Randolph, J. F. (2018). Gender-affirming hormone therapy for transgender females. Clinical Obstetrics and Gynecology, 61(4), 705–721. [DOI:10.1097/GRF.0000000000000396]:
The use of progesterone, or progestins, to enhance breast development is controversial and not based on any reliable evidence. Although there are many anecdotal reports of breast growth with the addition of such agents to estrogen therapy in transwomen, no objective clinical trials are available to provide guidance on choice of medication, dose, duration, or response rate. Extrapolation from the experience in inducing breast growth in adolescent girls with absent or delayed pubertal development suggests that simultaneous initial administration of progestins with estrogen may result in abnormal and limited growth due to the simultaneous induction of ductal proliferation and terminal lobular differentiation. It is therefore recommended to initiate breast growth with estrogen alone until stability is reached with a consideration for trial of progesterone/progestin at that time. The risks of long-term progesterone/progestin therapy are unknown in transwomen. […] In view of the known course of development in normal puberty, and a description of abnormal breast growth with the early addition of progestins, it seems prudent to hold off on adding progesterone/progestin therapy until initial estrogen-induced ductal growth is complete.
Donaldson et al. (2019)
Donaldson, M., Kriström, B., Ankarberg-Lindgren, C., Verlinde, S., van Alfen-van der Velden, J., Gawlik, A., van Gelder, M. M. H. J., & Sas, T. (2019). Optimal pubertal induction in girls with Turner syndrome using either oral or transdermal estradiol: a proposed modern strategy. Hormone Research in Paediatrics, 91(3), 153–163. [DOI:10.1159/000500050]:
In contrast to the Cincinnati guidelines [12], advice from gynaecology and reproductive endocrine colleagues indicated that oral progesterone should not be given pre-emptively after 2 years, or automatically at the time of the first breakthrough bleed. Instead, to allow maximum time for uterine and breast development with unopposed estrogen, it was recommended that pubertal staging and where possible pelvic ultrasound examination should be carried out at the time of bleeding so that uterine size and endometrial thickness could be determined. In cases where the endometrium is still thin and the uterus relatively small, progesterone treatment should be deferred, but introduced if ultrasound shows a mature uterus with thick endometrium.
Heath & Wynne (2019)
Heath, R. A., & Wynne, K. (2019). [Chapter 6:] Children and Adolescents. In Heath, R. A., & Wynne, K. A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (pp. 87–106). Santa Barbara: Praeger/ABC-CLIO. [Google Books]:
In young people assigned female at birth, the first sign of estrogen exposure is usually the development of breast buds that slowly progress toward a mature breast size and shape. The beginning of breast development coincides with a growth spurt and an increase in body fat. Hair develops gradually in the armpits and pubic region. The menstrual cycle indicated by monthly periods usually starts around two years later, after breast growth is well underway. During puberty, progesterone is not made until the ovaries have started to produce eggs, at a point when breast development has finished.
Heath, R. A., & Wynne, K. (2019). [Chapter 7:] Hormone and Surgical Therapies for Adults. In Heath, R. A., & Wynne, K. A Guide to Transgender Health: State-of-the-art Information for Gender-Affirming People and Their Supporters (pp. 107–146). Santa Barbara: Praeger/ABC-CLIO. [Google Books]:
Progesterone is not involved in breast development in cisgender females. The levels of progesterone only increase late in puberty when breast development is complete.28 Progesterone is therefore not included in the initiating hormonal regimens for young cisgender women who have not spontaneously entered puberty.29 Neither does the current available evidence suggest that progestins enhance breast development for transgender women. However, there is insufficient high-quality evidence to form an absolute conclusion regarding the usefulness of progestins, and some Internet resources and clinical services still recommend their use.
These excerpts are somewhat inaccurate though—menarche occurs on average during Tanner breast stage 4, and in the first year after menarche a minority of cycles are ovulatory, resulting in significant although intermittent progesterone exposure (Aly, 2020).
Iwamoto et al. (2019)
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 2042018819871166. [DOI:10.1177/2042018819871166]:
Pubertal data in people assigned female at birth (AFAB) (e.g. girls with Turner syndrome) argue for delaying progesterone as it causes ductal differentiation and may interfere with optimal breast development.68
Crowley & Pitteloud (2020)
Crowley, W. F., & Pitteloud, N. (2020). Approach to the patient with delayed puberty. UpToDate. [Google Scholar] [URL]:
The initial estradiol doses used are below those required to induce menstruation. We add cyclic progestin therapy after two years of estradiol or when breakthrough bleeding occurs on unopposed estradiol. Our first choice for progestin therapy is oral micronized progesterone 200 mg days 1 to 12 of the calendar month. The progestin should not be added until there is substantial breast development that is not solely confined to the areolae and full contour breast growth has plateaued, because premature initiation of progestin therapy can compromise ultimate breast growth.
Naseem, Lokman, & Fitzgerald (2021)
Naseem, H., Lokman, M., & Fitzgerald, C. (2021). Management of congenital hypogonadotropic hypogonadism in females. Human Fertility, advance online publcation. [DOI:10.1080/14647273.2021.1998929]:
The aims of pubertal induction in CHH, are to achieve timely secondary sex characteristics which include breast and uterine development, to attain projected final height, acquire peak bone mass, optimise reproductive potential and maintain psychological well-being. It is important to recognise that randomised controlled trials on hormonal treatment in CHH are scarce, and data on clinical observational studies are also limited. Primary variables to determine the dose of hormone replacement therapy are age, patient satisfaction and clinical response.
Most breast development occurs in the two years of unopposed oestrogen prior to menarche. It has been stated that progesterone should be deferred until after adequate development of breast and uterus are achieved or with first breakthrough bleeding (Shifren, Gass, & NAMS Recommendations for Clinical Care of Midlife Women Working Group, 2014). However, if at first breakthrough bleed, B4 breast development or mature sonographic uterine dimensions have not been achieved, a rational approach is to slightly reduce oestradiol dose whilst continuing to defer progesterone treatment. Cyclical progestogens found in pre-packed preparations for post-menopausal women can be used or prescribed separately for 12 to 14 days in each cycle.
The combined oral contraceptive pill’s (COCP) relatively higher dose of oestrogen and progestogen in early puberty would disrupt breast development causing breast hypoplasia, leading to early epiphyseal closure and reduced bone mass (Matthews et al., 2017). Although COCP is commonly used as HRT after puberty, its use is not endorsed by the European Consensus Guidelines for CHH, which instead recommends oestradiol-based HRT (Boehm et al., 2015). Evidence suggests that COCP does not provide optimal oestrogen replacement in young women postpuberty and the use of COCP represents inadequate treatment in these women (O’Donnell et al., 2012; Swee et al., 2019).
Federici et al. (2022)
Federici, S., Goggi, G., Quinton, R., Giovanelli, L., Persani, L., Cangiano, B., & Bonomi, M. (2022). New and Consolidated Therapeutic Options for Pubertal Induction in Hypogonadism: In-depth Review of the Literature. Endocrine Reviews, 43(5), 824–851. [DOI:10.1210/endrev/bnab043]:
Induction of puberty in females
In females, adequate maturation of secondary sex characteristics is achieved with estrogen alone, while the main role of progesterone is to prevent endometrial hyperplasia. Indeed, clinical experience suggests that premature treatment with a progestogen may be deleterious to both final breast and probably also final uterine maturation.
In summary, treatment should be initiated with a starting dose of approximately 10% of the adult replacement dose and increased (by increments of of 1.5 to 2-fold) every 6 months over a 2 to 3-year period (or less in older adolescents or young women). After at least 2 years of unopposed oestrogen, or if more than one episode of significant breakthrough bleeding occurs, it is necessary to consider a progestin to induce withdrawal bleeding, but only if adult breast and uterine conformation has been achieved. We recommend that, if symptoms of endometrial hyperplasia develop when breasts or uterus are not yet fully developed, then a slight reduction in 17β-E2 dose should be considered instead of introducing the progestin, although we acknowledge the lack of direct evidence. However, in recognition that physiologically progesterone levels only rise substantially in the late stages of puberty (174), when ovulatory cycling becomes effective, and that it plays a role in both the breast and the uterus (175), it seems to us that following this approach that aims to recapitulate normal physiology represents the path of lowest risk.
Lucien et al. (2022)
Lucien, J. N., Ortega, M. T., Calvert, M. E., Smith, C., White, X., Rogers, H., Mosley, B., Agrawal, R., Drude, A., McGee, C., George, M., Brown, A., Downey, K., Wild, C., Njunge, A., Kuzmiak, C. M., Zava, D., Zava, T., Pollard, J., Francis, J., Beery, B. L., Harlin, M., Gonzalez, G. R., & Shaw, N. D. (2022). The Launch of A Girl’s First Period Study: Demystifying Reproductive Hormone Profiles in Adolescent Girls. Journal of Pediatric and Adolescent Gynecology, 35(4), 420–425. [DOI:10.1016/j.jpag.2021.12.018]:
Does exposure to low levels of [progesterone (P4)], as occurs before menarche, during anovulatory cycles with some degree of follicle luteinization, and during early, immature ovulatory cycles play an important role in normal breast development during puberty? This question has important clinical implications as hormone replacement during puberty does not typically include low-dose P4; rather, it is conducted using a staggered approach of estrogen-only therapy followed by the addition of full adult doses of exogenous P4 only after 2 years or when breakthrough bleeding occurs.27 This is done to avoid development of tubular breasts, although there are limited data linking early P4 exposure to suboptimal breast development.28
Rothman & Iwamoto (2022)
Rothman, M. S., & Iwamoto, S. J. (2022). Feminizing Gender-Affirming Hormone Therapy: Special Considerations for Older Adults. In Davis, T. F. (Ed.). A Case-Based Guide to Clinical Endocrinology (pp. 513–523). Cham: Springer International Publishing. [DOI:10.1007/978-3-030-84367-0_58]:
What Role Does Progesterone Have in Breast Development and Feminizing GAHT?
Although progesterone does not appear in feminizing GAHT recommendations in WPATH or the Endocrine Society clinical practice guidelines, many patients have questions surrounding its role in breast growth. We lack evidence for beneft from progesterone [6] in transfeminine patients although its use has been advocated for by some [7]. Much of what we know about progesterone’s risks and benefts come from data in cisgender female populations. In cisgender girls, early use of progesterone may lead to premature ductal differentiation and may be suboptimal for breast growth (e.g., Turner syndrome) [8]. […] Studies exploring progesterone’s impact on breast development have not found a difference in breast growth or referrals for breast augmentation surgery when progestins are added to estrogen treatment relative to estrogen alone [6]. In JD’s case, we would encourage her to start with estrogen and spironolactone and then reassess goals for breast growth after 1–2 years on this regimen.
\ No newline at end of file
diff --git a/transfemscience.org/articles/progestogens-sexual-desire/index.html b/transfemscience.org/articles/progestogens-sexual-desire/index.html
index afbbc519..765c9624 100644
--- a/transfemscience.org/articles/progestogens-sexual-desire/index.html
+++ b/transfemscience.org/articles/progestogens-sexual-desire/index.html
@@ -1 +1 @@
-The Influence of Progesterone and Other Progestogens on Sexual Desire and Function - Transfeminine Science
The Influence of Progesterone and Other Progestogens on Sexual Desire and Function
By Aly | First published January 9, 2020 | Last modified March 22, 2024
Abstract / TL;DR
Many transfeminine people believe on the basis of anecdotes that progesterone increases sexual desire. A review was conducted to assess the available evidence for the possibility that progesterone and/or progestogens increase sexual desire. Both animal studies as well as clinical studies with progesterone and progestins were considered. Progesterone has shown no indication of stimulatory effects on sexual desire or behavior in humans and non-human primates. High doses of progestogens have however been found to inhibit sexual desire in humans and non-human primates. More research is necessary to further characterize the role of progesterone and other progestogens in sexual desire in humans. In any case, available evidence does not support a positive role of these hormonal agents in sexual desire. Instead, sexual desire is more related to androgens and estrogens in men and women, respectively.
Introduction
Based on anecdotal experience, some people in the transfeminine community believe that progesterone has positive effects on sexual desire. This is such that even memes about progesterone and libido have been made (Reddit). In addition to anecdotes, some researchers have theorized that progesterone may be positively involved in sexual desire in cisgender women (Regan, 1999). Some have also argued that the neurosteroid metabolites of progesterone like allopregnanolone and pregnanolone may provide special benefits on sexual desire that are not shared with progestins. I’ve reviewed the available evidence and literature to assess whether progesterone or other progestogens indeed improve sexual desire function in cisgender women and might be useful for this in transfeminine people.
Animal Studies
Correlational
Progesterone levels are strongly and consistently negatively associated with female sexual desire and behavior in most animal species (Roney, 2015). This includes in monkeys and apes (Roney, 2015).
Experimental
Non-Human Primates
Progesterone, both alone and when added to an estrogen, has been shown to inhibit sexual behavior and receptivity in female monkeys in many studies (Luttge, 1971; Baum et al., 1976; Baum et al., 1977a; Michael et al., 1967). This inhibition is often marked. Likewise, subcutaneous implants of progesterone were shown to suppress sexual behavior in testosterone-treated castrated male monkeys (Zumpe, Clancy, & Michael, 1997). As the male monkeys were castrated and treated with testosterone, this was not due to decreased testosterone levels (which remained unchanged), and could only have been due to the other effects of progesterone. Similar findings have been made for the progestin medroxyprogesterone acetate (MPA) (Michael & Zumpe, 1993; Zumpe et al., 1991; Zumpe & Michael, 1988).
The following excerpt from a review is notable on the topic of progesterone and sexual behavior in primates (Baum, 1983):
The early work of Ball (1941) and of Michael et al. (1968) showed that progesterone acts in the female rhesus monkey to reduce the likelihood of copulation. In contrast to non primate females, no facilitatory action of progesterone on any aspect of feminine sexuality has ever been convincingly demonstrated in primates.
One study in female monkeys found that MPA inhibited sexual behavior while progesterone did not (Pazol et al., 2004). However, MPA is a much more potent progestogen than progesterone, and the progesterone levels were very low (2 ng/mL). Hence, the progesterone levels in this study may have been too low for adequate physiological effect, which might explain the conflicting finding compared to other studies. Another study found increased sexual receptivity in castrated female monkeys treated with a combination of an estrogen and parenteral progesterone (Baum et al., 1977b). However, this was relative to a baseline of castration, and the changes may have only been due to the estrogen. Indeed, the same researchers reported decreased sexual receptivity with the addition of progesterone to estrogen in other studies (Baum et al., 1976; Baum et al., 1977a).
Mifepristone, a progesterone receptor antagonist, showed no apparent effect on sexual behavior in either female or male monkeys (Nadler et al., 1985).
Rodents
A notable exception when it comes to progesterone and sexual behavior in animals is female rodents, in which progesterone is stimulatory and induces lordosis (Frye, 2007). Allopregnanolone is involved in the facilitatory effects of progesterone on female sexual behavior in rodents, but has mixed effects, showing either augmentation or inhibition depending on the specifics (King, 2013; Frye, 2007). However, progesterone stimulates sexual behavior in female rodents only acutely, and is actually inhibitory with continuous administration (Pazol et al., 2004; Pazol et al., 2006). Studies in estrogen-primed intact male rats have found that progesterone given systemically does not induce lordosis (Ward et al., 1977). Interestingly however, lordosis was found to be induced in some male rats when progesterone was injected directly into a specific part of the hypothalamus (Ward et al., 1977). The prosexual effects of progesterone in female rodents may be mediated via non-genomic mechanisms, for instance actions of progesterone’s neurosteroid metabolites (Delville, 1991).
In addition to female rodents, research has found that progesterone is involved in normal sexual behavior in male rats (Witt et al., 1995; Andersen & Tufik, 2006). In male rats, castration greatly decreases levels of both testosterone and progesterone and markedly reduces sexual behavior (Witt et al., 1995). Low-dose progesterone administration, resulting in low but physiological male-range levels of progesterone, significantly restores sexual behavior (Witt et al., 1995). Additionally, when testosterone is concomitantly administered, progesterone augments the improvement caused by testosterone and is required for sexual behavior to be fully restored (Witt et al., 1995). The effects are mimicked by progestins like promegestone and blocked by the progesterone receptor antagonist mifepristone, suggesting that they are due specifically to activation of the progesterone receptor (Witt et al., 1995).
However, the findings in male rats probably do not apply to primates. As already described, mifepristone, in contrast to male rats, has no influence on sexual behavior in male monkeys (Nadler et al., 1985). Similarly, a study in castrated male monkeys given testosterone in combination with a relatively low dose of progesterone—resulting in roughly doubled progesterone levels relative to normal male levels—found no difference in sexual behavior compared to castrated male monkeys given testosterone alone (Buhl, Jensen, & Phoenix, 1978). Hence, neither progesterone receptor antagonism nor restoration of progesterone levels following castration seems to influence sexual behavior in male primates. It’s also notable that unlike in male rodents, castration doesn’t appear to reduce progesterone levels in either male monkeys (Zumpe, Clancy, & Michael, 1997) or humans (Waxman et al., 2013). As such, even if the rodent findings did apply to humans, castrated men have normal male-range progesterone levels and would in theory have all the progesterone needed to maintain sexual desire.
The findings on male-range progesterone levels and sexual behavior in male rats are also in contrast to research with higher, supraphysiological levels of progesterone (Hawley & Mosura, 2019). This degree of progesterone exposure has been shown to inhibit sexual behavior in males of most or all species examined, notably including male rats (Hawley & Mosura, 2019). This is the case regardless of influence on testosterone levels (Hawley & Mosura, 2019).
Other Species
Progestogens are used to inhibit heat in female dogs and cats (Neumann, 1978).
Sexual desire is progressively decreased in women during pregnancy, a state characterized by very high levels of estrogens, progesterone, and prolactin (Regan et al., 2003; Gałązka et al., 2014). However, many confounding factors exist (e.g., the other hormonal changes, or being pregnant), and hence causation of the changes can’t necessarily be attributed to progesterone. The observations do suggest that very high levels of an estrogen and progesterone may be unlikely to increase sexual desire however.
Experimental
Progestins
A 2013 meta-analysis reported that combined birth control pills, containing both ethinylestradiol and a progestogen, result in either unchanged or increased sexual desire in most women (85%) and decreased sexual desire in a small percentage of women (15%) (Pastor et al., 2013). However, changes in sexual desire seem to be related to the ethinylestradiol component rather than to the progestin component; decreased sexual desire was only associated with birth control pills containing low-dose estrogen (20 μg ethinylestradiol) (Pastor et al., 2013). The fact that combined birth control pills infrequently decrease sexual desire in women is notable because these medications suppress ovulation and hence completely prevent the increase in progesterone levels during the luteal phase. As such, progesterone levels, and by extension circulating levels of allopregnanolone and pregnanolone, remain very low at all times. This suggests little or no importance of the neurosteroid metabolites of progesterone for sexual desire in women.
The fact that combined birth control pills don’t generally affect sexual desire in women is also potentially surprising because total and free testosterone levels are greatly decreased by them due to the ethinylestradiol component (Zimmerman et al., 2014). This is, however, in accordance with accumulating evidence suggesting that physiological levels of androgens in women have little or no involvement in sexual desire (Cappelletti & Wallen, 2016). Rather than progesterone or androgens, what seems to be important for sexual desire in women is estradiol, especially acutely at periovulatory levels (Cappelletti & Wallen, 2016).
A small minority of women treated with depot medroxyprogesterone acetate (DMPA) alone for birth control report decreased libido and only 2.3% cite it as a reason for discontinuation (Schaffir, 2006). A study of 80 women treated with DMPA alone for birth control monitored sexual function and found no difference from baseline to 4 months of therapy (Schaffir, 2006). Findings have been similar for subcutaneous birth control implants with nomegestrol acetate and levonorgestrel (Schaffir, 2006). A randomized controlled trial of 150 women given combined birth control pills, progestin-only birth control pills, and placebo found no difference in sexual desire or activity between progestin-only pills and placebo (Schaffir, 2006).
Interestingly, a 2018 meta-analysis reported a higher standardized mean difference (SMD) for improvement of sexual desire/function in studies with an estrogen plus a progestogen relative to studies using an estrogen alone (Javadivala et al., 2019). However, there was differing methodology (e.g., sexual measures) and high heterogeneity across studies, as well as a limited number of included studies (particularly of estrogen only). These issues make conclusions on the basis of the differing SMDs problematic. Indeed, the finding is contradicted by individual studies that have included both estrogen only and estrogen plus progestogen groups (see the sources above).
Besides progestogenic activity, it has been suggested that androgenic progestins, like most of the testosterone derivatives, might provide better effects on sexual desire/function than non-androgenic progestins (Graziottin & Serafini, 2011). However, aside from the case of the atypical agent tibolone (Biglia et al., 2010), this has not actually been assessed or shown in any clinical trials (Schaffir, 2006).
The addition of dydrogesterone to hormone therapy for transgender women to improve sexual desire showed no effect, whereas transdermal testosterone was effective (Kronawitter et al., 2009).
A 1975 study published as an abstract with little additional information claimed that some premenopausal women with “frigidity” had high progesterone levels and that treatment with levonorgestrel alone to suppress progesterone levels could increase libido in these women (Kristoffersen & Lebech, 1975). This is very low-quality information however.
Progesterone
Available clinical studies of progesterone and sexual desire in women have observed no changes (Worsley et al., 2016). Neither 200 mg/day oral progesterone nor 10 mg/day oral MPA had any influence on mood or libido in estrogen-treated postmenopausal women in a randomized controlled trial (Saadat et al., 2002). Likewise, treatment of pharmacologically hypogonadal women with 400 mg/day vaginal progesterone suppositories alone did not significantly alter any domain of sexual function in another randomized controlled trial (Schmidt et al., 2009). Treatment of women with subcutaneous implants of progesterone alone for gynecological disorders has been reported to decrease sexual desire in an uncontrolled clinical study (Regan, 1999). Conversely, a retrospective chart review of 137 women with severe premenstrual syndrome treated during the luteal phase with 100–200+ mg/day vaginal progesterone suppositories alone for birth control reported that decreased libido was a side effect in 1 woman (0.7%) (Dalton, Guthrie, & Dalton, 1987).
One study assessed the influence on mood and libido of estradiol plus cyclic 200 mg/day oral progesterone in premenopausal women and estradiol plus cyclic 200 mg/day vaginal progesterone in perimenopausal women (Nappi et al., 2004). The premenopausal women were suffering from menstrual irregularities, depression, and sexual dysfunction, while the perimenopausal women were presumably experiencing menopausal symptoms. A significant improvement in libido, sexual arousal, and pain was observed with therapy in the premenopausal women who had a concomitant improvement in anxiety, irritability, and appetite, but not in those who remained depressed. The findings in the perimenopausal women were similar; the hormonal therapy improved sexual desire only in those who experienced improvement in anxiety, panic, and tension, but not in those in whom depression remained. The findings of this study, although interesting, are unfortunately confounded by the lack of an estradiol-only control group, and so can’t necessarily be attributed to progesterone. Moreover, the findings are confounded by the presence and alleviation of the mood symptoms, anxiety, and other problems in the women; it’s possible that estradiol and progesterone did not increase sexual desire themselves but rather it was the improvement of the other symptoms that produced the changes.
Another randomized controlled trial found that an estrogen plus 1.25 mg/day methyltestosterone and 100 mg/day oral progesterone significantly improved sexual desire and function relative to placebo in postmenopausal women (Blümel et al., 2008). However, this study was confounded by the presence of methyltestosterone and the lack of a no-progesterone control group.
Transdermal progesterone cream at a dosage of 32 mg/day had no influence on sexual desire or other parameters in postmenopausal women (Wren et al., 2003; Worsley et al., 2016).
A recent chapter in a book on progestogens included the following passage (Nadjafi-Triebsch, 2015):
Progesterone improves sexual performance in [male] rats [13]. Consequently, progesterone should also improve sexual performance in men due to similarities to human neuro-endocrine mechanisms. Indeed, patients receiving progesterone reported more frequent morning erections and distinctively improved sexual performance.
However, no source or other information (e.g., study design, route/dosage, etc.) was provided. The claim was probably based on anecdotal clinical experience. In addition, the chapter makes some problematic statements like “progesterone prevents prostate cancer” and “progesterone treats autism”, along with ample reference to low-quality unpublished clinical experience, that seriously hurt its credibility.
High-Dose Progestogens in Men
High-dose progestogen monotherapy with cyproterone acetate (CPA) or MPA is widely used to suppress sexual desire and function in men with sexual deviance, including hypersexuality, paraphilias, and sex offenses (Wiki; Codispoti, 2008). This therapy results in strong sexual suppression and is very effective. Its efficacy is thought to be due to suppression of testosterone levels. However, there is some evidence that progestogenic activity in the brain itself and/or increased prolactin levels secondary to progestogenic activity may also be involved (Zumpe, Clancy, & Michael, 1997; Rastrelli, Corona, & Maggi, 2015; Wiki). Although estrogens suppress testosterone levels and increase prolactin levels similarly to progestogens and have likewise been used in the treatment of male sexual deviance (Wiki; Wiki), they are also known to have beneficial effects on sexual desire in both women and men (Wallen et al., 1984; Wibowo, Schellhammer, & Wasserug, 2011; Cappelletti & Wallen, 2016; Santoro et al., 2016; Gilbert et al., 2017). In relation to this, estrogens may not result in the same degree of suppression of sexual desire and function as progestogens. As with CPA and MPA, progesterone alone by intramuscular injection has been found to substantially suppress sexual desire and function in men (Wiki; Regan, 1999).
Conclusion
Taken together, there appears to be little or no evidence that progesterone or other progestogens improve sexual desire or function in humans. Instead, the available evidence suggests that progestogens are likely to result in either unchanged or decreased sexual desire and function. Decreased sexual desire/function may be especially likely at high doses. In any case, more research on progestogens and sexual desire, particularly with non-oral progesterone, is warranted.
References
Alexander, J. L., Kotz, K., Dennerstein, L., Kutner, S. J., Wallen, K., & Notelovitz, M. (2004). The effects of postmenopausal hormone therapies on female sexual functioning: a review of double-blind, randomized controlled trials. Menopause, 11(6), 749–765. [DOI:10.1097/01.gme.0000142887.31811.97]
Andersen, M. L., & Tufik, S. (2006). Does male sexual behavior require progesterone? Brain Research Reviews, 51(1), 136–143. [DOI:10.1016/j.brainresrev.2005.10.005]
Baum, M. J., Everitt, B. J., Herbert, J., Keverne, E. B., & de Greef, W. J. (1976). Reduction of sexual interaction in rhesus monkeys by a vaginal action of progesterone. Nature, 263(5578), 606–608. [DOI:10.1038/263606a0]
Baum, M. J., Everitt, B. J., Herbert, J., & Keverne, E. B. (1977). Hormonal basis of proceptivity and receptivity in female primates. Archives of Sexual Behavior, 6(3), 173–192. [DOI:10.1007/bf01541126]
Baum, M., Keverne, E., Everitt, B., Herbert, J., & De Greef, W. (1977). Effects of progesterone and estradiol on sexual attractivity of female rhesus monkeys. Physiology & Behavior, 18(4), 659–670. [DOI:10.1016/0031-9384(77)90064-6]
Baum, M. J. (1983). Hormonal Modulation of Sexuality in Female Primates. BioScience, 33(9), 578–582. [DOI:10.2307/1309209]
Biglia, N., Maffei, S., Lello, S., & Nappi, R. E. (2010). Tibolone in postmenopausal women: a review based on recent randomised controlled clinical trials. Gynecological Endocrinology, 26(11), 804–814. [DOI:10.3109/09513590.2010.495437]
Blümel, J. E., Del Pino, M., Aprikian, D., Vallejo, S., Sarrá, S., & Castelo-Branco, C. (2008). Effect of androgens combined with hormone therapy on quality of life in post-menopausal women with sexual dysfunction. Gynecological Endocrinology, 24(12), 691–695. [DOI:10.1080/09513590802454919]
Buhl, A. (1978). Influence of progesterone on sexual behavior in castrated male rhesus monkeys treated with testosterone. Hormones and Behavior, 10(3), 258–263. [DOI:10.1016/0018-506x(78)90070-3]
Cappelletti, M., & Wallen, K. (2016). Increasing women’s sexual desire: The comparative effectiveness of estrogens and androgens. Hormones and Behavior, 78, 178–193. [DOI:10.1016/j.yhbeh.2015.11.003]
Castelo-Branco, C., Cancelo, M. J., & Chedraui, P. (2007). Female sexual dysfunction in postmenopausal women. Expert Opinion on Therapeutic Patents, 17(6), 639–647. [DOI:10.1517/13543776.17.6.639]
Çayan, F., Dilek, U., Pata, Ö., & Dilek, S. (2008). Comparison of the Effects of Hormone Therapy Regimens, Oral and Vaginal Estradiol, Estradiol+Drospirenone and Tibolone, on Sexual Function in Healthy Postmenopausal Women. The Journal of Sexual Medicine, 5(1), 132–138. [DOI:10.1111/j.1743-6109.2007.00635.x]
Codispoti, V. L. (2008). Pharmacology of Sexually Compulsive Behavior. Psychiatric Clinics of North America, 31(4), 671–679. [DOI:10.1016/j.psc.2008.06.002]
Dalton, M. E., Guthrie, K. A., & Dalton, K. (1987). A preliminary study into the efficacy of progesterone suppositories as a contraceptive for women with severe premenstrual syndrome. The British Journal of Family Planning, 13(3), 87–89. [Google Scholar] [PDF]
Davis, S. R., Guay, A. T., Shifren, J. L., & Mazer, N. A. (2004). Endocrine Aspects of Female Sexual Dysfunction. The Journal of Sexual Medicine, 1(1), 82–86. [DOI:10.1111/j.1743-6109.2004.10112.x]
Delville, Y. (1991). Progesterone-facilitated sexual receptivity: A review of arguments supporting a nongenomic mechanism. Neuroscience & Biobehavioral Reviews, 15(3), 407–414. [DOI:10.1016/s0149-7634(05)80033-8]
Dennerstein, L., Burrows, G. D., Wood, C., & Hyman, G. (1980). Hormones and sexuality: effect of estrogen and progestogen. Obstetrics & Gynecology, 56(3), 316–322. [Google Scholar] [PubMed] [URL] [PDF]
Frye, C. A. (2007). Progestins influence motivation, reward, conditioning, stress, and/or response to drugs of abuse. Pharmacology Biochemistry and Behavior, 86(2), 209–219. [DOI:10.1016/j.pbb.2006.07.033]
Gałązka, I., Drosdzol-Cop, A., Naworska, B., Czajkowska, M., & Skrzypulec-Plinta, V. (2015). Changes in the Sexual Function During Pregnancy. The Journal of Sexual Medicine, 12(2), 445–454. [DOI:10.1111/jsm.12747]
Gilbert, D. C., Duong, T., Kynaston, H. G., Alhasso, A. A., Cafferty, F. H., Rosen, S. D., Kanaga-Sundaram, S., Dixit, S., Laniado, M., Madaan, S., Collins, G., Pope, A., Welland, A., Nankivell, M., Wassersug, R., Parmar, M. K., Langley, R. E., & Abel, P. D. (2016). Quality-of-life outcomes from the Prostate Adenocarcinoma: TransCutaneous Hormones (PATCH) trial evaluating luteinising hormone-releasing hormone agonists versus transdermal oestradiol for androgen suppression in advanced prostate cancer. BJU International, 119(5), 667–675. [DOI:10.1111/bju.13687]
Graziottin, A., & Serafini, A. (2011). Medical Treatments for Sexual Problems in Women. In Mulhall, J. P., Incrocci, L., Goldstein, I., & Rosen, R. (Eds.). Cancer and Sexual Health (pp. 627–641). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-60761-916-1_40]
Hawley, W. R., & Mosura, D. E. (2019). Effects of progesterone treatment during adulthood on consummatory and motivational aspects of sexual behavior in male rats. Behavioural Pharmacology, 30(7), 617–622. [DOI:10.1097/fbp.0000000000000490]
Holder, M. K., & Mong, J. A. (2017). The Role of Ovarian Hormones and the Medial Amygdala in Sexual Motivation. Current Sexual Health Reports, 9(4), 262–270. [DOI:10.1007/s11930-017-0131-4]
Javadivala, Z., Merghati-Khoei, E., Asghari Jafarabadi, M., Allahverdipour, H., Mirghafourvand, M., Nadrian, H., & Kouzekanani, K. (2018). Efficacy of pharmacological and non-pharmacological interventions on low sexual interest/arousal of peri- and post-menopausal women: a meta-analysis. Sexual and Relationship Therapy, 34(2), 242–270. [DOI:10.1080/14681994.2018.1446515]
Jones, B. C., Hahn, A. C., & DeBruine, L. M. (2019). Ovulation, Sex Hormones, and Women’s Mating Psychology. Trends in Cognitive Sciences, 23(1), 51–62. [DOI:10.1016/j.tics.2018.10.008]
King, S. R. (2012). Neurosteroids and the Nervous System. In King, S. R. Neurosteroids and the Nervous System (pp. 1–122). New York: Springer New York. [DOI:10.1007/978-1-4614-5559-2_1]
Kovalevsky, G. (2005). Female Sexual Dysfunction and Use of Hormone Therapy in Postmenopausal Women. Seminars in Reproductive Medicine, 23(2), 180–187. [DOI:10.1055/s-2005-869486]
Kristoffersen, K., & Lebech, P. E. (1975). May Hyperprogesteronaemia Cause Frigidity? Acta Obstetricia et Gynecologica Scandinavica, 54(S47), 42–42. [DOI:10.3109/00016347509158744]
Kronawitter, D., Gooren, L. J., Zollver, H., Oppelt, P. G., Beckmann, M. W., Dittrich, R., & Mueller, A. (2009). Effects of transdermal testosterone or oral dydrogesterone on hypoactive sexual desire disorder in transsexual women: results of a pilot study. European Journal of Endocrinology, 161(2), 363–368. [DOI:10.1530/eje-09-0265]
Luttge, W. G. (1971). The role of gonadal hormones in the sexual behavior of the rhesus monkey and human: A literature survey. Archives of Sexual Behavior, 1(1), 61–88. [DOI:10.1007/bf01540937]
Michael, R. P., Saayman, G. S., & Zumpe, D. (1967). Inhibition of sexual receptivity by progesterone in rhesus monkeys. Journal of Endocrinology, 39(2), 309–310. [DOI:10.1677/joe.0.0390309]
Michael, R. P., & Zumpe, D. (1993). Medroxyprogesterone acetate decreases the sexual activity of male cynomolgus monkeys (Macaca fascicularis): An action on the brain? Physiology & Behavior, 53(4), 783–788. [DOI:10.1016/0031-9384(93)90189-m]
Modelska, K., & Milián, M. L. (2004). Treatment of female sexual dysfunction in postmenopausal women—What is the evidence? Reviews in Gynaecological Practice, 4(2), 121–132. [DOI:10.1016/j.rigp.2004.01.001]
Myers, L. S., Dixen, J., Morrissette, D., Carmichael, M., & Davidson, J. M. (1990). Effects of Estrogen, Androgen, and Progestin on Sexual Psychophysiology and Behavior in Postmenopausal Women. The Journal of Clinical Endocrinology & Metabolism, 70(4), 1124–1131. [DOI:10.1210/jcem-70-4-1124]
Nadjafi-Triebsch, C. (2015). Progestogens in Non-gynecological Indications. In Carp, H. J. A. (Ed.). Progestogens in Obstetrics and Gynecology (pp. 191–202). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-14385-9_14]
Nadler, R. D., Roth-Meyer, C., & Baulieu, E.-E. (1985). Behavioral and Endocrine Consequences of Long-Term Antiprogesterone (RU 486) Administration to Cynomolgus Monkeys: Preliminary Results. In Baulieu, E.-E., & Segal, S. J. (Eds.). The Antiprogestin Steroid RU 486 and Human Fertility Control (pp. 169–177). Boston: Springer US. [DOI:10.1007/978-1-4684-1242-0_14]
Nappi, R. E., Sommacal, A., Sances, G., Abbiati, I., Ferdeghini, F., Vercesi, C., Albani, F., & Polatti, F. (2004). Progesterone supplementation on mood and sexual well-being. Gynecological Endocrinology, 18(Suppl 1 [Abstracts of the 11th World Congress of Gynecological Endocrinology, Florence, Italy, February 26-29, 2004]), 133–134 (abstract no. S9). [Google Scholar] [PubMed] [URL] [PDF]
Neumann, F. (1978). The physiological action of progesterone and the pharmacological effects of progestogens – a short review. Postgraduate Medical Journal, 54(Suppl 2), 11–24. [Google Scholar] [PubMed] [PDF]
Pastor, Z., Holla, K., & Chmel, R. (2013). The influence of combined oral contraceptives on female sexual desire: A systematic review. The European Journal of Contraception & Reproductive Health Care, 18(1), 27–43. [DOI:10.3109/13625187.2012.728643]
Pazol, K., Wilson, M. E., & Wallen, K. (2004). Medroxyprogesterone Acetate Antagonizes the Effects of Estrogen Treatment on Social and Sexual Behavior in Female Macaques. The Journal of Clinical Endocrinology & Metabolism, 89(6), 2998–3006. [DOI:10.1210/jc.2003-032086]
Pazol, K., Northcutt, K. V., Wilson, M. E., & Wallen, K. (2006). Medroxyprogesterone acetate acutely facilitates and sequentially inhibits sexual behavior in female rats. Hormones and Behavior, 49(1), 105–113. [DOI:10.1016/j.yhbeh.2005.05.008]
Rastrelli, G., Corona, G., & Maggi, M. (2015). The role of prolactin in andrology: what is new? Reviews in Endocrine and Metabolic Disorders, 16(3), 233–248. [DOI:10.1007/s11154-015-9322-3]
Regan, P. C. (1999). Hormonal correlates and causes of sexual desire: A review. The Canadian Journal of Human Sexuality, 8(1), 1–16. [Google Scholar] [URL] [PDF 1] [PDF 2]
Regan, P. C., Lyle, J. L., Otto, A. L., & Joshi, A. (2003). Pregnancy and changes in female sexual desire: a review. Social Behavior and Personality, 31(6), 603–611. [DOI:10.2224/sbp.2003.31.6.603]
Roney, J. R., & Simmons, Z. L. (2013). Hormonal predictors of sexual motivation in natural menstrual cycles. Hormones and Behavior, 63(4), 636–645. [DOI:10.1016/j.yhbeh.2013.02.013]
Roney, J. R. (2014). An Evolutionary Functional Analysis of the Hormonal Predictors of Women’s Sexual Motivation. In Shackelford, T. K., & Hansen, R. D. (Eds.). The Evolution of Sexuality (pp. 99–121). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-09384-0_6]
Roney, J. R., & Simmons, Z. L. (2016). Within-cycle fluctuations in progesterone negatively predict changes in both in-pair and extra-pair desire among partnered women. Hormones and Behavior, 81, 45–52. [DOI:10.1016/j.yhbeh.2016.03.008]
Roney, J. R. (2019). Evolutionary Perspectives on Hypoactive Sexual Desire Disorder in Women. Current Sexual Health Reports, 11(4), 243–250. [DOI:10.1007/s11930-019-00223-w]
Saadat, P., Boostanfar, R., Poysky, J., Munevar, C., Stanczyk, F., Buckwalter, G., & Roy, S. (2002). A randomized prospective study comparing the effects of micronized progesterone and medroxyprogesterone acetate on mood and libido. Fertility and Sterility, 77(Suppl 3 [Abstracts of the 50th Annual Meeting of the Pacific Coast Reproductive Society. April 17-21, 2002. Rancho Mirage, California, USA]), S21–S21 (abstract no. P-28). [Google Scholar] [PubMed] [DOI:10.1016/s0015-0282(02)03053-4]
Sandhu, K. S., Melman, A., & Mikhail, M. S. (2011). Impact of Hormones on Female Sexual Function and Dysfunction. Female Pelvic Medicine & Reconstructive Surgery, 17(1), 8–16. [DOI:10.1097/spv.0b013e318204491f]
Santoro, N., Worsley, R., Miller, K. K., Parish, S. J., & Davis, S. R. (2016). Role of Estrogens and Estrogen-Like Compounds in Female Sexual Function and Dysfunction. The Journal of Sexual Medicine, 13(3), 305–316. [DOI:10.1016/j.jsxm.2015.11.015]
Savolainen-Peltonen, H., Hautamäki, H., Tuomikoski, P., Ylikorkala, O., & Mikkola, T. S. (2014). Health-related quality of life in women with or without hot flashes. Menopause, 21(7), 732–739. [DOI:10.1097/gme.0000000000000120]
Schaffir, J. (2006). Hormonal Contraception and Sexual Desire: A Critical Review. Journal of Sex & Marital Therapy, 32(4), 305–314. [DOI:10.1080/00926230600666311]
Schmidt, P. J., Steinberg, E. M., Negro, P. P., Haq, N., Gibson, C., & Rubinow, D. R. (2008). Pharmacologically Induced Hypogonadism and Sexual Function in Healthy Young Women and Men. Neuropsychopharmacology, 34(3), 565–576. [DOI:10.1038/npp.2008.24]
Sherwin, B. B. (1991). The Impact of Different Doses of Estrogen and Progestin on Mood and Sexual Behavior in Postmenopausal Women. The Journal of Clinical Endocrinology & Metabolism, 72(2), 336–343. [DOI:10.1210/jcem-72-2-336]
Shirazi, T. N., Self, H., Dawood, K., Rosenfield, K. A., Penke, L., Carré, J. M., Ortiz, T., & Puts, D. A. (2019). Hormonal predictors of women’s sexual motivation. Evolution and Human Behavior, 40(3), 336–344. [DOI:10.1016/j.evolhumbehav.2019.02.002]
Wallen, K. (1984). Periovulatory changes in female sexual behavior and patterns of ovarian steroid secretion in group-living rhesus monkeys. Hormones and Behavior, 18(4), 431–450. [DOI:10.1016/0018-506x(84)90028-x]
Ward, I. L., Franck, J. E., & Crowley, W. R. (1977). Central progesterone induces female sexual behavior in estrogen-primed intact male rats. Journal of Comparative and Physiological Psychology, 91(6), 1417–1423. [DOI:10.1037/h0077419]
Waxman, J. H., Wass, J. A., Hendry, W. F., Whitfield, H. N., Bary, P., Besser, G. M., Malpas, J. S., & Oliver, R. T. (1983). Treatment of Advanced Prostatic Cancer with Buserelin, an Analogue of Gonadotrophin Releasing Hormone. British Journal of Urology, 55(6), 737–742. [DOI:10.1111/j.1464-410x.1983.tb03416.x]
Wibowo, E., Schellhammer, P., & Wassersug, R. J. (2011). Role of Estrogen in Normal Male Function: Clinical Implications for Patients With Prostate Cancer on Androgen Deprivation Therapy. Journal of Urology, 185(1), 17–23. [DOI:10.1016/j.juro.2010.08.094]
Witt, D. M., Young, L. J., & Crews, D. (1995). Progesterone modulation of androgen-dependent sexual behavior in male rats. Physiology & Behavior, 57(2), 307–313. [DOI:10.1016/0031-9384(94)00247-3]
Worsley, R., Santoro, N., Miller, K. K., Parish, S. J., & Davis, S. R. (2016). Hormones and Female Sexual Dysfunction: Beyond Estrogens and Androgens—Findings From the Fourth International Consultation on Sexual Medicine. The Journal of Sexual Medicine, 13(3), 283–290. [DOI:10.1016/j.jsxm.2015.12.014]
Wren, B. G., Champion, S. M., Willetts, K., Manga, R. Z., & Eden, J. A. (2003). Transdermal progesterone and its effect on vasomotor symptoms, blood lipid levels, bone metabolic markers, moods, and quality of life for postmenopausal women. Menopause, 10(1), 13–18. [DOI:10.1097/00042192-200310010-00004]
Zimmerman, Y., Eijkemans, M. J., Coelingh Bennink, H. J., Blankenstein, M. A., & Fauser, B. C. (2013). The effect of combined oral contraception on testosterone levels in healthy women: a systematic review and meta-analysis. Human Reproduction Update, 20(1), 76–105. [DOI:10.1093/humupd/dmt038]
Zumpe, D., & Michael, R. P. (1988). Effects of medroxyprogesterone acetate on plasma testosterone and sexual behavior in male cynomolgus monkeys (Macaca fascicularis). Physiology & Behavior, 42(4), 343–349. [DOI:10.1016/0031-9384(88)90275-2]
Zumpe, D. (1991). Medroxyprogesterone acetate, aggression, and sexual behavior in male cynomolgus monkeys (Macaca fascicularis). Hormones and Behavior, 25(3), 394–409. [DOI:10.1016/0018-506x(91)90010-f]
Zumpe, D., Clancy, A. N., & Michael, R. P. (1997). Effects of Progesterone on the Sexual Behavior of Castrated, Testosterone-Treated Male Cynomolgus Monkeys (Macaca Fascicularis). Physiology & Behavior, 62(1), 61–67. [DOI:10.1016/s0031-9384(97)00135-2]
\ No newline at end of file
+The Influence of Progesterone and Other Progestogens on Sexual Desire and Function - Transfeminine Science
The Influence of Progesterone and Other Progestogens on Sexual Desire and Function
By Aly | First published January 9, 2020 | Last modified March 22, 2024
Abstract / TL;DR
Many transfeminine people believe on the basis of anecdotes that progesterone increases sexual desire. A review was conducted to assess the available evidence for the possibility that progesterone and/or progestogens increase sexual desire. Both animal studies as well as clinical studies with progesterone and progestins were considered. Progesterone has shown no indication of stimulatory effects on sexual desire or behavior in humans and non-human primates. High doses of progestogens have however been found to inhibit sexual desire in humans and non-human primates. More research is necessary to further characterize the role of progesterone and other progestogens in sexual desire in humans. In any case, available evidence does not support a positive role of these hormonal agents in sexual desire. Instead, sexual desire is more related to androgens and estrogens in men and women, respectively.
Introduction
Based on anecdotal experience, some people in the transfeminine community believe that progesterone has positive effects on sexual desire. This is such that even memes about progesterone and libido have been made (Reddit). In addition to anecdotes, some researchers have theorized that progesterone may be positively involved in sexual desire in cisgender women (Regan, 1999). Some have also argued that the neurosteroid metabolites of progesterone like allopregnanolone and pregnanolone may provide special benefits on sexual desire that are not shared with progestins. I’ve reviewed the available evidence and literature to assess whether progesterone or other progestogens indeed improve sexual desire function in cisgender women and might be useful for this in transfeminine people.
Animal Studies
Correlational
Progesterone levels are strongly and consistently negatively associated with female sexual desire and behavior in most animal species (Roney, 2015). This includes in monkeys and apes (Roney, 2015).
Experimental
Non-Human Primates
Progesterone, both alone and when added to an estrogen, has been shown to inhibit sexual behavior and receptivity in female monkeys in many studies (Luttge, 1971; Baum et al., 1976; Baum et al., 1977a; Michael et al., 1967). This inhibition is often marked. Likewise, subcutaneous implants of progesterone were shown to suppress sexual behavior in testosterone-treated castrated male monkeys (Zumpe, Clancy, & Michael, 1997). As the male monkeys were castrated and treated with testosterone, this was not due to decreased testosterone levels (which remained unchanged), and could only have been due to the other effects of progesterone. Similar findings have been made for the progestin medroxyprogesterone acetate (MPA) (Michael & Zumpe, 1993; Zumpe et al., 1991; Zumpe & Michael, 1988).
The following excerpt from a review is notable on the topic of progesterone and sexual behavior in primates (Baum, 1983):
The early work of Ball (1941) and of Michael et al. (1968) showed that progesterone acts in the female rhesus monkey to reduce the likelihood of copulation. In contrast to non primate females, no facilitatory action of progesterone on any aspect of feminine sexuality has ever been convincingly demonstrated in primates.
One study in female monkeys found that MPA inhibited sexual behavior while progesterone did not (Pazol et al., 2004). However, MPA is a much more potent progestogen than progesterone, and the progesterone levels were very low (2 ng/mL). Hence, the progesterone levels in this study may have been too low for adequate physiological effect, which might explain the conflicting finding compared to other studies. Another study found increased sexual receptivity in castrated female monkeys treated with a combination of an estrogen and parenteral progesterone (Baum et al., 1977b). However, this was relative to a baseline of castration, and the changes may have only been due to the estrogen. Indeed, the same researchers reported decreased sexual receptivity with the addition of progesterone to estrogen in other studies (Baum et al., 1976; Baum et al., 1977a).
Mifepristone, a progesterone receptor antagonist, showed no apparent effect on sexual behavior in either female or male monkeys (Nadler et al., 1985).
Rodents
A notable exception when it comes to progesterone and sexual behavior in animals is female rodents, in which progesterone is stimulatory and induces lordosis (Frye, 2007). Allopregnanolone is involved in the facilitatory effects of progesterone on female sexual behavior in rodents, but has mixed effects, showing either augmentation or inhibition depending on the specifics (King, 2013; Frye, 2007). However, progesterone stimulates sexual behavior in female rodents only acutely, and is actually inhibitory with continuous administration (Pazol et al., 2004; Pazol et al., 2006). Studies in estrogen-primed intact male rats have found that progesterone given systemically does not induce lordosis (Ward et al., 1977). Interestingly however, lordosis was found to be induced in some male rats when progesterone was injected directly into a specific part of the hypothalamus (Ward et al., 1977). The prosexual effects of progesterone in female rodents may be mediated via non-genomic mechanisms, for instance actions of progesterone’s neurosteroid metabolites (Delville, 1991).
In addition to female rodents, research has found that progesterone is involved in normal sexual behavior in male rats (Witt et al., 1995; Andersen & Tufik, 2006). In male rats, castration greatly decreases levels of both testosterone and progesterone and markedly reduces sexual behavior (Witt et al., 1995). Low-dose progesterone administration, resulting in low but physiological male-range levels of progesterone, significantly restores sexual behavior (Witt et al., 1995). Additionally, when testosterone is concomitantly administered, progesterone augments the improvement caused by testosterone and is required for sexual behavior to be fully restored (Witt et al., 1995). The effects are mimicked by progestins like promegestone and blocked by the progesterone receptor antagonist mifepristone, suggesting that they are due specifically to activation of the progesterone receptor (Witt et al., 1995).
However, the findings in male rats probably do not apply to primates. As already described, mifepristone, in contrast to male rats, has no influence on sexual behavior in male monkeys (Nadler et al., 1985). Similarly, a study in castrated male monkeys given testosterone in combination with a relatively low dose of progesterone—resulting in roughly doubled progesterone levels relative to normal male levels—found no difference in sexual behavior compared to castrated male monkeys given testosterone alone (Buhl, Jensen, & Phoenix, 1978). Hence, neither progesterone receptor antagonism nor restoration of progesterone levels following castration seems to influence sexual behavior in male primates. It’s also notable that unlike in male rodents, castration doesn’t appear to reduce progesterone levels in either male monkeys (Zumpe, Clancy, & Michael, 1997) or humans (Waxman et al., 2013). As such, even if the rodent findings did apply to humans, castrated men have normal male-range progesterone levels and would in theory have all the progesterone needed to maintain sexual desire.
The findings on male-range progesterone levels and sexual behavior in male rats are also in contrast to research with higher, supraphysiological levels of progesterone (Hawley & Mosura, 2019). This degree of progesterone exposure has been shown to inhibit sexual behavior in males of most or all species examined, notably including male rats (Hawley & Mosura, 2019). This is the case regardless of influence on testosterone levels (Hawley & Mosura, 2019).
Other Species
Progestogens are used to inhibit heat in female dogs and cats (Neumann, 1978).
Sexual desire is progressively decreased in women during pregnancy, a state characterized by very high levels of estrogens, progesterone, and prolactin (Regan et al., 2003; Gałązka et al., 2014). However, many confounding factors exist (e.g., the other hormonal changes, or being pregnant), and hence causation of the changes can’t necessarily be attributed to progesterone. The observations do suggest that very high levels of an estrogen and progesterone may be unlikely to increase sexual desire however.
Experimental
Progestins
A 2013 meta-analysis reported that combined birth control pills, containing both ethinylestradiol and a progestogen, result in either unchanged or increased sexual desire in most women (85%) and decreased sexual desire in a small percentage of women (15%) (Pastor et al., 2013). However, changes in sexual desire seem to be related to the ethinylestradiol component rather than to the progestin component; decreased sexual desire was only associated with birth control pills containing low-dose estrogen (20 μg ethinylestradiol) (Pastor et al., 2013). The fact that combined birth control pills infrequently decrease sexual desire in women is notable because these medications suppress ovulation and hence completely prevent the increase in progesterone levels during the luteal phase. As such, progesterone levels, and by extension circulating levels of allopregnanolone and pregnanolone, remain very low at all times. This suggests little or no importance of the neurosteroid metabolites of progesterone for sexual desire in women.
The fact that combined birth control pills don’t generally affect sexual desire in women is also potentially surprising because total and free testosterone levels are greatly decreased by them due to the ethinylestradiol component (Zimmerman et al., 2014). This is, however, in accordance with accumulating evidence suggesting that physiological levels of androgens in women have little or no involvement in sexual desire (Cappelletti & Wallen, 2016). Rather than progesterone or androgens, what seems to be important for sexual desire in women is estradiol, especially acutely at periovulatory levels (Cappelletti & Wallen, 2016).
A small minority of women treated with depot medroxyprogesterone acetate (DMPA) alone for birth control report decreased libido and only 2.3% cite it as a reason for discontinuation (Schaffir, 2006). A study of 80 women treated with DMPA alone for birth control monitored sexual function and found no difference from baseline to 4 months of therapy (Schaffir, 2006). Findings have been similar for subcutaneous birth control implants with nomegestrol acetate and levonorgestrel (Schaffir, 2006). A randomized controlled trial of 150 women given combined birth control pills, progestin-only birth control pills, and placebo found no difference in sexual desire or activity between progestin-only pills and placebo (Schaffir, 2006).
Interestingly, a 2018 meta-analysis reported a higher standardized mean difference (SMD) for improvement of sexual desire/function in studies with an estrogen plus a progestogen relative to studies using an estrogen alone (Javadivala et al., 2019). However, there was differing methodology (e.g., sexual measures) and high heterogeneity across studies, as well as a limited number of included studies (particularly of estrogen only). These issues make conclusions on the basis of the differing SMDs problematic. Indeed, the finding is contradicted by individual studies that have included both estrogen only and estrogen plus progestogen groups (see the sources above).
Besides progestogenic activity, it has been suggested that androgenic progestins, like most of the testosterone derivatives, might provide better effects on sexual desire/function than non-androgenic progestins (Graziottin & Serafini, 2011). However, aside from the case of the atypical agent tibolone (Biglia et al., 2010), this has not actually been assessed or shown in any clinical trials (Schaffir, 2006).
The addition of dydrogesterone to hormone therapy for transgender women to improve sexual desire showed no effect, whereas transdermal testosterone was effective (Kronawitter et al., 2009).
A 1975 study published as an abstract with little additional information claimed that some premenopausal women with “frigidity” had high progesterone levels and that treatment with levonorgestrel alone to suppress progesterone levels could increase libido in these women (Kristoffersen & Lebech, 1975). This is very low-quality information however.
Progesterone
Available clinical studies of progesterone and sexual desire in women have observed no changes (Worsley et al., 2016). Neither 200 mg/day oral progesterone nor 10 mg/day oral MPA had any influence on mood or libido in estrogen-treated postmenopausal women in a randomized controlled trial (Saadat et al., 2002). Likewise, treatment of pharmacologically hypogonadal women with 400 mg/day vaginal progesterone suppositories alone did not significantly alter any domain of sexual function in another randomized controlled trial (Schmidt et al., 2009). Treatment of women with subcutaneous implants of progesterone alone for gynecological disorders has been reported to decrease sexual desire in an uncontrolled clinical study (Regan, 1999). Conversely, a retrospective chart review of 137 women with severe premenstrual syndrome treated during the luteal phase with 100–200+ mg/day vaginal progesterone suppositories alone for birth control reported that decreased libido was a side effect in 1 woman (0.7%) (Dalton, Guthrie, & Dalton, 1987).
One study assessed the influence on mood and libido of estradiol plus cyclic 200 mg/day oral progesterone in premenopausal women and estradiol plus cyclic 200 mg/day vaginal progesterone in perimenopausal women (Nappi et al., 2004). The premenopausal women were suffering from menstrual irregularities, depression, and sexual dysfunction, while the perimenopausal women were presumably experiencing menopausal symptoms. A significant improvement in libido, sexual arousal, and pain was observed with therapy in the premenopausal women who had a concomitant improvement in anxiety, irritability, and appetite, but not in those who remained depressed. The findings in the perimenopausal women were similar; the hormonal therapy improved sexual desire only in those who experienced improvement in anxiety, panic, and tension, but not in those in whom depression remained. The findings of this study, although interesting, are unfortunately confounded by the lack of an estradiol-only control group, and so can’t necessarily be attributed to progesterone. Moreover, the findings are confounded by the presence and alleviation of the mood symptoms, anxiety, and other problems in the women; it’s possible that estradiol and progesterone did not increase sexual desire themselves but rather it was the improvement of the other symptoms that produced the changes.
Another randomized controlled trial found that an estrogen plus 1.25 mg/day methyltestosterone and 100 mg/day oral progesterone significantly improved sexual desire and function relative to placebo in postmenopausal women (Blümel et al., 2008). However, this study was confounded by the presence of methyltestosterone and the lack of a no-progesterone control group.
Transdermal progesterone cream at a dosage of 32 mg/day had no influence on sexual desire or other parameters in postmenopausal women (Wren et al., 2003; Worsley et al., 2016).
A recent chapter in a book on progestogens included the following passage (Nadjafi-Triebsch, 2015):
Progesterone improves sexual performance in [male] rats [13]. Consequently, progesterone should also improve sexual performance in men due to similarities to human neuro-endocrine mechanisms. Indeed, patients receiving progesterone reported more frequent morning erections and distinctively improved sexual performance.
However, no source or other information (e.g., study design, route/dosage, etc.) was provided. The claim was probably based on anecdotal clinical experience. In addition, the chapter makes some problematic statements like “progesterone prevents prostate cancer” and “progesterone treats autism”, along with ample reference to low-quality unpublished clinical experience, that seriously hurt its credibility.
High-Dose Progestogens in Men
High-dose progestogen monotherapy with cyproterone acetate (CPA) or MPA is widely used to suppress sexual desire and function in men with sexual deviance, including hypersexuality, paraphilias, and sex offenses (Wiki; Codispoti, 2008). This therapy results in strong sexual suppression and is very effective. Its efficacy is thought to be due to suppression of testosterone levels. However, there is some evidence that progestogenic activity in the brain itself and/or increased prolactin levels secondary to progestogenic activity may also be involved (Zumpe, Clancy, & Michael, 1997; Rastrelli, Corona, & Maggi, 2015; Wiki). Although estrogens suppress testosterone levels and increase prolactin levels similarly to progestogens and have likewise been used in the treatment of male sexual deviance (Wiki; Wiki), they are also known to have beneficial effects on sexual desire in both women and men (Wallen et al., 1984; Wibowo, Schellhammer, & Wasserug, 2011; Cappelletti & Wallen, 2016; Santoro et al., 2016; Gilbert et al., 2017). In relation to this, estrogens may not result in the same degree of suppression of sexual desire and function as progestogens. As with CPA and MPA, progesterone alone by intramuscular injection has been found to substantially suppress sexual desire and function in men (Wiki; Regan, 1999).
Conclusion
Taken together, there appears to be little or no evidence that progesterone or other progestogens improve sexual desire or function in humans. Instead, the available evidence suggests that progestogens are likely to result in either unchanged or decreased sexual desire and function. Decreased sexual desire/function may be especially likely at high doses. In any case, more research on progestogens and sexual desire, particularly with non-oral progesterone, is warranted.
References
Alexander, J. L., Kotz, K., Dennerstein, L., Kutner, S. J., Wallen, K., & Notelovitz, M. (2004). The effects of postmenopausal hormone therapies on female sexual functioning: a review of double-blind, randomized controlled trials. Menopause, 11(6), 749–765. [DOI:10.1097/01.gme.0000142887.31811.97]
Andersen, M. L., & Tufik, S. (2006). Does male sexual behavior require progesterone? Brain Research Reviews, 51(1), 136–143. [DOI:10.1016/j.brainresrev.2005.10.005]
Baum, M. J., Everitt, B. J., Herbert, J., Keverne, E. B., & de Greef, W. J. (1976). Reduction of sexual interaction in rhesus monkeys by a vaginal action of progesterone. Nature, 263(5578), 606–608. [DOI:10.1038/263606a0]
Baum, M. J., Everitt, B. J., Herbert, J., & Keverne, E. B. (1977). Hormonal basis of proceptivity and receptivity in female primates. Archives of Sexual Behavior, 6(3), 173–192. [DOI:10.1007/bf01541126]
Baum, M., Keverne, E., Everitt, B., Herbert, J., & De Greef, W. (1977). Effects of progesterone and estradiol on sexual attractivity of female rhesus monkeys. Physiology & Behavior, 18(4), 659–670. [DOI:10.1016/0031-9384(77)90064-6]
Baum, M. J. (1983). Hormonal Modulation of Sexuality in Female Primates. BioScience, 33(9), 578–582. [DOI:10.2307/1309209]
Biglia, N., Maffei, S., Lello, S., & Nappi, R. E. (2010). Tibolone in postmenopausal women: a review based on recent randomised controlled clinical trials. Gynecological Endocrinology, 26(11), 804–814. [DOI:10.3109/09513590.2010.495437]
Blümel, J. E., Del Pino, M., Aprikian, D., Vallejo, S., Sarrá, S., & Castelo-Branco, C. (2008). Effect of androgens combined with hormone therapy on quality of life in post-menopausal women with sexual dysfunction. Gynecological Endocrinology, 24(12), 691–695. [DOI:10.1080/09513590802454919]
Buhl, A. (1978). Influence of progesterone on sexual behavior in castrated male rhesus monkeys treated with testosterone. Hormones and Behavior, 10(3), 258–263. [DOI:10.1016/0018-506x(78)90070-3]
Cappelletti, M., & Wallen, K. (2016). Increasing women’s sexual desire: The comparative effectiveness of estrogens and androgens. Hormones and Behavior, 78, 178–193. [DOI:10.1016/j.yhbeh.2015.11.003]
Castelo-Branco, C., Cancelo, M. J., & Chedraui, P. (2007). Female sexual dysfunction in postmenopausal women. Expert Opinion on Therapeutic Patents, 17(6), 639–647. [DOI:10.1517/13543776.17.6.639]
Çayan, F., Dilek, U., Pata, Ö., & Dilek, S. (2008). Comparison of the Effects of Hormone Therapy Regimens, Oral and Vaginal Estradiol, Estradiol+Drospirenone and Tibolone, on Sexual Function in Healthy Postmenopausal Women. The Journal of Sexual Medicine, 5(1), 132–138. [DOI:10.1111/j.1743-6109.2007.00635.x]
Codispoti, V. L. (2008). Pharmacology of Sexually Compulsive Behavior. Psychiatric Clinics of North America, 31(4), 671–679. [DOI:10.1016/j.psc.2008.06.002]
Dalton, M. E., Guthrie, K. A., & Dalton, K. (1987). A preliminary study into the efficacy of progesterone suppositories as a contraceptive for women with severe premenstrual syndrome. The British Journal of Family Planning, 13(3), 87–89. [Google Scholar] [PDF]
Davis, S. R., Guay, A. T., Shifren, J. L., & Mazer, N. A. (2004). Endocrine Aspects of Female Sexual Dysfunction. The Journal of Sexual Medicine, 1(1), 82–86. [DOI:10.1111/j.1743-6109.2004.10112.x]
Delville, Y. (1991). Progesterone-facilitated sexual receptivity: A review of arguments supporting a nongenomic mechanism. Neuroscience & Biobehavioral Reviews, 15(3), 407–414. [DOI:10.1016/s0149-7634(05)80033-8]
Dennerstein, L., Burrows, G. D., Wood, C., & Hyman, G. (1980). Hormones and sexuality: effect of estrogen and progestogen. Obstetrics & Gynecology, 56(3), 316–322. [Google Scholar] [PubMed] [URL] [PDF]
Frye, C. A. (2007). Progestins influence motivation, reward, conditioning, stress, and/or response to drugs of abuse. Pharmacology Biochemistry and Behavior, 86(2), 209–219. [DOI:10.1016/j.pbb.2006.07.033]
Gałązka, I., Drosdzol-Cop, A., Naworska, B., Czajkowska, M., & Skrzypulec-Plinta, V. (2015). Changes in the Sexual Function During Pregnancy. The Journal of Sexual Medicine, 12(2), 445–454. [DOI:10.1111/jsm.12747]
Gilbert, D. C., Duong, T., Kynaston, H. G., Alhasso, A. A., Cafferty, F. H., Rosen, S. D., Kanaga-Sundaram, S., Dixit, S., Laniado, M., Madaan, S., Collins, G., Pope, A., Welland, A., Nankivell, M., Wassersug, R., Parmar, M. K., Langley, R. E., & Abel, P. D. (2016). Quality-of-life outcomes from the Prostate Adenocarcinoma: TransCutaneous Hormones (PATCH) trial evaluating luteinising hormone-releasing hormone agonists versus transdermal oestradiol for androgen suppression in advanced prostate cancer. BJU International, 119(5), 667–675. [DOI:10.1111/bju.13687]
Graziottin, A., & Serafini, A. (2011). Medical Treatments for Sexual Problems in Women. In Mulhall, J. P., Incrocci, L., Goldstein, I., & Rosen, R. (Eds.). Cancer and Sexual Health (pp. 627–641). Totowa, New Jersey: Humana Press. [DOI:10.1007/978-1-60761-916-1_40]
Hawley, W. R., & Mosura, D. E. (2019). Effects of progesterone treatment during adulthood on consummatory and motivational aspects of sexual behavior in male rats. Behavioural Pharmacology, 30(7), 617–622. [DOI:10.1097/fbp.0000000000000490]
Holder, M. K., & Mong, J. A. (2017). The Role of Ovarian Hormones and the Medial Amygdala in Sexual Motivation. Current Sexual Health Reports, 9(4), 262–270. [DOI:10.1007/s11930-017-0131-4]
Javadivala, Z., Merghati-Khoei, E., Asghari Jafarabadi, M., Allahverdipour, H., Mirghafourvand, M., Nadrian, H., & Kouzekanani, K. (2018). Efficacy of pharmacological and non-pharmacological interventions on low sexual interest/arousal of peri- and post-menopausal women: a meta-analysis. Sexual and Relationship Therapy, 34(2), 242–270. [DOI:10.1080/14681994.2018.1446515]
Jones, B. C., Hahn, A. C., & DeBruine, L. M. (2019). Ovulation, Sex Hormones, and Women’s Mating Psychology. Trends in Cognitive Sciences, 23(1), 51–62. [DOI:10.1016/j.tics.2018.10.008]
King, S. R. (2012). Neurosteroids and the Nervous System. In King, S. R. Neurosteroids and the Nervous System (pp. 1–122). New York: Springer New York. [DOI:10.1007/978-1-4614-5559-2_1]
Kovalevsky, G. (2005). Female Sexual Dysfunction and Use of Hormone Therapy in Postmenopausal Women. Seminars in Reproductive Medicine, 23(2), 180–187. [DOI:10.1055/s-2005-869486]
Kristoffersen, K., & Lebech, P. E. (1975). May Hyperprogesteronaemia Cause Frigidity? Acta Obstetricia et Gynecologica Scandinavica, 54(S47), 42–42. [DOI:10.3109/00016347509158744]
Kronawitter, D., Gooren, L. J., Zollver, H., Oppelt, P. G., Beckmann, M. W., Dittrich, R., & Mueller, A. (2009). Effects of transdermal testosterone or oral dydrogesterone on hypoactive sexual desire disorder in transsexual women: results of a pilot study. European Journal of Endocrinology, 161(2), 363–368. [DOI:10.1530/eje-09-0265]
Luttge, W. G. (1971). The role of gonadal hormones in the sexual behavior of the rhesus monkey and human: A literature survey. Archives of Sexual Behavior, 1(1), 61–88. [DOI:10.1007/bf01540937]
Michael, R. P., Saayman, G. S., & Zumpe, D. (1967). Inhibition of sexual receptivity by progesterone in rhesus monkeys. Journal of Endocrinology, 39(2), 309–310. [DOI:10.1677/joe.0.0390309]
Michael, R. P., & Zumpe, D. (1993). Medroxyprogesterone acetate decreases the sexual activity of male cynomolgus monkeys (Macaca fascicularis): An action on the brain? Physiology & Behavior, 53(4), 783–788. [DOI:10.1016/0031-9384(93)90189-m]
Modelska, K., & Milián, M. L. (2004). Treatment of female sexual dysfunction in postmenopausal women—What is the evidence? Reviews in Gynaecological Practice, 4(2), 121–132. [DOI:10.1016/j.rigp.2004.01.001]
Myers, L. S., Dixen, J., Morrissette, D., Carmichael, M., & Davidson, J. M. (1990). Effects of Estrogen, Androgen, and Progestin on Sexual Psychophysiology and Behavior in Postmenopausal Women. The Journal of Clinical Endocrinology & Metabolism, 70(4), 1124–1131. [DOI:10.1210/jcem-70-4-1124]
Nadjafi-Triebsch, C. (2015). Progestogens in Non-gynecological Indications. In Carp, H. J. A. (Ed.). Progestogens in Obstetrics and Gynecology (pp. 191–202). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-14385-9_14]
Nadler, R. D., Roth-Meyer, C., & Baulieu, E.-E. (1985). Behavioral and Endocrine Consequences of Long-Term Antiprogesterone (RU 486) Administration to Cynomolgus Monkeys: Preliminary Results. In Baulieu, E.-E., & Segal, S. J. (Eds.). The Antiprogestin Steroid RU 486 and Human Fertility Control (pp. 169–177). Boston: Springer US. [DOI:10.1007/978-1-4684-1242-0_14]
Nappi, R. E., Sommacal, A., Sances, G., Abbiati, I., Ferdeghini, F., Vercesi, C., Albani, F., & Polatti, F. (2004). Progesterone supplementation on mood and sexual well-being. Gynecological Endocrinology, 18(Suppl 1 [Abstracts of the 11th World Congress of Gynecological Endocrinology, Florence, Italy, February 26-29, 2004]), 133–134 (abstract no. S9). [Google Scholar] [PubMed] [URL] [PDF]
Neumann, F. (1978). The physiological action of progesterone and the pharmacological effects of progestogens – a short review. Postgraduate Medical Journal, 54(Suppl 2), 11–24. [Google Scholar] [PubMed] [PDF]
Pastor, Z., Holla, K., & Chmel, R. (2013). The influence of combined oral contraceptives on female sexual desire: A systematic review. The European Journal of Contraception & Reproductive Health Care, 18(1), 27–43. [DOI:10.3109/13625187.2012.728643]
Pazol, K., Wilson, M. E., & Wallen, K. (2004). Medroxyprogesterone Acetate Antagonizes the Effects of Estrogen Treatment on Social and Sexual Behavior in Female Macaques. The Journal of Clinical Endocrinology & Metabolism, 89(6), 2998–3006. [DOI:10.1210/jc.2003-032086]
Pazol, K., Northcutt, K. V., Wilson, M. E., & Wallen, K. (2006). Medroxyprogesterone acetate acutely facilitates and sequentially inhibits sexual behavior in female rats. Hormones and Behavior, 49(1), 105–113. [DOI:10.1016/j.yhbeh.2005.05.008]
Rastrelli, G., Corona, G., & Maggi, M. (2015). The role of prolactin in andrology: what is new? Reviews in Endocrine and Metabolic Disorders, 16(3), 233–248. [DOI:10.1007/s11154-015-9322-3]
Regan, P. C. (1999). Hormonal correlates and causes of sexual desire: A review. The Canadian Journal of Human Sexuality, 8(1), 1–16. [Google Scholar] [URL] [PDF 1] [PDF 2]
Regan, P. C., Lyle, J. L., Otto, A. L., & Joshi, A. (2003). Pregnancy and changes in female sexual desire: a review. Social Behavior and Personality, 31(6), 603–611. [DOI:10.2224/sbp.2003.31.6.603]
Roney, J. R., & Simmons, Z. L. (2013). Hormonal predictors of sexual motivation in natural menstrual cycles. Hormones and Behavior, 63(4), 636–645. [DOI:10.1016/j.yhbeh.2013.02.013]
Roney, J. R. (2014). An Evolutionary Functional Analysis of the Hormonal Predictors of Women’s Sexual Motivation. In Shackelford, T. K., & Hansen, R. D. (Eds.). The Evolution of Sexuality (pp. 99–121). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-09384-0_6]
Roney, J. R., & Simmons, Z. L. (2016). Within-cycle fluctuations in progesterone negatively predict changes in both in-pair and extra-pair desire among partnered women. Hormones and Behavior, 81, 45–52. [DOI:10.1016/j.yhbeh.2016.03.008]
Roney, J. R. (2019). Evolutionary Perspectives on Hypoactive Sexual Desire Disorder in Women. Current Sexual Health Reports, 11(4), 243–250. [DOI:10.1007/s11930-019-00223-w]
Saadat, P., Boostanfar, R., Poysky, J., Munevar, C., Stanczyk, F., Buckwalter, G., & Roy, S. (2002). A randomized prospective study comparing the effects of micronized progesterone and medroxyprogesterone acetate on mood and libido. Fertility and Sterility, 77(Suppl 3 [Abstracts of the 50th Annual Meeting of the Pacific Coast Reproductive Society. April 17-21, 2002. Rancho Mirage, California, USA]), S21–S21 (abstract no. P-28). [Google Scholar] [PubMed] [DOI:10.1016/s0015-0282(02)03053-4]
Sandhu, K. S., Melman, A., & Mikhail, M. S. (2011). Impact of Hormones on Female Sexual Function and Dysfunction. Female Pelvic Medicine & Reconstructive Surgery, 17(1), 8–16. [DOI:10.1097/spv.0b013e318204491f]
Santoro, N., Worsley, R., Miller, K. K., Parish, S. J., & Davis, S. R. (2016). Role of Estrogens and Estrogen-Like Compounds in Female Sexual Function and Dysfunction. The Journal of Sexual Medicine, 13(3), 305–316. [DOI:10.1016/j.jsxm.2015.11.015]
Savolainen-Peltonen, H., Hautamäki, H., Tuomikoski, P., Ylikorkala, O., & Mikkola, T. S. (2014). Health-related quality of life in women with or without hot flashes. Menopause, 21(7), 732–739. [DOI:10.1097/gme.0000000000000120]
Schaffir, J. (2006). Hormonal Contraception and Sexual Desire: A Critical Review. Journal of Sex & Marital Therapy, 32(4), 305–314. [DOI:10.1080/00926230600666311]
Schmidt, P. J., Steinberg, E. M., Negro, P. P., Haq, N., Gibson, C., & Rubinow, D. R. (2008). Pharmacologically Induced Hypogonadism and Sexual Function in Healthy Young Women and Men. Neuropsychopharmacology, 34(3), 565–576. [DOI:10.1038/npp.2008.24]
Sherwin, B. B. (1991). The Impact of Different Doses of Estrogen and Progestin on Mood and Sexual Behavior in Postmenopausal Women. The Journal of Clinical Endocrinology & Metabolism, 72(2), 336–343. [DOI:10.1210/jcem-72-2-336]
Shirazi, T. N., Self, H., Dawood, K., Rosenfield, K. A., Penke, L., Carré, J. M., Ortiz, T., & Puts, D. A. (2019). Hormonal predictors of women’s sexual motivation. Evolution and Human Behavior, 40(3), 336–344. [DOI:10.1016/j.evolhumbehav.2019.02.002]
Wallen, K. (1984). Periovulatory changes in female sexual behavior and patterns of ovarian steroid secretion in group-living rhesus monkeys. Hormones and Behavior, 18(4), 431–450. [DOI:10.1016/0018-506x(84)90028-x]
Ward, I. L., Franck, J. E., & Crowley, W. R. (1977). Central progesterone induces female sexual behavior in estrogen-primed intact male rats. Journal of Comparative and Physiological Psychology, 91(6), 1417–1423. [DOI:10.1037/h0077419]
Waxman, J. H., Wass, J. A., Hendry, W. F., Whitfield, H. N., Bary, P., Besser, G. M., Malpas, J. S., & Oliver, R. T. (1983). Treatment of Advanced Prostatic Cancer with Buserelin, an Analogue of Gonadotrophin Releasing Hormone. British Journal of Urology, 55(6), 737–742. [DOI:10.1111/j.1464-410x.1983.tb03416.x]
Wibowo, E., Schellhammer, P., & Wassersug, R. J. (2011). Role of Estrogen in Normal Male Function: Clinical Implications for Patients With Prostate Cancer on Androgen Deprivation Therapy. Journal of Urology, 185(1), 17–23. [DOI:10.1016/j.juro.2010.08.094]
Witt, D. M., Young, L. J., & Crews, D. (1995). Progesterone modulation of androgen-dependent sexual behavior in male rats. Physiology & Behavior, 57(2), 307–313. [DOI:10.1016/0031-9384(94)00247-3]
Worsley, R., Santoro, N., Miller, K. K., Parish, S. J., & Davis, S. R. (2016). Hormones and Female Sexual Dysfunction: Beyond Estrogens and Androgens—Findings From the Fourth International Consultation on Sexual Medicine. The Journal of Sexual Medicine, 13(3), 283–290. [DOI:10.1016/j.jsxm.2015.12.014]
Wren, B. G., Champion, S. M., Willetts, K., Manga, R. Z., & Eden, J. A. (2003). Transdermal progesterone and its effect on vasomotor symptoms, blood lipid levels, bone metabolic markers, moods, and quality of life for postmenopausal women. Menopause, 10(1), 13–18. [DOI:10.1097/00042192-200310010-00004]
Zimmerman, Y., Eijkemans, M. J., Coelingh Bennink, H. J., Blankenstein, M. A., & Fauser, B. C. (2013). The effect of combined oral contraception on testosterone levels in healthy women: a systematic review and meta-analysis. Human Reproduction Update, 20(1), 76–105. [DOI:10.1093/humupd/dmt038]
Zumpe, D., & Michael, R. P. (1988). Effects of medroxyprogesterone acetate on plasma testosterone and sexual behavior in male cynomolgus monkeys (Macaca fascicularis). Physiology & Behavior, 42(4), 343–349. [DOI:10.1016/0031-9384(88)90275-2]
Zumpe, D. (1991). Medroxyprogesterone acetate, aggression, and sexual behavior in male cynomolgus monkeys (Macaca fascicularis). Hormones and Behavior, 25(3), 394–409. [DOI:10.1016/0018-506x(91)90010-f]
Zumpe, D., Clancy, A. N., & Michael, R. P. (1997). Effects of Progesterone on the Sexual Behavior of Castrated, Testosterone-Treated Male Cynomolgus Monkeys (Macaca Fascicularis). Physiology & Behavior, 62(1), 61–67. [DOI:10.1016/s0031-9384(97)00135-2]
\ No newline at end of file
diff --git a/transfemscience.org/articles/progestogens-transfem-excerpts/index.html b/transfemscience.org/articles/progestogens-transfem-excerpts/index.html
index 1c152489..dabd54b1 100644
--- a/transfemscience.org/articles/progestogens-transfem-excerpts/index.html
+++ b/transfemscience.org/articles/progestogens-transfem-excerpts/index.html
@@ -1 +1 @@
-Sources/Excerpts: Perspectives and Opinions of Clinicians and Researchers Concerning the Use of Progestogens in Transfeminine Hormone Therapy - Transfeminine Science
Sources/Excerpts: Perspectives and Opinions of Clinicians and Researchers Concerning the Use of Progestogens in Transfeminine Hormone Therapy
By Sam | First published April 18, 2020 | Last modified October 5, 2020
Oriel (2000)
Oriel, K. A. (2000). Clinical update: Medical care of transsexual patients. Journal of the Gay and Lesbian Medical Association, 4(4), 185–194. [DOI:10.1023/a:1026563806480]:
The use of progesterone to augment breast development is controversial in physicians treating MTF transsexuals. When deciding on a hormone regimen, prescribers should remember that it is estrogen that causes the serious side effects, so the lowest effective dose should be used.
[…] Progesterone is the third and optional component of the MTF regimen. Although some physicians working with transgendered patients do not prescribe progesterone, others argue feminization involves the actions of both estrogen and progesterone (31). Medroxyprogesterone is a weak antiandrogen, and testosterone suppression may be accomplished with lower doses of estrogen. Medroxyprogesterone is less androgenic than norethindrone and norgestrel. Progesterone is important for breast development (24). Unlike estrogen, progesterone does not carry the risk of thromboembolism, prolactinoma, and myocardial infarction. Although it has been noted that doses of 20–40 mg per day are required to suppress luteinizing hormone (20), medroxyprogesterone acetate may become androgenic at higher doses, so others (5) recommend 10 mg a day. Micronized progesterone (Prometrium) is advantageous because it has a more favorable side effect profile (anxiety and irritability) than medroxyprogesterone. It is also less androgenic when higher progesterone doses are needed, but is more costly.
Dittrich et al. (2005)
Dittrich, R., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M. W., & Mueller, A. (2005). Endocrine treatment of male-to-female transsexuals using gonadotropin-releasing hormone agonist. Experimental and Clinical Endocrinology & Diabetes, 113(10), 586–592. [DOI:10.1055/s-2005-865900]:
Ethinyl oestradiol was no more effective in comparison with the oestradiol-17β valerate administered in the regimen studied here. Similar efficacy in terms of feminisation, e. g., increases in breast size, were achieved in the present study group compared to prior reported treatment regimes using cyproterone acetate and ethinyl oestradiol. The use of a progestin as part of the endocrine treatment regime of male-to-female transsexuals is still controversial; however, advantages in only a couple of patients with abnormal psychological irritability and mammary tenderness could be observed (Michel et al., 2001).
Gooren, Giltay, & Bunck (2008)
Gooren, L. J., Giltay, E. J., & Bunck, M. C. (2008). Long-term treatment of transsexuals with cross-sex hormones: extensive personal experience. The Journal of Clinical Endocrinology & Metabolism, 93(1), 19–25. [DOI:10.1210/jc.2007-1809]:
There is no evidence that progestagens add to the feminization process of M2F. In female reproductive endocrinology, progesterone prepares the uterus for conception and the breasts for lactation. Some patients strongly believe that progestagens are a necessary addition to estrogens in their feminization process. But this is not the case, and progestagens may have side effects, such as salt/water retention leading to elevated blood pressure or venous varicosis. In the large-scale study of postmenopausal hormone use in women, the combination of estrogens and progestagens appeared to be associated with a higher incidence of breast cancer (4) and cardiovascular disease.
Gooren (2011)
Gooren, L. J. (2011). Care of transsexual persons. New England Journal of Medicine, 364(13), 1251–1257. [DOI:10.1056/nejmcp1008161]:
Although progestins suppress androgen production, they have no role in the feminization of the body and may have harmful metabolic effects; consequently, progestins should be discontinued after orchiectomy.24 In postmenopausal women, progestins combined with estrogens increase the risk of breast cancer.25
Wierckx, Gooren, T’Sjoen (2014)
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical review: breast development in trans women receiving cross-sex hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]:
With regard to the hormonal impact on female breast development, the classic view is that estrogens induce proliferation, whereas progestins cause differentiation in female breast development [2,3]. It is widely assumed that progestins have no significant role in the formation of the volume of the breasts, the reason why it is not included in endocrine treatment of girls with Turner and juvenile trans women persons [1].
[…] Although there is undeniably a role of progesterone in breast development and lactation, it is uncertain whether the treatment of progestogens adds much to the volume of the breasts, the concern of trans women. […] In the medical profession and even more in the transsexual community, there has been an ongoing debate for many years on the potential benefit of adding progestogens to estrogen use in trans women’s hormonal treatment, especially concerning its role in the volume/size breast development and maintenance. Progestagen treatment is also often requested by trans women themselves as it is their perception that their treatment should closely mimic hormonal treatment of hypogonadal women requiring hormone treatment. However, for the latter group, addition of progestogens has a different relevance, specifically the modifying estrogen effects on the uterus potentially inducing cancerous development.
[…] Few studies have investigated the effects of cross-sex hormone treatment on breast volume. Meyer et al. [17] investigated breast growth in 52 trans women during cross-sex hormone treatment. Notably, 41 trans women received cross-sex hormone treatment with a median of 26.4 months before inclusion in the study. Different estrogen regimes (ethinyl estradiol [EE], conjugated estrogen, or both) were analyzed, and 15 trans women of their sample (28%) additionally received a progestational agent. No difference in breast size was observed between trans women who received progestogens compared with the others.
[…] The available evidence does not provide support for better effects on breast size of adding progestogens to cross-sex hormone administration in trans women as suggested by some authors [14,18,48–51]. However, it should be said that the quality and amount of available evidence are extremely poor and hamper any firm conclusion at this moment.
[…] Our knowledge concerning the natural history and effects of different cross-sex hormone therapies including progestogens on breast development in trans women is extremely sparse and based on low quality of evidence. This prevents us from drawing any firm conclusion at this moment and demonstrates the need for further research to clarify these important clinical questions.
Deutsch (2016)
Deutsch, M. B. (Ed.). (2016). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition. Center of Excellence for Transgender Health, Department of Family and Community Medicine, University of California, San Francisco. [URL] [PDF]:
There have been no well-designed studies of the role of progestagens in feminizing hormone regimens. Many transgender women and providers alike report an anecdotal improved breast and/or areolar development, mood, or libido with the use of progestagens.[17,18] There is no evidence to suggest that using progestagens in the setting of transgender care are harmful. In reality some patients may respond favorably to progestagens while others may find negative effects on mood. While progestagens have some anti-androgen effect through central blockade of gonadotropins, there is also a theoretical risk of a direct androgenizing effect of progestagens. This class includes micronized bioidentical progesterone (Prometrium) as well as a number of synthetic progestins. The most commonly used synthetic progestin in the context of transgender care is the oral medroxyprogesterone acetate (Provera).
While concerns exist from the Women’s Health Initiative (WHI) regarding risks of cardiovascular disease and breast cancer in the setting of medroxyprogesterone use, these concerns likely do not apply in the context of transgender care for several reasons. First, the transgender women may be at lower risk of breast cancer than non-transgender women. Second, this arm of the WHI involved the use of conjugated equine estrogens in combination with medroxyprogesterone in a sample of menopausal women, some of whom were as long as 10 years post-menopausal at the time of hormone initiation. Third, while statistically significant, the clinical significance of the findings in the WHI was subtle at best. The study aimed to evaluate the role of menopausal hormone therapy in the prevention of chronic disease. The actual findings in the conjugated equine estrogen plus medroxyprogesterone group were an excess absolute risk per 10 000 person-years of 7 more cardiac events events, 8 more strokes, 8 more pulmonary emboli, and 8 more invasive breast cancers, with no change in overall mortality.[19] As such this arm of the WHI was stopped early, and it was concluded that combined menopausal hormone therapy is not indicated for prevention of chronic disease.
In the setting of gender-affirming care, there are numerous differences to the findings of the WHI: populations tend to be younger, equine estrogens are not used, and the emphasis is on genderaffirming interventions which have numerous benefits on mental health and quality of life, rather than prevention. Considering these differences in demographics and goals of therapy, extremely modest increase in overall risk, and lack of difference in mortality, as well as more recent reassuring data with other forms of estrogen, the risks of using progestagens in transgender women are likely minimal or even absent (Grading: NT O M). Injected depomedroxyprogesterone acetate (Depo-Provera®) is less commonly used in transgender women. Other synthetic progestins may be used as necessitated by formulary limitations; some evidence suggests that norpregnane derived progestins (norethindrone, norgestrel) may have an increased risk of venous thromboembolism.[20]
Tangpricha & den Heijer (2017)
Tangpricha, V., & den Heijer, M. (2017). Oestrogen and anti-androgen therapy for transgender women. The Lancet Diabetes & Endocrinology, 5(4), 291–300. [DOI:10.1016/S2213-8587(16)30319-9]:
Some patients request progesterone for enhanced breast growth. However, there have not been any well designed studies to assess the eff ectiveness of progesterone to improve breast development. Results of studies of progesterone combined with oestrogen in postmenopausal cis-gender women—ie, women who are not transgender—suggest that progesterone combined with oestrogen might be associated with an increased risk of cardiovascular disease.21 In fact, in a population-based study of premenopausal cis-gender women, taking oral contraceptives including progesterone with or without oestrogen was associated with increased risk of thromboembolism.22
Hembree et al. (2017)
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society* Clinical Practice Guideline [2nd Version]. The Journal of Clinical Endocrinology & Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]:
In the future, we need more rigorous evaluations of the effectiveness and safety of endocrine and surgical protocols. Specifically, endocrine treatment protocols for GD/gender incongruence should include the careful assessment of the following: (1) the effects of prolonged delay of puberty in adolescents on bone health, gonadal function, and the brain (including effects on cognitive, emotional, social, and sexual development); (2) the effects of treatment in adults on sex hormone levels; (3) the requirement for and the effects of progestins and other agents used to suppress endogenous sex steroids during treatment; and (4) the risks and benefits of gender-affirming hormone treatment in older transgender people.
[…] Although the time course of breast development in transgender females has been studied (150), precise information about other changes induced by sex hormones is lacking (141). There is a great deal of variability among individuals, as evidenced during pubertal development. We all know that a major concern for transgender females is breast development. If we work with estrogens, the result will be often not what the transgender female expects. Alternatively, there are transgender females who report an anecdotal improved breast development, mood, or sexual desire with the use of progestogens. However, there have been no well-designed studies of the role of progestogens in feminizing hormone regimens, so the question is still open. Our knowledge concerning the natural history and effects of different cross-sex hormone therapies on breast development in transgender females is extremely sparse and based on the low quality of evidence. Current evidence does not indicate that progestogens enhance breast development in transgender females, nor does evidence prove the absence of such an effect. This prevents us from drawing any firm conclusion at this moment and demonstrates the need for further research to clarify these important clinical questions (162).
Coxon & Seal (2018)
Coxon, J., & Seal, L. (2018). Hormone management of trans women. Trends in Urology & Men’s Health, 9(6), 10–14. [DOI:10.1002/tre.663]:
Progesterone is recommended by some gender clinics internationally, but rarely in the UK. Some trans women strongly believe that it should be added, to enhance breast development. A meta-analysis found no additional benefit for breast development when comparing progesterone plus oestrogen to oestrogen alone.17 We advise patients that oestrogen-only hormone therapy is the best and safest option, as progesterone is not produced during the breast development phase of physiological female puberty, and trans women do not have endometria to protect. At a cellular level, progesterone reverses oestrogen induced cell proliferation and, more importantly, evidence in cis women has shown that adding progesterone to oestrogen therapy is associated with increased risk of cardiovascular disease and breast cancer.18
Prior (2019)
Prior, J. C. (2019). Progesterone Is Important for Transgender Women’s Therapy—Applying Evidence for the Benefits of Progesterone in Ciswomen. The Journal of Clinical Endocrinology & Metabolism, 104(4), 1181–1186. [DOI:10.1210/jc.2018-01777]:
Oral micronized progesterone, a fundamental ovarian steroid, molecularly identical to the natural hormone, should be added to E2 for transgender women based on physiology and emerging evidence of the importance of progesterone with E2 for ciswomen’s bone and likely cardiovascular health. Progesterone will probably prevent at least some of the negative cardiovascular system and bone health effects reported in transgender women on current long-term, E/E2-only, or E/E2 antiandrogen CHT. Progesterone will also aid antiandrogen effects through different pathways than spironolactone or cyproterone acetate and may promote feminine physiological breast maturation, while also aiding disturbed sleep and perhaps decreasing anxiety. It may also facilitate transgender women’s acceptance of physiological (rather than high) E2 doses ideally delivered transdermally. Evidence is mounting that ciswomen’s lifelong health is enhanced by sufficient P4 (normally ovulatory) within regular estradiol-sufficient monthly menstrual cycles. I believe it is time that we now follow current guidelines and provide transgender women with these P4 or progesterone benefits in their CHT.
Iwamoto et al. (2019)
Iwamoto, S. J., T’Sjoen, G., Safer, J. D., Davidge-Pitts, C. J., Wierman, M. E., Glodowski, M. B., & Rothman, M. S. (2019). Letter to the Editor: “Progesterone Is Important for Transgender Women’s Therapy—Applying Evidence for the Benefits of Progesterone in Ciswomen”. The Journal of Clinical Endocrinology & Metabolism, 104(8), 3127–3128. [DOI:10.1210/jc.2019-00249]:
We respect Dr. Jerilynn C. Prior’s many contributions to endocrinology and transgender (trans) health. Yet, we have multiple concerns about her Perspective(1) which is limited to her clinical experience, assumptions taken from in vitro and animal data, and selected data from cisgender (cis) women, whose female gender identity aligns with their natal sex.
[…] Prior asserts that P4 with estradiol (E2) will optimize breast maturation and size, but P4 causes ductal differentiation in puberty and has not been shown to enhance breast development in trans women(2). Prior argues that bone mineral density (BMD) improves with P4. However, selected studies from her meta-analysis were limited by significant heterogeneity of hormone dose, race and range of years after menopause(3). Although Prior states that low BMD is a major risk for trans women “on long-term” hormones, the major concerns are low baseline BMD before pharmacologic gender-affirming hormone therapy (GAHT) and monitoring after gonadectomy and cessation of GAHT(4). Recent ten-year GAHT data (without P4) showed gains in lumbar spine Z-scores of trans women(4).
We also object to Prior’s assumption that P4 will decrease breast cancer risk in trans women based on the French E3N self-administered questionnaire study in cis women. Randomized controlled trial (RCT) data from the Women’s Health Initiative (WHI) demonstrated increased invasive breast cancer during the intervention, postintervention and cumulative follow-up in the conjugated equine estrogen (CEE)+medroxyprogesterone (MPA) group compared to CEE alone(5).
Lastly, Prior’s statement of cardiovascular benefits with progestins conflicts with WHI data which revealed increased coronary heart disease in women ≥20 years past menopause onset and higher risk of stroke and blood clots in the CEE+MPA group during cumulative follow-up(5). Recent data raise concerns for higher risks of blood clots and stroke among trans women on GAHT compared to reference women and men(6).
[…] To conclude that oral micronized P4 will be positive for bone and cardiovascular health based on selected data in menopausal cis women is premature and potentially dangerous. Further, we know that clinical assumptions based on observational data, murine studies, cell models and personal experience can lead to risks to patients. Our transgender patients deserve the benefits of rigorous RCT data so we can provide evidence-based recommendations regarding the risks and benefits of GAHT.
Cheung et al. (2019)
Cheung, A. S., Wynne, K., Erasmus, J., Murray, S., & Zajac, J. D. (2019). Position statement on the hormonal management of adult transgender and gender diverse individuals. Medical Journal of Australia, 211(3), 127–133. [DOI:10.5694/mja2.50259]:
Despite anecdotal reports that progestins increase breast growth, no data support their use. Healthy postmenopausal women who received estradiol with progestins had increased risk of coronary heart disease compared with placebo34 (not reported with estradiol alone42). Progestins can also increase risk of thrombosis, bloating, nausea and weight gain and are not recommended.10 Cyproterone acetate, a commonly used anti-androgen agent, has progestogenic effects.
Iwamoto et al. (2019)
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 2042018819871166. [DOI:10.1177/2042018819871166]:
Alternative anti-androgen therapies exist that have not yet been substantiated for routine use by supporting research. Progesterone is discussed in the breast development section, but we do not recommend its routine use in TW until more rigorous studies demonstrate potential benefits outweigh any risks.40
[…] The impact of various combinations of different oestrogens, anti-androgens and progestogens on breast development was previously reviewed by Wierckx and colleagues,67 who concluded that current evidence did not support or refute any enhancement of breast development with progestogens. Pubertal data in people assigned female at birth (AFAB) (e.g. girls with Turner syndrome) argue for delaying progesterone as it causes ductal differentiation and may interfere with optimal breast development.68 Due to unsatisfactory effects of GAHT alone on breast growth and development, 60–70% of all TW sought additional surgical breast augmentation.67 Articles on breast development in TW report inconclusive results, possibly due to differences in methodology and the lack of prospective RCTs. Prior69 recently published on the potential benefits of progesterone use on breast development (among other conclusions) in TW but the perspective was limited to clinical experience, assumptions taken from in vitro and animal data, and selected data from cis women.40 It would be worthwhile for future studies to use volume measurements for examining hormonally induced breast development in TW over a longer follow-up period in a large cohort, which may result in the identification of predictive markers for breast development.62 Additionally, rigorous prospective RCTs of oestrogen and oestrogen plus progesterone may provide more conclusive data to guide routine use of progesterone for breast development.
T’Sjoen et al. (2020)
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine Position Statement “Assessment and Hormonal Management in Adolescent and Adult Trans People, with Attention for Sexual Function and Satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]:
To induce (more) feminine secondary sex characteristics and/or to reduce the masculine ones, estrogens and/or antiandrogens can be used. The debate on the use of progesterone is ongoing, caused by a lack of data.3 A wide range of estrogenic compounds exists, of which 17beta-estradiol (oral 2–6 mg/d or transdermal) represents the treatment of choice. Estrogen dosage should be adjusted to maintain serum estradiol at the level for premenopausal women (100–200 pg/mL), although timing of treatment intake may affect blood concentrations. The most commonly used antiandrogen drug in Europe is cyproterone acetate (oral 10–50 mg, once daily), a progestin with antiandrogenic properties. GnRHa are also effective in reducing testosterone levels with a low risk of adverse effects.3
Reisman (2020)
Reisman, T. (2020). Breast imaging in transgender individuals. In Legato, M. J. (Ed.). The Plasticity of Sex: The Molecular Biology and Clinical Features of Genomic Sex, Gender Identity and Sexual Behavior (pp. 187–205). London: Academic Press. [DOI:10.1016/B978-0-12-815968-2.00016-5]:
While the role of estrogen in breast growth has been most clearly established, other hormones have also been implicated in breast development. In his 1958 study, Lyon demonstrated in oophorectomized, adrenalectomized, and hypophysectomized rodents that estrogen, growth hormone, and corticosteroids are all required for ductal develoment.10 Progesterone does not appear to be important for either early pubertal duct development or breast growth. However, with increasing levels throughout pregnancy, progesterone is reported to mediate the lobular alveolar development facilitating lactation.
\ No newline at end of file
+Sources/Excerpts: Perspectives and Opinions of Clinicians and Researchers Concerning the Use of Progestogens in Transfeminine Hormone Therapy - Transfeminine Science
Sources/Excerpts: Perspectives and Opinions of Clinicians and Researchers Concerning the Use of Progestogens in Transfeminine Hormone Therapy
By Sam | First published April 18, 2020 | Last modified October 5, 2020
Oriel (2000)
Oriel, K. A. (2000). Clinical update: Medical care of transsexual patients. Journal of the Gay and Lesbian Medical Association, 4(4), 185–194. [DOI:10.1023/a:1026563806480]:
The use of progesterone to augment breast development is controversial in physicians treating MTF transsexuals. When deciding on a hormone regimen, prescribers should remember that it is estrogen that causes the serious side effects, so the lowest effective dose should be used.
[…] Progesterone is the third and optional component of the MTF regimen. Although some physicians working with transgendered patients do not prescribe progesterone, others argue feminization involves the actions of both estrogen and progesterone (31). Medroxyprogesterone is a weak antiandrogen, and testosterone suppression may be accomplished with lower doses of estrogen. Medroxyprogesterone is less androgenic than norethindrone and norgestrel. Progesterone is important for breast development (24). Unlike estrogen, progesterone does not carry the risk of thromboembolism, prolactinoma, and myocardial infarction. Although it has been noted that doses of 20–40 mg per day are required to suppress luteinizing hormone (20), medroxyprogesterone acetate may become androgenic at higher doses, so others (5) recommend 10 mg a day. Micronized progesterone (Prometrium) is advantageous because it has a more favorable side effect profile (anxiety and irritability) than medroxyprogesterone. It is also less androgenic when higher progesterone doses are needed, but is more costly.
Dittrich et al. (2005)
Dittrich, R., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M. W., & Mueller, A. (2005). Endocrine treatment of male-to-female transsexuals using gonadotropin-releasing hormone agonist. Experimental and Clinical Endocrinology & Diabetes, 113(10), 586–592. [DOI:10.1055/s-2005-865900]:
Ethinyl oestradiol was no more effective in comparison with the oestradiol-17β valerate administered in the regimen studied here. Similar efficacy in terms of feminisation, e. g., increases in breast size, were achieved in the present study group compared to prior reported treatment regimes using cyproterone acetate and ethinyl oestradiol. The use of a progestin as part of the endocrine treatment regime of male-to-female transsexuals is still controversial; however, advantages in only a couple of patients with abnormal psychological irritability and mammary tenderness could be observed (Michel et al., 2001).
Gooren, Giltay, & Bunck (2008)
Gooren, L. J., Giltay, E. J., & Bunck, M. C. (2008). Long-term treatment of transsexuals with cross-sex hormones: extensive personal experience. The Journal of Clinical Endocrinology & Metabolism, 93(1), 19–25. [DOI:10.1210/jc.2007-1809]:
There is no evidence that progestagens add to the feminization process of M2F. In female reproductive endocrinology, progesterone prepares the uterus for conception and the breasts for lactation. Some patients strongly believe that progestagens are a necessary addition to estrogens in their feminization process. But this is not the case, and progestagens may have side effects, such as salt/water retention leading to elevated blood pressure or venous varicosis. In the large-scale study of postmenopausal hormone use in women, the combination of estrogens and progestagens appeared to be associated with a higher incidence of breast cancer (4) and cardiovascular disease.
Gooren (2011)
Gooren, L. J. (2011). Care of transsexual persons. New England Journal of Medicine, 364(13), 1251–1257. [DOI:10.1056/nejmcp1008161]:
Although progestins suppress androgen production, they have no role in the feminization of the body and may have harmful metabolic effects; consequently, progestins should be discontinued after orchiectomy.24 In postmenopausal women, progestins combined with estrogens increase the risk of breast cancer.25
Wierckx, Gooren, T’Sjoen (2014)
Wierckx, K., Gooren, L., & T’Sjoen, G. (2014). Clinical review: breast development in trans women receiving cross-sex hormones. The Journal of Sexual Medicine, 11(5), 1240–1247. [DOI:10.1111/jsm.12487]:
With regard to the hormonal impact on female breast development, the classic view is that estrogens induce proliferation, whereas progestins cause differentiation in female breast development [2,3]. It is widely assumed that progestins have no significant role in the formation of the volume of the breasts, the reason why it is not included in endocrine treatment of girls with Turner and juvenile trans women persons [1].
[…] Although there is undeniably a role of progesterone in breast development and lactation, it is uncertain whether the treatment of progestogens adds much to the volume of the breasts, the concern of trans women. […] In the medical profession and even more in the transsexual community, there has been an ongoing debate for many years on the potential benefit of adding progestogens to estrogen use in trans women’s hormonal treatment, especially concerning its role in the volume/size breast development and maintenance. Progestagen treatment is also often requested by trans women themselves as it is their perception that their treatment should closely mimic hormonal treatment of hypogonadal women requiring hormone treatment. However, for the latter group, addition of progestogens has a different relevance, specifically the modifying estrogen effects on the uterus potentially inducing cancerous development.
[…] Few studies have investigated the effects of cross-sex hormone treatment on breast volume. Meyer et al. [17] investigated breast growth in 52 trans women during cross-sex hormone treatment. Notably, 41 trans women received cross-sex hormone treatment with a median of 26.4 months before inclusion in the study. Different estrogen regimes (ethinyl estradiol [EE], conjugated estrogen, or both) were analyzed, and 15 trans women of their sample (28%) additionally received a progestational agent. No difference in breast size was observed between trans women who received progestogens compared with the others.
[…] The available evidence does not provide support for better effects on breast size of adding progestogens to cross-sex hormone administration in trans women as suggested by some authors [14,18,48–51]. However, it should be said that the quality and amount of available evidence are extremely poor and hamper any firm conclusion at this moment.
[…] Our knowledge concerning the natural history and effects of different cross-sex hormone therapies including progestogens on breast development in trans women is extremely sparse and based on low quality of evidence. This prevents us from drawing any firm conclusion at this moment and demonstrates the need for further research to clarify these important clinical questions.
Deutsch (2016)
Deutsch, M. B. (Ed.). (2016). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition. Center of Excellence for Transgender Health, Department of Family and Community Medicine, University of California, San Francisco. [URL] [PDF]:
There have been no well-designed studies of the role of progestagens in feminizing hormone regimens. Many transgender women and providers alike report an anecdotal improved breast and/or areolar development, mood, or libido with the use of progestagens.[17,18] There is no evidence to suggest that using progestagens in the setting of transgender care are harmful. In reality some patients may respond favorably to progestagens while others may find negative effects on mood. While progestagens have some anti-androgen effect through central blockade of gonadotropins, there is also a theoretical risk of a direct androgenizing effect of progestagens. This class includes micronized bioidentical progesterone (Prometrium) as well as a number of synthetic progestins. The most commonly used synthetic progestin in the context of transgender care is the oral medroxyprogesterone acetate (Provera).
While concerns exist from the Women’s Health Initiative (WHI) regarding risks of cardiovascular disease and breast cancer in the setting of medroxyprogesterone use, these concerns likely do not apply in the context of transgender care for several reasons. First, the transgender women may be at lower risk of breast cancer than non-transgender women. Second, this arm of the WHI involved the use of conjugated equine estrogens in combination with medroxyprogesterone in a sample of menopausal women, some of whom were as long as 10 years post-menopausal at the time of hormone initiation. Third, while statistically significant, the clinical significance of the findings in the WHI was subtle at best. The study aimed to evaluate the role of menopausal hormone therapy in the prevention of chronic disease. The actual findings in the conjugated equine estrogen plus medroxyprogesterone group were an excess absolute risk per 10 000 person-years of 7 more cardiac events events, 8 more strokes, 8 more pulmonary emboli, and 8 more invasive breast cancers, with no change in overall mortality.[19] As such this arm of the WHI was stopped early, and it was concluded that combined menopausal hormone therapy is not indicated for prevention of chronic disease.
In the setting of gender-affirming care, there are numerous differences to the findings of the WHI: populations tend to be younger, equine estrogens are not used, and the emphasis is on genderaffirming interventions which have numerous benefits on mental health and quality of life, rather than prevention. Considering these differences in demographics and goals of therapy, extremely modest increase in overall risk, and lack of difference in mortality, as well as more recent reassuring data with other forms of estrogen, the risks of using progestagens in transgender women are likely minimal or even absent (Grading: NT O M). Injected depomedroxyprogesterone acetate (Depo-Provera®) is less commonly used in transgender women. Other synthetic progestins may be used as necessitated by formulary limitations; some evidence suggests that norpregnane derived progestins (norethindrone, norgestrel) may have an increased risk of venous thromboembolism.[20]
Tangpricha & den Heijer (2017)
Tangpricha, V., & den Heijer, M. (2017). Oestrogen and anti-androgen therapy for transgender women. The Lancet Diabetes & Endocrinology, 5(4), 291–300. [DOI:10.1016/S2213-8587(16)30319-9]:
Some patients request progesterone for enhanced breast growth. However, there have not been any well designed studies to assess the eff ectiveness of progesterone to improve breast development. Results of studies of progesterone combined with oestrogen in postmenopausal cis-gender women—ie, women who are not transgender—suggest that progesterone combined with oestrogen might be associated with an increased risk of cardiovascular disease.21 In fact, in a population-based study of premenopausal cis-gender women, taking oral contraceptives including progesterone with or without oestrogen was associated with increased risk of thromboembolism.22
Hembree et al. (2017)
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society* Clinical Practice Guideline [2nd Version]. The Journal of Clinical Endocrinology & Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]:
In the future, we need more rigorous evaluations of the effectiveness and safety of endocrine and surgical protocols. Specifically, endocrine treatment protocols for GD/gender incongruence should include the careful assessment of the following: (1) the effects of prolonged delay of puberty in adolescents on bone health, gonadal function, and the brain (including effects on cognitive, emotional, social, and sexual development); (2) the effects of treatment in adults on sex hormone levels; (3) the requirement for and the effects of progestins and other agents used to suppress endogenous sex steroids during treatment; and (4) the risks and benefits of gender-affirming hormone treatment in older transgender people.
[…] Although the time course of breast development in transgender females has been studied (150), precise information about other changes induced by sex hormones is lacking (141). There is a great deal of variability among individuals, as evidenced during pubertal development. We all know that a major concern for transgender females is breast development. If we work with estrogens, the result will be often not what the transgender female expects. Alternatively, there are transgender females who report an anecdotal improved breast development, mood, or sexual desire with the use of progestogens. However, there have been no well-designed studies of the role of progestogens in feminizing hormone regimens, so the question is still open. Our knowledge concerning the natural history and effects of different cross-sex hormone therapies on breast development in transgender females is extremely sparse and based on the low quality of evidence. Current evidence does not indicate that progestogens enhance breast development in transgender females, nor does evidence prove the absence of such an effect. This prevents us from drawing any firm conclusion at this moment and demonstrates the need for further research to clarify these important clinical questions (162).
Coxon & Seal (2018)
Coxon, J., & Seal, L. (2018). Hormone management of trans women. Trends in Urology & Men’s Health, 9(6), 10–14. [DOI:10.1002/tre.663]:
Progesterone is recommended by some gender clinics internationally, but rarely in the UK. Some trans women strongly believe that it should be added, to enhance breast development. A meta-analysis found no additional benefit for breast development when comparing progesterone plus oestrogen to oestrogen alone.17 We advise patients that oestrogen-only hormone therapy is the best and safest option, as progesterone is not produced during the breast development phase of physiological female puberty, and trans women do not have endometria to protect. At a cellular level, progesterone reverses oestrogen induced cell proliferation and, more importantly, evidence in cis women has shown that adding progesterone to oestrogen therapy is associated with increased risk of cardiovascular disease and breast cancer.18
Prior (2019)
Prior, J. C. (2019). Progesterone Is Important for Transgender Women’s Therapy—Applying Evidence for the Benefits of Progesterone in Ciswomen. The Journal of Clinical Endocrinology & Metabolism, 104(4), 1181–1186. [DOI:10.1210/jc.2018-01777]:
Oral micronized progesterone, a fundamental ovarian steroid, molecularly identical to the natural hormone, should be added to E2 for transgender women based on physiology and emerging evidence of the importance of progesterone with E2 for ciswomen’s bone and likely cardiovascular health. Progesterone will probably prevent at least some of the negative cardiovascular system and bone health effects reported in transgender women on current long-term, E/E2-only, or E/E2 antiandrogen CHT. Progesterone will also aid antiandrogen effects through different pathways than spironolactone or cyproterone acetate and may promote feminine physiological breast maturation, while also aiding disturbed sleep and perhaps decreasing anxiety. It may also facilitate transgender women’s acceptance of physiological (rather than high) E2 doses ideally delivered transdermally. Evidence is mounting that ciswomen’s lifelong health is enhanced by sufficient P4 (normally ovulatory) within regular estradiol-sufficient monthly menstrual cycles. I believe it is time that we now follow current guidelines and provide transgender women with these P4 or progesterone benefits in their CHT.
Iwamoto et al. (2019)
Iwamoto, S. J., T’Sjoen, G., Safer, J. D., Davidge-Pitts, C. J., Wierman, M. E., Glodowski, M. B., & Rothman, M. S. (2019). Letter to the Editor: “Progesterone Is Important for Transgender Women’s Therapy—Applying Evidence for the Benefits of Progesterone in Ciswomen”. The Journal of Clinical Endocrinology & Metabolism, 104(8), 3127–3128. [DOI:10.1210/jc.2019-00249]:
We respect Dr. Jerilynn C. Prior’s many contributions to endocrinology and transgender (trans) health. Yet, we have multiple concerns about her Perspective(1) which is limited to her clinical experience, assumptions taken from in vitro and animal data, and selected data from cisgender (cis) women, whose female gender identity aligns with their natal sex.
[…] Prior asserts that P4 with estradiol (E2) will optimize breast maturation and size, but P4 causes ductal differentiation in puberty and has not been shown to enhance breast development in trans women(2). Prior argues that bone mineral density (BMD) improves with P4. However, selected studies from her meta-analysis were limited by significant heterogeneity of hormone dose, race and range of years after menopause(3). Although Prior states that low BMD is a major risk for trans women “on long-term” hormones, the major concerns are low baseline BMD before pharmacologic gender-affirming hormone therapy (GAHT) and monitoring after gonadectomy and cessation of GAHT(4). Recent ten-year GAHT data (without P4) showed gains in lumbar spine Z-scores of trans women(4).
We also object to Prior’s assumption that P4 will decrease breast cancer risk in trans women based on the French E3N self-administered questionnaire study in cis women. Randomized controlled trial (RCT) data from the Women’s Health Initiative (WHI) demonstrated increased invasive breast cancer during the intervention, postintervention and cumulative follow-up in the conjugated equine estrogen (CEE)+medroxyprogesterone (MPA) group compared to CEE alone(5).
Lastly, Prior’s statement of cardiovascular benefits with progestins conflicts with WHI data which revealed increased coronary heart disease in women ≥20 years past menopause onset and higher risk of stroke and blood clots in the CEE+MPA group during cumulative follow-up(5). Recent data raise concerns for higher risks of blood clots and stroke among trans women on GAHT compared to reference women and men(6).
[…] To conclude that oral micronized P4 will be positive for bone and cardiovascular health based on selected data in menopausal cis women is premature and potentially dangerous. Further, we know that clinical assumptions based on observational data, murine studies, cell models and personal experience can lead to risks to patients. Our transgender patients deserve the benefits of rigorous RCT data so we can provide evidence-based recommendations regarding the risks and benefits of GAHT.
Cheung et al. (2019)
Cheung, A. S., Wynne, K., Erasmus, J., Murray, S., & Zajac, J. D. (2019). Position statement on the hormonal management of adult transgender and gender diverse individuals. Medical Journal of Australia, 211(3), 127–133. [DOI:10.5694/mja2.50259]:
Despite anecdotal reports that progestins increase breast growth, no data support their use. Healthy postmenopausal women who received estradiol with progestins had increased risk of coronary heart disease compared with placebo34 (not reported with estradiol alone42). Progestins can also increase risk of thrombosis, bloating, nausea and weight gain and are not recommended.10 Cyproterone acetate, a commonly used anti-androgen agent, has progestogenic effects.
Iwamoto et al. (2019)
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 2042018819871166. [DOI:10.1177/2042018819871166]:
Alternative anti-androgen therapies exist that have not yet been substantiated for routine use by supporting research. Progesterone is discussed in the breast development section, but we do not recommend its routine use in TW until more rigorous studies demonstrate potential benefits outweigh any risks.40
[…] The impact of various combinations of different oestrogens, anti-androgens and progestogens on breast development was previously reviewed by Wierckx and colleagues,67 who concluded that current evidence did not support or refute any enhancement of breast development with progestogens. Pubertal data in people assigned female at birth (AFAB) (e.g. girls with Turner syndrome) argue for delaying progesterone as it causes ductal differentiation and may interfere with optimal breast development.68 Due to unsatisfactory effects of GAHT alone on breast growth and development, 60–70% of all TW sought additional surgical breast augmentation.67 Articles on breast development in TW report inconclusive results, possibly due to differences in methodology and the lack of prospective RCTs. Prior69 recently published on the potential benefits of progesterone use on breast development (among other conclusions) in TW but the perspective was limited to clinical experience, assumptions taken from in vitro and animal data, and selected data from cis women.40 It would be worthwhile for future studies to use volume measurements for examining hormonally induced breast development in TW over a longer follow-up period in a large cohort, which may result in the identification of predictive markers for breast development.62 Additionally, rigorous prospective RCTs of oestrogen and oestrogen plus progesterone may provide more conclusive data to guide routine use of progesterone for breast development.
T’Sjoen et al. (2020)
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine Position Statement “Assessment and Hormonal Management in Adolescent and Adult Trans People, with Attention for Sexual Function and Satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]:
To induce (more) feminine secondary sex characteristics and/or to reduce the masculine ones, estrogens and/or antiandrogens can be used. The debate on the use of progesterone is ongoing, caused by a lack of data.3 A wide range of estrogenic compounds exists, of which 17beta-estradiol (oral 2–6 mg/d or transdermal) represents the treatment of choice. Estrogen dosage should be adjusted to maintain serum estradiol at the level for premenopausal women (100–200 pg/mL), although timing of treatment intake may affect blood concentrations. The most commonly used antiandrogen drug in Europe is cyproterone acetate (oral 10–50 mg, once daily), a progestin with antiandrogenic properties. GnRHa are also effective in reducing testosterone levels with a low risk of adverse effects.3
Reisman (2020)
Reisman, T. (2020). Breast imaging in transgender individuals. In Legato, M. J. (Ed.). The Plasticity of Sex: The Molecular Biology and Clinical Features of Genomic Sex, Gender Identity and Sexual Behavior (pp. 187–205). London: Academic Press. [DOI:10.1016/B978-0-12-815968-2.00016-5]:
While the role of estrogen in breast growth has been most clearly established, other hormones have also been implicated in breast development. In his 1958 study, Lyon demonstrated in oophorectomized, adrenalectomized, and hypophysectomized rodents that estrogen, growth hormone, and corticosteroids are all required for ductal develoment.10 Progesterone does not appear to be important for either early pubertal duct development or breast growth. However, with increasing levels throughout pregnancy, progesterone is reported to mediate the lobular alveolar development facilitating lactation.
\ No newline at end of file
diff --git a/transfemscience.org/articles/puberty-blockers/index.html b/transfemscience.org/articles/puberty-blockers/index.html
index 01c23068..6067e55e 100644
--- a/transfemscience.org/articles/puberty-blockers/index.html
+++ b/transfemscience.org/articles/puberty-blockers/index.html
@@ -1 +1 @@
-Puberty Blockers: A Review of GnRH Analogues in Transgender Youth - Transfeminine Science
Puberty Blockers: A Review of GnRH Analogues in Transgender Youth
By Mitzi | First published January 30, 2022 | Last modified January 31, 2022
Abstract / TL;DR
Puberty blockers are medications used to pause puberty in both cisgender and transgender youth. For the latter, significant evidence suggests that they improve well-being, psychological functioning, and risk of suicidality, both during puberty and in later life. Their effects are reversible upon discontinuation. Current evidence does not suggest any negative impact on cognitive development, IQ, or fertility. A minor impact on bone density may exist, affecting primarily transgender girls, but little high quality data is available. Based on limited data, prescribers may wish to consider calcium supplementation in transgender teens receiving puberty blockers, and may wish to prefer transdermal delivery over oral estrogens in transgender girls starting hormone therapy in order to optimise bone density outcomes. There is a lack of evidence supporting the common belief that most children grow out of gender dysphoria (“desistance”), as widely cited data describing the rate at which this happens appears highly unreliable. Puberty blockers are difficult to access, and many Western countries have sharply restricted their use recently, in a trend condemned by numerous medical associations. Randomised controlled trials on puberty blockers can likely never be performed, but nonetheless, there is clear evidence they offer significant benefit, and have relatively minor risks.
Introduction
Puberty blockers, also known as gonadotropin-releasing hormone (GnRH) analogues, were introduced for medical use in the 1980s (Swerdloff & Heber, 1983). Originally developed to supersede other therapies in the treatment of prostate cancer, they were soon adapted for paediatric use, revolutionising the treatment of precocious puberty: a rare condition in which puberty begins before the age of 8 (in natal girls) or 9 (in natal boys). Precocious puberty is associated with several negative consequences, such as short stature, teasing, bullying, and worse mental health outcomes. By reversibly pausing puberty for several years in children with this condition, outcomes are often significantly improved, and puberty blockers remain the mainstay treatment for this condition several decades later.
In the 1990s, puberty blockers began to be used in transgender adolescents, as a way of pausing their unwanted puberty, and giving them more time to consider their future (Cohen-Kettenis & van Goozen, 1998). The protocol for this, originally develped by the Dutch VUmc clinic, has sometimes been referred to as the “Dutch Method.” Cohen-Kettenis et al. (2011) published a study following one such Dutch patient 22 years later. Since then, the use of puberty blockers has increased tremendously with the increase in patients seeking transgender healthcare.
Recently, puberty blockers have been the subject of controversy, with legal proceedings seeking to prohibit their use across several countries. Notably, their use was temporarily stopped in the United Kingdom in December 2020 following a ruling in the Bell v. Tavistock case, which was appealed in 2021. Also in 2021, Arkansas became the first U.S. state to make it illegal for doctors to prescribe puberty blockers, with several other states pursuing similar legislation. Critics express concern about the safety of puberty blockers, their reversibility, and effectiveness.
This article seeks to review the literature on the use and safety of puberty blockers in transgender youth, examining their safety, and arguments for and against their use in a comprehensive way. While rarely, alternative medications like the progestin medroxyprogesterone acetate have been used for this, this article mainly focuses on GnRH agonists: by far the most widely used class of medication for puberty blockade, and what’s most commonly colloquially referred to as “puberty blockers.”
Mechanism of Action
GnRH is a naturally occurring hormone in humans responsible for the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the pituitary gland. Through this mechanism, the body produces its gonadal estrogen and testosterone. GnRH agonists bind to the GnRH receptor and activate it, causing it to be continuously stimulated. This causes an initial increase of LH and FSH, then over the course of several weeks, causes the pituitary gland to become desensitised, pausing the natural sex hormone production for the duration the medication is taken. When the medication is stopped, its effect is reversed, with normal sex hormone production resuming about a week after the medication clears the body (Cedrin-Durnerin et al., 2000).
GnRH agonists are prescribed as an injection administered every one to six months, a surgically implanted pellet once per year, or a nasal spray administered two to three times per day. A short-acting daily injection exists, but is not used for puberty blockade in clinical practise. Common examples of GnRH agonists include leuprorelin (Lupron; Eligard), triptorelin (Decapeptyl), goserelin (Zoladex), histrelin (Supprelin LA), nafarelin (Synarel) and buserelin (Suprefact).
Like GnRH agonists, GnRH antagonists bind to the GnRH receptor, however, they do not stimulate it. Instead, they compete with the body’s own GnRH, rendering it ineffective. As a result, they achieve similar effects without causing an initial increase in hormone levels. Also unlike GnRH agonists, oral formulations of GnRH antagonists exist, allowing some of them to be taken as a daily pill. Common examples include elagolix (Orilissa), degarelix (Firmagon), cetrorelix (Cetrotide), ganirelix (Orgalutran; Antagon) and relugolix (Orgovyx; Relumina). Unfortunately, being much newer drugs, GnRH antagonists are not normally used as puberty blockers at the moment.
In gender dysphoric youth, GnRH agonists are prescribed after the onset of puberty. GnRH agonists are not prescribed to children who have not yet started puberty, but may be started at any point during puberty to pause further changes (Hembree et al., 2017).
Quality of Evidence
In medicine, the gold standard for evidence is the randomised controlled trial, or RCT. In a nutshell, participants are randomly assigned into two or more treatment groups (arms), such that the only difference between arms is the treatment they receive. Commonly, one group receives a placebo, while another receives the treatment being studied. The ideal RCT is blinded, meaning neither participants nor investigators of the study know which group is receiving which treatment. No such trials have been performed with puberty blockers, giving rise to concerns that there could be insufficient evidence available for their use.
Unfortunately, RCTs may not be practically possible for puberty blockers, and are unlikely to ever be performed. A good summary of the reasons for this is provided by Giordano & Holm (2020):
There are two main practical problems that preclude conducting a RCT.
First, patients who approach clinics for help because of distress caused by the first signs of puberty will be unlikely to accept to be a part of a RCT. Medications are needed within a relatively short period of time, at pain of treatment being less effective or ineffective. Recruitment would thus be hard if not impossible.
Second, the ideal RCT is either double blind, i.e. neither researchers nor participants know who gets the active drug, or it assesses outcomes using blinded observers when treatment allocation cannot be hidden from participants. Blinding is necessary in order to reduce bias in outcome assessments. But, a RCT of puberty delay could not maintain blinding. Because GnRHa are effective in delaying puberty it would soon become evident to participants, researchers and outcome assessors who was in the active treatment arm and who was not. This breakdown of blinding would mean that there would be potential bias in the outcome assessments, both in relation to biological and psychological outcomes. It would also mean that participants allocated to the non-treatment arm of the study would be likely to either withdraw from the study at a much higher rate than in the treatment arm introducing potential bias, and/or be more likely not to adhere to the trial but seek puberty delaying treatment outside of the trial thereby adding a confounder.
Mul et al. (2001) ran into this problem conducting a similar study on teens with precocious puberty:
In the original study design a third arm with untreated children was scheduled as a control group. It was decided to omit this control group from the study design after it appeared that the parents of all patients who were randomized in the untreated control group refused further participation in the study as GnRHa treatment could be obtained elsewhere.
Besides practical limitations, such RCTs are likely to be unethical. Evidence suggests withholding puberty blockers may cause lasting harm in itself. To knowingly cause such harm to the control group of an RCT is likely to be morally unacceptable, and such an RCT would be unlikely to receive approval from an ethical review board.
This is not to say that studies evaluating such outcomes don’t exist at all: for example, while not randomised or blinded, Costa et al. (2015) compares 101 patients receiving psychological support and puberty blockers to 100 patients receiving psychological support alone. The results of this study are further outlined below.
As a result of these limitations, this article mainly cites cohort studies, making the argument that sufficient other high-quality studies exist to reach well-supported conclusions: a practise sometimes required in other areas of medicine as well. Because this is the only way we can practically evaluate puberty blockers and RCTs are likely impossible, it seems disingenuous to make the claim that lack of RCTs equate to lack of evidence around puberty blockers, as this standard of evidence can never be met, and the claim ignores a substantial existing body of literature.
Suicidality and Well-being
A significant body of evidence associates the use of puberty blockers in those who want such treatment with improved psychological well-being: the primary argument for their use.
While different studies use different methodologies, three standardised psychological questionnaires are typically used to evaluate well-being: the Children’s Global Assessment Scale (CGAS), the Child Behavior Checklist (CBCL), and the Youth Self-Report (YSR). All three are aimed at evaluating psychological functioning and problematic behaviour: typically, the CGAS is administered by a clinician, the CBCL is filled out by a parent or guardian, and the YSR is filled out by a child themselves. It’s important to note that scores in these assessments are known to markedly worsen in adolescence in general, with the onset of psychological difficulties and self-harm often appearing during puberty (Verhuist et al., 2003; Nock et al., 2013; Morey et al., 2017; Jung et al., 2018).
One of the largest studies to investigate well-being to date has been Turban et al. (2020). It surveyed 20,619 American transgender adults. 3,494 (16.9%) reported that they ever wanted to receive puberty blockers. Of those, only 89 received them. In total, 75.3% of those who received puberty blockers reported ever experiencing suicidal thoughts, compared to 90.2% of those who did not. After controlling for demographic variables like income, family support, and education level, puberty blockers remained significantly associated with decreased odds of lifetime suicidal ideation.
A similarly large survey by Green et al. (2021) included 11,914 Americans aged 13–24 who identified as transgender or nonbinary. The study compares those who received hormone therapy or puberty blockers to those who wished to receive them, but didn’t. It finds that in those who received treatment, rates of depression, suicidal ideation, and suicide attempts were significantly lower. This remained true of those aged 13–17, who were significantly more likely to receive puberty blockers specifically.
Costa et al. (2015) studied 201 gender dysphoric adolescents who presented at the British Tavistock and Portman NHS Gender Identity Development Service. Of them, half were considered eligible for puberty blockers immediately, receiving them in addition to psychological support. The other half were not considered immediately eligible for puberty blockers, citing reasons such as psychiatric problems or conflicts with parents and siblings. These patients received only psychological support for the following 18 months. All patients’ global psychological functioning was assessed using the CGAS questionnaire. Both groups showed significantly improved psychological functioning with psychological support, but the group receiving only psychological support stalled and showed no further improvement towards the end of the study, while those receiving puberty blockers continued to show greater improvement. The authors point out that the eventual CGAS score of the group receiving puberty blockers coincided almost perfectly with those found in a sample of children/adolescents without observed psychological/psychiatric symptoms.
Figure 1: CGAS scores of psychological functioning in transgender teens receiving puberty blockers and psychological support, compared to those receiving psychological support alone in Costa et al. (2015).
A later study at the same British gender identity clinic, Carmichael et al. (2021), received widespread media coverage in the United Kingdom following its mixed findings. It followed 44 gender dysphoric adolescents who received puberty blockers. CGAS scores were higher than the 2015 study at baseline, and showed slower and more modest improvement. The study reached contradictory conclusions, with improvements reported in some questionnaires, but not others, even for comparable measurements. Interestingly, in some of the researchers’ measures of well-being, social acceptance, and self-perception, adolescents themselves reported significant improvements, while their parents reported almost no improvement. The study characterises participants’ overall experiences with puberty blockers as positive, but is difficult to draw any conclusions from.
De Vries et al. (2011) and de Vries et al. (2014) investigate the psychological outcomes of the same cohort of transgender adolescents receiving puberty blockers at the VUmc gender clinic in the Netherlands. Both investigate psychological outcomes in a range of tests, with the 2014 study providing long-term follow-up many years after puberty blockers, and after gender reassignment surgery. In the studied cohort, psychological functioning improves and depression decreases over time, as evidenced by standardised tests, including CGAS scores. Significant improvements in well-being are reported both during treatment with puberty blockers, and in the years after, with hormone therapy and surgery. Unlike Carmichael et al., CBCL and YSR scores improve.
Van der Miesen et al. (2020) charts psychological well-being across 3 groups of Dutch adolescents: 272 transgender adolescents who haven’t yet received puberty blockers, 178 adolescents receiving puberty blockers, and 651 cisgender adolescents from the general population. The study finds poorer psychological functioning in those before treatment, while psychological functioning and well-being is similar to cisgender adolescents in those receiving pubertal suppression. These findings are in line with Costa et al. (2015), which noted that those receiving puberty blockers reached CGAS scores comparable to the general (age-matched) population.
Figure 2: Percentages of teens who report suicidality in van der Miesen et al. (2020). Suicidality was defined as endorsing the statement “I deliberately try to hurt or kill myself” or “I think about killing myself.” Suicidality among Dutch transgender youth has not significantly changed over time, making cohort differences unlikely (Arnoldussen et al., 2020).
In addition to the studies listed above, several smaller, less focused studies have also assessed the well-being of transgender adolescents receiving puberty blockers and reported overall positive experiences (Khatchadourian et al., 2014; Achille et al., 2020; Kuper et al., 2020). No studies report a decline in psychological functioning or notably negative psychological outcomes with the use of puberty blockers.
In combination, this strongly suggests that puberty blockers improve well-being and psychological functioning in children who experience gender dysphoria. In addition, it suggests that inappropriately withholding them may lead to worse later-life outcomes, such as increased suicidality.
Counterintuitively, several of the studies listed do note that puberty blockers don’t reduce gender dysphoria (de Vries et al., 2011; Carmichael et al., 2021). It’s important to be aware that this finding refers to the wish to transition, rather than psychological well-being. The finding is based on the Utrecht Gender Dysphoria Scale questionnaire (de Vries et al., 2006). To illustrate, the version for transmasculine youth asks patients whether they endorse such statements as “I prefer to behave like a boy”, “I wish I had been born as a boy”, “I hate having breasts”, and “every time someone treats me like a girl, I feel hurt.”
When studies note that puberty blockers don’t reduce gender dysphoria, this means children don’t stop identifying as transgender after receiving puberty blockers. They continue to want to transition. De Vries et al. (2011) points out this is the expected outcome:
As expected, puberty suppression did not result in an amelioration of gender dysphoria. Previous studies have shown that only gender reassignment consisting of cross-sex hormone treatment and surgery may end the actual gender dysphoria. None of the gender dysphoric adolescents in this study renounced their wish for gender reassignment during puberty suppression. This finding supports earlier studies showing that young adolescents who had been carefully diagnosed show persisting gender dysphoria into late adolescence or young adulthood
Fertility
Unlike hormone therapy, no risk of permanent infertility is believed to exist with the use of puberty blockers. Several long-term follow-up studies of patients treated with puberty blockers have found normal fertility. Among others, Feuillan et al. (1999), Heger et al. (1999), Heger et al. (2006) and Lazar et al. (2014) find no indication of impaired fertility in patients treated with puberty blockers for precocious puberty. In the years and decades following their treatments, the several hundred patients in these studies are found to conceive normally without an increased need for assisted reproductive technology, and with uneventful pregnancies. Despite several decades of use, no reports exist in literature of permanent infertility linked to puberty blockers. Interestingly, transgender populations do have higher rates of sperm abnormalities than cisgender populations, before any medical treatment has taken place (Li et al., 2018; Rodriguez-Wallberg et al., 2021).
In contrast, hormone therapy may cause permanent infertility (Hembree et al., 2017; Cheng et al., 2019). If fertility preservation has not been accessed before beginning treatment, puberty blockers must be stopped to do so, ideally before hormone therapy begins. When puberty blockers are stopped, unwanted sex characteristics continue to develop. Transgender people may find this extremely distressing, which may be one reason for them to not pursue fertility preservation.
No data exists on the exact length of time for which puberty blockers need to be stopped before full fertility is restored, and it likely varies depending on the age puberty blockers were initiated. Bertelloni et al. (2000) found spermarche took place between 0.7 to 3 years after discontinuation of puberty blockers in boys treated for precocious puberty. Barnard et al. (2019) report on the case of a single transgender patient who had been receiving puberty blockers for 6 months, from the age of 17. Three months after the last dose of monthly leuprorelin, no viable sperm sample could be produced. Five months after, their sample was viable.
Regardless of this, transgender individuals are extremely unlikely to use fertility preservation, with some estimates suggesting utilisation rates below 5% in North America (Chen et al., 2017; Nahata et al., 2017). In one piece of research, Pang et al. (2020) questioned 102 transgender Australian teens on their reasons for declining fertility preservation. The following statistics were gathered:
Figure 3: Australian transgender teens’ reasons for declining fertility preservation in Pang et al. (2020).
As such, there is no evidence-based reason to believe puberty blockers could cause infertility, with fertility returning when they are discontinued. However, due to low discontinuation rates for puberty blockers and low fertility preservation rates, those who start puberty blockers and persist are unlikely to have biological children. Clinical guidelines recommend that adolescents seeking puberty blockers should be counselled on options for fertility preservation, and parents should be involved in this (Hembree et al., 2017).
Bone Density
Bone density is a measure of the amount of bone mineral present in bone tissue. Bone density is measured using imaging techniques, such as DEXA scans: a type of X-ray. It is used to predict patients’ risk of breaking bones. The clinical terms for low bone density are osteopenia, and in more severe cases, osteoporosis, which is common among the elderly. For the purposes of this review, the most relevant measurement of bone density is the z-score, which expresses a patient’s bone density in comparison to other people of the same age and sex. A z-score of 0 indicates bone density equal to the general population. Small deviations, such as -0.2, may not always be relevant, but z-scores below -1 may be cause for concern.
There are concerns around the effects of puberty blockers on bone health. Puberty is a critical time for the accrual of bone density: a process largely driven by sex hormones. This process is delayed in those receiving puberty blockers, leading to temporary lower bone density and z-scores compared to peers going through puberty normally. While these short-term z-scores are not particularly relevant, long-term outcomes are very important: the question becomes what z-scores look like in the long term, into adulthood, and whether the use of puberty blockers has any impact on later-life fracture risk. Literature on this is uncertain.
Klink et al. (2015) finds that in both trans girls and trans boys, z-scores are lower both before treatment, and after long-term follow-up. The study suggests a small negative effect on final bone density from the use of puberty blockers, although many measurements fail to reach statistical significance. The study records notably lower final z-scores for trans girls than trans boys.
Vlot et al. (2017) finds that 2 years after beginning hormone therapy, z-scores were returning towards normal. In trans boys, final z-scores were negligibly lower, while in trans girls, the effect was much more pronounced, with meaningfully lower z-scores both before and after treatment.
In line with this, Schagen et al. (2020) finds that in its cohort, final z-scores normalised after 3 years of hormone therapy for trans boys, while they remained meaningfully low both before and after treatment for trans girls.
Figure 4: Bone Mineral Apparent Density (BMAD) of the lumbar spine across multiple studies, relative to sex assigned at birth. Three measurements are taken in each study: the initiation of puberty blockers, the initiation of hormone therapy, and one measurement after several years of hormone therapy. A z-score below -1 is commonly considered to be clinically relevant osteopenia, while a score below -2.5 is considered to be osteoporosis. The figure illustrates that trans girls tend to have significantly lower bone density before, during, and after treatment, while this is not the case for trans boys. Trans girls also tend to receive puberty blockers for a longer time.
Guaraldi et al. (2016) find in their literature review that in those receiving puberty blockers for precocious puberty, bone mineral density is lower than that of untreated peers during treatment with puberty blockers, then typically recovers when puberty is initiated, with long-term follow-up showing little difference to the general population. Combined with the results of trans boys, this suggests that not puberty blockers themselves, but rather, subsequent suboptimal hormone therapy in trans girls could potentially be to blame for their more pronounced negative outcomes.
The hormone therapy prescribed to trans girls in the listed studies may be suboptimal in several ways. To begin with, all three use very low adult maintenance dosages of no more than 2 mg oral estradiol in transfeminine patients. Such a dose is likely to produce serum estradiol levels of roughly 50 pg/ml on average: below the average estradiol exposure of cis women (Aly, 2018; Aly, 2020). Many clinical guidelines recommend higher levels, which some research suggests could have a small positive effect on final bone density compared to lower dosages (Roux, 1997; Riggs et al., 2012). Indeed, the authors of all three studies themselves note their doses were low and may have been inadequate for optimal bone density.
As a final confounding factor, none of the studies control for lifestyle factors associated with lower bone density, such as exercise, smoking, vitamin D, and calcium intake. These factors have a significant effect on bone density. Transgender people are more likely to smoke, less likely to exercise, and less likely to consume adequate calcium, both as teens and as adults (Jones et al., 2018; Kidd, Dolezal & Bockting, 2018). This is believed to be the reason transgender people of all ages tend to have lower bone density before any treatment is initiated. Without controlling for these factors, which may distort the available data significantly, it’s difficult to draw confident conclusions from these studies, and a causal link between the use of puberty blockers and lower final bone density remains unproven. If such a link does exist, the effect seems unlikely to be dramatic, and unlikely to outweigh the benefits of puberty blockers.
In a noteworthy study, Antoniazzi et al. (2003) report that in those receiving puberty blockers for precocious puberty, bone mineral density is better preserved through calcium supplementation. Calcium intake is often inadequate in transgender youth (Lee et al., 2020), and therefore warrants further study for improving their bone mineral density. Alongside calcium, lifestyle interventions, the use of transdermal instead of oral estrogens, and the avoidance of subphysiological adult dosages of estradiol could all potentially improve bone-related outcomes over current clinical practise.
IQ and Cognitive Development
One possible concern is the impact of puberty blockers on IQ and cognitive development. Very little research on the subject exists, with commonly cited critical studies investigating sheep rather than humans (Hough et al., 2017), or being case studies of a single patient (Schneider et al., 2017). Only two larger studies investigate this:
Staphorsius et al. (2015), the only study to investigate this in a transgender population, evaluated performance in the standardised Tower of London test, as well as IQ scores. The study found no significant differences in executive functioning between the two groups. IQ was slightly lower in transgender girls receiving puberty suppression than the control group, but the same was not true in a statistically relevant way of transgender boys. Age differences, lifestyle factors, and a very low sample size may all explain these differences.
Wojniusz et al. (2016) assessed 15 girls suffering from precocious puberty and treated with a puberty blocker. The 15 girls were compared with 15 age-matched controls. Both groups showed similar IQ scores.
Neither study has very many participants, records baseline cognitive performance, or controls for confounding factors. As such, very few conclusions can be drawn from them. Decades of clinical experience with the use of puberty blockers in children suggests it’s unlikely any particularly dramatic effect on IQ exists, but without much larger, higher quality studies, no conclusion on this can be reached, and further research is needed.
Desistance
Discontinuation rates for patients on puberty blockers are very low, with fewer than 5% of teens typically stopping them without going on to hormone therapy (Wiepjes et al., 2018; Brik et al., 2020; Kuper et al., 2020). A potential concern is that this could mean puberty blockers put children on an almost guaranteed path towards gender transition, when they might otherwise change their minds.
Surprisingly, while a commonly held belief suggests most gender dysphoric children will grow out of it without treatment at a later age, little convincing evidence supports this claim. While existing studies report desistance rates ranging from 43% (Wallien & Cohen-Kettenis, 2008) to 88% (Drummond et al., 2008), they often contain significant methodological issues.
Historically, in the 20th century, a transgender identity was viewed as a negative outcome: it was something for a patient to be cured of, for example through aversion therapy. Since then, a cultural shift towards transgender people has taken place. Older studies into desistance rates are often reflective of this. As an example, Kosky (1987) describes eight boys who were hospitalised in a psychiatric unit for displaying effeminate behaviour and cross-dressing, where they received intensive treatment aimed at curing them. Today, these behaviours are more accepted, and they are not necessarily viewed as the same thing as a transgender gender identity. Clearly, a study like this cannot be used to estimate the desistance rates of today’s gender dysphoric children.
Other studies describing the 1960s through 1980s are similar (Bakwin, 1968; Lebovitz, 1972; Zuger, 1978; Money & Russo, 1979; Davenport, 1986). Many predate the DSM-III, and thus the existence of formal diagnostic criteria. Few studied self-reported gender identity: instead, they tend to study gender non-conforming behaviour, such as cross-dressing, that doesn’t necessarily constitute a transgender identity. Many of them try to discourage patients as a core part of their treatment, sometimes in ways that are now banned across much of the world as conversion therapy. Combined with a drastically changed society, extrapolating modern transgender desistance rates from these studies is unreasonable.
A small number of more recent studies do exist. The highest desistance rates found in modern literature are approximately 88%, reported by three frequently cited Canadian studies: Drummond et al. (2008), Drummond et al. (2018), and Singh, Bradley & Zucker (2021). Unfortunately, these studies appear to be at a high risk of bias: calling their credibility into question, the clinic in which they took place was closed in 2015, amidst allegations of conversion therapy. An independent review found that it “cannot state that the clinic does not practice reparative approaches.” In the review, many children and their parents report feeling the clinic was invasive and intimidating to them. Some instances include:
Assessments are described as intrusive and even traumatic by some, who described feeling “poked and prodded”. One way mirror and multiple observers create discomfort. Many questions were felt to be irrelevant, unnecessarily intrusive (particularly those regarding sexual fantasies), especially when asked without context, rationale, and what seems to be inadequate or even absent informed consent. Also, it is unclear whether any potential benefit of this line of questioning to the patient was explained. Parents of younger clients report their child appearing to be and later reporting feeling they were very uncomfortable with the way they were asked about their gender variance “as if my child was not okay as a person.” One parent described feeling “dismissed” when she spoke to clinicians about this.
Patients reported feeling intimidated to question Dr. Zucker regarding their concerns and were not offered the opportunity to decline. Multiple informants commented on this.
Chart documentation revealed statements reflecting that the diversity of gender expression and variance are not accepted equally. One example is of a child for whom all gender and body dysphoria had resolved and multiple informants indicated sustained good mood and satisfaction with social and academic functioning. Despite this, the parents of the child were advised at discharge to encourage the child to spend more time with cisgendered boys because he had effeminate speech and mannerisms. These were not goals of the client or family.
This may explain why these studies find a much higher desistance rate than other modern literature, and makes them very unlikely to be representative figures. As an alternative possibility, Steensma & Cohen-Kettenis (2018) suggest that differences in the local social climate regarding gender variance may have also been an important contributing factor.
Steensma et al. (2011) and Steensma et al. (2013) set out to investigate factors that could contribute to the persistence or desistance of gender dysphoria in children. The 2011 study reports a desistance rate of 45%, while the 2013 study reports 73%. The figures have been criticised because all children lost to follow-up are assumed to have desisted, which may or may not have inflated their number. More importantly however, in Steensma & Cohen-Kettenis (2018), the authors themselves argue they’ve been cited out of context, and their figures can’t be used to extrapolate desistance rates:
Unlike what is suggested, we have not studied the gender identities of the children. Instead we have studied the persistence and desistence of children’s distress caused by the gender incongruence they experience to the point that they seek clinical assistance. […] Using the term desistence in this way does not imply anything about the identity of the desisters. The children could still be hesitating, searching, fluctuating, or exploring with regard to their gender experience and expression, and trying to figure out how they wanted to live. Apparently, they no longer desired some form of gender-affirming treatment at that point in their lives.
Again, because of the purpose and the design of this study we did not report prevalence numbers in the sample under study. Furthermore, the sample in the 2013 study did not include children in the younger age spectrum of the referred population to the Amsterdam clinic. Reporting prevalence of persistence and/or desistance in this sample would therefore not be reliable.
The only other modern study into persistence rates has been Wallien & Cohen-Kettenis (2008). The study appears to be of higher quality and provides the most convincing estimate available: a 27% persistence rate and a 43% desistance rate over the course of (on average) 10 years. The remaining 30% of participants were lost to follow-up.
Several further problems cast doubt on the data presented in all of these studies, including Wallien & Cohen-Kettenis (2008). Firstly: children are diagnosed using DSM-III and DSM-IV criteria, which are dated by today’s standards. In these older criteria, gender identity was not a diagnostic requirement: a child could be diagnosed with a gender identity disorder for a range of gender non-conforming behaviours, without themselves identifying as a different gender, or experiencing distress with their gender role or sex characteristics (Temple Newhook et al., 2018).
Strikingly, with the exception of Steensma et al. (2011), all studies include a significant number of children who never actually met then-current DSM diagnostic criteria for Gender Identity Disorder: in the case of Wallien & Cohen-Kettenis (2008), a quarter of all participants. These participants have been assumed to be transgender for the purposes of extrapolating desistance rates, but held a diagnosis of Gender Identity Disorder Not Otherwise Specified: a broad category described by the DSM as representing those who may not necessarily seek medical transition, but may transiently cross-dress, be preoccupied with castration, or be intersex and experience gender dysphoria. These participants’ exact circumstances are not described by the researchers, but both Wallien & Cohen-Kettenis and Steensma et al. report in their studies that all, or nearly all persisters met DSM diagnostic criteria, while only about half of desisters did.
Outside of this, there is a lack of long-term follow-up. A gender dysphoric child might desist in transitioning during their teens, but go on to transition in adulthood, for example because of peer pressure or lack of parental acceptance. Whether this happens at any significant rate has not been studied.
The studies do suggest that for an unknown percentage of children, gender dysphoria will resolve over time, but the high desistance rates often cited as an established fact don’t appear to be supported by evidence. Concerns that puberty blockers cause children to transition when they otherwise would’ve aged out of gender dysphoria appear misplaced: children whose dysphoria persisted were much more likely to have met the diagnostic criteria to receive puberty blockers.
Current literature on this will likely soon be superseded by higher-quality data, with several very large, well-funded studies into gender dysphoric youth now underway the United States (Olson-Kennedy et al., 2019), Australia (Tollit et al., 2019) and the United Kingdom (Kennedy et al., 2019).
Desistance and persistence rates can suggest a binary view, and should be seen in a greater context. Because children’s needs change over time, a hypothetical child might feel uncertain about their gender, possibly even receive puberty blockers, and then later decide they do not wish to transition. In such a case, puberty blockers may have met their needs at the time, and were not automatically harmful or regrettable, particularly due to their reversible nature. Neither being transgender, nor being cisgender should be seen as a negative outcome. In their critical commentary, Temple Newhook et al. (2018) write that it is important to respect children’s wishes and autonomy, and move away from the question of, “How should children’s gender identities develop over time?” toward a more useful question: “How should children best be supported as their gender identity develops?”
Regret
In light of persistence and desistance rates, it makes sense to ask how patients themselves feel about their treatment with puberty blockers, and whether they regret receiving them. Limited research exists on the subject:
A large retrospective review of the medical files of all 6,793 patients treated at the Dutch VUmc clinic between 1972 and 2015 found that 14 patients (0.2%) regretted their treatment in total. This included patients who received puberty suppression, hormone therapy, and/or surgery. Notably, 5 of them regretted their treatment because of a lack of social acceptance (Wiepjes et al., 2018).
De Vries et al. (2014), found none of the 55 transgender patients they followed regretted receiving puberty blockers, hormone therapy, or surgery. Psychological well-being continued to improve in their cohort, both with puberty blockers, hormone therapy, and later gender reassignment surgery.
Vrouenraets et al. (2016) interviewed 13 adolescents who had been seen at a Dutch gender identity clinic, twelve of whom had received puberty blockers. Asked about long-term risks, most responded that they were significantly outweighed by puberty blockers allowing them to live a more happy life. Quotes from the interviewed children in the study include:
The possible long-term consequences are incomparable with the unhappy feeling that you have and will keep having if you don’t receive treatment with puberty suppression. (trans boy; age: 18)
It isn’t a choice, even though a lot of people think that. Well, actually it is a choice: living a happy life or living an unhappy life. (trans girl; age: 14)
They also comment on the increasing attention to transgender people in media, with one child saying:
Thanks to media coverage I learned that gender dysphoria exists; that someone can have these feelings and that you can get treatment for it. Beforehand I thought I was the only one like this. (trans boy; age: 18)
Ease of Access
While large geographic differences exist, on the whole, access to puberty blockers is often difficult.
In the United States, Turban et al. (2020) found that access to puberty blockers was associated with a greater household income, noting that the annual cost of them ranges from $4,000 to $25,000 and insurance coverage was unavailable to many. It also found that transgender teens were less likely to receive puberty blockers if they did not identify as heterosexual or binary. Of those who received puberty blockers, 60% reported traveling <25 miles for gender-affirming care, 29% travelled between 25 and 100 miles, and 11% travelled >100 miles. As of 2021, several states are pursuing regulation banning the use of puberty blockers, with Arkansas having become the first to pass such a law. Several large professional bodies representing thousands of medical experts have condemned this type of regulation (American Academy of Child and Adolescent Psychiatry, 2019; American Medical Association, 2021; Endocrine Society, 2021).
In the United Kingdom, waiting lists as long as 4 years or more exist for initial intake appointments for puberty blockers. Legislative changes in light of Bell v. Tavistock complicated access dramatically: in the nine months between the ruling and its reversal, no under-17s received puberty blockers under the public healthcare sytem, and reports described the care of adolescents over 16, who were not affected by the judgement, being discontinued as well. Restrictions in light of Bell v. Tavistock were condemned by WPATH, EPATH, USPATH, AsiaPATH, CPATH, AusPATH, and PATHA, the leading medical associations for transgender health, who released a statement saying they believe it will cause significant harm to the affected patients (WPATH, 2020), as well as Amnesty International UK and Liberty (Amnesty International UK, 2020).
In Finland, new prescriber guidelines for treating gender dysphoric teens were released in 2020 (Society for Evidence Based Medicine, 2021). They broke with WPATH guidelines, instead recommending that gender dysphoric teens receive psychosocial support and psychotherapy as a first-line treatment, and discouraging the use of puberty blockers, with the addition of much stricter criteria for their use. The Finnish health authority has stated that these recommendations will not be revised until further research is available.
A similar trend of increasing wait times and difficult access holds in many other countries, with the process to receive puberty blockers sometimes taking up to several years. Because of their time-sensitive nature in preventing unwanted permanent changes, long-term outcomes are likely to be worse with slower treatment. Some evidence supports this: for example, one study found that reducing treatment wait times led to reduced depression and anxiety compared to historical controls (Dahlgren Allen et al., 2021).
Conclusion
Unknowns exist around puberty blockers in transgender youth, but their risks seem to be relatively minor based on available research, while clear evidence associates their use with improved well-being, psychological functioning, and reduced suicidality.
Based on parallels from research in cisgender teens treated for precocious puberty, as well as limited studies and clinical experience with transgender teens, it’s unlikely that puberty suppression has a dramatic negative effect on children’s final bone density, lifetime fracture risk, IQ, or cognitive development when prescribed in line with medical guidelines. However, there is insufficient evidence to determine whether or not they have any impact at all.
Although not supported by conclusive evidence, the use of puberty blockers may have a modest negative impact on bone density. This could be related to the use of puberty blockers themselves, but could also be related to suboptimal hormone therapy regimens after their use, particularly in transgender girls, as well as lifestyle factors. Studies investigating this suffer from significant methodological issues, and a definitive causal link remains unproven. Based on limited evidence, prescribers may wish to consider calcium supplementation in transgender teens receiving puberty blockers, and may wish to avoid oral estrogens in transgender girls beginning hormone therapy.
Compared to their cisgender peers, transgender adolescents who take puberty blockers are less likely to choose to have biological children, but puberty blockers do not permanently affect fertility.
Widely cited statistics around children growing out of gender dysphoria (“desistance”) as they grow older are based on highly unreliable data. Surprisingly, based on current evidence, we cannot reasonably guess the rate at which this happens. Regardless, desistance rates are not an argument for or against the use of puberty blockers. It is important to respect children’s wishes and autonomy, and to find the best way to support them as their gender identity develops, without imposing the idea that either a transgender or a cisgender gender identity is a bad outcome.
Very few patients who receive puberty blockers experience regret. In broader context, for the small minority of adult transgender patients who report feeling regret after undergoing hormone therapy or surgery, a common reason for that is a lack of social acceptance.
More high-quality research is urgently needed in this field. In particular, the effects of puberty blockers on IQ and cognitive development, bone outcomes, and desistance remain understudied subjects. Randomised controlled trials on puberty blockers are not available, and likely cannot be performed for both practical and ethical reasons. This should not be seen as a reason to discard all other research on the subject, or to label their use as experimental, as it is a standard of evidence that can never be met.
Puberty blockers are extremely difficult for patients to access in many countries, including the United States, the United Kingdom, and parts of Europe. Several countries have recently banned their use, or further restricted it significantly. This review provides further evidence supporting WPATH, EPATH, USPATH, AsiaPATH, CPATH, AusAPTH, PATHA, the Endocrine Society, the American Academy of Child and Adolescent Psychiatry, and the American Medical Association in condemning recent attempts to bar transgender teens from receiving gender-affirming care, including puberty blockers. To better support gender dysphoric children, barriers of access should instead be reduced where possible.
References
Achille, C., Taggart, T., Eaton, N. R., Osipoff, J., Tafuri, K., Lane, A., & Wilson, T. A. (2020). Longitudinal impact of gender-affirming endocrine intervention on the mental health and well-being of transgender youths: preliminary results. International Journal of Pediatric Endocrinology, 2020, 8. [DOI:10.1186/s13633-020-00078-2]
Antoniazzi, F., Zamboni, G., Bertoldo, F., Lauriola, S., Mengarda, F., Pietrobelli, A., & Tatò, L. (2003). Bone mass at final height in precocious puberty after gonadotropin-releasing hormone agonist with and without calcium supplementation. The Journal of Clinical Endocrinology and Metabolism, 88(3), 1096–1101. [DOI:10.1210/jc.2002-021154]
Arnoldussen, M., Steensma, T. D., Popma, A., van der Miesen, A., Twisk, J., & de Vries, A. (2020). Re-evaluation of the Dutch approach: are recently referred transgender youth different compared to earlier referrals? European Child & Adolescent Psychiatry, 29(6), 803–811. [DOI:10.1007/s00787-019-01394-6]
Bakwin, H. (1968). Deviant gender-role behavior in children: relation to homosexuality. Pediatrics, 41(3), 620–629. [PubMed] [DOI:10.1542/peds.41.3.620]
Barake, M., Klibanski, A., & Tritos, N. A. (2014). Effects of recombinant human growth hormone therapy on bone mineral density in adults with growth hormone deficiency: a meta-analysis. The Journal of Clinical Endocrinology and Metabolism, 99(3), 852–860. [DOI:10.1210/jc.2013-3921]
Barake, M., Arabi, A., Nakhoul, N., El-Hajj Fuleihan, G., El Ghandour, S., Klibanski, A., & Tritos, N. A. (2018). Effects of growth hormone therapy on bone density and fracture risk in age-related osteoporosis in the absence of growth hormone deficiency: a systematic review and meta-analysis. Endocrine, 59(1), 39–49. [DOI:10.1007/s12020-017-1440-0]
Barnard, E. P., Dhar, C. P., Rothenberg, S. S., Menke, M. N., Witchel, S. F., Montano, G. T., Orwig, K. E., & Valli-Pulaski, H. (2019). Fertility Preservation Outcomes in Adolescent and Young Adult Feminizing Transgender Patients. Pediatrics, 144(3), e20183943. [DOI:10.1542/peds.2018-3943]
Bertelloni, S., Baroncelli, G. I., Ferdeghini, M., Menchini-Fabris, F., & Saggese, G. (2000). Final height, gonadal function and bone mineral density of adolescent males with central precocious puberty after therapy with gonadotropin-releasing hormone analogues. European Journal of Pediatrics, 159(5), 369–374. [DOI:10.1007/s004310051289]
Brik, T., Vrouenraets, L., de Vries, M. C., & Hannema, S. E. (2020). Trajectories of Adolescents Treated with Gonadotropin-Releasing Hormone Analogues for Gender Dysphoria. Archives of Sexual Behavior, 49(7), 2611–2618. [DOI:10.1007/s10508-020-01660-8]
Carmichael, P., Butler, G., Masic, U., Cole, T. J., De Stavola, B. L., Davidson, S., Skageberg, E. M., Khadr, S., & Viner, R. M. (2021). Short-term outcomes of pubertal suppression in a selected cohort of 12 to 15 year old young people with persistent gender dysphoria in the UK. PloS One, 16(2), e0243894. [DOI:10.1371/journal.pone.0243894]
Cedrin-Durnerin, I., Bidart, J. M., Robert, P., Wolf, J. P., Uzan, M., & Hugues, J. N. (2000). Consequences on gonadotrophin secretion of an early discontinuation of gonadotrophin-releasing hormone agonist administration in short-term protocol for in-vitro fertilization. Human Reproduction, 15(5), 1009–1014. [DOI:10.1093/humrep/15.5.1009]
Chen, D., Simons, L., Johnson, E. K., Lockart, B. A., & Finlayson, C. (2017). Fertility Preservation for Transgender Adolescents. The Journal of Adolescent Health, 61(1), 120–123. [DOI:10.1016/j.jadohealth.2017.01.022]
Cheng, P. J., Pastuszak, A. W., Myers, J. B., Goodwin, I. A., & Hotaling, J. M. (2019). Fertility concerns of the transgender patient. Translational Andrology and Urology, 8(3), 209–218. [DOI:10.21037/tau.2019.05.09]
Cohen-Kettenis, P. T., & van Goozen, S. H. (1998). Pubertal delay as an aid in diagnosis and treatment of a transsexual adolescent. European Child & Adolescent Psychiatry, 7(4), 246–248. [DOI:10.1007/s007870050073]
Cohen-Kettenis, P. T., Schagen, S. E., Steensma, T. D., de Vries, A. L., & Delemarre-van de Waal, H. A. (2011). Puberty suppression in a gender-dysphoric adolescent: a 22-year follow-up. Archives of Sexual Behavior, 40(4), 843–847. [DOI:10.1007/s10508-011-9758-9]
Costa, R., Dunsford, M., Skagerberg, E., Holt, V., Carmichael, P., & Colizzi, M. (2015). Psychological Support, Puberty Suppression, and Psychosocial Functioning in Adolescents with Gender Dysphoria. The Journal of Sexual Medicine, 12(11), 2206–2214. [DOI:10.1111/jsm.13034]
Dahlgren Allen, S., Tollit, M. A., McDougall, R., Eade, D., Hoq, M., & Pang, K. C. (2021). A Waitlist Intervention for Transgender Young People and Psychosocial Outcomes. Pediatrics, 148(2), e2020042762. [DOI:10.1542/peds.2020-042762]
Davenport, C. W. (1986). A follow-up study of 10 feminine boys. Archives of Sexual Behavior, 15(6), 511–517. [DOI:10.1007/BF01542316]
Davenport M. L. (2010). Approach to the patient with Turner syndrome. The Journal of Clinical Endocrinology and Metabolism, 95(4), 1487–1495. [DOI:10.1210/jc.2009-0926]
de Vries, A. L. C., Cohen-Kettenis, P. T., & Delemarre-van de Waal, H. (2006). Clinical Management of Gender Dysphoria in Adolescents. International Journal of Transgenderism, 9(4), 83–94. [DOI:10.1300/J485v09n03_04]
de Vries, A. L., Steensma, T. D., Doreleijers, T. A., & Cohen-Kettenis, P. T. (2011). Puberty suppression in adolescents with gender identity disorder: a prospective follow-up study. The Journal of Sexual Medicine, 8(8), 2276–2283. [DOI:10.1111/j.1743-6109.2010.01943.x]
de Vries, A. L., McGuire, J. K., Steensma, T. D., Wagenaar, E. C., Doreleijers, T. A., & Cohen-Kettenis, P. T. (2014). Young adult psychological outcome after puberty suppression and gender reassignment. Pediatrics, 134(4), 696–704. [DOI:10.1542/peds.2013-2958]
Drummond, K. D., Bradley, S. J., Peterson-Badali, M., & Zucker, K. J. (2008). A follow-up study of girls with gender identity disorder. Developmental Psychology, 44(1), 34–45. [DOI:10.1037/0012-1649.44.1.34]
Drummond, K. D., Bradley, S. J., Peterson-Badali, M., VanderLaan, D. P., & Zucker, K. J. (2018). Behavior Problems and Psychiatric Diagnoses in Girls with Gender Identity Disorder: A Follow-Up Study. Journal of Sex & Marital Therapy, 44(2), 172–187. [DOI:10.1080/0092623X.2017.1340382]
Ekbote, V. H., Khadilkar, V. V., Khadilkar, A. V., Mughal, Z., Chiplonkar, S. A., Palande, S. A., Phanse-Gupte, S. S., Patwardhan, V. G., & Shilvant, D. S. (2015). Relationship of insulin-like growth factor 1 and bone parameters in 7–15 years old apparently, healthy Indian children. Indian Journal of Endocrinology and Metabolism, 19(6), 770–774. [DOI:10.4103/2230-8210.167549]
Feuillan, P. P., Jones, J. V., Barnes, K., Oerter-Klein, K., & Cutler, G. B., Jr. (1999). Reproductive axis after discontinuation of gonadotropin-releasing hormone analog treatment of girls with precocious puberty: long term follow-up comparing girls with hypothalamic hamartoma to those with idiopathic precocious puberty. The Journal of Clinical Endocrinology and Metabolism, 84(1), 44–49. [DOI:10.1210/jcem.84.1.5409]
Giordano, S., & Holm, S. (2020). Is puberty delaying treatment ‘experimental treatment’? International Journal of Transgender Health, 21(2), 113–121. [DOI:10.1080/26895269.2020.1747768]
Green, A. E., DeChants, J. P., Price, M. N., & Davis, C. K. (2022). Association of Gender-Affirming Hormone Therapy With Depression, Thoughts of Suicide, and Attempted Suicide Among Transgender and Nonbinary Youth. Journal of Adolescent Health, 70(4), 643–649. [DOI:10.1016/j.jadohealth.2021.10.036]
Guaraldi, F., Beccuti, G., Gori, D., & Ghizzoni, L. (2016). MANAGEMENT OF ENDOCRINE DISEASE: Long-term outcomes of the treatment of central precocious puberty. European Journal of Endocrinology, 174(3), R79–R87. [DOI:10.1530/EJE-15-0590]
Heger, S., Partsch, C. J., & Sippell, W. G. (1999). Long-term outcome after depot gonadotropin-releasing hormone agonist treatment of central precocious puberty: final height, body proportions, body composition, bone mineral density, and reproductive function. The Journal of Clinical Endocrinology and Metabolism, 84(12), 4583–4590. [DOI:10.1210/jcem.84.12.6203]
Heger, S., Müller, M., Ranke, M., Schwarz, H. P., Waldhauser, F., Partsch, C. J., & Sippell, W. G. (2006). Long-term GnRH agonist treatment for female central precocious puberty does not impair reproductive function. Molecular and Cellular Endocrinology, 254–255, 217–220. [DOI:10.1016/j.mce.2006.04.012]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658]
Hough, D., Bellingham, M., Haraldsen, I. R., McLaughlin, M., Robinson, J. E., Solbakk, A. K., & Evans, N. P. (2017). A reduction in long-term spatial memory persists after discontinuation of peripubertal GnRH agonist treatment in sheep. Psychoneuroendocrinology, 77, 1–8. [DOI:10.1016/j.psyneuen.2016.11.029]
Isotton, A. L., Wender, M. C., Casagrande, A., Rollin, G., & Czepielewski, M. A. (2012). Effects of oral and transdermal estrogen on IGF1, IGFBP3, IGFBP1, serum lipids, and glucose in patients with hypopituitarism during GH treatment: a randomized study. European Journal of Endocrinology, 166(2), 207–213. [DOI:10.1530/EJE-11-0560]
Jones, B. A., Haycraft, E., Bouman, W. P., & Arcelus, J. (2018). The Levels and Predictors of Physical Activity Engagement Within the Treatment-Seeking Transgender Population: A Matched Control Study. Journal of Physical Activity & Health, 15(2), 99–107. [DOI:10.1123/jpah.2017-0298]
Jung, K. Y., Kim, T., Hwang, S. Y., Lee, T. R., Yoon, H., Shin, T. G., Sim, M. S., Cha, W. C., & Jeon, H. J. (2018). Deliberate Self-harm among Young People Begins to Increase at the Very Early Age: a Nationwide Study. Journal of Korean Medical Science, 33(30), e191. [DOI:10.3346/jkms.2018.33.e191]
Kennedy, E., Spinner, L., Lane, C., Stynes, H., Ranieri, V., Carmichael, P., Omar, R., Vickerstaff, V., Hunter, R., Wright, T., Senior, R., Butler, G., Baron-Cohen, S., Young, B., & King, M. (2021). Longitudinal Outcomes of Gender Identity in Children (LOGIC): protocol for a prospective longitudinal cohort study of children referred to the UK gender identity development service. BMJ Open, 11(9), e045628. [DOI:10.1136/bmjopen-2020-045628]
Khatchadourian, K., Amed, S., & Metzger, D. L. (2014). Clinical management of youth with gender dysphoria in Vancouver. The Journal of Pediatrics, 164(4), 906–911. [DOI:10.1016/j.jpeds.2013.10.068]
Kidd, J. D., Dolezal, C., & Bockting, W. O. (2018). The Relationship Between Tobacco Use and Legal Document Gender-Marker Change, Hormone Use, and Gender-Affirming Surgery in a United States Sample of Trans-Feminine and Trans-Masculine Individuals: Implications for Cardiovascular Health. LGBT Health, 5(7), 401–411. [DOI:10.1089/lgbt.2018.0103]
Klink, D., Caris, M., Heijboer, A., van Trotsenburg, M., & Rotteveel, J. (2015). Bone mass in young adulthood following gonadotropin-releasing hormone analog treatment and cross-sex hormone treatment in adolescents with gender dysphoria. The Journal of Clinical Endocrinology and Metabolism, 100(2), E270–E275. [DOI:10.1210/jc.2014-2439]
Kosky, R. J. (1987). Gender-disordered children: does inpatient treatment help? The Medical Journal of Australia, 146(11), 565–569. [DOI:10.5694/j.1326-5377.1987.tb120415.x]
Kuper, L. E., Stewart, S., Preston, S., Lau, M., & Lopez, X. (2020). Body Dissatisfaction and Mental Health Outcomes of Youth on Gender-Affirming Hormone Therapy. Pediatrics, 145(4), e20193006. [DOI:10.1542/peds.2019-3006]
Lazar, L., Meyerovitch, J., de Vries, L., Phillip, M., & Lebenthal, Y. (2014). Treated and untreated women with idiopathic precocious puberty: long-term follow-up and reproductive outcome between the third and fifth decades. Clinical Endocrinology, 80(4), 570–576. [DOI:10.1111/cen.12319]
Lebovitz, P. S. (1972). Feminine behavior in boys: aspects of its outcome. The American Journal of Psychiatry, 128(10), 1283–1289. [DOI:10.1176/ajp.128.10.1283]
Lee, J. Y., Finlayson, C., Olson-Kennedy, J., Garofalo, R., Chan, Y. M., Glidden, D. V., & Rosenthal, S. M. (2020). Low Bone Mineral Density in Early Pubertal Transgender/Gender Diverse Youth: Findings From the Trans Youth Care Study. Journal of the Endocrine Society, 4(9), bvaa065. [DOI:10.1210/jendso/bvaa065]
Li, K., Rodriguez, D., Gabrielsen, J. S., Centola, G. M., & Tanrikut, C. (2018). Sperm cryopreservation of transgender individuals: trends and findings in the past decade. Andrology, 6(6), 860–864. [DOI:10.1111/andr.12527]
Lindsey, R. C., & Mohan, S. (2016). Skeletal effects of growth hormone and insulin-like growth factor-I therapy. Molecular and Cellular Endocrinology, 432, 44–55. [DOI:10.1016/j.mce.2015.09.017]
Locatelli, V., & Bianchi, V. E. (2014). Effect of GH/IGF-1 on Bone Metabolism and Osteoporsosis. International Journal of Endocrinology, 2014, 235060. [DOI:10.1155/2014/235060]
Money, J., & Russo, A. J. (1979). Homosexual outcome of discordant gender identity/role in childhood: Longitudinal follow-up. Journal of Pediatric Psychology, 4(1), 29–41. [DOI:10.1093/jpepsy/4.1.29]
Morey, Y., Mellon, D., Dailami, N., Verne, J., & Tapp, A. (2017). Adolescent self-harm in the community: an update on prevalence using a self-report survey of adolescents aged 13–18 in England. Journal of Public Health, 39(1), 58–64. [DOI:10.1093/pubmed/fdw010]
Mul, D., Versluis-den Bieman, H. J., Slijper, F. M., Oostdijk, W., Waelkens, J. J., & Drop, S. L. (2001). Psychological assessments before and after treatment of early puberty in adopted children. Acta Paediatrica, 90(9), 965–971. [DOI:10.1080/080352501316978011]
Nahata, L., Tishelman, A. C., Caltabellotta, N. M., & Quinn, G. P. (2017). Low Fertility Preservation Utilization Among Transgender Youth. The Journal of Adolescent Health, 61(1), 40–44. [DOI:10.1016/j.jadohealth.2016.12.012]
Nock, M. K., Green, J. G., Hwang, I., McLaughlin, K. A., Sampson, N. A., Zaslavsky, A. M., & Kessler, R. C. (2013). Prevalence, correlates, and treatment of lifetime suicidal behavior among adolescents: results from the National Comorbidity Survey Replication Adolescent Supplement. JAMA Psychiatry, 70(3), 300–310. [DOI:10.1001/2013.jamapsychiatry.55]
Olson-Kennedy, J., Chan, Y. M., Garofalo, R., Spack, N., Chen, D., Clark, L., Ehrensaft, D., Hidalgo, M., Tishelman, A., & Rosenthal, S. (2019). Impact of Early Medical Treatment for Transgender Youth: Protocol for the Longitudinal, Observational Trans Youth Care Study. JMIR Research Protocols, 8(7), e14434. [DOI:10.2196/14434]
Pang, K. C., Peri, A., Chung, H. E., Telfer, M., Elder, C. V., Grover, S., & Jayasinghe, Y. (2020). Rates of Fertility Preservation Use Among Transgender Adolescents. JAMA Pediatrics, 174(9), 890–891. [DOI:10.1001/jamapediatrics.2020.0264]
Riggs, M. M., Bennetts, M., van der Graaf, P. H., & Martin, S. W. (2012). Integrated pharmacometrics and systems pharmacology model-based analyses to guide GnRH receptor modulator development for management of endometriosis. CPT: Pharmacometrics & Systems Pharmacology, 1(10), e11. [DOI:10.1038/psp.2012.10]
Rodriguez-Wallberg, K. A., Häljestig, J., Arver, S., Johansson, A., & Lundberg, F. E. (2021). Sperm quality in transgender women before or after gender affirming hormone therapy—A prospective cohort study. Andrology, 9(6), 1773–1780. [DOI:10.1111/andr.12999]
Roux, C. (1997). Estrogen therapy in postmenopausal osteoporosis. What we know and what we don’t. Revue du Rhumatisme (English Ed.), 64(6), 402–409. [Google Scholar] [PubMed] [PDF]
Schagen, S., Wouters, F. M., Cohen-Kettenis, P. T., Gooren, L. J., & Hannema, S. E. (2020). Bone Development in Transgender Adolescents Treated With GnRH Analogues and Subsequent Gender-Affirming Hormones. The Journal of Clinical Endocrinology and Metabolism, 105(12), e4252–e4263. [DOI:10.1210/clinem/dgaa604]
Schneider, M. A., Spritzer, P. M., Soll, B., Fontanari, A., Carneiro, M., Tovar-Moll, F., Costa, A. B., da Silva, D. C., Schwarz, K., Anes, M., Tramontina, S., & Lobato, M. (2017). Brain Maturation, Cognition and Voice Pattern in a Gender Dysphoria Case under Pubertal Suppression. Frontiers in Human Neuroscience, 11, 528. [DOI:10.3389/fnhum.2017.00528]
Singh, D., Bradley, S. J., & Zucker, K. J. (2021). A Follow-Up Study of Boys With Gender Identity Disorder. Frontiers in Psychiatry, 12, 632784. [DOI:10.3389/fpsyt.2021.632784]
Southmayd, E. A., & De Souza, M. J. (2017). A summary of the influence of exogenous estrogen administration across the lifespan on the GH/IGF-1 axis and implications for bone health. Growth Hormone & IGF Research, 32, 2–13. [DOI:10.1016/j.ghir.2016.09.001]
Staphorsius, A. S., Kreukels, B. P., Cohen-Kettenis, P. T., Veltman, D. J., Burke, S. M., Schagen, S. E., Wouters, F. M., Delemarre-van de Waal, H. A., & Bakker, J. (2015). Puberty suppression and executive functioning: An fMRI-study in adolescents with gender dysphoria. Psychoneuroendocrinology, 56, 190–199. [DOI:10.1016/j.psyneuen.2015.03.007]
Steensma, T. D., Biemond, R., de Boer, F., & Cohen-Kettenis, P. T. (2011). Desisting and persisting gender dysphoria after childhood: a qualitative follow-up study. Clinical Child Psychology and Psychiatry, 16(4), 499–516. [DOI:10.1177/1359104510378303]
Steensma, T. D., McGuire, J. K., Kreukels, B. P., Beekman, A. J., & Cohen-Kettenis, P. T. (2013). Factors associated with desistence and persistence of childhood gender dysphoria: a quantitative follow-up study. Journal of the American Academy of Child and Adolescent Psychiatry, 52(6), 582–590. [DOI:10.1016/j.jaac.2013.03.016]
Steensma, T. D., & Cohen-Kettenis, P. T. (2018). A critical commentary on “A critical commentary on follow-up studies and “desistence” theories about transgender and gender non-conforming children”. International Journal of Transgenderism, 19(2), 225–230. [DOI:10.1080/15532739.2018.1468292]
Swerdloff, R. S., & Heber, D. (1983). Superactive gonadotropin-releasing hormone agonists. Annual Review of Medicine, 34, 491–500. [DOI:10.1146/annurev.me.34.020183.002423]
Temple Newhook, J., Pyne, J., Winters, K., Feder, S., Holmes, C., Tosh, J., Sinnott, M.-L., Jamieson, A., & Pickett, S. (2018). A critical commentary on follow-up studies and “desistance” theories about transgender and gender-nonconforming children. International Journal of Transgenderism, 19(2), 212–224. [DOI:10.1080/15532739.2018.1456390]
Tollit, M. A., Pace, C. C., Telfer, M., Hoq, M., Bryson, J., Fulkoski, N., Cooper, C., & Pang, K. C. (2019). What are the health outcomes of trans and gender diverse young people in Australia? Study protocol for the Trans20 longitudinal cohort study. BMJ Open, 9(11), e032151. [DOI:10.1136/bmjopen-2019-032151]
Turban, J. L., King, D., Carswell, J. M., & Keuroghlian, A. S. (2020). Pubertal Suppression for Transgender Youth and Risk of Suicidal Ideation. Pediatrics, 145(2), e20191725. [DOI:10.1542/peds.2019-1725]
van der Miesen, A., Steensma, T. D., de Vries, A., Bos, H., & Popma, A. (2020). Psychological Functioning in Transgender Adolescents Before and After Gender-Affirmative Care Compared With Cisgender General Population Peers. The Journal of Adolescent Health, 66(6), 699–704. [DOI:10.1016/j.jadohealth.2019.12.018]
Verhulst, F. C., Achenbach, T. M., van der Ende, J., Erol, N., Lambert, M. C., Leung, P. W., Silva, M. A., Zilber, N., & Zubrick, S. R. (2003). Comparisons of problems reported by youths from seven countries. The American Journal of Psychiatry, 160(8), 1479–1485. [DOI:10.1176/appi.ajp.160.8.1479]
Vlot, M. C., Klink, D. T., den Heijer, M., Blankenstein, M. A., Rotteveel, J., & Heijboer, A. C. (2017). Effect of pubertal suppression and cross-sex hormone therapy on bone turnover markers and bone mineral apparent density (BMAD) in transgender adolescents. Bone, 95, 11–19. [DOI:10.1016/j.bone.2016.11.008]
Vrouenraets, L. J., Fredriks, A. M., Hannema, S. E., Cohen-Kettenis, P. T., & de Vries, M. C. (2016). Perceptions of Sex, Gender, and Puberty Suppression: A Qualitative Analysis of Transgender Youth. Archives of Sexual Behavior, 45(7), 1697–1703. [DOI:10.1007/s10508-016-0764-9]
Wallien, M. S., & Cohen-Kettenis, P. T. (2008). Psychosexual outcome of gender-dysphoric children. Journal of the American Academy of Child and Adolescent Psychiatry, 47(12), 1413–1423. [DOI:10.1097/CHI.0b013e31818956b9]
Wiepjes, C. M., Nota, N. M., de Blok, C., Klaver, M., de Vries, A., Wensing-Kruger, S. A., de Jongh, R. T., Bouman, M. B., Steensma, T. D., Cohen-Kettenis, P., Gooren, L., Kreukels, B., & den Heijer, M. (2018). The Amsterdam Cohort of Gender Dysphoria Study (1972–2015): Trends in Prevalence, Treatment, and Regrets. The Journal of Sexual Medicine, 15(4), 582–590. [DOI:10.1016/j.jsxm.2018.01.016]
Wojniusz, S., Callens, N., Sütterlin, S., Andersson, S., De Schepper, J., Gies, I., Vanbesien, J., De Waele, K., Van Aken, S., Craen, M., Vögele, C., Cools, M., & Haraldsen, I. R. (2016). Cognitive, Emotional, and Psychosocial Functioning of Girls Treated with Pharmacological Puberty Blockage for Idiopathic Central Precocious Puberty. Frontiers in Psychology, 7, 1053. [DOI:10.3389/fpsyg.2016.01053]
Zaiem, F., Alahdab, F., Al Nofal, A., Murad, M. H., & Javed, A. (2017). Oral Versus Transdermal Estrogen In Turner Syndrome: A Systematic Review And Meta-Analysis. Endocrine Practice, 23(4), 408–421. [DOI:10.4158/ep161622.or]
Zuger, B. (1978). Effeminate behavior present in boys from childhood: ten additional years of follow-up. Comprehensive Psychiatry, 19(4), 363–369. [DOI:10.1016/0010-440x(78)90019-6]
\ No newline at end of file
+Puberty Blockers: A Review of GnRH Analogues in Transgender Youth - Transfeminine Science
Puberty Blockers: A Review of GnRH Analogues in Transgender Youth
By Mitzi | First published January 30, 2022 | Last modified January 31, 2022
Abstract / TL;DR
Puberty blockers are medications used to pause puberty in both cisgender and transgender youth. For the latter, significant evidence suggests that they improve well-being, psychological functioning, and risk of suicidality, both during puberty and in later life. Their effects are reversible upon discontinuation. Current evidence does not suggest any negative impact on cognitive development, IQ, or fertility. A minor impact on bone density may exist, affecting primarily transgender girls, but little high quality data is available. Based on limited data, prescribers may wish to consider calcium supplementation in transgender teens receiving puberty blockers, and may wish to prefer transdermal delivery over oral estrogens in transgender girls starting hormone therapy in order to optimise bone density outcomes. There is a lack of evidence supporting the common belief that most children grow out of gender dysphoria (“desistance”), as widely cited data describing the rate at which this happens appears highly unreliable. Puberty blockers are difficult to access, and many Western countries have sharply restricted their use recently, in a trend condemned by numerous medical associations. Randomised controlled trials on puberty blockers can likely never be performed, but nonetheless, there is clear evidence they offer significant benefit, and have relatively minor risks.
Introduction
Puberty blockers, also known as gonadotropin-releasing hormone (GnRH) analogues, were introduced for medical use in the 1980s (Swerdloff & Heber, 1983). Originally developed to supersede other therapies in the treatment of prostate cancer, they were soon adapted for paediatric use, revolutionising the treatment of precocious puberty: a rare condition in which puberty begins before the age of 8 (in natal girls) or 9 (in natal boys). Precocious puberty is associated with several negative consequences, such as short stature, teasing, bullying, and worse mental health outcomes. By reversibly pausing puberty for several years in children with this condition, outcomes are often significantly improved, and puberty blockers remain the mainstay treatment for this condition several decades later.
In the 1990s, puberty blockers began to be used in transgender adolescents, as a way of pausing their unwanted puberty, and giving them more time to consider their future (Cohen-Kettenis & van Goozen, 1998). The protocol for this, originally develped by the Dutch VUmc clinic, has sometimes been referred to as the “Dutch Method.” Cohen-Kettenis et al. (2011) published a study following one such Dutch patient 22 years later. Since then, the use of puberty blockers has increased tremendously with the increase in patients seeking transgender healthcare.
Recently, puberty blockers have been the subject of controversy, with legal proceedings seeking to prohibit their use across several countries. Notably, their use was temporarily stopped in the United Kingdom in December 2020 following a ruling in the Bell v. Tavistock case, which was appealed in 2021. Also in 2021, Arkansas became the first U.S. state to make it illegal for doctors to prescribe puberty blockers, with several other states pursuing similar legislation. Critics express concern about the safety of puberty blockers, their reversibility, and effectiveness.
This article seeks to review the literature on the use and safety of puberty blockers in transgender youth, examining their safety, and arguments for and against their use in a comprehensive way. While rarely, alternative medications like the progestin medroxyprogesterone acetate have been used for this, this article mainly focuses on GnRH agonists: by far the most widely used class of medication for puberty blockade, and what’s most commonly colloquially referred to as “puberty blockers.”
Mechanism of Action
GnRH is a naturally occurring hormone in humans responsible for the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the pituitary gland. Through this mechanism, the body produces its gonadal estrogen and testosterone. GnRH agonists bind to the GnRH receptor and activate it, causing it to be continuously stimulated. This causes an initial increase of LH and FSH, then over the course of several weeks, causes the pituitary gland to become desensitised, pausing the natural sex hormone production for the duration the medication is taken. When the medication is stopped, its effect is reversed, with normal sex hormone production resuming about a week after the medication clears the body (Cedrin-Durnerin et al., 2000).
GnRH agonists are prescribed as an injection administered every one to six months, a surgically implanted pellet once per year, or a nasal spray administered two to three times per day. A short-acting daily injection exists, but is not used for puberty blockade in clinical practise. Common examples of GnRH agonists include leuprorelin (Lupron; Eligard), triptorelin (Decapeptyl), goserelin (Zoladex), histrelin (Supprelin LA), nafarelin (Synarel) and buserelin (Suprefact).
Like GnRH agonists, GnRH antagonists bind to the GnRH receptor, however, they do not stimulate it. Instead, they compete with the body’s own GnRH, rendering it ineffective. As a result, they achieve similar effects without causing an initial increase in hormone levels. Also unlike GnRH agonists, oral formulations of GnRH antagonists exist, allowing some of them to be taken as a daily pill. Common examples include elagolix (Orilissa), degarelix (Firmagon), cetrorelix (Cetrotide), ganirelix (Orgalutran; Antagon) and relugolix (Orgovyx; Relumina). Unfortunately, being much newer drugs, GnRH antagonists are not normally used as puberty blockers at the moment.
In gender dysphoric youth, GnRH agonists are prescribed after the onset of puberty. GnRH agonists are not prescribed to children who have not yet started puberty, but may be started at any point during puberty to pause further changes (Hembree et al., 2017).
Quality of Evidence
In medicine, the gold standard for evidence is the randomised controlled trial, or RCT. In a nutshell, participants are randomly assigned into two or more treatment groups (arms), such that the only difference between arms is the treatment they receive. Commonly, one group receives a placebo, while another receives the treatment being studied. The ideal RCT is blinded, meaning neither participants nor investigators of the study know which group is receiving which treatment. No such trials have been performed with puberty blockers, giving rise to concerns that there could be insufficient evidence available for their use.
Unfortunately, RCTs may not be practically possible for puberty blockers, and are unlikely to ever be performed. A good summary of the reasons for this is provided by Giordano & Holm (2020):
There are two main practical problems that preclude conducting a RCT.
First, patients who approach clinics for help because of distress caused by the first signs of puberty will be unlikely to accept to be a part of a RCT. Medications are needed within a relatively short period of time, at pain of treatment being less effective or ineffective. Recruitment would thus be hard if not impossible.
Second, the ideal RCT is either double blind, i.e. neither researchers nor participants know who gets the active drug, or it assesses outcomes using blinded observers when treatment allocation cannot be hidden from participants. Blinding is necessary in order to reduce bias in outcome assessments. But, a RCT of puberty delay could not maintain blinding. Because GnRHa are effective in delaying puberty it would soon become evident to participants, researchers and outcome assessors who was in the active treatment arm and who was not. This breakdown of blinding would mean that there would be potential bias in the outcome assessments, both in relation to biological and psychological outcomes. It would also mean that participants allocated to the non-treatment arm of the study would be likely to either withdraw from the study at a much higher rate than in the treatment arm introducing potential bias, and/or be more likely not to adhere to the trial but seek puberty delaying treatment outside of the trial thereby adding a confounder.
Mul et al. (2001) ran into this problem conducting a similar study on teens with precocious puberty:
In the original study design a third arm with untreated children was scheduled as a control group. It was decided to omit this control group from the study design after it appeared that the parents of all patients who were randomized in the untreated control group refused further participation in the study as GnRHa treatment could be obtained elsewhere.
Besides practical limitations, such RCTs are likely to be unethical. Evidence suggests withholding puberty blockers may cause lasting harm in itself. To knowingly cause such harm to the control group of an RCT is likely to be morally unacceptable, and such an RCT would be unlikely to receive approval from an ethical review board.
This is not to say that studies evaluating such outcomes don’t exist at all: for example, while not randomised or blinded, Costa et al. (2015) compares 101 patients receiving psychological support and puberty blockers to 100 patients receiving psychological support alone. The results of this study are further outlined below.
As a result of these limitations, this article mainly cites cohort studies, making the argument that sufficient other high-quality studies exist to reach well-supported conclusions: a practise sometimes required in other areas of medicine as well. Because this is the only way we can practically evaluate puberty blockers and RCTs are likely impossible, it seems disingenuous to make the claim that lack of RCTs equate to lack of evidence around puberty blockers, as this standard of evidence can never be met, and the claim ignores a substantial existing body of literature.
Suicidality and Well-being
A significant body of evidence associates the use of puberty blockers in those who want such treatment with improved psychological well-being: the primary argument for their use.
While different studies use different methodologies, three standardised psychological questionnaires are typically used to evaluate well-being: the Children’s Global Assessment Scale (CGAS), the Child Behavior Checklist (CBCL), and the Youth Self-Report (YSR). All three are aimed at evaluating psychological functioning and problematic behaviour: typically, the CGAS is administered by a clinician, the CBCL is filled out by a parent or guardian, and the YSR is filled out by a child themselves. It’s important to note that scores in these assessments are known to markedly worsen in adolescence in general, with the onset of psychological difficulties and self-harm often appearing during puberty (Verhuist et al., 2003; Nock et al., 2013; Morey et al., 2017; Jung et al., 2018).
One of the largest studies to investigate well-being to date has been Turban et al. (2020). It surveyed 20,619 American transgender adults. 3,494 (16.9%) reported that they ever wanted to receive puberty blockers. Of those, only 89 received them. In total, 75.3% of those who received puberty blockers reported ever experiencing suicidal thoughts, compared to 90.2% of those who did not. After controlling for demographic variables like income, family support, and education level, puberty blockers remained significantly associated with decreased odds of lifetime suicidal ideation.
A similarly large survey by Green et al. (2021) included 11,914 Americans aged 13–24 who identified as transgender or nonbinary. The study compares those who received hormone therapy or puberty blockers to those who wished to receive them, but didn’t. It finds that in those who received treatment, rates of depression, suicidal ideation, and suicide attempts were significantly lower. This remained true of those aged 13–17, who were significantly more likely to receive puberty blockers specifically.
Costa et al. (2015) studied 201 gender dysphoric adolescents who presented at the British Tavistock and Portman NHS Gender Identity Development Service. Of them, half were considered eligible for puberty blockers immediately, receiving them in addition to psychological support. The other half were not considered immediately eligible for puberty blockers, citing reasons such as psychiatric problems or conflicts with parents and siblings. These patients received only psychological support for the following 18 months. All patients’ global psychological functioning was assessed using the CGAS questionnaire. Both groups showed significantly improved psychological functioning with psychological support, but the group receiving only psychological support stalled and showed no further improvement towards the end of the study, while those receiving puberty blockers continued to show greater improvement. The authors point out that the eventual CGAS score of the group receiving puberty blockers coincided almost perfectly with those found in a sample of children/adolescents without observed psychological/psychiatric symptoms.
Figure 1: CGAS scores of psychological functioning in transgender teens receiving puberty blockers and psychological support, compared to those receiving psychological support alone in Costa et al. (2015).
A later study at the same British gender identity clinic, Carmichael et al. (2021), received widespread media coverage in the United Kingdom following its mixed findings. It followed 44 gender dysphoric adolescents who received puberty blockers. CGAS scores were higher than the 2015 study at baseline, and showed slower and more modest improvement. The study reached contradictory conclusions, with improvements reported in some questionnaires, but not others, even for comparable measurements. Interestingly, in some of the researchers’ measures of well-being, social acceptance, and self-perception, adolescents themselves reported significant improvements, while their parents reported almost no improvement. The study characterises participants’ overall experiences with puberty blockers as positive, but is difficult to draw any conclusions from.
De Vries et al. (2011) and de Vries et al. (2014) investigate the psychological outcomes of the same cohort of transgender adolescents receiving puberty blockers at the VUmc gender clinic in the Netherlands. Both investigate psychological outcomes in a range of tests, with the 2014 study providing long-term follow-up many years after puberty blockers, and after gender reassignment surgery. In the studied cohort, psychological functioning improves and depression decreases over time, as evidenced by standardised tests, including CGAS scores. Significant improvements in well-being are reported both during treatment with puberty blockers, and in the years after, with hormone therapy and surgery. Unlike Carmichael et al., CBCL and YSR scores improve.
Van der Miesen et al. (2020) charts psychological well-being across 3 groups of Dutch adolescents: 272 transgender adolescents who haven’t yet received puberty blockers, 178 adolescents receiving puberty blockers, and 651 cisgender adolescents from the general population. The study finds poorer psychological functioning in those before treatment, while psychological functioning and well-being is similar to cisgender adolescents in those receiving pubertal suppression. These findings are in line with Costa et al. (2015), which noted that those receiving puberty blockers reached CGAS scores comparable to the general (age-matched) population.
Figure 2: Percentages of teens who report suicidality in van der Miesen et al. (2020). Suicidality was defined as endorsing the statement “I deliberately try to hurt or kill myself” or “I think about killing myself.” Suicidality among Dutch transgender youth has not significantly changed over time, making cohort differences unlikely (Arnoldussen et al., 2020).
In addition to the studies listed above, several smaller, less focused studies have also assessed the well-being of transgender adolescents receiving puberty blockers and reported overall positive experiences (Khatchadourian et al., 2014; Achille et al., 2020; Kuper et al., 2020). No studies report a decline in psychological functioning or notably negative psychological outcomes with the use of puberty blockers.
In combination, this strongly suggests that puberty blockers improve well-being and psychological functioning in children who experience gender dysphoria. In addition, it suggests that inappropriately withholding them may lead to worse later-life outcomes, such as increased suicidality.
Counterintuitively, several of the studies listed do note that puberty blockers don’t reduce gender dysphoria (de Vries et al., 2011; Carmichael et al., 2021). It’s important to be aware that this finding refers to the wish to transition, rather than psychological well-being. The finding is based on the Utrecht Gender Dysphoria Scale questionnaire (de Vries et al., 2006). To illustrate, the version for transmasculine youth asks patients whether they endorse such statements as “I prefer to behave like a boy”, “I wish I had been born as a boy”, “I hate having breasts”, and “every time someone treats me like a girl, I feel hurt.”
When studies note that puberty blockers don’t reduce gender dysphoria, this means children don’t stop identifying as transgender after receiving puberty blockers. They continue to want to transition. De Vries et al. (2011) points out this is the expected outcome:
As expected, puberty suppression did not result in an amelioration of gender dysphoria. Previous studies have shown that only gender reassignment consisting of cross-sex hormone treatment and surgery may end the actual gender dysphoria. None of the gender dysphoric adolescents in this study renounced their wish for gender reassignment during puberty suppression. This finding supports earlier studies showing that young adolescents who had been carefully diagnosed show persisting gender dysphoria into late adolescence or young adulthood
Fertility
Unlike hormone therapy, no risk of permanent infertility is believed to exist with the use of puberty blockers. Several long-term follow-up studies of patients treated with puberty blockers have found normal fertility. Among others, Feuillan et al. (1999), Heger et al. (1999), Heger et al. (2006) and Lazar et al. (2014) find no indication of impaired fertility in patients treated with puberty blockers for precocious puberty. In the years and decades following their treatments, the several hundred patients in these studies are found to conceive normally without an increased need for assisted reproductive technology, and with uneventful pregnancies. Despite several decades of use, no reports exist in literature of permanent infertility linked to puberty blockers. Interestingly, transgender populations do have higher rates of sperm abnormalities than cisgender populations, before any medical treatment has taken place (Li et al., 2018; Rodriguez-Wallberg et al., 2021).
In contrast, hormone therapy may cause permanent infertility (Hembree et al., 2017; Cheng et al., 2019). If fertility preservation has not been accessed before beginning treatment, puberty blockers must be stopped to do so, ideally before hormone therapy begins. When puberty blockers are stopped, unwanted sex characteristics continue to develop. Transgender people may find this extremely distressing, which may be one reason for them to not pursue fertility preservation.
No data exists on the exact length of time for which puberty blockers need to be stopped before full fertility is restored, and it likely varies depending on the age puberty blockers were initiated. Bertelloni et al. (2000) found spermarche took place between 0.7 to 3 years after discontinuation of puberty blockers in boys treated for precocious puberty. Barnard et al. (2019) report on the case of a single transgender patient who had been receiving puberty blockers for 6 months, from the age of 17. Three months after the last dose of monthly leuprorelin, no viable sperm sample could be produced. Five months after, their sample was viable.
Regardless of this, transgender individuals are extremely unlikely to use fertility preservation, with some estimates suggesting utilisation rates below 5% in North America (Chen et al., 2017; Nahata et al., 2017). In one piece of research, Pang et al. (2020) questioned 102 transgender Australian teens on their reasons for declining fertility preservation. The following statistics were gathered:
Figure 3: Australian transgender teens’ reasons for declining fertility preservation in Pang et al. (2020).
As such, there is no evidence-based reason to believe puberty blockers could cause infertility, with fertility returning when they are discontinued. However, due to low discontinuation rates for puberty blockers and low fertility preservation rates, those who start puberty blockers and persist are unlikely to have biological children. Clinical guidelines recommend that adolescents seeking puberty blockers should be counselled on options for fertility preservation, and parents should be involved in this (Hembree et al., 2017).
Bone Density
Bone density is a measure of the amount of bone mineral present in bone tissue. Bone density is measured using imaging techniques, such as DEXA scans: a type of X-ray. It is used to predict patients’ risk of breaking bones. The clinical terms for low bone density are osteopenia, and in more severe cases, osteoporosis, which is common among the elderly. For the purposes of this review, the most relevant measurement of bone density is the z-score, which expresses a patient’s bone density in comparison to other people of the same age and sex. A z-score of 0 indicates bone density equal to the general population. Small deviations, such as -0.2, may not always be relevant, but z-scores below -1 may be cause for concern.
There are concerns around the effects of puberty blockers on bone health. Puberty is a critical time for the accrual of bone density: a process largely driven by sex hormones. This process is delayed in those receiving puberty blockers, leading to temporary lower bone density and z-scores compared to peers going through puberty normally. While these short-term z-scores are not particularly relevant, long-term outcomes are very important: the question becomes what z-scores look like in the long term, into adulthood, and whether the use of puberty blockers has any impact on later-life fracture risk. Literature on this is uncertain.
Klink et al. (2015) finds that in both trans girls and trans boys, z-scores are lower both before treatment, and after long-term follow-up. The study suggests a small negative effect on final bone density from the use of puberty blockers, although many measurements fail to reach statistical significance. The study records notably lower final z-scores for trans girls than trans boys.
Vlot et al. (2017) finds that 2 years after beginning hormone therapy, z-scores were returning towards normal. In trans boys, final z-scores were negligibly lower, while in trans girls, the effect was much more pronounced, with meaningfully lower z-scores both before and after treatment.
In line with this, Schagen et al. (2020) finds that in its cohort, final z-scores normalised after 3 years of hormone therapy for trans boys, while they remained meaningfully low both before and after treatment for trans girls.
Figure 4: Bone Mineral Apparent Density (BMAD) of the lumbar spine across multiple studies, relative to sex assigned at birth. Three measurements are taken in each study: the initiation of puberty blockers, the initiation of hormone therapy, and one measurement after several years of hormone therapy. A z-score below -1 is commonly considered to be clinically relevant osteopenia, while a score below -2.5 is considered to be osteoporosis. The figure illustrates that trans girls tend to have significantly lower bone density before, during, and after treatment, while this is not the case for trans boys. Trans girls also tend to receive puberty blockers for a longer time.
Guaraldi et al. (2016) find in their literature review that in those receiving puberty blockers for precocious puberty, bone mineral density is lower than that of untreated peers during treatment with puberty blockers, then typically recovers when puberty is initiated, with long-term follow-up showing little difference to the general population. Combined with the results of trans boys, this suggests that not puberty blockers themselves, but rather, subsequent suboptimal hormone therapy in trans girls could potentially be to blame for their more pronounced negative outcomes.
The hormone therapy prescribed to trans girls in the listed studies may be suboptimal in several ways. To begin with, all three use very low adult maintenance dosages of no more than 2 mg oral estradiol in transfeminine patients. Such a dose is likely to produce serum estradiol levels of roughly 50 pg/ml on average: below the average estradiol exposure of cis women (Aly, 2018; Aly, 2020). Many clinical guidelines recommend higher levels, which some research suggests could have a small positive effect on final bone density compared to lower dosages (Roux, 1997; Riggs et al., 2012). Indeed, the authors of all three studies themselves note their doses were low and may have been inadequate for optimal bone density.
As a final confounding factor, none of the studies control for lifestyle factors associated with lower bone density, such as exercise, smoking, vitamin D, and calcium intake. These factors have a significant effect on bone density. Transgender people are more likely to smoke, less likely to exercise, and less likely to consume adequate calcium, both as teens and as adults (Jones et al., 2018; Kidd, Dolezal & Bockting, 2018). This is believed to be the reason transgender people of all ages tend to have lower bone density before any treatment is initiated. Without controlling for these factors, which may distort the available data significantly, it’s difficult to draw confident conclusions from these studies, and a causal link between the use of puberty blockers and lower final bone density remains unproven. If such a link does exist, the effect seems unlikely to be dramatic, and unlikely to outweigh the benefits of puberty blockers.
In a noteworthy study, Antoniazzi et al. (2003) report that in those receiving puberty blockers for precocious puberty, bone mineral density is better preserved through calcium supplementation. Calcium intake is often inadequate in transgender youth (Lee et al., 2020), and therefore warrants further study for improving their bone mineral density. Alongside calcium, lifestyle interventions, the use of transdermal instead of oral estrogens, and the avoidance of subphysiological adult dosages of estradiol could all potentially improve bone-related outcomes over current clinical practise.
IQ and Cognitive Development
One possible concern is the impact of puberty blockers on IQ and cognitive development. Very little research on the subject exists, with commonly cited critical studies investigating sheep rather than humans (Hough et al., 2017), or being case studies of a single patient (Schneider et al., 2017). Only two larger studies investigate this:
Staphorsius et al. (2015), the only study to investigate this in a transgender population, evaluated performance in the standardised Tower of London test, as well as IQ scores. The study found no significant differences in executive functioning between the two groups. IQ was slightly lower in transgender girls receiving puberty suppression than the control group, but the same was not true in a statistically relevant way of transgender boys. Age differences, lifestyle factors, and a very low sample size may all explain these differences.
Wojniusz et al. (2016) assessed 15 girls suffering from precocious puberty and treated with a puberty blocker. The 15 girls were compared with 15 age-matched controls. Both groups showed similar IQ scores.
Neither study has very many participants, records baseline cognitive performance, or controls for confounding factors. As such, very few conclusions can be drawn from them. Decades of clinical experience with the use of puberty blockers in children suggests it’s unlikely any particularly dramatic effect on IQ exists, but without much larger, higher quality studies, no conclusion on this can be reached, and further research is needed.
Desistance
Discontinuation rates for patients on puberty blockers are very low, with fewer than 5% of teens typically stopping them without going on to hormone therapy (Wiepjes et al., 2018; Brik et al., 2020; Kuper et al., 2020). A potential concern is that this could mean puberty blockers put children on an almost guaranteed path towards gender transition, when they might otherwise change their minds.
Surprisingly, while a commonly held belief suggests most gender dysphoric children will grow out of it without treatment at a later age, little convincing evidence supports this claim. While existing studies report desistance rates ranging from 43% (Wallien & Cohen-Kettenis, 2008) to 88% (Drummond et al., 2008), they often contain significant methodological issues.
Historically, in the 20th century, a transgender identity was viewed as a negative outcome: it was something for a patient to be cured of, for example through aversion therapy. Since then, a cultural shift towards transgender people has taken place. Older studies into desistance rates are often reflective of this. As an example, Kosky (1987) describes eight boys who were hospitalised in a psychiatric unit for displaying effeminate behaviour and cross-dressing, where they received intensive treatment aimed at curing them. Today, these behaviours are more accepted, and they are not necessarily viewed as the same thing as a transgender gender identity. Clearly, a study like this cannot be used to estimate the desistance rates of today’s gender dysphoric children.
Other studies describing the 1960s through 1980s are similar (Bakwin, 1968; Lebovitz, 1972; Zuger, 1978; Money & Russo, 1979; Davenport, 1986). Many predate the DSM-III, and thus the existence of formal diagnostic criteria. Few studied self-reported gender identity: instead, they tend to study gender non-conforming behaviour, such as cross-dressing, that doesn’t necessarily constitute a transgender identity. Many of them try to discourage patients as a core part of their treatment, sometimes in ways that are now banned across much of the world as conversion therapy. Combined with a drastically changed society, extrapolating modern transgender desistance rates from these studies is unreasonable.
A small number of more recent studies do exist. The highest desistance rates found in modern literature are approximately 88%, reported by three frequently cited Canadian studies: Drummond et al. (2008), Drummond et al. (2018), and Singh, Bradley & Zucker (2021). Unfortunately, these studies appear to be at a high risk of bias: calling their credibility into question, the clinic in which they took place was closed in 2015, amidst allegations of conversion therapy. An independent review found that it “cannot state that the clinic does not practice reparative approaches.” In the review, many children and their parents report feeling the clinic was invasive and intimidating to them. Some instances include:
Assessments are described as intrusive and even traumatic by some, who described feeling “poked and prodded”. One way mirror and multiple observers create discomfort. Many questions were felt to be irrelevant, unnecessarily intrusive (particularly those regarding sexual fantasies), especially when asked without context, rationale, and what seems to be inadequate or even absent informed consent. Also, it is unclear whether any potential benefit of this line of questioning to the patient was explained. Parents of younger clients report their child appearing to be and later reporting feeling they were very uncomfortable with the way they were asked about their gender variance “as if my child was not okay as a person.” One parent described feeling “dismissed” when she spoke to clinicians about this.
Patients reported feeling intimidated to question Dr. Zucker regarding their concerns and were not offered the opportunity to decline. Multiple informants commented on this.
Chart documentation revealed statements reflecting that the diversity of gender expression and variance are not accepted equally. One example is of a child for whom all gender and body dysphoria had resolved and multiple informants indicated sustained good mood and satisfaction with social and academic functioning. Despite this, the parents of the child were advised at discharge to encourage the child to spend more time with cisgendered boys because he had effeminate speech and mannerisms. These were not goals of the client or family.
This may explain why these studies find a much higher desistance rate than other modern literature, and makes them very unlikely to be representative figures. As an alternative possibility, Steensma & Cohen-Kettenis (2018) suggest that differences in the local social climate regarding gender variance may have also been an important contributing factor.
Steensma et al. (2011) and Steensma et al. (2013) set out to investigate factors that could contribute to the persistence or desistance of gender dysphoria in children. The 2011 study reports a desistance rate of 45%, while the 2013 study reports 73%. The figures have been criticised because all children lost to follow-up are assumed to have desisted, which may or may not have inflated their number. More importantly however, in Steensma & Cohen-Kettenis (2018), the authors themselves argue they’ve been cited out of context, and their figures can’t be used to extrapolate desistance rates:
Unlike what is suggested, we have not studied the gender identities of the children. Instead we have studied the persistence and desistence of children’s distress caused by the gender incongruence they experience to the point that they seek clinical assistance. […] Using the term desistence in this way does not imply anything about the identity of the desisters. The children could still be hesitating, searching, fluctuating, or exploring with regard to their gender experience and expression, and trying to figure out how they wanted to live. Apparently, they no longer desired some form of gender-affirming treatment at that point in their lives.
Again, because of the purpose and the design of this study we did not report prevalence numbers in the sample under study. Furthermore, the sample in the 2013 study did not include children in the younger age spectrum of the referred population to the Amsterdam clinic. Reporting prevalence of persistence and/or desistance in this sample would therefore not be reliable.
The only other modern study into persistence rates has been Wallien & Cohen-Kettenis (2008). The study appears to be of higher quality and provides the most convincing estimate available: a 27% persistence rate and a 43% desistance rate over the course of (on average) 10 years. The remaining 30% of participants were lost to follow-up.
Several further problems cast doubt on the data presented in all of these studies, including Wallien & Cohen-Kettenis (2008). Firstly: children are diagnosed using DSM-III and DSM-IV criteria, which are dated by today’s standards. In these older criteria, gender identity was not a diagnostic requirement: a child could be diagnosed with a gender identity disorder for a range of gender non-conforming behaviours, without themselves identifying as a different gender, or experiencing distress with their gender role or sex characteristics (Temple Newhook et al., 2018).
Strikingly, with the exception of Steensma et al. (2011), all studies include a significant number of children who never actually met then-current DSM diagnostic criteria for Gender Identity Disorder: in the case of Wallien & Cohen-Kettenis (2008), a quarter of all participants. These participants have been assumed to be transgender for the purposes of extrapolating desistance rates, but held a diagnosis of Gender Identity Disorder Not Otherwise Specified: a broad category described by the DSM as representing those who may not necessarily seek medical transition, but may transiently cross-dress, be preoccupied with castration, or be intersex and experience gender dysphoria. These participants’ exact circumstances are not described by the researchers, but both Wallien & Cohen-Kettenis and Steensma et al. report in their studies that all, or nearly all persisters met DSM diagnostic criteria, while only about half of desisters did.
Outside of this, there is a lack of long-term follow-up. A gender dysphoric child might desist in transitioning during their teens, but go on to transition in adulthood, for example because of peer pressure or lack of parental acceptance. Whether this happens at any significant rate has not been studied.
The studies do suggest that for an unknown percentage of children, gender dysphoria will resolve over time, but the high desistance rates often cited as an established fact don’t appear to be supported by evidence. Concerns that puberty blockers cause children to transition when they otherwise would’ve aged out of gender dysphoria appear misplaced: children whose dysphoria persisted were much more likely to have met the diagnostic criteria to receive puberty blockers.
Current literature on this will likely soon be superseded by higher-quality data, with several very large, well-funded studies into gender dysphoric youth now underway the United States (Olson-Kennedy et al., 2019), Australia (Tollit et al., 2019) and the United Kingdom (Kennedy et al., 2019).
Desistance and persistence rates can suggest a binary view, and should be seen in a greater context. Because children’s needs change over time, a hypothetical child might feel uncertain about their gender, possibly even receive puberty blockers, and then later decide they do not wish to transition. In such a case, puberty blockers may have met their needs at the time, and were not automatically harmful or regrettable, particularly due to their reversible nature. Neither being transgender, nor being cisgender should be seen as a negative outcome. In their critical commentary, Temple Newhook et al. (2018) write that it is important to respect children’s wishes and autonomy, and move away from the question of, “How should children’s gender identities develop over time?” toward a more useful question: “How should children best be supported as their gender identity develops?”
Regret
In light of persistence and desistance rates, it makes sense to ask how patients themselves feel about their treatment with puberty blockers, and whether they regret receiving them. Limited research exists on the subject:
A large retrospective review of the medical files of all 6,793 patients treated at the Dutch VUmc clinic between 1972 and 2015 found that 14 patients (0.2%) regretted their treatment in total. This included patients who received puberty suppression, hormone therapy, and/or surgery. Notably, 5 of them regretted their treatment because of a lack of social acceptance (Wiepjes et al., 2018).
De Vries et al. (2014), found none of the 55 transgender patients they followed regretted receiving puberty blockers, hormone therapy, or surgery. Psychological well-being continued to improve in their cohort, both with puberty blockers, hormone therapy, and later gender reassignment surgery.
Vrouenraets et al. (2016) interviewed 13 adolescents who had been seen at a Dutch gender identity clinic, twelve of whom had received puberty blockers. Asked about long-term risks, most responded that they were significantly outweighed by puberty blockers allowing them to live a more happy life. Quotes from the interviewed children in the study include:
The possible long-term consequences are incomparable with the unhappy feeling that you have and will keep having if you don’t receive treatment with puberty suppression. (trans boy; age: 18)
It isn’t a choice, even though a lot of people think that. Well, actually it is a choice: living a happy life or living an unhappy life. (trans girl; age: 14)
They also comment on the increasing attention to transgender people in media, with one child saying:
Thanks to media coverage I learned that gender dysphoria exists; that someone can have these feelings and that you can get treatment for it. Beforehand I thought I was the only one like this. (trans boy; age: 18)
Ease of Access
While large geographic differences exist, on the whole, access to puberty blockers is often difficult.
In the United States, Turban et al. (2020) found that access to puberty blockers was associated with a greater household income, noting that the annual cost of them ranges from $4,000 to $25,000 and insurance coverage was unavailable to many. It also found that transgender teens were less likely to receive puberty blockers if they did not identify as heterosexual or binary. Of those who received puberty blockers, 60% reported traveling <25 miles for gender-affirming care, 29% travelled between 25 and 100 miles, and 11% travelled >100 miles. As of 2021, several states are pursuing regulation banning the use of puberty blockers, with Arkansas having become the first to pass such a law. Several large professional bodies representing thousands of medical experts have condemned this type of regulation (American Academy of Child and Adolescent Psychiatry, 2019; American Medical Association, 2021; Endocrine Society, 2021).
In the United Kingdom, waiting lists as long as 4 years or more exist for initial intake appointments for puberty blockers. Legislative changes in light of Bell v. Tavistock complicated access dramatically: in the nine months between the ruling and its reversal, no under-17s received puberty blockers under the public healthcare sytem, and reports described the care of adolescents over 16, who were not affected by the judgement, being discontinued as well. Restrictions in light of Bell v. Tavistock were condemned by WPATH, EPATH, USPATH, AsiaPATH, CPATH, AusPATH, and PATHA, the leading medical associations for transgender health, who released a statement saying they believe it will cause significant harm to the affected patients (WPATH, 2020), as well as Amnesty International UK and Liberty (Amnesty International UK, 2020).
In Finland, new prescriber guidelines for treating gender dysphoric teens were released in 2020 (Society for Evidence Based Medicine, 2021). They broke with WPATH guidelines, instead recommending that gender dysphoric teens receive psychosocial support and psychotherapy as a first-line treatment, and discouraging the use of puberty blockers, with the addition of much stricter criteria for their use. The Finnish health authority has stated that these recommendations will not be revised until further research is available.
A similar trend of increasing wait times and difficult access holds in many other countries, with the process to receive puberty blockers sometimes taking up to several years. Because of their time-sensitive nature in preventing unwanted permanent changes, long-term outcomes are likely to be worse with slower treatment. Some evidence supports this: for example, one study found that reducing treatment wait times led to reduced depression and anxiety compared to historical controls (Dahlgren Allen et al., 2021).
Conclusion
Unknowns exist around puberty blockers in transgender youth, but their risks seem to be relatively minor based on available research, while clear evidence associates their use with improved well-being, psychological functioning, and reduced suicidality.
Based on parallels from research in cisgender teens treated for precocious puberty, as well as limited studies and clinical experience with transgender teens, it’s unlikely that puberty suppression has a dramatic negative effect on children’s final bone density, lifetime fracture risk, IQ, or cognitive development when prescribed in line with medical guidelines. However, there is insufficient evidence to determine whether or not they have any impact at all.
Although not supported by conclusive evidence, the use of puberty blockers may have a modest negative impact on bone density. This could be related to the use of puberty blockers themselves, but could also be related to suboptimal hormone therapy regimens after their use, particularly in transgender girls, as well as lifestyle factors. Studies investigating this suffer from significant methodological issues, and a definitive causal link remains unproven. Based on limited evidence, prescribers may wish to consider calcium supplementation in transgender teens receiving puberty blockers, and may wish to avoid oral estrogens in transgender girls beginning hormone therapy.
Compared to their cisgender peers, transgender adolescents who take puberty blockers are less likely to choose to have biological children, but puberty blockers do not permanently affect fertility.
Widely cited statistics around children growing out of gender dysphoria (“desistance”) as they grow older are based on highly unreliable data. Surprisingly, based on current evidence, we cannot reasonably guess the rate at which this happens. Regardless, desistance rates are not an argument for or against the use of puberty blockers. It is important to respect children’s wishes and autonomy, and to find the best way to support them as their gender identity develops, without imposing the idea that either a transgender or a cisgender gender identity is a bad outcome.
Very few patients who receive puberty blockers experience regret. In broader context, for the small minority of adult transgender patients who report feeling regret after undergoing hormone therapy or surgery, a common reason for that is a lack of social acceptance.
More high-quality research is urgently needed in this field. In particular, the effects of puberty blockers on IQ and cognitive development, bone outcomes, and desistance remain understudied subjects. Randomised controlled trials on puberty blockers are not available, and likely cannot be performed for both practical and ethical reasons. This should not be seen as a reason to discard all other research on the subject, or to label their use as experimental, as it is a standard of evidence that can never be met.
Puberty blockers are extremely difficult for patients to access in many countries, including the United States, the United Kingdom, and parts of Europe. Several countries have recently banned their use, or further restricted it significantly. This review provides further evidence supporting WPATH, EPATH, USPATH, AsiaPATH, CPATH, AusAPTH, PATHA, the Endocrine Society, the American Academy of Child and Adolescent Psychiatry, and the American Medical Association in condemning recent attempts to bar transgender teens from receiving gender-affirming care, including puberty blockers. To better support gender dysphoric children, barriers of access should instead be reduced where possible.
References
Achille, C., Taggart, T., Eaton, N. R., Osipoff, J., Tafuri, K., Lane, A., & Wilson, T. A. (2020). Longitudinal impact of gender-affirming endocrine intervention on the mental health and well-being of transgender youths: preliminary results. International Journal of Pediatric Endocrinology, 2020, 8. [DOI:10.1186/s13633-020-00078-2]
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Antoniazzi, F., Zamboni, G., Bertoldo, F., Lauriola, S., Mengarda, F., Pietrobelli, A., & Tatò, L. (2003). Bone mass at final height in precocious puberty after gonadotropin-releasing hormone agonist with and without calcium supplementation. The Journal of Clinical Endocrinology and Metabolism, 88(3), 1096–1101. [DOI:10.1210/jc.2002-021154]
Arnoldussen, M., Steensma, T. D., Popma, A., van der Miesen, A., Twisk, J., & de Vries, A. (2020). Re-evaluation of the Dutch approach: are recently referred transgender youth different compared to earlier referrals? European Child & Adolescent Psychiatry, 29(6), 803–811. [DOI:10.1007/s00787-019-01394-6]
Bakwin, H. (1968). Deviant gender-role behavior in children: relation to homosexuality. Pediatrics, 41(3), 620–629. [PubMed] [DOI:10.1542/peds.41.3.620]
Barake, M., Klibanski, A., & Tritos, N. A. (2014). Effects of recombinant human growth hormone therapy on bone mineral density in adults with growth hormone deficiency: a meta-analysis. The Journal of Clinical Endocrinology and Metabolism, 99(3), 852–860. [DOI:10.1210/jc.2013-3921]
Barake, M., Arabi, A., Nakhoul, N., El-Hajj Fuleihan, G., El Ghandour, S., Klibanski, A., & Tritos, N. A. (2018). Effects of growth hormone therapy on bone density and fracture risk in age-related osteoporosis in the absence of growth hormone deficiency: a systematic review and meta-analysis. Endocrine, 59(1), 39–49. [DOI:10.1007/s12020-017-1440-0]
Barnard, E. P., Dhar, C. P., Rothenberg, S. S., Menke, M. N., Witchel, S. F., Montano, G. T., Orwig, K. E., & Valli-Pulaski, H. (2019). Fertility Preservation Outcomes in Adolescent and Young Adult Feminizing Transgender Patients. Pediatrics, 144(3), e20183943. [DOI:10.1542/peds.2018-3943]
Bertelloni, S., Baroncelli, G. I., Ferdeghini, M., Menchini-Fabris, F., & Saggese, G. (2000). Final height, gonadal function and bone mineral density of adolescent males with central precocious puberty after therapy with gonadotropin-releasing hormone analogues. European Journal of Pediatrics, 159(5), 369–374. [DOI:10.1007/s004310051289]
Brik, T., Vrouenraets, L., de Vries, M. C., & Hannema, S. E. (2020). Trajectories of Adolescents Treated with Gonadotropin-Releasing Hormone Analogues for Gender Dysphoria. Archives of Sexual Behavior, 49(7), 2611–2618. [DOI:10.1007/s10508-020-01660-8]
Carmichael, P., Butler, G., Masic, U., Cole, T. J., De Stavola, B. L., Davidson, S., Skageberg, E. M., Khadr, S., & Viner, R. M. (2021). Short-term outcomes of pubertal suppression in a selected cohort of 12 to 15 year old young people with persistent gender dysphoria in the UK. PloS One, 16(2), e0243894. [DOI:10.1371/journal.pone.0243894]
Cedrin-Durnerin, I., Bidart, J. M., Robert, P., Wolf, J. P., Uzan, M., & Hugues, J. N. (2000). Consequences on gonadotrophin secretion of an early discontinuation of gonadotrophin-releasing hormone agonist administration in short-term protocol for in-vitro fertilization. Human Reproduction, 15(5), 1009–1014. [DOI:10.1093/humrep/15.5.1009]
Chen, D., Simons, L., Johnson, E. K., Lockart, B. A., & Finlayson, C. (2017). Fertility Preservation for Transgender Adolescents. The Journal of Adolescent Health, 61(1), 120–123. [DOI:10.1016/j.jadohealth.2017.01.022]
Cheng, P. J., Pastuszak, A. W., Myers, J. B., Goodwin, I. A., & Hotaling, J. M. (2019). Fertility concerns of the transgender patient. Translational Andrology and Urology, 8(3), 209–218. [DOI:10.21037/tau.2019.05.09]
Cohen-Kettenis, P. T., & van Goozen, S. H. (1998). Pubertal delay as an aid in diagnosis and treatment of a transsexual adolescent. European Child & Adolescent Psychiatry, 7(4), 246–248. [DOI:10.1007/s007870050073]
Cohen-Kettenis, P. T., Schagen, S. E., Steensma, T. D., de Vries, A. L., & Delemarre-van de Waal, H. A. (2011). Puberty suppression in a gender-dysphoric adolescent: a 22-year follow-up. Archives of Sexual Behavior, 40(4), 843–847. [DOI:10.1007/s10508-011-9758-9]
Costa, R., Dunsford, M., Skagerberg, E., Holt, V., Carmichael, P., & Colizzi, M. (2015). Psychological Support, Puberty Suppression, and Psychosocial Functioning in Adolescents with Gender Dysphoria. The Journal of Sexual Medicine, 12(11), 2206–2214. [DOI:10.1111/jsm.13034]
Dahlgren Allen, S., Tollit, M. A., McDougall, R., Eade, D., Hoq, M., & Pang, K. C. (2021). A Waitlist Intervention for Transgender Young People and Psychosocial Outcomes. Pediatrics, 148(2), e2020042762. [DOI:10.1542/peds.2020-042762]
Davenport, C. W. (1986). A follow-up study of 10 feminine boys. Archives of Sexual Behavior, 15(6), 511–517. [DOI:10.1007/BF01542316]
Davenport M. L. (2010). Approach to the patient with Turner syndrome. The Journal of Clinical Endocrinology and Metabolism, 95(4), 1487–1495. [DOI:10.1210/jc.2009-0926]
de Vries, A. L. C., Cohen-Kettenis, P. T., & Delemarre-van de Waal, H. (2006). Clinical Management of Gender Dysphoria in Adolescents. International Journal of Transgenderism, 9(4), 83–94. [DOI:10.1300/J485v09n03_04]
de Vries, A. L., Steensma, T. D., Doreleijers, T. A., & Cohen-Kettenis, P. T. (2011). Puberty suppression in adolescents with gender identity disorder: a prospective follow-up study. The Journal of Sexual Medicine, 8(8), 2276–2283. [DOI:10.1111/j.1743-6109.2010.01943.x]
de Vries, A. L., McGuire, J. K., Steensma, T. D., Wagenaar, E. C., Doreleijers, T. A., & Cohen-Kettenis, P. T. (2014). Young adult psychological outcome after puberty suppression and gender reassignment. Pediatrics, 134(4), 696–704. [DOI:10.1542/peds.2013-2958]
Drummond, K. D., Bradley, S. J., Peterson-Badali, M., & Zucker, K. J. (2008). A follow-up study of girls with gender identity disorder. Developmental Psychology, 44(1), 34–45. [DOI:10.1037/0012-1649.44.1.34]
Drummond, K. D., Bradley, S. J., Peterson-Badali, M., VanderLaan, D. P., & Zucker, K. J. (2018). Behavior Problems and Psychiatric Diagnoses in Girls with Gender Identity Disorder: A Follow-Up Study. Journal of Sex & Marital Therapy, 44(2), 172–187. [DOI:10.1080/0092623X.2017.1340382]
Ekbote, V. H., Khadilkar, V. V., Khadilkar, A. V., Mughal, Z., Chiplonkar, S. A., Palande, S. A., Phanse-Gupte, S. S., Patwardhan, V. G., & Shilvant, D. S. (2015). Relationship of insulin-like growth factor 1 and bone parameters in 7–15 years old apparently, healthy Indian children. Indian Journal of Endocrinology and Metabolism, 19(6), 770–774. [DOI:10.4103/2230-8210.167549]
Feuillan, P. P., Jones, J. V., Barnes, K., Oerter-Klein, K., & Cutler, G. B., Jr. (1999). Reproductive axis after discontinuation of gonadotropin-releasing hormone analog treatment of girls with precocious puberty: long term follow-up comparing girls with hypothalamic hamartoma to those with idiopathic precocious puberty. The Journal of Clinical Endocrinology and Metabolism, 84(1), 44–49. [DOI:10.1210/jcem.84.1.5409]
Giordano, S., & Holm, S. (2020). Is puberty delaying treatment ‘experimental treatment’? International Journal of Transgender Health, 21(2), 113–121. [DOI:10.1080/26895269.2020.1747768]
Green, A. E., DeChants, J. P., Price, M. N., & Davis, C. K. (2022). Association of Gender-Affirming Hormone Therapy With Depression, Thoughts of Suicide, and Attempted Suicide Among Transgender and Nonbinary Youth. Journal of Adolescent Health, 70(4), 643–649. [DOI:10.1016/j.jadohealth.2021.10.036]
Guaraldi, F., Beccuti, G., Gori, D., & Ghizzoni, L. (2016). MANAGEMENT OF ENDOCRINE DISEASE: Long-term outcomes of the treatment of central precocious puberty. European Journal of Endocrinology, 174(3), R79–R87. [DOI:10.1530/EJE-15-0590]
Heger, S., Partsch, C. J., & Sippell, W. G. (1999). Long-term outcome after depot gonadotropin-releasing hormone agonist treatment of central precocious puberty: final height, body proportions, body composition, bone mineral density, and reproductive function. The Journal of Clinical Endocrinology and Metabolism, 84(12), 4583–4590. [DOI:10.1210/jcem.84.12.6203]
Heger, S., Müller, M., Ranke, M., Schwarz, H. P., Waldhauser, F., Partsch, C. J., & Sippell, W. G. (2006). Long-term GnRH agonist treatment for female central precocious puberty does not impair reproductive function. Molecular and Cellular Endocrinology, 254–255, 217–220. [DOI:10.1016/j.mce.2006.04.012]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658]
Hough, D., Bellingham, M., Haraldsen, I. R., McLaughlin, M., Robinson, J. E., Solbakk, A. K., & Evans, N. P. (2017). A reduction in long-term spatial memory persists after discontinuation of peripubertal GnRH agonist treatment in sheep. Psychoneuroendocrinology, 77, 1–8. [DOI:10.1016/j.psyneuen.2016.11.029]
Isotton, A. L., Wender, M. C., Casagrande, A., Rollin, G., & Czepielewski, M. A. (2012). Effects of oral and transdermal estrogen on IGF1, IGFBP3, IGFBP1, serum lipids, and glucose in patients with hypopituitarism during GH treatment: a randomized study. European Journal of Endocrinology, 166(2), 207–213. [DOI:10.1530/EJE-11-0560]
Jones, B. A., Haycraft, E., Bouman, W. P., & Arcelus, J. (2018). The Levels and Predictors of Physical Activity Engagement Within the Treatment-Seeking Transgender Population: A Matched Control Study. Journal of Physical Activity & Health, 15(2), 99–107. [DOI:10.1123/jpah.2017-0298]
Jung, K. Y., Kim, T., Hwang, S. Y., Lee, T. R., Yoon, H., Shin, T. G., Sim, M. S., Cha, W. C., & Jeon, H. J. (2018). Deliberate Self-harm among Young People Begins to Increase at the Very Early Age: a Nationwide Study. Journal of Korean Medical Science, 33(30), e191. [DOI:10.3346/jkms.2018.33.e191]
Kennedy, E., Spinner, L., Lane, C., Stynes, H., Ranieri, V., Carmichael, P., Omar, R., Vickerstaff, V., Hunter, R., Wright, T., Senior, R., Butler, G., Baron-Cohen, S., Young, B., & King, M. (2021). Longitudinal Outcomes of Gender Identity in Children (LOGIC): protocol for a prospective longitudinal cohort study of children referred to the UK gender identity development service. BMJ Open, 11(9), e045628. [DOI:10.1136/bmjopen-2020-045628]
Khatchadourian, K., Amed, S., & Metzger, D. L. (2014). Clinical management of youth with gender dysphoria in Vancouver. The Journal of Pediatrics, 164(4), 906–911. [DOI:10.1016/j.jpeds.2013.10.068]
Kidd, J. D., Dolezal, C., & Bockting, W. O. (2018). The Relationship Between Tobacco Use and Legal Document Gender-Marker Change, Hormone Use, and Gender-Affirming Surgery in a United States Sample of Trans-Feminine and Trans-Masculine Individuals: Implications for Cardiovascular Health. LGBT Health, 5(7), 401–411. [DOI:10.1089/lgbt.2018.0103]
Klink, D., Caris, M., Heijboer, A., van Trotsenburg, M., & Rotteveel, J. (2015). Bone mass in young adulthood following gonadotropin-releasing hormone analog treatment and cross-sex hormone treatment in adolescents with gender dysphoria. The Journal of Clinical Endocrinology and Metabolism, 100(2), E270–E275. [DOI:10.1210/jc.2014-2439]
Kosky, R. J. (1987). Gender-disordered children: does inpatient treatment help? The Medical Journal of Australia, 146(11), 565–569. [DOI:10.5694/j.1326-5377.1987.tb120415.x]
Kuper, L. E., Stewart, S., Preston, S., Lau, M., & Lopez, X. (2020). Body Dissatisfaction and Mental Health Outcomes of Youth on Gender-Affirming Hormone Therapy. Pediatrics, 145(4), e20193006. [DOI:10.1542/peds.2019-3006]
Lazar, L., Meyerovitch, J., de Vries, L., Phillip, M., & Lebenthal, Y. (2014). Treated and untreated women with idiopathic precocious puberty: long-term follow-up and reproductive outcome between the third and fifth decades. Clinical Endocrinology, 80(4), 570–576. [DOI:10.1111/cen.12319]
Lebovitz, P. S. (1972). Feminine behavior in boys: aspects of its outcome. The American Journal of Psychiatry, 128(10), 1283–1289. [DOI:10.1176/ajp.128.10.1283]
Lee, J. Y., Finlayson, C., Olson-Kennedy, J., Garofalo, R., Chan, Y. M., Glidden, D. V., & Rosenthal, S. M. (2020). Low Bone Mineral Density in Early Pubertal Transgender/Gender Diverse Youth: Findings From the Trans Youth Care Study. Journal of the Endocrine Society, 4(9), bvaa065. [DOI:10.1210/jendso/bvaa065]
Li, K., Rodriguez, D., Gabrielsen, J. S., Centola, G. M., & Tanrikut, C. (2018). Sperm cryopreservation of transgender individuals: trends and findings in the past decade. Andrology, 6(6), 860–864. [DOI:10.1111/andr.12527]
Lindsey, R. C., & Mohan, S. (2016). Skeletal effects of growth hormone and insulin-like growth factor-I therapy. Molecular and Cellular Endocrinology, 432, 44–55. [DOI:10.1016/j.mce.2015.09.017]
Locatelli, V., & Bianchi, V. E. (2014). Effect of GH/IGF-1 on Bone Metabolism and Osteoporsosis. International Journal of Endocrinology, 2014, 235060. [DOI:10.1155/2014/235060]
Money, J., & Russo, A. J. (1979). Homosexual outcome of discordant gender identity/role in childhood: Longitudinal follow-up. Journal of Pediatric Psychology, 4(1), 29–41. [DOI:10.1093/jpepsy/4.1.29]
Morey, Y., Mellon, D., Dailami, N., Verne, J., & Tapp, A. (2017). Adolescent self-harm in the community: an update on prevalence using a self-report survey of adolescents aged 13–18 in England. Journal of Public Health, 39(1), 58–64. [DOI:10.1093/pubmed/fdw010]
Mul, D., Versluis-den Bieman, H. J., Slijper, F. M., Oostdijk, W., Waelkens, J. J., & Drop, S. L. (2001). Psychological assessments before and after treatment of early puberty in adopted children. Acta Paediatrica, 90(9), 965–971. [DOI:10.1080/080352501316978011]
Nahata, L., Tishelman, A. C., Caltabellotta, N. M., & Quinn, G. P. (2017). Low Fertility Preservation Utilization Among Transgender Youth. The Journal of Adolescent Health, 61(1), 40–44. [DOI:10.1016/j.jadohealth.2016.12.012]
Nock, M. K., Green, J. G., Hwang, I., McLaughlin, K. A., Sampson, N. A., Zaslavsky, A. M., & Kessler, R. C. (2013). Prevalence, correlates, and treatment of lifetime suicidal behavior among adolescents: results from the National Comorbidity Survey Replication Adolescent Supplement. JAMA Psychiatry, 70(3), 300–310. [DOI:10.1001/2013.jamapsychiatry.55]
Olson-Kennedy, J., Chan, Y. M., Garofalo, R., Spack, N., Chen, D., Clark, L., Ehrensaft, D., Hidalgo, M., Tishelman, A., & Rosenthal, S. (2019). Impact of Early Medical Treatment for Transgender Youth: Protocol for the Longitudinal, Observational Trans Youth Care Study. JMIR Research Protocols, 8(7), e14434. [DOI:10.2196/14434]
Pang, K. C., Peri, A., Chung, H. E., Telfer, M., Elder, C. V., Grover, S., & Jayasinghe, Y. (2020). Rates of Fertility Preservation Use Among Transgender Adolescents. JAMA Pediatrics, 174(9), 890–891. [DOI:10.1001/jamapediatrics.2020.0264]
Riggs, M. M., Bennetts, M., van der Graaf, P. H., & Martin, S. W. (2012). Integrated pharmacometrics and systems pharmacology model-based analyses to guide GnRH receptor modulator development for management of endometriosis. CPT: Pharmacometrics & Systems Pharmacology, 1(10), e11. [DOI:10.1038/psp.2012.10]
Rodriguez-Wallberg, K. A., Häljestig, J., Arver, S., Johansson, A., & Lundberg, F. E. (2021). Sperm quality in transgender women before or after gender affirming hormone therapy—A prospective cohort study. Andrology, 9(6), 1773–1780. [DOI:10.1111/andr.12999]
Roux, C. (1997). Estrogen therapy in postmenopausal osteoporosis. What we know and what we don’t. Revue du Rhumatisme (English Ed.), 64(6), 402–409. [Google Scholar] [PubMed] [PDF]
Schagen, S., Wouters, F. M., Cohen-Kettenis, P. T., Gooren, L. J., & Hannema, S. E. (2020). Bone Development in Transgender Adolescents Treated With GnRH Analogues and Subsequent Gender-Affirming Hormones. The Journal of Clinical Endocrinology and Metabolism, 105(12), e4252–e4263. [DOI:10.1210/clinem/dgaa604]
Schneider, M. A., Spritzer, P. M., Soll, B., Fontanari, A., Carneiro, M., Tovar-Moll, F., Costa, A. B., da Silva, D. C., Schwarz, K., Anes, M., Tramontina, S., & Lobato, M. (2017). Brain Maturation, Cognition and Voice Pattern in a Gender Dysphoria Case under Pubertal Suppression. Frontiers in Human Neuroscience, 11, 528. [DOI:10.3389/fnhum.2017.00528]
Singh, D., Bradley, S. J., & Zucker, K. J. (2021). A Follow-Up Study of Boys With Gender Identity Disorder. Frontiers in Psychiatry, 12, 632784. [DOI:10.3389/fpsyt.2021.632784]
Southmayd, E. A., & De Souza, M. J. (2017). A summary of the influence of exogenous estrogen administration across the lifespan on the GH/IGF-1 axis and implications for bone health. Growth Hormone & IGF Research, 32, 2–13. [DOI:10.1016/j.ghir.2016.09.001]
Staphorsius, A. S., Kreukels, B. P., Cohen-Kettenis, P. T., Veltman, D. J., Burke, S. M., Schagen, S. E., Wouters, F. M., Delemarre-van de Waal, H. A., & Bakker, J. (2015). Puberty suppression and executive functioning: An fMRI-study in adolescents with gender dysphoria. Psychoneuroendocrinology, 56, 190–199. [DOI:10.1016/j.psyneuen.2015.03.007]
Steensma, T. D., Biemond, R., de Boer, F., & Cohen-Kettenis, P. T. (2011). Desisting and persisting gender dysphoria after childhood: a qualitative follow-up study. Clinical Child Psychology and Psychiatry, 16(4), 499–516. [DOI:10.1177/1359104510378303]
Steensma, T. D., McGuire, J. K., Kreukels, B. P., Beekman, A. J., & Cohen-Kettenis, P. T. (2013). Factors associated with desistence and persistence of childhood gender dysphoria: a quantitative follow-up study. Journal of the American Academy of Child and Adolescent Psychiatry, 52(6), 582–590. [DOI:10.1016/j.jaac.2013.03.016]
Steensma, T. D., & Cohen-Kettenis, P. T. (2018). A critical commentary on “A critical commentary on follow-up studies and “desistence” theories about transgender and gender non-conforming children”. International Journal of Transgenderism, 19(2), 225–230. [DOI:10.1080/15532739.2018.1468292]
Swerdloff, R. S., & Heber, D. (1983). Superactive gonadotropin-releasing hormone agonists. Annual Review of Medicine, 34, 491–500. [DOI:10.1146/annurev.me.34.020183.002423]
Temple Newhook, J., Pyne, J., Winters, K., Feder, S., Holmes, C., Tosh, J., Sinnott, M.-L., Jamieson, A., & Pickett, S. (2018). A critical commentary on follow-up studies and “desistance” theories about transgender and gender-nonconforming children. International Journal of Transgenderism, 19(2), 212–224. [DOI:10.1080/15532739.2018.1456390]
Tollit, M. A., Pace, C. C., Telfer, M., Hoq, M., Bryson, J., Fulkoski, N., Cooper, C., & Pang, K. C. (2019). What are the health outcomes of trans and gender diverse young people in Australia? Study protocol for the Trans20 longitudinal cohort study. BMJ Open, 9(11), e032151. [DOI:10.1136/bmjopen-2019-032151]
Turban, J. L., King, D., Carswell, J. M., & Keuroghlian, A. S. (2020). Pubertal Suppression for Transgender Youth and Risk of Suicidal Ideation. Pediatrics, 145(2), e20191725. [DOI:10.1542/peds.2019-1725]
van der Miesen, A., Steensma, T. D., de Vries, A., Bos, H., & Popma, A. (2020). Psychological Functioning in Transgender Adolescents Before and After Gender-Affirmative Care Compared With Cisgender General Population Peers. The Journal of Adolescent Health, 66(6), 699–704. [DOI:10.1016/j.jadohealth.2019.12.018]
Verhulst, F. C., Achenbach, T. M., van der Ende, J., Erol, N., Lambert, M. C., Leung, P. W., Silva, M. A., Zilber, N., & Zubrick, S. R. (2003). Comparisons of problems reported by youths from seven countries. The American Journal of Psychiatry, 160(8), 1479–1485. [DOI:10.1176/appi.ajp.160.8.1479]
Vlot, M. C., Klink, D. T., den Heijer, M., Blankenstein, M. A., Rotteveel, J., & Heijboer, A. C. (2017). Effect of pubertal suppression and cross-sex hormone therapy on bone turnover markers and bone mineral apparent density (BMAD) in transgender adolescents. Bone, 95, 11–19. [DOI:10.1016/j.bone.2016.11.008]
Vrouenraets, L. J., Fredriks, A. M., Hannema, S. E., Cohen-Kettenis, P. T., & de Vries, M. C. (2016). Perceptions of Sex, Gender, and Puberty Suppression: A Qualitative Analysis of Transgender Youth. Archives of Sexual Behavior, 45(7), 1697–1703. [DOI:10.1007/s10508-016-0764-9]
Wallien, M. S., & Cohen-Kettenis, P. T. (2008). Psychosexual outcome of gender-dysphoric children. Journal of the American Academy of Child and Adolescent Psychiatry, 47(12), 1413–1423. [DOI:10.1097/CHI.0b013e31818956b9]
Wiepjes, C. M., Nota, N. M., de Blok, C., Klaver, M., de Vries, A., Wensing-Kruger, S. A., de Jongh, R. T., Bouman, M. B., Steensma, T. D., Cohen-Kettenis, P., Gooren, L., Kreukels, B., & den Heijer, M. (2018). The Amsterdam Cohort of Gender Dysphoria Study (1972–2015): Trends in Prevalence, Treatment, and Regrets. The Journal of Sexual Medicine, 15(4), 582–590. [DOI:10.1016/j.jsxm.2018.01.016]
Wojniusz, S., Callens, N., Sütterlin, S., Andersson, S., De Schepper, J., Gies, I., Vanbesien, J., De Waele, K., Van Aken, S., Craen, M., Vögele, C., Cools, M., & Haraldsen, I. R. (2016). Cognitive, Emotional, and Psychosocial Functioning of Girls Treated with Pharmacological Puberty Blockage for Idiopathic Central Precocious Puberty. Frontiers in Psychology, 7, 1053. [DOI:10.3389/fpsyg.2016.01053]
Zaiem, F., Alahdab, F., Al Nofal, A., Murad, M. H., & Javed, A. (2017). Oral Versus Transdermal Estrogen In Turner Syndrome: A Systematic Review And Meta-Analysis. Endocrine Practice, 23(4), 408–421. [DOI:10.4158/ep161622.or]
Zuger, B. (1978). Effeminate behavior present in boys from childhood: ten additional years of follow-up. Comprehensive Psychiatry, 19(4), 363–369. [DOI:10.1016/0010-440x(78)90019-6]
\ No newline at end of file
diff --git a/transfemscience.org/articles/serms-transfem-suppl/index.html b/transfemscience.org/articles/serms-transfem-suppl/index.html
index 1239c7a1..06385ffe 100644
--- a/transfemscience.org/articles/serms-transfem-suppl/index.html
+++ b/transfemscience.org/articles/serms-transfem-suppl/index.html
@@ -1 +1 @@
-Sources/Excerpts: A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy - Transfeminine Science
Sources/Excerpts: A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy
By Lain | First published October 5, 2019 | Last modified June 3, 2021
Preface
This page is a sources and excerpts supplement to the main article which can be found here.
Maximov, Lee, & Jordan (2013)
Maximov, P. Y., Lee, T. M., & Jordan, V. C. (2013). The Discovery and Development of Selective Estrogen Receptor Modulators (SERMs) for Clinical Practice. Current Clinical Pharmacology, 8(2), 135–155. [DOI:10.2174/1574884711308020006]:
However, although these SERMs have many benefits, they also have some potentially serious adverse effects, such as thromboembolic disorders and, in the case of tamoxifen, uterine cancer. These adverse effects represent a major concern given that long-term therapy is required to prevent osteoporosis or prevent and treat breast cancer.
The search for the ‘ideal’ SERM, which would have estrogenic effects on bone and serum lipids, neutral effects on the uterus, and antiestrogenic effects on breast tissue, but none of the adverse effects associated with current therapies, is currently under way.
Note: Serum lipids are like HDL/LDL cholesterol.
There are two ERs referred to as α and β [82-84]. Each receptor protein is encoded on different chromosomes, and have homology as members of the steroid receptor superfamily. There are distinct patterns of distribution and distinct and subtle differences in structure and ligand binding affinity [85]. The ratio of ERα and ERβ at a target site may be an additional dimension for tissue modulation. A high ERα: ERβ ratio correlates well with high levels of cellular proliferation whereas the predominance of functional ERβ over ERα correlates with repression of proliferation [86-89]. Indeed, the ratio of ERs in normal and neoplasic breast tissue could be important for the long-term success of chemoprevention with SERMs.
Kraichely and co-workers [97] demonstrated the important observation that agonists for ERα and ERβ produce subtle quantitative differences with the interaction of members of the SRC family (SRC 1, 2 and 3) and that the coactivator can enhance ligand affinity for the ER.
Evidence has now accumulated to document that the broad spectrum of ligands that bind to the ER can create a broad range of ER complexes that are either fully estrogenic or antiestrogenic at a particular target site [100]. Thus a mechanistic model of estrogen action and antiestrogen action (Fig. 2) has emerged based on the shape of the ligand that programs the complex for future action.
Thus for effective gene transcription, programmed and targeted by the shape and phosphorylation status of the ER and coactivators, a dynamic and cyclic process of remodeling capacity is required for transcriptional assembly [103] that is immediately followed by the routine destruction of transcription complexes by the proteasome. Estrogen and SERM-ER complexes have distinct accumulation patterns in the target cell nucleus [104,105] because they are destroyed at different rates.
Raloxifene lacks estrogenic activity in the uterus and has not demonstrated tamoxifen-like effects in the uterus either histopathologically or ultrasonographically [116], but it has been associated with adverse effects such as VTE and vasomotor symptoms, including hot flushes.
Perhaps the reason for this difference in effect is related to differences on the agents’ effect on inflammation as the agents influence C-reactive protein (CRP) differently. Estrogen and tibolone increase levels of CRP [192], raloxifene and arzoxifene have no effect on CRP levels, and lasofoxifene decreases CRP levels [55]. All decrease LDL levels.
Brzozowski et al. (1997)
Brzozowski, A. M., Pike, A. C., Dauter, Z., Hubbard, R. E., Bonn, T., Engström, O., Ohman, L., Greene, G. L., Gustafsson, J. A., & Carlquist, M. (1997). Molecular basis of agonism and antagonism in the oestrogen receptor. Nature, 389(6652), 753–758. [DOI:10.1038/39645]:
The overall architecture of the ER LBD (helices H3–H12) is similar to that seen in the crystal structures of other nuclear receptor LBDs6–8, and emphasizes the universal nature of this fold within this receptor superfamily9. The LBD is folded into a three-layered antiparallel a-helical sandwich comprising a central core layer of three helices (H5/6, H9 and H10) sandwiched between two additional layers of helices (H1–4 and H7, H8, H11). This helical arrangement creates a ‘wedge-shaped’ molecular scaffold that maintains a sizeable ligandbinding cavity at the narrower end of the domain. The remaining secondary structural elements, a small two-stranded antiparallel β-sheet (S1 and S2) and H12, are located at this ligand-binding portion of the molecule, and flank the main three-layered motif (Fig. 1a).
E2 binds diagonally across the cavity between H11, H3 and H6 and adopts a low-energy conformation. The phenolic hydroxyl of the A-ring (O3; see Fig. 2c for atom numbering) nestles between H3 and H6 and makes direct hydrogen bonds to the carboxylate of Glu 353, the guanidinium group of Arg 394, and a water molecule. The 17-β hydroxyl (O17) of the Dring makes a single hydrogen bond with His 524 in H11. The remainder of the molecule participates in a number of hydrophobic contactsthat are concentrated overthe A, A/B interface and D-rings.
The ER is unique among the steroid receptors in its ability to embrace a wide variety of non-steroidal compounds. Although the ‘pincer-like’ arrangement around the A-ring imposes an absolute requirement on effective ligands to contain an aromatic ring, the remainder of the binding pocket can accept a number of different hydrophobic groups12,13. This overall promiscuity can be attributed to the size of the cavity, which has a probe accessible volume (450 A˚3) nearly twice that of E2’s molecular volume (245 A˚3). The length and breadth of the E2 skeleton is well matched by the receptor, but there are large unoccupied cavities opposite the α face of the B-ring and the β face of the C-ring (Fig. 2a). The positions of these preformed cavities are similar to those predicted from binding studies12.
Note: This may be why things like bisphenol A (BPA) and other random chemicals bind to the estrogen receptor.
Rosano et al. (2011)
Rosano, C., Stec-Martyna, E., Lappano, R., & Maggiolini, M. (2011). Structure-Based Approach for the Discovery of Novel Selective Estrogen Receptor Modulators. Current Medicinal Chemistry, 18(8), 1188–1194. [DOI:10.2174/092986711795029645]:
These two zinc fingers are responsible for the binding of ERα to the estrogen response elements (EREs) located in the promoter regions of target genes. The P-box sequence is identical in the two ER isoforms, suggesting that ERα and ERβ bind EREs with similar specificity and affinity.
Pure anti-estrogen development has also included a further class of molecules represented by the inhibitors of aromatase enzymatic activity, which increases estrogen availability at tissue levels converting androgens into estrogens.
4OHT binds to ERs within the same binding pocket that recognises E2 and produces a nuclear complex which decreases DNA synthesis and inhibits the action of estrogens [24]. Moreover, the complex between ER and 4OHT recruits co-repressor proteins like NCoR and SMRT, which contribute to the antagonist effects of 4OHT [78].
In particular, the position of H12 shifts from the ligand binding pocket to cover a part of the co-activator binding groove, which is formed by residues belonging to H3, H4 and H5 [24]. The receptor recognition of 4OHT is dictated by the phenolic “A ring” and the bulky side chain.
[…] whilst the bulky side chain protruding from the binding cavity displaces H12 and allows a conformation that inhibits the coactivator recruitment.
Next, the long hydrophobic side chain of RAL makes extensive contacts with the hydrophobic residues of the receptor allowing the displacement of H12 similar to 4OHT.
Comparable to other SERMs, Lasofoxifene diverts the receptor from its agonistbound conformation by displacing the C-terminal AF-2 helix 12 into the site at which the LXXLL motif of coactivator proteins would otherwise be able to bind. LAS achieves this effect by occupying the space normally filled by residue Leu540, as well as by modulating the conformation of residues of helix 11.
In addition to these features of SERM recognition, a large alkyl “pendant” side chain, terminating in a pyrollidine head group, threads its way from the ligand-binding pocket out toward the surface of the protein, where it directly interferes with the correct positioning of the AF-2 helix.
Nilsson & Gustafsson (2010)
Nilsson, S., & Gustafsson, J. (2010). Estrogen Receptors: Therapies Targeted to Receptor Subtypes. Clinical Pharmacology & Therapeutics, 89(1), 44–55. [DOI:10.1038/clpt.2010.226]:
Like other members of the nuclear receptor family, the ERs are best known as transcription factors, regulating the transcriptional activity of target genes in cells in response to the natural hormone 17β-estradiol (E2) or to synthetic nonhormonal agonists and antagonists1 (Figure 1). In more recent years, we have also learned that the effects of E2 can have a very rapid onset, within seconds to minutes. These effects may be mediated by membrane-localized ERα and ERβ, respectively (after exposure to ligand stimuli), or by membrane receptors that are genetically and structurally unrelated to ERα and ERβ, for example, GPR30 (G-protein coupled receptor 30).
An increase in the expression of ERβ relative to ERα was shown to inhibit the ERα response to E2, silence the partial ERα-mediated agonism of tamoxifen, and potentiate its antagonism.10 In mice, the level of expression of genes induced by E2 in bone were, on average, 85% higher in the absence of ERβ than in its presence, thereby suggesting that ERβ has an opposing effect on ERα-mediated gene transcription.11
In some organs, ERα and ERβ are expressed at similar levels, sometimes in different cell types within the same organ, whereas in others, one or the other subtype predominates. ERα is predominantly expressed in the uterus, prostate (stroma), ovary (theca cells), testes (Leydig cells), epididymis, bone, breast, liver, kidney, white adipose tissue, and various regions of the brain. ERβ is predominantly expressed in the colon, prostate (epithelium), testis, ovary (granulosa cells), bone marrow, salivary gland, vascular endothelium, lung, bladder, and certain regions of the brain (Figure 3).1,4
In summary, the metabolic findings in knockout mice and in aromatase-deficient or ERα-deficient patients, and the use of ER subtype-selective ligands, suggest a major role for ERα in the maintenance of metabolic control. ERβ may, however, have an opposite effect, especially when unopposed, resulting in a perturbed energy balance and an increased risk for the development of diabetes. Unopposed ERβ activity is a possible explanation for the diabetic phenotype of the male patient with homozygous loss of ERα function. Both ER subtypes are reported to have roles in the prevention of atherosclerotic lesion development, although seemingly through different mechanisms.
SERMs such as tamoxifen and raloxifene and the pure antagonist ICI 182,780, however, antagonize the neuroprotective effects of E2 after experimentally induced ischemia.36
Note: It seems likely to me that in general oral raloxifene would not cause gynoid fat distribution due to its ERα antagonist and ERα-mediated anti-lipolytic pathways in subcutaneous fat – I was wrong.
Studies in the aromatase/αERKO recombinant mouse model confirmed the important role of ERα in the development of the mammary gland. Treatment of ovariectomized rats with the ERβ-selective agonist ERB-041 resulted in no observable effects on rat mammary tissue, suggesting that ERβ-selective agonists are nonmammotrophic.1
In summary, estrogens are of fundamental importance in the development of the mammary gland, and ERα is the primary ER subtype mediating the effects of estrogens. ERβ, however, may play an important role in controlling the mammotrophic effects of ERα and may be an important novel target for endocrine therapy in breast cancer.
In summary, tissues of the urogenital tract are dependent on estrogens for proper growth, development, and functional integrity. ERα is uterotrophic and mediates uterine proliferative responses to E2. It is also the ER subtype that plays the most important role in the feedback on the hypothalamic–pituitary–gonadal axis and the regulation of LH and follicle-stimulating hormone levels. ERβ has an important role in folliculogenesis and ovulation and may have very important therapeutic implications in the diseased prostate, as a drug target in benign prostatic hyperplasia and in both castrate-responsive and castrateresistant prostate cancer.
Greene (2000)
Greene, G. (2000). Comparative Structural Analysis of ERa and ERb Bound to Selective Estrogen Agonists and Antagonists. Chicago: The University of Chicago. [Google Scholar] [URL] [PDF]:
The structures of the LBD complexed with E2 and RAL show that, although both ligands bind at the same site within the core of the LBD (Brzozowski et al. 1997), each of these ligands induces a different conform”ation of helix 12. Whereas helix 12 in the E2-LBD complex packs against helices 3, 5/6, and 11 in a conformation that has been observed for the corresponding helix in other agonist-bound NR LBD structures, helix 12 in the RAL-LBD complex is bound in a hydrophobic groove composed of residues from helices 3 and 5. This alternative orientation of helix 12 partially buries residues in the groove that are necessary for AF-2 activity, suggesting that RAL and possibly other antagonists block AF-2 function by disrupting the topography of the AF-2 surface.
Shiau et al. (1998)
Shiau, A. K., Barstad, D., Loria, P. M., Cheng, L., Kushner, P. J., Agard, D. A., & Greene, G. L. (1998). The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell, 95(7), 927–937. [DOI:10.1016/s0092-8674(00)81717-1]:
Several proteins, including SRC-1/N-CoA1 (Onate et al., 1995; Kamei et al., 1996), GRIP1/TIF2/N-CoA2 (Hong et al., 1996; Voegel et al., 1996; Torchia et al., 1997;), p/CIP/RAC3/ACTR/AIB1 (Anzick et al., 1997; Chen et al., 1997; Li et al., 1997; Torchia et al., 1997), and CBP/ p300 (Hanstein et al., 1996), associate in a ligand-dependent manner with the ERa. These proteins have been classified as transcriptional coactivators because they enhance ligand-dependent transcriptional activation by the ERa as well as by several other NRs (Horwitz et al., 1996; Glass et al., 1997). SRC-1 and GRIP1 bind to the agonist-bound LBDs of both the human thyroid receptor b (TRb) and human ERa using the putative AF-2 interaction surface (Feng et al., 1998). Members of the p160 family of coactivators, such as SRC-1 and GRIP1, as well as other coactivators, recognize agonist-bound NR LBDs through a short signature sequence motif, LXXLL (where L is leucine and X is any amino acid), known as the NR box (Le Douarin et al., 1996; Heery et al., 1997; Torchia et al., 1997; Ding et al., 1998). Mutagenesis studies indicate that the affinity of coactivators for NR LBDs is determined principally, if not exclusively, by these NR boxes (Le Douarin et al., 1996; Heery et al., 1997; Torchia et al., 1997; Ding et al., 1998).
Rambhatla, Mills, & Rajfer (2016)
Rambhatla, A., Mills, J. N., & Rajfer, J. (2016). The Role of Estrogen Modulators in Male Hypogonadism and Infertility. Reviews in Urology, 18(2), 66–72. [PubMed] [PubMed Central] [DOI:10.3909/riu0711]:
To circumvent the inhibition of LH and FSH from the pituitary by exogenous testosterone, a common strategy used by male reproductive and sexual medicine specialists, in which the goal is to increase testosterone levels while maintaining spermatogenesis, is to use SERMs and/or AIs instead of exogenous testosterone.
Clomiphene citrate works as an estrogen antagonist at the level of the pituitary gland and thus stimulates the release of LH and FSH, which in turn drives both the steroidogenic and spermatogenic functions of the testes.
Other studies have also found that aside from improvement in serum testosterone levels, clomiphene therapy also leads to significant improvement in bone mineral density, as well as androgen deficiency in the aging male (ADAM) scores without any significant adverse events.
Tamoxifen citrate is another oral SERM that was approved in the 1970s for the treatment of breast cancer. It has tissue specific action and acts as an estrogen receptor blocker in breast tissue and exhibits agonistic properties in the bone and uterus.9 Although its use is primarily in women, in men’s health it is used off-label and acts as an estrogen antagonist in the hypothalamus and pituitary gland.23 Because of its mechanism of action, tamoxifen results in the inhibition of the negative feedback of estrogen at the hypothalamus and pituitary gland, and results in the release of LH and FSH, which in turn increases testosterone biosynthesis and “stimulates” spermatogenesis.
Tsourdi and colleagues23 looked at the effects of three SERMs—tamoxifen, toremifine, and raloxifene—on the hypothalamic-pituitary axis in men with oligospermia. They found that after 3 months of treatment with each of these SERMs, there was a statistically significant increase in serum gonadotropins, testosterone, and semen parameters.
Tamoxifen, given its estrogen antagonist properties in the breast, as well as AIs, has also been used in the treatment of gynecomastia. Much of the data stem from boys with pubertal gynecomastia and in men with prostate cancer on antiandrogen therapy; there is evidence that these medications are effective in the treatment of gynecomastia.43-45 Their use in the treatment of gynecomastia induced by exogenous testosterone therapy is largely anecdotal and not evidence based.46 However, the manipulation of estrogen levels in men may not be without consequences. We know that estrogen receptors are present throughout the body and play a role in bone health, body composition, cardiovascular well being, libido and sexual function, and testicular steroidogenesis and spermatogenesis.47-49 Although a high estradiol level may have a negative impact on fertility, a level that is excessively low may not be desirable either.
Kunath et al. (2012)
Kunath, F., Keck, B., Antes, G., Wullich, B., & Meerpohl, J. J. (2012). Tamoxifen for the management of breast events induced by non-steroidal antiandrogens in patients with prostate cancer: a systematic review. BMC Medicine, 10(1), 96. [DOI:10.1186/1741-7015-10-96]:
Our results suggest that tamoxifen has a beneficial effect if compared to no treatment for the prevention of breast events. However, not all patients need prophylaxis to prevent the development of breast events induced by non-steroidal antiandrogen therapy [26,27], and not all patients with gynecomastia require treatment [28]. Therefore, a patient-oriented, pragmatic approach appears reasonable. This approach was also proposed by van Poppel and by Di Lorenzo et al. [14,15,27]. Before starting non-steroidal antiandrogen treatment (either with non-steroidal monotherapy or in combination with LHRH analogues), patients should be informed about the likelihood of breast events and about possible prophylactic therapy options. As recommended earlier by Di Lorenzo et al. [14,15], we also suggest that the physician could wait for the occurrence of breast events in selected patients. Prophylaxis should be started only if the patient is afraid of developing gynecomastia or breast pain.
Jover-Mengual et al. (2017)
Jover-Mengual, T., Castelló-Ruiz, M., Burguete, M. C., Jorques, M., López-Morales, M. A., Aliena-Valero, A., Jurado-Rodríguez, A., Pérez, S., Centeno, J. M., Miranda, F. J., Alborch, E., Torregrosa, G., & Salom, J. B. (2017). Molecular mechanisms mediating the neuroprotective role of the selective estrogen receptor modulator, bazedoxifene, in acute ischemic stroke: A comparative study with 17β-estradiol. The Journal of Steroid Biochemistry and Molecular Biology, 171, 296–304. [DOI:10.1016/j.jsbmb.2017.05.001]:
Note: This shows some variances and similarities in how various estrogen receptor pathways in the brain of mice are with estradiol versus bazedoxifene.
Simpkins et al. [2] provided the first evidence that estrogens (specifically 17β-estradiol, E2) exerted neuroprotective effects in the now widely used rodent model of AIS, namely the middle cerebral artery occlusion (MCAO) model. The authors suggested that estrogens may be a useful therapy to protect neurons against the damaging effects of stroke.
Bazedoxifene acetate (BZA) is the first of the third-generation SERMs approved for the treatment of postmenopausal women at risk for, or presenting with, osteoporosis in Europe and Japan [21]. Due to its favorable preclinical effects, BZA has been selected to combine with conjugated estrogens (CE) resulting in CE/BZA as a new progestin-free hormone therapy option for alleviating estrogen deficiency symptoms in postmenopausal women [22]. As for neural tissue, BZA had been reported to prevent neuronal loss in the hippocampus of rats exposed to excitotoxic kainic acid [23], and to decrease the inflammatory response of astrocytes exposed to lipopolysaccharide [24].
Both BZA and E2 significantly counteracted the ischemia-induced downregulation of ERα mRNA expression in the ipsilateral hemisphere.
As for the effect of the estrogenic compounds, BZA significantly counteracted the ischemia-induced downregulation of ERβ mRNA expression, while E2 was without significant effect.
As for the effect of the estrogenic compounds, neither BZA nor E2 had a significant effect on GPER mRNA expression in the ipsilateral hemisphere.
BZA but not E2 significantly attenuated the ischemia-induced endogenous phosphorylation of ERK1 in the ipsilateral hemisphere (Fig. 6A and B). While p-ERK2 levels were significantly reduced in the ipsilateral hemisphere in animals treated with BZA, there was no significant change in animals treated with E2.
The hypothesis that the neuroprotective mechanism of the SERM, tamoxifen, could be due to its ability to attenuate apoptotic cell death was first suggested and verified by Wakade et al. [11], and subsequently confirmed by Zou et al. [17]. By contrast, the SERM, raloxifene, has been recently reported to have no effect on the hypoxia-induced increase of caspase-3 activity in cultured hippocampal cells [20]. Therefore, our results lend support to the view that, as in the case of E2, SERMs (specifically BZA) are effective anti-apoptotic drugs in stroke.
Wibowo et al. (2016)
Wibowo, E., Pollock, P. A., Hollis, N., & Wassersug, R. J. (2016). Tamoxifen in men: a review of adverse events. Andrology, 4(5), 776–788. [DOI:10.1111/andr.12197]:
Five RCTs have investigated the effect of tamoxifen on men with infertility, and the ages of the participants ranged between 18 and 44 years old (unspecified in two studies). These men (n = 321) were treated daily with 20 or 30 mg tamoxifen for 3–6 months. Three studies reported minimal or no side effects (AinMelk et al., 1987; Maier & Hienert, 1990; Cakan et al., 2009), one study (Kotoulas et al., 1994) reported three men lost their libido, and another study (Maier & Hienert, 1988) reported gastrointestinal events. From these studies, only three men stopped tamoxifen treatment, but the reason was not specified.
Studies in women indicate that genetic variants of cytochrome P450 enzymes may be associated with the presence and severity of tamoxifen AEs. For example, one study on postmenopausal women with ER-positive breast cancer reported that women with CYP2D6 phenotypes, that either are poor or intermediate metabolizer of tamoxifen, have increased risk of developing hot flashes (Regan et al., 2012). In another study, women who are carriers of a variant of CYP3A4 are more likely to experience severe hot flashes (Baxter et al., 2014). However, to date, it remains unclear whether cytochrome P450 enzymes genetic variations can influence the incidences and severity of such AEs in men.
Other AEs also seems to occur more frequently in women than in men including gastrointestinal problems, cardiovascular events, musculoskeletal problems, fatigue, and mood disturbances.
The mechanism(s) to account for the sex difference in tamoxifen AE is/are not understood. However, if this sex difference is real, one possible factor is a difference in activity of cytochrome P450 enzymes that metabolize tamoxifen in the liver (Anderson, 2008; Schmetzer & Florcken, 2012; Spoletini et al., 2012), and thus may influence the bioavailability of more potent tamoxifen metabolites. Furthermore, there may also be sex difference in the distribution of ERs in various organs, as already documented for different brain areas (Kruijver et al., 2003). However, one needs to keep in mind that the low incidence of reported AEs in men on tamoxifen may also be as a result of the fact that these AEs are not thoroughly evaluated in many studies, especially in the non-RCTs. A rigorous assessment of tamoxifen AEs in male populations has yet to be undertaken.
Kuiper et al. (1997)
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Häggblad, J., Nilsson, S., & Gustafsson, J. (1997). Comparison of the Ligand Binding Specificity and Transcript Tissue Distribution of Estrogen Receptors α and β. Endocrinology, 138(3), 863–870. [DOI:10.1210/endo.138.3.4979]
Note: Only skimmed – looked at relative binding affinities (RBAs) and noted no raloxifene – going to search for other new papers with more info on RBAs.
Rabe et al. (2016)
Rabe, T., Bruyniks, N., Merkle, E., Hadji, P., Kuhl, H., Ahrendt, H. J., Albring, C., Bitzer, J., Egarter, C., Kiesel, L., König, K., Merki Feld, G., Mueck, A. O., Sänger, N., Windler, E. (2016). Selective Estrogen Receptor Modulators–an Update (Joint Statement by the German Society for Gynecological Endocrinology and Reproductive Medicine [DGGEF] and the German Professional Association of Gynecologists [BVF]). Journal für Reproduktionsmedizin und Endokrinologie-Journal of Reproductive Medicine and Endocrinology, 12(4), 287–317. [Google Scholar] [URL] [PDF]:
It is probable that due to the binding of the SERMs to the ER certain co-factors are activated which, either as co-activators or as co-repressors, lead to different gene activations and deactivations in target tissues. Alternative explanations relate to a different affinity to ER and ER or activation of ER and ER, respectively.
Receptor Distribution Both ERs are expressed in various tissue types, but there are some notable differences in their expression pattern [4]: – ERα: endometrium, breast cancer cells, stromal cells of the ovary and hypothalamus [5] – ERβ: kidney, brain, bone, heart, lung, intestinal mucosa, prostate, and endothelial cells [6]
Polymorphism: Various polymorphisms of ERα- and ERβ-gene have been described.
Various ligands differ in their affinity for the alpha and beta isoforms of the estrogen receptor: – 17-estradiol binds equally well to both receptors – Estrone and raloxifene preferentially bind to the receptor alpha (ERα) – Estriol and genistein preferentially bind to the beta receptor (ERβ)
Selective estrogen receptor modulators preferably bind to the α- or β-subtype of the receptor. In addition, the different estrogen receptor combinations can react differently to the different ligands, thus tissue-specific agonist and antagonist effects arise [7]. The ratio of the concentration of α- to β-subtype plays a role in certain diseases.
The concept of selective estrogen receptor modulators is based on the ability to influence the interaction with other ER proteins such as transcriptional co-activators or co-repressors, based on the conformational changes in the ligand-receptor complex which is different between SERMs and estrogen and also different for different SERMs. Since the ratio of co-activators and co-repressors is different in various tissues [9], the same ligand may act as an agonist in one tissue (provided that the co-activators predominate) and as an antagonist in other tissues (where co-repressors dominate).
Note: Also worth pointing out that it’s tamoxifene’s metabolites that are the active ingredients and should probably have been included here.
Tamoxifene induces the recruitment of co-repressors to target gene promoters in mammary cells. In endometrial cells, tamoxifene, in contrast to raloxifene, acts like an estrogen by stimulating the recruitment of co-activators to a subset of genes.
Raloxifene induces the recruitment of co-repressors to target gene promoters in mammary cells [9].
Raloxifene: Non-genomic mechanisms of endothelial nitric oxide synthase activation by the selective estrogen receptor modulator raloxifene have been found by Simoncini et al (2002) [29]. This pathway might be important to understand the different effects of SERMs on the cardiovascular system.
[Tamoxifen:] Transcription of E-responsive genes(s) is attenuated because AF2 is inactive, and co-activator binding is attenuated by the T-ER complex; partial agonist activity results from AF1, which remains active in the T-ER complex.
[New SERM:] Afimoxifene: (4-hydroxytamoxifene) is a selective estrogen receptor modulator which is the active metabolite of tamoxifene [66]. Afimoxifene is a transdermal gel formulation and is being developed by Ascend Therapeutics, Inc. under the trademark TamoGel. Afimoxifene has completed a phase II clinical trial for the treatment of cyclical mastalgia [198].
Gruber et al. (2002)
Gruber, C. J., Tschugguel, W., Schneeberger, C., & Huber, J. C. (2002). Production and actions of estrogens. New England Journal of Medicine, 346(5), 340–352. [DOI:10.1056/NEJMra000471]:
Pickar, Boucher, & Morgenstern (2018)
Pickar, J. H., Boucher, M., & Morgenstern, D. (2018). Tissue selective estrogen complex (TSEC): a review. Menopause, 25(9), 1033–1045. [DOI:10.1097/gme.0000000000001095]:
The combination of estrogens and a SERM is sometimes referred to as a tissue selective estrogen complex (TSEC), although this term is not formally recognized by health authorities.
The question then arose, in the late 1990s, as to whether a combination of SERMs and estrogens might produce estrogen agonist and antagonist effects distinct from that of either component alone and come closer to achieving that goal.54
A key finding from the preclinical “proof of concept” investigation in ovariectomized rats was that BZA could completely counter CE’s stimulatory effects on the endometrium (based on uterine wet weight) when adequately dosed without attenuating CE’s beneficial effects on vasomotor instability, lipids, or bone,54 which supported clinical evaluation of this combination.
In a clinical trial of raloxifene 60 mg/d combined with oral 17β-estradiol 1 mg/d (n = 61) compared with raloxifene alone (n = 62) in postmenopausal women with prior HT use, the TSEC combination significantly (P < 0.001) reduced the frequency of VMS compared with baseline and with raloxifene alone.
It may be possible to create a TSEC combination that uses 17β-estradiol rather than CE in combination with a SERM; however, results cannot be assumed to be comparable to those with CE/BZA without clinical testing. Due to the unique profile of each TSEC, each individual TSEC combination needs to be evaluated separately in clinical studies to identify appropriate doses and assess safety and efficacy.
Liu, Han, & Smith (2013)
Liu, S., Han, S. J., & Smith, C. L. (2013). Cooperative Activation of Gene Expression by Agonists and Antagonists Mediated by Estrogen Receptor Heteroligand Dimer Complexes. Molecular Pharmacology, 83(5), 1066–1077. [DOI:10.1124/mol.112.084228]:
Moreover, antagonist-bound ERs adopt a distinct conformation that enables them to preferentially interact with corepressors rather than coactivators (Huang et al., 2010), thereby reinforcing their negative regulatory properties.
We demonstrate that ER agonist/antagonist combined treatment can cooperatively activate gene expression through an ER-HLD complex consisting of one receptor monomer bound to agonist and another occupied by antagonist. This cooperative activation of gene expression can be enhanced by an SRC-3 coactivator, and requires both ligand-bound subunits to bind to DNA and both AF-1 domains within the ER-HLD for maximal transcriptional activity. Moreover, ER-HLD complexes can activate transcription in the context of a natural promoter, and taken together, these findings demonstrate that ERs bound to different classes of ligands can form dimers that promote gene expression.
Crystal structures of the ERα LBDs bound to either E2 or raloxifene reveal that both ligands bind to the same pocket, and that the overall homodimeric arrangement is the same regardless of whether the LBD is agonist- or antagonist-bound.
Relative to costimulation by agonist/antagonist, the AF-1 domain is important for the partial agonist activity of SERMs such as tamoxifen (McInerney et al., 1996), and SRC-1 can bind to both the AF-1 and AF-2 domains of ERα through the coactivator’s Q-rich region and LXXLL motifs, respectively (Mérot et al., 2004). This raises the possibility that SRC-3 interacts with ER-HLDs through different AF domains on distinct subunits [e.g., AF-2 on agonist-bound ER(GSCKV) and AF-1 on antagonist-bound ER-G521R] to induce maximal transcriptional activity.
Thornton (2007)
Thornton, J. (2007). Effect of estrogens on skin aging and the potential role of SERMs. Clinical Interventions in Aging, 2(3), 283–297. [DOI:10.2147/cia.s798]:
A number of studies have shown that estrogens have many important beneficial and protective roles in skin physiology (reviewed in Thornton 2002, 2005). They have been shown to accelerate cutaneous wound healing (Ashcroft and Ashworth 2003), while a significant number of women notice an improvement in inflammatory skin disorders such as psoriasis during pregnancy (Dunna and Finlay 1989; Boyd et al 1996; Raychaudhuri et al 2003). Estrogens also offer some degree of protection against skin photoaging (Weinstock 1994; Tsukahara et al 2001, 2004) and epidemiological studies indicate that the mortality rates from both non-melanoma skin cancers (Weinstock 1994) and melanoma (Miller and MacNeil 1997) are significantly lower in women.
Many women report a sudden onset of skin aging several months after menopausal symptoms begin. The menopause causes hypoestrogenism, accelerating age-related deterioration, which results in thinner skin, an increase in number and depth of wrinkles, increased skin dryness, and decreased skin firmness and elasticity (Brincat 2000). Hormone replacement therapy (HRT) has been shown to increase epidermal hydration, skin elasticity, skin thickness (Sator et al 2001), and also reduces skin wrinkles (Phillips et al 2001). Furthermore, the content and quality of collagen and the level of vascularization is enhanced (Brincat et al 1987).
More recently, specific antibodies have demonstrated ERβ, but not ERα is expressed by dermal fibroblasts in the papillary dermis of human scalp skin in both sexes (Thornton et al 2003), whereas primary cultures of human dermal fibroblasts from female skin have been shown to express both mRNA and protein for ERα and ERβ (Haczynski et al 2002).
In addition, in female hair follicles the phytoestrogen, genistein inhibits hair shaft elongation to a similar extent as 17β-estradiol. Since genistein preferentially binds to ERβ, this opens the possibility that the inhibition of hair growth in response to 17β-estradiol may be mediated via ERβ rather than ERα (Nelson 2006). Therefore the development of selective estrogen receptor ligands may provide important clinical applications for the prevention and treatment of disorders of hair growth.
A histopathological assessment of rat skin following subcutaneous administration of tamoxifen observed that tamoxifen treatment resulted in the appearance of abnormal hair follicles, epidermal atrophy and increased dermal fibrosis, particularly around the hair follicles (Inaloz et al 2002). There have been reports of tamoxifen treatment causing diffuse thinning of the hair with moderate receding of the frontal hair line (Ayoub et al 1997) and the development of alopecia on the crown, which was reversed when treatment was stopped.
We have recently reported that tamoxifen alone has no effect on human hair shaft elongation in organ culture, suggesting it is not an estrogen agonist (Nelson 2006). However, a 10-fold excess of tamoxifen incubated in combination with 17β-estradiol eliminated the inhibitory effect of 17β-estradiol, suggesting that tamoxifen acts as an antagonist of estrogen in the female scalp hair follicle (Nelson 2006).
Another study has shown that raloxfine has a stronger positive stimulatory effect on collagen biosynthesis than 17β-estradiol (Surazynski et al 2003) and that in contrast to estradiol, raloxifene inhibits the expression of MMP-9.
Mauvais-Jarvis, Clegg, & Hevener (2013)
Mauvais-Jarvis, F., Clegg, D. J., & Hevener, A. L. (2013). The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews, 34(3), 309–338. [DOI:10.1210/er.2012-1055]:
However, tamoxifen treatment is associated with an increased risk of developing fatty liver (steatosis) (337–339), with approximately 43% of breast cancer patients treated with tamoxifen developing hepatic steatosis (339). The exact mechanism by which this occurs is still unclear.
In the hypothalamus, tamoxifen appears to act as an ER agonist.
In OVX mice, raloxifene reversed OVX-induced increases in food intake, body weight, fat mass, and hyperleptinemia to an extent similar to that of E2. This suggests that in rodents, raloxifene acts as an ER agonist in hypothalamic neurons and fat (346). In postmenopausal women, raloxifene prevents the shift from android to gynoid fat distribution, increases in abdominal adiposity (347), as well as total increases in adiposity (348). The absence of significant effect on body weight in women (349) could be due to the concomitant increase in lean mass and water after raloxifene treatment (350).
Nonetheless, in healthy and diabetic postmenopausal women, short-term raloxifene treatment did not impact fasting glucose, glucose tolerance, or indices of-cell function and sensitivity (351, 352). However, it decreased hepatic insulin extraction and, as a result, increased insulin half-life (352). Additionally, in the Multiple Outcomes of Raloxifene Evaluation trial, 3-year raloxifene treatment of postmenopausal women with or without type 2 diabetes reduced total cholesterol and LDL-cholesterol but had no effect on glycemic control compared to placebo (349). Thus, there is no argument for an effect of raloxifene in improving glucose homeostasis in postmenopausal women with type 2 diabetes. In fact, women with a previous history of hypertriglyceridemia who receive oral estrogen therapy are at risk for clinically relevant progression in this existing risk factor during raloxifene therapy.
In preclinical studies, the TSEC partnering bazedoxifene with CEE seemed to provide these benefits. Thus far, the effect of TSEC partnering bazedoxifene on glucose and energy metabolism is unknown.
Tommaselli et al. (2006)
Tommaselli, G. A., Di Carlo, C., Di Spiezio Sardo, A., Bifulco, G., Cirillo, D., Guida, M., Capasso, R., & Nappi, C. (2006). Serum leptin levels and body composition in postmenopausal women treated with tibolone and raloxifene. Menopause, 13(4), 660–668. [DOI:10.1097/01.gme.0000227335.27996.d8]:
At the end of the study, untreated women showed increased total and percentage of fat mass and increased serum leptin levels compared with baseline values, whereas women treated with HT showed no significant changes. These data seem to indicate that the main determinant of leptin levels in postmenopausal women is body fat content, whereas hypoestrogenism does not seem to influence these levels.
Women in group C showed no significant changes in fat mass, in total and at all areas evaluated, after 1 year of treatment with raloxifene (Fig. 1A-D). No significant changes were observed in lean mass, both total and at all areas evaluated (Fig. 2A-D). The same was observed for the percentage of fat mass (Fig. 3A-D).
We did not detect significant changes in serum leptin levels in subjects in group B and C throughout the study (group B: 16.3 [4.2/39.8] ng/mL vs 17.0 [5.6 /28.7] ng/mL; group C: 15.1 [6.7/26.5] ng/mL vs 17.5 [6.7/29.8] ng/mL).
The present study, although hampered by the limited sample size, seems to confirm that postmenopausal hypoestrogenism leads to increased fat content and suggests that both raloxifene and tibolone may have a role in preventing the changes of body composition induced by postmenopausal hypoestrogenism.
Francucci et al. (2005)
Francucci, C. M., Pantaleo, D., Iori, N., Camilletti, A., Massi, F., & Boscaro, M. (2005). Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women. Journal of Endocrinological Investigation, 28(9), 623–631. [DOI:10.1007/bf03347261]:
For the first time, our results suggest that RLX, most likely because of its estrogen-agonist action, might prevent the shift to a more central fat dis- tribution associated with menopause. In fact, we have found in RLX-users, in relation to controls, no substantial changes in body weight during the 12 months of treatment, a significant lower fat mass in trunk and abdominal region (ROI-L2-4) and a trend towards higher fat depots in legs. At variance, after 1 yr we observed in untreated post-menopausal women a not significant increase of body weight and fat mass in central and trunk region with a decrease in legs. Therefore, RLX could have, similar to the estrogens, different effects on several regional fat tissues. In fact, in pre-menopausal women the gluteo-femoral adipocytes have a low lipolytic activity with tendency towards larger fat cells in the femoral region, while the adipocytes in the central fat depot have a high basal lipolysis with smaller fat cells (32). Besides, the estrogens would seem to enhance the lipoprotein-lipase (LPL) activity, the key enzyme in the regulation of fatty acid deposi- tion, of gluteofemoral fat cells and to decrease the visceral LPL activity (33).
Jacobsen et al. (2010)
Jacobsen, D. E., Samson, M. M., Emmelot-Vonk, M. H., & Verhaar, H. J. (2010). Raloxifene and body composition and muscle strength in postmenopausal women: a randomized, double-blind, placebo-controlled trial. European Journal of Endocrinology, 162(2), 371–376. [DOI:10.1530/eje-09-0619]:
Repeated measurement analysis showed there to be a significant increase in FFM at 3, 6, and 12 months in the raloxifene group (P=0.05) as well as an increase in TBW (P=0.02; Table 2). At 12 months, the mean increase in FFM was 0.83 (2.4) kg in the raloxifene group compared with 0.03 (1.5) kg in the placebo group (P=0.05), and the mean increase in TBW was 0.6 (1.8) litres compared with a decrease of 0.06 (1.1) litres in the placebo group (P=0.02).
In our study, we found an increase in the FFM after supplementation with raloxifene, which could be partly explained by the increase in the TBW content, a known effect of raloxifene supplementation. However, the increase in the TBW content could not explain the whole increase in the FFM, and also the amount of physical activity was comparable between both groups during the study. This means that there is also a real change in body composition with an increase in lean body mass. The results of our study are comparable with some other studies, but in most of these studies there were problems with the design of the study (no blinding, no placebo group, too small groups, and a too short intervention period).
The increase in FFM was not accompanied by an increase in muscle strength and muscle power in our study. Muscle strength is a key factor in maintaining independence in elderly people. There are no other studies concerning raloxifene and muscle strength or power. Studies concerning HRT (estrogen and progestagens) show conflicting results: two randomized controlled trials with no effect (38, 39) and three randomized controlled trials with significant increase in muscle strength (11–13).
Komm & Mirkin (2014)
Komm, B. S., & Mirkin, S. (2014). An overview of current and emerging SERMs. The Journal of Steroid Biochemistry and Molecular Biology, 143, 207–222. [DOI:10.1016/j.jsbmb.2014.03.003]:
Raloxifene binds with high affinity to ERα, with 46% relative binding affinity for human ERα compared with 17β-estradiol, and with a 26% relative binding affinity for rat ERβ compared with 17β-estradiol
Note: A lot more on this article but this was the only relevant thing I really cared about at the time.
Smith & O’Malley (2004)
Smith, C. L., & O’Malley, B. W. (2004). Coregulator Function: A Key to Understanding Tissue Specificity of Selective Receptor Modulators. Endocrine Reviews, 25(1), 45–71. [DOI:10.1210/er.2003-0023]:
For instance, in the absence of endogenous estrogens, tamoxifen frequently exhibits weak estrogenic activity, such as modest stimulation of uterine wet weight and bone density in ovariectomized rats, whereas in the presence of estradiol, it can serve as an antiestrogen, inhibiting responses to a level corresponding to the comparatively modest agonist activity of tamoxifen itself.
However, because neither tamoxifen nor raloxifene possesses significant estrogen-like activity in the CNS, there is clearly a market for other SERMs to fill this niche.
It is also noteworthy that SERMs likely exist in nature. For instance, the estrone metabolite, Δ8,9- dehydroestrone sulfate suppresses hot flushes in postmenopausal women, an estrogenic action. However, it is unable to significantly affect certain other parameters associated with estrogenic responses such as total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol (23).
[…] raloxifene and 4HT block the ligand-activated AF-2 domain and particularly in the case of 4HT, leave AF-1 able to initiate gene expression.
As a result, 4HT and raloxifene stimulate gene expression in some, but not all, contexts, and these antiestrogens are therefore classified as SERMs.
For ER, 4HT and raloxifene also inhibit its AF-2 domain, and due to the relatively poor AF-1 activity of the receptor, these ligands generally block ER transcriptional activity measured on EREs (61, 78, 79).
It should be noted that 4HT and raloxifene also exert effects on the transcriptional activity of both ER and ER tethered to DNA indirectly through interaction with other transcription factors such as activator protein 1 (AP-1) and Sp1 (80, 81). In this context, the agonist activities of these ligands also is apparent in a cell-specific manner.
For ERE-dependent gene expression, tamoxifen is a partial agonist of ER but is generally unable to stimulate ER transcriptional activity (61, 171, 173). Conversely, when assessing ER activity on AP-1 containing reporter genes, tamoxifen will stimulate ER and ER transcriptional activity.
Carneiro, de Cassia de Maio Dardes, & Haidar (2012)
Carneiro, A. L., de Cassia de Maio Dardes, R., & Haidar, M. A. (2012). Estrogens plus raloxifene on endometrial safety and menopausal symptoms—semisystematic review. Menopause, 19(7), 830–834. [DOI:10.1097/gme.0b013e31824a74ce]:
Most of the studies found a benefit profile with association of RLX and E on women’s quality of life, satisfaction with the treatment, and vaginal dryness. Nevertheless, different qualityof-life scales and visual scales were used to access mainly vasomotor symptoms, which could limit further comparison.
Arevalo et al. (2010)
Arevalo, M. A., Santos-Galindo, M., Lagunas, N., Azcoitia, I., & Garcia-Segura, L. M. (2010). Selective estrogen receptor modulators as brain therapeutic agents. Journal of Molecular Endocrinology, 46(1), R1–R9. [DOI:10.1677/jme-10-0122]:
The neuroprotective actions of tamoxifen and raloxifene, two SERMs that are currently used in clinical practice for the treatment of breast cancer and osteoporosis, have been assessed in different experimental models of neural dysfunction. These include animal models of traumatic injury of the central nervous system and peripheral nerves, stroke, multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, cognitive decline, and mood disorders (Fig. 1). This section is a succinct description of the main findings of these experimental studies, including the limited available information from human studies.
In spite of the neuroprotective actions of tamoxifen in different forms of neural injury, it is unclear whether this molecule may have some benefits for cognition in humans. Indeed, several studies suggest an increased risk of cognitive impairment in women receiving tamoxifen for the treatment of breast cancer, including worse performance in visual memory, word fluency, immediate verbal memory, visuospatial ability, and processing speed tasks (Paganini-Hill & Clark 2000, Shilling et al. 2003, Palmer et al. 2008, Phillips et al. 2010, Schilder et al. 2010). However, other studies have not detected a significant effect of tamoxifen on cognition (Debess et al. 2010).
In contrast to the potential negative effects of tamoxifen on cognition, the results of the multiple outcomes of raloxifene evaluation randomized trial suggest that raloxifene prevents cognitive decline in postmenopausal women (Yaffe et al. 2005). The results of a recent randomized, double-blind, placebo-controlled trial also suggest that raloxifene improves verbal memory in late postmenopausal women ( Jacobsen et al. 2010). In addition, raloxifene treatment enhances brain activation during performance on a face-encoding paradigm and during recognition of familiar items in healthy elderly men.
In agreement with this possibility, we have recently observed that both tamoxifen and raloxifene improve hippocampus-dependent memory in androgen-deprived male rats (N Lagunas, I Calmarza-Font, D Grassi & LM Garcia-Segura, unpublished observations).
Tamoxifen reduces amphetamine-induced manic-like behavioral alterations in rats (Einat et al. 2007) and reduces acute manic episodes in women with bipolar affective disorder (Kulkarni et al. 2006, Zarate et al. 2007). Raloxifene reduces anxiety behavior, assessed in the elevated plus maze test, on ovariectomized rats (Walf & Frye 2010) and decreases anxiety (Strickler et al. 2000, Florio et al. 2001) and depression (Carranza-Lira et al. 2004, Grigoriadis et al. 2005, Sugiyama et al. 2007) in postmenopausal women. Recent clinical studies also suggest the potential application of raloxifene hydrochloride (120 mg/day oral) for the treatment of postmenopausal women with schizophrenia.
Other neuroprotective mechanisms of SERMs are mediated by classical ERs, since they are inhibited by ER antagonists, such as ICI 182 780 (Zhang et al. 2009). The neuroprotective signaling of SERMs may involve the activation of kinases, such as mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and Akt (Du et al. 2004, Lee et al. 2009a,b), and the phosphorylation of CREB (Sharma et al. 2007) or the inhibition of nuclear factor (NF)-kB-induced transcription (Cerciat et al. 2010). Through these mechanisms, SERMs control synaptic transmission and the expression of molecules involved in the regulation of cell death, oxidative stress, and inflammation.
Raloxifene may also regulate opiate and GABAergic neurotransmission by the modulation of the levels of b-endorphin and neuroactive steroids respectively. Chronic raloxifene administration to postmeInopausal women increases plasma levels of b-endorphin and tetrahydroprogesterone (allopregnanolone), an anxiolytic metabolite of progesterone that modulates GABAA receptors.
These findings suggest that raloxifene may regulate synaptic function by the modulation of local levels of neuroactive substances within the brain.
In general, there is still poor knowledge of the precise molecular targets of SERMs in the nervous system. Although some key molecules have been identified, such as MAPK, PI3K/Akt, CREB, and NF-kB, the molecular mechanisms involved in the neuroprotective actions of SERMs should be investigated with more detail in the different cellular populations of the nervous system.
Lawrence et al. (2004)
Lawrence, S. E., Arnold Faught, K., Vethamuthu, J., & Lawson, M. L. (2004). Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. The Journal of Pediatrics, 145(1), 71–76. [DOI:10.1016/j.jpeds.2004.03.057]:
Results: Mean (SD) age of treated subjects was 14.6 (1.5) years with gynecomastia duration of 28.3 (16.4) months. Mean reduction in breast nodule diameter was 2.1 cm (95% CI 1.7, 2.7, P < .0001) after treatment with tamoxifen and 2.5 cm (95% CI 1.7, 3.3, P < .0001) with raloxifene. Some improvement was seen in 86% of patients receiving tamoxifen and in 91% receiving raloxifene, but a greater proportion had a significant decrease (>50%) with raloxifene (86%) than tamoxifen (41%). No side effects were seen in any patients.
Surazynski et al. (2003)
Surazynski, A., Jarzabek, K., Haczynski, J., Laudanski, P., Palka, J., & Wolczynski, S. (2003). Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. International Journal of Molecular Medicine, 12(5), 803–809. [DOI:10.3892/ijmm.12.5.803]:
Raloxifene had stronger positive stimulative effects on collagen biosynthesis, through different biochemical mechanisms, than estradiol in human skin fibroblasts and might reverse some of the postmenopausal changes in skin or connective tissue. Increase of collagen synthesis induced by raloxifene may be activated by both estrogen receptor dependent and independent pathways such as up-regulation of estrogen receptors, up-regulation of IGF receptor, transcriptional regulation of collagen genes by estrogen receptor-raloxifene complex, increasing of prolidase activity or finally by inhibition of MMP-9 expression.
\ No newline at end of file
+Sources/Excerpts: A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy - Transfeminine Science
Sources/Excerpts: A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy
By Lain | First published October 5, 2019 | Last modified June 3, 2021
Preface
This page is a sources and excerpts supplement to the main article which can be found here.
Maximov, Lee, & Jordan (2013)
Maximov, P. Y., Lee, T. M., & Jordan, V. C. (2013). The Discovery and Development of Selective Estrogen Receptor Modulators (SERMs) for Clinical Practice. Current Clinical Pharmacology, 8(2), 135–155. [DOI:10.2174/1574884711308020006]:
However, although these SERMs have many benefits, they also have some potentially serious adverse effects, such as thromboembolic disorders and, in the case of tamoxifen, uterine cancer. These adverse effects represent a major concern given that long-term therapy is required to prevent osteoporosis or prevent and treat breast cancer.
The search for the ‘ideal’ SERM, which would have estrogenic effects on bone and serum lipids, neutral effects on the uterus, and antiestrogenic effects on breast tissue, but none of the adverse effects associated with current therapies, is currently under way.
Note: Serum lipids are like HDL/LDL cholesterol.
There are two ERs referred to as α and β [82-84]. Each receptor protein is encoded on different chromosomes, and have homology as members of the steroid receptor superfamily. There are distinct patterns of distribution and distinct and subtle differences in structure and ligand binding affinity [85]. The ratio of ERα and ERβ at a target site may be an additional dimension for tissue modulation. A high ERα: ERβ ratio correlates well with high levels of cellular proliferation whereas the predominance of functional ERβ over ERα correlates with repression of proliferation [86-89]. Indeed, the ratio of ERs in normal and neoplasic breast tissue could be important for the long-term success of chemoprevention with SERMs.
Kraichely and co-workers [97] demonstrated the important observation that agonists for ERα and ERβ produce subtle quantitative differences with the interaction of members of the SRC family (SRC 1, 2 and 3) and that the coactivator can enhance ligand affinity for the ER.
Evidence has now accumulated to document that the broad spectrum of ligands that bind to the ER can create a broad range of ER complexes that are either fully estrogenic or antiestrogenic at a particular target site [100]. Thus a mechanistic model of estrogen action and antiestrogen action (Fig. 2) has emerged based on the shape of the ligand that programs the complex for future action.
Thus for effective gene transcription, programmed and targeted by the shape and phosphorylation status of the ER and coactivators, a dynamic and cyclic process of remodeling capacity is required for transcriptional assembly [103] that is immediately followed by the routine destruction of transcription complexes by the proteasome. Estrogen and SERM-ER complexes have distinct accumulation patterns in the target cell nucleus [104,105] because they are destroyed at different rates.
Raloxifene lacks estrogenic activity in the uterus and has not demonstrated tamoxifen-like effects in the uterus either histopathologically or ultrasonographically [116], but it has been associated with adverse effects such as VTE and vasomotor symptoms, including hot flushes.
Perhaps the reason for this difference in effect is related to differences on the agents’ effect on inflammation as the agents influence C-reactive protein (CRP) differently. Estrogen and tibolone increase levels of CRP [192], raloxifene and arzoxifene have no effect on CRP levels, and lasofoxifene decreases CRP levels [55]. All decrease LDL levels.
Brzozowski et al. (1997)
Brzozowski, A. M., Pike, A. C., Dauter, Z., Hubbard, R. E., Bonn, T., Engström, O., Ohman, L., Greene, G. L., Gustafsson, J. A., & Carlquist, M. (1997). Molecular basis of agonism and antagonism in the oestrogen receptor. Nature, 389(6652), 753–758. [DOI:10.1038/39645]:
The overall architecture of the ER LBD (helices H3–H12) is similar to that seen in the crystal structures of other nuclear receptor LBDs6–8, and emphasizes the universal nature of this fold within this receptor superfamily9. The LBD is folded into a three-layered antiparallel a-helical sandwich comprising a central core layer of three helices (H5/6, H9 and H10) sandwiched between two additional layers of helices (H1–4 and H7, H8, H11). This helical arrangement creates a ‘wedge-shaped’ molecular scaffold that maintains a sizeable ligandbinding cavity at the narrower end of the domain. The remaining secondary structural elements, a small two-stranded antiparallel β-sheet (S1 and S2) and H12, are located at this ligand-binding portion of the molecule, and flank the main three-layered motif (Fig. 1a).
E2 binds diagonally across the cavity between H11, H3 and H6 and adopts a low-energy conformation. The phenolic hydroxyl of the A-ring (O3; see Fig. 2c for atom numbering) nestles between H3 and H6 and makes direct hydrogen bonds to the carboxylate of Glu 353, the guanidinium group of Arg 394, and a water molecule. The 17-β hydroxyl (O17) of the Dring makes a single hydrogen bond with His 524 in H11. The remainder of the molecule participates in a number of hydrophobic contactsthat are concentrated overthe A, A/B interface and D-rings.
The ER is unique among the steroid receptors in its ability to embrace a wide variety of non-steroidal compounds. Although the ‘pincer-like’ arrangement around the A-ring imposes an absolute requirement on effective ligands to contain an aromatic ring, the remainder of the binding pocket can accept a number of different hydrophobic groups12,13. This overall promiscuity can be attributed to the size of the cavity, which has a probe accessible volume (450 A˚3) nearly twice that of E2’s molecular volume (245 A˚3). The length and breadth of the E2 skeleton is well matched by the receptor, but there are large unoccupied cavities opposite the α face of the B-ring and the β face of the C-ring (Fig. 2a). The positions of these preformed cavities are similar to those predicted from binding studies12.
Note: This may be why things like bisphenol A (BPA) and other random chemicals bind to the estrogen receptor.
Rosano et al. (2011)
Rosano, C., Stec-Martyna, E., Lappano, R., & Maggiolini, M. (2011). Structure-Based Approach for the Discovery of Novel Selective Estrogen Receptor Modulators. Current Medicinal Chemistry, 18(8), 1188–1194. [DOI:10.2174/092986711795029645]:
These two zinc fingers are responsible for the binding of ERα to the estrogen response elements (EREs) located in the promoter regions of target genes. The P-box sequence is identical in the two ER isoforms, suggesting that ERα and ERβ bind EREs with similar specificity and affinity.
Pure anti-estrogen development has also included a further class of molecules represented by the inhibitors of aromatase enzymatic activity, which increases estrogen availability at tissue levels converting androgens into estrogens.
4OHT binds to ERs within the same binding pocket that recognises E2 and produces a nuclear complex which decreases DNA synthesis and inhibits the action of estrogens [24]. Moreover, the complex between ER and 4OHT recruits co-repressor proteins like NCoR and SMRT, which contribute to the antagonist effects of 4OHT [78].
In particular, the position of H12 shifts from the ligand binding pocket to cover a part of the co-activator binding groove, which is formed by residues belonging to H3, H4 and H5 [24]. The receptor recognition of 4OHT is dictated by the phenolic “A ring” and the bulky side chain.
[…] whilst the bulky side chain protruding from the binding cavity displaces H12 and allows a conformation that inhibits the coactivator recruitment.
Next, the long hydrophobic side chain of RAL makes extensive contacts with the hydrophobic residues of the receptor allowing the displacement of H12 similar to 4OHT.
Comparable to other SERMs, Lasofoxifene diverts the receptor from its agonistbound conformation by displacing the C-terminal AF-2 helix 12 into the site at which the LXXLL motif of coactivator proteins would otherwise be able to bind. LAS achieves this effect by occupying the space normally filled by residue Leu540, as well as by modulating the conformation of residues of helix 11.
In addition to these features of SERM recognition, a large alkyl “pendant” side chain, terminating in a pyrollidine head group, threads its way from the ligand-binding pocket out toward the surface of the protein, where it directly interferes with the correct positioning of the AF-2 helix.
Nilsson & Gustafsson (2010)
Nilsson, S., & Gustafsson, J. (2010). Estrogen Receptors: Therapies Targeted to Receptor Subtypes. Clinical Pharmacology & Therapeutics, 89(1), 44–55. [DOI:10.1038/clpt.2010.226]:
Like other members of the nuclear receptor family, the ERs are best known as transcription factors, regulating the transcriptional activity of target genes in cells in response to the natural hormone 17β-estradiol (E2) or to synthetic nonhormonal agonists and antagonists1 (Figure 1). In more recent years, we have also learned that the effects of E2 can have a very rapid onset, within seconds to minutes. These effects may be mediated by membrane-localized ERα and ERβ, respectively (after exposure to ligand stimuli), or by membrane receptors that are genetically and structurally unrelated to ERα and ERβ, for example, GPR30 (G-protein coupled receptor 30).
An increase in the expression of ERβ relative to ERα was shown to inhibit the ERα response to E2, silence the partial ERα-mediated agonism of tamoxifen, and potentiate its antagonism.10 In mice, the level of expression of genes induced by E2 in bone were, on average, 85% higher in the absence of ERβ than in its presence, thereby suggesting that ERβ has an opposing effect on ERα-mediated gene transcription.11
In some organs, ERα and ERβ are expressed at similar levels, sometimes in different cell types within the same organ, whereas in others, one or the other subtype predominates. ERα is predominantly expressed in the uterus, prostate (stroma), ovary (theca cells), testes (Leydig cells), epididymis, bone, breast, liver, kidney, white adipose tissue, and various regions of the brain. ERβ is predominantly expressed in the colon, prostate (epithelium), testis, ovary (granulosa cells), bone marrow, salivary gland, vascular endothelium, lung, bladder, and certain regions of the brain (Figure 3).1,4
In summary, the metabolic findings in knockout mice and in aromatase-deficient or ERα-deficient patients, and the use of ER subtype-selective ligands, suggest a major role for ERα in the maintenance of metabolic control. ERβ may, however, have an opposite effect, especially when unopposed, resulting in a perturbed energy balance and an increased risk for the development of diabetes. Unopposed ERβ activity is a possible explanation for the diabetic phenotype of the male patient with homozygous loss of ERα function. Both ER subtypes are reported to have roles in the prevention of atherosclerotic lesion development, although seemingly through different mechanisms.
SERMs such as tamoxifen and raloxifene and the pure antagonist ICI 182,780, however, antagonize the neuroprotective effects of E2 after experimentally induced ischemia.36
Note: It seems likely to me that in general oral raloxifene would not cause gynoid fat distribution due to its ERα antagonist and ERα-mediated anti-lipolytic pathways in subcutaneous fat – I was wrong.
Studies in the aromatase/αERKO recombinant mouse model confirmed the important role of ERα in the development of the mammary gland. Treatment of ovariectomized rats with the ERβ-selective agonist ERB-041 resulted in no observable effects on rat mammary tissue, suggesting that ERβ-selective agonists are nonmammotrophic.1
In summary, estrogens are of fundamental importance in the development of the mammary gland, and ERα is the primary ER subtype mediating the effects of estrogens. ERβ, however, may play an important role in controlling the mammotrophic effects of ERα and may be an important novel target for endocrine therapy in breast cancer.
In summary, tissues of the urogenital tract are dependent on estrogens for proper growth, development, and functional integrity. ERα is uterotrophic and mediates uterine proliferative responses to E2. It is also the ER subtype that plays the most important role in the feedback on the hypothalamic–pituitary–gonadal axis and the regulation of LH and follicle-stimulating hormone levels. ERβ has an important role in folliculogenesis and ovulation and may have very important therapeutic implications in the diseased prostate, as a drug target in benign prostatic hyperplasia and in both castrate-responsive and castrateresistant prostate cancer.
Greene (2000)
Greene, G. (2000). Comparative Structural Analysis of ERa and ERb Bound to Selective Estrogen Agonists and Antagonists. Chicago: The University of Chicago. [Google Scholar] [URL] [PDF]:
The structures of the LBD complexed with E2 and RAL show that, although both ligands bind at the same site within the core of the LBD (Brzozowski et al. 1997), each of these ligands induces a different conform”ation of helix 12. Whereas helix 12 in the E2-LBD complex packs against helices 3, 5/6, and 11 in a conformation that has been observed for the corresponding helix in other agonist-bound NR LBD structures, helix 12 in the RAL-LBD complex is bound in a hydrophobic groove composed of residues from helices 3 and 5. This alternative orientation of helix 12 partially buries residues in the groove that are necessary for AF-2 activity, suggesting that RAL and possibly other antagonists block AF-2 function by disrupting the topography of the AF-2 surface.
Shiau et al. (1998)
Shiau, A. K., Barstad, D., Loria, P. M., Cheng, L., Kushner, P. J., Agard, D. A., & Greene, G. L. (1998). The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell, 95(7), 927–937. [DOI:10.1016/s0092-8674(00)81717-1]:
Several proteins, including SRC-1/N-CoA1 (Onate et al., 1995; Kamei et al., 1996), GRIP1/TIF2/N-CoA2 (Hong et al., 1996; Voegel et al., 1996; Torchia et al., 1997;), p/CIP/RAC3/ACTR/AIB1 (Anzick et al., 1997; Chen et al., 1997; Li et al., 1997; Torchia et al., 1997), and CBP/ p300 (Hanstein et al., 1996), associate in a ligand-dependent manner with the ERa. These proteins have been classified as transcriptional coactivators because they enhance ligand-dependent transcriptional activation by the ERa as well as by several other NRs (Horwitz et al., 1996; Glass et al., 1997). SRC-1 and GRIP1 bind to the agonist-bound LBDs of both the human thyroid receptor b (TRb) and human ERa using the putative AF-2 interaction surface (Feng et al., 1998). Members of the p160 family of coactivators, such as SRC-1 and GRIP1, as well as other coactivators, recognize agonist-bound NR LBDs through a short signature sequence motif, LXXLL (where L is leucine and X is any amino acid), known as the NR box (Le Douarin et al., 1996; Heery et al., 1997; Torchia et al., 1997; Ding et al., 1998). Mutagenesis studies indicate that the affinity of coactivators for NR LBDs is determined principally, if not exclusively, by these NR boxes (Le Douarin et al., 1996; Heery et al., 1997; Torchia et al., 1997; Ding et al., 1998).
Rambhatla, Mills, & Rajfer (2016)
Rambhatla, A., Mills, J. N., & Rajfer, J. (2016). The Role of Estrogen Modulators in Male Hypogonadism and Infertility. Reviews in Urology, 18(2), 66–72. [PubMed] [PubMed Central] [DOI:10.3909/riu0711]:
To circumvent the inhibition of LH and FSH from the pituitary by exogenous testosterone, a common strategy used by male reproductive and sexual medicine specialists, in which the goal is to increase testosterone levels while maintaining spermatogenesis, is to use SERMs and/or AIs instead of exogenous testosterone.
Clomiphene citrate works as an estrogen antagonist at the level of the pituitary gland and thus stimulates the release of LH and FSH, which in turn drives both the steroidogenic and spermatogenic functions of the testes.
Other studies have also found that aside from improvement in serum testosterone levels, clomiphene therapy also leads to significant improvement in bone mineral density, as well as androgen deficiency in the aging male (ADAM) scores without any significant adverse events.
Tamoxifen citrate is another oral SERM that was approved in the 1970s for the treatment of breast cancer. It has tissue specific action and acts as an estrogen receptor blocker in breast tissue and exhibits agonistic properties in the bone and uterus.9 Although its use is primarily in women, in men’s health it is used off-label and acts as an estrogen antagonist in the hypothalamus and pituitary gland.23 Because of its mechanism of action, tamoxifen results in the inhibition of the negative feedback of estrogen at the hypothalamus and pituitary gland, and results in the release of LH and FSH, which in turn increases testosterone biosynthesis and “stimulates” spermatogenesis.
Tsourdi and colleagues23 looked at the effects of three SERMs—tamoxifen, toremifine, and raloxifene—on the hypothalamic-pituitary axis in men with oligospermia. They found that after 3 months of treatment with each of these SERMs, there was a statistically significant increase in serum gonadotropins, testosterone, and semen parameters.
Tamoxifen, given its estrogen antagonist properties in the breast, as well as AIs, has also been used in the treatment of gynecomastia. Much of the data stem from boys with pubertal gynecomastia and in men with prostate cancer on antiandrogen therapy; there is evidence that these medications are effective in the treatment of gynecomastia.43-45 Their use in the treatment of gynecomastia induced by exogenous testosterone therapy is largely anecdotal and not evidence based.46 However, the manipulation of estrogen levels in men may not be without consequences. We know that estrogen receptors are present throughout the body and play a role in bone health, body composition, cardiovascular well being, libido and sexual function, and testicular steroidogenesis and spermatogenesis.47-49 Although a high estradiol level may have a negative impact on fertility, a level that is excessively low may not be desirable either.
Kunath et al. (2012)
Kunath, F., Keck, B., Antes, G., Wullich, B., & Meerpohl, J. J. (2012). Tamoxifen for the management of breast events induced by non-steroidal antiandrogens in patients with prostate cancer: a systematic review. BMC Medicine, 10(1), 96. [DOI:10.1186/1741-7015-10-96]:
Our results suggest that tamoxifen has a beneficial effect if compared to no treatment for the prevention of breast events. However, not all patients need prophylaxis to prevent the development of breast events induced by non-steroidal antiandrogen therapy [26,27], and not all patients with gynecomastia require treatment [28]. Therefore, a patient-oriented, pragmatic approach appears reasonable. This approach was also proposed by van Poppel and by Di Lorenzo et al. [14,15,27]. Before starting non-steroidal antiandrogen treatment (either with non-steroidal monotherapy or in combination with LHRH analogues), patients should be informed about the likelihood of breast events and about possible prophylactic therapy options. As recommended earlier by Di Lorenzo et al. [14,15], we also suggest that the physician could wait for the occurrence of breast events in selected patients. Prophylaxis should be started only if the patient is afraid of developing gynecomastia or breast pain.
Jover-Mengual et al. (2017)
Jover-Mengual, T., Castelló-Ruiz, M., Burguete, M. C., Jorques, M., López-Morales, M. A., Aliena-Valero, A., Jurado-Rodríguez, A., Pérez, S., Centeno, J. M., Miranda, F. J., Alborch, E., Torregrosa, G., & Salom, J. B. (2017). Molecular mechanisms mediating the neuroprotective role of the selective estrogen receptor modulator, bazedoxifene, in acute ischemic stroke: A comparative study with 17β-estradiol. The Journal of Steroid Biochemistry and Molecular Biology, 171, 296–304. [DOI:10.1016/j.jsbmb.2017.05.001]:
Note: This shows some variances and similarities in how various estrogen receptor pathways in the brain of mice are with estradiol versus bazedoxifene.
Simpkins et al. [2] provided the first evidence that estrogens (specifically 17β-estradiol, E2) exerted neuroprotective effects in the now widely used rodent model of AIS, namely the middle cerebral artery occlusion (MCAO) model. The authors suggested that estrogens may be a useful therapy to protect neurons against the damaging effects of stroke.
Bazedoxifene acetate (BZA) is the first of the third-generation SERMs approved for the treatment of postmenopausal women at risk for, or presenting with, osteoporosis in Europe and Japan [21]. Due to its favorable preclinical effects, BZA has been selected to combine with conjugated estrogens (CE) resulting in CE/BZA as a new progestin-free hormone therapy option for alleviating estrogen deficiency symptoms in postmenopausal women [22]. As for neural tissue, BZA had been reported to prevent neuronal loss in the hippocampus of rats exposed to excitotoxic kainic acid [23], and to decrease the inflammatory response of astrocytes exposed to lipopolysaccharide [24].
Both BZA and E2 significantly counteracted the ischemia-induced downregulation of ERα mRNA expression in the ipsilateral hemisphere.
As for the effect of the estrogenic compounds, BZA significantly counteracted the ischemia-induced downregulation of ERβ mRNA expression, while E2 was without significant effect.
As for the effect of the estrogenic compounds, neither BZA nor E2 had a significant effect on GPER mRNA expression in the ipsilateral hemisphere.
BZA but not E2 significantly attenuated the ischemia-induced endogenous phosphorylation of ERK1 in the ipsilateral hemisphere (Fig. 6A and B). While p-ERK2 levels were significantly reduced in the ipsilateral hemisphere in animals treated with BZA, there was no significant change in animals treated with E2.
The hypothesis that the neuroprotective mechanism of the SERM, tamoxifen, could be due to its ability to attenuate apoptotic cell death was first suggested and verified by Wakade et al. [11], and subsequently confirmed by Zou et al. [17]. By contrast, the SERM, raloxifene, has been recently reported to have no effect on the hypoxia-induced increase of caspase-3 activity in cultured hippocampal cells [20]. Therefore, our results lend support to the view that, as in the case of E2, SERMs (specifically BZA) are effective anti-apoptotic drugs in stroke.
Wibowo et al. (2016)
Wibowo, E., Pollock, P. A., Hollis, N., & Wassersug, R. J. (2016). Tamoxifen in men: a review of adverse events. Andrology, 4(5), 776–788. [DOI:10.1111/andr.12197]:
Five RCTs have investigated the effect of tamoxifen on men with infertility, and the ages of the participants ranged between 18 and 44 years old (unspecified in two studies). These men (n = 321) were treated daily with 20 or 30 mg tamoxifen for 3–6 months. Three studies reported minimal or no side effects (AinMelk et al., 1987; Maier & Hienert, 1990; Cakan et al., 2009), one study (Kotoulas et al., 1994) reported three men lost their libido, and another study (Maier & Hienert, 1988) reported gastrointestinal events. From these studies, only three men stopped tamoxifen treatment, but the reason was not specified.
Studies in women indicate that genetic variants of cytochrome P450 enzymes may be associated with the presence and severity of tamoxifen AEs. For example, one study on postmenopausal women with ER-positive breast cancer reported that women with CYP2D6 phenotypes, that either are poor or intermediate metabolizer of tamoxifen, have increased risk of developing hot flashes (Regan et al., 2012). In another study, women who are carriers of a variant of CYP3A4 are more likely to experience severe hot flashes (Baxter et al., 2014). However, to date, it remains unclear whether cytochrome P450 enzymes genetic variations can influence the incidences and severity of such AEs in men.
Other AEs also seems to occur more frequently in women than in men including gastrointestinal problems, cardiovascular events, musculoskeletal problems, fatigue, and mood disturbances.
The mechanism(s) to account for the sex difference in tamoxifen AE is/are not understood. However, if this sex difference is real, one possible factor is a difference in activity of cytochrome P450 enzymes that metabolize tamoxifen in the liver (Anderson, 2008; Schmetzer & Florcken, 2012; Spoletini et al., 2012), and thus may influence the bioavailability of more potent tamoxifen metabolites. Furthermore, there may also be sex difference in the distribution of ERs in various organs, as already documented for different brain areas (Kruijver et al., 2003). However, one needs to keep in mind that the low incidence of reported AEs in men on tamoxifen may also be as a result of the fact that these AEs are not thoroughly evaluated in many studies, especially in the non-RCTs. A rigorous assessment of tamoxifen AEs in male populations has yet to be undertaken.
Kuiper et al. (1997)
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Häggblad, J., Nilsson, S., & Gustafsson, J. (1997). Comparison of the Ligand Binding Specificity and Transcript Tissue Distribution of Estrogen Receptors α and β. Endocrinology, 138(3), 863–870. [DOI:10.1210/endo.138.3.4979]
Note: Only skimmed – looked at relative binding affinities (RBAs) and noted no raloxifene – going to search for other new papers with more info on RBAs.
Rabe et al. (2016)
Rabe, T., Bruyniks, N., Merkle, E., Hadji, P., Kuhl, H., Ahrendt, H. J., Albring, C., Bitzer, J., Egarter, C., Kiesel, L., König, K., Merki Feld, G., Mueck, A. O., Sänger, N., Windler, E. (2016). Selective Estrogen Receptor Modulators–an Update (Joint Statement by the German Society for Gynecological Endocrinology and Reproductive Medicine [DGGEF] and the German Professional Association of Gynecologists [BVF]). Journal für Reproduktionsmedizin und Endokrinologie-Journal of Reproductive Medicine and Endocrinology, 12(4), 287–317. [Google Scholar] [URL] [PDF]:
It is probable that due to the binding of the SERMs to the ER certain co-factors are activated which, either as co-activators or as co-repressors, lead to different gene activations and deactivations in target tissues. Alternative explanations relate to a different affinity to ER and ER or activation of ER and ER, respectively.
Receptor Distribution Both ERs are expressed in various tissue types, but there are some notable differences in their expression pattern [4]: – ERα: endometrium, breast cancer cells, stromal cells of the ovary and hypothalamus [5] – ERβ: kidney, brain, bone, heart, lung, intestinal mucosa, prostate, and endothelial cells [6]
Polymorphism: Various polymorphisms of ERα- and ERβ-gene have been described.
Various ligands differ in their affinity for the alpha and beta isoforms of the estrogen receptor: – 17-estradiol binds equally well to both receptors – Estrone and raloxifene preferentially bind to the receptor alpha (ERα) – Estriol and genistein preferentially bind to the beta receptor (ERβ)
Selective estrogen receptor modulators preferably bind to the α- or β-subtype of the receptor. In addition, the different estrogen receptor combinations can react differently to the different ligands, thus tissue-specific agonist and antagonist effects arise [7]. The ratio of the concentration of α- to β-subtype plays a role in certain diseases.
The concept of selective estrogen receptor modulators is based on the ability to influence the interaction with other ER proteins such as transcriptional co-activators or co-repressors, based on the conformational changes in the ligand-receptor complex which is different between SERMs and estrogen and also different for different SERMs. Since the ratio of co-activators and co-repressors is different in various tissues [9], the same ligand may act as an agonist in one tissue (provided that the co-activators predominate) and as an antagonist in other tissues (where co-repressors dominate).
Note: Also worth pointing out that it’s tamoxifene’s metabolites that are the active ingredients and should probably have been included here.
Tamoxifene induces the recruitment of co-repressors to target gene promoters in mammary cells. In endometrial cells, tamoxifene, in contrast to raloxifene, acts like an estrogen by stimulating the recruitment of co-activators to a subset of genes.
Raloxifene induces the recruitment of co-repressors to target gene promoters in mammary cells [9].
Raloxifene: Non-genomic mechanisms of endothelial nitric oxide synthase activation by the selective estrogen receptor modulator raloxifene have been found by Simoncini et al (2002) [29]. This pathway might be important to understand the different effects of SERMs on the cardiovascular system.
[Tamoxifen:] Transcription of E-responsive genes(s) is attenuated because AF2 is inactive, and co-activator binding is attenuated by the T-ER complex; partial agonist activity results from AF1, which remains active in the T-ER complex.
[New SERM:] Afimoxifene: (4-hydroxytamoxifene) is a selective estrogen receptor modulator which is the active metabolite of tamoxifene [66]. Afimoxifene is a transdermal gel formulation and is being developed by Ascend Therapeutics, Inc. under the trademark TamoGel. Afimoxifene has completed a phase II clinical trial for the treatment of cyclical mastalgia [198].
Gruber et al. (2002)
Gruber, C. J., Tschugguel, W., Schneeberger, C., & Huber, J. C. (2002). Production and actions of estrogens. New England Journal of Medicine, 346(5), 340–352. [DOI:10.1056/NEJMra000471]:
Pickar, Boucher, & Morgenstern (2018)
Pickar, J. H., Boucher, M., & Morgenstern, D. (2018). Tissue selective estrogen complex (TSEC): a review. Menopause, 25(9), 1033–1045. [DOI:10.1097/gme.0000000000001095]:
The combination of estrogens and a SERM is sometimes referred to as a tissue selective estrogen complex (TSEC), although this term is not formally recognized by health authorities.
The question then arose, in the late 1990s, as to whether a combination of SERMs and estrogens might produce estrogen agonist and antagonist effects distinct from that of either component alone and come closer to achieving that goal.54
A key finding from the preclinical “proof of concept” investigation in ovariectomized rats was that BZA could completely counter CE’s stimulatory effects on the endometrium (based on uterine wet weight) when adequately dosed without attenuating CE’s beneficial effects on vasomotor instability, lipids, or bone,54 which supported clinical evaluation of this combination.
In a clinical trial of raloxifene 60 mg/d combined with oral 17β-estradiol 1 mg/d (n = 61) compared with raloxifene alone (n = 62) in postmenopausal women with prior HT use, the TSEC combination significantly (P < 0.001) reduced the frequency of VMS compared with baseline and with raloxifene alone.
It may be possible to create a TSEC combination that uses 17β-estradiol rather than CE in combination with a SERM; however, results cannot be assumed to be comparable to those with CE/BZA without clinical testing. Due to the unique profile of each TSEC, each individual TSEC combination needs to be evaluated separately in clinical studies to identify appropriate doses and assess safety and efficacy.
Liu, Han, & Smith (2013)
Liu, S., Han, S. J., & Smith, C. L. (2013). Cooperative Activation of Gene Expression by Agonists and Antagonists Mediated by Estrogen Receptor Heteroligand Dimer Complexes. Molecular Pharmacology, 83(5), 1066–1077. [DOI:10.1124/mol.112.084228]:
Moreover, antagonist-bound ERs adopt a distinct conformation that enables them to preferentially interact with corepressors rather than coactivators (Huang et al., 2010), thereby reinforcing their negative regulatory properties.
We demonstrate that ER agonist/antagonist combined treatment can cooperatively activate gene expression through an ER-HLD complex consisting of one receptor monomer bound to agonist and another occupied by antagonist. This cooperative activation of gene expression can be enhanced by an SRC-3 coactivator, and requires both ligand-bound subunits to bind to DNA and both AF-1 domains within the ER-HLD for maximal transcriptional activity. Moreover, ER-HLD complexes can activate transcription in the context of a natural promoter, and taken together, these findings demonstrate that ERs bound to different classes of ligands can form dimers that promote gene expression.
Crystal structures of the ERα LBDs bound to either E2 or raloxifene reveal that both ligands bind to the same pocket, and that the overall homodimeric arrangement is the same regardless of whether the LBD is agonist- or antagonist-bound.
Relative to costimulation by agonist/antagonist, the AF-1 domain is important for the partial agonist activity of SERMs such as tamoxifen (McInerney et al., 1996), and SRC-1 can bind to both the AF-1 and AF-2 domains of ERα through the coactivator’s Q-rich region and LXXLL motifs, respectively (Mérot et al., 2004). This raises the possibility that SRC-3 interacts with ER-HLDs through different AF domains on distinct subunits [e.g., AF-2 on agonist-bound ER(GSCKV) and AF-1 on antagonist-bound ER-G521R] to induce maximal transcriptional activity.
Thornton (2007)
Thornton, J. (2007). Effect of estrogens on skin aging and the potential role of SERMs. Clinical Interventions in Aging, 2(3), 283–297. [DOI:10.2147/cia.s798]:
A number of studies have shown that estrogens have many important beneficial and protective roles in skin physiology (reviewed in Thornton 2002, 2005). They have been shown to accelerate cutaneous wound healing (Ashcroft and Ashworth 2003), while a significant number of women notice an improvement in inflammatory skin disorders such as psoriasis during pregnancy (Dunna and Finlay 1989; Boyd et al 1996; Raychaudhuri et al 2003). Estrogens also offer some degree of protection against skin photoaging (Weinstock 1994; Tsukahara et al 2001, 2004) and epidemiological studies indicate that the mortality rates from both non-melanoma skin cancers (Weinstock 1994) and melanoma (Miller and MacNeil 1997) are significantly lower in women.
Many women report a sudden onset of skin aging several months after menopausal symptoms begin. The menopause causes hypoestrogenism, accelerating age-related deterioration, which results in thinner skin, an increase in number and depth of wrinkles, increased skin dryness, and decreased skin firmness and elasticity (Brincat 2000). Hormone replacement therapy (HRT) has been shown to increase epidermal hydration, skin elasticity, skin thickness (Sator et al 2001), and also reduces skin wrinkles (Phillips et al 2001). Furthermore, the content and quality of collagen and the level of vascularization is enhanced (Brincat et al 1987).
More recently, specific antibodies have demonstrated ERβ, but not ERα is expressed by dermal fibroblasts in the papillary dermis of human scalp skin in both sexes (Thornton et al 2003), whereas primary cultures of human dermal fibroblasts from female skin have been shown to express both mRNA and protein for ERα and ERβ (Haczynski et al 2002).
In addition, in female hair follicles the phytoestrogen, genistein inhibits hair shaft elongation to a similar extent as 17β-estradiol. Since genistein preferentially binds to ERβ, this opens the possibility that the inhibition of hair growth in response to 17β-estradiol may be mediated via ERβ rather than ERα (Nelson 2006). Therefore the development of selective estrogen receptor ligands may provide important clinical applications for the prevention and treatment of disorders of hair growth.
A histopathological assessment of rat skin following subcutaneous administration of tamoxifen observed that tamoxifen treatment resulted in the appearance of abnormal hair follicles, epidermal atrophy and increased dermal fibrosis, particularly around the hair follicles (Inaloz et al 2002). There have been reports of tamoxifen treatment causing diffuse thinning of the hair with moderate receding of the frontal hair line (Ayoub et al 1997) and the development of alopecia on the crown, which was reversed when treatment was stopped.
We have recently reported that tamoxifen alone has no effect on human hair shaft elongation in organ culture, suggesting it is not an estrogen agonist (Nelson 2006). However, a 10-fold excess of tamoxifen incubated in combination with 17β-estradiol eliminated the inhibitory effect of 17β-estradiol, suggesting that tamoxifen acts as an antagonist of estrogen in the female scalp hair follicle (Nelson 2006).
Another study has shown that raloxfine has a stronger positive stimulatory effect on collagen biosynthesis than 17β-estradiol (Surazynski et al 2003) and that in contrast to estradiol, raloxifene inhibits the expression of MMP-9.
Mauvais-Jarvis, Clegg, & Hevener (2013)
Mauvais-Jarvis, F., Clegg, D. J., & Hevener, A. L. (2013). The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews, 34(3), 309–338. [DOI:10.1210/er.2012-1055]:
However, tamoxifen treatment is associated with an increased risk of developing fatty liver (steatosis) (337–339), with approximately 43% of breast cancer patients treated with tamoxifen developing hepatic steatosis (339). The exact mechanism by which this occurs is still unclear.
In the hypothalamus, tamoxifen appears to act as an ER agonist.
In OVX mice, raloxifene reversed OVX-induced increases in food intake, body weight, fat mass, and hyperleptinemia to an extent similar to that of E2. This suggests that in rodents, raloxifene acts as an ER agonist in hypothalamic neurons and fat (346). In postmenopausal women, raloxifene prevents the shift from android to gynoid fat distribution, increases in abdominal adiposity (347), as well as total increases in adiposity (348). The absence of significant effect on body weight in women (349) could be due to the concomitant increase in lean mass and water after raloxifene treatment (350).
Nonetheless, in healthy and diabetic postmenopausal women, short-term raloxifene treatment did not impact fasting glucose, glucose tolerance, or indices of-cell function and sensitivity (351, 352). However, it decreased hepatic insulin extraction and, as a result, increased insulin half-life (352). Additionally, in the Multiple Outcomes of Raloxifene Evaluation trial, 3-year raloxifene treatment of postmenopausal women with or without type 2 diabetes reduced total cholesterol and LDL-cholesterol but had no effect on glycemic control compared to placebo (349). Thus, there is no argument for an effect of raloxifene in improving glucose homeostasis in postmenopausal women with type 2 diabetes. In fact, women with a previous history of hypertriglyceridemia who receive oral estrogen therapy are at risk for clinically relevant progression in this existing risk factor during raloxifene therapy.
In preclinical studies, the TSEC partnering bazedoxifene with CEE seemed to provide these benefits. Thus far, the effect of TSEC partnering bazedoxifene on glucose and energy metabolism is unknown.
Tommaselli et al. (2006)
Tommaselli, G. A., Di Carlo, C., Di Spiezio Sardo, A., Bifulco, G., Cirillo, D., Guida, M., Capasso, R., & Nappi, C. (2006). Serum leptin levels and body composition in postmenopausal women treated with tibolone and raloxifene. Menopause, 13(4), 660–668. [DOI:10.1097/01.gme.0000227335.27996.d8]:
At the end of the study, untreated women showed increased total and percentage of fat mass and increased serum leptin levels compared with baseline values, whereas women treated with HT showed no significant changes. These data seem to indicate that the main determinant of leptin levels in postmenopausal women is body fat content, whereas hypoestrogenism does not seem to influence these levels.
Women in group C showed no significant changes in fat mass, in total and at all areas evaluated, after 1 year of treatment with raloxifene (Fig. 1A-D). No significant changes were observed in lean mass, both total and at all areas evaluated (Fig. 2A-D). The same was observed for the percentage of fat mass (Fig. 3A-D).
We did not detect significant changes in serum leptin levels in subjects in group B and C throughout the study (group B: 16.3 [4.2/39.8] ng/mL vs 17.0 [5.6 /28.7] ng/mL; group C: 15.1 [6.7/26.5] ng/mL vs 17.5 [6.7/29.8] ng/mL).
The present study, although hampered by the limited sample size, seems to confirm that postmenopausal hypoestrogenism leads to increased fat content and suggests that both raloxifene and tibolone may have a role in preventing the changes of body composition induced by postmenopausal hypoestrogenism.
Francucci et al. (2005)
Francucci, C. M., Pantaleo, D., Iori, N., Camilletti, A., Massi, F., & Boscaro, M. (2005). Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women. Journal of Endocrinological Investigation, 28(9), 623–631. [DOI:10.1007/bf03347261]:
For the first time, our results suggest that RLX, most likely because of its estrogen-agonist action, might prevent the shift to a more central fat dis- tribution associated with menopause. In fact, we have found in RLX-users, in relation to controls, no substantial changes in body weight during the 12 months of treatment, a significant lower fat mass in trunk and abdominal region (ROI-L2-4) and a trend towards higher fat depots in legs. At variance, after 1 yr we observed in untreated post-menopausal women a not significant increase of body weight and fat mass in central and trunk region with a decrease in legs. Therefore, RLX could have, similar to the estrogens, different effects on several regional fat tissues. In fact, in pre-menopausal women the gluteo-femoral adipocytes have a low lipolytic activity with tendency towards larger fat cells in the femoral region, while the adipocytes in the central fat depot have a high basal lipolysis with smaller fat cells (32). Besides, the estrogens would seem to enhance the lipoprotein-lipase (LPL) activity, the key enzyme in the regulation of fatty acid deposi- tion, of gluteofemoral fat cells and to decrease the visceral LPL activity (33).
Jacobsen et al. (2010)
Jacobsen, D. E., Samson, M. M., Emmelot-Vonk, M. H., & Verhaar, H. J. (2010). Raloxifene and body composition and muscle strength in postmenopausal women: a randomized, double-blind, placebo-controlled trial. European Journal of Endocrinology, 162(2), 371–376. [DOI:10.1530/eje-09-0619]:
Repeated measurement analysis showed there to be a significant increase in FFM at 3, 6, and 12 months in the raloxifene group (P=0.05) as well as an increase in TBW (P=0.02; Table 2). At 12 months, the mean increase in FFM was 0.83 (2.4) kg in the raloxifene group compared with 0.03 (1.5) kg in the placebo group (P=0.05), and the mean increase in TBW was 0.6 (1.8) litres compared with a decrease of 0.06 (1.1) litres in the placebo group (P=0.02).
In our study, we found an increase in the FFM after supplementation with raloxifene, which could be partly explained by the increase in the TBW content, a known effect of raloxifene supplementation. However, the increase in the TBW content could not explain the whole increase in the FFM, and also the amount of physical activity was comparable between both groups during the study. This means that there is also a real change in body composition with an increase in lean body mass. The results of our study are comparable with some other studies, but in most of these studies there were problems with the design of the study (no blinding, no placebo group, too small groups, and a too short intervention period).
The increase in FFM was not accompanied by an increase in muscle strength and muscle power in our study. Muscle strength is a key factor in maintaining independence in elderly people. There are no other studies concerning raloxifene and muscle strength or power. Studies concerning HRT (estrogen and progestagens) show conflicting results: two randomized controlled trials with no effect (38, 39) and three randomized controlled trials with significant increase in muscle strength (11–13).
Komm & Mirkin (2014)
Komm, B. S., & Mirkin, S. (2014). An overview of current and emerging SERMs. The Journal of Steroid Biochemistry and Molecular Biology, 143, 207–222. [DOI:10.1016/j.jsbmb.2014.03.003]:
Raloxifene binds with high affinity to ERα, with 46% relative binding affinity for human ERα compared with 17β-estradiol, and with a 26% relative binding affinity for rat ERβ compared with 17β-estradiol
Note: A lot more on this article but this was the only relevant thing I really cared about at the time.
Smith & O’Malley (2004)
Smith, C. L., & O’Malley, B. W. (2004). Coregulator Function: A Key to Understanding Tissue Specificity of Selective Receptor Modulators. Endocrine Reviews, 25(1), 45–71. [DOI:10.1210/er.2003-0023]:
For instance, in the absence of endogenous estrogens, tamoxifen frequently exhibits weak estrogenic activity, such as modest stimulation of uterine wet weight and bone density in ovariectomized rats, whereas in the presence of estradiol, it can serve as an antiestrogen, inhibiting responses to a level corresponding to the comparatively modest agonist activity of tamoxifen itself.
However, because neither tamoxifen nor raloxifene possesses significant estrogen-like activity in the CNS, there is clearly a market for other SERMs to fill this niche.
It is also noteworthy that SERMs likely exist in nature. For instance, the estrone metabolite, Δ8,9- dehydroestrone sulfate suppresses hot flushes in postmenopausal women, an estrogenic action. However, it is unable to significantly affect certain other parameters associated with estrogenic responses such as total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol (23).
[…] raloxifene and 4HT block the ligand-activated AF-2 domain and particularly in the case of 4HT, leave AF-1 able to initiate gene expression.
As a result, 4HT and raloxifene stimulate gene expression in some, but not all, contexts, and these antiestrogens are therefore classified as SERMs.
For ER, 4HT and raloxifene also inhibit its AF-2 domain, and due to the relatively poor AF-1 activity of the receptor, these ligands generally block ER transcriptional activity measured on EREs (61, 78, 79).
It should be noted that 4HT and raloxifene also exert effects on the transcriptional activity of both ER and ER tethered to DNA indirectly through interaction with other transcription factors such as activator protein 1 (AP-1) and Sp1 (80, 81). In this context, the agonist activities of these ligands also is apparent in a cell-specific manner.
For ERE-dependent gene expression, tamoxifen is a partial agonist of ER but is generally unable to stimulate ER transcriptional activity (61, 171, 173). Conversely, when assessing ER activity on AP-1 containing reporter genes, tamoxifen will stimulate ER and ER transcriptional activity.
Carneiro, de Cassia de Maio Dardes, & Haidar (2012)
Carneiro, A. L., de Cassia de Maio Dardes, R., & Haidar, M. A. (2012). Estrogens plus raloxifene on endometrial safety and menopausal symptoms—semisystematic review. Menopause, 19(7), 830–834. [DOI:10.1097/gme.0b013e31824a74ce]:
Most of the studies found a benefit profile with association of RLX and E on women’s quality of life, satisfaction with the treatment, and vaginal dryness. Nevertheless, different qualityof-life scales and visual scales were used to access mainly vasomotor symptoms, which could limit further comparison.
Arevalo et al. (2010)
Arevalo, M. A., Santos-Galindo, M., Lagunas, N., Azcoitia, I., & Garcia-Segura, L. M. (2010). Selective estrogen receptor modulators as brain therapeutic agents. Journal of Molecular Endocrinology, 46(1), R1–R9. [DOI:10.1677/jme-10-0122]:
The neuroprotective actions of tamoxifen and raloxifene, two SERMs that are currently used in clinical practice for the treatment of breast cancer and osteoporosis, have been assessed in different experimental models of neural dysfunction. These include animal models of traumatic injury of the central nervous system and peripheral nerves, stroke, multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, cognitive decline, and mood disorders (Fig. 1). This section is a succinct description of the main findings of these experimental studies, including the limited available information from human studies.
In spite of the neuroprotective actions of tamoxifen in different forms of neural injury, it is unclear whether this molecule may have some benefits for cognition in humans. Indeed, several studies suggest an increased risk of cognitive impairment in women receiving tamoxifen for the treatment of breast cancer, including worse performance in visual memory, word fluency, immediate verbal memory, visuospatial ability, and processing speed tasks (Paganini-Hill & Clark 2000, Shilling et al. 2003, Palmer et al. 2008, Phillips et al. 2010, Schilder et al. 2010). However, other studies have not detected a significant effect of tamoxifen on cognition (Debess et al. 2010).
In contrast to the potential negative effects of tamoxifen on cognition, the results of the multiple outcomes of raloxifene evaluation randomized trial suggest that raloxifene prevents cognitive decline in postmenopausal women (Yaffe et al. 2005). The results of a recent randomized, double-blind, placebo-controlled trial also suggest that raloxifene improves verbal memory in late postmenopausal women ( Jacobsen et al. 2010). In addition, raloxifene treatment enhances brain activation during performance on a face-encoding paradigm and during recognition of familiar items in healthy elderly men.
In agreement with this possibility, we have recently observed that both tamoxifen and raloxifene improve hippocampus-dependent memory in androgen-deprived male rats (N Lagunas, I Calmarza-Font, D Grassi & LM Garcia-Segura, unpublished observations).
Tamoxifen reduces amphetamine-induced manic-like behavioral alterations in rats (Einat et al. 2007) and reduces acute manic episodes in women with bipolar affective disorder (Kulkarni et al. 2006, Zarate et al. 2007). Raloxifene reduces anxiety behavior, assessed in the elevated plus maze test, on ovariectomized rats (Walf & Frye 2010) and decreases anxiety (Strickler et al. 2000, Florio et al. 2001) and depression (Carranza-Lira et al. 2004, Grigoriadis et al. 2005, Sugiyama et al. 2007) in postmenopausal women. Recent clinical studies also suggest the potential application of raloxifene hydrochloride (120 mg/day oral) for the treatment of postmenopausal women with schizophrenia.
Other neuroprotective mechanisms of SERMs are mediated by classical ERs, since they are inhibited by ER antagonists, such as ICI 182 780 (Zhang et al. 2009). The neuroprotective signaling of SERMs may involve the activation of kinases, such as mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and Akt (Du et al. 2004, Lee et al. 2009a,b), and the phosphorylation of CREB (Sharma et al. 2007) or the inhibition of nuclear factor (NF)-kB-induced transcription (Cerciat et al. 2010). Through these mechanisms, SERMs control synaptic transmission and the expression of molecules involved in the regulation of cell death, oxidative stress, and inflammation.
Raloxifene may also regulate opiate and GABAergic neurotransmission by the modulation of the levels of b-endorphin and neuroactive steroids respectively. Chronic raloxifene administration to postmeInopausal women increases plasma levels of b-endorphin and tetrahydroprogesterone (allopregnanolone), an anxiolytic metabolite of progesterone that modulates GABAA receptors.
These findings suggest that raloxifene may regulate synaptic function by the modulation of local levels of neuroactive substances within the brain.
In general, there is still poor knowledge of the precise molecular targets of SERMs in the nervous system. Although some key molecules have been identified, such as MAPK, PI3K/Akt, CREB, and NF-kB, the molecular mechanisms involved in the neuroprotective actions of SERMs should be investigated with more detail in the different cellular populations of the nervous system.
Lawrence et al. (2004)
Lawrence, S. E., Arnold Faught, K., Vethamuthu, J., & Lawson, M. L. (2004). Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. The Journal of Pediatrics, 145(1), 71–76. [DOI:10.1016/j.jpeds.2004.03.057]:
Results: Mean (SD) age of treated subjects was 14.6 (1.5) years with gynecomastia duration of 28.3 (16.4) months. Mean reduction in breast nodule diameter was 2.1 cm (95% CI 1.7, 2.7, P < .0001) after treatment with tamoxifen and 2.5 cm (95% CI 1.7, 3.3, P < .0001) with raloxifene. Some improvement was seen in 86% of patients receiving tamoxifen and in 91% receiving raloxifene, but a greater proportion had a significant decrease (>50%) with raloxifene (86%) than tamoxifen (41%). No side effects were seen in any patients.
Surazynski et al. (2003)
Surazynski, A., Jarzabek, K., Haczynski, J., Laudanski, P., Palka, J., & Wolczynski, S. (2003). Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. International Journal of Molecular Medicine, 12(5), 803–809. [DOI:10.3892/ijmm.12.5.803]:
Raloxifene had stronger positive stimulative effects on collagen biosynthesis, through different biochemical mechanisms, than estradiol in human skin fibroblasts and might reverse some of the postmenopausal changes in skin or connective tissue. Increase of collagen synthesis induced by raloxifene may be activated by both estrogen receptor dependent and independent pathways such as up-regulation of estrogen receptors, up-regulation of IGF receptor, transcriptional regulation of collagen genes by estrogen receptor-raloxifene complex, increasing of prolidase activity or finally by inhibition of MMP-9 expression.
\ No newline at end of file
diff --git a/transfemscience.org/articles/serms-transfem/index.html b/transfemscience.org/articles/serms-transfem/index.html
index a904558e..88c33680 100644
--- a/transfemscience.org/articles/serms-transfem/index.html
+++ b/transfemscience.org/articles/serms-transfem/index.html
@@ -1 +1 @@
-A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy - Transfeminine Science
As with my other articles, this one was borne out of a personal need to understand. Like many transfeminine non-binary individuals, my desired gender-affirming hormone therapy (GAHT) outcome is a minimization of breast development with effective feminization elsewhere. From Aly’s excellent resource on non-binary hormone therapy (Aly, 2019) came my knowledge of raloxifene and a desire to fully understand as precisely as possible its actions. I hope this article is illuminative to others undergoing similar experiences and that together we can build an even greater understanding in creating ourselves.
Executive Summary
I encourage everyone that wishes a deeper understanding to read the sections below and for those that desire more knowledge to read the supplement page provided.
Selective estrogen receptor modulators (SERMs) work by blocking or activating different estrogen receptors at different levels. Since different tissues have different ratios of these estrogen receptors SERMs act like estrogens in some tissues and block estrogens in other tissues.
The main SERM up for consideration for transgender GAHT is raloxifene as of the two widely available, generic SERMs, it has the best safety profile and most relevant research.
Raloxifene acts largely antiestrogenically in breast tissue. It helps to prevent and treat breast cancer. It seems to act estrogenically in various tissues of interest to transgender individuals. It produces a gynoid (feminine) fat distribution, it causes estrogenic like actions on skin, and helps prevent osteoporosis by maintaining bone mineral density. Further it seems to have some estrogenic actions in the brain.
A TSEC of estradiol and raloxifene may warrant further study for transfeminine individuals. The reasons for discontinuation of research of combined estradiol and raloxifene were due to endometrial thickening and as transfeminine individuals (intersex individuals excepted) do not have an endometrium this is not a cause for concern.
Lastly, this article would be remiss if not to end with a major word of caution. There is to my knowledge, no published research on SERMs in transgender individuals. There is no long term research on raloxifene or tamoxifen on natal men. Finally, the mechanism of action of SERMs in general is that they partially suppress various types of estrogen receptor activity. They are far away from being fully characterized and it may be the case that the subtle ways in which they differ from estradiol may cause unforeseen long term consequences to transfeminine individuals.
That being said, they are an exciting frontier in the area of non-binary transfeminine gender-affirming hormone therapy (GAHT).
Introduction
While many people that begin a path of transfeminine GAHT do so with the desire to fully develop breasts there exists a sizable number of people, many on the non-binary transfeminine spectrum who wish to medically feminize themselves with little breast development. Because of this, raloxifene, a selective estrogen receptor modulator (SERM), has become talked about as a potential way to achieve this. A second-generation SERM, it was developed to prevent and treat breast cancer and (in the U.S.) prevent osteoporosis. In this article, I will review the estrogen receptor, the mechanism of action of SERMs and how they work in various relevant tissues, as well as discuss a few tissue-selective estrogen complexes (TSEC) that may be of interest.
Background
For an extremely in-depth review of the estrogen receptors, how they form, and the compounds that bind to them, I recommend Ascenzi, Bocedi, & Marino (2006).
Estrogen Receptors
Estrogens are compounds that cause changes in cells by binding to various estrogen receptors. These receptors are complexes consisting of proteins and other molecules. This binding causes a conformational change (change in shape) in the receptor that then results in some change in cell activity.
The main types of estrogen receptors are estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) as well as G protein-coupled estrogen receptor (GPER). The classical way in which estrogens were thought to have an impact is via ERα and ERβ. These are part of the nuclear receptor family. They exist on the nuclear membrane and once bound to an estrogen these receptors form a complicated biological machine that then causes a change in gene expression.
This estrogen receptor complex comes to exist due to the estrogen receptor containing many binding domains and on these domains other proteins or molecules can bind that then cause the estrogen receptor complex to be either activated, repressed, or destroyed.
In addition to the classic method of estrogen activation, there exists also GPER activity. G protein-coupled receptors are a huge super family of receptors that are membrane bound (they exist inside the cell membrane) and GPER receptors cause non genomic changes that can result in rapid actions within the cell as there is not a delay of having to generate RNA and then proteins. While there is some research into the role of SERMs with GPERs, much of the focus of research and this article will concern ourselves with classic estrogen receptor activity.
Selective Estrogen Receptor Modulators (SERMs)
Estradiol is the classic molecule that binds to the various estrogen receptors. Estradiol is an agonist of the estrogen receptors in that it activates them to action. In contrast, there exist full estrogen receptor antagonists such as fulvestrant (ICI-182,780), a compound that completely blocks the estrogen receptors from activating. SERMs are molecules that bind to the various estrogen receptors in a way that is at least partially different from estradiol. This means that in some contexts it is an antagonist (antiestrogenic) and in others it is an agonist (estrogenic) of the estrogen receptors. SERMs work by binding to ERα and ERβ and GPER at relatively different rates compared to estradiol and acting as either a full or partial antagonist at the receptor. Because different tissues have different ratios of expression of these receptors, the subsequent result is that SERMs act like estradiol in some tissues and act to suppress estradiol like actions in others.
At a molecular level, this occurs because a SERM is a different shape than estradiol and when it binds to the estrogen receptors its shape and the subsequent change in shape of the estrogen receptor may physically prevent (sterically inhibit) another compound or compounds that would need to bind to the estrogen receptor as well to activate its activity.
Originally developed for the treatment of breast cancer, the currently developed SERMs all display some similar properties. They act antiestrogenically in breast tissue, with many acting estrogenically on cholesterol and bones, and thus have found use as a preventative measure for osteoporosis in postmenopausal women. The first SERM to become available was tamoxifen in 1978, with second-generation SERM raloxifene becoming available in 1997 (in the U.S.). More recently, there have been a number of new third-generation SERMs developed, such as bazedoxifene, lasofoxifene, and ospemifene. However, as these are still under patent they are expensive and therefore do not currently make good candidates for GAHT.
Tissue-Selective Estrogen Complexes (TSECs)
Tissue-selective estrogen complexes (TSECs) are a combination of a SERM with an estrogen. The goal in their development was to create a unique mixture of estrogenic and antiestrogenic effects that would combine the antiestrogenic effects of some SERMs in the breasts (to prevent breast cancer) and endometrium (to prevent endometrial cancer), while maintaining the positive effects of estrogens in postmenopausal women such as improved bone density, cardiovascular health and weight maintenance. The only currently approved TSEC is a combination of conjugated equine estrogens (CEE) with bazedoxifene (CEE/BZA). An abandoned TSEC, estradiol/raloxifene, will also be considered in this article – as its reasons for abandonment are largely irrelevant to transfeminine people undergoing GAHT and has a number of advantages over CEE/BZA such as availability and lower risk of complications for transfeminine individuals.
Safety Studies
The two generically available SERMs, raloxifene and tamoxifen, have both undergone long-term, large-scale studies, specifically the Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) (Davis et al., 2013) study for tamoxifen, showing that its largest risk is endometrial cancer. However, it has significant risk of other serious side effects such as reduced cognition, liver toxicity, and various negative cardiovascular issues (fatty liver disease, deep vein thrombosis, etc…).
Raloxifene similarly had the Multiple Outcomes of Raloxifene Evaluation (MORE) (Ettinger, 1999) and Raloxifene Use for The Heart (RUTH) (Wenger et al., 2002) studies conducted and together they had the Study of Tamoxifen and Raloxifene (STAR) (Vogel et al., 2006) study, which shows that while it still has an increased risk of deep vein thrombosis, it is lower than tamoxifen and it does not have the other negative side effects associated with tamoxifen.
Tissue-Specific Effects of SERMs
In Breasts
Both tamoxifen and raloxifene have been shown to act antiestrogenically in the breast, and indeed this is the main reason for their development as drugs. They are both shown to decrease the risk of breast cancer in natal women and both have been shown to be effective in treating gynecomastia (male breast growth) (Lawrence et al., 2004; Kunath et al., 2012). The exact mechanism of action of both raloxifene and tamoxifen in the breast is still not entirely known. While they induce estrogen receptor changes, there are also several pathways that are not via the estrogen receptor they are involved with that may mediate their anti-breast cancer action and subsequently their anti-breast development activity.
In Fat
Changes in fat distribution and metabolism are a major aspect of GAHT (see Lain (2019) for an in-depth look on changes in fat distribution and metabolism with GAHT). Gynoid fat distribution occurs via both peripheral activity often by the leptin–ghrelin hormone system, and raloxifene seems to act estrogenically on the leptin-ghrelin hormone system helping to prevent abdominal adiposity (Tommaselli et al., 2006; Mauvais-Jarvis, Clegg, & Hevener, 2013). It also appears to act estrogenically directly in fat tissue (adipose tissue) (Francucci et al., 2005), again helping to increase subcutaneous fat around the thighs and buttocks that is a primary characteristic of gynoid fat distribution. In addition in postmenopausal women, it is found to reduce total cholesterol and LDL cholesterol. However, it does not appear to impact glycemic index, meaning that it does not help prevent postmenopausal type 2 diabetes. It is unknown how exactly this may map to transfeminine individuals.
In Muscle
While muscle mass is strongly mediated via testosterone (see Lain (2019), a review on muscle mass and testosterone), there also seems to be a correlation between estrogen and muscle mass. Postmenopausal natal women undergo significant sarcopenia (age-related muscle mass loss) that can be prevented or minimized via estrogen GAHT. Similar to estrogens, raloxifene also helps increase muscle mass in postmenopausal women and prevent loss of muscle mass (Jacobsen et al., 2010). Because the exact mechanism of action by which the estrogen receptors mediates muscle strength is unknown, it is hard to understand how this translates to transfeminine individuals. However, it does point to estrogenic action in muscle tissue.
In Skin
In classic transfeminine hormone therapy, the softening of skin is often one of the first noticeable effects. In addition in postmenopausal women, a lack of estrogen can cause thinner skin and decreased elasticity, which is preventable via GAHT. In studies, tamoxifen appears to cause abnormal hair follicles, skin atrophy, and even possibly alopecia (hair loss) developing on the crown of the head; this and direct evidence suggests that tamoxifen is an antagonist of the estrogen receptor in hair follicles (Thornton, 2007). However, there is some evidence to suggest that raloxifene has a positive estrogenic effect on collagen biosynthesis in human skin (Surazynski et al., 2003).
In Testes
SERMs such as raloxifene and tamoxifen have been found to cause an increase in serum gonadotropins (luteinizing hormone and follicle stimulating hormone) as well as a concurrent increase in testosterone (Rambhatla, Mills, & Rajfer, 2016). This highlights the importance of using an antiandrogen in conjunction with a SERM in GAHT.
In the Brain
The estrogen receptors have significant impact on brain function. Estradiol GAHT has been associated with antidepressant effects in postmenopausal women and estrogen receptors mediate significant changes in the hypothalamus related to maintaining body fat percentage and hunger. SERMs in general appear to act on the brain both via the estrogen receptor as well as via non-estrogen-receptor-mediated pathways.
Tamoxifen in animal models has neuroprotective effects for a number of modes of neural dysfunction including traumatic injury, Parkinson’s disease, Alzheimer’s disease, and mania. However, in some trials, it is also associated with cognitive impairment, including visual memory, verbal memory, visuospatial ability, and speed processing tasks (Arevalo et al., 2010).
Raloxifene does not appear to have negative cognitive implications as tamoxifen does. However, it is shown to act at least partially estrogenically in the hypothalamus, to reduce anxiety in ovariectomized rats, and to decrease anxiety and depression in postmenopausal women. It may also regulate opioid and GABAergic activity by modulating endorphin levels and has been shown to positively impact schizophrenia (Arevalo et al., 2010).
SERMs in general and raloxifene in particular seem to have some estrogenic like actions in the brain, though it does not seem to be especially significant (Smith & O’Malley, 2004). However, it is still very far from understood all of its actions in the brain as estrogen receptors are found throughout.
Tissue-Specific Effects of TSECs
Up till now, we have only talked about individual compounds. However, TSECs came to be developed by the thinking that by combining an estrogen with a SERM you could get a different effect profile. The only currently medically approved TSEC is CEE/BZA. Specifically, the goal for TSECs was to gain many of the beneficial aspects of estrogen GAHT in postmenopausal natal women while blocking the increased risks of breast cancer and endometrial thickening associated with it. In studies of ovariectomized rats, bazedoxifene was found to maintain its antiestrogenic activity in the endometrium without attenuating the estrogen’s positive effects in movement, fat or bone (Pickar, Boucher, & Morgenstern, 2018).
CEE/BZA while approved for postmenopausal women presents a potentially higher risk for transgender individuals due to its increased risk of deep vein thrombosis, and is less affordable being unavailable as a generic formulation. That being the case another TSEC – a combination of raloxifene and estradiol – has had a number of clinical studies performed on it. It was ultimately abandoned by the medical community due to causing endometrial thickening, again something not typically of concern to transfeminine individuals undergoing GAHT. In the studies performed, raloxifene plus estradiol seemed to reduce many postmenopausal symptoms, such as hot flashes, vaginal dryness, and overall treatment satisfaction, compared to just raloxifene (Carneiro, de Cassia de Maio Dardes, & Haidar, 2012).
Discussion
The primary driver of interest in using SERMs in GAHT is their antiestrogenic effects in the breasts with potential for feminization in other tissues. Of the two affordable and available SERMs, raloxifene by far seems to be the one that has the best safety profile, while also being most effective in the literal research we have on preventing breast growth.
That being said, TSECs seem to be a potentially untapped area of research. Raloxifene and estradiol, while potentially unsafe for those with uteruses, may be well-tolerated by those without. Research into the potential efficacy of raloxifene plus estradiol both in preventing breast growth and relative rate and degree of feminization otherwise versus estradiol alone and raloxifene alone could potentially yield extremely interesting results for transfeminine individuals seeking GAHT that includes minimal breast development.
Ultimately, SERMs, and how different they are from estradiol, is still far from being understood. While we have some knowledge that they are generally safe long-term, this information applies pretty much exclusively to postmenopausal natal women, though there is some indication that tamoxifen has a low incidence of adverse effects in natal men (Wibowo et al., 2016). Because estrogen receptors and their actions in a wide variety of tissues are extremely complex and multifactorial, this means that each SERM and TSEC must be evaluated extremely carefully across these tissues to fully understand it.
References
Arevalo, M. A., Santos-Galindo, M., Lagunas, N., Azcoitia, I., & Garcia-Segura, L. M. (2010). Selective estrogen receptor modulators as brain therapeutic agents. Journal of Molecular Endocrinology, 46(1), R1–R9. [DOI:10.1677/jme-10-0122]
Ascenzi, P., Bocedi, A., & Marino, M. (2006). Structure–function relationship of estrogen receptor α and β: Impact on human health. Molecular Aspects of Medicine, 27(4), 299–402. [DOI:10.1016/j.mam.2006.07.001]
Carneiro, A. L., de Cassia de Maio Dardes, R., & Haidar, M. A. (2012). Estrogens plus raloxifene on endometrial safety and menopausal symptoms—semisystematic review. Menopause, 19(7), 830–834. [DOI:10.1097/gme.0b013e31824a74ce]
Davies, C., Pan, H., Godwin, J., Gray, R., Arriagada, R., Raina, V., Abraham, M., Alencar, V. H., Badran, A., Bonfill, X., Bradbury, J., Clarke, M., Collins, R., Davis, S. R., Delmestri, A., Forbes, J. F., Haddad, P., Hou, M., Inbar, M., Khaled, H., Kielanowska, J., Kwan, W., Mathew, B. S., Mittra, I., Müller, B., Nicolucci, A., Peralta, O., Pernas, F., Petruzelka, L., Pienkowski, T., Radhika, R., Rajan, B., Rubach, M. T., Tort, S., Urrútia, G., Valentini, M., Wang, Y., & Peto, R. (2013). Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. The Lancet, 381(9869), 805–816. [DOI:10.1016/s0140-6736(12)61963-1]
Ettinger, B. (1999). Reduction of Vertebral Fracture Risk in Postmenopausal Women With Osteoporosis Treated With Raloxifene: Results From a 3-Year Randomized Clinical Trial. JAMA, 282(7), 637–645. [DOI:10.1001/jama.282.7.637]
Francucci, C. M., Pantaleo, D., Iori, N., Camilletti, A., Massi, F., & Boscaro, M. (2005). Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women. Journal of Endocrinological Investigation, 28(9), 623–631. [DOI:10.1007/bf03347261]
Jacobsen, D. E., Samson, M. M., Emmelot-Vonk, M. H., & Verhaar, H. J. (2010). Raloxifene and body composition and muscle strength in postmenopausal women: a randomized, double-blind, placebo-controlled trial. European Journal of Endocrinology, 162(2), 371–376. [DOI:10.1530/eje-09-0619]
Kunath, F., Keck, B., Antes, G., Wullich, B., & Meerpohl, J. J. (2012). Tamoxifen for the management of breast events induced by non-steroidal antiandrogens in patients with prostate cancer: a systematic review. BMC Medicine, 10(1), 96. [DOI:10.1186/1741-7015-10-96]
Lawrence, S. E., Arnold Faught, K., Vethamuthu, J., & Lawson, M. L. (2004). Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. The Journal of Pediatrics, 145(1), 71–76. [DOI:10.1016/j.jpeds.2004.03.057]
Mauvais-Jarvis, F., Clegg, D. J., & Hevener, A. L. (2013). The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews, 34(3), 309–338. [DOI:10.1210/er.2012-1055]
Pickar, J. H., Boucher, M., & Morgenstern, D. (2018). Tissue selective estrogen complex (TSEC): a review. Menopause, 25(9), 1033–1045. [DOI:10.1097/gme.0000000000001095]
Rambhatla, A., Mills, J. N., & Rajfer, J. (2016). The Role of Estrogen Modulators in Male Hypogonadism and Infertility. Reviews in Urology, 18(2), 66–72. [PubMed] [PubMed Central] [DOI:10.3909/riu0711]
Smith, C. L., & O’Malley, B. W. (2004). Coregulator Function: A Key to Understanding Tissue Specificity of Selective Receptor Modulators. Endocrine Reviews, 25(1), 45–71. [DOI:10.1210/er.2003-0023]
Surazynski, A., Jarzabek, K., Haczynski, J., Laudanski, P., Palka, J., & Wolczynski, S. (2003). Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. International Journal of Molecular Medicine, 12(5), 803–809. [DOI:10.3892/ijmm.12.5.803]
Thornton, J. (2007). Effect of estrogens on skin aging and the potential role of SERMs. Clinical Interventions in Aging, 2(3), 283–297. [DOI:10.2147/cia.s798]
Tommaselli, G. A., Di Carlo, C., Di Spiezio Sardo, A., Bifulco, G., Cirillo, D., Guida, M., Capasso, R., & Nappi, C. (2006). Serum leptin levels and body composition in postmenopausal women treated with tibolone and raloxifene. Menopause, 13(4), 660–668. [DOI:10.1097/01.gme.0000227335.27996.d8]
Vogel, V. G., Costantino, J. P., Wickerham, D. L., Cronin, W. M., Cecchini, R. S., Atkins, J. N., Bevers, T. B., Fehrenbacher, L., Pajon, E. R., Jr, Wade, J. L., 3rd, Robidoux, A., Margolese, R. G., James, J., Lippman, S. M., Runowicz, C. D., Ganz, P. A., Reis, S. E., McCaskill-Stevens, W., Ford, L. G., Jordan, V. C., … National Surgical Adjuvant Breast and Bowel Project (NSABP) (2006). Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA, 295(23), 2727–2741. [DOI:10.1001/jama.295.23.joc60074]
Wenger, N. K., Barrett-Connor, E., Collins, P., Grady, D., Kornitzer, M., Mosca, L., Sashegyi, A., Baygani, S. K., Anderson, P. W., & Moscarelli, E. (2002). Baseline characteristics of participants in the Raloxifene Use for The Heart (RUTH) trial. The American Journal of Cardiology, 90(11), 1204–1210. [DOI:10.1016/s0002-9149(02)02835-7]
Wibowo, E., Pollock, P. A., Hollis, N., & Wassersug, R. J. (2016). Tamoxifen in men: a review of adverse events. Andrology, 4(5), 776–788. [DOI:10.1111/andr.12197]
\ No newline at end of file
+A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy - Transfeminine Science
As with my other articles, this one was borne out of a personal need to understand. Like many transfeminine non-binary individuals, my desired gender-affirming hormone therapy (GAHT) outcome is a minimization of breast development with effective feminization elsewhere. From Aly’s excellent resource on non-binary hormone therapy (Aly, 2019) came my knowledge of raloxifene and a desire to fully understand as precisely as possible its actions. I hope this article is illuminative to others undergoing similar experiences and that together we can build an even greater understanding in creating ourselves.
Executive Summary
I encourage everyone that wishes a deeper understanding to read the sections below and for those that desire more knowledge to read the supplement page provided.
Selective estrogen receptor modulators (SERMs) work by blocking or activating different estrogen receptors at different levels. Since different tissues have different ratios of these estrogen receptors SERMs act like estrogens in some tissues and block estrogens in other tissues.
The main SERM up for consideration for transgender GAHT is raloxifene as of the two widely available, generic SERMs, it has the best safety profile and most relevant research.
Raloxifene acts largely antiestrogenically in breast tissue. It helps to prevent and treat breast cancer. It seems to act estrogenically in various tissues of interest to transgender individuals. It produces a gynoid (feminine) fat distribution, it causes estrogenic like actions on skin, and helps prevent osteoporosis by maintaining bone mineral density. Further it seems to have some estrogenic actions in the brain.
A TSEC of estradiol and raloxifene may warrant further study for transfeminine individuals. The reasons for discontinuation of research of combined estradiol and raloxifene were due to endometrial thickening and as transfeminine individuals (intersex individuals excepted) do not have an endometrium this is not a cause for concern.
Lastly, this article would be remiss if not to end with a major word of caution. There is to my knowledge, no published research on SERMs in transgender individuals. There is no long term research on raloxifene or tamoxifen on natal men. Finally, the mechanism of action of SERMs in general is that they partially suppress various types of estrogen receptor activity. They are far away from being fully characterized and it may be the case that the subtle ways in which they differ from estradiol may cause unforeseen long term consequences to transfeminine individuals.
That being said, they are an exciting frontier in the area of non-binary transfeminine gender-affirming hormone therapy (GAHT).
Introduction
While many people that begin a path of transfeminine GAHT do so with the desire to fully develop breasts there exists a sizable number of people, many on the non-binary transfeminine spectrum who wish to medically feminize themselves with little breast development. Because of this, raloxifene, a selective estrogen receptor modulator (SERM), has become talked about as a potential way to achieve this. A second-generation SERM, it was developed to prevent and treat breast cancer and (in the U.S.) prevent osteoporosis. In this article, I will review the estrogen receptor, the mechanism of action of SERMs and how they work in various relevant tissues, as well as discuss a few tissue-selective estrogen complexes (TSEC) that may be of interest.
Background
For an extremely in-depth review of the estrogen receptors, how they form, and the compounds that bind to them, I recommend Ascenzi, Bocedi, & Marino (2006).
Estrogen Receptors
Estrogens are compounds that cause changes in cells by binding to various estrogen receptors. These receptors are complexes consisting of proteins and other molecules. This binding causes a conformational change (change in shape) in the receptor that then results in some change in cell activity.
The main types of estrogen receptors are estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) as well as G protein-coupled estrogen receptor (GPER). The classical way in which estrogens were thought to have an impact is via ERα and ERβ. These are part of the nuclear receptor family. They exist on the nuclear membrane and once bound to an estrogen these receptors form a complicated biological machine that then causes a change in gene expression.
This estrogen receptor complex comes to exist due to the estrogen receptor containing many binding domains and on these domains other proteins or molecules can bind that then cause the estrogen receptor complex to be either activated, repressed, or destroyed.
In addition to the classic method of estrogen activation, there exists also GPER activity. G protein-coupled receptors are a huge super family of receptors that are membrane bound (they exist inside the cell membrane) and GPER receptors cause non genomic changes that can result in rapid actions within the cell as there is not a delay of having to generate RNA and then proteins. While there is some research into the role of SERMs with GPERs, much of the focus of research and this article will concern ourselves with classic estrogen receptor activity.
Selective Estrogen Receptor Modulators (SERMs)
Estradiol is the classic molecule that binds to the various estrogen receptors. Estradiol is an agonist of the estrogen receptors in that it activates them to action. In contrast, there exist full estrogen receptor antagonists such as fulvestrant (ICI-182,780), a compound that completely blocks the estrogen receptors from activating. SERMs are molecules that bind to the various estrogen receptors in a way that is at least partially different from estradiol. This means that in some contexts it is an antagonist (antiestrogenic) and in others it is an agonist (estrogenic) of the estrogen receptors. SERMs work by binding to ERα and ERβ and GPER at relatively different rates compared to estradiol and acting as either a full or partial antagonist at the receptor. Because different tissues have different ratios of expression of these receptors, the subsequent result is that SERMs act like estradiol in some tissues and act to suppress estradiol like actions in others.
At a molecular level, this occurs because a SERM is a different shape than estradiol and when it binds to the estrogen receptors its shape and the subsequent change in shape of the estrogen receptor may physically prevent (sterically inhibit) another compound or compounds that would need to bind to the estrogen receptor as well to activate its activity.
Originally developed for the treatment of breast cancer, the currently developed SERMs all display some similar properties. They act antiestrogenically in breast tissue, with many acting estrogenically on cholesterol and bones, and thus have found use as a preventative measure for osteoporosis in postmenopausal women. The first SERM to become available was tamoxifen in 1978, with second-generation SERM raloxifene becoming available in 1997 (in the U.S.). More recently, there have been a number of new third-generation SERMs developed, such as bazedoxifene, lasofoxifene, and ospemifene. However, as these are still under patent they are expensive and therefore do not currently make good candidates for GAHT.
Tissue-Selective Estrogen Complexes (TSECs)
Tissue-selective estrogen complexes (TSECs) are a combination of a SERM with an estrogen. The goal in their development was to create a unique mixture of estrogenic and antiestrogenic effects that would combine the antiestrogenic effects of some SERMs in the breasts (to prevent breast cancer) and endometrium (to prevent endometrial cancer), while maintaining the positive effects of estrogens in postmenopausal women such as improved bone density, cardiovascular health and weight maintenance. The only currently approved TSEC is a combination of conjugated equine estrogens (CEE) with bazedoxifene (CEE/BZA). An abandoned TSEC, estradiol/raloxifene, will also be considered in this article – as its reasons for abandonment are largely irrelevant to transfeminine people undergoing GAHT and has a number of advantages over CEE/BZA such as availability and lower risk of complications for transfeminine individuals.
Safety Studies
The two generically available SERMs, raloxifene and tamoxifen, have both undergone long-term, large-scale studies, specifically the Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) (Davis et al., 2013) study for tamoxifen, showing that its largest risk is endometrial cancer. However, it has significant risk of other serious side effects such as reduced cognition, liver toxicity, and various negative cardiovascular issues (fatty liver disease, deep vein thrombosis, etc…).
Raloxifene similarly had the Multiple Outcomes of Raloxifene Evaluation (MORE) (Ettinger, 1999) and Raloxifene Use for The Heart (RUTH) (Wenger et al., 2002) studies conducted and together they had the Study of Tamoxifen and Raloxifene (STAR) (Vogel et al., 2006) study, which shows that while it still has an increased risk of deep vein thrombosis, it is lower than tamoxifen and it does not have the other negative side effects associated with tamoxifen.
Tissue-Specific Effects of SERMs
In Breasts
Both tamoxifen and raloxifene have been shown to act antiestrogenically in the breast, and indeed this is the main reason for their development as drugs. They are both shown to decrease the risk of breast cancer in natal women and both have been shown to be effective in treating gynecomastia (male breast growth) (Lawrence et al., 2004; Kunath et al., 2012). The exact mechanism of action of both raloxifene and tamoxifen in the breast is still not entirely known. While they induce estrogen receptor changes, there are also several pathways that are not via the estrogen receptor they are involved with that may mediate their anti-breast cancer action and subsequently their anti-breast development activity.
In Fat
Changes in fat distribution and metabolism are a major aspect of GAHT (see Lain (2019) for an in-depth look on changes in fat distribution and metabolism with GAHT). Gynoid fat distribution occurs via both peripheral activity often by the leptin–ghrelin hormone system, and raloxifene seems to act estrogenically on the leptin-ghrelin hormone system helping to prevent abdominal adiposity (Tommaselli et al., 2006; Mauvais-Jarvis, Clegg, & Hevener, 2013). It also appears to act estrogenically directly in fat tissue (adipose tissue) (Francucci et al., 2005), again helping to increase subcutaneous fat around the thighs and buttocks that is a primary characteristic of gynoid fat distribution. In addition in postmenopausal women, it is found to reduce total cholesterol and LDL cholesterol. However, it does not appear to impact glycemic index, meaning that it does not help prevent postmenopausal type 2 diabetes. It is unknown how exactly this may map to transfeminine individuals.
In Muscle
While muscle mass is strongly mediated via testosterone (see Lain (2019), a review on muscle mass and testosterone), there also seems to be a correlation between estrogen and muscle mass. Postmenopausal natal women undergo significant sarcopenia (age-related muscle mass loss) that can be prevented or minimized via estrogen GAHT. Similar to estrogens, raloxifene also helps increase muscle mass in postmenopausal women and prevent loss of muscle mass (Jacobsen et al., 2010). Because the exact mechanism of action by which the estrogen receptors mediates muscle strength is unknown, it is hard to understand how this translates to transfeminine individuals. However, it does point to estrogenic action in muscle tissue.
In Skin
In classic transfeminine hormone therapy, the softening of skin is often one of the first noticeable effects. In addition in postmenopausal women, a lack of estrogen can cause thinner skin and decreased elasticity, which is preventable via GAHT. In studies, tamoxifen appears to cause abnormal hair follicles, skin atrophy, and even possibly alopecia (hair loss) developing on the crown of the head; this and direct evidence suggests that tamoxifen is an antagonist of the estrogen receptor in hair follicles (Thornton, 2007). However, there is some evidence to suggest that raloxifene has a positive estrogenic effect on collagen biosynthesis in human skin (Surazynski et al., 2003).
In Testes
SERMs such as raloxifene and tamoxifen have been found to cause an increase in serum gonadotropins (luteinizing hormone and follicle stimulating hormone) as well as a concurrent increase in testosterone (Rambhatla, Mills, & Rajfer, 2016). This highlights the importance of using an antiandrogen in conjunction with a SERM in GAHT.
In the Brain
The estrogen receptors have significant impact on brain function. Estradiol GAHT has been associated with antidepressant effects in postmenopausal women and estrogen receptors mediate significant changes in the hypothalamus related to maintaining body fat percentage and hunger. SERMs in general appear to act on the brain both via the estrogen receptor as well as via non-estrogen-receptor-mediated pathways.
Tamoxifen in animal models has neuroprotective effects for a number of modes of neural dysfunction including traumatic injury, Parkinson’s disease, Alzheimer’s disease, and mania. However, in some trials, it is also associated with cognitive impairment, including visual memory, verbal memory, visuospatial ability, and speed processing tasks (Arevalo et al., 2010).
Raloxifene does not appear to have negative cognitive implications as tamoxifen does. However, it is shown to act at least partially estrogenically in the hypothalamus, to reduce anxiety in ovariectomized rats, and to decrease anxiety and depression in postmenopausal women. It may also regulate opioid and GABAergic activity by modulating endorphin levels and has been shown to positively impact schizophrenia (Arevalo et al., 2010).
SERMs in general and raloxifene in particular seem to have some estrogenic like actions in the brain, though it does not seem to be especially significant (Smith & O’Malley, 2004). However, it is still very far from understood all of its actions in the brain as estrogen receptors are found throughout.
Tissue-Specific Effects of TSECs
Up till now, we have only talked about individual compounds. However, TSECs came to be developed by the thinking that by combining an estrogen with a SERM you could get a different effect profile. The only currently medically approved TSEC is CEE/BZA. Specifically, the goal for TSECs was to gain many of the beneficial aspects of estrogen GAHT in postmenopausal natal women while blocking the increased risks of breast cancer and endometrial thickening associated with it. In studies of ovariectomized rats, bazedoxifene was found to maintain its antiestrogenic activity in the endometrium without attenuating the estrogen’s positive effects in movement, fat or bone (Pickar, Boucher, & Morgenstern, 2018).
CEE/BZA while approved for postmenopausal women presents a potentially higher risk for transgender individuals due to its increased risk of deep vein thrombosis, and is less affordable being unavailable as a generic formulation. That being the case another TSEC – a combination of raloxifene and estradiol – has had a number of clinical studies performed on it. It was ultimately abandoned by the medical community due to causing endometrial thickening, again something not typically of concern to transfeminine individuals undergoing GAHT. In the studies performed, raloxifene plus estradiol seemed to reduce many postmenopausal symptoms, such as hot flashes, vaginal dryness, and overall treatment satisfaction, compared to just raloxifene (Carneiro, de Cassia de Maio Dardes, & Haidar, 2012).
Discussion
The primary driver of interest in using SERMs in GAHT is their antiestrogenic effects in the breasts with potential for feminization in other tissues. Of the two affordable and available SERMs, raloxifene by far seems to be the one that has the best safety profile, while also being most effective in the literal research we have on preventing breast growth.
That being said, TSECs seem to be a potentially untapped area of research. Raloxifene and estradiol, while potentially unsafe for those with uteruses, may be well-tolerated by those without. Research into the potential efficacy of raloxifene plus estradiol both in preventing breast growth and relative rate and degree of feminization otherwise versus estradiol alone and raloxifene alone could potentially yield extremely interesting results for transfeminine individuals seeking GAHT that includes minimal breast development.
Ultimately, SERMs, and how different they are from estradiol, is still far from being understood. While we have some knowledge that they are generally safe long-term, this information applies pretty much exclusively to postmenopausal natal women, though there is some indication that tamoxifen has a low incidence of adverse effects in natal men (Wibowo et al., 2016). Because estrogen receptors and their actions in a wide variety of tissues are extremely complex and multifactorial, this means that each SERM and TSEC must be evaluated extremely carefully across these tissues to fully understand it.
References
Aly. (2019). An Exploration of Possibilities for Hormone Therapy in Non-Binary Transfeminine People. Transfeminine Science. [URL]
Arevalo, M. A., Santos-Galindo, M., Lagunas, N., Azcoitia, I., & Garcia-Segura, L. M. (2010). Selective estrogen receptor modulators as brain therapeutic agents. Journal of Molecular Endocrinology, 46(1), R1–R9. [DOI:10.1677/jme-10-0122]
Ascenzi, P., Bocedi, A., & Marino, M. (2006). Structure–function relationship of estrogen receptor α and β: Impact on human health. Molecular Aspects of Medicine, 27(4), 299–402. [DOI:10.1016/j.mam.2006.07.001]
Carneiro, A. L., de Cassia de Maio Dardes, R., & Haidar, M. A. (2012). Estrogens plus raloxifene on endometrial safety and menopausal symptoms—semisystematic review. Menopause, 19(7), 830–834. [DOI:10.1097/gme.0b013e31824a74ce]
Davies, C., Pan, H., Godwin, J., Gray, R., Arriagada, R., Raina, V., Abraham, M., Alencar, V. H., Badran, A., Bonfill, X., Bradbury, J., Clarke, M., Collins, R., Davis, S. R., Delmestri, A., Forbes, J. F., Haddad, P., Hou, M., Inbar, M., Khaled, H., Kielanowska, J., Kwan, W., Mathew, B. S., Mittra, I., Müller, B., Nicolucci, A., Peralta, O., Pernas, F., Petruzelka, L., Pienkowski, T., Radhika, R., Rajan, B., Rubach, M. T., Tort, S., Urrútia, G., Valentini, M., Wang, Y., & Peto, R. (2013). Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. The Lancet, 381(9869), 805–816. [DOI:10.1016/s0140-6736(12)61963-1]
Ettinger, B. (1999). Reduction of Vertebral Fracture Risk in Postmenopausal Women With Osteoporosis Treated With Raloxifene: Results From a 3-Year Randomized Clinical Trial. JAMA, 282(7), 637–645. [DOI:10.1001/jama.282.7.637]
Francucci, C. M., Pantaleo, D., Iori, N., Camilletti, A., Massi, F., & Boscaro, M. (2005). Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women. Journal of Endocrinological Investigation, 28(9), 623–631. [DOI:10.1007/bf03347261]
Jacobsen, D. E., Samson, M. M., Emmelot-Vonk, M. H., & Verhaar, H. J. (2010). Raloxifene and body composition and muscle strength in postmenopausal women: a randomized, double-blind, placebo-controlled trial. European Journal of Endocrinology, 162(2), 371–376. [DOI:10.1530/eje-09-0619]
Kunath, F., Keck, B., Antes, G., Wullich, B., & Meerpohl, J. J. (2012). Tamoxifen for the management of breast events induced by non-steroidal antiandrogens in patients with prostate cancer: a systematic review. BMC Medicine, 10(1), 96. [DOI:10.1186/1741-7015-10-96]
Lain. (2019). On the Impact of Sex Hormones on Fat Metabolism with an Eye Towards Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Lain. (2019). Sources/Excerpts: A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Lain. (2019). The Relationship Between the Endocrine System and Muscle Mass. Transfeminine Science. [URL]
Lawrence, S. E., Arnold Faught, K., Vethamuthu, J., & Lawson, M. L. (2004). Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia. The Journal of Pediatrics, 145(1), 71–76. [DOI:10.1016/j.jpeds.2004.03.057]
Mauvais-Jarvis, F., Clegg, D. J., & Hevener, A. L. (2013). The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews, 34(3), 309–338. [DOI:10.1210/er.2012-1055]
Pickar, J. H., Boucher, M., & Morgenstern, D. (2018). Tissue selective estrogen complex (TSEC): a review. Menopause, 25(9), 1033–1045. [DOI:10.1097/gme.0000000000001095]
Rambhatla, A., Mills, J. N., & Rajfer, J. (2016). The Role of Estrogen Modulators in Male Hypogonadism and Infertility. Reviews in Urology, 18(2), 66–72. [PubMed] [PubMed Central] [DOI:10.3909/riu0711]
Smith, C. L., & O’Malley, B. W. (2004). Coregulator Function: A Key to Understanding Tissue Specificity of Selective Receptor Modulators. Endocrine Reviews, 25(1), 45–71. [DOI:10.1210/er.2003-0023]
Surazynski, A., Jarzabek, K., Haczynski, J., Laudanski, P., Palka, J., & Wolczynski, S. (2003). Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. International Journal of Molecular Medicine, 12(5), 803–809. [DOI:10.3892/ijmm.12.5.803]
Thornton, J. (2007). Effect of estrogens on skin aging and the potential role of SERMs. Clinical Interventions in Aging, 2(3), 283–297. [DOI:10.2147/cia.s798]
Tommaselli, G. A., Di Carlo, C., Di Spiezio Sardo, A., Bifulco, G., Cirillo, D., Guida, M., Capasso, R., & Nappi, C. (2006). Serum leptin levels and body composition in postmenopausal women treated with tibolone and raloxifene. Menopause, 13(4), 660–668. [DOI:10.1097/01.gme.0000227335.27996.d8]
Vogel, V. G., Costantino, J. P., Wickerham, D. L., Cronin, W. M., Cecchini, R. S., Atkins, J. N., Bevers, T. B., Fehrenbacher, L., Pajon, E. R., Jr, Wade, J. L., 3rd, Robidoux, A., Margolese, R. G., James, J., Lippman, S. M., Runowicz, C. D., Ganz, P. A., Reis, S. E., McCaskill-Stevens, W., Ford, L. G., Jordan, V. C., … National Surgical Adjuvant Breast and Bowel Project (NSABP) (2006). Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA, 295(23), 2727–2741. [DOI:10.1001/jama.295.23.joc60074]
Wenger, N. K., Barrett-Connor, E., Collins, P., Grady, D., Kornitzer, M., Mosca, L., Sashegyi, A., Baygani, S. K., Anderson, P. W., & Moscarelli, E. (2002). Baseline characteristics of participants in the Raloxifene Use for The Heart (RUTH) trial. The American Journal of Cardiology, 90(11), 1204–1210. [DOI:10.1016/s0002-9149(02)02835-7]
Wibowo, E., Pollock, P. A., Hollis, N., & Wassersug, R. J. (2016). Tamoxifen in men: a review of adverse events. Andrology, 4(5), 776–788. [DOI:10.1111/andr.12197]
\ No newline at end of file
diff --git a/transfemscience.org/articles/shbg-unimportant-suppl/index.html b/transfemscience.org/articles/shbg-unimportant-suppl/index.html
index e2fc3c31..be3867f7 100644
--- a/transfemscience.org/articles/shbg-unimportant-suppl/index.html
+++ b/transfemscience.org/articles/shbg-unimportant-suppl/index.html
@@ -1 +1 @@
-Supplement: The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy - Transfeminine Science
Supplement: The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy
By Aly | First published July 8, 2020 | Last modified March 14, 2023
Preface
This article is a supplement to the article here. It was originally just for calculation of free sex hormone levels but I decided to add some other content to it as well.
Calculation of Free Hormone Levels
Spreadsheet Calculator (Mazer, 2009)
A researcher developed and published a “user-friendly” spreadsheet that can be used to calculate free and bioavailable levels of several steroid hormones (Mazer, 2009). This spreadsheet approach is analogous to how free hormone levels are calculated with actual conventional blood work. Total hormone levels and levels of plasma proteins like albumin and SHBG are taken as inputs by the spreadsheet, and free and bioavailable hormone levels are given as outputs.
The spreadsheet is supplementary material for Mazer (2009) and is behind a paywall. Because of this, I’ve uploaded a copy of the original spreadsheet here (Microsoft Excel or XLS format).
If you’re curious how SHBG may be influencing your free estradiol percentage, you can use the spreadsheet to get an estimate. If you don’t have albumin, CBG, or cortisol values, you can use the default input values in the spreadsheet. If you don’t have other input values (e.g., estrone or SHBG), you can input representative values that are sensible for your scenario. It should be noted that calculated free hormone levels are only estimates and hence can be inaccurate. In any case, they are generally fairly close to the values that would be obtained with actual measurement. Use of default input values as opposed to real measured numbers may further contribute to inaccuracy.
Some Experimentation with the Calculator
Here are the results of some experimentation I did with the calculator:
Table: Relationships between SHBG levels and calculated free estradiol fraction at fixed estradiol levels:
SHBG
Estradiol fixed ≤1,000 pg/mL
Estradiol fixed 20,000 pg/mL
Level
Change a
Free E2 fraction
Change a
Free E2 fraction
Change a
0 nmol/L
0.0×
3.26%
+77.2%
3.26%
+37.0%
25 nmol/L
0.5×
2.36%
+28.3%
2.78%
+16.8%
50 nmol/L
1.0×
1.84%
0%
2.38%
0%
75 nmol/L
1.5×
1.50%
–18.5%
2.04%
–14.3%
100 nmol/L
2.0×
1.27%
–31.0%
1.77%
–25.6%
125 nmol/L
2.5×
1.10%
–40.2%
1.54%
–35.3%
150 nmol/L
3.0×
0.97%
–47.3%
1.36%
–42.9%
200 nmol/L
4.0×
0.79%
–57.1%
1.08%
–54.6%
250 nmol/L
5.0×
0.66%
–64.1%
0.89%
–62.6%
300 nmol/L
6.0×
0.57%
–69.0%
0.75%
–68.5%
350 nmol/L
7.0×
0.50%
–72.8%
0.65%
–72.7%
400 nmol/L
8.0×
0.44%
–76.1%
0.57%
–76.1%
a Change relative to a reasonable non-pregnancy physiological value (specifically 50 nmol/L for SHBG, 1.84% for free E2 at a fixed level of ≤1,000 pg/mL, 2.38% for free E2 at a fixed level of 20,000 pg/mL).
Androgen levels were set to female levels, estrone levels were set to be the same as estradiol levels, and all other inputs besides SHBG and total estradiol levels were left as the defaults. There was very little variation in free estradiol fractions with different estradiol levels at and below 1,000 pg/mL for each given level of SHBG (hence why the table says “Estradiol fixed ≤1,000 pg/mL”).
The estradiol levels fixed to ≤1,000 pg/mL are intended to represent typical therapeutic circumstances while the estradiol levels fixed to 20,000 pg/mL are supposed to represent late pregnancy.
Note that since estradiol induces SHBG production, SHBG levels are strongly correlated with estradiol levels. Generally speaking, when estradiol is low, SHBG will also be low, and when estradiol is high, SHBG will also be high. Hence, having highly divergent SHBG and estradiol levels as in the table would be very unusual and is physiologically unrealistic. It is only explored here as a thought experiment.
Note again that these free estradiol numbers are calculated and hence are only estimates.
Other Papers on Calculation of Free Hormone Levels
Another tool for calculating free estradiol and testosterone can be found here. Various other free testosterone calculators also exist on the web (Google Search).
Additional Literature on SHBG and Free Estradiol
Additional SHBG and Free Estradiol Clinical Studies
Some more good studies on SHBG and free estradiol that weren’t discussed in the main article:
A case report of a young woman with estrogen insensitivity syndrome (EIS) (i.e., defective ERα) suggests that the ERα is the specific estrogen receptor that is responsible for increased SHBG production and levels with estrogens (Quaynor et al., 2013). Due to her EIS and lack of negative feedback on the hypothalamus–pituitary–gonadal axis, the woman had estradiol levels of as high as 3,500 pg/mL. In spite of this however, her SHBG levels remained less than 50 nmol/L. During pregnancy, at the point in the second trimester at which estradiol levels reach 3,000 pg/mL, SHBG levels are normally around 300 nmol/L on average (a 6-fold increase from a pre-pregnancy baseline of about 50 nmol/L).
References
Ben-Rafael, Z., Mastroianni, L., Meloni, F., Strauss, J. F., & Flickinger, G. L. (1986). Changes in serum sex hormone-binding globulin, free estradiol, and testosterone during gonadotropin treatment. Fertility and Sterility, 46(4), 593–598. [DOI:10.1016/s0015-0282(16)49633-0]
Carlström, K., Pschera, H., & Lunell, N. (1988). Serum levels of oestrogens, progesterone, follicle-stimulating hormone and sex-hormone-binding globulin during simultaneous vaginal administration of 17β-oestradiol and progesterone in the pre- and post-menopause. Maturitas, 10(4), 307–316. [DOI:10.1016/0378-5122(88)90066-7]
Elaut, E., De Cuypere, G., De Sutter, P., Gijs, L., Van Trotsenburg, M., Heylens, G., Kaufman, J., Rubens, R., & T’Sjoen, G. (2008). Hypoactive sexual desire in transsexual women: prevalence and association with testosterone levels. European Journal of Endocrinology, 158(3), 393–399. [DOI:10.1530/eje-07-0511]
Jasonni, V., Bulletti, C., Naldi, S., Ciotti, P., Di Cosmo, D., Lazzaretto, R., & Flamigni, C. (1988). Biological and endocrine aspects of transdermal 17β-oestradiol administration in post-menopausal women. Maturitas, 10(4), 263–270. [DOI:10.1016/0378-5122(88)90062-x]
Lapauw, B., Taes, Y., Simoens, S., Van Caenegem, E., Weyers, S., Goemaere, S., Toye, K., Kaufman, J., & T’Sjoen, G. G. (2008). Body composition, volumetric and areal bone parameters in male-to-female transsexual persons. Bone, 43(6), 1016–1021. [DOI:10.1016/j.bone.2008.09.001]
Mazer, N. A. (2009). A novel spreadsheet method for calculating the free serum concentrations of testosterone, dihydrotestosterone, estradiol, estrone and cortisol: With illustrative examples from male and female populations. Steroids, 74(6), 512–519. [DOI:10.1016/j.steroids.2009.01.008]
Nelson, M. D., Szczepaniak, L. S., Wei, J., Szczepaniak, E., Sánchez, F. J., Vilain, E., Stern, J. H., Bergman, R. N., Bairey Merz, C. N., & Clegg, D. J. (2016). Transwomen and the Metabolic Syndrome: Is Orchiectomy Protective? Transgender Health, 1(1), 165–171. [DOI:10.1089/trgh.2016.0016]
Odlind, V., Elamsson, K., Englund, D. E., Victor, A., & Johansson, E. D. (1982). Effects of oestradiol on sex hormone binding globulin. Acta Endocrinologica, 101(2), 248–253. [DOI:10.1530/acta.0.1010248] [PDF]
Quaynor, S. D., Stradtman, E. W., Kim, H., Shen, Y., Chorich, L. P., Schreihofer, D. A., & Layman, L. C. (2013). Delayed Puberty and Estrogen Resistance in a Woman with Estrogen Receptor α Variant. New England Journal of Medicine, 369(2), 164–171. [DOI:10.1056/nejmoa1303611]
Rinaldi, S., Geay, A., Déchaud, H., Biessy, C., Zeleniuch-Jacquotte, A., Akhmedkhanov, A., Shore, R. E., Riboli, E., Toniolo, P., & Kaaks, R. (2002). Validity of free testosterone and free estradiol determinations in serum samples from postmenopausal women by theoretical calculations. Cancer Epidemiology and Prevention Biomarkers, 11(10), 1065–1071. [Google Scholar] [PubMed] [URL]
Vermeulen, A., Verdonck, L., & Kaufman, J. M. (1999). A critical evaluation of simple methods for the estimation of free testosterone in serum. The Journal of Clinical Endocrinology & Metabolism, 84(10), 3666–3672. [DOI:10.1210/jcem.84.10.6079]
\ No newline at end of file
+Supplement: The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy - Transfeminine Science
Supplement: The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy
By Aly | First published July 8, 2020 | Last modified March 14, 2023
Preface
This article is a supplement to the article here. It was originally just for calculation of free sex hormone levels but I decided to add some other content to it as well.
Calculation of Free Hormone Levels
Spreadsheet Calculator (Mazer, 2009)
A researcher developed and published a “user-friendly” spreadsheet that can be used to calculate free and bioavailable levels of several steroid hormones (Mazer, 2009). This spreadsheet approach is analogous to how free hormone levels are calculated with actual conventional blood work. Total hormone levels and levels of plasma proteins like albumin and SHBG are taken as inputs by the spreadsheet, and free and bioavailable hormone levels are given as outputs.
The spreadsheet is supplementary material for Mazer (2009) and is behind a paywall. Because of this, I’ve uploaded a copy of the original spreadsheet here (Microsoft Excel or XLS format).
If you’re curious how SHBG may be influencing your free estradiol percentage, you can use the spreadsheet to get an estimate. If you don’t have albumin, CBG, or cortisol values, you can use the default input values in the spreadsheet. If you don’t have other input values (e.g., estrone or SHBG), you can input representative values that are sensible for your scenario. It should be noted that calculated free hormone levels are only estimates and hence can be inaccurate. In any case, they are generally fairly close to the values that would be obtained with actual measurement. Use of default input values as opposed to real measured numbers may further contribute to inaccuracy.
Some Experimentation with the Calculator
Here are the results of some experimentation I did with the calculator:
Table: Relationships between SHBG levels and calculated free estradiol fraction at fixed estradiol levels:
SHBG
Estradiol fixed ≤1,000 pg/mL
Estradiol fixed 20,000 pg/mL
Level
Change a
Free E2 fraction
Change a
Free E2 fraction
Change a
0 nmol/L
0.0×
3.26%
+77.2%
3.26%
+37.0%
25 nmol/L
0.5×
2.36%
+28.3%
2.78%
+16.8%
50 nmol/L
1.0×
1.84%
0%
2.38%
0%
75 nmol/L
1.5×
1.50%
–18.5%
2.04%
–14.3%
100 nmol/L
2.0×
1.27%
–31.0%
1.77%
–25.6%
125 nmol/L
2.5×
1.10%
–40.2%
1.54%
–35.3%
150 nmol/L
3.0×
0.97%
–47.3%
1.36%
–42.9%
200 nmol/L
4.0×
0.79%
–57.1%
1.08%
–54.6%
250 nmol/L
5.0×
0.66%
–64.1%
0.89%
–62.6%
300 nmol/L
6.0×
0.57%
–69.0%
0.75%
–68.5%
350 nmol/L
7.0×
0.50%
–72.8%
0.65%
–72.7%
400 nmol/L
8.0×
0.44%
–76.1%
0.57%
–76.1%
a Change relative to a reasonable non-pregnancy physiological value (specifically 50 nmol/L for SHBG, 1.84% for free E2 at a fixed level of ≤1,000 pg/mL, 2.38% for free E2 at a fixed level of 20,000 pg/mL).
Androgen levels were set to female levels, estrone levels were set to be the same as estradiol levels, and all other inputs besides SHBG and total estradiol levels were left as the defaults. There was very little variation in free estradiol fractions with different estradiol levels at and below 1,000 pg/mL for each given level of SHBG (hence why the table says “Estradiol fixed ≤1,000 pg/mL”).
The estradiol levels fixed to ≤1,000 pg/mL are intended to represent typical therapeutic circumstances while the estradiol levels fixed to 20,000 pg/mL are supposed to represent late pregnancy.
Note that since estradiol induces SHBG production, SHBG levels are strongly correlated with estradiol levels. Generally speaking, when estradiol is low, SHBG will also be low, and when estradiol is high, SHBG will also be high. Hence, having highly divergent SHBG and estradiol levels as in the table would be very unusual and is physiologically unrealistic. It is only explored here as a thought experiment.
Note again that these free estradiol numbers are calculated and hence are only estimates.
Other Papers on Calculation of Free Hormone Levels
Another tool for calculating free estradiol and testosterone can be found here. Various other free testosterone calculators also exist on the web (Google Search).
Additional Literature on SHBG and Free Estradiol
Additional SHBG and Free Estradiol Clinical Studies
Some more good studies on SHBG and free estradiol that weren’t discussed in the main article:
A case report of a young woman with estrogen insensitivity syndrome (EIS) (i.e., defective ERα) suggests that the ERα is the specific estrogen receptor that is responsible for increased SHBG production and levels with estrogens (Quaynor et al., 2013). Due to her EIS and lack of negative feedback on the hypothalamus–pituitary–gonadal axis, the woman had estradiol levels of as high as 3,500 pg/mL. In spite of this however, her SHBG levels remained less than 50 nmol/L. During pregnancy, at the point in the second trimester at which estradiol levels reach 3,000 pg/mL, SHBG levels are normally around 300 nmol/L on average (a 6-fold increase from a pre-pregnancy baseline of about 50 nmol/L).
References
Aly. (2020). The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Ben-Rafael, Z., Mastroianni, L., Meloni, F., Strauss, J. F., & Flickinger, G. L. (1986). Changes in serum sex hormone-binding globulin, free estradiol, and testosterone during gonadotropin treatment. Fertility and Sterility, 46(4), 593–598. [DOI:10.1016/s0015-0282(16)49633-0]
Carlström, K., Pschera, H., & Lunell, N. (1988). Serum levels of oestrogens, progesterone, follicle-stimulating hormone and sex-hormone-binding globulin during simultaneous vaginal administration of 17β-oestradiol and progesterone in the pre- and post-menopause. Maturitas, 10(4), 307–316. [DOI:10.1016/0378-5122(88)90066-7]
Elaut, E., De Cuypere, G., De Sutter, P., Gijs, L., Van Trotsenburg, M., Heylens, G., Kaufman, J., Rubens, R., & T’Sjoen, G. (2008). Hypoactive sexual desire in transsexual women: prevalence and association with testosterone levels. European Journal of Endocrinology, 158(3), 393–399. [DOI:10.1530/eje-07-0511]
Jasonni, V., Bulletti, C., Naldi, S., Ciotti, P., Di Cosmo, D., Lazzaretto, R., & Flamigni, C. (1988). Biological and endocrine aspects of transdermal 17β-oestradiol administration in post-menopausal women. Maturitas, 10(4), 263–270. [DOI:10.1016/0378-5122(88)90062-x]
Lapauw, B., Taes, Y., Simoens, S., Van Caenegem, E., Weyers, S., Goemaere, S., Toye, K., Kaufman, J., & T’Sjoen, G. G. (2008). Body composition, volumetric and areal bone parameters in male-to-female transsexual persons. Bone, 43(6), 1016–1021. [DOI:10.1016/j.bone.2008.09.001]
Mazer, N. A. (2009). A novel spreadsheet method for calculating the free serum concentrations of testosterone, dihydrotestosterone, estradiol, estrone and cortisol: With illustrative examples from male and female populations. Steroids, 74(6), 512–519. [DOI:10.1016/j.steroids.2009.01.008]
Nelson, M. D., Szczepaniak, L. S., Wei, J., Szczepaniak, E., Sánchez, F. J., Vilain, E., Stern, J. H., Bergman, R. N., Bairey Merz, C. N., & Clegg, D. J. (2016). Transwomen and the Metabolic Syndrome: Is Orchiectomy Protective? Transgender Health, 1(1), 165–171. [DOI:10.1089/trgh.2016.0016]
Odlind, V., Elamsson, K., Englund, D. E., Victor, A., & Johansson, E. D. (1982). Effects of oestradiol on sex hormone binding globulin. Acta Endocrinologica, 101(2), 248–253. [DOI:10.1530/acta.0.1010248] [PDF]
Quaynor, S. D., Stradtman, E. W., Kim, H., Shen, Y., Chorich, L. P., Schreihofer, D. A., & Layman, L. C. (2013). Delayed Puberty and Estrogen Resistance in a Woman with Estrogen Receptor α Variant. New England Journal of Medicine, 369(2), 164–171. [DOI:10.1056/nejmoa1303611]
Rinaldi, S., Geay, A., Déchaud, H., Biessy, C., Zeleniuch-Jacquotte, A., Akhmedkhanov, A., Shore, R. E., Riboli, E., Toniolo, P., & Kaaks, R. (2002). Validity of free testosterone and free estradiol determinations in serum samples from postmenopausal women by theoretical calculations. Cancer Epidemiology and Prevention Biomarkers, 11(10), 1065–1071. [Google Scholar] [PubMed] [URL]
Vermeulen, A., Verdonck, L., & Kaufman, J. M. (1999). A critical evaluation of simple methods for the estimation of free testosterone in serum. The Journal of Clinical Endocrinology & Metabolism, 84(10), 3666–3672. [DOI:10.1210/jcem.84.10.6079]
\ No newline at end of file
diff --git a/transfemscience.org/articles/shbg-unimportant/index.html b/transfemscience.org/articles/shbg-unimportant/index.html
index 6b928bb2..8f48e72e 100644
--- a/transfemscience.org/articles/shbg-unimportant/index.html
+++ b/transfemscience.org/articles/shbg-unimportant/index.html
@@ -1 +1 @@
-The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy - Transfeminine Science
The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy
By Aly | First published July 10, 2020 | Last modified March 25, 2023
Abstract / TL;DR
Sex hormones such as testosterone and estradiol bind to blood proteins like albumin and SHBG. This limits their biological activity by reducing their free fractions. Androgens decrease SHBG production while estrogens increase SHBG production. Hence, testosterone and estradiol can influence their own free fractions. Due to robust inactivation in the liver, testosterone and estradiol have relatively small influences on SHBG levels under normal physiological circumstances. At very high levels however, they can considerably influence SHBG levels. During pregnancy, when there are massive increases in estradiol levels (e.g., 100-fold), a maximal 5- to 10-fold elevation in SHBG levels occurs. Although large increases in SHBG levels can strongly limit the biological activity of testosterone, the situation with estradiol is different. In late pregnancy, the percentage of estradiol that is free appears to be decreased only to around 60% of that of non-pregnancy. Earlier in pregnancy, when estradiol levels are lower, the free fractions of estradiol are reduced to a lesser extent. At typical therapeutic levels of estradiol in transfeminine hormone therapy (<200 pg/mL), the limiting influence of SHBG on free estradiol is minimal. Oral estradiol has a greater influence on SHBG production than non-oral estradiol and may be a different case however. In any case, consequent lesser activity of oral estradiol is only theoretical, and available clinical studies so far haven’t reported important therapeutic differences relative to non-oral estradiol. Although SHBG may reduce free estradiol fractions in some contexts, only relatively low estradiol levels (<50 pg/mL) appear to be needed for maximal feminization and breast development in cisgender females and transfeminine people. In conclusion, the influence of SHBG on the effectiveness of estradiol isn’t something that should be a major source of concern in transfeminine hormone therapy.
Binding of Sex Hormones to Blood Proteins
Sex hormones bind to proteins in the blood called plasma proteins. This is a phenomenon known as plasma protein binding. In the case of androgens and estrogens, the plasma proteins they bind to are mainly albumin and sex hormone-binding globulin (SHBG). Plasma protein binding serves to prevent sex hormones from interacting with their target cells and hence from binding to and activating their receptors (Hammond, 2016). This is because plasma proteins are too large and lipid-insoluble to cross the lipid-rich cell membrane. As a result, they’re unable to diffuse through capillaries to exit the circulation and enter into tissues or to be taken up into cells. When the sex hormone is bound to plasma protein, it can’t reach target cells either. Hence, plasma protein binding limits the biological activity of sex hormones (Hammond, 2016). Binding to plasma proteins also serves to extend the biological half-lives of sex hormones. This is because protein-bound sex hormone is likewise unavailable for metabolism and elimination, processes that depend on cellular uptake.
There is only a single sex hormone binding site per molecule of SHBG (Moore & Bulbrook, 1988), whereas albumin has six binding sites for different substrates (Pardridge, 1988). Androgens and estradiol have high affinity for SHBG (nM) and low affinity for albumin (μM) (Moore & Bulbrook, 1988; Hammond, 2016). However, albumin levels are several orders of magnitude higher than SHBG levels (μM vs. nM), so this serves to balance out the fractions of sex hormone bound to each protein (Hammond, 2016). Androgens have higher affinities for SHBG than do estradiol or other estrogens. Estradiol has only about 10 to 20% of the affinity of dihydrotestosterone (DHT) and 33 to 50% of the affinity of testosterone for SHBG (Anderson, 1974; Ojasoo & Raynaud, 1978; Pugeat, Dunn, Nisula, 1981). As such, testosterone and DHT bind more strongly to SHBG than does estradiol.
The vast majority of sex hormone content in the blood is bound to plasma proteins; at any given time more than 97% of the testosterone, estradiol, and progesterone in the blood is plasma protein-bound (Strauss & FitzGerald, 2019). The fraction of sex hormone that isn’t bound to plasma proteins is known as the free or unbound fraction. This is the fraction that is available for diffusion into cells and hence is considered to be biologically active (Hammond, 2016). Total levels refer to both free/unbound and bound hormone. Bioavailable levels include both albumin-bound and free hormone levels. Due to their relatively weak affinity for albumin, sex hormones bound to albumin may to some extent be biologically active—hence the “bioavailable” descriptor (Nguyen et al., 2008). However, more research is needed to fully elucidate the biological activity of albumin-bound sex hormone fractions.
The relative calculated free and bound percentages of estradiol, testosterone, and DHT to albumin, SHBG, and another plasma protein known as corticosteroid-binding globulin (CBG) (only binds small fractions of the androgens and has no binding to estradiol) are shown in the table below.
Free sex hormone levels and percentages are often calculated from levels of total sex hormone, albumin, SHBG, and CBG with validated mathematical models constructed from data of published studies. This is because free sex hormone levels are usually very low (pM range) and are difficult to measure with routine blood testing methods. While generally in the vicinity of the true values, calculated results may not always be fully accurate (Rosner, 2015; Goldman et al., 2017; Handelsman, 2017; Keevil & Adaway, 2019). As such, measured levels, when feasible, are preferable.
Testosterone, DHT, and estradiol are strongly inactivated by the liver and have relatively weak effects in this part of the body under normal circumstances. As a result, they have much less relative impact on SHBG production than do synthetic hormonal agents. Accordingly, SHBG levels change only slightly over the course of the menstrual cycle in women despite substantial fluctuations in estradiol levels (Freymann et al., 1977b; Plymate et al., 1985; Schijf et al., 1993; Braunstein et al., 2011; Rothman et al., 2011; Fanelli et al., 2013; Rezaii et al., 2017). In one study, SHBG levels increased by about 6 to 13% (+2.9–5.3 nmol/L) going from the follicular phase to the luteal phase of the cycle (Braunstein et al., 2011). There is additionally only a small decrease in SHBG levels attributable to the sharp decline in estradiol with menopause (Burger et al., 2000; Guthrie et al., 2004). Nonetheless, estradiol therapy can more considerably influence the production of SHBG and other liver proteins as well under specific conditions (Kuhl, 1998). This is due to 1) use of oral estradiol, which because of the first pass through the liver has a greater impact on estrogen-sensitive liver synthesis than non-oral routes (Kuhl, 2005); and 2) use of high estradiol doses, for instance typical injectable doses. The table below shows SHBG increases from various studies with different estrogen routes, doses, and types.
Table 2: Relative increases in SHBG levels with some different estrogenic exposures:
a Estimated typical estradiol levels from various sources (e.g., Aly, 2020; Wiki). b Due to differences in molecular weight, EV has about 75% of the amount of estradiol as regular estradiol. Hence, 6 mg/day EV is approximately equivalent to 4.5 mg/day E2.
The influence of estradiol on SHBG levels is most relevant to pregnancy, when estradiol levels increase to far higher levels than usual. In late pregnancy, estradiol levels are generally around 15,000 to 25,000 pg/mL on average (Graphs; Troisi et al., 2003; Adamcová et al., 2018). These estradiol levels are on the order of 100-fold higher than normal menstrual cycle levels. In parallel with the massive increases in estradiol levels, SHBG levels increase by about 5- to 10-fold by late pregnancy (Anderson, 1974; Hammond, 2017). The dose–response curve of estrogens on SHBG production shows saturation, with most of the increase in SHBG levels happening at lower estradiol levels as well as limits to how much SHBG levels can be increased (Mean, Pellaton, & Magrini, 1977; O’Leary et al., 1991; Kerlan et al., 1994; Kuhl, 1999). The graphs below show SHBG levels throughout pregnancy.
Figure 1: SHBG and total estradiol levels during pregnancy in women (O’Leary et al., 1991). The lines are the mean and/or 95th percentile levels while the points are individual measurements.
Figure 2: Total sex hormone and SHBG levels during pregnancy in women (Kerlan et al., 1994).
Effects of SHBG Increase on Free Sex Hormone Levels
Changes in SHBG levels result in changes in SHBG-bound and free sex hormone levels. Aside from DHT, estradiol and testosterone are the hormones of the greatest interest in this regard.
SHBG Increase and Free Testosterone
EE-containing birth control pills, with their 4-fold increase in SHBG levels, substantially decrease the free percentage of testosterone (Graham et al., 2007; Zimmerman et al., 2014). In one study, an EE-containing birth control pill decreased the free testosterone fraction from 2.45% to 0.78% (a 3.2-fold decrease or to 32% of baseline) (Graham et al., 2007). Due to concomitant suppression of testosterone production and hence reduced total testosterone levels, free testosterone levels decreased from 0.89 pg/mL to 0.18 pg/mL (a 5-fold decrease, to 20% of baseline) (Graham et al., 2007). The influence of EE on SHBG levels contributes significantly to the antiandrogenic effects of EE-containing birth control pills, which are taken advantage of therapeutically to treat acne and hirsutism in women.
During pregnancy, testosterone levels increase to as much as 150 ng/dL (around 5-fold higher than non-pregnancy levels) (McClamrock, 2007). The increase in SHBG production during pregnancy serves an important function in that the higher SHBG levels neutralize the biological activity of the increased testosterone levels (Hammond, 2017). In one study, the free testosterone fraction was 6-fold lower in late pregnancy than in non-pregnant women (0.23% vs. 1.36%—or to 17% of non-pregnancy) (Dunn, Nisula, & Rodbard, 1981). Hence, despite substantial increases in total testosterone levels during pregnancy, free testosterone levels and by extension androgenic action in the body change minimally (Barini, Liberale, & Menini, 1993; Schuijt et al., 2019). A case report of marked hyperandrogenism due to severe SHBG deficiency in a pregnant woman evidences the role of SHBG in limiting the androgenic actions of testosterone during this time (Hogeveen et al., 2002; Hammond, 2017).
SHBG Increase and Free Estradiol
Endogenous and Non-Oral Estradiol
The research indicates that increases in SHBG levels and by extension decreases in the free estradiol fraction are minimal with physiological levels of estradiol (e.g., <200 pg/mL). This is the case whether the estradiol is endogenous or exogenous in origin—so long as it is taken non-orally. Such conclusions are based on both calculated and measured studies of free estradiol (e.g., Freymann et al., 1977b).
Increases in SHBG levels and decreases in the free estradiol fraction become more significant with supraphysiological levels of estradiol however, for instance during pregnancy and with very-high-dose estradiol therapy. Studies on changes in free estradiol with high doses of estradiol are few. This is especially true in the case of measured as opposed to calculated free estradiol. In any case, one can look at pregnancy to gain insight on the question of free estradiol with high estradiol levels. Moreover, due to the very high estradiol levels in pregnancy, free estradiol is more amenable to measurement during this time. Accordingly, multiple studies of measured free estradiol in pregnancy are available.
Although free estradiol percentages during pregnancy certainly decrease, the increases in estradiol are far from neutralized by SHBG. Hence, the situation with free estradiol in pregnancy is very different from that of testosterone. This is illustrated in the following excerpt (Rubinow et al., 2002):
Pregnancy is accompanied by a slow but sustained rise in the plasma levels of many steroid and peptide hormones and is followed by a precipitous drop in their levels over the first few days after delivery. By the third trimester of pregnancy, plasma progesterone levels average approximately 150 ng/ml and estradiol levels range from 10 to 15 ng/ml. These amounts represent a 10- and 50-fold increase, respectively, of maximum menstrual cycle levels (Tulchinsky et al., 1972). Although only a small fraction of these steroids are unbound, the amount of “free” (and thus biologically active) progesterone and estrogen also undergo similarly large increases during pregnancy (Heidrich et al., 1994).
In the study by Heidrich and colleagues cited in the excerpt, total estradiol levels at the time of delivery were 21,500 pg/mL and measured free estradiol levels were 232 pg/mL, with a resultant free estradiol fraction of 1.08% (Heidrich et al., 1994). For context, the free estradiol percentage in non-pregnant women ranges from 1.5 to 2.1% with RIA, while actual free estradiol levels are 0.30 to 4.1 pg/mL with RIA and 0.40 to 5.9 pg/mL with LC–MS/MS (Nakamoto, 2016). Hence, in this study free estradiol levels in late pregnancy were around 50-fold higher than maximal non-pregnancy levels.
Due to variable methodology, the findings of a single study may not be representative. As such, the table below provides measured free estradiol percentages in late pregnancy from several studies.
Table 3: Measured free estradiol percentages in late pregnancy (mean ± SD) (Perry et al., 1987):
As can be seen in the table, the free estradiol fraction in late pregnancy ranges from about 0.7 to 1.5%. Results for the free estradiol fraction from studies using calculated free estradiol levels in late pregnancy rather than measured levels are similar to measured findings, although sometimes a bit lower in comparison (e.g., 0.5%) (Dunn, Nisula, & Rodbard, 1981; Campino et al., 2001). The measured free estradiol percentage in late pregnancy can be cautiously compared to the fraction of 1.5 to 2.1% in non-pregnant women. Using middle values from these ranges, the free estradiol fraction in late pregnancy may be somewhere around 60% of that of non-pregnancy. This estimate is quite close to the actual findings of a study, which observed a decrease in the measured free estradiol percentage to 55% of that of non-pregnancy (Freymann et al., 1977a; Freymann et al., 1977b).
In contrast to estradiol, the free percentages of estrone and estriol are not different in late pregnancy when compared to non-pregnancy (Tulchinsky & Chopra, 1973; Steingold et al., 1987). This is attributable to the much lower affinities of estrone and estriol for SHBG relative to estradiol (Kuhl, 2005).
Studies have also assessed free estradiol fractions earlier in pregnancy, which might in theory differ from late pregnancy. The results of a study that measured free estradiol throughout pregnancy are shown in the table below (Freymann et al., 1977a; Freymann et al., 1977b).
In similar studies by another group of researchers, free estradiol fractions were measured in earlier pregnancy (weeks 7–16) and were found to be lower than those obtained by Freymann and colleagues (Bernstein et al., 1986; Depue et al., 1987; Bernstein et al., 1988). The free estradiol percentage was about 0.9 or 1.0% at 10 weeks and about 0.7% at 12 weeks (Bernstein et al., 1986; Depue et al., 1987; Bernstein et al., 1988). Hence, as with the results of Freymann and colleagues, the free estradiol fraction decreased as pregnancy progressed. The figure below provides a visualization of the findings.
Figure 3: Changes in total and free estradiol levels (pg/mL), free estradiol fraction (%), and SHBG binding capacity (μg/dL) during weeks 7 to 16 of pregnancy in women (Bernstein et al., 1986).
Free estradiol during pregnancy can also be calculated using total estradiol levels and SHBG levels. I roughly calculated the free estradiol fraction during pregnancy using the data from O’Leary et al. (1991) and a published calculator spreadsheet by Mazer (2009) (Aly, 2020). The results are shown below.
Figure 4: Average measured total estradiol and SHBG levels (O’Leary et al., 1991) and calculated free estradiol percentage (Mazer, 2009) throughout pregnancy in women. Another version of this graph scaled to only the first trimester of pregnancy (when estradiol levels are typically ≤2,000 pg/mL) is also provided (Graph).
The free estradiol fractions in the figure are merely rough estimations and hence should be given conservative consideration. In any case, they are similar to the findings of the available studies on measured free estradiol in earlier pregnancy just discussed—for instance in magnitude (relative to Bernstein et al.) and pattern of change throughout pregnancy (relative to both Bernstein et al. and Freymann et al.). As such, these calculated values offer a plausible and interesting model.
To summarize, there are profound increases in total estradiol levels and proportionally lower but still substantial increases in SHBG levels during pregnancy. In accordance with the marked increase in SHBG levels, the free estradiol fraction progressively decreases over the course of pregnancy. Studies are conflicting on the exact degrees to which free estradiol percentages decrease. In any case, the possibilities for the free estradiol fraction by late pregnancy range from about 0.5 to 1.5%. These figures can be compared to non-pregnancy free estradiol percentages of 1.5 to 2.1%. This may correspond to a maximal decrease in the free estradiol fraction in late pregnancy to around 60% of non-pregnancy. At the greatest extreme, the decrease may be to around 25% of non-pregnancy. Conversely, in earlier pregnancy, when estradiol levels are lower, free estradiol percentages are higher.
Despite the decreases in the free estradiol fraction during pregnancy, there are profound increases in free estradiol levels that parallel the massive increases in total estradiol. As such, the increase in estradiol levels during pregnancy markedly exceeds the limiting influences of the simultaneously elevated SHBG levels. For this reason, pregnancy is a profoundly hyperestrogenic state.
SHBG doesn’t impact estradiol like it does testosterone during pregnancy because the proportional increases in estradiol levels relative to SHBG levels are far greater in comparison and because of the relatively lower affinity of estradiol for SHBG. In general, it’s not possible for SHBG to limit the activity of estradiol in the way that it can with testosterone due to the inherent requirement for substantially increased SHBG production of much more highly increased estradiol levels.
Oral Estradiol
Oral estradiol may differ from non-oral estradiol when it comes to the issue of free estradiol. This is because oral estradiol undergoes a first pass that results in greater estradiol levels in the liver relative to the circulation. As a result, oral estradiol has disproportionate liver effects and increases SHBG levels to a proportionally greater extent than non-oral estradiol. Hence, the greater SHBG increases with oral estradiol may result in lower free estradiol fractions than with non-oral estradiol.
While this is probable, it is more difficult to determine the precise magnitudes of the differences between oral and non-oral estradiol in terms of free estradiol. Some data are available however. Clinical studies of low-dose oral estradiol in menopausal cisgender women have reported the limiting influence of the SHBG increase on calculated free estradiol to be modest (Nilsson, Holst, & von Schoultz, 1984; Nachtigall et al., 2000). Likewise, oral estradiol appears to have similar effectiveness for menopausal symptoms when compared to non-oral estradiol (Wiki; 2nd paragraph). Studies of higher doses of oral estradiol that provide data on SHBG or free estradiol levels are rare. In any case, a few studies by one group found that 6 mg/day oral estradiol valerate (a dose equivalent to approximately 4.5 mg/day oral estradiol) increased SHBG levels by about 2.5- to 3.0-fold in transgender women (Dittrich et al., 2005; Mueller et al., 2005; Mueller et al., 2006). Using the numbers from one of the studies for total estradiol and SHBG levels, it can be roughly calculated (Mazer, 2009) that the free estradiol fraction may have decreased from around 2.1% to 1.2% (a 43% reduction). Analogously, a study using oral conjugated estrogens (CEEs; Premarin) at a dose that increased SHBG levels by 2.3-fold reported that the calculated free estradiol percentage was 40% lower relative to an equivalent dose of transdermal estradiol (in terms of total estradiol levels) (Shifren et al., 2007). These findings suggest a non-trivial reduction in the free estradiol fraction with typical doses of oral estradiol in transfeminine people. Consequently, it’s possible that oral estradiol could be to a certain degree less potent at the same total estradiol levels relative to non-oral estradiol.
It’s important to be clear that it’s also not a certainty however. Levels of estrone are much higher with oral estradiol than with non-oral estradiol (~5-fold) (Kuhl, 2005), and estrone, although far less potent than estradiol, has significant intrinsic estrogenic activity similarly to estradiol (Kuhl, 2005). The degree to which estrone might add to the estrogenic activity of estradiol, if at all, is uncertain. In any case, it’s within the realm of possibility that estrone could contribute significantly to the estrogenic activity of oral estradiol (Pande et al., 2019). This additional estrogenic exposure could potentially serve to offset the impact of the higher SHBG levels and reduced free estradiol fractions that occur with oral estradiol. Further research is needed to evaluate such a possibility however. As another consideration, the higher SHBG levels with oral estradiol can be expected to reduce the free testosterone fraction in addition to that of estradiol (and to an even greater extent in comparison). This is important as testosterone suppression is a key therapeutic effect of estradiol in transfeminine people and the main justified reason for use of higher estradiol levels. Due to possibilities like these and the fact that free levels of hormones only theoretically represent their biological activity, it shouldn’t necessarily be assumed that oral estradiol is less potent or efficacious than non-oral estradiol. Only further clinical studies comparing oral estradiol to non-oral estradiol will be able to clarify this question.
Relevance for Transfeminine Hormone Therapy
Some have concerns that SHBG may substantially limit the effectiveness of estradiol and thereby hinder feminization and/or breast development. Some have even claimed that high levels of estradiol may be less effective than lower levels as a result of SHBG increases at higher levels. Before even touching on SHBG however, such notions are likely to be misguided. This is because low estradiol levels (<50 pg/mL) are known to be fully effective in terms of feminization and breast development. This is evidenced by normal and induced puberty in cisgender girls (Aly, 2020), as well as by the excellent secondary sexual development of women with complete androgen insensitivity syndrome (CAIS) (Aly, 2020; Wiki). No evidence exists at this time to indicate that higher estradiol levels are necessary or beneficial in terms of feminization or breast development (Nolan & Cheung, 2020). Available studies in fact suggest no relationship between estradiol levels and breast development in transfeminine people at typical therapeutic levels of estradiol (e.g., 50–200 pg/mL) (de Blok et al., 2017; Meyer et al., 2020; de Blok et al., 2020). This is in accordance with the concept of the maximal effect of estradiol on feminization and breast development being established at lower estradiol levels. Hence, besides the use of higher estradiol levels for testosterone suppression in transfeminine people, concerns about incomplete feminizing efficacy of estradiol consequent to inadequate estrogenic exposure have little basis.
If SHBG is nonetheless explored however, the research indicates that the role of SHBG in restricting free estradiol, and hence presumably the biological activity of estradiol, is only so considerable. Within physiological non-pregnancy ranges for estradiol (e.g., <200 pg/mL), changes in SHBG levels and free estradiol fractions due to endogenous or non-oral estradiol are minimal. Very high estradiol levels have greater influence on SHBG production than normal physiological levels however. During pregnancy, with the massive increases in estradiol and resultant 5- to 10-fold maximal elevation in SHBG levels, the free estradiol percentage may be decreased to around 60% of that of non-pregnancy. But actual free estradiol levels are nonetheless profoundly increased in pregnancy. Moreover, increases in SHBG levels and decreases in free estradiol fraction earlier in pregnancy are lower than in late pregnancy. Even with among the highest estradiol levels that would normally be encountered with non-oral estradiol therapy, the decreases in the free estradiol fraction due to SHBG are likely to be modest. The impact of such a reduction could easily be negated by a slightly greater estradiol dose.
While the preceding is applicable to non-oral estradiol, oral estradiol has a greater influence on SHBG production in comparison and hence the higher SHBG levels with oral estradiol could result in more significant limitation of free estradiol than with non-oral estradiol. The notion that this reduction in free estradiol corresponds to a decrease in the activity or potency of oral estradiol is only a theoretical possibility however. Therapeutically, oral estradiol has shown itself to be very effective. The decreases in free estradiol percentage with low-dose oral estradiol seem to be small. In addition, while no direct comparisons exist this time, higher doses of oral estradiol seem to show similar testosterone suppression as non-oral estradiol (Wiki; Graphs). Besides testosterone suppression, available studies have found no differences between oral and non-oral estradiol in terms of outcomes like breast development or feminization (Sam, 2020). As such, the differences between oral and non-oral estradiol in terms SHBG levels and free estradiol fraction may be of little therapeutic importance.
Aside from decreasing free estradiol fractions, increased SHBG levels also decrease free testosterone fractions to an even greater extent. This is advantageous in the case of transfeminine people.
Taken together, lower free estradiol due to increased SHBG levels, whether with non-oral or oral estradiol, isn’t something that should be a major source of concern in transfeminine hormone therapy.
Supplementary Material
See here for supplementary material for this article, including a spreadsheet and other calculators that can be used to estimate free hormone levels (e.g., Mazer, 2009).
References
Adamcová, K., Kolátorová, L., Škodová, T., Šimková, M., Pařízek, A., Stárka, L., & Dušková, M. (2018). Steroid hormone levels in the peripartum period – differences caused by fetal sex and delivery type. Physiological Research, 67(Suppl 3), S489–S497. [DOI:10.33549/physiolres.934019]
Anderson, P. J., Hancock, K. W., & Oakey, R. E. (1985). Non-protein-bound oestradiol and progesterone in human peripheral plasma before labour and delivery. Journal of Endocrinology, 104(1), 7–15. [DOI:10.1677/joe.0.1040007]
Barini, A., Liberale, I., & Menini, E. (1993). Simultaneous Determination of Free Testosterone and Testosterone Bound to Non-Sex-Hormone-Binding Globulin by Equilibrium Dialysis. Clinical Chemistry, 39(6), 936–941. [DOI:10.1093/clinchem/39.6.936]
Bernstein, L., Depue, R. H., Ross, R. K., Judd, H. L., Pike, M. C., & Henderson, B. E. (1986). Higher maternal levels of free estradiol in first compared to second pregnancy: early gestational differences. Journal of the National Cancer Institute, 76(6), 1035–1039. [DOI:10.1093/jnci/76.6.1035]
Bernstein, L., Pike, M., Depue, R., Ross, R., Moore, J., & Henderson, B. (1988). Maternal hormone levels in early gestation of cryptorchid males: a case-control study. British Journal of Cancer, 58(3), 379–381. [DOI:10.1038/bjc.1988.223]
Bland, L. B., Garzotto, M., DeLoughery, T. G., Ryan, C. W., Schuff, K. G., Wersinger, E. M., Lemmon, D., & Beer, T. M. (2005). Phase II study of transdermal estradiol in androgen-independent prostate carcinoma. Cancer, 103(4), 717–723. [DOI:10.1002/cncr.20857]
Braunstein, G. D., Reitz, R. E., Buch, A., Schnell, D., & Caulfield, M. P. (2011). Testosterone Reference Ranges in Normally Cycling Healthy Premenopausal Women. The Journal of Sexual Medicine, 8(10), 2924–2934. [DOI:10.1111/j.1743-6109.2011.02380.x]
Burger, H. G., Dudley, E. C., Cui, J., Dennerstein, L., & Hopper, J. L. (2000). A Prospective Longitudinal Study of Serum Testosterone, Dehydroepiandrosterone Sulfate, and Sex Hormone-Binding Globulin Levels through the Menopause Transition. The Journal of Clinical Endocrinology & Metabolism, 85(8), 2832–2838. [DOI:10.1210/jcem.85.8.6740]
Campino, C., Torres, C., Rioseco, A., Poblete, A., Pugin, E., Valdés, V., Catalán, S., Belmar, C., & Serón-Ferré, M. (2001). Plasma prolactin/oestradiol ratio at 38 weeks gestation predicts the duration of lactational amenorrhoea. Human Reproduction, 16(12), 2540–2545. [DOI:10.1093/humrep/16.12.2540]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de Blok, C. J., Dijkman, B. A., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K. M., & den Heijer, M. (2020). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
Depue, R. H., Bernstein, L., Ross, R. K., Judd, H. L., & Henderson, B. E. (1987). Hyperemesis gravidarum in relation to estradiol levels, pregnancy outcome, and other maternal factors: A seroepidemiologic study. American Journal of Obstetrics and Gynecology, 156(5), 1137–1141. [DOI:10.1016/0002-9378(87)90126-8]
Derra, C. (1981). Hormonprofile unter Östrogen- und Antiandrogentherapie bei Patienten mit Prostatakarzinom: Östradiolundecylat versus Cyproteronacetat. [Hormone Profiles under Estrogen and Antiandrogen Therapy in Patients with Prostate Cancer: Estradiol Undecylate versus Cyproterone Acetate.] (Doctoral dissertation, University of Mainz.) [Google Scholar] [WorldCat] [PDF] [Translation]
Dittrich, R., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Mueller, A. (2005). Endocrine Treatment of Male-to-Female Transsexuals Using Gonadotropin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 113(10), 586–592. [DOI:10.1055/s-2005-865900]
Dunn, J. F., Nisula, B. C., & Rodbard, D. (1981). Transport of Steroid Hormones: Binding of 21 Endogenous Steroids to Both Testosterone-Binding Globulin and Corticosteroid-Binding Globulin in Human Plasma. The Journal of Clinical Endocrinology & Metabolism, 53(1), 58–68. [DOI:10.1210/jcem-53-1-58]
Fåhraeus, L., & Larsson-Cohn, U. (1982). Oestrogens, gonadotrophins and SHBG during oral and cutaneous administration of oestradiol-17β to menopausal women. Acta Endocrinologica, 101(4), 592–596. [DOI:10.1530/acta.0.1010592]
Fanelli, F., Gambineri, A., Belluomo, I., Repaci, A., Di Lallo, V. D., Di Dalmazi, G., Mezzullo, M., Prontera, O., Cuomo, G., Zanotti, L., Paccapelo, A., Morselli-Labate, A. M., Pagotto, U., & Pasquali, R. (2013). Androgen Profiling by Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS) in Healthy Normal-Weight Ovulatory and Anovulatory Late Adolescent and Young Women. The Journal of Clinical Endocrinology & Metabolism, 98(7), 3058–3067. [DOI:10.1210/jc.2013-1381]
Freymann, E., Hubl, W., Büchner, M., & Belleée, H. (1977). Eine spezifische, radioimmunologische Bestimmung des Plasmaöstradiols ohne Chromatographie im Zyklus und in der Schwangerschaft und die Bestimmung des freien, nichtproteingebundenen Anteils mittels Dialyse. [A specific radioimmunologi determination of plasma estradiol without chromatography during the cycle and in pregnancy and determination of the free non-protein-bound fraction using dialysis.] Zentralblatt für Gynäkologie, 99(6), 321–329. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF]
Freymann, E., Hubl, W., Büchner, M., & Rohde, W. (1977). Plasma levels of apparent free estradiol during pregnancy. Endokrinologie, 69(2), 269–271. [Google Scholar] [PubMed] [PDF]
Gibney, J., Johannsson, G., Leung, K., & Ho, K. K. (2005). Comparison of the Metabolic Effects of Raloxifene and Oral Estrogen in Postmenopausal and Growth Hormone-Deficient Women. The Journal of Clinical Endocrinology & Metabolism, 90(7), 3897–3903. [DOI:10.1210/jc.2005-0173]
Goldman, A. L., Bhasin, S., Wu, F. C., Krishna, M., Matsumoto, A. M., & Jasuja, R. (2017). A Reappraisal of Testosterone’s Binding in Circulation: Physiological and Clinical Implications. Endocrine Reviews, 38(4), 302–324. [DOI:10.1210/er.2017-00025]
Graham, C. A., Bancroft, J., Doll, H. A., Greco, T., & Tanner, A. (2007). Does oral contraceptive-induced reduction in free testosterone adversely affect the sexuality or mood of women? Psychoneuroendocrinology, 32(3), 246–255. [DOI:10.1016/j.psyneuen.2006.12.011]
Guthrie, J., Dennerstein, L., Taffe, J., Lehert, P., & Burger, H. (2004). The menopausal transition: a 9-year prospective population-based study. The Melbourne Women’s Midlife Health Project. Climacteric, 7(4), 375–389. [DOI:10.1080/13697130400012163]
Hammond, G. L., Nisker, J. A., Jones, L. A., & Siiteri, P. K. (1980). Estimation of the percentage of free steroid in undiluted serum by centrifugal ultrafiltration-dialysis. Journal of Biological Chemistry, 255(11), 5023–5026. [DOI:10.1016/s0021-9258(19)70742-x]
Hammond, G. L. (2016). Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. Journal of Endocrinology, 230(1), R13–R25. [DOI:10.1530/joe-16-0070]
Hammond, G. L. (2017). Sex Hormone-Binding Globulin and the Metabolic Syndrome. In Winters, S. J., & Huhtaniemi, I. T. (Eds.). Male Hypogonadism: Basic, Clinical and Therapeutic Principles (pp. 305–324). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-53298-1_15]
Handelsman, D. J. (2017). Free Testosterone: Pumping up the Tires or Ending the Free Ride? Endocrine Reviews, 38(4), 297–301. [DOI:10.1210/er.2017-00171]
Heidrich, A., Schleyer, M., Spingler, H., Albert, P., Knoche, M., Fritze, J., & Lanczik, M. (1994). Postpartum blues: Relationship between not-protein bound steroid hormones in plasma and postpartum mood changes. Journal of Affective Disorders, 30(2), 93–98. [DOI:10.1016/0165-0327(94)90036-1]
Heubner, A., Brockerhoff, P., Kreienberg, R., Grill, H., Rathgen, G., & Pollow, K. (1987). The influence of various dosages of megestrol acetate on SHBG, CBG and lipoprotein patterns. Journal of Steroid Biochemistry, 28(Suppl 1), 214S–214S (abstract no. 6). [DOI:10.1016/0022-4731(87)91680-3]
Hogeveen, K. N., Cousin, P., Pugeat, M., Dewailly, D., Soudan, B., & Hammond, G. L. (2002). Human sex hormone-binding globulin variants associated with hyperandrogenism and ovarian dysfunction. The Journal of Clinical Investigation, 109(7), 973–981. [DOI:10.1172/JCI14060]
Keevil, B. G., & Adaway, J. (2019). Assessment of free testosterone concentration. The Journal of Steroid Biochemistry and Molecular Biology, 190, 207–211. [DOI:10.1016/j.jsbmb.2019.04.008]
Kerlan, V., Nahoul, K., Martelot, M., & Bercovici, J. (1994). Longitudinal study of maternal plasma bioavailable testosterone and androstanediol glucuronide levels during pregnancy. Clinical Endocrinology, 40(2), 263–267. [DOI:10.1111/j.1365-2265.1994.tb02478.x]
Krause, A., Sinnecker, G., Hiort, O., Thamm, B., & Hoepffner, W. (2004). Applicability of the SHBG Androgen Sensitivity Test in the Differential Diagnosis of 46,XY Gonadal Dysgenesis, True Hermaphroditism, and Androgen Insensitivity Syndrome. Experimental and Clinical Endocrinology & Diabetes, 112(5), 236–240. [DOI:10.1055/s-2004-817969]
Kronawitter, D., Gooren, L. J., Zollver, H., Oppelt, P. G., Beckmann, M. W., Dittrich, R., & Mueller, A. (2009). Effects of transdermal testosterone or oral dydrogesterone on hypoactive sexual desire disorder in transsexual women: results of a pilot study. European Journal of Endocrinology, 161(2), 363–368. [DOI:10.1530/eje-09-0265] [Table]
Kuhl, H. (1997). Metabolische Effekte der Östrogene und Gestagene. [Metabolic Effects of Estrogens and Progestogens.] Der Gynäkologe, 30(4), 357–369. [DOI:10.1007/pl00003042]
Kuhl, H. (1998). Adverse effects of estrogen treatment: natural vs. synthetic estrogens. In Lippert, T. H., Mueck, A. O., & Ginsburg, J. (Eds.). Sex Steroids and the Cardiovascular System: The Proceedings of the 1st Interdisciplinary Workshop, Tuebingen, Germany, October 1996. Parthenon Publishing Group, New York, London (pp. 201–210). London/New York: Parthenon. [Google Scholar] [Google Books] [PDF]
Kuhl, H. (1999). Hormonal contraception. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 363–407). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_18]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Lundgren, S., Lønning, P., Utaaker, E., Aakvaag, A., & Kvinnsland, S. (1990). Influence of progestins on serum hormone levels in postmenopausal women with advanced breast cancer—I. General findings. Journal of Steroid Biochemistry, 36(1–2), 99–104. [DOI:10.1016/0022-4731(90)90118-c]
Lundgren, S., & Lønning, P. (1990). Influence of progestins on serum hormone levels in postmenopausal women with advanced breast cancer—II. A differential effect of megestrol acetate and medroxyprogesterone acetate on serum estrone sulfate and sex hormone binding globulin. Journal of Steroid Biochemistry, 36(1–2), 105–109. [DOI:10.1016/0022-4731(90)90119-d]
Mazer, N. A. (2009). A novel spreadsheet method for calculating the free serum concentrations of testosterone, dihydrotestosterone, estradiol, estrone and cortisol: With illustrative examples from male and female populations. Steroids, 74(6), 512–519. [DOI:10.1016/j.steroids.2009.01.008] [Spreadsheet]
McClamrock, H. D. (2007). Pregnancy-Related Androgen Excess. In Azziz, R., Nestler, J. E., & Dewailly, D. (Eds.). Androgen Excess Disorders in Women: Polycystic Ovary Syndrome and Other Disorders, 2nd Edition (Contemporary Endocrinology) (pp. 107–119). Totowa, New Jersey: Humana Press. [Google Books] [DOI:10.1007/978-1-59745-179-6_9]
Mean, F., Pellaton, M., & Magrini, G. (1977). Study on the binding of dihydrotestosterone, testosterone and oestradiol with sex hormone binding globulin. Clinica Chimica Acta, 80(1), 171–180. [DOI:10.1016/0009-8981(77)90276-5]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463] [PDF]
Moore, J. W., & Bulbrook, R. D. (1988). The epidemiology and function of sex hormone-binding globulin. Oxford Reviews of Reproductive Biology, 10, 180–236. [Google Scholar] [PubMed] [PDF]
Mueller, A., Dittrich, R., Binder, H., Kuehnel, W., Maltaris, T., Hoffmann, I., & Beckmann, M. W. (2005). High dose estrogen treatment increases bone mineral density in male-to-female transsexuals receiving gonadotropin-releasing hormone agonist in the absence of testosterone. European Journal of Endocrinology, 153(1), 107–113. [DOI:10.1530/eje.1.01943]
Mueller, A., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Dittrich, R. (2006). Effects on the Male Endocrine System of Long-term Treatment with Gonadotropin-releasing Hormone Agonists and Estrogens in Male-to-Female Transsexuals. Hormone and Metabolic Research, 38(3), 183–187. [DOI:10.1055/s-2006-925198]
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body composition and bone mineral density in male-to-female transsexuals during cross-sex hormone therapy using gonadotrophin-releasing hormone agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074] [Table]
Nachtigall, L. E., Raju, U., Banerjee, S., Wan, L., & Levitz, M. (2000). Serum Estradiol-Binding Profiles in Postmenopausal Women Undergoing Three Common Estrogen Replacement Therapies. Menopause, 7(4), 243–250. [DOI:10.1097/00042192-200007040-00006]
Nakamoto, J. (2016). Endocrine Testing. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition (pp. 2655–2688.e1). Philadelphia: Saunders/Elsevier. [Google Books] [DOI:10.1016/B978-0-323-18907-1.00154-2]
Nguyen, T. D. T., Dolomie-Fagour, L., Georges, A., & Corcuff, J. B. (2008). Dosage des stéroïdes sexuels sériques: quelle place pour l’estradiol biodisponible? [What about bioavailable estradiol?] Annales de Biologie Clinique, 66(5), 493–497. [DOI:10.1684/abc.2008.0259]
Nilsson, B., Holst, J., & Schoultz, B. (1984). Serum levels of unbound 17β-oestradiol during oral and percutaneous postmenopausal replacement therapy. British Journal of Obstetrics and Gynaecology, 91(10), 1031–1036. [DOI:10.1111/j.1471-0528.1984.tb03683.x]
Nolan, B. J., & Cheung, A. S. (2021). Relationship Between Serum Estradiol Concentrations and Clinical Outcomes in Transgender Individuals Undergoing Feminizing Hormone Therapy: A Narrative Review. Transgender Health, 6(3), 125–131. [DOI:10.1089/trgh.2020.0077]
O’Leary, P., Boyne, P., Flett, P., Beilby, J., & James, I. (1991). Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clinical Chemistry, 37(5), 667–672. [DOI:10.1093/clinchem/37.5.667]
Odlind, V., Milsom, I., Persson, I., & Victor, A. (2002). Can changes in sex hormone binding globulin predict the risk of venous thromboembolism with combined oral contraceptive pills? Acta Obstetricia et Gynecologica Scandinavica, 81(6), 482–490. [DOI:10.1034/j.1600-0412.2002.810603.x]
Ojasoo, T., & Raynaud, J. P. (1978). Unique steroid congeners for receptor studies. Cancer Research, 38(11 Part 2), 4186–4198. [Google Scholar] [PubMed] [URL]
Pande, P., Fleck, S. C., Twaddle, N. C., Churchwell, M. I., Doerge, D. R., & Teeguarden, J. G. (2019). Comparative estrogenicity of endogenous, environmental and dietary estrogens in pregnant women II: Total estrogenicity calculations accounting for competitive protein and receptor binding and potency. Food and Chemical Toxicology, 125, 341–353. [DOI:10.1016/j.fct.2018.12.013]
Pardridge, W. M. (1988). Selective delivery of sex steroid hormones to tissues by albumin and by sex hormone-binding globulin. Oxford Reviews of Reproductive Biology, 10, 237–292. [Google Scholar] [PubMed] [PDF]
Perry, L., Wathen, N., & Chard, T. (1987). Saliva Levels of Oestradiol and Progesterone in Relation to Non-Protein-Bound Concentrations in Blood During Late Pregnancy. Hormone and Metabolic Research, 19(9), 444–447. [DOI:10.1055/s-2007-1011848]
Plymate, S. R., Moore, D. E., Cheng, C. Y., Bardin, C. W., Southworth, M. B., & Levinski, M. J. (1985). Sex Hormone-Binding Globulin Changes during the Menstrual Cycle. The Journal of Clinical Endocrinology & Metabolism, 61(5), 993–996. [DOI:10.1210/jcem-61-5-993]
Pugeat, M. M., Dunn, J. F., & Nisula, B. C. (1981). Transport of Steroid Hormones: Interaction of 70 Drugs with Testosterone-Binding Globulin and Corticosteroid-Binding Globulin in Human Plasma. The Journal of Clinical Endocrinology & Metabolism, 53(1), 69–75. [DOI:10.1210/jcem-53-1-69]
Rezaii, T., Gustafsson, T. P., Axelson, M., Zamani, L., Ernberg, M., Hirschberg, A. L., & Carlström, K. A. (2017). Circulating androgens and SHBG during the normal menstrual cycle in two ethnic populations. Scandinavian Journal of Clinical and Laboratory Investigation, 77(3), 184–189. [DOI:10.1080/00365513.2017.1286685]
Ropponen, A., Aittomäki, K., Vihma, V., Tikkanen, M. J., & Ylikorkala, O. (2005). Effects of Oral and Transdermal Estradiol Administration on Levels of Sex Hormone-Binding Globulin in Postmenopausal Women with and without a History of Intrahepatic Cholestasis of Pregnancy. The Journal of Clinical Endocrinology & Metabolism, 90(6), 3431–3434. [DOI:10.1210/jc.2005-0352]
Rosner, W. (2015). Free estradiol and sex hormone-binding globulin. Steroids, 99, 113–116. [DOI:10.1016/j.steroids.2014.08.005]
Rothman, M. S., Carlson, N. E., Xu, M., Wang, C., Swerdloff, R., Lee, P., Goh, V. H., Ridgway, E. C., & Wierman, M. E. (2011). Reexamination of testosterone, dihydrotestosterone, estradiol and estrone levels across the menstrual cycle and in postmenopausal women measured by liquid chromatography–tandem mass spectrometry. Steroids, 76(1–2), 177–182. [DOI:10.1016/j.steroids.2010.10.010]
Rubinow, D. R., Schmidt, P. J., Roca, C. A., & Daly, R. C. (2002). Gonadal Hormones and Behavior in Women: Concentrations versus Context. In Pfaff, D. W., Arnold, A. P., Etgen, A. M., Fahrbach, S. E., & Rubin, R. T. (Eds.). Hormones, Brain and Behavior, Volume 5 (pp. 37–73). Amsterdam: Academic Press. [Google Books] [DOI:10.1016/B978-012532104-4/50086-X]
Ruokonen, A., Alén, M., Bolton, N., & Vihko, R. (1985). Response of serum testosterone and its precursor steroids, SHBG and CBG to anabolic steroid and testosterone self-administration in man. Journal of Steroid Biochemistry, 23(1), 33–38. [DOI:10.1016/0022-4731(85)90257-2]
Schijf, C. P., van der Mooren, M. J., Doesburg, W. H., Thomas, C. M., & Rolland, R. (1993). Differences in serum lipids, lipoproteins, sex hormone binding globulin and testosterone between the follicular and the luteal phase of the menstrual cycle. Acta Endocrinologica, 129(2), 130–133. [DOI:10.1530/acta.0.1290130]
Schuijt, M. P., Sweep, C. G., van der Steen, R., Olthaar, A. J., Stikkelbroeck, N. M., Ross, H. A., & van Herwaarden, A. E. (2019). Validity of free testosterone calculation in pregnant women. Endocrine Connections, 8(6), 672–679. [DOI:10.1530/ec-19-0110]
Shifren, J. L., Desindes, S., McIlwain, M., Doros, G., & Mazer, N. A. (2007). A randomized, open-label, crossover study comparing the effects of oral versus transdermal estrogen therapy on serum androgens, thyroid hormones, and adrenal hormones in naturally menopausal women. Menopause, 14(6), 985–994. [DOI:10.1097/gme.0b013e31803867a]
Shifren, J. L., Rifai, N., Desindes, S., McIlwain, M., Doros, G., & Mazer, N. A. (2008). A Comparison of the Short-Term Effects of Oral Conjugated Equine Estrogens Versus Transdermal Estradiol on C-Reactive Protein, Other Serum Markers of Inflammation, and Other Hepatic Proteins in Naturally Menopausal Women. The Journal of Clinical Endocrinology & Metabolism, 93(5), 1702–1710. [DOI:10.1210/jc.2007-2193]
Smith, K., Galazi, M., Openshaw, M. R., Wilson, P., Sarker, S. J., O’Brien, N., Alifrangis, C., Stebbing, J., & Shamash, J. (2020). The Use of Transdermal Estrogen in Castrate-resistant, Steroid-refractory Prostate Cancer. Clinical Genitourinary Cancer, 18(3), e217–e223. [DOI:10.1016/j.clgc.2019.09.019]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., & Pousette, A. (1988). Single drug polyestradiol phosphate therapy in prostatic cancer. American Journal of Clinical Oncology, 11(Suppl 2), S101–S103. [DOI:10.1097/00000421-198801102-00024] [PDF]
Steingold, K. A., Pardridge, W. M., Judd, H. L., & Chaudhuri, G. (1987). The effects of membrane permeability and binding by human serum proteins on steroid influx into the rabbit uterus. American Journal of Obstetrics and Gynecology, 157(6), 1543–1549. [DOI:10.1016/s0002-9378(87)80260-0]
Strauss, J. F., & FitzGerald, G. A. (2019). Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 75–114.e7). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/b978-0-323-47912-7.00004-4]
Troisi, R., Potischman, N., Roberts, J. M., Harger, G., Markovic, N., Cole, B., Lykins, D., Siiteri, P., & Hoover, R. N. (2003). Correlation of serum hormone concentrations in maternal and umbilical cord samples. Cancer Epidemiology, Biomarkers & Prevention, 12(5), 452–456. [Google Scholar] [PubMed] [URL]
Tulchinsky, D., & Chopra, I. J. (1973). Competitive Ligand-Binding Assay for Measurement of Sex Hormone-Binding Globulin (SHBG). The Journal of Clinical Endocrinology & Metabolism, 37(6), 873–881. [DOI:10.1210/jcem-37-6-873]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
Zimmerman, Y., Eijkemans, M. J., Coelingh Bennink, H. J., Blankenstein, M. A., & Fauser, B. C. (2013). The effect of combined oral contraception on testosterone levels in healthy women: a systematic review and meta-analysis. Human Reproduction Update, 20(1), 76–105. [DOI:10.1093/humupd/dmt038]
\ No newline at end of file
+The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy - Transfeminine Science
The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy
By Aly | First published July 10, 2020 | Last modified March 25, 2023
Abstract / TL;DR
Sex hormones such as testosterone and estradiol bind to blood proteins like albumin and SHBG. This limits their biological activity by reducing their free fractions. Androgens decrease SHBG production while estrogens increase SHBG production. Hence, testosterone and estradiol can influence their own free fractions. Due to robust inactivation in the liver, testosterone and estradiol have relatively small influences on SHBG levels under normal physiological circumstances. At very high levels however, they can considerably influence SHBG levels. During pregnancy, when there are massive increases in estradiol levels (e.g., 100-fold), a maximal 5- to 10-fold elevation in SHBG levels occurs. Although large increases in SHBG levels can strongly limit the biological activity of testosterone, the situation with estradiol is different. In late pregnancy, the percentage of estradiol that is free appears to be decreased only to around 60% of that of non-pregnancy. Earlier in pregnancy, when estradiol levels are lower, the free fractions of estradiol are reduced to a lesser extent. At typical therapeutic levels of estradiol in transfeminine hormone therapy (<200 pg/mL), the limiting influence of SHBG on free estradiol is minimal. Oral estradiol has a greater influence on SHBG production than non-oral estradiol and may be a different case however. In any case, consequent lesser activity of oral estradiol is only theoretical, and available clinical studies so far haven’t reported important therapeutic differences relative to non-oral estradiol. Although SHBG may reduce free estradiol fractions in some contexts, only relatively low estradiol levels (<50 pg/mL) appear to be needed for maximal feminization and breast development in cisgender females and transfeminine people. In conclusion, the influence of SHBG on the effectiveness of estradiol isn’t something that should be a major source of concern in transfeminine hormone therapy.
Binding of Sex Hormones to Blood Proteins
Sex hormones bind to proteins in the blood called plasma proteins. This is a phenomenon known as plasma protein binding. In the case of androgens and estrogens, the plasma proteins they bind to are mainly albumin and sex hormone-binding globulin (SHBG). Plasma protein binding serves to prevent sex hormones from interacting with their target cells and hence from binding to and activating their receptors (Hammond, 2016). This is because plasma proteins are too large and lipid-insoluble to cross the lipid-rich cell membrane. As a result, they’re unable to diffuse through capillaries to exit the circulation and enter into tissues or to be taken up into cells. When the sex hormone is bound to plasma protein, it can’t reach target cells either. Hence, plasma protein binding limits the biological activity of sex hormones (Hammond, 2016). Binding to plasma proteins also serves to extend the biological half-lives of sex hormones. This is because protein-bound sex hormone is likewise unavailable for metabolism and elimination, processes that depend on cellular uptake.
There is only a single sex hormone binding site per molecule of SHBG (Moore & Bulbrook, 1988), whereas albumin has six binding sites for different substrates (Pardridge, 1988). Androgens and estradiol have high affinity for SHBG (nM) and low affinity for albumin (μM) (Moore & Bulbrook, 1988; Hammond, 2016). However, albumin levels are several orders of magnitude higher than SHBG levels (μM vs. nM), so this serves to balance out the fractions of sex hormone bound to each protein (Hammond, 2016). Androgens have higher affinities for SHBG than do estradiol or other estrogens. Estradiol has only about 10 to 20% of the affinity of dihydrotestosterone (DHT) and 33 to 50% of the affinity of testosterone for SHBG (Anderson, 1974; Ojasoo & Raynaud, 1978; Pugeat, Dunn, Nisula, 1981). As such, testosterone and DHT bind more strongly to SHBG than does estradiol.
The vast majority of sex hormone content in the blood is bound to plasma proteins; at any given time more than 97% of the testosterone, estradiol, and progesterone in the blood is plasma protein-bound (Strauss & FitzGerald, 2019). The fraction of sex hormone that isn’t bound to plasma proteins is known as the free or unbound fraction. This is the fraction that is available for diffusion into cells and hence is considered to be biologically active (Hammond, 2016). Total levels refer to both free/unbound and bound hormone. Bioavailable levels include both albumin-bound and free hormone levels. Due to their relatively weak affinity for albumin, sex hormones bound to albumin may to some extent be biologically active—hence the “bioavailable” descriptor (Nguyen et al., 2008). However, more research is needed to fully elucidate the biological activity of albumin-bound sex hormone fractions.
The relative calculated free and bound percentages of estradiol, testosterone, and DHT to albumin, SHBG, and another plasma protein known as corticosteroid-binding globulin (CBG) (only binds small fractions of the androgens and has no binding to estradiol) are shown in the table below.
Free sex hormone levels and percentages are often calculated from levels of total sex hormone, albumin, SHBG, and CBG with validated mathematical models constructed from data of published studies. This is because free sex hormone levels are usually very low (pM range) and are difficult to measure with routine blood testing methods. While generally in the vicinity of the true values, calculated results may not always be fully accurate (Rosner, 2015; Goldman et al., 2017; Handelsman, 2017; Keevil & Adaway, 2019). As such, measured levels, when feasible, are preferable.
Testosterone, DHT, and estradiol are strongly inactivated by the liver and have relatively weak effects in this part of the body under normal circumstances. As a result, they have much less relative impact on SHBG production than do synthetic hormonal agents. Accordingly, SHBG levels change only slightly over the course of the menstrual cycle in women despite substantial fluctuations in estradiol levels (Freymann et al., 1977b; Plymate et al., 1985; Schijf et al., 1993; Braunstein et al., 2011; Rothman et al., 2011; Fanelli et al., 2013; Rezaii et al., 2017). In one study, SHBG levels increased by about 6 to 13% (+2.9–5.3 nmol/L) going from the follicular phase to the luteal phase of the cycle (Braunstein et al., 2011). There is additionally only a small decrease in SHBG levels attributable to the sharp decline in estradiol with menopause (Burger et al., 2000; Guthrie et al., 2004). Nonetheless, estradiol therapy can more considerably influence the production of SHBG and other liver proteins as well under specific conditions (Kuhl, 1998). This is due to 1) use of oral estradiol, which because of the first pass through the liver has a greater impact on estrogen-sensitive liver synthesis than non-oral routes (Kuhl, 2005); and 2) use of high estradiol doses, for instance typical injectable doses. The table below shows SHBG increases from various studies with different estrogen routes, doses, and types.
Table 2: Relative increases in SHBG levels with some different estrogenic exposures:
a Estimated typical estradiol levels from various sources (e.g., Aly, 2020; Wiki). b Due to differences in molecular weight, EV has about 75% of the amount of estradiol as regular estradiol. Hence, 6 mg/day EV is approximately equivalent to 4.5 mg/day E2.
The influence of estradiol on SHBG levels is most relevant to pregnancy, when estradiol levels increase to far higher levels than usual. In late pregnancy, estradiol levels are generally around 15,000 to 25,000 pg/mL on average (Graphs; Troisi et al., 2003; Adamcová et al., 2018). These estradiol levels are on the order of 100-fold higher than normal menstrual cycle levels. In parallel with the massive increases in estradiol levels, SHBG levels increase by about 5- to 10-fold by late pregnancy (Anderson, 1974; Hammond, 2017). The dose–response curve of estrogens on SHBG production shows saturation, with most of the increase in SHBG levels happening at lower estradiol levels as well as limits to how much SHBG levels can be increased (Mean, Pellaton, & Magrini, 1977; O’Leary et al., 1991; Kerlan et al., 1994; Kuhl, 1999). The graphs below show SHBG levels throughout pregnancy.
Figure 1: SHBG and total estradiol levels during pregnancy in women (O’Leary et al., 1991). The lines are the mean and/or 95th percentile levels while the points are individual measurements.
Figure 2: Total sex hormone and SHBG levels during pregnancy in women (Kerlan et al., 1994).
Effects of SHBG Increase on Free Sex Hormone Levels
Changes in SHBG levels result in changes in SHBG-bound and free sex hormone levels. Aside from DHT, estradiol and testosterone are the hormones of the greatest interest in this regard.
SHBG Increase and Free Testosterone
EE-containing birth control pills, with their 4-fold increase in SHBG levels, substantially decrease the free percentage of testosterone (Graham et al., 2007; Zimmerman et al., 2014). In one study, an EE-containing birth control pill decreased the free testosterone fraction from 2.45% to 0.78% (a 3.2-fold decrease or to 32% of baseline) (Graham et al., 2007). Due to concomitant suppression of testosterone production and hence reduced total testosterone levels, free testosterone levels decreased from 0.89 pg/mL to 0.18 pg/mL (a 5-fold decrease, to 20% of baseline) (Graham et al., 2007). The influence of EE on SHBG levels contributes significantly to the antiandrogenic effects of EE-containing birth control pills, which are taken advantage of therapeutically to treat acne and hirsutism in women.
During pregnancy, testosterone levels increase to as much as 150 ng/dL (around 5-fold higher than non-pregnancy levels) (McClamrock, 2007). The increase in SHBG production during pregnancy serves an important function in that the higher SHBG levels neutralize the biological activity of the increased testosterone levels (Hammond, 2017). In one study, the free testosterone fraction was 6-fold lower in late pregnancy than in non-pregnant women (0.23% vs. 1.36%—or to 17% of non-pregnancy) (Dunn, Nisula, & Rodbard, 1981). Hence, despite substantial increases in total testosterone levels during pregnancy, free testosterone levels and by extension androgenic action in the body change minimally (Barini, Liberale, & Menini, 1993; Schuijt et al., 2019). A case report of marked hyperandrogenism due to severe SHBG deficiency in a pregnant woman evidences the role of SHBG in limiting the androgenic actions of testosterone during this time (Hogeveen et al., 2002; Hammond, 2017).
SHBG Increase and Free Estradiol
Endogenous and Non-Oral Estradiol
The research indicates that increases in SHBG levels and by extension decreases in the free estradiol fraction are minimal with physiological levels of estradiol (e.g., <200 pg/mL). This is the case whether the estradiol is endogenous or exogenous in origin—so long as it is taken non-orally. Such conclusions are based on both calculated and measured studies of free estradiol (e.g., Freymann et al., 1977b).
Increases in SHBG levels and decreases in the free estradiol fraction become more significant with supraphysiological levels of estradiol however, for instance during pregnancy and with very-high-dose estradiol therapy. Studies on changes in free estradiol with high doses of estradiol are few. This is especially true in the case of measured as opposed to calculated free estradiol. In any case, one can look at pregnancy to gain insight on the question of free estradiol with high estradiol levels. Moreover, due to the very high estradiol levels in pregnancy, free estradiol is more amenable to measurement during this time. Accordingly, multiple studies of measured free estradiol in pregnancy are available.
Although free estradiol percentages during pregnancy certainly decrease, the increases in estradiol are far from neutralized by SHBG. Hence, the situation with free estradiol in pregnancy is very different from that of testosterone. This is illustrated in the following excerpt (Rubinow et al., 2002):
Pregnancy is accompanied by a slow but sustained rise in the plasma levels of many steroid and peptide hormones and is followed by a precipitous drop in their levels over the first few days after delivery. By the third trimester of pregnancy, plasma progesterone levels average approximately 150 ng/ml and estradiol levels range from 10 to 15 ng/ml. These amounts represent a 10- and 50-fold increase, respectively, of maximum menstrual cycle levels (Tulchinsky et al., 1972). Although only a small fraction of these steroids are unbound, the amount of “free” (and thus biologically active) progesterone and estrogen also undergo similarly large increases during pregnancy (Heidrich et al., 1994).
In the study by Heidrich and colleagues cited in the excerpt, total estradiol levels at the time of delivery were 21,500 pg/mL and measured free estradiol levels were 232 pg/mL, with a resultant free estradiol fraction of 1.08% (Heidrich et al., 1994). For context, the free estradiol percentage in non-pregnant women ranges from 1.5 to 2.1% with RIA, while actual free estradiol levels are 0.30 to 4.1 pg/mL with RIA and 0.40 to 5.9 pg/mL with LC–MS/MS (Nakamoto, 2016). Hence, in this study free estradiol levels in late pregnancy were around 50-fold higher than maximal non-pregnancy levels.
Due to variable methodology, the findings of a single study may not be representative. As such, the table below provides measured free estradiol percentages in late pregnancy from several studies.
Table 3: Measured free estradiol percentages in late pregnancy (mean ± SD) (Perry et al., 1987):
As can be seen in the table, the free estradiol fraction in late pregnancy ranges from about 0.7 to 1.5%. Results for the free estradiol fraction from studies using calculated free estradiol levels in late pregnancy rather than measured levels are similar to measured findings, although sometimes a bit lower in comparison (e.g., 0.5%) (Dunn, Nisula, & Rodbard, 1981; Campino et al., 2001). The measured free estradiol percentage in late pregnancy can be cautiously compared to the fraction of 1.5 to 2.1% in non-pregnant women. Using middle values from these ranges, the free estradiol fraction in late pregnancy may be somewhere around 60% of that of non-pregnancy. This estimate is quite close to the actual findings of a study, which observed a decrease in the measured free estradiol percentage to 55% of that of non-pregnancy (Freymann et al., 1977a; Freymann et al., 1977b).
In contrast to estradiol, the free percentages of estrone and estriol are not different in late pregnancy when compared to non-pregnancy (Tulchinsky & Chopra, 1973; Steingold et al., 1987). This is attributable to the much lower affinities of estrone and estriol for SHBG relative to estradiol (Kuhl, 2005).
Studies have also assessed free estradiol fractions earlier in pregnancy, which might in theory differ from late pregnancy. The results of a study that measured free estradiol throughout pregnancy are shown in the table below (Freymann et al., 1977a; Freymann et al., 1977b).
In similar studies by another group of researchers, free estradiol fractions were measured in earlier pregnancy (weeks 7–16) and were found to be lower than those obtained by Freymann and colleagues (Bernstein et al., 1986; Depue et al., 1987; Bernstein et al., 1988). The free estradiol percentage was about 0.9 or 1.0% at 10 weeks and about 0.7% at 12 weeks (Bernstein et al., 1986; Depue et al., 1987; Bernstein et al., 1988). Hence, as with the results of Freymann and colleagues, the free estradiol fraction decreased as pregnancy progressed. The figure below provides a visualization of the findings.
Figure 3: Changes in total and free estradiol levels (pg/mL), free estradiol fraction (%), and SHBG binding capacity (μg/dL) during weeks 7 to 16 of pregnancy in women (Bernstein et al., 1986).
Free estradiol during pregnancy can also be calculated using total estradiol levels and SHBG levels. I roughly calculated the free estradiol fraction during pregnancy using the data from O’Leary et al. (1991) and a published calculator spreadsheet by Mazer (2009) (Aly, 2020). The results are shown below.
Figure 4: Average measured total estradiol and SHBG levels (O’Leary et al., 1991) and calculated free estradiol percentage (Mazer, 2009) throughout pregnancy in women. Another version of this graph scaled to only the first trimester of pregnancy (when estradiol levels are typically ≤2,000 pg/mL) is also provided (Graph).
The free estradiol fractions in the figure are merely rough estimations and hence should be given conservative consideration. In any case, they are similar to the findings of the available studies on measured free estradiol in earlier pregnancy just discussed—for instance in magnitude (relative to Bernstein et al.) and pattern of change throughout pregnancy (relative to both Bernstein et al. and Freymann et al.). As such, these calculated values offer a plausible and interesting model.
To summarize, there are profound increases in total estradiol levels and proportionally lower but still substantial increases in SHBG levels during pregnancy. In accordance with the marked increase in SHBG levels, the free estradiol fraction progressively decreases over the course of pregnancy. Studies are conflicting on the exact degrees to which free estradiol percentages decrease. In any case, the possibilities for the free estradiol fraction by late pregnancy range from about 0.5 to 1.5%. These figures can be compared to non-pregnancy free estradiol percentages of 1.5 to 2.1%. This may correspond to a maximal decrease in the free estradiol fraction in late pregnancy to around 60% of non-pregnancy. At the greatest extreme, the decrease may be to around 25% of non-pregnancy. Conversely, in earlier pregnancy, when estradiol levels are lower, free estradiol percentages are higher.
Despite the decreases in the free estradiol fraction during pregnancy, there are profound increases in free estradiol levels that parallel the massive increases in total estradiol. As such, the increase in estradiol levels during pregnancy markedly exceeds the limiting influences of the simultaneously elevated SHBG levels. For this reason, pregnancy is a profoundly hyperestrogenic state.
SHBG doesn’t impact estradiol like it does testosterone during pregnancy because the proportional increases in estradiol levels relative to SHBG levels are far greater in comparison and because of the relatively lower affinity of estradiol for SHBG. In general, it’s not possible for SHBG to limit the activity of estradiol in the way that it can with testosterone due to the inherent requirement for substantially increased SHBG production of much more highly increased estradiol levels.
Oral Estradiol
Oral estradiol may differ from non-oral estradiol when it comes to the issue of free estradiol. This is because oral estradiol undergoes a first pass that results in greater estradiol levels in the liver relative to the circulation. As a result, oral estradiol has disproportionate liver effects and increases SHBG levels to a proportionally greater extent than non-oral estradiol. Hence, the greater SHBG increases with oral estradiol may result in lower free estradiol fractions than with non-oral estradiol.
While this is probable, it is more difficult to determine the precise magnitudes of the differences between oral and non-oral estradiol in terms of free estradiol. Some data are available however. Clinical studies of low-dose oral estradiol in menopausal cisgender women have reported the limiting influence of the SHBG increase on calculated free estradiol to be modest (Nilsson, Holst, & von Schoultz, 1984; Nachtigall et al., 2000). Likewise, oral estradiol appears to have similar effectiveness for menopausal symptoms when compared to non-oral estradiol (Wiki; 2nd paragraph). Studies of higher doses of oral estradiol that provide data on SHBG or free estradiol levels are rare. In any case, a few studies by one group found that 6 mg/day oral estradiol valerate (a dose equivalent to approximately 4.5 mg/day oral estradiol) increased SHBG levels by about 2.5- to 3.0-fold in transgender women (Dittrich et al., 2005; Mueller et al., 2005; Mueller et al., 2006). Using the numbers from one of the studies for total estradiol and SHBG levels, it can be roughly calculated (Mazer, 2009) that the free estradiol fraction may have decreased from around 2.1% to 1.2% (a 43% reduction). Analogously, a study using oral conjugated estrogens (CEEs; Premarin) at a dose that increased SHBG levels by 2.3-fold reported that the calculated free estradiol percentage was 40% lower relative to an equivalent dose of transdermal estradiol (in terms of total estradiol levels) (Shifren et al., 2007). These findings suggest a non-trivial reduction in the free estradiol fraction with typical doses of oral estradiol in transfeminine people. Consequently, it’s possible that oral estradiol could be to a certain degree less potent at the same total estradiol levels relative to non-oral estradiol.
It’s important to be clear that it’s also not a certainty however. Levels of estrone are much higher with oral estradiol than with non-oral estradiol (~5-fold) (Kuhl, 2005), and estrone, although far less potent than estradiol, has significant intrinsic estrogenic activity similarly to estradiol (Kuhl, 2005). The degree to which estrone might add to the estrogenic activity of estradiol, if at all, is uncertain. In any case, it’s within the realm of possibility that estrone could contribute significantly to the estrogenic activity of oral estradiol (Pande et al., 2019). This additional estrogenic exposure could potentially serve to offset the impact of the higher SHBG levels and reduced free estradiol fractions that occur with oral estradiol. Further research is needed to evaluate such a possibility however. As another consideration, the higher SHBG levels with oral estradiol can be expected to reduce the free testosterone fraction in addition to that of estradiol (and to an even greater extent in comparison). This is important as testosterone suppression is a key therapeutic effect of estradiol in transfeminine people and the main justified reason for use of higher estradiol levels. Due to possibilities like these and the fact that free levels of hormones only theoretically represent their biological activity, it shouldn’t necessarily be assumed that oral estradiol is less potent or efficacious than non-oral estradiol. Only further clinical studies comparing oral estradiol to non-oral estradiol will be able to clarify this question.
Relevance for Transfeminine Hormone Therapy
Some have concerns that SHBG may substantially limit the effectiveness of estradiol and thereby hinder feminization and/or breast development. Some have even claimed that high levels of estradiol may be less effective than lower levels as a result of SHBG increases at higher levels. Before even touching on SHBG however, such notions are likely to be misguided. This is because low estradiol levels (<50 pg/mL) are known to be fully effective in terms of feminization and breast development. This is evidenced by normal and induced puberty in cisgender girls (Aly, 2020), as well as by the excellent secondary sexual development of women with complete androgen insensitivity syndrome (CAIS) (Aly, 2020; Wiki). No evidence exists at this time to indicate that higher estradiol levels are necessary or beneficial in terms of feminization or breast development (Nolan & Cheung, 2020). Available studies in fact suggest no relationship between estradiol levels and breast development in transfeminine people at typical therapeutic levels of estradiol (e.g., 50–200 pg/mL) (de Blok et al., 2017; Meyer et al., 2020; de Blok et al., 2020). This is in accordance with the concept of the maximal effect of estradiol on feminization and breast development being established at lower estradiol levels. Hence, besides the use of higher estradiol levels for testosterone suppression in transfeminine people, concerns about incomplete feminizing efficacy of estradiol consequent to inadequate estrogenic exposure have little basis.
If SHBG is nonetheless explored however, the research indicates that the role of SHBG in restricting free estradiol, and hence presumably the biological activity of estradiol, is only so considerable. Within physiological non-pregnancy ranges for estradiol (e.g., <200 pg/mL), changes in SHBG levels and free estradiol fractions due to endogenous or non-oral estradiol are minimal. Very high estradiol levels have greater influence on SHBG production than normal physiological levels however. During pregnancy, with the massive increases in estradiol and resultant 5- to 10-fold maximal elevation in SHBG levels, the free estradiol percentage may be decreased to around 60% of that of non-pregnancy. But actual free estradiol levels are nonetheless profoundly increased in pregnancy. Moreover, increases in SHBG levels and decreases in free estradiol fraction earlier in pregnancy are lower than in late pregnancy. Even with among the highest estradiol levels that would normally be encountered with non-oral estradiol therapy, the decreases in the free estradiol fraction due to SHBG are likely to be modest. The impact of such a reduction could easily be negated by a slightly greater estradiol dose.
While the preceding is applicable to non-oral estradiol, oral estradiol has a greater influence on SHBG production in comparison and hence the higher SHBG levels with oral estradiol could result in more significant limitation of free estradiol than with non-oral estradiol. The notion that this reduction in free estradiol corresponds to a decrease in the activity or potency of oral estradiol is only a theoretical possibility however. Therapeutically, oral estradiol has shown itself to be very effective. The decreases in free estradiol percentage with low-dose oral estradiol seem to be small. In addition, while no direct comparisons exist this time, higher doses of oral estradiol seem to show similar testosterone suppression as non-oral estradiol (Wiki; Graphs). Besides testosterone suppression, available studies have found no differences between oral and non-oral estradiol in terms of outcomes like breast development or feminization (Sam, 2020). As such, the differences between oral and non-oral estradiol in terms SHBG levels and free estradiol fraction may be of little therapeutic importance.
Aside from decreasing free estradiol fractions, increased SHBG levels also decrease free testosterone fractions to an even greater extent. This is advantageous in the case of transfeminine people.
Taken together, lower free estradiol due to increased SHBG levels, whether with non-oral or oral estradiol, isn’t something that should be a major source of concern in transfeminine hormone therapy.
Supplementary Material
See here for supplementary material for this article, including a spreadsheet and other calculators that can be used to estimate free hormone levels (e.g., Mazer, 2009).
References
Adamcová, K., Kolátorová, L., Škodová, T., Šimková, M., Pařízek, A., Stárka, L., & Dušková, M. (2018). Steroid hormone levels in the peripartum period – differences caused by fetal sex and delivery type. Physiological Research, 67(Suppl 3), S489–S497. [DOI:10.33549/physiolres.934019]
Aly. (2020). A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Aly. (2020). Hormone Levels During Normal Puberty in Cisgender Girls. Transfeminine Science. [URL]
Aly. (2020). Supplement: The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Anderson, P. J., Hancock, K. W., & Oakey, R. E. (1985). Non-protein-bound oestradiol and progesterone in human peripheral plasma before labour and delivery. Journal of Endocrinology, 104(1), 7–15. [DOI:10.1677/joe.0.1040007]
Barini, A., Liberale, I., & Menini, E. (1993). Simultaneous Determination of Free Testosterone and Testosterone Bound to Non-Sex-Hormone-Binding Globulin by Equilibrium Dialysis. Clinical Chemistry, 39(6), 936–941. [DOI:10.1093/clinchem/39.6.936]
Bernstein, L., Depue, R. H., Ross, R. K., Judd, H. L., Pike, M. C., & Henderson, B. E. (1986). Higher maternal levels of free estradiol in first compared to second pregnancy: early gestational differences. Journal of the National Cancer Institute, 76(6), 1035–1039. [DOI:10.1093/jnci/76.6.1035]
Bernstein, L., Pike, M., Depue, R., Ross, R., Moore, J., & Henderson, B. (1988). Maternal hormone levels in early gestation of cryptorchid males: a case-control study. British Journal of Cancer, 58(3), 379–381. [DOI:10.1038/bjc.1988.223]
Bland, L. B., Garzotto, M., DeLoughery, T. G., Ryan, C. W., Schuff, K. G., Wersinger, E. M., Lemmon, D., & Beer, T. M. (2005). Phase II study of transdermal estradiol in androgen-independent prostate carcinoma. Cancer, 103(4), 717–723. [DOI:10.1002/cncr.20857]
Braunstein, G. D., Reitz, R. E., Buch, A., Schnell, D., & Caulfield, M. P. (2011). Testosterone Reference Ranges in Normally Cycling Healthy Premenopausal Women. The Journal of Sexual Medicine, 8(10), 2924–2934. [DOI:10.1111/j.1743-6109.2011.02380.x]
Burger, H. G., Dudley, E. C., Cui, J., Dennerstein, L., & Hopper, J. L. (2000). A Prospective Longitudinal Study of Serum Testosterone, Dehydroepiandrosterone Sulfate, and Sex Hormone-Binding Globulin Levels through the Menopause Transition. The Journal of Clinical Endocrinology & Metabolism, 85(8), 2832–2838. [DOI:10.1210/jcem.85.8.6740]
Campino, C., Torres, C., Rioseco, A., Poblete, A., Pugin, E., Valdés, V., Catalán, S., Belmar, C., & Serón-Ferré, M. (2001). Plasma prolactin/oestradiol ratio at 38 weeks gestation predicts the duration of lactational amenorrhoea. Human Reproduction, 16(12), 2540–2545. [DOI:10.1093/humrep/16.12.2540]
de Blok, C. J., Klaver, M., Wiepjes, C. M., Nota, N. M., Heijboer, A. C., Fisher, A. D., Schreiner, T., T’Sjoen, G., & den Heijer, M. (2017). Breast Development in Transwomen After 1 Year of Cross-Sex Hormone Therapy: Results of a Prospective Multicenter Study. The Journal of Clinical Endocrinology & Metabolism, 103(2), 532–538. [DOI:10.1210/jc.2017-01927]
de Blok, C. J., Dijkman, B. A., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K. M., & den Heijer, M. (2020). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
Depue, R. H., Bernstein, L., Ross, R. K., Judd, H. L., & Henderson, B. E. (1987). Hyperemesis gravidarum in relation to estradiol levels, pregnancy outcome, and other maternal factors: A seroepidemiologic study. American Journal of Obstetrics and Gynecology, 156(5), 1137–1141. [DOI:10.1016/0002-9378(87)90126-8]
Derra, C. (1981). Hormonprofile unter Östrogen- und Antiandrogentherapie bei Patienten mit Prostatakarzinom: Östradiolundecylat versus Cyproteronacetat. [Hormone Profiles under Estrogen and Antiandrogen Therapy in Patients with Prostate Cancer: Estradiol Undecylate versus Cyproterone Acetate.] (Doctoral dissertation, University of Mainz.) [Google Scholar] [WorldCat] [PDF] [Translation]
Dittrich, R., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Mueller, A. (2005). Endocrine Treatment of Male-to-Female Transsexuals Using Gonadotropin-Releasing Hormone Agonist. Experimental and Clinical Endocrinology & Diabetes, 113(10), 586–592. [DOI:10.1055/s-2005-865900]
Dunn, J. F., Nisula, B. C., & Rodbard, D. (1981). Transport of Steroid Hormones: Binding of 21 Endogenous Steroids to Both Testosterone-Binding Globulin and Corticosteroid-Binding Globulin in Human Plasma. The Journal of Clinical Endocrinology & Metabolism, 53(1), 58–68. [DOI:10.1210/jcem-53-1-58]
Fåhraeus, L., & Larsson-Cohn, U. (1982). Oestrogens, gonadotrophins and SHBG during oral and cutaneous administration of oestradiol-17β to menopausal women. Acta Endocrinologica, 101(4), 592–596. [DOI:10.1530/acta.0.1010592]
Fanelli, F., Gambineri, A., Belluomo, I., Repaci, A., Di Lallo, V. D., Di Dalmazi, G., Mezzullo, M., Prontera, O., Cuomo, G., Zanotti, L., Paccapelo, A., Morselli-Labate, A. M., Pagotto, U., & Pasquali, R. (2013). Androgen Profiling by Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS) in Healthy Normal-Weight Ovulatory and Anovulatory Late Adolescent and Young Women. The Journal of Clinical Endocrinology & Metabolism, 98(7), 3058–3067. [DOI:10.1210/jc.2013-1381]
Freymann, E., Hubl, W., Büchner, M., & Belleée, H. (1977). Eine spezifische, radioimmunologische Bestimmung des Plasmaöstradiols ohne Chromatographie im Zyklus und in der Schwangerschaft und die Bestimmung des freien, nichtproteingebundenen Anteils mittels Dialyse. [A specific radioimmunologi determination of plasma estradiol without chromatography during the cycle and in pregnancy and determination of the free non-protein-bound fraction using dialysis.] Zentralblatt für Gynäkologie, 99(6), 321–329. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF]
Freymann, E., Hubl, W., Büchner, M., & Rohde, W. (1977). Plasma levels of apparent free estradiol during pregnancy. Endokrinologie, 69(2), 269–271. [Google Scholar] [PubMed] [PDF]
Gibney, J., Johannsson, G., Leung, K., & Ho, K. K. (2005). Comparison of the Metabolic Effects of Raloxifene and Oral Estrogen in Postmenopausal and Growth Hormone-Deficient Women. The Journal of Clinical Endocrinology & Metabolism, 90(7), 3897–3903. [DOI:10.1210/jc.2005-0173]
Goldman, A. L., Bhasin, S., Wu, F. C., Krishna, M., Matsumoto, A. M., & Jasuja, R. (2017). A Reappraisal of Testosterone’s Binding in Circulation: Physiological and Clinical Implications. Endocrine Reviews, 38(4), 302–324. [DOI:10.1210/er.2017-00025]
Graham, C. A., Bancroft, J., Doll, H. A., Greco, T., & Tanner, A. (2007). Does oral contraceptive-induced reduction in free testosterone adversely affect the sexuality or mood of women? Psychoneuroendocrinology, 32(3), 246–255. [DOI:10.1016/j.psyneuen.2006.12.011]
Guthrie, J., Dennerstein, L., Taffe, J., Lehert, P., & Burger, H. (2004). The menopausal transition: a 9-year prospective population-based study. The Melbourne Women’s Midlife Health Project. Climacteric, 7(4), 375–389. [DOI:10.1080/13697130400012163]
Hammond, G. L., Nisker, J. A., Jones, L. A., & Siiteri, P. K. (1980). Estimation of the percentage of free steroid in undiluted serum by centrifugal ultrafiltration-dialysis. Journal of Biological Chemistry, 255(11), 5023–5026. [DOI:10.1016/s0021-9258(19)70742-x]
Hammond, G. L. (2016). Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. Journal of Endocrinology, 230(1), R13–R25. [DOI:10.1530/joe-16-0070]
Hammond, G. L. (2017). Sex Hormone-Binding Globulin and the Metabolic Syndrome. In Winters, S. J., & Huhtaniemi, I. T. (Eds.). Male Hypogonadism: Basic, Clinical and Therapeutic Principles (pp. 305–324). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-53298-1_15]
Handelsman, D. J. (2017). Free Testosterone: Pumping up the Tires or Ending the Free Ride? Endocrine Reviews, 38(4), 297–301. [DOI:10.1210/er.2017-00171]
Heidrich, A., Schleyer, M., Spingler, H., Albert, P., Knoche, M., Fritze, J., & Lanczik, M. (1994). Postpartum blues: Relationship between not-protein bound steroid hormones in plasma and postpartum mood changes. Journal of Affective Disorders, 30(2), 93–98. [DOI:10.1016/0165-0327(94)90036-1]
Heubner, A., Brockerhoff, P., Kreienberg, R., Grill, H., Rathgen, G., & Pollow, K. (1987). The influence of various dosages of megestrol acetate on SHBG, CBG and lipoprotein patterns. Journal of Steroid Biochemistry, 28(Suppl 1), 214S–214S (abstract no. 6). [DOI:10.1016/0022-4731(87)91680-3]
Hogeveen, K. N., Cousin, P., Pugeat, M., Dewailly, D., Soudan, B., & Hammond, G. L. (2002). Human sex hormone-binding globulin variants associated with hyperandrogenism and ovarian dysfunction. The Journal of Clinical Investigation, 109(7), 973–981. [DOI:10.1172/JCI14060]
Keevil, B. G., & Adaway, J. (2019). Assessment of free testosterone concentration. The Journal of Steroid Biochemistry and Molecular Biology, 190, 207–211. [DOI:10.1016/j.jsbmb.2019.04.008]
Kerlan, V., Nahoul, K., Martelot, M., & Bercovici, J. (1994). Longitudinal study of maternal plasma bioavailable testosterone and androstanediol glucuronide levels during pregnancy. Clinical Endocrinology, 40(2), 263–267. [DOI:10.1111/j.1365-2265.1994.tb02478.x]
Krause, A., Sinnecker, G., Hiort, O., Thamm, B., & Hoepffner, W. (2004). Applicability of the SHBG Androgen Sensitivity Test in the Differential Diagnosis of 46,XY Gonadal Dysgenesis, True Hermaphroditism, and Androgen Insensitivity Syndrome. Experimental and Clinical Endocrinology & Diabetes, 112(5), 236–240. [DOI:10.1055/s-2004-817969]
Kronawitter, D., Gooren, L. J., Zollver, H., Oppelt, P. G., Beckmann, M. W., Dittrich, R., & Mueller, A. (2009). Effects of transdermal testosterone or oral dydrogesterone on hypoactive sexual desire disorder in transsexual women: results of a pilot study. European Journal of Endocrinology, 161(2), 363–368. [DOI:10.1530/eje-09-0265] [Table]
Kuhl, H. (1997). Metabolische Effekte der Östrogene und Gestagene. [Metabolic Effects of Estrogens and Progestogens.] Der Gynäkologe, 30(4), 357–369. [DOI:10.1007/pl00003042]
Kuhl, H. (1998). Adverse effects of estrogen treatment: natural vs. synthetic estrogens. In Lippert, T. H., Mueck, A. O., & Ginsburg, J. (Eds.). Sex Steroids and the Cardiovascular System: The Proceedings of the 1st Interdisciplinary Workshop, Tuebingen, Germany, October 1996. Parthenon Publishing Group, New York, London (pp. 201–210). London/New York: Parthenon. [Google Scholar] [Google Books] [PDF]
Kuhl, H. (1999). Hormonal contraception. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 363–407). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_18]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Lundgren, S., Lønning, P., Utaaker, E., Aakvaag, A., & Kvinnsland, S. (1990). Influence of progestins on serum hormone levels in postmenopausal women with advanced breast cancer—I. General findings. Journal of Steroid Biochemistry, 36(1–2), 99–104. [DOI:10.1016/0022-4731(90)90118-c]
Lundgren, S., & Lønning, P. (1990). Influence of progestins on serum hormone levels in postmenopausal women with advanced breast cancer—II. A differential effect of megestrol acetate and medroxyprogesterone acetate on serum estrone sulfate and sex hormone binding globulin. Journal of Steroid Biochemistry, 36(1–2), 105–109. [DOI:10.1016/0022-4731(90)90119-d]
Mazer, N. A. (2009). A novel spreadsheet method for calculating the free serum concentrations of testosterone, dihydrotestosterone, estradiol, estrone and cortisol: With illustrative examples from male and female populations. Steroids, 74(6), 512–519. [DOI:10.1016/j.steroids.2009.01.008] [Spreadsheet]
McClamrock, H. D. (2007). Pregnancy-Related Androgen Excess. In Azziz, R., Nestler, J. E., & Dewailly, D. (Eds.). Androgen Excess Disorders in Women: Polycystic Ovary Syndrome and Other Disorders, 2nd Edition (Contemporary Endocrinology) (pp. 107–119). Totowa, New Jersey: Humana Press. [Google Books] [DOI:10.1007/978-1-59745-179-6_9]
Mean, F., Pellaton, M., & Magrini, G. (1977). Study on the binding of dihydrotestosterone, testosterone and oestradiol with sex hormone binding globulin. Clinica Chimica Acta, 80(1), 171–180. [DOI:10.1016/0009-8981(77)90276-5]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463] [PDF]
Moore, J. W., & Bulbrook, R. D. (1988). The epidemiology and function of sex hormone-binding globulin. Oxford Reviews of Reproductive Biology, 10, 180–236. [Google Scholar] [PubMed] [PDF]
Mueller, A., Dittrich, R., Binder, H., Kuehnel, W., Maltaris, T., Hoffmann, I., & Beckmann, M. W. (2005). High dose estrogen treatment increases bone mineral density in male-to-female transsexuals receiving gonadotropin-releasing hormone agonist in the absence of testosterone. European Journal of Endocrinology, 153(1), 107–113. [DOI:10.1530/eje.1.01943]
Mueller, A., Binder, H., Cupisti, S., Hoffmann, I., Beckmann, M., & Dittrich, R. (2006). Effects on the Male Endocrine System of Long-term Treatment with Gonadotropin-releasing Hormone Agonists and Estrogens in Male-to-Female Transsexuals. Hormone and Metabolic Research, 38(3), 183–187. [DOI:10.1055/s-2006-925198]
Mueller, A., Zollver, H., Kronawitter, D., Oppelt, P. G., Claassen, T., Hoffmann, I., Beckmann, M. W., & Dittrich, R. (2011). Body composition and bone mineral density in male-to-female transsexuals during cross-sex hormone therapy using gonadotrophin-releasing hormone agonist. Experimental and Clinical Endocrinology & Diabetes, 119(2), 95–100. [DOI:10.1055/s-0030-1255074] [Table]
Nachtigall, L. E., Raju, U., Banerjee, S., Wan, L., & Levitz, M. (2000). Serum Estradiol-Binding Profiles in Postmenopausal Women Undergoing Three Common Estrogen Replacement Therapies. Menopause, 7(4), 243–250. [DOI:10.1097/00042192-200007040-00006]
Nakamoto, J. (2016). Endocrine Testing. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition (pp. 2655–2688.e1). Philadelphia: Saunders/Elsevier. [Google Books] [DOI:10.1016/B978-0-323-18907-1.00154-2]
Nguyen, T. D. T., Dolomie-Fagour, L., Georges, A., & Corcuff, J. B. (2008). Dosage des stéroïdes sexuels sériques: quelle place pour l’estradiol biodisponible? [What about bioavailable estradiol?] Annales de Biologie Clinique, 66(5), 493–497. [DOI:10.1684/abc.2008.0259]
Nilsson, B., Holst, J., & Schoultz, B. (1984). Serum levels of unbound 17β-oestradiol during oral and percutaneous postmenopausal replacement therapy. British Journal of Obstetrics and Gynaecology, 91(10), 1031–1036. [DOI:10.1111/j.1471-0528.1984.tb03683.x]
Nolan, B. J., & Cheung, A. S. (2021). Relationship Between Serum Estradiol Concentrations and Clinical Outcomes in Transgender Individuals Undergoing Feminizing Hormone Therapy: A Narrative Review. Transgender Health, 6(3), 125–131. [DOI:10.1089/trgh.2020.0077]
O’Leary, P., Boyne, P., Flett, P., Beilby, J., & James, I. (1991). Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clinical Chemistry, 37(5), 667–672. [DOI:10.1093/clinchem/37.5.667]
Odlind, V., Milsom, I., Persson, I., & Victor, A. (2002). Can changes in sex hormone binding globulin predict the risk of venous thromboembolism with combined oral contraceptive pills? Acta Obstetricia et Gynecologica Scandinavica, 81(6), 482–490. [DOI:10.1034/j.1600-0412.2002.810603.x]
Ojasoo, T., & Raynaud, J. P. (1978). Unique steroid congeners for receptor studies. Cancer Research, 38(11 Part 2), 4186–4198. [Google Scholar] [PubMed] [URL]
Pande, P., Fleck, S. C., Twaddle, N. C., Churchwell, M. I., Doerge, D. R., & Teeguarden, J. G. (2019). Comparative estrogenicity of endogenous, environmental and dietary estrogens in pregnant women II: Total estrogenicity calculations accounting for competitive protein and receptor binding and potency. Food and Chemical Toxicology, 125, 341–353. [DOI:10.1016/j.fct.2018.12.013]
Pardridge, W. M. (1988). Selective delivery of sex steroid hormones to tissues by albumin and by sex hormone-binding globulin. Oxford Reviews of Reproductive Biology, 10, 237–292. [Google Scholar] [PubMed] [PDF]
Perry, L., Wathen, N., & Chard, T. (1987). Saliva Levels of Oestradiol and Progesterone in Relation to Non-Protein-Bound Concentrations in Blood During Late Pregnancy. Hormone and Metabolic Research, 19(9), 444–447. [DOI:10.1055/s-2007-1011848]
Plymate, S. R., Moore, D. E., Cheng, C. Y., Bardin, C. W., Southworth, M. B., & Levinski, M. J. (1985). Sex Hormone-Binding Globulin Changes during the Menstrual Cycle. The Journal of Clinical Endocrinology & Metabolism, 61(5), 993–996. [DOI:10.1210/jcem-61-5-993]
Pugeat, M. M., Dunn, J. F., & Nisula, B. C. (1981). Transport of Steroid Hormones: Interaction of 70 Drugs with Testosterone-Binding Globulin and Corticosteroid-Binding Globulin in Human Plasma. The Journal of Clinical Endocrinology & Metabolism, 53(1), 69–75. [DOI:10.1210/jcem-53-1-69]
Rezaii, T., Gustafsson, T. P., Axelson, M., Zamani, L., Ernberg, M., Hirschberg, A. L., & Carlström, K. A. (2017). Circulating androgens and SHBG during the normal menstrual cycle in two ethnic populations. Scandinavian Journal of Clinical and Laboratory Investigation, 77(3), 184–189. [DOI:10.1080/00365513.2017.1286685]
Ropponen, A., Aittomäki, K., Vihma, V., Tikkanen, M. J., & Ylikorkala, O. (2005). Effects of Oral and Transdermal Estradiol Administration on Levels of Sex Hormone-Binding Globulin in Postmenopausal Women with and without a History of Intrahepatic Cholestasis of Pregnancy. The Journal of Clinical Endocrinology & Metabolism, 90(6), 3431–3434. [DOI:10.1210/jc.2005-0352]
Rosner, W. (2015). Free estradiol and sex hormone-binding globulin. Steroids, 99, 113–116. [DOI:10.1016/j.steroids.2014.08.005]
Rothman, M. S., Carlson, N. E., Xu, M., Wang, C., Swerdloff, R., Lee, P., Goh, V. H., Ridgway, E. C., & Wierman, M. E. (2011). Reexamination of testosterone, dihydrotestosterone, estradiol and estrone levels across the menstrual cycle and in postmenopausal women measured by liquid chromatography–tandem mass spectrometry. Steroids, 76(1–2), 177–182. [DOI:10.1016/j.steroids.2010.10.010]
Rubinow, D. R., Schmidt, P. J., Roca, C. A., & Daly, R. C. (2002). Gonadal Hormones and Behavior in Women: Concentrations versus Context. In Pfaff, D. W., Arnold, A. P., Etgen, A. M., Fahrbach, S. E., & Rubin, R. T. (Eds.). Hormones, Brain and Behavior, Volume 5 (pp. 37–73). Amsterdam: Academic Press. [Google Books] [DOI:10.1016/B978-012532104-4/50086-X]
Ruokonen, A., Alén, M., Bolton, N., & Vihko, R. (1985). Response of serum testosterone and its precursor steroids, SHBG and CBG to anabolic steroid and testosterone self-administration in man. Journal of Steroid Biochemistry, 23(1), 33–38. [DOI:10.1016/0022-4731(85)90257-2]
Sam. (2020). A Comparison of Oral and Transdermal Estradiol in Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Schijf, C. P., van der Mooren, M. J., Doesburg, W. H., Thomas, C. M., & Rolland, R. (1993). Differences in serum lipids, lipoproteins, sex hormone binding globulin and testosterone between the follicular and the luteal phase of the menstrual cycle. Acta Endocrinologica, 129(2), 130–133. [DOI:10.1530/acta.0.1290130]
Schuijt, M. P., Sweep, C. G., van der Steen, R., Olthaar, A. J., Stikkelbroeck, N. M., Ross, H. A., & van Herwaarden, A. E. (2019). Validity of free testosterone calculation in pregnant women. Endocrine Connections, 8(6), 672–679. [DOI:10.1530/ec-19-0110]
Shifren, J. L., Desindes, S., McIlwain, M., Doros, G., & Mazer, N. A. (2007). A randomized, open-label, crossover study comparing the effects of oral versus transdermal estrogen therapy on serum androgens, thyroid hormones, and adrenal hormones in naturally menopausal women. Menopause, 14(6), 985–994. [DOI:10.1097/gme.0b013e31803867a]
Shifren, J. L., Rifai, N., Desindes, S., McIlwain, M., Doros, G., & Mazer, N. A. (2008). A Comparison of the Short-Term Effects of Oral Conjugated Equine Estrogens Versus Transdermal Estradiol on C-Reactive Protein, Other Serum Markers of Inflammation, and Other Hepatic Proteins in Naturally Menopausal Women. The Journal of Clinical Endocrinology & Metabolism, 93(5), 1702–1710. [DOI:10.1210/jc.2007-2193]
Smith, K., Galazi, M., Openshaw, M. R., Wilson, P., Sarker, S. J., O’Brien, N., Alifrangis, C., Stebbing, J., & Shamash, J. (2020). The Use of Transdermal Estrogen in Castrate-resistant, Steroid-refractory Prostate Cancer. Clinical Genitourinary Cancer, 18(3), e217–e223. [DOI:10.1016/j.clgc.2019.09.019]
Stege, R., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., & Pousette, A. (1988). Single drug polyestradiol phosphate therapy in prostatic cancer. American Journal of Clinical Oncology, 11(Suppl 2), S101–S103. [DOI:10.1097/00000421-198801102-00024] [PDF]
Steingold, K. A., Pardridge, W. M., Judd, H. L., & Chaudhuri, G. (1987). The effects of membrane permeability and binding by human serum proteins on steroid influx into the rabbit uterus. American Journal of Obstetrics and Gynecology, 157(6), 1543–1549. [DOI:10.1016/s0002-9378(87)80260-0]
Strauss, J. F., & FitzGerald, G. A. (2019). Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 75–114.e7). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/b978-0-323-47912-7.00004-4]
Troisi, R., Potischman, N., Roberts, J. M., Harger, G., Markovic, N., Cole, B., Lykins, D., Siiteri, P., & Hoover, R. N. (2003). Correlation of serum hormone concentrations in maternal and umbilical cord samples. Cancer Epidemiology, Biomarkers & Prevention, 12(5), 452–456. [Google Scholar] [PubMed] [URL]
Tulchinsky, D., & Chopra, I. J. (1973). Competitive Ligand-Binding Assay for Measurement of Sex Hormone-Binding Globulin (SHBG). The Journal of Clinical Endocrinology & Metabolism, 37(6), 873–881. [DOI:10.1210/jcem-37-6-873]
von Schoultz, B., Carlström, K., Collste, L., Eriksson, A., Henriksson, P., Pousette, Å., & Stege, R. (1989). Estrogen therapy and liver function—metabolic effects of oral and parenteral administration. The Prostate, 14(4), 389–395. [DOI:10.1002/pros.2990140410]
Zimmerman, Y., Eijkemans, M. J., Coelingh Bennink, H. J., Blankenstein, M. A., & Fauser, B. C. (2013). The effect of combined oral contraception on testosterone levels in healthy women: a systematic review and meta-analysis. Human Reproduction Update, 20(1), 76–105. [DOI:10.1093/humupd/dmt038]
\ No newline at end of file
diff --git a/transfemscience.org/articles/spiro-testosterone/index.html b/transfemscience.org/articles/spiro-testosterone/index.html
index e7030205..32b04974 100644
--- a/transfemscience.org/articles/spiro-testosterone/index.html
+++ b/transfemscience.org/articles/spiro-testosterone/index.html
@@ -1 +1 @@
-A Review of Studies on Spironolactone and Testosterone Suppression in Cisgender Men, Cisgender Women, and Transfeminine People - Transfeminine Science
A Review of Studies on Spironolactone and Testosterone Suppression in Cisgender Men, Cisgender Women, and Transfeminine People
By Aly | First published December 19, 2018 | Last modified August 23, 2025
Abstract / TL;DR
Spironolactone is an antiandrogen used in transfeminine hormone therapy which is especially employed in the United States. It is widely considered to act as an androgen receptor antagonist and as an androgen synthesis inhibitor, both blocking the actions of testosterone and lowering testosterone levels in transfeminine people. A literature search was conducted to review studies assessing the influence of spironolactone on testosterone levels in cisgender men, cisgender women, and transfeminine people. The results of these studies were mixed, but in most studies spironolactone showed no apparent influence on testosterone levels. These findings suggest that spironolactone has inconsistent and limited effects on testosterone levels. Moreover, these data, as well as studies of estradiol alone, indicate that estradiol is mainly responsible for lowered testosterone levels when the combination of estradiol and spironolactone is used for hormone therapy in transfeminine people. Besides testosterone suppression, spironolactone also acts as a direct antagonist of the androgen receptor, and this importantly contributes to its antiandrogenic efficacy as well. However, studies in cisgender women suggest that spironolactone is a relatively weak androgen receptor antagonist, and is likely best-suited for blocking relatively low testosterone levels. Taken together, the antiandrogenic effectiveness of spironolactone in transfeminine people appears to be limited. Other antiandrogenic approaches may be more effective in transfeminine people, and may be considered instead or as alternatives to spironolactone in those in whom testosterone levels with estradiol plus spironolactone remain inadequately suppressed.
Introduction
Spironolactone, also known by its major brand name Aldactone, is an antiandrogen which is commonly used in transfeminine hormone therapy. It is used in combination with estrogen in transfeminine people to help reduce the effects of testosterone. Spironolactone is used in transfeminine hormone therapy particularly in the United States, where another antiandrogen, cyproterone acetate (CPA; brand name Androcur), is unavailable. Conversely, CPA is the main antiandrogen used in transfeminine people in Europe and most of the rest of the world. Another type of medication, gonadotropin-releasing hormone (GnRH) agonists, are the major antiandrogens used in certain places like the United Kingdom. The combination of estradiol with CPA or a GnRH agonist in transfeminine people consistently suppresses testosterone levels into the normal female range (<50 ng/dL or <1.8 nmol/L) (Aly, 2018; Aly, 2019). Hence, both CPA and GnRH agonists are very effective antiandrogens in transfeminine people.
Spironolactone acts as an androgen receptor antagonist, but is also known to function as an androgen synthesis inhibitor. As an example, spironolactone has been shown in preclinical research to inhibit several enzymes involved in gonadal and adrenal androgen production, including CYP17A1 (17α-hydroxylase/17,20-lyase) among others, and to substantially decrease concentrations of androgens in these studies (Loriaux et al., 1976; Callan, 1988; McMullen & Van Herle, 1993). However, the steroid synthesis inhibition of spironolactone appears to only occur at very high doses and concentrations of spironolactone (Loriaux et al., 1976; McMullen & Van Herle, 1993). For example, spironolactone is used at 10- to 20-fold smaller doses by body weight in humans than in animal studies that have demonstrated substantial steroid synthesis inhibition with the agent (McMullen & Van Herle, 1993).
A widespread notion in the transgender community, as well as in the transgender health community and in the medical literature, is that spironolactone decreases testosterone levels and that this is a major part of how it works as an antiandrogen in transfeminine people. In actuality however, the clinical evidence to support this notion appears to be limited, and available data from studies appear to be highly conflicting. The purpose of this article is to review the available clinical studies on spironolactone and testosterone levels in cisgender men, cisgender women, and transfeminine people in order to help elucidate whether and to what extent spironolactone lowers testosterone levels in humans. In addition, the role of androgen receptor blockade in the antiandrogenic effects of spironolactone is briefly reviewed.
A total of 22 studies of spironolactone and sex hormone levels in cisgender males were identified (Table 1). These studies assessed pre-treatment versus post-treatment hormone levels with spironolactone, hormone levels with spironolactone versus a comparator group, or both. Within the identified studies, testosterone levels were not significantly changed in 12 of 22 studies (55%), decreased in 4 of 22 (18%) studies, increased in 1 of 22 (4.5%) studies, and mixed or unknown (e.g. divergences in changes of total versus free testosterone levels or didn’t actually report testosterone levels) in 4 of 22 (18%) studies. Most of the studies were very small (fewer than 10 people), with several exceptions. The studies were of highly variable lengths, with some being several days and others lasting for weeks or months. Few of the studies were RCTs. Most of the studies were very old, with a majority published in the 1970s and the rest published in the 1980s and 1990s. In relation to the preceding, the quality of data was limited.
Table 1: Studies of sex hormone levels with spironolactone alone in cisgender males:
Treatment and subjects
Findings
Source(s)
100 mg/day for 2 weeks in 7 healthy men (23–34 years)
T significantly decreased and LH significantly increased. No significant change in E1, E2, or E3. No change urinary total T excretion but significantly increased urinary total E excretion (including of E1 (7.72 to 10.54 µg/24 hrs), E2 (2.60 to 3.34 ug/24 hours), E3 (7.69 to 11.75 µg/24 hrs)). Slightly but significantly decreased excretion of 17-KS in urine.
400 mg/day for 5 days in 6 healthy men (21–33 years)
Significant increase in P4 and 17α-OHP (approximately doubled) for whole duration. Small and transient increases in LH (+20%) and FSH on the 2nd but not on the 3rd or 5th days (only other days measured). No significant changes in T, E2, or PRL. E2 and PRL non-significantly increased (+56% and +34% on the 5th day, respectively).
100 or 400 mg/day spironolactone for 8 weeks in 7 orchiectomized men (46–78 years) with metastatic prostate cancer
T, A4, and DHEA significantly decreased with both doses of spironolactone and of similar magnitude between doses. Influence more apparent after 2–3 weeks of treatment.
5 mg/kg/day for 1 week (275 mg/day for a 55 kg person) in 7 boys with delayed puberty (14–16 years)
Significant increase in LH (+60%) and non-significant increase in FSH (+60%); individual responses for FSH variable. Increased P4 and 17α-OHP. T and E2 not actually reported.
Initially 400 mg/day for 12 weeks; dosage later decreased in some due to hypotension (range 150–400 mg/day) in 5 men and 5 women (3 premenopausal, 2 postmenopausal) with normal or low renin hypertension
P4 and 17α-OHP increased by 2 to 4 times compared to pre-treatment and post-treatment. T, E2, LH, FSH, PRL, and 17-KS all unchanged.
200–400 mg/day for 4–13 months (mean 7 months) in 6 men with hypertension (35–61 years; mean 47 years) vs. 10 untreated male controls with hypertension (mean age 45 years)
Significantly greater LH and E2 (30 pg/mL vs. 13 pg/mL; +130%), significantly lower T (440 ng/dL vs. 270 ng/dL; –38%), no difference in FSH. Also, significantly greater metabolic clearance rate of T, significantly greater rate of peripheral conversion (conversion ratio and transfer constant) of T into E2, non-significantly greater metabolic clearance rate of E2, no difference in blood production rate of T, and significantly greater blood production rate of E2.
200–400 mg/day (mean 330 mg/day) for 20–27 days in 5 gonadally intact men (50–76 years) with prostate cancer
P4 increased significantly from 0.25 ± 0.10 ng/mL (mean ± SD) to maximum of 1.3 ± 0.31 ng/mL by 20 days (increase of 5.2-fold or 420%). T decreased significantly from 427 ± 74.3 ng/dL to 200 ± 80.3 ng/dL (–53.2%). No significant change in E2, LH, or FSH.
200 mg/day for 21 days in 4 healthy men (26–35 years)
No change in total T or E2. Unbound T and E2 slightly but significantly increased. Thought to be due to a direct interaction of spironolactone metabolites with the plasma protein binding of T and E2. But not due to binding to SHBG as T binding to SHBG was not significantly altered.
75–150 mg/day for 12 weeks in 6 men with essential hypertension (28–64 years; mean 48 years)
E1 significantly increased. E2 small, gradual, non-significant increase. T, LH, and PRL not significantly changed. PRL responses to TRH normal/not significantly changed.
150–300 mg/day for 40 weeks in 2 men with idiopathic hyperaldosteronism (23 and 44 years)
E1 increased. E2 fluctuated. E2 increased by 10-fold in one person by 16 weeks and this was associated with gynecomastia. T, LH, and PRL not altered significantly.
200 mg/day for 10 days (n=5) vs. placebo (n=5) in 10 healthy men (18–31 years) (RCT)
Significantly greater urinary A4, urinary EC, and urinary total E excretion. Differences in T, E2, LH, and FSH as well as urinary DHEA, LH, and FSH not significant. Examination of interaction between treatment and time showed significant changes in T, LH, and urinary DHEA. Concluded that there was a transient rise in T and urine DHEA for 2–4 days followed by increase in LH and normalization of T and DHEA excretion after 4–10 days.
300 mg/day for 7 days (n=5) vs. 200 mg/day triamterene (n=5) in 10 normal young men with diet-induced hyperaldosteronism (14 days of a diet modifying electrolyte intake)
P4, 17α-OHP, unchanged. T near-but-non-significantly decreased (704.6 ± 55.5 ng/dL (mean ± SEM) to 508.4 ± 45.9 ng/dL on day 6; p < 0.10). Also assessed endogenous corticosteroids.
100 mg/day for 3 months in treatment group of 47 men (age 60–80 years) with BPH; control group of 58 healthy men without BPH (also age 60–80 years)
In spiro/BPH group, T decreased from 650 ng/dL to 290 ng/dL and DHT decreased from 450 ng/dL to 150 ng/dL. In control/non-BPH group, T was 280 ng/dL and DHT was 90 ng/dL. P4, E2, and LH increased in spiro/BPH group. FSH also assessed. The authors stated that prostate gland can be a source of androgen production, implying that BPH can produce elevated androgen levels and that spironolactone can normalize elevated androgen levels in the condition.
150 mg/m2/day for 5 days in 6 boys with irregular puberty (11–13 years)
No significant changes in T or urinary 17-KS excretion, elevated LH (by 600%—likely typo of “60%” (?)), and slightly increased FSH (from 0.75 ng/mL to 0.86 ng/mL).
25–400 mg/day (median 100 mg/day) for 12 months in 32 males (59%) of a group of 54 males (17–64 years; mean 44 years) with non-alcoholic liver disease requiring liver transplantation vs. 469 healthy male controls (mean 31 years) with normal liver function
Significantly decreased T with spironolactone in men with moderate-severity liver disease but not with low- or high-severity liver disease. SHBG not influenced by spironolactone dosage. No influence on gonadotropin responses to GnRH stimulation.
Although the quality of these studies is limited, the findings of the studies, which are mixed but are overall more suggestive against spironolactone reducing testosterone levels than it doing so, are in notable contrast to similar studies of CPA and testosterone suppression in cisgender men that were published in the 1970s and 1980s. These studies consistently found that CPA suppressed testosterone levels by 40 to 70% on average (Aly, 2019). Subsequently, the findings were replicated in several more modern studies of CPA in cisgender men and transfeminine people, which likewise found that the drug given alone consistently suppressed testosterone levels by about 45 to 65% on average (Aly, 2019).
Spironolactone in Cisgender Women
Spironolactone has a long history of use in cisgender women in the treatment of androgen-dependent skin and hair conditions like acne, hirsutism, scalp hair loss, and hyperandrogenism (due to e.g. polycystic ovary syndrome (PCOS)). It has been used at similar doses for androgen-dependent conditions in cisgender women as it has in transfeminine people (e.g., 50–200 mg/day most typically). There are many dozens of studies of spironolactone as an antiandrogen in cisgender women (e.g., PubMed). Instead of attempting to individually review all of these studies, the present article will discuss the findings of several papers that have themselves reviewed substantial numbers of these studies and have summarized available findings on testosterone levels with spironolactone.
Callan (1988) reviewed the literature on spironolactone for treatment of acne and hirsutism in cisgender women and found that some clinical studies reported decreased levels of testosterone and/or other androgens with spironolactone (4 studies cited) whereas other studies reported no change in androgen levels (4 studies cited). The author cited several studies to support the claim that androgen receptor antagonism with spironolactone is more clinically important than any influence it has on androgen production (5 studies cited). For instance, clinical benefits against acne and hirsutism occurred with spironolactone both before androgen levels decrease as well as when androgen levels do not decrease.
McMullen & Van Herle (1993) reviewed 19 studies of spironolactone for treatment of androgen-dependent conditions in cisgender women, with a majority of these studies reporting long-term hormone levels. Most of the studies were open-label and uncontrolled, with only five studies having a control group and only two studies being double-blind placebo-controlled trials. Changes in hormone levels across studies were very heterogenous, with the majority of changes not reaching statistical significance. Only 1 of 7 (14%) studies found a decrease in DHEA-S levels. The review concluded that a clinically significant change in adrenal androgen levels with spironolactone in cisgender women was not supported. Conversely, testosterone levels were decreased with spironolactone in 13 of 16 (81%) of studies. However, in the only two RCTs, there were no differences in testosterone levels with spironolactone versus in the placebo control groups. As such, the review concluded that the decreased testosterone levels with spironolactone in cisgender women reported in many of the non-RCT studies may not actually be a real phenomenon. As with Callan (1988), the review noted that the major mechanism of action of spironolactone as an antiandrogen is likely to be androgen receptor blockade.
Bradstreet et al. (2007) cited and discussed a Cochrane review of spironolactone for treatment of acne and/or hirsutism in cisgender women (Farquhar et al., 2003). Cochrane reviews are rigorous high-quality systematic reviews of all of the available RCTs for a given medical intervention. The Cochrane review identified 19 RCTs, with 9 included in the review, 8 excluded due to methodological issues (e.g., with randomization), and two others which were described as “awaiting assessment” (Farquhar et al., 2003). Bradstreet and colleagues noted per the Cochrane review that spironolactone at a dosage of 100 mg/day had little influence on levels of DHEA, DHEA-S, or testosterone in the trials evaluated and said that this is because its mechanism of action as an antiandrogen is androgen receptor antagonism (Bradstreet et al., 2007). The Cochrane review itself did not discuss changes in androgen or testosterone levels with spironolactone in aggregate. An update of the Cochrane review was published in 2009, but with no new studies found and with the findings unchanged (Brown et al., 2009).
Layton et al. (2017) was a hybrid systematic review of spironolactone for acne in cisgender women. In a table discussing the mechanism of action of spironolactone and other antiandrogens for acne, the authors stated that “Data from over 50 articles reporting effects [of spironolactone] on serum androgens are equivocal” (i.e., ambiguous, uncertain, questionable) (Layton et al., 2017). The review further noted that inhibition of androgen synthesis by spironolactone in humans may be unlikely at therapeutic doses and may occur instead only at supraphysiological doses (with Menard et al. (1979) cited in support of these claims, presumably related to the very high doses required) (Layton et al., 2017).
Rozner et al. (2019) reviewed clinical studies of the endocrine effects of spironolactone in cisgender women to assess whether it is safe to use in women with past or present breast cancer receiving endocrine therapy. The review included 18 studies with 465 women (mostly having androgen-dependent conditions) assessing the influence of spironolactone on sex hormone levels. The assessed studies included retrospective cohort studies, case–control studies, and RCTs. Of the included studies, 10 (56%) studies (with 179 women) found no change in testosterone levels with spironolactone, 8 (44%) studies (with 253 women) found a decrease, and 1 (6%) study (with 33 women) found an increase in free but not total testosterone levels. Changes in levels of DHEA-S, androstenedione, and estrogen were also assessed and findings were similar, with no changes observed in majorities of studies for these hormones. The review concluded that there is no significant change in levels of androgens, estrogen, or gonadotropins with spironolactone in cisgender women.
Almalki et al. (2020) conducted a systematic review and network meta-analysis of RCTs on the comparative efficacy of several types of medications (statins, metformin, spironolactone, and combined birth control pills) on reducing testosterone levels in cisgender women specifically with PCOS. Nine RCTs including 613 women were included for all of the medications. The meta-analysis concluded that the statin atorvastatin was more effective than the other included medications in reducing testosterone levels. Only two of the included RCTs employed spironolactone, one of which was with spironolactone alone (n=34) versus metformin (n=35) (Ganie et al., 2004) and the other of which was with spironolactone plus metformin (n=62) versus spironolactone alone (n=51) versus metformin alone (n=56) (Ganie et al., 2013). Both of the included trials found that spironolactone alone significantly decreased testosterone levels in pre-treatment versus post-treatment comparisons (Ganie et al., 2004; Ganie et al., 2013). No trials of spironolactone versus placebo controls were included.
Taken together, the available studies of spironolactone and testosterone levels in cisgender women with androgen-dependent conditions are highly inconsistent and mixed, but with numerous studies finding no significant changes in testosterone levels. The reasons for the findings being so mixed are unclear, but may relate to study methodology and quality. Findings in this population seem particularly notable as regulation of the hypothalamic–pituitary–gonadal (HPG) axis by androgens in women is minimal to negligible, in turn making it such that androgen receptor antagonists will have little effect of upregulating gonadal sex hormone production as they can in cisgender men and transfeminine people. As a result, there is less homeostatic interference that could influence findings in evaluating the steroid synthesis inhibition of spironolactone in this sex, and hence these studies may provide a clearer picture of steroid synthesis inhibition as a possible clinical effect of spironolactone. However, as the findings are still so mixed, the results seem inconclusive. In any case, only a limited effect at best seems clear.
Spironolactone Alone in Transfeminine People
Only one study of spironolactone alone (without estrogen) and sex hormone levels in transfeminine people was identified (Table 2). It was conducted by Louis Gooren and colleagues of the Dutch Center of Expertise on Gender Dysphoria (CEGD) at the Vrije Universiteit Medical Center (VUMC) in Amsterdam, Netherlands in the 1980s. The study compared levels of testosterone, DHT, estradiol, LH, FSH, and prolactin before and after treatment with 200 mg/day spironolactone for 6 weeks in 6 young pre-hormone-therapy transfeminine people. It found slightly but significantly increased testosterone levels, increased prolactin levels, and no change in levels of estradiol, DHT, LH, or FSH.
Table 2: Studies of sex hormone levels with spironolactone alone in transfeminine people:
Treatment and subjects
Findings
Source(s)
200 mg/day for 6 weeks in 6 pre-hormone therapy transfeminine people (21–39 years)
T (mean ± SEM) increased significantly from 17.2 ± 0.8 nmol/L (496 ± 20 ng/dL) to 20.6 ± 1.7 nmol/L (594 ± 50 ng/dL) (+19.8%). No change in E2 (90 ± 20 pmol/L [25 ± 5.0 pg/mL] vs. 100 ± 30 pmol/L [27 ± 8.2 pg/mL] or 80 ± 20 pmol/L [22 ± 5.4 pg/mL]) or DHT (1.7 ± 0.8 nmol/L [49 ± 20 ng/dL] vs. 1.8 ± 0.9 nmol/L [52 ± 30 ng/dL]). LH, FSH, and GnRH-stimulated LH and FSH unchanged. PRL and TRH-stimulated PRL increased.
Abbreviations: T = testosterone; E2 = estradiol; DHT = dihydrotestosterone; LH = luteinizing hormone; FSH = follicle-stimulating hormone; GnRH = gonadotropin-releasing hormone; PRL = prolactin; TRH = thyrotropin-releasing hormone.
The fact that this study was done by the CEGD is notable as this institute is among the most prolific research centers on transgender hormone therapy in the world (Bakker, 2021), and, while they evaluated spironolactone as well as nilutamide as antiandrogens in studies in transfeminine people in the 1980s and 1990s (Wiki), the group ultimately settled on using only CPA instead. This was probably related to the lack of testosterone suppression with spironolactone and pure androgen receptor antagonists like nilutamide, as the researchers have touched on in other publications (e.g., Gooren, 1999).
Estrogen Plus Spironolactone in Transfeminine People
Eleven studies of the combination of estrogen and spironolactone and sex hormone levels in transfeminine people were identified (Table 3). The first study was conducted by Jerilynn Prior and colleagues in Canada in the 1980s. Subsequent studies were conducted over 25 years later by groups in the United States, Australia, Israel, and Thailand. All of the studies were retrospective chart reviews or prospective non-randomized studies, with the exception of a single RCT.
Table 3: Studies of testosterone levels with estrogen plus spironolactone in transfeminine people:
Treatment and subjects
Findings
Source(s)
Oral CEEs (0.625–5 mg/day cyclically—3 of 4 weeks per month), oral MPA (10–20 mg/day cyclically—3 of 4 weeks per month—or continuously—”if gonadotrophins increased or to aid in T reduction or breast development”), and spironolactone (100–600 mg/day continuously) for 12 months in 27 transfeminine people who had been on “high-dose” E alone for an extended duration (Group 1) and 23 transfeminine people who were pre-hormone-therapy (Group 2), or 50 transfeminine people total, at Vancouver General Hospital.
T decreased in Group 1 from mean 169 ng/dL to 87.4 ng/dL (–48.2%) and in Group 2 from mean 642 ng/dL to 49.2 ng/dL (–92.3%). In the groups combined, T following treatment would be mean 69.8 ng/dL. Per authors, spironolactone was intended to help reduce T and facilitate feminization while MPA was intended to help suppress gonadotropins and T and improve breast development. However, authors emphasized the decrease in T as being due to spironolactone despite inclusion of MPA, without data provided to substantiate this.
Sublingual estradiol (4 mg/day—2 mg b.i.d.) (n=14), transdermal estradiol patch (100 μg/day) (n=1), or injectable estradiol valerate (20 mg/2 weeks) (n=1) with spironolactone (100–200 mg/day) for 6 months in 16 transfeminine people at an LGBT community health center in Los Angeles, California.
T was median 405 ng/dL at baseline and 42 ng/dL after 6 months (–89.6%). Free T was median 11.4 ng/dL at baseline and 0.8 ng/dL at 6 months (–93.0%). 10 of 15 (66.7%) had total T in female range and 14 of 15 (93.3%) had free T in female range.
Oral E2 (1–8 mg/day) with or without spironolactone (200 mg/day) (n=61), finasteride (5 mg/day) (n=49), and/or MPA (2.5–10 mg/day) (n=38) for 0.3 to 10.5 years (mean 4.3 ± 3.1 years) in 156 transfeminine people at Albany Medical Center.
Oral E2 dose-dependently and substantially but incompletely suppressed T. Relative to E2 alone (at equivalent E2 levels), E2 plus spironolactone had no significant influence on T (+10.6 ± 16 ng/dL (mean ± SE); p = 0.5) and no greater likelihood of achieving better T suppression (<100 ng/dL) (OR = 0.75; 95% CI = 0.44–1.29). T levels with E2 alone were mean ~80 ng/dL and with E2 plus spironolactone were mean ~95 ng/dL per own re-analysis. Finasteride was also associated with greater T levels. MPA helped with T suppression in some (71% of subjects). More discussion and re-analysis including graphs (Aly, 2019).
Oral E2 (0.5–10 mg/day) (n=67) or oral CEEs (0.625–5 mg/day) (n=12) and spironolactone (25–400 mg/day; mean/median 145 mg/day) for 12 months in 98 transfeminine people at Boston Medical Center.
Combined E and spironolactone decreased T from median 385 ng/dL to 130 ng/dL (–66.2%). E alone vs. E and spironolactone not reported. No significant influence of spironolactone dosage on T. Incomplete suppression of T (>50 ng/dL) in all but the lowest quartile (25%) of individuals.
Oral EV (4–6 mg/day; median 5–6 mg/day) (88.3%) or transdermal E2 (11.7%) alone or in combination with CPA (25–50 mg/day; median 50 mg/day) or spironolactone (87.5–200 mg/day; median 100 mg/day) for 0.9 to 2.6 years (median 1.5 years) in 80 transfeminine people at two gender clinics in Melbourne, Australia.
T was median 10.5 nmol/L (303 ng/dL) with E2 only, 2.0 nmol/L (58 ng/dL) with E2 plus spironolactone, and 0.8 nmol/L (23 ng/dL) with E2 plus CPA. 90% of those on E2 plus CPA and 40% of those on E2 plus spironolactone had T of <2 nmol/L (<58 ng/dL). T significantly lower with E2 plus CPA compared to E2 plus spironolactone and E2 alone. T with E2 plus spironolactone lower than with E2 alone but non-significantly. No significant differences between groups in age, hormone therapy duration, or E2 dosage or levels. Graph that visually summarizes the results.
Sublingual estradiol (2–12 mg/day) and spironolactone (100–200 mg/day) with or without sublingual MPA (5–10 mg/day) or injectable MPA (150 mg/3 months) for 3.4 ± 1.7 years in 92 transfeminine people at Rhode Island Hospital.
T (mean ± SD) was 215 ± 29 ng/dL with E2 plus spironolactone and 79 ± 18 ng/dL with E2 plus spironolactone and MPA.
Oral E2 (2–8 mg/day) (84.2%) or other E forms (15.8%) with spironolactone (80.4%; n=107) or without spironolactone (19.6%) for more than 6 months in 133 transfeminine people at three clinics in Dallas, Texas.
T decreased from median 367 ng/dL (95% range 175–731 ng/dL) (n=70) at baseline to median 55 ng/dL (95% range 3–709 ng/dL) (n=131) in whole group (80.4% taking spironolactone). 65 of 133 (49%) had adequate T suppression (presumably <50 or <60 ng/dL) in whole group. T with E2 plus spironolactone at 25–75 mg/day (n=15) was mean 129.4 ng/dL (range <3—611 ng/dL), at 100–175 mg/day (n=61) was mean 180.4 ng/dL (range <3–1137 ng/dL), and at 200–300 mg/day (n=31) was mean 170.1 ng/dL (range <3–798 ng/dL). In the whole E2 plus spironolactone group (n=107), T would be mean 170.3 ng/dL.
Oral E2 (2–8 mg/day), transdermal E2 gel (2.5–5 mg/day), or transdermal E2 patches (50–200 μg/day) plus spironolactone (50–200 mg/day) (n=16), CPA (10–100 mg/day) (n=41), or a GnRH agonist (n=10) for 12 months in 67 transfeminine people at Tel Aviv-Sourasky Medical Center in Israel.
With spironolactone, T (mean ± SD) decreased from 15.2 ± 8.1 nmol/L (438 ± 230 ng/dL) at baseline to 10.2 ± 5.7 nmol/L (294 ± 164 ng/dL) at 3 months (–32.9%), 3.5 ± 1.2 nmol/L (100 ± 35 ng/dL) at 6 months (–77.0%), and 4 ± 7.1 nmol/L (120 ± 200 ng/dL) at 12 months (–73.7%). T was in the female range (<1.8 nmol/L [52 ng/dL]) at all follow-ups after baseline for both CPA and GnRH agonist (–92.0% to –96.4%).
Oral EV 4 mg/day plus spironolactone (100 mg/day) (n=26) or CPA (25 mg/day) (n=26) for 12 weeks in 52 transfeminine people at two clinics in Bangkok, Thailand (RCT).
With intention-to-treat analysis, T decreased with E2 plus spironolactone from median 645.0 ng/dL (IQR 466.7−1027.7 ng/dL) to 468.3 ng/dL (IQR 287.0−765.4 ng/dL) (–27.4%) and with E2 plus CPA from 655.5 ng/dL (402.6−872.7 ng/dL) to 9.3 ng/dL (IQR 5.5−310.4 ng/dL) (–98.6%). Adequate suppression of testosterone (<50 ng/dL) was achieved by 4 of 26 (15%) in the E2 plus spironolactone group and by 18 of 26 (69%) in the E2 plus CPA group. Study also assessed and reported E2, SHBG, and PRL levels.
E2 (sublingual, transdermal, or injectable) with spironolactone (n=39) or without spironolactone (n=37) for 12 months in 93 transfeminine people at two LGBTQ-oriented clinics in Seattle, Washington and Iowa City, Iowa.
T was median 11 to 18 ng/dL in different estradiol groups without spironolactone and median 10 to 12 ng/dL in different estradiol groups with spironolactone. T was significantly lower with spironolactone only for sublingual E2 group (median 11 ng/dL (IQR 6–35 ng/dL) [n=27] vs. median 18 ng/dL (IQR 13–205 ng/dL) [n=16]) and not for transdermal or injectable E2 groups.
Oral E2 (4–12 mg/day, median 6 mg/day) (n=27) or injectable EV (2–5 mg/week, median 4 mg/week) (n=6) with spironolactone (n=31) or without spironolactone (n=2) for median 6.2 months (range 0.6–28.2 months) (time on optimized E2 dose specifically) in 33 transfeminine people at Maine Medical Center.
T was median 13.0 ng/dL (range 2.7–559 ng/dL) for whole group (93.9% taking spironolactone). 28 of 33 (84.8%) of whole group had female-range T (<50 ng/dL). However, in earlier studies by the same group, similar T suppression with E2 alone was reported (Reardon et al., 2013; Spratt et al., 2014).
The data on the testosterone levels with estrogen plus spironolactone in transfeminine people from the 11 studies in the table can be roughly summarized. Some studies reported mean testosterone levels and some reported median testosterone levels, so these cases must be considered separately. In terms of reported mean testosterone levels across studies (4 studies), the median value of these study averages would be about 171 ng/dL and the range of study averages would be about 95 to 215 ng/dL. In terms of reported median testosterone levels across studies (7 studies), the median value of these study medians would be about 55 ng/dL and the range of study medians would be about 11 to 468 ng/dL. One study had to be excluded due to concomitant use of the progestogen medroxyprogesterone acetate (MPA) in all individuals (Prior, Vigna, & Watson, 1989; Prior et al., 1986). Insights from the preceding results include large variability in testosterone levels across studies and mean testosterone levels being much higher than median testosterone levels. Limitations of the preceding values include lack of equivalent estrogen and spironolactone dosages and levels across studies, lack of equivalent durations of hormone therapy across studies, lack of equivalent testosterone blood-testing methodologies across studies, lack of equivalent transfeminine patient samples, and, in the case of the study median testosterone values, two of the studies notably having almost all but not all individuals on spironolactone (80 and 94% rather than 100%). These limitations likely underlie the large variability in reported values across studies. In any case, these results suggest that estrogen plus spironolactone results in variably inadequate testosterone suppression in most transfeminine people, which is in notable major contrast to testosterone suppression with estrogen plus CPA or a GnRH agonist in transfeminine people.
Individual findings of the studies include inadequate testosterone suppression with estradiol plus spironolactone in most transfeminine people (Leinung et al., 2018; Liang et al., 2018; Jain, Kwan, & Forcier, 2019; Sofer et al., 2020; Burinkul et al., 2021), no difference in testosterone suppression with spironolactone versus without spironolactone (Leinung et al., 2018), lack of notable influence of spironolactone dosage on testosterone suppression (Liang et al., 2018; SoRelle et al., 2019), and inferior testosterone suppression with estradiol plus spironolactone compared to estradiol plus CPA or a GnRH agonist in transfeminine people (Angus et al., 2019; Sofer et al., 2020; Burinkul et al., 2021). Conversely, some studies have found adequate or near-adequate testosterone suppression with estradiol plus spironolactone in most or almost all transfeminine people (Deutsch, Bhakri, & Kubicek, 2015; Angus et al., 2019; SoRelle et al., 2019; Cirrincione et al., 2021; Pappas et al., 2021), and some studies have found indications of greater testosterone suppression with spironolactone versus without spironolactone (Angus et al., 2019; Cirrincione et al., 2021). On the other hand, some studies using estradiol alone without any antiandrogen at physiological estradiol levels (<200 pg/mL) have reported adequate testosterone suppression similarly to the preceding estradiol plus spironolactone studies (Reardon et al., 2013; Spratt et al., 2014; Cirrincione et al., 2021). One study was confounded by the concomitant use of MPA, which is known to suppress testosterone levels on its own, and hence reliable conclusions cannot not be drawn from this study (Prior, Vigna, & Watson, 1989; Prior et al., 1986). Indeed, it is notable that this study found lower mean testosterone levels with estrogen and spironolactone than any other study did. A couple of studies found that testosterone levels progressively decline with time (particularly over the first 12 months) with estradiol plus spironolactone in most transfeminine people (Liang et al., 2018; Sofer et al., 2020). Whether the decreases in testosterone levels with time were more related to estradiol or to spironolactone is unclear, though estradiol seems more likely (e.g., Wiki).
Taken together, the findings of available studies on estradiol plus spironolactone and testosterone suppression in transfeminine people are highly variable and mixed, although overall more studies support spironolactone having poor or no testosterone-suppressing effectiveness. The reasons underlying the differences in findings on testosterone suppression between studies are unclear, but contributing factors may include varying estradiol doses, routes, and levels, durations of hormone therapy, differing laboratory assays of testosterone levels, and other differences in study methodologies, as well as limitations in study and evidence quality. In any case, the conflicting nature of the findings is in major contrast to the almost invariably strong to maximal testosterone suppression in studies of estradiol plus CPA and estradiol plus GnRH agonists in transfeminine people.
Spironolactone, Androgen Receptor Antagonism, and Clinical Antiandrogenic Effectiveness
The clinical antiandrogenic effectiveness of spironolactone in cisgender women with androgen-dependent skin and hair conditions, like acne, hirsutism, and scalp hair loss, is well-established (Brown et al., 2009; van Zuuren & Fedorowicz, 2016; Layton et al., 2017; Barrionuevo et al., 2018; James, Jamerson, & Aguh, 2022; Wang et al., 2023). Conversely, the clinical antiandrogenic efficacy of spironolactone in transfeminine people has been very limitedly assessed to date and is largely unknown (Angus et al., 2021). Spironolactone does not appear to be very effective for decreasing testosterone levels in either cisgender women or transfeminine people based on the findings of the present review. However, spironolactone is a competitive antagonist of the androgen receptor in addition to its actions a weak androgen synthesis inhibitor, and hence it also directly blocks androgens from mediating their effects in the body (Loriaux et al., 1976; McMullen & Van Herle, 1993). Based on studies in populations besides transfeminine people, for instance cisgender women (discussed above) and cisgender boys with gonadotropin-independent precocious puberty (e.g., Holland, 1991), in which spironolactone has not decreased testosterone levels but has nonetheless been effective as an antiandrogen, the androgen receptor blockade of spironolactone is likely to be its main mechanism of action as an antiandrogen and may account for most or all of its therapeutic antiandrogenic effectiveness.
However, while spironolactone is clearly effective as an androgen receptor antagonist, it appears to be a relatively weak androgen receptor blocker at typical doses used in cisgender women and transfeminine people. Numerous publications in the literature describe spironolactone as being only a weak androgen receptor antagonist (Wiki; Wiki). In relation to this, animal studies have found that spironolactone is a far less potent androgen receptor antagonist than other antiandrogens like CPA, flutamide, and bicalutamide (Bonne & Raynaud, 1974; Hecker, Hasan, & Neumann, 1980; Sivelle, Underwood, & Jelly, 1982; Weissmann et al., 1985; Labrie et al., 1987; Snyder, Winneker, & Batzold, 1989 [Table]; Yamasaki et al., 2004 [Graph]). Moreover, in cisgender women, the population in which spironolactone is most widely used as an antiandrogen, testosterone levels are relatively low, on average about 20-fold lower than in cisgender men (around 30 ng/dL on average compared to about 600 ng/dL on average, respectively) (Aly, 2018). However, many cisgender women with androgen-dependent conditions have PCOS, which is associated with limitedly elevated testosterone levels (e.g., perhaps around 60 ng/dL on average) (Aly, 2018). The typical therapeutic dose range of spironolactone in cisgender women with androgen-dependent conditions is 50 to 200 mg/day, in which its effectiveness may be assumed to be dose-dependent, and this is roughly the same general dosage range used in transfeminine people (though up to 300–400 mg/day may be used and are allowed for by guidelines) (Aly, 2018; Aly, 2020).
A relatively small amount of dose-ranging data on spironolactone in cisgender women with androgen-dependent conditions exists, but in any case substantiates its dose-dependent effectiveness across its clinically used dose range (partially reviewed in Hammerstein (1990) and Shaw (1996)). One study compared spironolactone at doses of 50 to 200 mg/day with placebo for treatment of acne in cisgender women and reported progressive increases in effectiveness with spironolactone up to the 200 mg/day dosage (Goodfellow et al., 1984). Similarly, another study found that progressively increasing the dosage of spironolactone from 100 mg/day, to 150 mg/day, and up to 200 mg/day, resulted in increased effectiveness in the treatment of acne in cisgender women (Charny, Choi, & James, 2017). Spironolactone has been reported to be effective in the treatment of hirsutism in cisgender women at a dosage of as low as 50 mg/day (Diamanti-Kandarakis, Tolis, & Duleba, 1995). However, even a dosage of 100 mg/day did not appear to be maximally effective for hirsutism in a study that compared different doses of spironolactone; effectiveness was near-significantly greater at a dosage of 200 mg/day relative to a dosage of 100 mg/day (30% ± 3% and 19% ± 8% (mean ± SEM) reduction in hair shaft diameter, respectively; p = 0.07) (Lobo et al., 1985). Levels of free testosterone in this study were unchanged, suggesting that the effects of spironolactone were purely due to androgen receptor blockade. Finally, a 2022 systematic review of spironolactone for treatment of androgen-related scalp hair loss in cisgender women reported that the drug was “largely ineffective” at doses of less than 100 mg/day, whereas doses of 100 to 200 mg/day were effective (James, Jamerson, & Aguh, 2022).
Aside from dose-ranging studies, the antiandrogenic efficacy of spironolactone can be evaluated by comparing it to more potent antiandrogenic regimens. A study found that spironolactone 100 mg/day was significantly inferior to flutamide, a substantially more potent androgen receptor antagonist, in improving androgen-dependent skin and hair symptoms in cisgender women (Cusan et al., 1994). However, in other studies, there were no significant differences between spironolactone 100 mg/day and flutamide for hirsutism (Erenus et al., 1994; Moghetti et al., 2000; Inal, Yildirim, & Taner, 2005; Karakurt et al., 2008). Spironolactone and flutamide were variably taken together with an ethinylestradiol-containing combined birth control pill in these studies, which is likely to have limited detection of differences in effectiveness. This is because these birth control pills considerably suppress total and free testosterone levels and hence have substantial antiandrogenic effects themselves (Zimmerman et al., 2014; Amiri et al., 2018). In a biochemical study, spironolactone 100 mg/day was numerically inferior to flutamide in reducing levels of prostate-specific antigen (PSA) in cisgender women (Negri et al., 2000). This is notable as PSA is a systemic biomarker of androgen action (Negri et al., 2000). However, the study had small sample sizes, and the differences between groups were not statistically significant (Negri et al., 2000). A case report of a cisgender woman with female pattern hair loss and normal androgen levels found that treatment with spironolactone 200 mg/day for 5 years failed to improve or halt progression of her hair loss, in spite of almost complete loss of secondary sexual hair, but switching to flutamide resulted in a considerable improvement in hair loss after 12 months (Yazdabadi & Sinclair, 2011 [Figure]). Besides comparison with flutamide, a study found that spironolactone 100 mg/day was inferior to spironolactone 100 mg/day plus finasteride, a 5α-reductase inhibitor and hence functional antiandrogen, for hirsutism in cisgender women (–36.6% vs. –51.3% in scores; p < 0.005) (Unlühizarci et al., 2002; Keleştimur et al., 2004).
The preceding findings suggest that the clinical antiandrogenic effectiveness of spironolactone in cisgender women is not maximal at a dosage of below at least 200 mg/day despite the relatively low testosterone levels in these individuals. Put another way, spironolactone at typical doses seems best-suited for blocking female-range levels of testosterone. As many transfeminine people do not achieve female-range testosterone levels with estradiol plus spironolactone therapy, and in fact often have testosterone levels well above the normal female range or even in the male range, spironolactone may not be fully effective as an antiandrogen at the typical doses used in transfeminine hormone therapy. Higher doses of spironolactone, like 300 to 400 mg/day, may be to some degree more effective.
Summary, Discussion, and Conclusions
Numerous studies have assessed the influence of spironolactone on testosterone levels in cisgender men, cisgender women, and transfeminine people. Although the quality of these studies has often been limited, the studies have revealed highly inconsistent influences of spironolactone on testosterone levels in these populations, with many studies finding no changes, some studies finding decreases, and a small number of studies finding increases. The findings of studies of spironolactone and testosterone levels are in notable contrast to those of studies with estrogens, progestogens like CPA, and GnRH agonists, which consistently show substantial decreases in testosterone levels. This has been the case even in studies of similarly low quality to those of some of the included spironolactone studies (e.g., many of those in cisgender men). The fact that in the available studies testosterone levels with spironolactone have usually been unchanged, but have sometimes been decreased and have rarely been decreased, seems to suggest that spironolactone may be a clinically significant inhibitor of steroid hormone synthesis, but that it is only a weakly efficacious one, and that its effects may be variable depending on the individual and other clinical circumstances. In any case, the conflicting findings warrant more research with higher-quality study designs, particularly RCTs that have with spironolactone versus without comparison groups.
The notion that spironolactone decreases testosterone levels in transfeminine people, and the use of spironolactone in transfeminine hormone therapy in general, appear to have originated from the papers on spironolactone in transfeminine people published by Dr. Jerilynn Prior and colleagues in the 1980s (Prior, Vigna, & Watson, 1989; Prior et al., 1986). In their study, transfeminine people who were either already on high-dose estrogen therapy with inadequate testosterone suppression or had not yet started hormone therapy were put on physiological-dose estrogen therapy in combination with 200 to 600 mg/day spironolactone. Cyclic or continuous administration of the progestogen MPA at an oral dose of 10 mg/day was also given to all of the individuals. The authors reported that despite the lower estrogen dosage, testosterone levels decreased, from 169 ng/dL to 87 ng/dL (–49%) in those who had already been on hormone therapy and to 49 ng/dL in those who were pre-hormone therapy. Prior and her colleagues concluded that spironolactone helps to decrease testosterone levels in transfeminine people and that it can be used as a safer alternative to high doses of estrogen for this purpose.
However, the concomitant use of MPA in the study is a major confounding factor in terms of their results. This is because MPA is a progestogen, and progestogens, like estrogens, are antigonadotropins which are able to robustly suppress testosterone levels on their own (Aly, 2018; Aly, 2019). Indeed, MPA alone has been shown to dose-dependently lower testosterone levels in cisgender men (Wiki), and at a dosage of 10 mg/day, has been shown to considerably suppress testosterone levels in transfeminine people when added to estradiol and spironolactone therapy (Jain, Kwan, & Forcier, 2019). Hence, MPA may have been, and likely was, responsible for the decreases in testosterone levels seen in the study, rather than spironolactone. This point was also notably raised by other researchers, who were unable to replicate Prior and colleagues’ results on spironolactone and testosterone levels in transfeminine people (Leinung et al., 2018). Strangely, Prior and colleagues concluded that spironolactone was responsible for the decreased testosterone levels in their study even though they noted in their papers that MPA was also given to help suppress testosterone levels (as well as to help improve breast development). The work of Prior and colleagues likely resulted in the prominent and long-standing, but poorly supported, notion that spironolactone decreases testosterone levels in transfeminine people. Subsequent studies assessing the hypothesis that spironolactone decreases testosterone levels in transfeminine people were not published until 25 years after Prior and colleagues’ studies, with several of these studies, though not all of them, failing to replicate the earlier findings of Prior and colleagues.
Many people do not realize the capacity of estradiol to substantially and even completely suppress testosterone, and many mistakenly assume that it is the antiandrogen—which is often spironolactone—that is mostly or fully responsible for the decrease in testosterone levels seen with estradiol and antiandrogen therapy in transfeminine people. It is certainly true that antiandrogens like CPA and GnRH agonists play an important role in testosterone suppression in transfeminine people. However, as evidenced by the present review of studies of testosterone suppression with spironolactone, it is not necessarily always the case that the antiandrogen plays a major role—or potentially even any role—in reducing testosterone levels. This is notably also not the case with certain other antiandrogens besides spironolactone, for instance pure androgen receptor antagonists like bicalutamide, which likewise do not decrease testosterone levels but instead can actually increase them (Aly, 2019; Wiki). Clinicians and transfeminine people attributing observations of testosterone decreases to spironolactone rather than to estradiol with estradiol and spironolactone therapy may also have played a role in the perception that spironolactone considerably decreases testosterone levels in transfeminine people.
Due to its relatively weak strength as an androgen receptor antagonist and its limited efficacy in lowering testosterone levels, spironolactone is likely to be a limitedly effective antiandrogen in transfeminine people. Additionally, spironolactone is likely to be less effective than other antiandrogenic approaches used in transfeminine hormone therapy which either more robustly block androgens or more substantially reduce testosterone levels, for instance CPA, other progestogens (e.g., MPA, non-oral progesterone), GnRH agonists (and antagonists), bicalutamide, and high-dose parenteral estradiol monotherapy. These approaches can be used in transfeminine people instead of or in addition to spironolactone, or could be considered when testosterone suppression is inadequate with estradiol and spironolactone.
More studies are needed to evaluate the influence of spironolactone on testosterone levels, especially RCTs that compare estradiol alone versus estradiol plus spironolactone in transfeminine people. More research is also needed to clarify why some studies find highly inadequate testosterone suppression with estradiol alone or estradiol plus spironolactone while other studies find excellent or satisfactory testosterone suppression with these regimens. In any case, available data overall suggest that spironolactone does not consistently suppress testosterone levels, and that estradiol plus spironolactone produces inadequate testosterone suppression in many transfeminine people. Moreover, available data suggest that spironolactone is a relatively weak androgen receptor antagonist at the typical clinical doses used in cisgender women and transfeminine people, and is able to block only relatively low or female-range testosterone levels. Hence, spironolactone may not be fully effective in blocking the testosterone it fails to suppress, and may be particularly unsuitable for transfeminine people with testosterone levels that are well above the normal female range. In any case, more research is similarly needed to assess the androgen receptor antagonism and clinical antiandrogenic effectiveness of spironolactone.
Updates
Update 1: Spironolactone for Adult Female Acne (SAFA) Trial
A large new phase 3 RCT, the Spironolactone for Adult Female Acne (SAFA) trial, was published in May 2023 and assessed the effectiveness of spironolactone in the treatment of acne in cisgender women:
Santer, M., Lawrence, M., Renz, S., Eminton, Z., Stuart, B., Sach, T. H., Pyne, S., Ridd, M. J., Francis, N., Soulsby, I., Thomas, K., Permyakova, N., Little, P., Muller, I., Nuttall, J., Griffiths, G., Thomas, K. S., & Layton, A. M. (2023). Effectiveness of spironolactone for women with acne vulgaris (SAFA) in England and Wales: pragmatic, multicentre, phase 3, double blind, randomised controlled trial. BMJ, 381, e074349. [DOI:10.1136/bmj-2022-074349]
The trial included a total of 342 women, including 176 treated with spironolactone and 166 in the placebo control group. The dose of spironolactone employed was 50 mg/day for the first 6 weeks and then 100 mg/day thereafter. The trial was 24 weeks (5.5 months) in duration. Women who might become pregnant were required to use a hormonal or barrier method of contraception.
Spironolactone significantly outperformed placebo in terms of improvement in mean Acne-QoL symptom scores (higher is better). Significant improvement was apparent within 12 weeks of treatment (+45% in scores with spironolactone, +38% with placebo) and was highest at 24 weeks (+61% in scores with spironolactone, +35% with placebo). There was no difference in the rates of women who reported improvement in acne scores at 12 weeks (72% with spironolactone, 68% with placebo), but there was a significant difference at 24 weeks (82% with spironolactone, 63% with placebo). In terms of the Investigator’s Global Assessment (IGA), treatment success at 12 weeks was 19% with spironolactone and 6% with placebo. Rates of hormonal contraceptive use in the spironolactone and placebo groups were not reported. Testosterone levels were also not reported. A small subset of the women had PCOS (15% in the spironolactone group, 23% in the placebo group).
Adverse effects occurred only slightly more often with spironolactone than with placebo (64% vs. 51%, p = 0.01). The only side effect that occurred significantly more often with spironolactone than with placebo was headache (20% vs. 12%; p = 0.02). However, a few other side effects trended towards occurring significantly more frequently with spironolactone than with placebo: “other” (17% vs. 11%; p = 0.06), dizziness/vertigo/lightheadness (19% vs. 12%; p = 0.07), vomiting/being sick (2% vs. 1%; p = 0.16), and polyuria (urinary frequency) (31% vs. 25%; p = 0.18). Rates of other potentially relevant side effects, like abdominal pain, breast enlargement, breast tenderness, drowsiness/sleepiness, fatigue/tiredness, menstrual irregularity, and reduced libido, were all not different between spironolactone and placebo. There were no serious adverse reactions in the trial. Rates of compliance were similar between the spironolactone and placebo groups, suggesting that spironolactone was well-tolerated.
This trial is the largest and most rigorous RCT of spironolactone in the treatment of androgen-dependent skin and hair conditions in cisgender women that has been conducted to date. Although spironolactone was found to be effective in this study and was about twice as effective as placebo in terms of Acne-QoL symptom scores and three times as effective as placebo in terms of IGA treatment success rates, the effectiveness of spironolactone was seemingly less than in previous clinical studies of spironolactone for acne. This may be related to the relatively low doses of spironolactone used in this study (50–100 mg/day), to the more rigorous and less-risk-of-bias design of the study (large phase 3 RCT), to a possibly too-short treatment duration (24 weeks/5.5 months), and to concomitant hormonal contraceptive use possibly blunting the degree of potential improvement. The latter is relevant as hormonal contraceptives containing ethinylestradiol provide a considerable improvement in acne via functional antiandrogenic effects all on their own. A final possibility however is that spironolactone is simply a less effective antiandrogen even in cisgender women than has been previously thought. On the other hand, similarly to findings in previous clinical studies, spironolactone was well-tolerated and produced few side effects.
Update 2: New Spironolactone and Testosterone Suppression Studies
The following new studies have additionally assessed and found inadequate testosterone suppression in transfeminine people treated with estradiol and spironolactone:
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (November 2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (November 2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Miro, E., Rizzone, K., Ho, T., Mark, B., Sullivan, E., & Cushman, D. (2024). 2024 AMSSM Research Podium Presentations: Testosterone Levels Among Transgender Women on Gender-affirming Hormone Therapy. Clinical Journal of Sports Medicine, 34(2), 152–152. [DOI:10.1097/JSM.0000000000001212]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery JCG, Zajac, J. D., & Cheung, A. S. (2025). Effect of Spironolactone and Cyproterone Acetate on Breast Growth in Transgender People: A Randomized Clinical Trial. The Journal of Clinical Endocrinology and Metabolism, 110(6), e1874–e1884. [DOI:10.1210/clinem/dgae650]
Angus et al. (2023/2025) and Yang et al. (2024) compared estradiol plus spironolactone to estradiol plus CPA and are described in-depth in a section of a different article located here. Yang et al. (2024) found that in addition to spironolactone resulting in much less testosterone suppression than CPA, it was also less effective than CPA as an antiandrogen on multiple clinical measures of demasculinization.
Update 3: Bonadonna et al. (2025)
In August 2025, the following conference abstract was published online:
Bonadonna, S., Amer, M., Foletti, F., Federici, S., Persani, L., Bonomi, M. (2025). Evaluation of Antiandrogen Therapy Effectiveness in Transgender individuals Assigned Male At Birth (AMAB). EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
It was an abstract for a retrospective observational study of spironolactone versus CPA, presumably in combination with estrogen, in 149 transfeminine people. The study found that testosterone and gonadotropin levels were significantly higher with spironolactone than with CPA. In addition, it found that spironolactone was associated with less suppression of libido and spontaneous erections than CPA. Conversely, there was no difference in waist–hip ratio between the groups. The authors concluded that spironolactone appears to be less effective than CPA as an antiandrogen in transfeminine people. The full study may be published in a journal article at some point in the future.
References
Abshagen, U., Spörl, S., Schöneshöfer, M., L’age, M., & Oelkers, W. (1978). Interference of spironolactone therapy with adrenal steroid metabolism in secondary hyperaldosteronism. [Zum Einfluß einer Spironolaktonbehandlung auf den adrenalen Steroidstoffwechsel bei sekundärem Hyperaldosteronismus.] Klinische Wochenschrift, 56(7), 341–349. [DOI:10.1007/bf01477394]
Aizawa, H., & Niimura, M. (1992). Oral Spironolactone Therapy in Male Patients with Rosacea. The Journal of Dermatology, 19(5), 293–297. [DOI:10.1111/j.1346-8138.1992.tb03227.x]
Allen, A. N., Jiao, R., Day, P., Pagels, P., Gimpel, N., & SoRelle, J. A. (2020). Dynamic Impact of Hormone Therapy on Laboratory Values in Transgender Patients over Time. The Journal of Applied Laboratory Medicine, 6(1), 27–40. [DOI:10.1093/jalm/jfaa192]
Almalki, H. H., Alshibani, T. M., Alhifany, A. A., & Almohammed, O. A. (2020). Comparative efficacy of statins, metformin, spironolactone and combined oral contraceptives in reducing testosterone levels in women with polycystic ovary syndrome: a network meta-analysis of randomized clinical trials. BMC Women’s Health, 20(1), 68. [DOI:10.1186/s12905-020-00919-5]
Amiri, M., Kabir, A., Nahidi, F., Shekofteh, M., & Ramezani Tehrani, F. (2018). Effects of combined oral contraceptives on the clinical and biochemical parameters of hyperandrogenism in patients with polycystic ovary syndrome: a systematic review and meta-analysis. The European Journal of Contraception & Reproductive Health Care, 23(1), 64–77. [DOI:10.1080/13625187.2018.1435779]
Angus, L., Leemaqz, S., Ooi, O., Cundill, P., Silberstein, N., Locke, P., Zajac, J. D., & Cheung, A. S. (2019). Cyproterone acetate or spironolactone in lowering testosterone concentrations for transgender individuals receiving oestradiol therapy. Endocrine Connections, 8(7), 935–940. [DOI:10.1530/ec-19-0272]
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2021). A systematic review of antiandrogens and feminization in transgender women. Clinical Endocrinology, 94(5), 743–752. [DOI:10.1111/cen.14329]
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery JCG, Zajac, J. D., & Cheung, A. S. (2025). Effect of Spironolactone and Cyproterone Acetate on Breast Growth in Transgender People: A Randomized Clinical Trial. The Journal of Clinical Endocrinology and Metabolism, 110(6), e1874–e1884. [DOI:10.1210/clinem/dgae650]
Baba, S. (1977). Antiandrogenic effect of spironolactone. The Japanese Journal of Urology, 68(12), 1184–1192. [DOI:10.5980/jpnjurol1928.68.12_1184]
Baba, S., Murai, M., Jitsukawa, S., Hata, M., & Tazaki, H. (1978). Antiandrogenic Effects of Spironolactone: Hormonal and Ultrastructural Studies in Dogs and Men. Journal of Urology, 119(3), 375–380. [DOI:10.1016/s0022-5347(17)57495-9]
Bakker, A. (2021). Een halve eeuw transgenderzorg aan de VU [Half a Century of Transgender Care at the VU]. Amsterdam: Boom. [Google Books] [WorldCat] [URL]
Barrionuevo, P., Nabhan, M., Altayar, O., Wang, Z., Erwin, P. J., Asi, N., Martin, K. A., & Murad, M. H. (2018). Treatment Options for Hirsutism: A Systematic Review and Network Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism, 103(4), 1258–1264. [DOI:10.1210/jc.2017-02052]
Bonadonna, S., Amer, M., Foletti, F., Federici, S., Persani, L., Bonomi, M. (2025). Evaluation of Antiandrogen Therapy Effectiveness in Transgender individuals Assigned Male At Birth (AMAB). EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
Bonne, C., & Raynaud, J. (1974). Mode of spironolactone anti-androgenic action: Inhibition of androstanolone binding to rat prostate androgen receptor. Molecular and Cellular Endocrinology, 2(1), 59–67. [DOI:10.1016/0303-7207(74)90012-4]
Bonzagni, A. F. (2014). Understanding the effects of long-term hormone therapy in transgender individuals being provided care at Boston Medical Center Endocrinology Clinic: a quality assurance project. (Doctoral dissertation, Boston University.) [Google Scholar] [URL] [PDF]
Bradstreet, J. J., Smith, S., Granpeesheh, D., El-Dahr, J. M., & Rossignol, D. (2007). Spironolactone might be a desirable immunologic and hormonal intervention in autism spectrum disorders. Medical Hypotheses, 68(5), 979–987. [DOI:10.1016/j.mehy.2006.10.015]
Brown, J., Farquhar, C., Lee, O., Toomath, R., & Jepson, R. G. (2003). Spironolactone versus placebo or in combination with steroids for hirsutism and/or acne. Cochrane Database of Systematic Reviews, 2003(4), CD000194. [DOI:10.1002/14651858.cd000194]
Brown, J., Farquhar, C., Lee, O., Toomath, R., & Jepson, R. G. (2009). Spironolactone versus placebo or in combination with steroids for hirsutism and/or acne. Cochrane Database of Systematic Reviews, 2009(2), CD000194. [DOI:10.1002/14651858.cd000194.pub2]
Burinkul, S., Panyakhamlerd, K., Suwan, A., Tuntiviriyapun, P., & Wainipitapong, S. (2021). Anti-Androgenic Effects Comparison Between Cyproterone Acetate and Spironolactone in Transgender Women: A Randomized Controlled Trial. The Journal of Sexual Medicine, 18(7), 1299–1307. [DOI:10.1016/j.jsxm.2021.05.003]
Callan, A. W. (1988). Spironolactone therapy in hirsutism and acne. Australasian Journal of Dermatology, 29(3), 135–139. [DOI:10.1111/j.1440-0960.1988.tb00385.x]
Caminos-Torres, R., MA, L., & Snyder, P. J. (1977). Gynecomastia and Semen Abnormalities Induced by Spironolactone in Normal Men. The Journal of Clinical Endocrinology & Metabolism, 45(2), 255–260. [DOI:10.1210/jcem-45-2-255]
Charny, J., Choi, J., & James, W. (2017). Spironolactone for the treatment of acne in women, a retrospective study of 110 patients. International Journal of Women’s Dermatology, 3(2), 111–115. [DOI:10.1016/j.ijwd.2016.12.002]
Cheung, A. S., Ooi, O., Davidoff, D., Leemaqz, S. Y., Cundill, P., Silberstein, N., Bretherton, I., Grossmann, M., & Zajac, J. D. (2018). Cyproterone vs spironolactone as anti-androgen therapy for transgender females receiving oestradiol therapy. Clinical Endocrinology, 89(S1) [Abstracts of the Endocrine Society of Australia’s Annual Scientific Meeting 2017], 14–14 (abstract no. 62). [Google Scholar] [DOI:10.1111/cen.13727] [URL 1] [URL 2] [PDF]
Cirrincione, L. R., Winston McPherson, G., Rongitsch, J., Sadilkova, K., Drees, J. C., Krasowski, M. D., Dickerson, J. A., & Greene, D. N. (2021). Sublingual Estradiol Is Associated with Higher Estrone Concentrations than Transdermal or Injectable Preparations in Transgender Women and Gender Nonbinary Adults. LGBT Health, 8(2), 125–132. [DOI:10.1089/lgbt.2020.0249]
Corvol, P., Mahoudeau, J. A., Valcke, J. C., Ménard, J., & Bricaire, H. (1976). Effets sexuels secondaires des spirolactones. Mécanismes possibles de l’action antiandrogène. [Sexual side-effects of spironolactones. Possible mechanisms of their anti-androgen action]. La Nouvelle Presse Medicale, 5(11), 691–694. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF]
Cusan, L., Dupont, A., Gomez, J., Tremblay, R. R., & Labrie, F. (1994). Comparison of flutamide and spironolactone in the treatment of hirsutism: a randomized controlled trial. Fertility and Sterility, 61(2), 281–287. [DOI:10.1016/s0015-0282(16)56518-2]
Deutsch, M. B., Bhakri, V., & Kubicek, K. (2015). Effects of Cross-Sex Hormone Treatment on Transgender Women and Men. Obstetrics & Gynecology, 125(3), 605–610. [DOI:10.1097/aog.0000000000000692]
Diamanti-Kandarakis, E., Tolis, G., & Duleba, A. J. (1995). Androgens and Therapeutic Aspects of Antiandrogens in Women. Journal of the Society for Gynecologic Investigation, 2(4), 577–592. [DOI:10.1177/107155769500200401]
Dymling, J., Nilsson, K. O., & Hökfelt, B. (1972). The effect of Soldactona® (canrenoate potassium) on plasma testosterone and androstenedione and urinary 17-ketosteroids and 17-hydroxycorticosteroids. Acta Endocrinologica, 70(1), 104–112. [DOI:10.1530/acta.0.0700104]
Dymling, J. F., & Hökfelt, B. (1973). Reduced plasma testosterone following spirolactone in man. Acta Endocrinologica, 73(Suppl 177) [9th Acta Endocrinologica Congress], 53–53 (abstract no. 53). [Google Scholar] [PDF 1] [PDF 2]
Dymling, J. F. (1978). The effect of spirolactone and spironolactone on plasma testosterone in man. In Addison, G. M., Asmussen, N. W., Corvol, P., Kloppernborg, P. W. C., Norman, N., Schroder, R., & Robertson, J. I. C. (Eds.). Aldosterone Antagonists in Clinical Medicine: Proceedings of the Searle Symposium, Nice, April 13–15, 1978 (pp. 297–302). Amsterdam: Excerpta Medica. [Google Scholar] [Google Books] [WorldCat] [PDF 1] [PDF 2]
Erbler, H. C. (1974). Suppression by the spironolactone metabolite canrenone of plasma testosterone in man. Naunyn-Schmiedeberg’s Archives of Pharmacology, 285(4), 403–406. [DOI:10.1007/bf00501468]
Erenus, M., Gürbüz, O., Durmuşoğlu, F., Demirçay, Z., & Pekin, S. (1994). Comparison of the efficacy of spironolactone versus flutamide in the treatment of hirsutism. Fertility and Sterility, 61(4), 613–616. [DOI:10.1016/s0015-0282(16)56634-5]
Ganie, M. A., Khurana, M. L., Eunice, M., Gulati, M., Dwivedi, S. N., & Ammini, A. C. (2004). Comparison of Efficacy of Spironolactone with Metformin in the Management of Polycystic Ovary Syndrome: An Open-Labeled Study. The Journal of Clinical Endocrinology & Metabolism, 89(6), 2756–2762. [DOI:10.1210/jc.2003-031780]
Ganie, M. A., Khurana, M. L., Nisar, S., Shah, P. A., Shah, Z. A., Kulshrestha, B., Gupta, N., Zargar, M. A., Wani, T. A., Mudasir, S., Mir, F. A., & Taing, S. (2013). Improved Efficacy of Low-Dose Spironolactone and Metformin Combination Than Either Drug Alone in the Management of Women With Polycystic Ovary Syndrome (PCOS): A Six-Month, Open-Label Randomized Study. The Journal of Clinical Endocrinology & Metabolism, 98(9), 3599–3607. [DOI:10.1210/jc.2013-1040]
Goodfellow, A., Alaghband-Zadeh, J., Carter, G., Cream, J., Holland, S., Scully, J., & Wise, P. (1984). Oral spironolactone improves acne vulgaris and reduces sebum excretion. British Journal of Dermatology, 111(2), 209–214. [DOI:10.1111/j.1365-2133.1984.tb04045.x]
Gooren, L. J., Veen, E. A., Kessel, H., Harmsen-Louman, W., & Wiegel, A. R. (1984). Prolactin secretion in the human male is increased by endogenous oestrogens and decreased by exogenous/endogenous androgens. International Journal of Andrology, 7(1), 53–60. [DOI:10.1111/j.1365-2605.1984.tb00759.x]
Gooren, L., Veen, E., Kessel, H., Harmsen-Louman, W., & Wiegel, A. (1984). Androgens in the Feedback Regulation of Gonadotropin Secretion in Men: Effects of Administration of Dihydrotestosterone to Eugonadal and Agonadal Subjects and of Spironolactone to Eugonadal Subjects. Andrologia, 16(4), 289–298. [DOI:10.1111/j.1439-0272.1984.tb00286.x]
Gooren, L. J. G. (1999). Hormonal Sex Reassignment. International Journal of Transgenderism, 3(3), 1–7. [Google Scholar] [URL 1] [URL 2]
Hammerstein, J. (1990). Antiandrogens: Clinical Aspects. In Orfanos, C. E., & Happle, R. (Eds.). Hair and Hair Diseases (pp. 827–886). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-74612-3_35]
Handelsman, D. J., Strasser, S., McDonald, J. A., Conway, A. J., & McCaughan, G. W. (1995). Hypothalamic-pituitary-testicular function in end-stage non-alcoholic liver disease before and after liver transplantation. Clinical Endocrinology, 43(3), 331–337. [DOI:10.1111/j.1365-2265.1995.tb02040.x]
Hecker, A., Hasan, S. H., & Neumann, F. (1980). Disturbances in sexual differentiation of rat foetuses following spironolactone treatment. Acta Endocrinologica, 95(4), 540–545. [DOI:10.1530/acta.0.0950540]
Holland, F. J. (1991). Gonadotropin-lndependent Precocious Puberty. Endocrinology and Metabolism Clinics of North America, 20(1), 191–210. [DOI:10.1016/s0889-8529(18)30288-3]
Horth, C., Lobo, P., Shelton, J., Asbury, M., Clarke, J., & Venning, G. (1977). Effects of spironolaetone on the plasma binding and unbound levels of testosterone and oestradiol in healthy men. Journal of Steroid Biochemistry, 8(12), xx–xx. [DOI:10.1016/0022-4731(77)90178-9]
Horth, C. E., Lobo, P. J., Shelton, J. R., Asbury, M. J., Clarke, J. M., & Venning, G. R. (1979). Effects of spironolactone on the plasma binding and unbound levels of testosterone and oestradiol in healthy men. In Klopper, A., Lerner, L., van der Molen, H. J., & Sciarra, F. (Eds.). Research on Steroids: Proceedings of the International Study Group for Steroid Hormones, Volume 8 / Research on Steroids Transactions of the Eighth Meeting of the International Study Group for Steroid Hormones (Proceedings of the Serono Symposia, Volume 21) (pp. 313–315). London/New York/San Francisco: Academic Press. [Google Scholar]
Huffman, D. H., Kampmann, J. P., Hignite, C. E., & Azarnoff, D. L. (1978). Gynecomastia induced in normal males by spironolactone. Clinical Pharmacology & Therapeutics, 24(4), 465–473. [DOI:10.1002/cpt1978244465]
Inal, M. M., Yildirim, Y., & Taner, C. E. (2005). Comparison of the clinical efficacy of flutamide and spironolactone plus Diane 35 in the treatment of idiopathic hirsutism: a randomized controlled study. Fertility and Sterility, 84(6), 1693–1697. [DOI:10.1016/j.fertnstert.2005.05.051]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
James, J. F., Jamerson, T. A., & Aguh, C. (2022). Efficacy and safety profile of oral spironolactone use for androgenic alopecia: A systematic review. Journal of the American Academy of Dermatology, 86(2), 425–429. [DOI:10.1016/j.jaad.2021.07.048]
Karakurt, F., Sahin, I., Güler, S., Demirbas, B., Culha, C., Serter, R., Aral, Y., & Bavbek, N. (2008). Comparison of the clinical efficacy of flutamide and spironolactone plus ethinyloestradiol/cyproterone acetate in the treatment of hirsutism: A randomised controlled study. Advances in Therapy, 25(4), 321–328. [DOI:10.1007/s12325-008-0039-5]
Keleştimur, F., Everest, H., Unlühizarci, K., Bayram, F., & Șahin, Y. (2004). A comparison between spironolactone and spironolactone plus finasteride in the treatment of hirsutism. European Journal of Endocrinology, 150(3), 351–354. [DOI:10.1530/eje.0.1500351]
Labrie, C., Cusan, L., Plante, M., Lapointe, S., & Labrie, F. (1987). Analysis of the androgenic activity of synthetic “progestins” currently used for the treatment of prostate cancer. Journal of Steroid Biochemistry, 28(4), 379–384. [DOI:10.1016/0022-4731(87)91054-5]
Layton, A. M., Eady, E. A., Whitehouse, H., Del Rosso, J. Q., Fedorowicz, Z., & van Zuuren, E. J. (2017). Oral Spironolactone for Acne Vulgaris in Adult Females: A Hybrid Systematic Review. American Journal of Clinical Dermatology, 18(2), 169–191. [DOI:10.1007/s40257-016-0245-x]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Liang, J. J., Jolly, D., Chan, K. J., & Safer, J. D. (2018). Testosterone Levels Achieved by Medically Treated Transgender Women in a United States Endocrinology Clinic Cohort. Endocrine Practice, 24(2), 135–142. [DOI:10.4158/ep-2017-0116]
Lobo, R. A., Shoupe, D., Serafini, P., Brinton, D., & Horton, R. (1985). The effects of two doses of spironolactone on serum androgens and anagen hair in hirsute women. Fertility and Sterility, 43(2), 200–205. [DOI:10.1016/s0015-0282(16)48373-1]
Loriaux, D. L. (1976). Spironolactone and Endocrine Dysfunction. Annals of Internal Medicine, 85(5), 630–630. [DOI:10.7326/0003-4819-85-5-630]
McMullen, G. R., & Van Herle, A. J. (1993). Hirsutism and the effectiveness of spironolactone in its management. Journal of Endocrinological Investigation, 16(11), 925–932. [DOI:10.1007/bf03348960]
Menard, R., Guenthner, T., Kon, H., & Gillette, J. (1979). Studies on the destruction of adrenal and testicular cytochrome P-450 by spironolactone. Requirement for the 7α-thio group and evidence for the loss of the heme and apoproteins of cytochrome P-450. Journal of Biological Chemistry, 254(5), 1726–1733. [DOI:10.1016/s0021-9258(17)37833-x]
Miro, E., Rizzone, K., Ho, T., Mark, B., Sullivan, E., & Cushman, D. (2024). 2024 AMSSM Research Podium Presentations: Testosterone Levels Among Transgender Women on Gender-affirming Hormone Therapy. Clinical Journal of Sports Medicine, 34(2), 152–152. [DOI:10.1097/JSM.0000000000001212]
Miyatake, A., Noma, K., Nakao, K., Morimoto, Y., & Yamamura, Y. (1978). Increased serum oestrone and oestradiol following spironolactone administration in hypertensive men. Clinical Endocrinology, 9(6), 523–533. [DOI:10.1111/j.1365-2265.1978.tb01510.x]
Moghetti, P., Tosi, F., Tosti, A., Negri, C., Misciali, C., Perrone, F., Caputo, M., Muggeo, M., & Castello, R. (2000). Comparison of Spironolactone, Flutamide, and Finasteride Efficacy in the Treatment of Hirsutism: A Randomized, Double Blind, Placebo-Controlled Trial. The Journal of Clinical Endocrinology & Metabolism, 85(1), 89–94. [DOI:10.1210/jcem.85.1.6245]
Negri, C., Tosi, F., Dorizzi, R., Fortunato, A., Spiazzi, G. G., Muggeo, M., Castello, R., & Moghetti, P. (2000). Antiandrogen Drugs Lower Serum Prostate-Specific Antigen (PSA) Levels in Hirsute Subjects: Evidence That Serum PSA Is a Marker of Androgen Action in Women. The Journal of Clinical Endocrinology & Metabolism, 85(1), 81–84. [DOI:10.1210/jcem.85.1.6230]
Pappas, I. I., Craig, W. Y., Spratt, L. V., & Spratt, D. I. (2020). Efficacy of Sex Steroid Therapy Without Progestin or GnRH Agonist for Gonadal Suppression in Adult Transgender Patients. The Journal of Clinical Endocrinology & Metabolism, 106(3), e1290–e1300. [DOI:10.1210/clinem/dgaa884]
Pappas, I. I., Craig, W., Spratt, L. V., & Spratt, D. I. (2020). Testosterone (T) and estradiol (E2) therapy alone can suppress gonadal function in transgender patients. Costas T. Lambrew Research Retreat 2020, 47. [Google Scholar] [URL] [PDF]
Pentikäinen, P. J., Pentikäinen, L. A., Huffman, D. H., & Azarnoff, D. L. (1974). The effect of spironolactone on plasma levels and excretion of testosterone and oestrogens in the urine in males. (A preliminary report). The Journal of International Medical Research, 2(6), 439–443. [Google Scholar] [PubMed] [PDF]
Prior, J. C., Vigna, Y. M., Watson, D., Diewold, P., & Robinow, O. (1986). Spironolactone in the presurgical therapy of male to female transsexuals: Philosophy and experience of the Vancouver Gender Dysphoria Clinic. Journal of Sex Information & Education Council of Canada, 1(1), 1–7. [Google Scholar] [PDF]
Prior, J. C., Vigna, Y. M., & Watson, D. (1989). Spironolactone with physiological female steroids for presurgical therapy of male-to-female transsexualism. Archives of Sexual Behavior, 18(1), 49–57. [DOI:10.1007/bf01579291]
Reardon, E., Farnell, P., Langzettel, J., & Spratt, D. (2013). Efficacy of single agent hormonal therapy in transgender patients to suppress endogenous hypothalamic-pituitary-gonadal (HPG) activity. Fertility and Sterility, 100(3 Suppl) [Scientific Program Supplement: Oral and Poster Session Abstracts 12 October 2013 - 17 October 2013], S333–S333 (abstract no. P-640). [Google Scholar] [DOI:10.1016/j.fertnstert.2013.07.914]
Rose, L. I. (1977). Pathophysiology of Spironolactone-Induced Gynecomastia. Annals of Internal Medicine, 87(4), 398–403. [DOI:10.7326/0003-4819-87-4-398]
Rozner, R. N., Freites-Martinez, A., Shapiro, J., Geer, E. B., Goldfarb, S., & Lacouture, M. E. (2018). Safety of 5α-reductase inhibitors and spironolactone in breast cancer patients receiving endocrine therapies. Breast Cancer Research and Treatment, 174(1), 15–26. [DOI:10.1007/s10549-018-4996-3]
Santen, R. J., Kulin, H. E., Loriaux, D. L., & Friend, J. (1976). Spironolactone Stimulation of Gonadotropin Secretion in Boys with Delayed Adolescence. The Journal of Clinical Endocrinology & Metabolism, 43(6), 1386–1390. [DOI:10.1210/jcem-43-6-1386]
Santer, M., Lawrence, M., Renz, S., Eminton, Z., Stuart, B., Sach, T. H., Pyne, S., Ridd, M. J., Francis, N., Soulsby, I., Thomas, K., Permyakova, N., Little, P., Muller, I., Nuttall, J., Griffiths, G., Thomas, K. S., & Layton, A. M. (2023). Effectiveness of spironolactone for women with acne vulgaris (SAFA) in England and Wales: pragmatic, multicentre, phase 3, double blind, randomised controlled trial. BMJ, 381, e074349. [DOI:10.1136/bmj-2022-074349]
Shaw, J. C. (1996). Antiandrogen and hormonal treatment of acne. Dermatologic Clinics, 14(4), 803–811. [DOI:10.1016/s0733-8635(05)70405-8]
Sivelle, P. C., Underwood, A. H., & Jelly, J. A. (1982). The effects of histamine H2 receptor antagonists on androgen action in vivo and dihydrotestosterone binding to the rat prostate androgen receptor in vitro. Biochemical Pharmacology, 31(5), 677–684. [DOI:10.1016/0006-2952(82)90449-x]
Skorodok, L. M., Savchenko, O. N., & Liss, V. L. (1983). Hypothalamo-hypophyseal-gonadal function in boys with irregular puberty. Neuroscience and Behavioral Physiology, 13(2), 141–145. [DOI:10.1007/bf01190800]
Snyder, B. W., Winneker, R. C., & Batzold, F. H. (1989). Endocrine profile of win 49596 in the rat: A novel androgen receptor antagonist. Journal of Steroid Biochemistry, 33(6), 1127–1132. [DOI:10.1016/0022-4731(89)90419-6]
Sofer, Y., Yaish, I., Yaron, M., Bach, M. Y., Stern, N., & Greenman, Y. (2020). Differential Endocrine and Metabolic Effects of Testosterone Suppressive Agents in Transgender Women. Endocrine Practice, 26(8), 883–890. [DOI:10.4158/ep-2020-0032]
SoRelle, J. A., Jiao, R., Gao, E., Veazey, J., Frame, I., Quinn, A. M., Day, P., Pagels, P., Gimpel, N., & Patel, K. (2019). Impact of Hormone Therapy on Laboratory Values in Transgender Patients. Clinical Chemistry, 65(1), 170–179. [DOI:10.1373/clinchem.2018.292730]
Spörl, S. (1978/1979). Über den Einfluß von Spironolacton (Aldactone®) auf den Stoffwechsel von Nebennierenrindenhormonen und von Testosteron bei Gesunden mit diätinduziertem Hyperaldosterismus. [On the influence of spironolactone (Aldactone®) on the metabolism of adrenocortical hormones and testosterone in healthy individuals with diet-induced hyperaldosteronism.] (Doctoral dissertation, Freie Universität, Berlin.) [Google Scholar] [Google Books] [WorldCat] [PDF]
Spratt, L. V., Reardon, E., Olshan, J. S., & Eimicke, T. (2014). Efficacy of Testosterone (T) or Estradiol (E2) Therapy without a GnRH Agonist or Progestin to Suppress Endogenous Gonadal Activity in Transsexual Patients. Endocrine Reviews, 35(Suppl 1) [Endocrine Society’s 96th Annual Meeting and Expo, June 21–24, 2014 – Chicago], ?–? (abstract no. OR42-2). [DOI:10.1093/edrv/35.supp.1] [URL]
Stewart, I., Spratt, L., Craig, W., Olshan, J., & Spratt, D. (2018). The efficacy of testosterone or estradiol therapy without a GnRH agonist or progestin to suppress endogenous gonadal activity in transgender patients. Fertility and Sterility, 110(4 Suppl) [Scientific Congress Supplement: Oral and Poster Session Abstracts 06 October 2018 - 10 October 2018], e21–e21 (abstract no. O-44). [Google Scholar] [DOI:10.1016/j.fertnstert.2018.07.075]
Stripp, B., Taylor, A. A., Bartter, F. C., Gillette, J. R., Loriaux, D. L., Easley, R., & Menard, R. H. (1975). Effect of Spironolactone on Sex Hormones in Man. The Journal of Clinical Endocrinology & Metabolism, 41(4), 777–781. [DOI:10.1210/jcem-41-4-777]
Taylor, A. A., Rollins, D., Snodgrass, W., & Bartter, F. (1976). Effect of spironolactone (S) on adrenal and gonadal steroids in man. Clinical Research, 24(3), A279–A279. [Google Scholar] [PDF]
Unlühizarci, K., Everest, H., Bayram, F., & Keleştimur, F. (2002). Comparison of spironolactone and spironolactone plus finasteride in the treatment of hirsutism. Fertility and Sterility, 78(6), 1331–1333. [DOI:10.1016/s0015-0282(02)04294-2]
Tidd, M. J., Horth, C. E., Ramsay, L. E., Shelton, J. R., & Palmer, R. F. (1978). Endocrine effects of spironolactone in man. Clinical Endocrinology, 9(5), 389–399. [DOI:10.1111/j.1365-2265.1978.tb03578.x]
van Zuuren, E., & Fedorowicz, Z. (2016). Interventions for hirsutism excluding laser and photoepilation therapy alone: abridged Cochrane systematic review including GRADE assessments. British Journal of Dermatology, 175(1), 45–61. [DOI:10.1111/bjd.14486]
Walsh, P. C., & Siiteri, P. K. (1975). Suppression of Plasma Androgens by Spironolactone in Castrated Men with Carcinoma of the Prostate. Journal of Urology, 114(2), 254–256. [DOI:10.1016/s0022-5347(17)67001-0]
Wang, C., Du, Y., Bi, L., Lin, X., Zhao, M., & Fan, W. (2023). The Efficacy and Safety of Oral and Topical Spironolactone in Androgenetic Alopecia Treatment: A Systematic Review. Clinical, Cosmetic and Investigational Dermatology, 16, 603–612. [DOI:10.2147/ccid.s398950]
Weissmann, A. (1985). Antiandrogenic Effects of Topically Applied Spironolactone on the Hamster Flank Organ. Archives of Dermatology, 121(1), 57–62. [DOI:10.1001/archderm.1985.01660010061019]
Yamasaki, K., Sawaki, M., Noda, S., Muroi, T., Takakura, S., Mitoma, H., Sakamoto, S., Nakai, M., & Yakabe, Y. (2004). Comparison of the Hershberger assay and androgen receptor binding assay of twelve chemicals. Toxicology, 195(2–3), 177–186. [DOI:10.1016/j.tox.2003.09.012]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Yazdabadi, A., & Sinclair, R. (2011). Treatment of female pattern hair loss with the androgen receptor antagonist flutamide. The Australasian Journal of Dermatology, 52(2), 132–134. [DOI:10.1111/j.1440-0960.2010.00735.x]
Zgliczynski, S., Baranowska, B., & Szymanowski, J. (1981). L’influence du spironolactone sur la concentration des gonatrophines et des hormones gonadiques dans l’hypertrophie prostatique. [The influence of spironolactone on the concentration of gonadotrophins and gonadal hormones in prostatic hypertrophy]. Journal d’Urologie, 87(9), 635–638. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF] [Translation]
Zimmerman, Y., Eijkemans, M. J., Coelingh Bennink, H. J., Blankenstein, M. A., & Fauser, B. C. (2013). The effect of combined oral contraception on testosterone levels in healthy women: a systematic review and meta-analysis. Human Reproduction Update, 20(1), 76–105. [DOI:10.1093/humupd/dmt038]
\ No newline at end of file
+A Review of Studies on Spironolactone and Testosterone Suppression in Cisgender Men, Cisgender Women, and Transfeminine People - Transfeminine Science
A Review of Studies on Spironolactone and Testosterone Suppression in Cisgender Men, Cisgender Women, and Transfeminine People
By Aly | First published December 19, 2018 | Last modified August 23, 2025
Abstract / TL;DR
Spironolactone is an antiandrogen used in transfeminine hormone therapy which is especially employed in the United States. It is widely considered to act as an androgen receptor antagonist and as an androgen synthesis inhibitor, both blocking the actions of testosterone and lowering testosterone levels in transfeminine people. A literature search was conducted to review studies assessing the influence of spironolactone on testosterone levels in cisgender men, cisgender women, and transfeminine people. The results of these studies were mixed, but in most studies spironolactone showed no apparent influence on testosterone levels. These findings suggest that spironolactone has inconsistent and limited effects on testosterone levels. Moreover, these data, as well as studies of estradiol alone, indicate that estradiol is mainly responsible for lowered testosterone levels when the combination of estradiol and spironolactone is used for hormone therapy in transfeminine people. Besides testosterone suppression, spironolactone also acts as a direct antagonist of the androgen receptor, and this importantly contributes to its antiandrogenic efficacy as well. However, studies in cisgender women suggest that spironolactone is a relatively weak androgen receptor antagonist, and is likely best-suited for blocking relatively low testosterone levels. Taken together, the antiandrogenic effectiveness of spironolactone in transfeminine people appears to be limited. Other antiandrogenic approaches may be more effective in transfeminine people, and may be considered instead or as alternatives to spironolactone in those in whom testosterone levels with estradiol plus spironolactone remain inadequately suppressed.
Introduction
Spironolactone, also known by its major brand name Aldactone, is an antiandrogen which is commonly used in transfeminine hormone therapy. It is used in combination with estrogen in transfeminine people to help reduce the effects of testosterone. Spironolactone is used in transfeminine hormone therapy particularly in the United States, where another antiandrogen, cyproterone acetate (CPA; brand name Androcur), is unavailable. Conversely, CPA is the main antiandrogen used in transfeminine people in Europe and most of the rest of the world. Another type of medication, gonadotropin-releasing hormone (GnRH) agonists, are the major antiandrogens used in certain places like the United Kingdom. The combination of estradiol with CPA or a GnRH agonist in transfeminine people consistently suppresses testosterone levels into the normal female range (<50 ng/dL or <1.8 nmol/L) (Aly, 2018; Aly, 2019). Hence, both CPA and GnRH agonists are very effective antiandrogens in transfeminine people.
Spironolactone acts as an androgen receptor antagonist, but is also known to function as an androgen synthesis inhibitor. As an example, spironolactone has been shown in preclinical research to inhibit several enzymes involved in gonadal and adrenal androgen production, including CYP17A1 (17α-hydroxylase/17,20-lyase) among others, and to substantially decrease concentrations of androgens in these studies (Loriaux et al., 1976; Callan, 1988; McMullen & Van Herle, 1993). However, the steroid synthesis inhibition of spironolactone appears to only occur at very high doses and concentrations of spironolactone (Loriaux et al., 1976; McMullen & Van Herle, 1993). For example, spironolactone is used at 10- to 20-fold smaller doses by body weight in humans than in animal studies that have demonstrated substantial steroid synthesis inhibition with the agent (McMullen & Van Herle, 1993).
A widespread notion in the transgender community, as well as in the transgender health community and in the medical literature, is that spironolactone decreases testosterone levels and that this is a major part of how it works as an antiandrogen in transfeminine people. In actuality however, the clinical evidence to support this notion appears to be limited, and available data from studies appear to be highly conflicting. The purpose of this article is to review the available clinical studies on spironolactone and testosterone levels in cisgender men, cisgender women, and transfeminine people in order to help elucidate whether and to what extent spironolactone lowers testosterone levels in humans. In addition, the role of androgen receptor blockade in the antiandrogenic effects of spironolactone is briefly reviewed.
A total of 22 studies of spironolactone and sex hormone levels in cisgender males were identified (Table 1). These studies assessed pre-treatment versus post-treatment hormone levels with spironolactone, hormone levels with spironolactone versus a comparator group, or both. Within the identified studies, testosterone levels were not significantly changed in 12 of 22 studies (55%), decreased in 4 of 22 (18%) studies, increased in 1 of 22 (4.5%) studies, and mixed or unknown (e.g. divergences in changes of total versus free testosterone levels or didn’t actually report testosterone levels) in 4 of 22 (18%) studies. Most of the studies were very small (fewer than 10 people), with several exceptions. The studies were of highly variable lengths, with some being several days and others lasting for weeks or months. Few of the studies were RCTs. Most of the studies were very old, with a majority published in the 1970s and the rest published in the 1980s and 1990s. In relation to the preceding, the quality of data was limited.
Table 1: Studies of sex hormone levels with spironolactone alone in cisgender males:
Treatment and subjects
Findings
Source(s)
100 mg/day for 2 weeks in 7 healthy men (23–34 years)
T significantly decreased and LH significantly increased. No significant change in E1, E2, or E3. No change urinary total T excretion but significantly increased urinary total E excretion (including of E1 (7.72 to 10.54 µg/24 hrs), E2 (2.60 to 3.34 ug/24 hours), E3 (7.69 to 11.75 µg/24 hrs)). Slightly but significantly decreased excretion of 17-KS in urine.
400 mg/day for 5 days in 6 healthy men (21–33 years)
Significant increase in P4 and 17α-OHP (approximately doubled) for whole duration. Small and transient increases in LH (+20%) and FSH on the 2nd but not on the 3rd or 5th days (only other days measured). No significant changes in T, E2, or PRL. E2 and PRL non-significantly increased (+56% and +34% on the 5th day, respectively).
100 or 400 mg/day spironolactone for 8 weeks in 7 orchiectomized men (46–78 years) with metastatic prostate cancer
T, A4, and DHEA significantly decreased with both doses of spironolactone and of similar magnitude between doses. Influence more apparent after 2–3 weeks of treatment.
5 mg/kg/day for 1 week (275 mg/day for a 55 kg person) in 7 boys with delayed puberty (14–16 years)
Significant increase in LH (+60%) and non-significant increase in FSH (+60%); individual responses for FSH variable. Increased P4 and 17α-OHP. T and E2 not actually reported.
Initially 400 mg/day for 12 weeks; dosage later decreased in some due to hypotension (range 150–400 mg/day) in 5 men and 5 women (3 premenopausal, 2 postmenopausal) with normal or low renin hypertension
P4 and 17α-OHP increased by 2 to 4 times compared to pre-treatment and post-treatment. T, E2, LH, FSH, PRL, and 17-KS all unchanged.
200–400 mg/day for 4–13 months (mean 7 months) in 6 men with hypertension (35–61 years; mean 47 years) vs. 10 untreated male controls with hypertension (mean age 45 years)
Significantly greater LH and E2 (30 pg/mL vs. 13 pg/mL; +130%), significantly lower T (440 ng/dL vs. 270 ng/dL; –38%), no difference in FSH. Also, significantly greater metabolic clearance rate of T, significantly greater rate of peripheral conversion (conversion ratio and transfer constant) of T into E2, non-significantly greater metabolic clearance rate of E2, no difference in blood production rate of T, and significantly greater blood production rate of E2.
200–400 mg/day (mean 330 mg/day) for 20–27 days in 5 gonadally intact men (50–76 years) with prostate cancer
P4 increased significantly from 0.25 ± 0.10 ng/mL (mean ± SD) to maximum of 1.3 ± 0.31 ng/mL by 20 days (increase of 5.2-fold or 420%). T decreased significantly from 427 ± 74.3 ng/dL to 200 ± 80.3 ng/dL (–53.2%). No significant change in E2, LH, or FSH.
200 mg/day for 21 days in 4 healthy men (26–35 years)
No change in total T or E2. Unbound T and E2 slightly but significantly increased. Thought to be due to a direct interaction of spironolactone metabolites with the plasma protein binding of T and E2. But not due to binding to SHBG as T binding to SHBG was not significantly altered.
75–150 mg/day for 12 weeks in 6 men with essential hypertension (28–64 years; mean 48 years)
E1 significantly increased. E2 small, gradual, non-significant increase. T, LH, and PRL not significantly changed. PRL responses to TRH normal/not significantly changed.
150–300 mg/day for 40 weeks in 2 men with idiopathic hyperaldosteronism (23 and 44 years)
E1 increased. E2 fluctuated. E2 increased by 10-fold in one person by 16 weeks and this was associated with gynecomastia. T, LH, and PRL not altered significantly.
200 mg/day for 10 days (n=5) vs. placebo (n=5) in 10 healthy men (18–31 years) (RCT)
Significantly greater urinary A4, urinary EC, and urinary total E excretion. Differences in T, E2, LH, and FSH as well as urinary DHEA, LH, and FSH not significant. Examination of interaction between treatment and time showed significant changes in T, LH, and urinary DHEA. Concluded that there was a transient rise in T and urine DHEA for 2–4 days followed by increase in LH and normalization of T and DHEA excretion after 4–10 days.
300 mg/day for 7 days (n=5) vs. 200 mg/day triamterene (n=5) in 10 normal young men with diet-induced hyperaldosteronism (14 days of a diet modifying electrolyte intake)
P4, 17α-OHP, unchanged. T near-but-non-significantly decreased (704.6 ± 55.5 ng/dL (mean ± SEM) to 508.4 ± 45.9 ng/dL on day 6; p < 0.10). Also assessed endogenous corticosteroids.
100 mg/day for 3 months in treatment group of 47 men (age 60–80 years) with BPH; control group of 58 healthy men without BPH (also age 60–80 years)
In spiro/BPH group, T decreased from 650 ng/dL to 290 ng/dL and DHT decreased from 450 ng/dL to 150 ng/dL. In control/non-BPH group, T was 280 ng/dL and DHT was 90 ng/dL. P4, E2, and LH increased in spiro/BPH group. FSH also assessed. The authors stated that prostate gland can be a source of androgen production, implying that BPH can produce elevated androgen levels and that spironolactone can normalize elevated androgen levels in the condition.
150 mg/m2/day for 5 days in 6 boys with irregular puberty (11–13 years)
No significant changes in T or urinary 17-KS excretion, elevated LH (by 600%—likely typo of “60%” (?)), and slightly increased FSH (from 0.75 ng/mL to 0.86 ng/mL).
25–400 mg/day (median 100 mg/day) for 12 months in 32 males (59%) of a group of 54 males (17–64 years; mean 44 years) with non-alcoholic liver disease requiring liver transplantation vs. 469 healthy male controls (mean 31 years) with normal liver function
Significantly decreased T with spironolactone in men with moderate-severity liver disease but not with low- or high-severity liver disease. SHBG not influenced by spironolactone dosage. No influence on gonadotropin responses to GnRH stimulation.
Although the quality of these studies is limited, the findings of the studies, which are mixed but are overall more suggestive against spironolactone reducing testosterone levels than it doing so, are in notable contrast to similar studies of CPA and testosterone suppression in cisgender men that were published in the 1970s and 1980s. These studies consistently found that CPA suppressed testosterone levels by 40 to 70% on average (Aly, 2019). Subsequently, the findings were replicated in several more modern studies of CPA in cisgender men and transfeminine people, which likewise found that the drug given alone consistently suppressed testosterone levels by about 45 to 65% on average (Aly, 2019).
Spironolactone in Cisgender Women
Spironolactone has a long history of use in cisgender women in the treatment of androgen-dependent skin and hair conditions like acne, hirsutism, scalp hair loss, and hyperandrogenism (due to e.g. polycystic ovary syndrome (PCOS)). It has been used at similar doses for androgen-dependent conditions in cisgender women as it has in transfeminine people (e.g., 50–200 mg/day most typically). There are many dozens of studies of spironolactone as an antiandrogen in cisgender women (e.g., PubMed). Instead of attempting to individually review all of these studies, the present article will discuss the findings of several papers that have themselves reviewed substantial numbers of these studies and have summarized available findings on testosterone levels with spironolactone.
Callan (1988) reviewed the literature on spironolactone for treatment of acne and hirsutism in cisgender women and found that some clinical studies reported decreased levels of testosterone and/or other androgens with spironolactone (4 studies cited) whereas other studies reported no change in androgen levels (4 studies cited). The author cited several studies to support the claim that androgen receptor antagonism with spironolactone is more clinically important than any influence it has on androgen production (5 studies cited). For instance, clinical benefits against acne and hirsutism occurred with spironolactone both before androgen levels decrease as well as when androgen levels do not decrease.
McMullen & Van Herle (1993) reviewed 19 studies of spironolactone for treatment of androgen-dependent conditions in cisgender women, with a majority of these studies reporting long-term hormone levels. Most of the studies were open-label and uncontrolled, with only five studies having a control group and only two studies being double-blind placebo-controlled trials. Changes in hormone levels across studies were very heterogenous, with the majority of changes not reaching statistical significance. Only 1 of 7 (14%) studies found a decrease in DHEA-S levels. The review concluded that a clinically significant change in adrenal androgen levels with spironolactone in cisgender women was not supported. Conversely, testosterone levels were decreased with spironolactone in 13 of 16 (81%) of studies. However, in the only two RCTs, there were no differences in testosterone levels with spironolactone versus in the placebo control groups. As such, the review concluded that the decreased testosterone levels with spironolactone in cisgender women reported in many of the non-RCT studies may not actually be a real phenomenon. As with Callan (1988), the review noted that the major mechanism of action of spironolactone as an antiandrogen is likely to be androgen receptor blockade.
Bradstreet et al. (2007) cited and discussed a Cochrane review of spironolactone for treatment of acne and/or hirsutism in cisgender women (Farquhar et al., 2003). Cochrane reviews are rigorous high-quality systematic reviews of all of the available RCTs for a given medical intervention. The Cochrane review identified 19 RCTs, with 9 included in the review, 8 excluded due to methodological issues (e.g., with randomization), and two others which were described as “awaiting assessment” (Farquhar et al., 2003). Bradstreet and colleagues noted per the Cochrane review that spironolactone at a dosage of 100 mg/day had little influence on levels of DHEA, DHEA-S, or testosterone in the trials evaluated and said that this is because its mechanism of action as an antiandrogen is androgen receptor antagonism (Bradstreet et al., 2007). The Cochrane review itself did not discuss changes in androgen or testosterone levels with spironolactone in aggregate. An update of the Cochrane review was published in 2009, but with no new studies found and with the findings unchanged (Brown et al., 2009).
Layton et al. (2017) was a hybrid systematic review of spironolactone for acne in cisgender women. In a table discussing the mechanism of action of spironolactone and other antiandrogens for acne, the authors stated that “Data from over 50 articles reporting effects [of spironolactone] on serum androgens are equivocal” (i.e., ambiguous, uncertain, questionable) (Layton et al., 2017). The review further noted that inhibition of androgen synthesis by spironolactone in humans may be unlikely at therapeutic doses and may occur instead only at supraphysiological doses (with Menard et al. (1979) cited in support of these claims, presumably related to the very high doses required) (Layton et al., 2017).
Rozner et al. (2019) reviewed clinical studies of the endocrine effects of spironolactone in cisgender women to assess whether it is safe to use in women with past or present breast cancer receiving endocrine therapy. The review included 18 studies with 465 women (mostly having androgen-dependent conditions) assessing the influence of spironolactone on sex hormone levels. The assessed studies included retrospective cohort studies, case–control studies, and RCTs. Of the included studies, 10 (56%) studies (with 179 women) found no change in testosterone levels with spironolactone, 8 (44%) studies (with 253 women) found a decrease, and 1 (6%) study (with 33 women) found an increase in free but not total testosterone levels. Changes in levels of DHEA-S, androstenedione, and estrogen were also assessed and findings were similar, with no changes observed in majorities of studies for these hormones. The review concluded that there is no significant change in levels of androgens, estrogen, or gonadotropins with spironolactone in cisgender women.
Almalki et al. (2020) conducted a systematic review and network meta-analysis of RCTs on the comparative efficacy of several types of medications (statins, metformin, spironolactone, and combined birth control pills) on reducing testosterone levels in cisgender women specifically with PCOS. Nine RCTs including 613 women were included for all of the medications. The meta-analysis concluded that the statin atorvastatin was more effective than the other included medications in reducing testosterone levels. Only two of the included RCTs employed spironolactone, one of which was with spironolactone alone (n=34) versus metformin (n=35) (Ganie et al., 2004) and the other of which was with spironolactone plus metformin (n=62) versus spironolactone alone (n=51) versus metformin alone (n=56) (Ganie et al., 2013). Both of the included trials found that spironolactone alone significantly decreased testosterone levels in pre-treatment versus post-treatment comparisons (Ganie et al., 2004; Ganie et al., 2013). No trials of spironolactone versus placebo controls were included.
Taken together, the available studies of spironolactone and testosterone levels in cisgender women with androgen-dependent conditions are highly inconsistent and mixed, but with numerous studies finding no significant changes in testosterone levels. The reasons for the findings being so mixed are unclear, but may relate to study methodology and quality. Findings in this population seem particularly notable as regulation of the hypothalamic–pituitary–gonadal (HPG) axis by androgens in women is minimal to negligible, in turn making it such that androgen receptor antagonists will have little effect of upregulating gonadal sex hormone production as they can in cisgender men and transfeminine people. As a result, there is less homeostatic interference that could influence findings in evaluating the steroid synthesis inhibition of spironolactone in this sex, and hence these studies may provide a clearer picture of steroid synthesis inhibition as a possible clinical effect of spironolactone. However, as the findings are still so mixed, the results seem inconclusive. In any case, only a limited effect at best seems clear.
Spironolactone Alone in Transfeminine People
Only one study of spironolactone alone (without estrogen) and sex hormone levels in transfeminine people was identified (Table 2). It was conducted by Louis Gooren and colleagues of the Dutch Center of Expertise on Gender Dysphoria (CEGD) at the Vrije Universiteit Medical Center (VUMC) in Amsterdam, Netherlands in the 1980s. The study compared levels of testosterone, DHT, estradiol, LH, FSH, and prolactin before and after treatment with 200 mg/day spironolactone for 6 weeks in 6 young pre-hormone-therapy transfeminine people. It found slightly but significantly increased testosterone levels, increased prolactin levels, and no change in levels of estradiol, DHT, LH, or FSH.
Table 2: Studies of sex hormone levels with spironolactone alone in transfeminine people:
Treatment and subjects
Findings
Source(s)
200 mg/day for 6 weeks in 6 pre-hormone therapy transfeminine people (21–39 years)
T (mean ± SEM) increased significantly from 17.2 ± 0.8 nmol/L (496 ± 20 ng/dL) to 20.6 ± 1.7 nmol/L (594 ± 50 ng/dL) (+19.8%). No change in E2 (90 ± 20 pmol/L [25 ± 5.0 pg/mL] vs. 100 ± 30 pmol/L [27 ± 8.2 pg/mL] or 80 ± 20 pmol/L [22 ± 5.4 pg/mL]) or DHT (1.7 ± 0.8 nmol/L [49 ± 20 ng/dL] vs. 1.8 ± 0.9 nmol/L [52 ± 30 ng/dL]). LH, FSH, and GnRH-stimulated LH and FSH unchanged. PRL and TRH-stimulated PRL increased.
Abbreviations: T = testosterone; E2 = estradiol; DHT = dihydrotestosterone; LH = luteinizing hormone; FSH = follicle-stimulating hormone; GnRH = gonadotropin-releasing hormone; PRL = prolactin; TRH = thyrotropin-releasing hormone.
The fact that this study was done by the CEGD is notable as this institute is among the most prolific research centers on transgender hormone therapy in the world (Bakker, 2021), and, while they evaluated spironolactone as well as nilutamide as antiandrogens in studies in transfeminine people in the 1980s and 1990s (Wiki), the group ultimately settled on using only CPA instead. This was probably related to the lack of testosterone suppression with spironolactone and pure androgen receptor antagonists like nilutamide, as the researchers have touched on in other publications (e.g., Gooren, 1999).
Estrogen Plus Spironolactone in Transfeminine People
Eleven studies of the combination of estrogen and spironolactone and sex hormone levels in transfeminine people were identified (Table 3). The first study was conducted by Jerilynn Prior and colleagues in Canada in the 1980s. Subsequent studies were conducted over 25 years later by groups in the United States, Australia, Israel, and Thailand. All of the studies were retrospective chart reviews or prospective non-randomized studies, with the exception of a single RCT.
Table 3: Studies of testosterone levels with estrogen plus spironolactone in transfeminine people:
Treatment and subjects
Findings
Source(s)
Oral CEEs (0.625–5 mg/day cyclically—3 of 4 weeks per month), oral MPA (10–20 mg/day cyclically—3 of 4 weeks per month—or continuously—”if gonadotrophins increased or to aid in T reduction or breast development”), and spironolactone (100–600 mg/day continuously) for 12 months in 27 transfeminine people who had been on “high-dose” E alone for an extended duration (Group 1) and 23 transfeminine people who were pre-hormone-therapy (Group 2), or 50 transfeminine people total, at Vancouver General Hospital.
T decreased in Group 1 from mean 169 ng/dL to 87.4 ng/dL (–48.2%) and in Group 2 from mean 642 ng/dL to 49.2 ng/dL (–92.3%). In the groups combined, T following treatment would be mean 69.8 ng/dL. Per authors, spironolactone was intended to help reduce T and facilitate feminization while MPA was intended to help suppress gonadotropins and T and improve breast development. However, authors emphasized the decrease in T as being due to spironolactone despite inclusion of MPA, without data provided to substantiate this.
Sublingual estradiol (4 mg/day—2 mg b.i.d.) (n=14), transdermal estradiol patch (100 μg/day) (n=1), or injectable estradiol valerate (20 mg/2 weeks) (n=1) with spironolactone (100–200 mg/day) for 6 months in 16 transfeminine people at an LGBT community health center in Los Angeles, California.
T was median 405 ng/dL at baseline and 42 ng/dL after 6 months (–89.6%). Free T was median 11.4 ng/dL at baseline and 0.8 ng/dL at 6 months (–93.0%). 10 of 15 (66.7%) had total T in female range and 14 of 15 (93.3%) had free T in female range.
Oral E2 (1–8 mg/day) with or without spironolactone (200 mg/day) (n=61), finasteride (5 mg/day) (n=49), and/or MPA (2.5–10 mg/day) (n=38) for 0.3 to 10.5 years (mean 4.3 ± 3.1 years) in 156 transfeminine people at Albany Medical Center.
Oral E2 dose-dependently and substantially but incompletely suppressed T. Relative to E2 alone (at equivalent E2 levels), E2 plus spironolactone had no significant influence on T (+10.6 ± 16 ng/dL (mean ± SE); p = 0.5) and no greater likelihood of achieving better T suppression (<100 ng/dL) (OR = 0.75; 95% CI = 0.44–1.29). T levels with E2 alone were mean ~80 ng/dL and with E2 plus spironolactone were mean ~95 ng/dL per own re-analysis. Finasteride was also associated with greater T levels. MPA helped with T suppression in some (71% of subjects). More discussion and re-analysis including graphs (Aly, 2019).
Oral E2 (0.5–10 mg/day) (n=67) or oral CEEs (0.625–5 mg/day) (n=12) and spironolactone (25–400 mg/day; mean/median 145 mg/day) for 12 months in 98 transfeminine people at Boston Medical Center.
Combined E and spironolactone decreased T from median 385 ng/dL to 130 ng/dL (–66.2%). E alone vs. E and spironolactone not reported. No significant influence of spironolactone dosage on T. Incomplete suppression of T (>50 ng/dL) in all but the lowest quartile (25%) of individuals.
Oral EV (4–6 mg/day; median 5–6 mg/day) (88.3%) or transdermal E2 (11.7%) alone or in combination with CPA (25–50 mg/day; median 50 mg/day) or spironolactone (87.5–200 mg/day; median 100 mg/day) for 0.9 to 2.6 years (median 1.5 years) in 80 transfeminine people at two gender clinics in Melbourne, Australia.
T was median 10.5 nmol/L (303 ng/dL) with E2 only, 2.0 nmol/L (58 ng/dL) with E2 plus spironolactone, and 0.8 nmol/L (23 ng/dL) with E2 plus CPA. 90% of those on E2 plus CPA and 40% of those on E2 plus spironolactone had T of <2 nmol/L (<58 ng/dL). T significantly lower with E2 plus CPA compared to E2 plus spironolactone and E2 alone. T with E2 plus spironolactone lower than with E2 alone but non-significantly. No significant differences between groups in age, hormone therapy duration, or E2 dosage or levels. Graph that visually summarizes the results.
Sublingual estradiol (2–12 mg/day) and spironolactone (100–200 mg/day) with or without sublingual MPA (5–10 mg/day) or injectable MPA (150 mg/3 months) for 3.4 ± 1.7 years in 92 transfeminine people at Rhode Island Hospital.
T (mean ± SD) was 215 ± 29 ng/dL with E2 plus spironolactone and 79 ± 18 ng/dL with E2 plus spironolactone and MPA.
Oral E2 (2–8 mg/day) (84.2%) or other E forms (15.8%) with spironolactone (80.4%; n=107) or without spironolactone (19.6%) for more than 6 months in 133 transfeminine people at three clinics in Dallas, Texas.
T decreased from median 367 ng/dL (95% range 175–731 ng/dL) (n=70) at baseline to median 55 ng/dL (95% range 3–709 ng/dL) (n=131) in whole group (80.4% taking spironolactone). 65 of 133 (49%) had adequate T suppression (presumably <50 or <60 ng/dL) in whole group. T with E2 plus spironolactone at 25–75 mg/day (n=15) was mean 129.4 ng/dL (range <3—611 ng/dL), at 100–175 mg/day (n=61) was mean 180.4 ng/dL (range <3–1137 ng/dL), and at 200–300 mg/day (n=31) was mean 170.1 ng/dL (range <3–798 ng/dL). In the whole E2 plus spironolactone group (n=107), T would be mean 170.3 ng/dL.
Oral E2 (2–8 mg/day), transdermal E2 gel (2.5–5 mg/day), or transdermal E2 patches (50–200 μg/day) plus spironolactone (50–200 mg/day) (n=16), CPA (10–100 mg/day) (n=41), or a GnRH agonist (n=10) for 12 months in 67 transfeminine people at Tel Aviv-Sourasky Medical Center in Israel.
With spironolactone, T (mean ± SD) decreased from 15.2 ± 8.1 nmol/L (438 ± 230 ng/dL) at baseline to 10.2 ± 5.7 nmol/L (294 ± 164 ng/dL) at 3 months (–32.9%), 3.5 ± 1.2 nmol/L (100 ± 35 ng/dL) at 6 months (–77.0%), and 4 ± 7.1 nmol/L (120 ± 200 ng/dL) at 12 months (–73.7%). T was in the female range (<1.8 nmol/L [52 ng/dL]) at all follow-ups after baseline for both CPA and GnRH agonist (–92.0% to –96.4%).
Oral EV 4 mg/day plus spironolactone (100 mg/day) (n=26) or CPA (25 mg/day) (n=26) for 12 weeks in 52 transfeminine people at two clinics in Bangkok, Thailand (RCT).
With intention-to-treat analysis, T decreased with E2 plus spironolactone from median 645.0 ng/dL (IQR 466.7−1027.7 ng/dL) to 468.3 ng/dL (IQR 287.0−765.4 ng/dL) (–27.4%) and with E2 plus CPA from 655.5 ng/dL (402.6−872.7 ng/dL) to 9.3 ng/dL (IQR 5.5−310.4 ng/dL) (–98.6%). Adequate suppression of testosterone (<50 ng/dL) was achieved by 4 of 26 (15%) in the E2 plus spironolactone group and by 18 of 26 (69%) in the E2 plus CPA group. Study also assessed and reported E2, SHBG, and PRL levels.
E2 (sublingual, transdermal, or injectable) with spironolactone (n=39) or without spironolactone (n=37) for 12 months in 93 transfeminine people at two LGBTQ-oriented clinics in Seattle, Washington and Iowa City, Iowa.
T was median 11 to 18 ng/dL in different estradiol groups without spironolactone and median 10 to 12 ng/dL in different estradiol groups with spironolactone. T was significantly lower with spironolactone only for sublingual E2 group (median 11 ng/dL (IQR 6–35 ng/dL) [n=27] vs. median 18 ng/dL (IQR 13–205 ng/dL) [n=16]) and not for transdermal or injectable E2 groups.
Oral E2 (4–12 mg/day, median 6 mg/day) (n=27) or injectable EV (2–5 mg/week, median 4 mg/week) (n=6) with spironolactone (n=31) or without spironolactone (n=2) for median 6.2 months (range 0.6–28.2 months) (time on optimized E2 dose specifically) in 33 transfeminine people at Maine Medical Center.
T was median 13.0 ng/dL (range 2.7–559 ng/dL) for whole group (93.9% taking spironolactone). 28 of 33 (84.8%) of whole group had female-range T (<50 ng/dL). However, in earlier studies by the same group, similar T suppression with E2 alone was reported (Reardon et al., 2013; Spratt et al., 2014).
The data on the testosterone levels with estrogen plus spironolactone in transfeminine people from the 11 studies in the table can be roughly summarized. Some studies reported mean testosterone levels and some reported median testosterone levels, so these cases must be considered separately. In terms of reported mean testosterone levels across studies (4 studies), the median value of these study averages would be about 171 ng/dL and the range of study averages would be about 95 to 215 ng/dL. In terms of reported median testosterone levels across studies (7 studies), the median value of these study medians would be about 55 ng/dL and the range of study medians would be about 11 to 468 ng/dL. One study had to be excluded due to concomitant use of the progestogen medroxyprogesterone acetate (MPA) in all individuals (Prior, Vigna, & Watson, 1989; Prior et al., 1986). Insights from the preceding results include large variability in testosterone levels across studies and mean testosterone levels being much higher than median testosterone levels. Limitations of the preceding values include lack of equivalent estrogen and spironolactone dosages and levels across studies, lack of equivalent durations of hormone therapy across studies, lack of equivalent testosterone blood-testing methodologies across studies, lack of equivalent transfeminine patient samples, and, in the case of the study median testosterone values, two of the studies notably having almost all but not all individuals on spironolactone (80 and 94% rather than 100%). These limitations likely underlie the large variability in reported values across studies. In any case, these results suggest that estrogen plus spironolactone results in variably inadequate testosterone suppression in most transfeminine people, which is in notable major contrast to testosterone suppression with estrogen plus CPA or a GnRH agonist in transfeminine people.
Individual findings of the studies include inadequate testosterone suppression with estradiol plus spironolactone in most transfeminine people (Leinung et al., 2018; Liang et al., 2018; Jain, Kwan, & Forcier, 2019; Sofer et al., 2020; Burinkul et al., 2021), no difference in testosterone suppression with spironolactone versus without spironolactone (Leinung et al., 2018), lack of notable influence of spironolactone dosage on testosterone suppression (Liang et al., 2018; SoRelle et al., 2019), and inferior testosterone suppression with estradiol plus spironolactone compared to estradiol plus CPA or a GnRH agonist in transfeminine people (Angus et al., 2019; Sofer et al., 2020; Burinkul et al., 2021). Conversely, some studies have found adequate or near-adequate testosterone suppression with estradiol plus spironolactone in most or almost all transfeminine people (Deutsch, Bhakri, & Kubicek, 2015; Angus et al., 2019; SoRelle et al., 2019; Cirrincione et al., 2021; Pappas et al., 2021), and some studies have found indications of greater testosterone suppression with spironolactone versus without spironolactone (Angus et al., 2019; Cirrincione et al., 2021). On the other hand, some studies using estradiol alone without any antiandrogen at physiological estradiol levels (<200 pg/mL) have reported adequate testosterone suppression similarly to the preceding estradiol plus spironolactone studies (Reardon et al., 2013; Spratt et al., 2014; Cirrincione et al., 2021). One study was confounded by the concomitant use of MPA, which is known to suppress testosterone levels on its own, and hence reliable conclusions cannot not be drawn from this study (Prior, Vigna, & Watson, 1989; Prior et al., 1986). Indeed, it is notable that this study found lower mean testosterone levels with estrogen and spironolactone than any other study did. A couple of studies found that testosterone levels progressively decline with time (particularly over the first 12 months) with estradiol plus spironolactone in most transfeminine people (Liang et al., 2018; Sofer et al., 2020). Whether the decreases in testosterone levels with time were more related to estradiol or to spironolactone is unclear, though estradiol seems more likely (e.g., Wiki).
Taken together, the findings of available studies on estradiol plus spironolactone and testosterone suppression in transfeminine people are highly variable and mixed, although overall more studies support spironolactone having poor or no testosterone-suppressing effectiveness. The reasons underlying the differences in findings on testosterone suppression between studies are unclear, but contributing factors may include varying estradiol doses, routes, and levels, durations of hormone therapy, differing laboratory assays of testosterone levels, and other differences in study methodologies, as well as limitations in study and evidence quality. In any case, the conflicting nature of the findings is in major contrast to the almost invariably strong to maximal testosterone suppression in studies of estradiol plus CPA and estradiol plus GnRH agonists in transfeminine people.
Spironolactone, Androgen Receptor Antagonism, and Clinical Antiandrogenic Effectiveness
The clinical antiandrogenic effectiveness of spironolactone in cisgender women with androgen-dependent skin and hair conditions, like acne, hirsutism, and scalp hair loss, is well-established (Brown et al., 2009; van Zuuren & Fedorowicz, 2016; Layton et al., 2017; Barrionuevo et al., 2018; James, Jamerson, & Aguh, 2022; Wang et al., 2023). Conversely, the clinical antiandrogenic efficacy of spironolactone in transfeminine people has been very limitedly assessed to date and is largely unknown (Angus et al., 2021). Spironolactone does not appear to be very effective for decreasing testosterone levels in either cisgender women or transfeminine people based on the findings of the present review. However, spironolactone is a competitive antagonist of the androgen receptor in addition to its actions a weak androgen synthesis inhibitor, and hence it also directly blocks androgens from mediating their effects in the body (Loriaux et al., 1976; McMullen & Van Herle, 1993). Based on studies in populations besides transfeminine people, for instance cisgender women (discussed above) and cisgender boys with gonadotropin-independent precocious puberty (e.g., Holland, 1991), in which spironolactone has not decreased testosterone levels but has nonetheless been effective as an antiandrogen, the androgen receptor blockade of spironolactone is likely to be its main mechanism of action as an antiandrogen and may account for most or all of its therapeutic antiandrogenic effectiveness.
However, while spironolactone is clearly effective as an androgen receptor antagonist, it appears to be a relatively weak androgen receptor blocker at typical doses used in cisgender women and transfeminine people. Numerous publications in the literature describe spironolactone as being only a weak androgen receptor antagonist (Wiki; Wiki). In relation to this, animal studies have found that spironolactone is a far less potent androgen receptor antagonist than other antiandrogens like CPA, flutamide, and bicalutamide (Bonne & Raynaud, 1974; Hecker, Hasan, & Neumann, 1980; Sivelle, Underwood, & Jelly, 1982; Weissmann et al., 1985; Labrie et al., 1987; Snyder, Winneker, & Batzold, 1989 [Table]; Yamasaki et al., 2004 [Graph]). Moreover, in cisgender women, the population in which spironolactone is most widely used as an antiandrogen, testosterone levels are relatively low, on average about 20-fold lower than in cisgender men (around 30 ng/dL on average compared to about 600 ng/dL on average, respectively) (Aly, 2018). However, many cisgender women with androgen-dependent conditions have PCOS, which is associated with limitedly elevated testosterone levels (e.g., perhaps around 60 ng/dL on average) (Aly, 2018). The typical therapeutic dose range of spironolactone in cisgender women with androgen-dependent conditions is 50 to 200 mg/day, in which its effectiveness may be assumed to be dose-dependent, and this is roughly the same general dosage range used in transfeminine people (though up to 300–400 mg/day may be used and are allowed for by guidelines) (Aly, 2018; Aly, 2020).
A relatively small amount of dose-ranging data on spironolactone in cisgender women with androgen-dependent conditions exists, but in any case substantiates its dose-dependent effectiveness across its clinically used dose range (partially reviewed in Hammerstein (1990) and Shaw (1996)). One study compared spironolactone at doses of 50 to 200 mg/day with placebo for treatment of acne in cisgender women and reported progressive increases in effectiveness with spironolactone up to the 200 mg/day dosage (Goodfellow et al., 1984). Similarly, another study found that progressively increasing the dosage of spironolactone from 100 mg/day, to 150 mg/day, and up to 200 mg/day, resulted in increased effectiveness in the treatment of acne in cisgender women (Charny, Choi, & James, 2017). Spironolactone has been reported to be effective in the treatment of hirsutism in cisgender women at a dosage of as low as 50 mg/day (Diamanti-Kandarakis, Tolis, & Duleba, 1995). However, even a dosage of 100 mg/day did not appear to be maximally effective for hirsutism in a study that compared different doses of spironolactone; effectiveness was near-significantly greater at a dosage of 200 mg/day relative to a dosage of 100 mg/day (30% ± 3% and 19% ± 8% (mean ± SEM) reduction in hair shaft diameter, respectively; p = 0.07) (Lobo et al., 1985). Levels of free testosterone in this study were unchanged, suggesting that the effects of spironolactone were purely due to androgen receptor blockade. Finally, a 2022 systematic review of spironolactone for treatment of androgen-related scalp hair loss in cisgender women reported that the drug was “largely ineffective” at doses of less than 100 mg/day, whereas doses of 100 to 200 mg/day were effective (James, Jamerson, & Aguh, 2022).
Aside from dose-ranging studies, the antiandrogenic efficacy of spironolactone can be evaluated by comparing it to more potent antiandrogenic regimens. A study found that spironolactone 100 mg/day was significantly inferior to flutamide, a substantially more potent androgen receptor antagonist, in improving androgen-dependent skin and hair symptoms in cisgender women (Cusan et al., 1994). However, in other studies, there were no significant differences between spironolactone 100 mg/day and flutamide for hirsutism (Erenus et al., 1994; Moghetti et al., 2000; Inal, Yildirim, & Taner, 2005; Karakurt et al., 2008). Spironolactone and flutamide were variably taken together with an ethinylestradiol-containing combined birth control pill in these studies, which is likely to have limited detection of differences in effectiveness. This is because these birth control pills considerably suppress total and free testosterone levels and hence have substantial antiandrogenic effects themselves (Zimmerman et al., 2014; Amiri et al., 2018). In a biochemical study, spironolactone 100 mg/day was numerically inferior to flutamide in reducing levels of prostate-specific antigen (PSA) in cisgender women (Negri et al., 2000). This is notable as PSA is a systemic biomarker of androgen action (Negri et al., 2000). However, the study had small sample sizes, and the differences between groups were not statistically significant (Negri et al., 2000). A case report of a cisgender woman with female pattern hair loss and normal androgen levels found that treatment with spironolactone 200 mg/day for 5 years failed to improve or halt progression of her hair loss, in spite of almost complete loss of secondary sexual hair, but switching to flutamide resulted in a considerable improvement in hair loss after 12 months (Yazdabadi & Sinclair, 2011 [Figure]). Besides comparison with flutamide, a study found that spironolactone 100 mg/day was inferior to spironolactone 100 mg/day plus finasteride, a 5α-reductase inhibitor and hence functional antiandrogen, for hirsutism in cisgender women (–36.6% vs. –51.3% in scores; p < 0.005) (Unlühizarci et al., 2002; Keleştimur et al., 2004).
The preceding findings suggest that the clinical antiandrogenic effectiveness of spironolactone in cisgender women is not maximal at a dosage of below at least 200 mg/day despite the relatively low testosterone levels in these individuals. Put another way, spironolactone at typical doses seems best-suited for blocking female-range levels of testosterone. As many transfeminine people do not achieve female-range testosterone levels with estradiol plus spironolactone therapy, and in fact often have testosterone levels well above the normal female range or even in the male range, spironolactone may not be fully effective as an antiandrogen at the typical doses used in transfeminine hormone therapy. Higher doses of spironolactone, like 300 to 400 mg/day, may be to some degree more effective.
Summary, Discussion, and Conclusions
Numerous studies have assessed the influence of spironolactone on testosterone levels in cisgender men, cisgender women, and transfeminine people. Although the quality of these studies has often been limited, the studies have revealed highly inconsistent influences of spironolactone on testosterone levels in these populations, with many studies finding no changes, some studies finding decreases, and a small number of studies finding increases. The findings of studies of spironolactone and testosterone levels are in notable contrast to those of studies with estrogens, progestogens like CPA, and GnRH agonists, which consistently show substantial decreases in testosterone levels. This has been the case even in studies of similarly low quality to those of some of the included spironolactone studies (e.g., many of those in cisgender men). The fact that in the available studies testosterone levels with spironolactone have usually been unchanged, but have sometimes been decreased and have rarely been decreased, seems to suggest that spironolactone may be a clinically significant inhibitor of steroid hormone synthesis, but that it is only a weakly efficacious one, and that its effects may be variable depending on the individual and other clinical circumstances. In any case, the conflicting findings warrant more research with higher-quality study designs, particularly RCTs that have with spironolactone versus without comparison groups.
The notion that spironolactone decreases testosterone levels in transfeminine people, and the use of spironolactone in transfeminine hormone therapy in general, appear to have originated from the papers on spironolactone in transfeminine people published by Dr. Jerilynn Prior and colleagues in the 1980s (Prior, Vigna, & Watson, 1989; Prior et al., 1986). In their study, transfeminine people who were either already on high-dose estrogen therapy with inadequate testosterone suppression or had not yet started hormone therapy were put on physiological-dose estrogen therapy in combination with 200 to 600 mg/day spironolactone. Cyclic or continuous administration of the progestogen MPA at an oral dose of 10 mg/day was also given to all of the individuals. The authors reported that despite the lower estrogen dosage, testosterone levels decreased, from 169 ng/dL to 87 ng/dL (–49%) in those who had already been on hormone therapy and to 49 ng/dL in those who were pre-hormone therapy. Prior and her colleagues concluded that spironolactone helps to decrease testosterone levels in transfeminine people and that it can be used as a safer alternative to high doses of estrogen for this purpose.
However, the concomitant use of MPA in the study is a major confounding factor in terms of their results. This is because MPA is a progestogen, and progestogens, like estrogens, are antigonadotropins which are able to robustly suppress testosterone levels on their own (Aly, 2018; Aly, 2019). Indeed, MPA alone has been shown to dose-dependently lower testosterone levels in cisgender men (Wiki), and at a dosage of 10 mg/day, has been shown to considerably suppress testosterone levels in transfeminine people when added to estradiol and spironolactone therapy (Jain, Kwan, & Forcier, 2019). Hence, MPA may have been, and likely was, responsible for the decreases in testosterone levels seen in the study, rather than spironolactone. This point was also notably raised by other researchers, who were unable to replicate Prior and colleagues’ results on spironolactone and testosterone levels in transfeminine people (Leinung et al., 2018). Strangely, Prior and colleagues concluded that spironolactone was responsible for the decreased testosterone levels in their study even though they noted in their papers that MPA was also given to help suppress testosterone levels (as well as to help improve breast development). The work of Prior and colleagues likely resulted in the prominent and long-standing, but poorly supported, notion that spironolactone decreases testosterone levels in transfeminine people. Subsequent studies assessing the hypothesis that spironolactone decreases testosterone levels in transfeminine people were not published until 25 years after Prior and colleagues’ studies, with several of these studies, though not all of them, failing to replicate the earlier findings of Prior and colleagues.
Many people do not realize the capacity of estradiol to substantially and even completely suppress testosterone, and many mistakenly assume that it is the antiandrogen—which is often spironolactone—that is mostly or fully responsible for the decrease in testosterone levels seen with estradiol and antiandrogen therapy in transfeminine people. It is certainly true that antiandrogens like CPA and GnRH agonists play an important role in testosterone suppression in transfeminine people. However, as evidenced by the present review of studies of testosterone suppression with spironolactone, it is not necessarily always the case that the antiandrogen plays a major role—or potentially even any role—in reducing testosterone levels. This is notably also not the case with certain other antiandrogens besides spironolactone, for instance pure androgen receptor antagonists like bicalutamide, which likewise do not decrease testosterone levels but instead can actually increase them (Aly, 2019; Wiki). Clinicians and transfeminine people attributing observations of testosterone decreases to spironolactone rather than to estradiol with estradiol and spironolactone therapy may also have played a role in the perception that spironolactone considerably decreases testosterone levels in transfeminine people.
Due to its relatively weak strength as an androgen receptor antagonist and its limited efficacy in lowering testosterone levels, spironolactone is likely to be a limitedly effective antiandrogen in transfeminine people. Additionally, spironolactone is likely to be less effective than other antiandrogenic approaches used in transfeminine hormone therapy which either more robustly block androgens or more substantially reduce testosterone levels, for instance CPA, other progestogens (e.g., MPA, non-oral progesterone), GnRH agonists (and antagonists), bicalutamide, and high-dose parenteral estradiol monotherapy. These approaches can be used in transfeminine people instead of or in addition to spironolactone, or could be considered when testosterone suppression is inadequate with estradiol and spironolactone.
More studies are needed to evaluate the influence of spironolactone on testosterone levels, especially RCTs that compare estradiol alone versus estradiol plus spironolactone in transfeminine people. More research is also needed to clarify why some studies find highly inadequate testosterone suppression with estradiol alone or estradiol plus spironolactone while other studies find excellent or satisfactory testosterone suppression with these regimens. In any case, available data overall suggest that spironolactone does not consistently suppress testosterone levels, and that estradiol plus spironolactone produces inadequate testosterone suppression in many transfeminine people. Moreover, available data suggest that spironolactone is a relatively weak androgen receptor antagonist at the typical clinical doses used in cisgender women and transfeminine people, and is able to block only relatively low or female-range testosterone levels. Hence, spironolactone may not be fully effective in blocking the testosterone it fails to suppress, and may be particularly unsuitable for transfeminine people with testosterone levels that are well above the normal female range. In any case, more research is similarly needed to assess the androgen receptor antagonism and clinical antiandrogenic effectiveness of spironolactone.
Updates
Update 1: Spironolactone for Adult Female Acne (SAFA) Trial
A large new phase 3 RCT, the Spironolactone for Adult Female Acne (SAFA) trial, was published in May 2023 and assessed the effectiveness of spironolactone in the treatment of acne in cisgender women:
Santer, M., Lawrence, M., Renz, S., Eminton, Z., Stuart, B., Sach, T. H., Pyne, S., Ridd, M. J., Francis, N., Soulsby, I., Thomas, K., Permyakova, N., Little, P., Muller, I., Nuttall, J., Griffiths, G., Thomas, K. S., & Layton, A. M. (2023). Effectiveness of spironolactone for women with acne vulgaris (SAFA) in England and Wales: pragmatic, multicentre, phase 3, double blind, randomised controlled trial. BMJ, 381, e074349. [DOI:10.1136/bmj-2022-074349]
The trial included a total of 342 women, including 176 treated with spironolactone and 166 in the placebo control group. The dose of spironolactone employed was 50 mg/day for the first 6 weeks and then 100 mg/day thereafter. The trial was 24 weeks (5.5 months) in duration. Women who might become pregnant were required to use a hormonal or barrier method of contraception.
Spironolactone significantly outperformed placebo in terms of improvement in mean Acne-QoL symptom scores (higher is better). Significant improvement was apparent within 12 weeks of treatment (+45% in scores with spironolactone, +38% with placebo) and was highest at 24 weeks (+61% in scores with spironolactone, +35% with placebo). There was no difference in the rates of women who reported improvement in acne scores at 12 weeks (72% with spironolactone, 68% with placebo), but there was a significant difference at 24 weeks (82% with spironolactone, 63% with placebo). In terms of the Investigator’s Global Assessment (IGA), treatment success at 12 weeks was 19% with spironolactone and 6% with placebo. Rates of hormonal contraceptive use in the spironolactone and placebo groups were not reported. Testosterone levels were also not reported. A small subset of the women had PCOS (15% in the spironolactone group, 23% in the placebo group).
Adverse effects occurred only slightly more often with spironolactone than with placebo (64% vs. 51%, p = 0.01). The only side effect that occurred significantly more often with spironolactone than with placebo was headache (20% vs. 12%; p = 0.02). However, a few other side effects trended towards occurring significantly more frequently with spironolactone than with placebo: “other” (17% vs. 11%; p = 0.06), dizziness/vertigo/lightheadness (19% vs. 12%; p = 0.07), vomiting/being sick (2% vs. 1%; p = 0.16), and polyuria (urinary frequency) (31% vs. 25%; p = 0.18). Rates of other potentially relevant side effects, like abdominal pain, breast enlargement, breast tenderness, drowsiness/sleepiness, fatigue/tiredness, menstrual irregularity, and reduced libido, were all not different between spironolactone and placebo. There were no serious adverse reactions in the trial. Rates of compliance were similar between the spironolactone and placebo groups, suggesting that spironolactone was well-tolerated.
This trial is the largest and most rigorous RCT of spironolactone in the treatment of androgen-dependent skin and hair conditions in cisgender women that has been conducted to date. Although spironolactone was found to be effective in this study and was about twice as effective as placebo in terms of Acne-QoL symptom scores and three times as effective as placebo in terms of IGA treatment success rates, the effectiveness of spironolactone was seemingly less than in previous clinical studies of spironolactone for acne. This may be related to the relatively low doses of spironolactone used in this study (50–100 mg/day), to the more rigorous and less-risk-of-bias design of the study (large phase 3 RCT), to a possibly too-short treatment duration (24 weeks/5.5 months), and to concomitant hormonal contraceptive use possibly blunting the degree of potential improvement. The latter is relevant as hormonal contraceptives containing ethinylestradiol provide a considerable improvement in acne via functional antiandrogenic effects all on their own. A final possibility however is that spironolactone is simply a less effective antiandrogen even in cisgender women than has been previously thought. On the other hand, similarly to findings in previous clinical studies, spironolactone was well-tolerated and produced few side effects.
Update 2: New Spironolactone and Testosterone Suppression Studies
The following new studies have additionally assessed and found inadequate testosterone suppression in transfeminine people treated with estradiol and spironolactone:
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (November 2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (November 2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Miro, E., Rizzone, K., Ho, T., Mark, B., Sullivan, E., & Cushman, D. (2024). 2024 AMSSM Research Podium Presentations: Testosterone Levels Among Transgender Women on Gender-affirming Hormone Therapy. Clinical Journal of Sports Medicine, 34(2), 152–152. [DOI:10.1097/JSM.0000000000001212]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery JCG, Zajac, J. D., & Cheung, A. S. (2025). Effect of Spironolactone and Cyproterone Acetate on Breast Growth in Transgender People: A Randomized Clinical Trial. The Journal of Clinical Endocrinology and Metabolism, 110(6), e1874–e1884. [DOI:10.1210/clinem/dgae650]
Angus et al. (2023/2025) and Yang et al. (2024) compared estradiol plus spironolactone to estradiol plus CPA and are described in-depth in a section of a different article located here. Yang et al. (2024) found that in addition to spironolactone resulting in much less testosterone suppression than CPA, it was also less effective than CPA as an antiandrogen on multiple clinical measures of demasculinization.
Update 3: Bonadonna et al. (2025)
In August 2025, the following conference abstract was published online:
Bonadonna, S., Amer, M., Foletti, F., Federici, S., Persani, L., Bonomi, M. (2025). Evaluation of Antiandrogen Therapy Effectiveness in Transgender individuals Assigned Male At Birth (AMAB). EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
It was an abstract for a retrospective observational study of spironolactone versus CPA, presumably in combination with estrogen, in 149 transfeminine people. The study found that testosterone and gonadotropin levels were significantly higher with spironolactone than with CPA. In addition, it found that spironolactone was associated with less suppression of libido and spontaneous erections than CPA. Conversely, there was no difference in waist–hip ratio between the groups. The authors concluded that spironolactone appears to be less effective than CPA as an antiandrogen in transfeminine people. The full study may be published in a journal article at some point in the future.
References
Abshagen, U., Spörl, S., Schöneshöfer, M., L’age, M., & Oelkers, W. (1978). Interference of spironolactone therapy with adrenal steroid metabolism in secondary hyperaldosteronism. [Zum Einfluß einer Spironolaktonbehandlung auf den adrenalen Steroidstoffwechsel bei sekundärem Hyperaldosteronismus.] Klinische Wochenschrift, 56(7), 341–349. [DOI:10.1007/bf01477394]
Aizawa, H., & Niimura, M. (1992). Oral Spironolactone Therapy in Male Patients with Rosacea. The Journal of Dermatology, 19(5), 293–297. [DOI:10.1111/j.1346-8138.1992.tb03227.x]
Allen, A. N., Jiao, R., Day, P., Pagels, P., Gimpel, N., & SoRelle, J. A. (2020). Dynamic Impact of Hormone Therapy on Laboratory Values in Transgender Patients over Time. The Journal of Applied Laboratory Medicine, 6(1), 27–40. [DOI:10.1093/jalm/jfaa192]
Almalki, H. H., Alshibani, T. M., Alhifany, A. A., & Almohammed, O. A. (2020). Comparative efficacy of statins, metformin, spironolactone and combined oral contraceptives in reducing testosterone levels in women with polycystic ovary syndrome: a network meta-analysis of randomized clinical trials. BMC Women’s Health, 20(1), 68. [DOI:10.1186/s12905-020-00919-5]
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). Analysis of Estradiol and Testosterone Levels with Oral Estradiol in Transfeminine People Based on Leinung et al. (2018). Transfeminine Science. [URL]
Aly. (2019). Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Clinical Guidelines with Information on Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Amiri, M., Kabir, A., Nahidi, F., Shekofteh, M., & Ramezani Tehrani, F. (2018). Effects of combined oral contraceptives on the clinical and biochemical parameters of hyperandrogenism in patients with polycystic ovary syndrome: a systematic review and meta-analysis. The European Journal of Contraception & Reproductive Health Care, 23(1), 64–77. [DOI:10.1080/13625187.2018.1435779]
Angus, L., Leemaqz, S., Ooi, O., Cundill, P., Silberstein, N., Locke, P., Zajac, J. D., & Cheung, A. S. (2019). Cyproterone acetate or spironolactone in lowering testosterone concentrations for transgender individuals receiving oestradiol therapy. Endocrine Connections, 8(7), 935–940. [DOI:10.1530/ec-19-0272]
Angus, L. M., Leemaqz, S., Zajac, J. D., & Cheung, A. S. (2023). A randomised controlled trial of spironolactone versus cyproterone in trans people commencing estradiol. AusPATH 2023 Symposium. [URL] [PDF] [Trans Health Research Blog Post]
Angus, L. M., Leemaqz, S. Y., Zajac, J. D., & Cheung, A. S. (2023). The effect of cyproterone and spironolactone on breast development in transgender women: a randomised controlled trial. ESA/SRB/ENSA 2023 ASM 26-29 November, Brisbane, 54–55 (abstract no. 132). [URL] [PDF] [Full Abstract Book] [Trans Health Research Blog Post]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2021). A systematic review of antiandrogens and feminization in transgender women. Clinical Endocrinology, 94(5), 743–752. [DOI:10.1111/cen.14329]
Angus, L. M., Leemaqz, S. Y., Kasielska-Trojan, A. K., Mikołajczyk, M., Doery JCG, Zajac, J. D., & Cheung, A. S. (2025). Effect of Spironolactone and Cyproterone Acetate on Breast Growth in Transgender People: A Randomized Clinical Trial. The Journal of Clinical Endocrinology and Metabolism, 110(6), e1874–e1884. [DOI:10.1210/clinem/dgae650]
Baba, S. (1977). Antiandrogenic effect of spironolactone. The Japanese Journal of Urology, 68(12), 1184–1192. [DOI:10.5980/jpnjurol1928.68.12_1184]
Baba, S., Murai, M., Jitsukawa, S., Hata, M., & Tazaki, H. (1978). Antiandrogenic Effects of Spironolactone: Hormonal and Ultrastructural Studies in Dogs and Men. Journal of Urology, 119(3), 375–380. [DOI:10.1016/s0022-5347(17)57495-9]
Bakker, A. (2021). Een halve eeuw transgenderzorg aan de VU [Half a Century of Transgender Care at the VU]. Amsterdam: Boom. [Google Books] [WorldCat] [URL]
Barrionuevo, P., Nabhan, M., Altayar, O., Wang, Z., Erwin, P. J., Asi, N., Martin, K. A., & Murad, M. H. (2018). Treatment Options for Hirsutism: A Systematic Review and Network Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism, 103(4), 1258–1264. [DOI:10.1210/jc.2017-02052]
Bonadonna, S., Amer, M., Foletti, F., Federici, S., Persani, L., Bonomi, M. (2025). Evaluation of Antiandrogen Therapy Effectiveness in Transgender individuals Assigned Male At Birth (AMAB). EPATH 6th Conference, September 4–6, 2025 in Hamburg Germany. [Abstract Book PDF] [PDF]
Bonne, C., & Raynaud, J. (1974). Mode of spironolactone anti-androgenic action: Inhibition of androstanolone binding to rat prostate androgen receptor. Molecular and Cellular Endocrinology, 2(1), 59–67. [DOI:10.1016/0303-7207(74)90012-4]
Bonzagni, A. F. (2014). Understanding the effects of long-term hormone therapy in transgender individuals being provided care at Boston Medical Center Endocrinology Clinic: a quality assurance project. (Doctoral dissertation, Boston University.) [Google Scholar] [URL] [PDF]
Bradstreet, J. J., Smith, S., Granpeesheh, D., El-Dahr, J. M., & Rossignol, D. (2007). Spironolactone might be a desirable immunologic and hormonal intervention in autism spectrum disorders. Medical Hypotheses, 68(5), 979–987. [DOI:10.1016/j.mehy.2006.10.015]
Brown, J., Farquhar, C., Lee, O., Toomath, R., & Jepson, R. G. (2003). Spironolactone versus placebo or in combination with steroids for hirsutism and/or acne. Cochrane Database of Systematic Reviews, 2003(4), CD000194. [DOI:10.1002/14651858.cd000194]
Brown, J., Farquhar, C., Lee, O., Toomath, R., & Jepson, R. G. (2009). Spironolactone versus placebo or in combination with steroids for hirsutism and/or acne. Cochrane Database of Systematic Reviews, 2009(2), CD000194. [DOI:10.1002/14651858.cd000194.pub2]
Burinkul, S., Panyakhamlerd, K., Suwan, A., Tuntiviriyapun, P., & Wainipitapong, S. (2021). Anti-Androgenic Effects Comparison Between Cyproterone Acetate and Spironolactone in Transgender Women: A Randomized Controlled Trial. The Journal of Sexual Medicine, 18(7), 1299–1307. [DOI:10.1016/j.jsxm.2021.05.003]
Callan, A. W. (1988). Spironolactone therapy in hirsutism and acne. Australasian Journal of Dermatology, 29(3), 135–139. [DOI:10.1111/j.1440-0960.1988.tb00385.x]
Caminos-Torres, R., MA, L., & Snyder, P. J. (1977). Gynecomastia and Semen Abnormalities Induced by Spironolactone in Normal Men. The Journal of Clinical Endocrinology & Metabolism, 45(2), 255–260. [DOI:10.1210/jcem-45-2-255]
Charny, J., Choi, J., & James, W. (2017). Spironolactone for the treatment of acne in women, a retrospective study of 110 patients. International Journal of Women’s Dermatology, 3(2), 111–115. [DOI:10.1016/j.ijwd.2016.12.002]
Cheung, A. S., Ooi, O., Davidoff, D., Leemaqz, S. Y., Cundill, P., Silberstein, N., Bretherton, I., Grossmann, M., & Zajac, J. D. (2018). Cyproterone vs spironolactone as anti-androgen therapy for transgender females receiving oestradiol therapy. Clinical Endocrinology, 89(S1) [Abstracts of the Endocrine Society of Australia’s Annual Scientific Meeting 2017], 14–14 (abstract no. 62). [Google Scholar] [DOI:10.1111/cen.13727] [URL 1] [URL 2] [PDF]
Cirrincione, L. R., Winston McPherson, G., Rongitsch, J., Sadilkova, K., Drees, J. C., Krasowski, M. D., Dickerson, J. A., & Greene, D. N. (2021). Sublingual Estradiol Is Associated with Higher Estrone Concentrations than Transdermal or Injectable Preparations in Transgender Women and Gender Nonbinary Adults. LGBT Health, 8(2), 125–132. [DOI:10.1089/lgbt.2020.0249]
Corvol, P., Mahoudeau, J. A., Valcke, J. C., Ménard, J., & Bricaire, H. (1976). Effets sexuels secondaires des spirolactones. Mécanismes possibles de l’action antiandrogène. [Sexual side-effects of spironolactones. Possible mechanisms of their anti-androgen action]. La Nouvelle Presse Medicale, 5(11), 691–694. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF]
Cusan, L., Dupont, A., Gomez, J., Tremblay, R. R., & Labrie, F. (1994). Comparison of flutamide and spironolactone in the treatment of hirsutism: a randomized controlled trial. Fertility and Sterility, 61(2), 281–287. [DOI:10.1016/s0015-0282(16)56518-2]
Deutsch, M. B., Bhakri, V., & Kubicek, K. (2015). Effects of Cross-Sex Hormone Treatment on Transgender Women and Men. Obstetrics & Gynecology, 125(3), 605–610. [DOI:10.1097/aog.0000000000000692]
Diamanti-Kandarakis, E., Tolis, G., & Duleba, A. J. (1995). Androgens and Therapeutic Aspects of Antiandrogens in Women. Journal of the Society for Gynecologic Investigation, 2(4), 577–592. [DOI:10.1177/107155769500200401]
Dymling, J., Nilsson, K. O., & Hökfelt, B. (1972). The effect of Soldactona® (canrenoate potassium) on plasma testosterone and androstenedione and urinary 17-ketosteroids and 17-hydroxycorticosteroids. Acta Endocrinologica, 70(1), 104–112. [DOI:10.1530/acta.0.0700104]
Dymling, J. F., & Hökfelt, B. (1973). Reduced plasma testosterone following spirolactone in man. Acta Endocrinologica, 73(Suppl 177) [9th Acta Endocrinologica Congress], 53–53 (abstract no. 53). [Google Scholar] [PDF 1] [PDF 2]
Dymling, J. F. (1978). The effect of spirolactone and spironolactone on plasma testosterone in man. In Addison, G. M., Asmussen, N. W., Corvol, P., Kloppernborg, P. W. C., Norman, N., Schroder, R., & Robertson, J. I. C. (Eds.). Aldosterone Antagonists in Clinical Medicine: Proceedings of the Searle Symposium, Nice, April 13–15, 1978 (pp. 297–302). Amsterdam: Excerpta Medica. [Google Scholar] [Google Books] [WorldCat] [PDF 1] [PDF 2]
Erbler, H. C. (1974). Suppression by the spironolactone metabolite canrenone of plasma testosterone in man. Naunyn-Schmiedeberg’s Archives of Pharmacology, 285(4), 403–406. [DOI:10.1007/bf00501468]
Erenus, M., Gürbüz, O., Durmuşoğlu, F., Demirçay, Z., & Pekin, S. (1994). Comparison of the efficacy of spironolactone versus flutamide in the treatment of hirsutism. Fertility and Sterility, 61(4), 613–616. [DOI:10.1016/s0015-0282(16)56634-5]
Ganie, M. A., Khurana, M. L., Eunice, M., Gulati, M., Dwivedi, S. N., & Ammini, A. C. (2004). Comparison of Efficacy of Spironolactone with Metformin in the Management of Polycystic Ovary Syndrome: An Open-Labeled Study. The Journal of Clinical Endocrinology & Metabolism, 89(6), 2756–2762. [DOI:10.1210/jc.2003-031780]
Ganie, M. A., Khurana, M. L., Nisar, S., Shah, P. A., Shah, Z. A., Kulshrestha, B., Gupta, N., Zargar, M. A., Wani, T. A., Mudasir, S., Mir, F. A., & Taing, S. (2013). Improved Efficacy of Low-Dose Spironolactone and Metformin Combination Than Either Drug Alone in the Management of Women With Polycystic Ovary Syndrome (PCOS): A Six-Month, Open-Label Randomized Study. The Journal of Clinical Endocrinology & Metabolism, 98(9), 3599–3607. [DOI:10.1210/jc.2013-1040]
Goodfellow, A., Alaghband-Zadeh, J., Carter, G., Cream, J., Holland, S., Scully, J., & Wise, P. (1984). Oral spironolactone improves acne vulgaris and reduces sebum excretion. British Journal of Dermatology, 111(2), 209–214. [DOI:10.1111/j.1365-2133.1984.tb04045.x]
Gooren, L. J., Veen, E. A., Kessel, H., Harmsen-Louman, W., & Wiegel, A. R. (1984). Prolactin secretion in the human male is increased by endogenous oestrogens and decreased by exogenous/endogenous androgens. International Journal of Andrology, 7(1), 53–60. [DOI:10.1111/j.1365-2605.1984.tb00759.x]
Gooren, L., Veen, E., Kessel, H., Harmsen-Louman, W., & Wiegel, A. (1984). Androgens in the Feedback Regulation of Gonadotropin Secretion in Men: Effects of Administration of Dihydrotestosterone to Eugonadal and Agonadal Subjects and of Spironolactone to Eugonadal Subjects. Andrologia, 16(4), 289–298. [DOI:10.1111/j.1439-0272.1984.tb00286.x]
Gooren, L. J. G. (1999). Hormonal Sex Reassignment. International Journal of Transgenderism, 3(3), 1–7. [Google Scholar] [URL 1] [URL 2]
Hammerstein, J. (1990). Antiandrogens: Clinical Aspects. In Orfanos, C. E., & Happle, R. (Eds.). Hair and Hair Diseases (pp. 827–886). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-74612-3_35]
Handelsman, D. J., Strasser, S., McDonald, J. A., Conway, A. J., & McCaughan, G. W. (1995). Hypothalamic-pituitary-testicular function in end-stage non-alcoholic liver disease before and after liver transplantation. Clinical Endocrinology, 43(3), 331–337. [DOI:10.1111/j.1365-2265.1995.tb02040.x]
Hecker, A., Hasan, S. H., & Neumann, F. (1980). Disturbances in sexual differentiation of rat foetuses following spironolactone treatment. Acta Endocrinologica, 95(4), 540–545. [DOI:10.1530/acta.0.0950540]
Holland, F. J. (1991). Gonadotropin-lndependent Precocious Puberty. Endocrinology and Metabolism Clinics of North America, 20(1), 191–210. [DOI:10.1016/s0889-8529(18)30288-3]
Horth, C., Lobo, P., Shelton, J., Asbury, M., Clarke, J., & Venning, G. (1977). Effects of spironolaetone on the plasma binding and unbound levels of testosterone and oestradiol in healthy men. Journal of Steroid Biochemistry, 8(12), xx–xx. [DOI:10.1016/0022-4731(77)90178-9]
Horth, C. E., Lobo, P. J., Shelton, J. R., Asbury, M. J., Clarke, J. M., & Venning, G. R. (1979). Effects of spironolactone on the plasma binding and unbound levels of testosterone and oestradiol in healthy men. In Klopper, A., Lerner, L., van der Molen, H. J., & Sciarra, F. (Eds.). Research on Steroids: Proceedings of the International Study Group for Steroid Hormones, Volume 8 / Research on Steroids Transactions of the Eighth Meeting of the International Study Group for Steroid Hormones (Proceedings of the Serono Symposia, Volume 21) (pp. 313–315). London/New York/San Francisco: Academic Press. [Google Scholar]
Huffman, D. H., Kampmann, J. P., Hignite, C. E., & Azarnoff, D. L. (1978). Gynecomastia induced in normal males by spironolactone. Clinical Pharmacology & Therapeutics, 24(4), 465–473. [DOI:10.1002/cpt1978244465]
Inal, M. M., Yildirim, Y., & Taner, C. E. (2005). Comparison of the clinical efficacy of flutamide and spironolactone plus Diane 35 in the treatment of idiopathic hirsutism: a randomized controlled study. Fertility and Sterility, 84(6), 1693–1697. [DOI:10.1016/j.fertnstert.2005.05.051]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
James, J. F., Jamerson, T. A., & Aguh, C. (2022). Efficacy and safety profile of oral spironolactone use for androgenic alopecia: A systematic review. Journal of the American Academy of Dermatology, 86(2), 425–429. [DOI:10.1016/j.jaad.2021.07.048]
Karakurt, F., Sahin, I., Güler, S., Demirbas, B., Culha, C., Serter, R., Aral, Y., & Bavbek, N. (2008). Comparison of the clinical efficacy of flutamide and spironolactone plus ethinyloestradiol/cyproterone acetate in the treatment of hirsutism: A randomised controlled study. Advances in Therapy, 25(4), 321–328. [DOI:10.1007/s12325-008-0039-5]
Keleştimur, F., Everest, H., Unlühizarci, K., Bayram, F., & Șahin, Y. (2004). A comparison between spironolactone and spironolactone plus finasteride in the treatment of hirsutism. European Journal of Endocrinology, 150(3), 351–354. [DOI:10.1530/eje.0.1500351]
Labrie, C., Cusan, L., Plante, M., Lapointe, S., & Labrie, F. (1987). Analysis of the androgenic activity of synthetic “progestins” currently used for the treatment of prostate cancer. Journal of Steroid Biochemistry, 28(4), 379–384. [DOI:10.1016/0022-4731(87)91054-5]
Layton, A. M., Eady, E. A., Whitehouse, H., Del Rosso, J. Q., Fedorowicz, Z., & van Zuuren, E. J. (2017). Oral Spironolactone for Acne Vulgaris in Adult Females: A Hybrid Systematic Review. American Journal of Clinical Dermatology, 18(2), 169–191. [DOI:10.1007/s40257-016-0245-x]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Liang, J. J., Jolly, D., Chan, K. J., & Safer, J. D. (2018). Testosterone Levels Achieved by Medically Treated Transgender Women in a United States Endocrinology Clinic Cohort. Endocrine Practice, 24(2), 135–142. [DOI:10.4158/ep-2017-0116]
Lobo, R. A., Shoupe, D., Serafini, P., Brinton, D., & Horton, R. (1985). The effects of two doses of spironolactone on serum androgens and anagen hair in hirsute women. Fertility and Sterility, 43(2), 200–205. [DOI:10.1016/s0015-0282(16)48373-1]
Loriaux, D. L. (1976). Spironolactone and Endocrine Dysfunction. Annals of Internal Medicine, 85(5), 630–630. [DOI:10.7326/0003-4819-85-5-630]
McMullen, G. R., & Van Herle, A. J. (1993). Hirsutism and the effectiveness of spironolactone in its management. Journal of Endocrinological Investigation, 16(11), 925–932. [DOI:10.1007/bf03348960]
Menard, R., Guenthner, T., Kon, H., & Gillette, J. (1979). Studies on the destruction of adrenal and testicular cytochrome P-450 by spironolactone. Requirement for the 7α-thio group and evidence for the loss of the heme and apoproteins of cytochrome P-450. Journal of Biological Chemistry, 254(5), 1726–1733. [DOI:10.1016/s0021-9258(17)37833-x]
Miro, E., Rizzone, K., Ho, T., Mark, B., Sullivan, E., & Cushman, D. (2024). 2024 AMSSM Research Podium Presentations: Testosterone Levels Among Transgender Women on Gender-affirming Hormone Therapy. Clinical Journal of Sports Medicine, 34(2), 152–152. [DOI:10.1097/JSM.0000000000001212]
Miyatake, A., Noma, K., Nakao, K., Morimoto, Y., & Yamamura, Y. (1978). Increased serum oestrone and oestradiol following spironolactone administration in hypertensive men. Clinical Endocrinology, 9(6), 523–533. [DOI:10.1111/j.1365-2265.1978.tb01510.x]
Moghetti, P., Tosi, F., Tosti, A., Negri, C., Misciali, C., Perrone, F., Caputo, M., Muggeo, M., & Castello, R. (2000). Comparison of Spironolactone, Flutamide, and Finasteride Efficacy in the Treatment of Hirsutism: A Randomized, Double Blind, Placebo-Controlled Trial. The Journal of Clinical Endocrinology & Metabolism, 85(1), 89–94. [DOI:10.1210/jcem.85.1.6245]
Negri, C., Tosi, F., Dorizzi, R., Fortunato, A., Spiazzi, G. G., Muggeo, M., Castello, R., & Moghetti, P. (2000). Antiandrogen Drugs Lower Serum Prostate-Specific Antigen (PSA) Levels in Hirsute Subjects: Evidence That Serum PSA Is a Marker of Androgen Action in Women. The Journal of Clinical Endocrinology & Metabolism, 85(1), 81–84. [DOI:10.1210/jcem.85.1.6230]
Pappas, I. I., Craig, W. Y., Spratt, L. V., & Spratt, D. I. (2020). Efficacy of Sex Steroid Therapy Without Progestin or GnRH Agonist for Gonadal Suppression in Adult Transgender Patients. The Journal of Clinical Endocrinology & Metabolism, 106(3), e1290–e1300. [DOI:10.1210/clinem/dgaa884]
Pappas, I. I., Craig, W., Spratt, L. V., & Spratt, D. I. (2020). Testosterone (T) and estradiol (E2) therapy alone can suppress gonadal function in transgender patients. Costas T. Lambrew Research Retreat 2020, 47. [Google Scholar] [URL] [PDF]
Pentikäinen, P. J., Pentikäinen, L. A., Huffman, D. H., & Azarnoff, D. L. (1974). The effect of spironolactone on plasma levels and excretion of testosterone and oestrogens in the urine in males. (A preliminary report). The Journal of International Medical Research, 2(6), 439–443. [Google Scholar] [PubMed] [PDF]
Prior, J. C., Vigna, Y. M., Watson, D., Diewold, P., & Robinow, O. (1986). Spironolactone in the presurgical therapy of male to female transsexuals: Philosophy and experience of the Vancouver Gender Dysphoria Clinic. Journal of Sex Information & Education Council of Canada, 1(1), 1–7. [Google Scholar] [PDF]
Prior, J. C., Vigna, Y. M., & Watson, D. (1989). Spironolactone with physiological female steroids for presurgical therapy of male-to-female transsexualism. Archives of Sexual Behavior, 18(1), 49–57. [DOI:10.1007/bf01579291]
Reardon, E., Farnell, P., Langzettel, J., & Spratt, D. (2013). Efficacy of single agent hormonal therapy in transgender patients to suppress endogenous hypothalamic-pituitary-gonadal (HPG) activity. Fertility and Sterility, 100(3 Suppl) [Scientific Program Supplement: Oral and Poster Session Abstracts 12 October 2013 - 17 October 2013], S333–S333 (abstract no. P-640). [Google Scholar] [DOI:10.1016/j.fertnstert.2013.07.914]
Rose, L. I. (1977). Pathophysiology of Spironolactone-Induced Gynecomastia. Annals of Internal Medicine, 87(4), 398–403. [DOI:10.7326/0003-4819-87-4-398]
Rozner, R. N., Freites-Martinez, A., Shapiro, J., Geer, E. B., Goldfarb, S., & Lacouture, M. E. (2018). Safety of 5α-reductase inhibitors and spironolactone in breast cancer patients receiving endocrine therapies. Breast Cancer Research and Treatment, 174(1), 15–26. [DOI:10.1007/s10549-018-4996-3]
Santen, R. J., Kulin, H. E., Loriaux, D. L., & Friend, J. (1976). Spironolactone Stimulation of Gonadotropin Secretion in Boys with Delayed Adolescence. The Journal of Clinical Endocrinology & Metabolism, 43(6), 1386–1390. [DOI:10.1210/jcem-43-6-1386]
Santer, M., Lawrence, M., Renz, S., Eminton, Z., Stuart, B., Sach, T. H., Pyne, S., Ridd, M. J., Francis, N., Soulsby, I., Thomas, K., Permyakova, N., Little, P., Muller, I., Nuttall, J., Griffiths, G., Thomas, K. S., & Layton, A. M. (2023). Effectiveness of spironolactone for women with acne vulgaris (SAFA) in England and Wales: pragmatic, multicentre, phase 3, double blind, randomised controlled trial. BMJ, 381, e074349. [DOI:10.1136/bmj-2022-074349]
Shaw, J. C. (1996). Antiandrogen and hormonal treatment of acne. Dermatologic Clinics, 14(4), 803–811. [DOI:10.1016/s0733-8635(05)70405-8]
Sivelle, P. C., Underwood, A. H., & Jelly, J. A. (1982). The effects of histamine H2 receptor antagonists on androgen action in vivo and dihydrotestosterone binding to the rat prostate androgen receptor in vitro. Biochemical Pharmacology, 31(5), 677–684. [DOI:10.1016/0006-2952(82)90449-x]
Skorodok, L. M., Savchenko, O. N., & Liss, V. L. (1983). Hypothalamo-hypophyseal-gonadal function in boys with irregular puberty. Neuroscience and Behavioral Physiology, 13(2), 141–145. [DOI:10.1007/bf01190800]
Snyder, B. W., Winneker, R. C., & Batzold, F. H. (1989). Endocrine profile of win 49596 in the rat: A novel androgen receptor antagonist. Journal of Steroid Biochemistry, 33(6), 1127–1132. [DOI:10.1016/0022-4731(89)90419-6]
Sofer, Y., Yaish, I., Yaron, M., Bach, M. Y., Stern, N., & Greenman, Y. (2020). Differential Endocrine and Metabolic Effects of Testosterone Suppressive Agents in Transgender Women. Endocrine Practice, 26(8), 883–890. [DOI:10.4158/ep-2020-0032]
SoRelle, J. A., Jiao, R., Gao, E., Veazey, J., Frame, I., Quinn, A. M., Day, P., Pagels, P., Gimpel, N., & Patel, K. (2019). Impact of Hormone Therapy on Laboratory Values in Transgender Patients. Clinical Chemistry, 65(1), 170–179. [DOI:10.1373/clinchem.2018.292730]
Spörl, S. (1978/1979). Über den Einfluß von Spironolacton (Aldactone®) auf den Stoffwechsel von Nebennierenrindenhormonen und von Testosteron bei Gesunden mit diätinduziertem Hyperaldosterismus. [On the influence of spironolactone (Aldactone®) on the metabolism of adrenocortical hormones and testosterone in healthy individuals with diet-induced hyperaldosteronism.] (Doctoral dissertation, Freie Universität, Berlin.) [Google Scholar] [Google Books] [WorldCat] [PDF]
Spratt, L. V., Reardon, E., Olshan, J. S., & Eimicke, T. (2014). Efficacy of Testosterone (T) or Estradiol (E2) Therapy without a GnRH Agonist or Progestin to Suppress Endogenous Gonadal Activity in Transsexual Patients. Endocrine Reviews, 35(Suppl 1) [Endocrine Society’s 96th Annual Meeting and Expo, June 21–24, 2014 – Chicago], ?–? (abstract no. OR42-2). [DOI:10.1093/edrv/35.supp.1] [URL]
Stewart, I., Spratt, L., Craig, W., Olshan, J., & Spratt, D. (2018). The efficacy of testosterone or estradiol therapy without a GnRH agonist or progestin to suppress endogenous gonadal activity in transgender patients. Fertility and Sterility, 110(4 Suppl) [Scientific Congress Supplement: Oral and Poster Session Abstracts 06 October 2018 - 10 October 2018], e21–e21 (abstract no. O-44). [Google Scholar] [DOI:10.1016/j.fertnstert.2018.07.075]
Stripp, B., Taylor, A. A., Bartter, F. C., Gillette, J. R., Loriaux, D. L., Easley, R., & Menard, R. H. (1975). Effect of Spironolactone on Sex Hormones in Man. The Journal of Clinical Endocrinology & Metabolism, 41(4), 777–781. [DOI:10.1210/jcem-41-4-777]
Taylor, A. A., Rollins, D., Snodgrass, W., & Bartter, F. (1976). Effect of spironolactone (S) on adrenal and gonadal steroids in man. Clinical Research, 24(3), A279–A279. [Google Scholar] [PDF]
Unlühizarci, K., Everest, H., Bayram, F., & Keleştimur, F. (2002). Comparison of spironolactone and spironolactone plus finasteride in the treatment of hirsutism. Fertility and Sterility, 78(6), 1331–1333. [DOI:10.1016/s0015-0282(02)04294-2]
Tidd, M. J., Horth, C. E., Ramsay, L. E., Shelton, J. R., & Palmer, R. F. (1978). Endocrine effects of spironolactone in man. Clinical Endocrinology, 9(5), 389–399. [DOI:10.1111/j.1365-2265.1978.tb03578.x]
van Zuuren, E., & Fedorowicz, Z. (2016). Interventions for hirsutism excluding laser and photoepilation therapy alone: abridged Cochrane systematic review including GRADE assessments. British Journal of Dermatology, 175(1), 45–61. [DOI:10.1111/bjd.14486]
Walsh, P. C., & Siiteri, P. K. (1975). Suppression of Plasma Androgens by Spironolactone in Castrated Men with Carcinoma of the Prostate. Journal of Urology, 114(2), 254–256. [DOI:10.1016/s0022-5347(17)67001-0]
Wang, C., Du, Y., Bi, L., Lin, X., Zhao, M., & Fan, W. (2023). The Efficacy and Safety of Oral and Topical Spironolactone in Androgenetic Alopecia Treatment: A Systematic Review. Clinical, Cosmetic and Investigational Dermatology, 16, 603–612. [DOI:10.2147/ccid.s398950]
Weissmann, A. (1985). Antiandrogenic Effects of Topically Applied Spironolactone on the Hamster Flank Organ. Archives of Dermatology, 121(1), 57–62. [DOI:10.1001/archderm.1985.01660010061019]
Yamasaki, K., Sawaki, M., Noda, S., Muroi, T., Takakura, S., Mitoma, H., Sakamoto, S., Nakai, M., & Yakabe, Y. (2004). Comparison of the Hershberger assay and androgen receptor binding assay of twelve chemicals. Toxicology, 195(2–3), 177–186. [DOI:10.1016/j.tox.2003.09.012]
Yang, W., Hong, T., Chang, X., Han, M., Gao, H., Pan, B., Zhao, Z., & Liu, Y. (2024). The efficacy of and user satisfaction with different antiandrogens in Chinese transgender women. International Journal of Transgender Health, advance online publication. [DOI:10.1080/26895269.2024.2323514]
Yazdabadi, A., & Sinclair, R. (2011). Treatment of female pattern hair loss with the androgen receptor antagonist flutamide. The Australasian Journal of Dermatology, 52(2), 132–134. [DOI:10.1111/j.1440-0960.2010.00735.x]
Zgliczynski, S., Baranowska, B., & Szymanowski, J. (1981). L’influence du spironolactone sur la concentration des gonatrophines et des hormones gonadiques dans l’hypertrophie prostatique. [The influence of spironolactone on the concentration of gonadotrophins and gonadal hormones in prostatic hypertrophy]. Journal d’Urologie, 87(9), 635–638. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF] [Translation]
Zimmerman, Y., Eijkemans, M. J., Coelingh Bennink, H. J., Blankenstein, M. A., & Fauser, B. C. (2013). The effect of combined oral contraception on testosterone levels in healthy women: a systematic review and meta-analysis. Human Reproduction Update, 20(1), 76–105. [DOI:10.1093/humupd/dmt038]
\ No newline at end of file
diff --git a/transfemscience.org/articles/spiro-visceral-fat/index.html b/transfemscience.org/articles/spiro-visceral-fat/index.html
index 17dcc321..10653c30 100644
--- a/transfemscience.org/articles/spiro-visceral-fat/index.html
+++ b/transfemscience.org/articles/spiro-visceral-fat/index.html
@@ -1 +1 @@
-Spironolactone and Claims About Increased Visceral Fat in Transfeminine People - Transfeminine Science
Spironolactone and Claims About Increased Visceral Fat in Transfeminine People
By Aly | First published October 25, 2020 | Last modified December 18, 2025
A claim has been originated by some in the online transgender community that the antiandrogen spironolactone increases visceral fat in transfeminine people and that this effect is irreversible. Visceral fat is a type of adipose tissue located in the intra-abdominal region which surrounds the internal organs (viscera) in that area. In excess, visceral fat causes the abdomen to look bloated and unattractive. The supposed phenomenon of visceral fat accumulation with spironolactone has sometimes been referred to by people in the transgender community as “spiro belly”. The claim is based on theory—specifically that spironolactone has been found to increase levels of the corticosteroid hormone cortisol due to its antimineralocorticoid activity and cortisol is known to increase visceral fat, which together imply that spironolactone might likewise be able to increase visceral fat. It is also based on claimed anecdotal observations of transfeminine people taking spironolactone, which are said to corroborate the hypothesis. Despite these claims however, there is no actual direct scientific or medical literature to support the idea that spironolactone increases visceral fat, and there is considerable evidence contradicting it.
The influence of spironolactone on cortisol levels in clinical studies is variable and the magnitude of effect is limited. Hence, the clinical significance of increased cortisol levels with spironolactone is uncertain. Moreover, cortisol is an agonist of the glucocorticoid receptor (thereby producing glucocorticoid effects) and of the mineralocorticoid receptor (thereby producing mineralocorticoid effects). As already touched on, spironolactone has potent antimineralocorticoid activity (that is, mineralocorticoid receptor antagonism). Hence, even if spironolactone did increase cortisol levels enough to potentially increase visceral fat, its antimineralocorticoid activity could modify the capacity of cortisol to produce this effect. In relation to this, there is accumulating research to suggest that spironolactone may actually decrease visceral fat via its antimineralocorticoid activity. Antimineralocorticoids like spironolactone show antiadipogenic (anti-fat-accumulation) effects in vitro (Caprio et al., 2007; Caprio et al., 2011) and have been shown to decrease visceral fat in animals (Karakurt, 2008; Armani et al., 2014; Mammi et al., 2016; Olatunji et al., 2018). It is possible that they may also be able to do so in humans. Here are some notable literature excerpts relevant to this topic (Infante et al., 2019; Giordano, Frontini, & Cinti, 2016):
A possible explanation for [MR antagonists reducing cardiovascular morbidity and mortality more in patients with abdominal obesity] may be that patients with heart failure and abdominal obesity have higher aldosterone concentrations due to excessive secretion of specific aldosterone-releasing factors from [visceral adipose tissue]. […] Several studies on murine models of genetic and diet-induced obesity have widely reported beneficial effects of MR antagonism in terms of metabolic outcomes, such as body weight, fat mass, adipose tissue inflammation, insulin sensitivity, and lipid metabolism (Armani, Cinti, et al., 2014; Armani, Marzolla, et al., 2014; Garg & Adler, 2012; Guo et al., 2008; Hirata et al., 2009). Nevertheless, data on the outcomes of MR pharmacological blockade for prevention and treatment of obesity and metabolic syndrome are still scarce in humans (Tirosh et al., 2010). Of note, Tanko et al. demonstrated that the powerful MR antagonist drospirenone, in combination with estradiol, leads to a significant reduction of central fat mass and central fat mass/peripheral fat mass ratio in healthy post-menopausal women (Tankó & Christiansen, 2005). Moreover, another study has reported that MR antagonists significantly reduce body mass index and visceral fat area in patients with primary aldosteronism after a 1-year treatment period (Karashima et al., 2016). […] In light of these data, MR antagonism may be a useful therapeutic tool for prevention and treatment of cardiometabolic derangements observed in metabolic syndrome, even though additional studies are deemed necessary to confirm its impact on larger clinical settings.
An anti-obesity drug whose primary mode of action is to induce browning should act predominantly on visceral fat, thereby directly counteracting the major cause of obesity-associated metabolic disorders. Accumulation of abdominal visceral fat is, to some extent, linked to increased local levels and/or activity of androgen and glucocorticoid steroid hormones145,146. These hormones are also ligands of the mineralocorticoid receptors, which are found on white and brown adipocytes and could have a role in abdominal visceral fat accumulation and BAT to WAT conversion147–151.** […] **In this context, mineralocorticoid receptor antagonism has been shown to protect mice from the adverse obesogenic and metabolic effects of a high-fat diet via conversion of a substantial amount of visceral and subcutaneous WAT into BAT153. Given that mineralocorticoid receptor antagonists are widely prescribed diuretics, used to manage chronic heart failure, hyperaldosteronism and female hirsutism154, patients receiving such drugs should also be assessed for weight loss and metabolic parameters to establish whether these compounds have anti-obesity properties.
A number of studies have assessed the influence of antimineralocorticoids like spironolactone and eplerenone (another antimineralocorticoid) on visceral fat in humans. Spironolactone (12.5–100 mg/day) and eplerenone (25–100 mg/day) decreased visceral fat in people with pathologically high levels of aldosterone (a major endogenous mineralocorticoid hormone) (Karashima et al., 2016). A study of cisgender girls with polycystic ovary syndrome (PCOS) found that a combination of spironolactone (50 mg/day), pioglitazone, and metformin decreased visceral fat (Diaz et al., 2018). However, this study was of course confounded by the other medications. In addition to the preceding studies, many other clinical studies (at least 10) have assessed and similarly found no indication of increased visceral or abdominal fat with spironolactone (25–200 mg/day) (as measured by visceral fat directly or by indirect related measures like waist circumference or waist–hip ratio) (Wild et al., 1991; Lovejoy et al., 1996; Ganie et al., 2004; Meyer, McGrath, & Teede, 2007; Karakurt et al., 2008; Vieira et al., 2012; Ganie et al., 2013; Harmanci et al., 2013; Leelaphiwat et al., 2015; Alpañés et al., 2017). I was not able to identify any studies assessing visceral fat with higher doses of spironolactone (>200 mg/day). Additional studies are also underway to assess the possibility that spironolactone could decrease visceral fat.
With regard to the anecdotal claims of spironolactone increasing visceral fat in transfeminine people, it’s important to note that anecdotes are unreliable and are considered to be the lowest form of evidence in medicine. This is for well-founded reasons—succinctly, anecdotes very often don’t hold up when rigorous studies are conducted. It’s probable that excess abdominal fat—a problem which afflicts many—has been misattributed to spironolactone rather than to the real causes in transfeminine people. It’s notable in this regard that androgens are known to increase visceral fat and that men have twice as much visceral fat as women on average (Blouin, Boivin, & Tchernof, 2008; Zerradi et al., 2014). It’s possible that many transfeminine people may have excess visceral fat due to prior androgen exposure and that this visceral fat may not fully reverse with hormone therapy. As we know, hormone therapy unfortunately isn’t able to reverse all established bodily sexual dimorphism.
Besides increased visceral fat, many other serious adverse effects with spironolactone have been claimed. However, these claimed adverse effects are likewise based on anecdotes and theory, and there is a lack of direct clinical evidence to support such side effects. In actuality, spironolactone even at high doses appears to be well-tolerated per studies and systematic reviews. The claimed side effects of spironolactone may actually largely be due to phenomena like nocebo and misattribution—which can be controlled for in systematic studies but not in the case of anecdotal observations.
To summarize, no research, animal or clinical, has found increased visceral fat with spironolactone, and there is accumulating evidence that spironolactone may cause the very opposite effect. More studies are needed to further characterize this possible benefit of spironolactone in humans however.
Update: Talathi et al. (2025)
The following clinical study of hormone therapy in transfeminine people was published by Talathi and colleagues in December 2025:
Talathi, R., Juhasz, V., Delgado, M., Quinaglia, T., Ghamari, A., Wang, M., Alhallak, I., Stinebaugh, S., Campbell, S., Stockman, S. L., Ozturk, M. A., Ahmadi, S. M., Looby, S. E., Lee, H., Poteat, T. C., Szczepaniak, L. S., Zanni, M. V., Neilan, T. G., & Toribio, M. (2025). Visceral adipose tissue and liver fat on 17-beta estradiol-dominant gender-affirming hormone therapy: A US-based cohort. The Journal of Clinical Endocrinology and Metabolism, online ahead of print. [DOI:10.1210/clinem/dgaf665]
It was a 12-month prospective observational study of hormone therapy with estradiol and an antiandrogen in 26 transfeminine people in the United States. The primary aim of the study was to assess the effects of feminizing hormone therapy on visceral and liver fat in transfeminine people. The individuals in the study were either newly or very recently initiating hormone therapy. The antiandrogen used was spironolactone in 24 of 26 (92%) individuals. Of the 26 people, 14 (56%) were also on a progestogen, which was bioidentical progesterone in all but one case. The spironolactone dose used was median 100 mg/day (IQR 50 to 100 mg/day) at the start of the study and was median 125 mg/day (IQR 100 to 200 mg/day) at the end of the study. As the dosage spread statistic was interquartile range (IQR), a subset of people in the study appear to have been on spironolactone doses in excess of 200 mg/day. Estradiol levels were 188 pg/mL (690 pmol/L) and testosterone levels were 16 ng/dL (0.55 nmol/L) at the end of the study and were both within the normal and acceptable female range.
Visceral fat was assessed via dual-energy X-ray absorptiometry (DEXA) and liver fat was assessed via magnetic resonance spectroscopy (MRS). After 12 months, visceral adipose tissue mass non-significantly decreased from 308 g to 250 g (–18.8%; p = 0.25) and visceral adipose tissue volume non-significantly decreased from 332 cm3 to 271 cm3 (–18.4%; p = 0.25). In addition, intrahepatic triglyceride content (i.e., liver fat) decreased significantly from 0.9% to 0.8% (–11%; p = 0.03). In contrast to the case of visceral and liver fat, total body fat mass and percentage both significantly increased (+2.8 kg (+6.2 lbs) and +2.8%, respectively). Body weight, body mass index (BMI), waist circumference, and waist–hip ratio (WHR) all did not significantly or importantly change. Other metabolic parameters were also reported.
There are some limitations of this study, such as it not having control or comparison groups, the sample size being small, the median spironolactone dose being on the lower side of the clinical range used in transfeminine people, and the spironolactone dose being variable and increased over the course of the study rather than fixed. In any case, the results of this study do not support the notion that spironolactone increases visceral fat in transfeminine people. Instead, there is a clear trend for visceral fat decreasing with hormone therapy including spironolactone in transfeminine people. This was even though total body fat (i.e., subcutaneous and visceral together) showed the opposite pattern and increased, which is notably an expected effect in line with feminization of fat distribution that occurs in conjunction with decreased muscle mass. Moreover, it was the case even though about 25% of the people in the study on spironolactone were treated with doses of 200 mg/day or more. The observed trends towards decreased visceral fat are in line with other studies of hormone therapy in transfeminine people that did not employ spironolactone, in which visceral fat was significantly decreased, and suggest that spironolactone-containing regimens may not differ in this regard from other regimens. Based on the findings of this study, transfeminine people can feel reassured about claims that spironolactone causes visceral fat accumulation and may instead more plausibly expect the opposite with such regimens.
References
Alpañés, M., Álvarez-Blasco, F., Fernández-Durán, E., Luque-Ramírez, M., & Escobar-Morreale, H. F. (2017). Combined oral contraceptives plus spironolactone compared with metformin in women with polycystic ovary syndrome: a one-year randomized clinical trial. European Journal of Endocrinology, 177(5), 399–408. [DOI:10.1530/eje-17-0516]
Armani, A., Cinti, F., Marzolla, V., Morgan, J., Cranston, G. A., Antelmi, A., Carpinelli, G., Canese, R., Pagotto, U., Quarta, C., Malorni, W., Matarrese, P., Marconi, M., Fabbri, A., Rosano, G., Cinti, S., Young, M. J., & Caprio, M. (2014). Mineralocorticoid receptor antagonism induces browning of white adipose tissue through impairment of autophagy and prevents adipocyte dysfunction in high‐fat‐diet‐fed mice. The FASEB Journal, 28(8), 3745–3757. [DOI:10.1096/fj.13-245415]
Blouin, K., Boivin, A., & Tchernof, A. (2008). Androgens and body fat distribution. The Journal of Steroid Biochemistry and Molecular Biology, 108(3–5), 272–280. [DOI:10.1016/j.jsbmb.2007.09.001]
Caprio, M., Fève, B., Claës, A., Viengchareun, S., Lombès, M., & Zennaro, M. (2007). Pivotal role of the mineralocorticoid receptor in corticosteroid‐induced adipogenesis. The FASEB Journal, 21(9), 2185–2194. [DOI:10.1096/fj.06-7970com]
Caprio, M., Antelmi, A., Chetrite, G., Muscat, A., Mammi, C., Marzolla, V., Fabbri, A., Zennaro, M., & Fève, B. (2011). Antiadipogenic Effects of the Mineralocorticoid Receptor Antagonist Drospirenone: Potential Implications for the Treatment of Metabolic Syndrome. Endocrinology, 152(1), 113–125. [DOI:10.1210/en.2010-0674]
Díaz, M., Gallego-Escuredo, J. M., López-Bermejo, A., de Zegher, F., Villarroya, F., & Ibáñez, L. (2018). Low-Dose Spironolactone-Pioglitazone-Metformin Normalizes Circulating Fetuin-A Concentrations in Adolescent Girls with Polycystic Ovary Syndrome. International Journal of Endocrinology, 2018, 4192940. [DOI:10.1155/2018/4192940]
Ganie, M. A., Khurana, M. L., Eunice, M., Gulati, M., Dwivedi, S. N., & Ammini, A. C. (2004). Comparison of Efficacy of Spironolactone with Metformin in the Management of Polycystic Ovary Syndrome: An Open-Labeled Study. The Journal of Clinical Endocrinology & Metabolism, 89(6), 2756–2762. [DOI:10.1210/jc.2003-031780]
Ganie, M. A., Khurana, M. L., Nisar, S., Shah, P. A., Shah, Z. A., Kulshrestha, B., Gupta, N., Zargar, M. A., Wani, T. A., Mudasir, S., Mir, F. A., & Taing, S. (2013). Improved Efficacy of Low-Dose Spironolactone and Metformin Combination Than Either Drug Alone in the Management of Women With Polycystic Ovary Syndrome (PCOS): A Six-Month, Open-Label Randomized Study. The Journal of Clinical Endocrinology & Metabolism, 98(9), 3599–3607. [DOI:10.1210/jc.2013-1040]
Giordano, A., Frontini, A., & Cinti, S. (2016). Convertible visceral fat as a therapeutic target to curb obesity. Nature Reviews Drug Discovery, 15(6), 405–424. [DOI:10.1038/nrd.2016.31]
Harmanci, A., Cinar, N., Bayraktar, M., & Yildiz, B. O. (2012). Oral contraceptive plus antiandrogen therapy and cardiometabolic risk in polycystic ovary syndrome. Clinical Endocrinology, 78(1), 120–125. [DOI:10.1111/j.1365-2265.2012.04466.x]
Infante, M., Armani, A., Marzolla, V., Fabbri, A., & Caprio, M. (2019). Adipocyte Mineralocorticoid Receptor. In Litwack, G. (Ed.). Aldosterone (Vitamins and Hormones, Volume 109) (pp. 189–209). Amsterdam: Elsevier Academic Press. [DOI:10.1016/bs.vh.2018.10.005]
Karakurt, F., Sahin, I., Güler, S., Demirbas, B., Culha, C., Serter, R., Aral, Y., & Bavbek, N. (2008). Comparison of the clinical efficacy of flutamide and spironolactone plus ethinyloestradiol/cyproterone acetate in the treatment of hirsutism: A randomised controlled study. Advances in Therapy, 25(4), 321–328. [DOI:10.1007/s12325-008-0039-5]
Karashima, S., Yoneda, T., Kometani, M., Ohe, M., Mori, S., Sawamura, T., Furukawa, K., Seta, T., Yamagishi, M., & Takeda, Y. (2015). Comparison of eplerenone and spironolactone for the treatment of primary aldosteronism. Hypertension Research, 39(3), 133–137. [DOI:10.1038/hr.2015.129]
Leelaphiwat, S., Jongwutiwes, T., Lertvikool, S., Tabcharoen, C., Sukprasert, M., Rattanasiri, S., & Weerakiet, S. (2014). Comparison of desogestrel/ethinyl estradiol plus spironolactone versus cyproterone acetate/ethinyl estradiol in the treatment of polycystic ovary syndrome: A randomized controlled trial. Journal of Obstetrics and Gynaecology Research, 41(3), 402–410. [DOI:10.1111/jog.12543]
Lovejoy, J. C., Bray, G. A., Bourgeois, M. O., Macchiavelli, R., Rood, J. C., Greeson, C., & Partington, C. (1996). Exogenous androgens influence body composition and regional body fat distribution in obese postmenopausal women–a clinical research center study. The Journal of Clinical Endocrinology & Metabolism, 81(6), 2198–2203. [DOI:10.1210/jcem.81.6.8964851]
Mammi, C., Marzolla, V., Armani, A., Feraco, A., Antelmi, A., Maslak, E., Chlopicki, S., Cinti, F., Hunt, H., Fabbri, A., & Caprio, M. (2016). A novel combined glucocorticoid-mineralocorticoid receptor selective modulator markedly prevents weight gain and fat mass expansion in mice fed a high-fat diet. International Journal of Obesity, 40(6), 964–972. [DOI:10.1038/ijo.2016.13]
Meyer, C., McGrath, B. P., & Teede, H. J. (2007). Effects of Medical Therapy on Insulin Resistance and the Cardiovascular System in Polycystic Ovary Syndrome. Diabetes Care, 30(3), 471–478. [DOI:10.2337/dc06-0618]
Olatunji, L. A., Adeyanju, O. A., Michael, O. S., Usman, T. O., Tostes, R. C., & Soladoye, A. O. (2019). Ameliorative effect of low-dose spironolactone on obesity and insulin resistance is through replenishment of estrogen in ovariectomized rats. Canadian Journal of Physiology and Pharmacology, 97(1), 65–74. [DOI:10.1139/cjpp-2018-0416]
Talathi, R., Juhasz, V., Delgado, M., Quinaglia, T., Ghamari, A., Wang, M., Alhallak, I., Stinebaugh, S., Campbell, S., Stockman, S. L., Ozturk, M. A., Ahmadi, S. M., Looby, S. E., Lee, H., Poteat, T. C., Szczepaniak, L. S., Zanni, M. V., Neilan, T. G., & Toribio, M. (2025). Visceral adipose tissue and liver fat on 17-beta estradiol-dominant gender-affirming hormone therapy: A US-based cohort. The Journal of Clinical Endocrinology and Metabolism, online ahead of print. [DOI:10.1210/clinem/dgaf665]
Vieira, C. S., Martins, W. P., Fernandes, J. B., Soares, G. M., dos Reis, R. M., de Sá, M. F., & Ferriani, R. A. (2012). The effects of 2 mg chlormadinone acetate/30 mcg ethinylestradiol, alone or combined with spironolactone, on cardiovascular risk markers in women with polycystic ovary syndrome. Contraception, 86(3), 268–275. [DOI:10.1016/j.contraception.2011.12.011]
Wild, R. A., Demers, L. M., Applebaum-Bowden, D., & Lenker, R. (1991). Hirsutism: Metabolic effects of two commonly used oral contraceptives and spironolactone. Contraception, 44(2), 113–124. [DOI:10.1016/0010-7824(91)90112-s]
Zerradi, M., Dereumetz, J., Boulet, M., & Tchernof, A. (2014). Androgens, body fat Distribution and Adipogenesis. Current Obesity Reports, 3(4), 396–403. [DOI:10.1007/s13679-014-0119-6]
\ No newline at end of file
+Spironolactone and Claims About Increased Visceral Fat in Transfeminine People - Transfeminine Science
Spironolactone and Claims About Increased Visceral Fat in Transfeminine People
By Aly | First published October 25, 2020 | Last modified December 18, 2025
A claim has been originated by some in the online transgender community that the antiandrogen spironolactone increases visceral fat in transfeminine people and that this effect is irreversible. Visceral fat is a type of adipose tissue located in the intra-abdominal region which surrounds the internal organs (viscera) in that area. In excess, visceral fat causes the abdomen to look bloated and unattractive. The supposed phenomenon of visceral fat accumulation with spironolactone has sometimes been referred to by people in the transgender community as “spiro belly”. The claim is based on theory—specifically that spironolactone has been found to increase levels of the corticosteroid hormone cortisol due to its antimineralocorticoid activity and cortisol is known to increase visceral fat, which together imply that spironolactone might likewise be able to increase visceral fat. It is also based on claimed anecdotal observations of transfeminine people taking spironolactone, which are said to corroborate the hypothesis. Despite these claims however, there is no actual direct scientific or medical literature to support the idea that spironolactone increases visceral fat, and there is considerable evidence contradicting it.
The influence of spironolactone on cortisol levels in clinical studies is variable and the magnitude of effect is limited. Hence, the clinical significance of increased cortisol levels with spironolactone is uncertain. Moreover, cortisol is an agonist of the glucocorticoid receptor (thereby producing glucocorticoid effects) and of the mineralocorticoid receptor (thereby producing mineralocorticoid effects). As already touched on, spironolactone has potent antimineralocorticoid activity (that is, mineralocorticoid receptor antagonism). Hence, even if spironolactone did increase cortisol levels enough to potentially increase visceral fat, its antimineralocorticoid activity could modify the capacity of cortisol to produce this effect. In relation to this, there is accumulating research to suggest that spironolactone may actually decrease visceral fat via its antimineralocorticoid activity. Antimineralocorticoids like spironolactone show antiadipogenic (anti-fat-accumulation) effects in vitro (Caprio et al., 2007; Caprio et al., 2011) and have been shown to decrease visceral fat in animals (Karakurt, 2008; Armani et al., 2014; Mammi et al., 2016; Olatunji et al., 2018). It is possible that they may also be able to do so in humans. Here are some notable literature excerpts relevant to this topic (Infante et al., 2019; Giordano, Frontini, & Cinti, 2016):
A possible explanation for [MR antagonists reducing cardiovascular morbidity and mortality more in patients with abdominal obesity] may be that patients with heart failure and abdominal obesity have higher aldosterone concentrations due to excessive secretion of specific aldosterone-releasing factors from [visceral adipose tissue]. […] Several studies on murine models of genetic and diet-induced obesity have widely reported beneficial effects of MR antagonism in terms of metabolic outcomes, such as body weight, fat mass, adipose tissue inflammation, insulin sensitivity, and lipid metabolism (Armani, Cinti, et al., 2014; Armani, Marzolla, et al., 2014; Garg & Adler, 2012; Guo et al., 2008; Hirata et al., 2009). Nevertheless, data on the outcomes of MR pharmacological blockade for prevention and treatment of obesity and metabolic syndrome are still scarce in humans (Tirosh et al., 2010). Of note, Tanko et al. demonstrated that the powerful MR antagonist drospirenone, in combination with estradiol, leads to a significant reduction of central fat mass and central fat mass/peripheral fat mass ratio in healthy post-menopausal women (Tankó & Christiansen, 2005). Moreover, another study has reported that MR antagonists significantly reduce body mass index and visceral fat area in patients with primary aldosteronism after a 1-year treatment period (Karashima et al., 2016). […] In light of these data, MR antagonism may be a useful therapeutic tool for prevention and treatment of cardiometabolic derangements observed in metabolic syndrome, even though additional studies are deemed necessary to confirm its impact on larger clinical settings.
An anti-obesity drug whose primary mode of action is to induce browning should act predominantly on visceral fat, thereby directly counteracting the major cause of obesity-associated metabolic disorders. Accumulation of abdominal visceral fat is, to some extent, linked to increased local levels and/or activity of androgen and glucocorticoid steroid hormones145,146. These hormones are also ligands of the mineralocorticoid receptors, which are found on white and brown adipocytes and could have a role in abdominal visceral fat accumulation and BAT to WAT conversion147–151.** […] **In this context, mineralocorticoid receptor antagonism has been shown to protect mice from the adverse obesogenic and metabolic effects of a high-fat diet via conversion of a substantial amount of visceral and subcutaneous WAT into BAT153. Given that mineralocorticoid receptor antagonists are widely prescribed diuretics, used to manage chronic heart failure, hyperaldosteronism and female hirsutism154, patients receiving such drugs should also be assessed for weight loss and metabolic parameters to establish whether these compounds have anti-obesity properties.
A number of studies have assessed the influence of antimineralocorticoids like spironolactone and eplerenone (another antimineralocorticoid) on visceral fat in humans. Spironolactone (12.5–100 mg/day) and eplerenone (25–100 mg/day) decreased visceral fat in people with pathologically high levels of aldosterone (a major endogenous mineralocorticoid hormone) (Karashima et al., 2016). A study of cisgender girls with polycystic ovary syndrome (PCOS) found that a combination of spironolactone (50 mg/day), pioglitazone, and metformin decreased visceral fat (Diaz et al., 2018). However, this study was of course confounded by the other medications. In addition to the preceding studies, many other clinical studies (at least 10) have assessed and similarly found no indication of increased visceral or abdominal fat with spironolactone (25–200 mg/day) (as measured by visceral fat directly or by indirect related measures like waist circumference or waist–hip ratio) (Wild et al., 1991; Lovejoy et al., 1996; Ganie et al., 2004; Meyer, McGrath, & Teede, 2007; Karakurt et al., 2008; Vieira et al., 2012; Ganie et al., 2013; Harmanci et al., 2013; Leelaphiwat et al., 2015; Alpañés et al., 2017). I was not able to identify any studies assessing visceral fat with higher doses of spironolactone (>200 mg/day). Additional studies are also underway to assess the possibility that spironolactone could decrease visceral fat.
With regard to the anecdotal claims of spironolactone increasing visceral fat in transfeminine people, it’s important to note that anecdotes are unreliable and are considered to be the lowest form of evidence in medicine. This is for well-founded reasons—succinctly, anecdotes very often don’t hold up when rigorous studies are conducted. It’s probable that excess abdominal fat—a problem which afflicts many—has been misattributed to spironolactone rather than to the real causes in transfeminine people. It’s notable in this regard that androgens are known to increase visceral fat and that men have twice as much visceral fat as women on average (Blouin, Boivin, & Tchernof, 2008; Zerradi et al., 2014). It’s possible that many transfeminine people may have excess visceral fat due to prior androgen exposure and that this visceral fat may not fully reverse with hormone therapy. As we know, hormone therapy unfortunately isn’t able to reverse all established bodily sexual dimorphism.
Besides increased visceral fat, many other serious adverse effects with spironolactone have been claimed. However, these claimed adverse effects are likewise based on anecdotes and theory, and there is a lack of direct clinical evidence to support such side effects. In actuality, spironolactone even at high doses appears to be well-tolerated per studies and systematic reviews. The claimed side effects of spironolactone may actually largely be due to phenomena like nocebo and misattribution—which can be controlled for in systematic studies but not in the case of anecdotal observations.
To summarize, no research, animal or clinical, has found increased visceral fat with spironolactone, and there is accumulating evidence that spironolactone may cause the very opposite effect. More studies are needed to further characterize this possible benefit of spironolactone in humans however.
Update: Talathi et al. (2025)
The following clinical study of hormone therapy in transfeminine people was published by Talathi and colleagues in December 2025:
Talathi, R., Juhasz, V., Delgado, M., Quinaglia, T., Ghamari, A., Wang, M., Alhallak, I., Stinebaugh, S., Campbell, S., Stockman, S. L., Ozturk, M. A., Ahmadi, S. M., Looby, S. E., Lee, H., Poteat, T. C., Szczepaniak, L. S., Zanni, M. V., Neilan, T. G., & Toribio, M. (2025). Visceral adipose tissue and liver fat on 17-beta estradiol-dominant gender-affirming hormone therapy: A US-based cohort. The Journal of Clinical Endocrinology and Metabolism, online ahead of print. [DOI:10.1210/clinem/dgaf665]
It was a 12-month prospective observational study of hormone therapy with estradiol and an antiandrogen in 26 transfeminine people in the United States. The primary aim of the study was to assess the effects of feminizing hormone therapy on visceral and liver fat in transfeminine people. The individuals in the study were either newly or very recently initiating hormone therapy. The antiandrogen used was spironolactone in 24 of 26 (92%) individuals. Of the 26 people, 14 (56%) were also on a progestogen, which was bioidentical progesterone in all but one case. The spironolactone dose used was median 100 mg/day (IQR 50 to 100 mg/day) at the start of the study and was median 125 mg/day (IQR 100 to 200 mg/day) at the end of the study. As the dosage spread statistic was interquartile range (IQR), a subset of people in the study appear to have been on spironolactone doses in excess of 200 mg/day. Estradiol levels were 188 pg/mL (690 pmol/L) and testosterone levels were 16 ng/dL (0.55 nmol/L) at the end of the study and were both within the normal and acceptable female range.
Visceral fat was assessed via dual-energy X-ray absorptiometry (DEXA) and liver fat was assessed via magnetic resonance spectroscopy (MRS). After 12 months, visceral adipose tissue mass non-significantly decreased from 308 g to 250 g (–18.8%; p = 0.25) and visceral adipose tissue volume non-significantly decreased from 332 cm3 to 271 cm3 (–18.4%; p = 0.25). In addition, intrahepatic triglyceride content (i.e., liver fat) decreased significantly from 0.9% to 0.8% (–11%; p = 0.03). In contrast to the case of visceral and liver fat, total body fat mass and percentage both significantly increased (+2.8 kg (+6.2 lbs) and +2.8%, respectively). Body weight, body mass index (BMI), waist circumference, and waist–hip ratio (WHR) all did not significantly or importantly change. Other metabolic parameters were also reported.
There are some limitations of this study, such as it not having control or comparison groups, the sample size being small, the median spironolactone dose being on the lower side of the clinical range used in transfeminine people, and the spironolactone dose being variable and increased over the course of the study rather than fixed. In any case, the results of this study do not support the notion that spironolactone increases visceral fat in transfeminine people. Instead, there is a clear trend for visceral fat decreasing with hormone therapy including spironolactone in transfeminine people. This was even though total body fat (i.e., subcutaneous and visceral together) showed the opposite pattern and increased, which is notably an expected effect in line with feminization of fat distribution that occurs in conjunction with decreased muscle mass. Moreover, it was the case even though about 25% of the people in the study on spironolactone were treated with doses of 200 mg/day or more. The observed trends towards decreased visceral fat are in line with other studies of hormone therapy in transfeminine people that did not employ spironolactone, in which visceral fat was significantly decreased, and suggest that spironolactone-containing regimens may not differ in this regard from other regimens. Based on the findings of this study, transfeminine people can feel reassured about claims that spironolactone causes visceral fat accumulation and may instead more plausibly expect the opposite with such regimens.
References
Alpañés, M., Álvarez-Blasco, F., Fernández-Durán, E., Luque-Ramírez, M., & Escobar-Morreale, H. F. (2017). Combined oral contraceptives plus spironolactone compared with metformin in women with polycystic ovary syndrome: a one-year randomized clinical trial. European Journal of Endocrinology, 177(5), 399–408. [DOI:10.1530/eje-17-0516]
Armani, A., Cinti, F., Marzolla, V., Morgan, J., Cranston, G. A., Antelmi, A., Carpinelli, G., Canese, R., Pagotto, U., Quarta, C., Malorni, W., Matarrese, P., Marconi, M., Fabbri, A., Rosano, G., Cinti, S., Young, M. J., & Caprio, M. (2014). Mineralocorticoid receptor antagonism induces browning of white adipose tissue through impairment of autophagy and prevents adipocyte dysfunction in high‐fat‐diet‐fed mice. The FASEB Journal, 28(8), 3745–3757. [DOI:10.1096/fj.13-245415]
Blouin, K., Boivin, A., & Tchernof, A. (2008). Androgens and body fat distribution. The Journal of Steroid Biochemistry and Molecular Biology, 108(3–5), 272–280. [DOI:10.1016/j.jsbmb.2007.09.001]
Caprio, M., Fève, B., Claës, A., Viengchareun, S., Lombès, M., & Zennaro, M. (2007). Pivotal role of the mineralocorticoid receptor in corticosteroid‐induced adipogenesis. The FASEB Journal, 21(9), 2185–2194. [DOI:10.1096/fj.06-7970com]
Caprio, M., Antelmi, A., Chetrite, G., Muscat, A., Mammi, C., Marzolla, V., Fabbri, A., Zennaro, M., & Fève, B. (2011). Antiadipogenic Effects of the Mineralocorticoid Receptor Antagonist Drospirenone: Potential Implications for the Treatment of Metabolic Syndrome. Endocrinology, 152(1), 113–125. [DOI:10.1210/en.2010-0674]
Díaz, M., Gallego-Escuredo, J. M., López-Bermejo, A., de Zegher, F., Villarroya, F., & Ibáñez, L. (2018). Low-Dose Spironolactone-Pioglitazone-Metformin Normalizes Circulating Fetuin-A Concentrations in Adolescent Girls with Polycystic Ovary Syndrome. International Journal of Endocrinology, 2018, 4192940. [DOI:10.1155/2018/4192940]
Ganie, M. A., Khurana, M. L., Eunice, M., Gulati, M., Dwivedi, S. N., & Ammini, A. C. (2004). Comparison of Efficacy of Spironolactone with Metformin in the Management of Polycystic Ovary Syndrome: An Open-Labeled Study. The Journal of Clinical Endocrinology & Metabolism, 89(6), 2756–2762. [DOI:10.1210/jc.2003-031780]
Ganie, M. A., Khurana, M. L., Nisar, S., Shah, P. A., Shah, Z. A., Kulshrestha, B., Gupta, N., Zargar, M. A., Wani, T. A., Mudasir, S., Mir, F. A., & Taing, S. (2013). Improved Efficacy of Low-Dose Spironolactone and Metformin Combination Than Either Drug Alone in the Management of Women With Polycystic Ovary Syndrome (PCOS): A Six-Month, Open-Label Randomized Study. The Journal of Clinical Endocrinology & Metabolism, 98(9), 3599–3607. [DOI:10.1210/jc.2013-1040]
Giordano, A., Frontini, A., & Cinti, S. (2016). Convertible visceral fat as a therapeutic target to curb obesity. Nature Reviews Drug Discovery, 15(6), 405–424. [DOI:10.1038/nrd.2016.31]
Harmanci, A., Cinar, N., Bayraktar, M., & Yildiz, B. O. (2012). Oral contraceptive plus antiandrogen therapy and cardiometabolic risk in polycystic ovary syndrome. Clinical Endocrinology, 78(1), 120–125. [DOI:10.1111/j.1365-2265.2012.04466.x]
Infante, M., Armani, A., Marzolla, V., Fabbri, A., & Caprio, M. (2019). Adipocyte Mineralocorticoid Receptor. In Litwack, G. (Ed.). Aldosterone (Vitamins and Hormones, Volume 109) (pp. 189–209). Amsterdam: Elsevier Academic Press. [DOI:10.1016/bs.vh.2018.10.005]
Karakurt, F., Sahin, I., Güler, S., Demirbas, B., Culha, C., Serter, R., Aral, Y., & Bavbek, N. (2008). Comparison of the clinical efficacy of flutamide and spironolactone plus ethinyloestradiol/cyproterone acetate in the treatment of hirsutism: A randomised controlled study. Advances in Therapy, 25(4), 321–328. [DOI:10.1007/s12325-008-0039-5]
Karashima, S., Yoneda, T., Kometani, M., Ohe, M., Mori, S., Sawamura, T., Furukawa, K., Seta, T., Yamagishi, M., & Takeda, Y. (2015). Comparison of eplerenone and spironolactone for the treatment of primary aldosteronism. Hypertension Research, 39(3), 133–137. [DOI:10.1038/hr.2015.129]
Leelaphiwat, S., Jongwutiwes, T., Lertvikool, S., Tabcharoen, C., Sukprasert, M., Rattanasiri, S., & Weerakiet, S. (2014). Comparison of desogestrel/ethinyl estradiol plus spironolactone versus cyproterone acetate/ethinyl estradiol in the treatment of polycystic ovary syndrome: A randomized controlled trial. Journal of Obstetrics and Gynaecology Research, 41(3), 402–410. [DOI:10.1111/jog.12543]
Lovejoy, J. C., Bray, G. A., Bourgeois, M. O., Macchiavelli, R., Rood, J. C., Greeson, C., & Partington, C. (1996). Exogenous androgens influence body composition and regional body fat distribution in obese postmenopausal women–a clinical research center study. The Journal of Clinical Endocrinology & Metabolism, 81(6), 2198–2203. [DOI:10.1210/jcem.81.6.8964851]
Mammi, C., Marzolla, V., Armani, A., Feraco, A., Antelmi, A., Maslak, E., Chlopicki, S., Cinti, F., Hunt, H., Fabbri, A., & Caprio, M. (2016). A novel combined glucocorticoid-mineralocorticoid receptor selective modulator markedly prevents weight gain and fat mass expansion in mice fed a high-fat diet. International Journal of Obesity, 40(6), 964–972. [DOI:10.1038/ijo.2016.13]
Meyer, C., McGrath, B. P., & Teede, H. J. (2007). Effects of Medical Therapy on Insulin Resistance and the Cardiovascular System in Polycystic Ovary Syndrome. Diabetes Care, 30(3), 471–478. [DOI:10.2337/dc06-0618]
Olatunji, L. A., Adeyanju, O. A., Michael, O. S., Usman, T. O., Tostes, R. C., & Soladoye, A. O. (2019). Ameliorative effect of low-dose spironolactone on obesity and insulin resistance is through replenishment of estrogen in ovariectomized rats. Canadian Journal of Physiology and Pharmacology, 97(1), 65–74. [DOI:10.1139/cjpp-2018-0416]
Talathi, R., Juhasz, V., Delgado, M., Quinaglia, T., Ghamari, A., Wang, M., Alhallak, I., Stinebaugh, S., Campbell, S., Stockman, S. L., Ozturk, M. A., Ahmadi, S. M., Looby, S. E., Lee, H., Poteat, T. C., Szczepaniak, L. S., Zanni, M. V., Neilan, T. G., & Toribio, M. (2025). Visceral adipose tissue and liver fat on 17-beta estradiol-dominant gender-affirming hormone therapy: A US-based cohort. The Journal of Clinical Endocrinology and Metabolism, online ahead of print. [DOI:10.1210/clinem/dgaf665]
Vieira, C. S., Martins, W. P., Fernandes, J. B., Soares, G. M., dos Reis, R. M., de Sá, M. F., & Ferriani, R. A. (2012). The effects of 2 mg chlormadinone acetate/30 mcg ethinylestradiol, alone or combined with spironolactone, on cardiovascular risk markers in women with polycystic ovary syndrome. Contraception, 86(3), 268–275. [DOI:10.1016/j.contraception.2011.12.011]
Wild, R. A., Demers, L. M., Applebaum-Bowden, D., & Lenker, R. (1991). Hirsutism: Metabolic effects of two commonly used oral contraceptives and spironolactone. Contraception, 44(2), 113–124. [DOI:10.1016/0010-7824(91)90112-s]
Zerradi, M., Dereumetz, J., Boulet, M., & Tchernof, A. (2014). Androgens, body fat Distribution and Adipogenesis. Current Obesity Reports, 3(4), 396–403. [DOI:10.1007/s13679-014-0119-6]
\ No newline at end of file
diff --git a/transfemscience.org/articles/sublingual-e2-transfem/index.html b/transfemscience.org/articles/sublingual-e2-transfem/index.html
index 356ceb0a..5a703b8e 100644
--- a/transfemscience.org/articles/sublingual-e2-transfem/index.html
+++ b/transfemscience.org/articles/sublingual-e2-transfem/index.html
@@ -1 +1 @@
-An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People - Transfeminine Science
An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People
By Sam | First published June 11, 2021 | Last modified August 14, 2025
Abstract / TL;DR
Sublingually-administered estradiol is an alternative route of administration to oral estradiol that has been used by a limited number of gender-affirming care providers internationally. We do not currently know if sublingual estradiol results in better, worse, or similar feminisation as other routes of administration because there is a paucity of clinical data in this area. There may be practical shortcomings associated with the sublingual route, however clinical experience suggest it to be effective and affordable when dosed correctly. Although much more research is clearly needed to properly characterise this route of administration, sublingual estradiol might have some advantageous properties and may be a useful alternative to oral estradiol for some transfeminine people.
Introduction
Although the most common way to administer medication in the form of pills or tablets is by the oral route, oral estradiol formulations can otherwise be taken sublingually or buccally (Kuhl, 2005). Sublingual administration is the administration of an oral pill or tablet by means of placing under the tongue to dissolve and be absorbed into the bloodstream. Buccal administration is similar and refers to placing the medication between the cheek and gums, where it also quickly dissolves and is absorbed (Gass et al., 2004; Bartlett & Maarschalk, 2012).
Many transfeminine people wonder or ask questions on online forums about the sublingual route of administration for estradiol. Some of the most common queries are “What doses of sublingual estradiol should I take?”, “How often should I take sublingual estradiol?”, “Is sublingual estradiol better than oral estradiol?” and so on.
Until very recently, published data about sublingual estradiol in transfeminine people was scarce, with only a very small number of relevant studies having considered it (eg: Jain, Kwan, & Forcier, 2019, Lim et al., 2019). Accordingly, most information about the sublingual route existed only in older studies of cisgender patient populations (Casper & Yen, 1981; Burnier et al., 1981; Price et al., 1997; Wren et al., 2003). In the last few years, there has been a renewed interest in sublingual estradiol within the literature, specifically with an eye towards gender-affirming hormone therapy. Consequently, many high-level publications including recent clinical guidelines and reviews now make reference to the sublingual route (Coleman et al., 2022; Sudhakar et al., 2023; Grock, Reema, & Ahern, 2024).
It is of note that, although the sublingual and buccal administration are distinct routes of administration, they are very similar to each other in how they are performed and in their pharmacology (Perloff, 1950; Chandrasekhara et al., 2002). As such, although the term “sublingual” has been ostensibly used in this literature review, much of the content here is applicable to buccal administration of estradiol as well.
Pharmacology of Sublingual Estradiol
While sublingual estradiol is not as widely used in clinical practice as oral estradiol and other formulations, a number of studies have examined its pharmacology. These studies include both samples of postmenopausal cisgender women and transfeminine people as well as other patient populations (Casper & Yen, 1981; Serhal & Craft, 1989; Cirrincione et al., 2021; Doll et al., 2022; Kariyawasam et al., 2025). Both oral estradiol and oral estradiol valerate tablets can be taken sublingually (Serhal, 1990).
After the administration of oral estradiol, the medication is heavily metabolised and inactivated into estrogen conjugates by the liver (Kuhl, 2005). In turn, these metabolites are gradually converted back into estradiol, which serves to prolong its half life (to approximately 13–20 hours) (Stanczyk, Archer, & Bhavnani, 2013). In contrast to oral estradiol, sublingual estradiol does not pass as extensively through the liver. Therefore, it does not undergo deactivation into clinically insignificant estrogen metabolites. Sublingually administered estradiol is absorbed rapidly into the bloodstream where it directly enters circulation. Consequently, it has greater bioavailability than oral estradiol, meaning that lower doses are needed to achieve similar area-under-the-curve (AUC) estradiol levels (Kuhl, 2005) (Figures 1 and 2). This is an advantage of sublingual estradiol over oral estradiol, as it allows for the use of lower doses. This in turn might reduce medication costs.
Because accidental swallowing of some of the estradiol seems probable, the sublingual route is, most likely, actually a combination of sublingual and oral delivery of estradiol (Lobo, 1987; Kuhl, 2005). A small pharamcokinetics study of transfeminine people reported that a single 1 mg dose of sublingual estradiol caused an average rise in the level of estradiol up to an average of 144 pg/mL (529 pmol/L) within one to two hours of administration. In contrast, a peak concentration of just 35 pg/mL (128 pmol/L) was found with the same dose of 1 mg administered orally (Doll et al., 2022). Thereafter, estradiol levels decreased rapidly. In another study, it was found that mean estradiol levels measured at any given point were 613 pmol/L (167 pg/mL) on sublingual estradiol (Kariyawasam et al., 2025). In this study, a wide range of doses were used and hence it is not possible to ascertain much about dose-specific peak concentrations. Similar findings have been reported in other studies of postmenopausal women, although a wide range of peak concentrations have been observed (Burnier et al., 1981; Price et al., 1997; Wren et al., 2003). Estradiol levels are found to rapidly rise on the order of about five to ten times that of the peak of oral estradiol, then rapidly decline, with an elimination half-life of only a few hours (Kuhl, 2005). Sublingual estradiol is somewhat analogous in this respect to intravenously administered estradiol, which also shows a rapid increase in estradiol levels and a very short elimination half-life (Kuhnz, Gansau, & Mahler, 1993). Another route of administration that is similar in this regard is intranasal administration (Devissaguet et al., 1999). Owing to the short half-life elimination of sublingual estradiol, it does not achieve as stable concentrations as other formulations do. This is a marked difference to other routes, such as oral estradiol, that produce much more stable hormone levels and that do not fluctuate as much over the course of the day. All these differences by themselves do not necessarily mean that sublingual estradiol is superior or inferior to oral estradiol (Safer, 2022; Sarvaideo, Doll, & Tangpricha, 2022). Nevertheless, they should be kept in mind when considering findings from relevant studies.
A range of estimates have been reported for the relative bioavailability of sublingual estradiol. One small randomised study of postmenopausal women found approximately 2.5-fold higher AUC levels of estradiol with sublingual estradiol than with the same doses of oral estradiol (Price et al., 1997). Other studies have reported relative bioavailability estimates for sublingual estradiol of up to five times that of oral estradiol (Pines et al., 1999). A study in marmoset monkeys found that the absolute bioavailability of sublingual estradiol was 10%; approximately twice that of conventional absolute bioavailability estimates of oral estradiol (5%, though with a wide range of 0.1 to 12%) (Kuhnz, Blode, & Zimmermann, 1993). Therefore, with respect to AUC levels of estradiol, the sublingual route appears to have between approximately two and five times higher estradiol levels compared to oral estradiol when given at the same doses. Based on these findings, approximately equivalent doses of sublingual estradiol can be derived (Table 1). It is notable that due to substantial interindividual variation in the metabolism of different forms of estradiol, these relative doses are unlikely to correspond to one another on a person-by-person basis. Measurement of circulating estradiol concentrations should always be used to guide dose titration.
Table 1: Approximately comparable doses of estradiol (E2) and estradiol valerate (EV) administered by the oral and sublingual routes in terms of total estradiol exposure (Price et al., 1997; Pines et al., 1999):
Low Dose
Moderate Dose
High Dose
Very-High Dose
Oral E2
2 mg/day
4 mg/day
8 mg/day
10 mg/day
Sublingual E2a
0.5–1 mg/day
1–2 mg/day
2–4 mg/day
2.5–5 mg/day
Oral EV
3 mg/day
6 mg/day
10 mg/day
12 mg/day
Sublingual EVa
0.75–1.5 mg/day
1.5–3 mg/day
2.5–5 mg/day
3–6 mg/day
a Range calculated by dividing oral doses by two and four to reflect differences in absolute bioavailability and rounding to the nearest 0.25 mg. * Bioidentical estradiol has wide interindividual variation in its pharmacology and the effects of doses are likely to vary significantly between individuals. EV has greater molecular weight and therefore contains less medication for the same amount/dose by weight. It should be noted that estimates for the relative bioavailability of EV are extrapolated from formulations with no valeric ester attached (i.e., E2).
Administration of Multiple Sublingual Doses Per Day
In order to compensate for the short half-life of sublingually administered estradiol, multiple doses of estrogens can be administered in smaller quantities per day to maintain hormone levels that are somewhat more consistent (Ahokas, Kaukoranta, & Aito, 1999; Yaish et al., 2023a; Cortez et al., 2024).
In one study of premenopausal women with high-dose estrogen therapy, 2 mg of sublingual estradiol was administered three or four times per day (a total of 6–8 mg/day), resulting in significantly more stable hormone levels than would be expected with a single dose per day (Serhal & Craft, 1989). This was replicated in another study where estradiol was administered three to eight times per day (Ahokas et al., 2001). Conversely, a third study investigating low-dose buccal estradiol found little difference between the “steady-state” estradiol concentrations with a once-daily and twice-daily 0.25 mg dose of buccal estradiol over a 12 hour observation period (Wren et al., 2003). These findings may indicate that sublingual and buccal estradiol needs to be taken at least thrice per day in order to achieve concentrations of estradiol that are more stable.
It would seem advantageous for transfeminine people using sublingual estradiol that sublingual estradiol is taken in divided doses throughout the day; perhaps ideally at least three or four times per day. For instance, instead of taking a 2 mg dose every 24 hours, it would be better to take four 0.5 mg doses in the space of 24 hours (as evenly spaced as practical). Administering sublingual estradiol multiple times throughout the day might be less convenient, but is likely to provide at least somewhat more balanced estradiol levels. The administration of multiple doses every day could be regarded as optimal for the use of sublingually administered estradiol.
Sublingually Administered Estradiol and Feminisation
The very short half-life of sublingually and buccally administered estradiol relative to other forms raises a few questions relating to its use in feminising hormone therapy. One of the most commonly asked questions on online forums is regarding which gender-affirming hormone therapy regimens might be most “effective” with respect to the feminising effects of estrogens. These include, but are not limited to, outcomes such as breast development and fat distribution.
In contrast to oral and trandermal estradiol, limited data exist describing the extent of feminisation with the sublingual route (Safer, 2022). A non-randomised study found that self-assessed Tanner stage after 6 months of treatment did not appear to be different in users of sublingual estradiol monotherapy as compared to users of oral estradiol plus 10 mg/day cyproterone acetate (Yaish et al., 2023a; Yaish et al., 2023b). However, since breast development itself was not measured objectively, these particular data are low-quality and prevent definitive conclusions either way about the superiority or inferiority of sublingual estradiol. The same study group reported that although there were similar increases in gynoid fat in the two arms, the oral estradiol group did show an increased amount of android fat as compared to the sublingual group (Yaish et al., 2025). On the other hand, a further complication of this study is the possible confounding by lack of concomitant antiandrogen therapy in the sublingual arm (Ruggles & Cheung, 2024; Yaish et al., 2024). Notably, progestogens like cyproterone acetate have been shown to be associated with weight gain (Lopez et al., 2016). This could explain the difference in android fat accumulation.
Oral estradiol and other non-oral forms of estradiol (such as transdermal administration) have not been found to differ in their effects on breast development or other feminising outcomes in transfeminine people or cisgender hypogonadal girls (Rosenfield et al., 2005; Shah et al., 2014; Klaver et al., 2018; de Blok et al., 2021, Tebbens et al., 2022). In consideration of this, differences in efficacy might not be expected for sublingual estradiol either. However, the use of supraphysiological doses of estrogens from the onset of therapy may stunt breast development and reduce final breast size in transfeminine people (Boogers et al., 2025). Because the use of sublingual estradiol results in estradiol concentrations that routinely achieve the supratherapeutic range, it is possible that this could have deleterious effects on breast development.
The fact that several gender clinics have employed sublingual estradiol for some time is encouraging (Deutsch, Bhakri, & Kubicek, 2015; Lim et al., 2019; Cirrincione et al., 2021). Nevertheless, as there is very limited data comparing the feminising efficacy of sublingual estradiol with objective measures, no firm conclusions about any differences in feminisation outcomes between sublingual estradiol and other routes of administration can currently be drawn. Hopefully, studies in the future will shed more light on this.
Testosterone Suppressing Efficacy of Sublingually Administered Estradiol
Another question that might be raised by the short half-life of sublingual estradiol is how it might compare to more conventional routes of administration in terms of its ability to suppress testosterone and other androgens.
Estrogens were first characterised for their use as antigonadotrophic antiandrogens in the 1940s in the form of oral synthetic estrogens, namely diethylstilbestrol (DES), to treat men with prostate cancer (Huggins & Hodges, 1941). Estrogens given in the form of oral ethinylestradiol (EE), long-acting estradiol esters, such as polyestradiol phosphate, and transdermal estradiol patches have been studied. Their efficacy for this indication is well established (Stege et al., 1996; Kohli, 2006; Sciarra et al., 2015). As data are more limited for testosterone suppression with estrogens in transfeminine people, these data are valuable for informing transfeminine hormone therapy. Since sublingual estradiol has never been used to treat prostatic cancer, no such data exist to show the ability of sublingual estradiol in this capacity.
Some studies have found that physiologic levels of estradiol (i.e., 100–200 pg/mL [367–734 pmol/L]) or slightly higher from non-sublingual estradiol alone result in rapid and near complete, if not complete, suppression of testosterone levels to the female range in many transfeminine people (Leinung, Feustel, & Joseph, 2018; Pappas et al., 2020; Misakian et al., 2025). Additionally, the Prostate Adenocarcinoma TransCutaneous Hormones (PATCH) study, a multicentre randomised controlled trial in the United Kingdom, showed that sustained median estradiol levels of between 215 to 250 pg/mL (789–918 pmol/L) from transdermal patches were similarly effective (~95%) to GnRH analogues in reducing testosterone levels to the castrate range (<50 ng/dL [<1.7 nmol/L]) (Langley et al., 2021). However, because sublingual estradiol differs in its pharmacokinetics to other forms of estradiol, it is plausible that this route of administration might result in sub-par suppression at doses with similar concentrations of estradiol.
A few studies have reported the extent of testosterone suppression under sublingual estradiol in transfeminine people. In a randomised controlled trial (RCT) comparing once-daily and twice-daily administration of 2 mg sublingual estradiol to 0.1 mcg/day transdermal estradiol with and without spironolactone, both of the sublingual arms were found to result in inferior testosterone suppression at 1-month and 6-month follow-up (Cortez et al., 2023; Cortez et al., 2024). The authors hypothesised that this could be due to the ability of high concentrations of estrone, which were seen with sublingual estradiol, to inhibit cooperative binding of the estrogen receptor. However, this notion is contradicted by studies comparing oral and transdermal administration of estradiol which have reported no difference in the ability of these formulations to suppress testosterone at equivalent doses (SoRelle et al., 2019; Salakphet et al., 2022; Slack et al., 2025). This is in spite of the large amount of estrone also known to be generated from oral estradiol. Another study of transfeminine people found that sublingual estradiol at a dose of 0.5 mg administered four times daily was able to suppress testosterone as well as oral estradiol in combination with low-dose cypoterone acetate (Yaish et al., 2023a; Yaish et al., 2023b). The use of the four times daily dosing regimen in this study may account for the difference in findings between these two studies in the ability to suppress testosterone. Sublingual estradiol has been studied in transfeminine people in combination with and without the low-dose use of the progestin medroxyprogesterone acetate (MPA) (Jain, Kwan, & Forcier, 2019). In this study, high rates of suppressed testosterone levels (ie: <50 ng/dL [<1.7 nmol/L]) were achieved by the transfeminine people who took sublingual estradiol with medroxyprogesterone acetate, showing that sublingual estradiol taken together with progestogens such as cyproterone acetate is viable for achieving effective testosterone suppression.
A possibility supported by some evidence from pharmacological studies of estradiol is that sustained estradiol levels may be more efficacious with respect to testosterone suppression than the frequent and short-lived peaks in estradiol concentrations that occur with the sublingual route. In some studies of both sublingual and intravenous administration, limited suppression of the gonadotropins (follicle-stimulating hormone and luteinising hormone) have been reported in women despite sufficiently elevated estradiol levels for several hours (Tsai & Yen, 1971; Burnier et al., 1981; Casper & Yen, 1981; Hoon et al., 1993). These studies are low quality and indirect since testosterone suppression itself was not measured and they were performed in cisgender women. Another problem is that all were single dose studies and their findings may not translate to multiple dosing. Nevertheless, these studies might suggest a mechanism by which sublingual estradiol is unable to fully suppress gonadal function in transfeminine people without the use of excessive doses that would lead to greater health risks or the additional use of other antiandrogens.
For the reasons above, transdermal patches, gels and parenteral estradiol esters, such as estradiol valerate, injected intramuscularly or subcutaneously are probably more reliable choices for estradiol monotherapy regimens. If sublingual estradiol is used as a single agent therapy, it would seem reasonable to suggest the use of many divided doses taken throughout the day, as this is probably more likely to be efficacious. Nevertheless, sublingual estradiol appears to be more effective in terms of testosterone suppression when used with concomitant antiandrogens.
Monitoring of Estradiol Levels with Sublingual Administration
A further consideration regarding the rapid changes in estradiol levels that occur with the use of sublingual estradiol is the relevance of monitoring of estradiol levels through bloodwork. Currently, consensus guidelines do not recommend a specific time for monitoring of the blood relative to the time of a last dose (Cheung et al., 2019; T’Sjoen et al., 2020; Coleman et al., 2022). This may be in part due to practical reasons, or because until very recently there were currently no robust data from randomised controlled trials to guide the specifics of dosing in transgender hormone therapy (Haupt et al., 2020). Nevertheless, because estradiol levels vary so significantly with sublingual estradiol, knowledge of how long after the last dose blood was drawn is important to ensure proper interpretation of laboratory results.
For instance, measuring hormone levels just after a dose of sublingual estradiol has been taken might lead to the misinterpretation that levels of estradiol are excessively high and that one’s dosage should be reduced to achieve a more sensible concentration of estradiol in the blood. In reality, this would be a misunderstanding caused by the pharmacology of sublingual estradiol as the point of measurement would be right around the time when estradiol levels are most likely to be at their highest. These estradiol levels would not be indicative of the average amount of exposure, which is the more accurate measure of overall estrogenicity. Similarly, on the opposite end of the scale, drawing blood just before the administration of a new dose might lead to the belief that estrogen levels are too low and, consequently, lead to the use of excessive doses of estrogens. The latter misinterpretation may be more common among people unfamiliar with the pharmacology of sublingual estradiol as levels of estradiol only remain very high in the first few hours after a dose of sublingual estradiol has been taken before falling rapidly.
A possible solution to the problem of rapidly changing hormone levels associated with the sublingual route might simply be to measure when estradiol levels are most likely to be closest to their average. In the case of sublingual estradiol, studies generally find this to be approximately four hours after the administration of a dose, although there is likely to be considerable variation between individuals (Kuhl, 2005). Nevertheless, this approach may give the most representative “snapshot” of overall estrogenic exposure and might help to avoid misleading laboratory data in users of sublingual estradiol.
Safety and Tolerability
Unfortunately, no long term safety data exist for sublingually administered estradiol in the same way that both oral and transdermal estradiol have been rigorously studied in menopausal women (Rovinski et al., 2018; CGHFBC, 2019). The published medical literature concerning the safety and tolerability of this route of administration leaves many questions unanswered.
Adverse Health Effects of Estrogens
With sufficient exposure, owing to their effects in the liver, estrogens are associated with an increased risk of blood clots (Kuhl, 2005). Additionally, under certain circumstances, estrogens can be associated with other cardiovascular complications (Anderson et al., 2004; Mikkola et al., 2005). Although the absolute risk is low in the short-term, these are the most significant health concerns associated with gender-affirming hormone therapy.
A retrospective cohort study in the United States found that the incidence of thromboembolism in transfeminine people with an average dose of 4 mg/day oral estradiol was approximately twice that of cisgender controls not taking hormone therapy after adjusting for confounders (HR 2.0, 95% CI 1.4–2.8 versus reference women) (Getahun et al., 2018). These increases in risks are much lower compared to regimens in transfeminine people in the past that included high doses of synthetic estrogens, however even such increases can significantly increase morbidity and mortality (Morimont, Dogné, & Douxfils, 2020). A 2021 meta-analysis reported an absolute incidence of VTE of 2% in transfeminine people receiving gender-affirming hormone therapy, although with significant between-study heterogeneity (Totaro et al., 2021).
Some studies have assessed the effects of sublingually administered estradiol on the liver (Pines et al., 1999; Lim et al., 2019). These data found similar effects on lipids and cholesterol to other estrogens. One line of evidence that indicates sublingual estradiol has greater hepatic impact than other non-oral forms such as trandermal estradiol is the significantly greater quantities of estrone and estrone sulphate that are generated by this route; a marker of estrogenic exposure in the liver (Burnier et al., 1981; Cirrincione et al., 2021). Intense estrogenic activation in the liver is the mechanism by which non-oral estradiol induces a hypercoagulable state at high doses (Kuhl, 2005). While a large body of research does exist concerning the short and long term health effects of estrogens, none of these studies have investigated sublingual or buccal estradiol (Oliver-Williams et al., 2019; Mishra et al., 2021). Given that oral estradiol has greater risks than non-oral estradiol, and that sublingual administration partially but not fully avoids first-pass metabolism, it may be the case that its own risk would be somewhere between the risk observed with oral estradiol and the risk observed with other conventional non-oral routes (such as transdermal estradiol). However, an ongoing prospective study reported that use of sublingual estradiol alone resulted in less favourable outcomes on some markers of coagulation in the liver as compared to oral estradiol and cyproterone acetate (Bar et al., 2024). These data are indirect, however could suggest that contrary to theoretical expectations, sublingual estradiol might be closer or even less favourable than the risk profile of oral estradiol.
Other adverse effects of estradiol include breast cancer and gallbladder disease. These risks are believed to be dose-dependent (Cummings et al., 1999; Liu et al., 2008). However, as with cardiovascular and thromboembolic complications, no data exist to describe the long-term risk in these other areas with sublingual formulations. In the interest of harm reduction and the balancing of the risks and benefits of gender-affirming hormone therapy, it would be advisable to limit doses of sublingual and buccal estradiol so that they are not excessive (i.e., <6 mg/day) (Jalal & Baldwin, 2023).
Non-compliance
A practical obstacle to the use of sublingual estradiol in transfeminine people is that it may be highly inconvenient to have to administer doses thrice, four times or perhaps even more often throughout the duration of a single day. It has been found in observational studies that, in general, the number of prescribed medications and doses per day are positively associated with patient non-compliance and the number of missed doses (Jin et al., 2008; Toh et al., 2014). These findings are especially of relevance to transfeminine people as, in most cases, we require decades of hormone therapy. While missing one dose from time to time may be of little consequence, missing doses repeatedly could be more problematic. Despite this, sublingual estradiol has been used in studies of transfeminine people where it has been administered up to four times daily (Yaish et al., 2023a).
In contrast to sublingual estradiol, the half-life of oral estradiol and transdermal gel is long enough to enable once-daily administration (Wiegratz et al., 2001; Potts & Lobo, 2005). In the case of transdermal patches and parenteral estradiol, these forms only have to be replenished every few days or after even longer intervals of time (Thurman et al., 2013; Wisner et al., 2015). Therefore, when considering the use of sublingual estradiol versus other forms, whether or not it would be practical or convenient to consistently take medication several times a day should probably also be an important consideration for transfeminine people. If not, then another formulation may be preferable for the person in question. This may be especially true for long term use.
Summary and Conclusions
Sublingual estradiol is different in its pharmacology to other routes. The main difference is that it is associated with a rapid rise and fall in estradiol levels. It has between two and four times the bioavailability of oral estradiol and hence provides the same total estradiol exposure at doses that are two to four times lower. This could be a particular advantage because sublingual estradiol, therefore, is cheaper than oral estradiol.
There is much less research investigating sublingual estradiol than other forms of estrogen. These forms, such as oral and transdermal estrogens, are widely used in the alleviation of the menopause and for other indications. Consequently, they have received much more attention and characterisation than sublingual estradiol has for transfeminine hormone therapy. However, studies are beginning to add our knowledge of sublingual estradiol. Clinical practice guidelines for transgender care, which historically did not make reference to the use of sublingual estradiol, have now begun to discuss it.
The evidence is inconclusive regarding whether sublingual estradiol results in better, worse, or the same feminisation when compared with other routes of administration. However, it is plausible that the supra-physiologic levels of estradiol that frequently occur with sublingual estradiol could be detrimental to breast development. It also seems that sublingual estradiol could result in lesser testosterone suppression when used as a single agent therapy as compared to other routes. Sublingual estradiol has, nonetheless, been shown to be effective with respect to testosterone suppression when paired with other antiandrogens. Care should be taken with sublingual estradiol when monitoring estradiol levels to ensure correct interpretation. In order to help minimise these potential problems, sublingual estradiol can be taken in multiple doses divided throughout the day.
The health risks of sublingual estradiol have not been quantified in large observational or randomised studies. Therefore, although the first pass effect in the liver is partially avoided, the cardiovascular risks associated with long-term sublingual estradiol remain unknown. Sublingual estradiol may also be inconvenient and other formulations can be used instead if preferred, particularly for more long-term therapy.
Taken together, although much more research is clearly needed to properly characterise sublingual estradiol in transfeminine hormone therapy, it might have some advantageous properties and may be a useful alternative to oral estradiol.
References
Ahokas, A., Kaukoranta, J., & Aito, M. (1999). Effect of oestradiol on postpartum depression. Psychopharmacology, 146(1), 108–110. [DOI:10.1007/s002130051095]
Ahokas, A., Kaukoranta, J., Wahlbeck, K., & Aito, M. (2001). Estrogen deficiency in severe postpartum depression: successful treatment with sublingual physiologic 17β-estradiol: a preliminary study. Journal of Clinical Psychiatry, 62(5), 332–336. [DOI:10.4088/jcp.v62n0504]
Anderson, G. L., Limacher, M., Assaf, A. R., Bassford, T., Beresford, S. A., Black, H., Bonds, D., Brunner, R., Brzyski, R., Caan, B., Chlebowski, R., Curb, D., Gass, M., Hays, J., Heiss, G., Hendrix, S., Howard, B. V., Hsia, J., Hubbell, A., Jackson, R., … & Women’s Health Initiative Steering Committee. (2004). Effects of Conjugated Equine Estrogen in Postmenopausal Women With Hysterectomy: The Women’s Health Initiative Randomized Controlled Trial. JAMA, 291(14), 1701–1712. [DOI:10.1001/jama.291.14.1701]
Bar, O. S., Yaish, I., Barzilai, M., Greenman, Y., Gindis, G., Moshe, Y., & Tordjman, K. (2024, May). Low dose sublingual estradiol decreases protein s, generating a potentially pro-thrombotic state: interim results of a controlled prospective pilot study of treatment-naive trans women. In Endocrine Abstracts (Vol. 99). Bioscientifica. [DOI:10.1089/trgh.2024.0105]
Bartlett, J. A., & van der Voort Maarschalk, K. (2012). Understanding the oral mucosal absorption and resulting clinical pharmacokinetics of asenapine. AAPS Pharmscitech, 13(4), 1110–1115. [DOI:10.1208/s12249-012-9839-7]
Boogers, L. S., Sardo Infirri, S. A., Bouchareb, A., Dijkman, B. A., Helder, D., de Blok, C. J., … & Hannema, S. E. (2025). Variations in Volume: Breast Size in Trans Women in Relation to Timing of Testosterone Suppression. The Journal of Clinical Endocrinology & Metabolism, 110(5), 1404-1410. [DOI:10.1210/clinem/dgae573]
Burnier, A. M., Martin, P. L., Yen, S. S., & Brooks, P. (1981). Sublingual absorption of micronized 17β-estradiol. American Journal of Obstetrics and Gynecology, 140(2), 146–150. [DOI:10.1016/0002-9378(81)90101-0]
Casper, R. F., & Yen, S. S. (1981). Rapid absorption of micronized estradiol-17β following sublingual administration. Obstetrics and Gynecology, 57(1), 62–64. [Google Scholar] [PubMed] [URL] [PDF]
Chandrasekhara, D. S., Ali, V., Prost, H. M., & Nader-Estekhari, S. (2002). Buccal estrogen in toothpaste study: systemic absorption of estradiol in postmenopausal or surgically menopausal women when administered as a component in toothpaste. Fertility and Sterility, 78(Suppl 1), S98–S98 (O-258). [DOI:10.1016/S0015-0282(02)03639-7]
Cheung, A. S., Wynne, K., Erasmus, J., Murray, S., & Zajac, J. D. (2019). Position statement on the hormonal management of adult transgender and gender diverse individuals. Medical Journal of Australia, 211(3), 127–133. [DOI:10.5694/mja2.50259]
Cirrincione, L. R., Winston McPherson, G., Rongitsch, J., Sadilkova, K., Drees, J. C., Krasowski, M. D., Dickerson, J. A., & Greene, D. N. (2021). Sublingual Estradiol Is Associated with Higher Estrone Concentrations than Transdermal or Injectable Preparations in Transgender Women and Gender Nonbinary Adults. LGBT Health, 8(2), 125–132. [DOI:10.1089/lgbt.2020.0249]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., De Vries, A. L., Deutsch, M. B., … & Arcelus, J. (2022). Standards of care for the health of transgender and gender diverse people, version 8. International Journal of Transgender Health, 23(sup1), S1-S259. [DOI:10.1080/26895269.2022.2100644]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/S0140-6736(19)31709-X]
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C., Fields, M., Gray, T., Guo, Z., Nicol, G., & Baranski, T. (2023). Effectiveness and Safety of Different Estradiol Regimens in Transgender Women (TREAT Study): Protocol for a Randomized Controlled Trial. JMIR Research Protocols, 12, e53092. [DOI:10.2196/53092]
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C. J., Fields, M. E., … & Baranski, T. (2024). Effectiveness and safety of different estradiol regimens in transgender females: a randomized controlled trial. Journal of the Endocrine Society, 8(8), bvae108. [DOI: 10.1210/jendso/bvae108]
Cummings, S. R., Eckert, S., Krueger, K. A., Grady, D., Powles, T. J., Cauley, J. A., … & Jordan, V. C. (1999). The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. JAMA, 281(23), 2189-2197. [DOI:10.1001/jama.281.23.2189]
de Blok, C., Dijkman, B., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K., & den Heijer, M. (2021). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
Deutsch, M. B., Bhakri, V., & Kubicek, K. (2015). Effects of cross-sex hormone treatment on transgender women and men. Obstetrics and Gynecology, 125(3), 605–610. [DOI:10.1097/AOG.0000000000000692]
Devissaguet, J. P., Brion, N., Lhote, O., & Deloffre, P. (1999). Pulsed estrogen therapy: pharmacokinetics of intranasal 17-beta-estradiol (S21400) in postmenopausal women and comparison with oral and transdermal formulations. European Journal of Drug Metabolism and Pharmacokinetics, 24(3), 265–271. [DOI:10.1007/BF03190030]
Doll, E. E., Gunsolus, I., Lamberton, N., Tangpricha, V., & Sarvaideo, J. L. (2020). Pharmacokinetics of Sublingual Versus Oral Estradiol in Transgender Women. Journal of the Endocrine Society, 4(Suppl 1), A1128–A1128 (SUN-LB9). [DOI:10.1210/jendso/bvaa046.2237]
Doll, E., Gunsolus, I., Thorgerson, A., Tangpricha, V., Lamberton, N., & Sarvaideo, J. L. (2022). Pharmacokinetics of Sublingual Versus Oral Estradiol in Transgender Women. Endocrine Practice, 28(3), 237–242. [DOI:10.1016/j.eprac.2021.11.081]
Fiet, J., Hermano, M., Witte, J., Villette, J. M., Haimart, M., Gourmel, B., Tabuteau, F., Rouffy, J., & Dreux, C. (1982). Post-menopausal concentrations of plasma oestradiol, oestrone, FSH and LH and of total urinary oestradiol and oestrone after a single sublingual dose of oestradiol-17β. Acta Endocrinologica, 101(1), 93–97. [DOI:10.1530/acta.0.1010093]
Gass, M. S., Rebar, R. W., Cuffie-Jackson, C., Cedars, M. I., Lobo, R. A., Shoupe, D., Judd, H. L., Buyalos, R. P., & Clisham, P. R. (2004). A short study in the treatment of hot flashes with buccal administration of 17-β estradiol. Maturitas, 49(2), 140–147. [DOI:10.1016/j.maturitas.2003.12.004]
Getahun, D., Nash, R., Flanders, W. D., Baird, T. C., Becerra-Culqui, T. A., Cromwell, L., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Silverberg, M. J., Safer, J., Slovis, J., Tangpricha, V., & Goodman, M. (2018). Cross-sex hormones and acute cardiovascular events in transgender persons: a cohort study. Annals of Internal Medicine, 169(4), 205–213. [DOI:10.7326/M17-2785]
Gindis, G., Yaish, I., Greenman, Y., Moshe, Y., Arbiv, M., Buch, A., Sofer, Y., Shefer, G., & Tordjman, K. (May 2023). Sublingual estradiol only, offers no apparent advantage over combined oral estradiol and cyproterone acetate, for Gender Affirming Hormone Therapy (GAHT) of treatment-naive transwomen: Results of a prospective pilot study. Endocrine Abstracts, 90 [25th European Congress of Endocrinology 2023, 13–16 May 2023, Istanbul, Turkey], 274–274 (abstract no. P182). [DOI:10.1530/endoabs.90.p182] [PDF]
Grock, S., Patel, R., & Ahern, S. (2024). Hormone Therapy for Transgender and Gender-Diverse Patients. In Genital Gender Affirming Surgery: An Illustrated Guide and Video Atlas (pp. 33-49). Cham: Springer Nature Switzerland. [DOI:10.1007/978-3-031-69997-9_4]
Haupt, C., Henke, M., Kutschmar, A., Hauser, B., Baldinger, S., Saenz, S. R., & Schreiber, G. (2020). Antiandrogen or estradiol treatment or both during hormone therapy in transitioning transgender women. Cochrane Database of Systematic Reviews, 11(11), CD013138. [DOI:10.1002/14651858.CD013138.pub2]
Hoon, T. J., Dawood, M. Y., Khan‐Dawood, F. S., Ramos, J., & Batenhorst, R. L. (1993). Bioequivalence of a 17β‐Estradiol Hydroxypropyl‐β‐Cyclodextrin Complex in Postmenopausal Women. The Journal of Clinical Pharmacology, 33(11), 1116–1121. [DOI:10.1002/j.1552-4604.1993.tb01949.x]
Huggins, C., & Hodges, C. V. (1941). Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Research, 1(4), 293–297. [DOI:10.3322/canjclin.22.4.232]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone acetate in Gender-Affirming therapy for Transwomen: results from a retrospective study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Jalal, E., & Baldwin, C. (2023). Supratherapeutic Estrogen Levels in Transgender Women Likely From Sublingual Estradiol. Journal of the Endocrine Society, 7(Suppl 1) [ENDO 2023 Abstracts Annual Meeting of the Endocrine Society], A1095–A1096 (abstract no. SAT391/bvad114.2062). [DOI:10.1210/jendso/bvad114.2062] [PDF]
Jin, J., Sklar, G. E., Oh, V. M. S., & Li, S. C. (2008). Factors affecting therapeutic compliance: A review from the patient’s perspective. Therapeutics and Clinical Risk Management, 4(1), 269–286. [DOI:10.2147/TCRM.S1458]
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2025). Comparison of estrone/estradiol ratio and levels in transfeminine individuals on different routes of estradiol. Transgender Health, 10(3), 261-268. [DOI:10.1089/trgh.2023.0138]
Klaver, M., de Blok, C. J. M., Wiepjes, C. M., Nota, N. M., Dekker, M. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/EJE-17-0496]
Kohli, M. (2006). Phase II study of transdermal estradiol in androgen‐independent prostate carcinoma. Cancer, 106(1), 234–235. [DOI:10.1002/cncr.21528]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhnz, W., Blode, H., & Zimmermann, H. (1993). Pharmacokinetics of exogenous natural and synthetic estrogens and antiestrogens. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 261–322). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_15]
Kuhnz, W., Gansau, C., & Mahler, M. (1993). Pharmacokinetics of estradiol, free and total estrone, in young women following single intravenous and oral administration of 17β-estradiol. Arzneimittelforschung, 43(9), 966–973. [Google Scholar] [PubMed] [PDF]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., … & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/S0140-6736(21)00100-8]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal treatment of transgender women with oral estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Lim, H. H., Jang, Y. H., Choi, G. Y., Lee, J. J., & Lee, E. S. (2019). Gender affirmative care of transgender people: a single center’s experience in Korea. Obstetrics & Gynecology Science, 62(1), 46–55. [DOI:10.5468/ogs.2019.62.1.46]
Liu, B., Beral, V., Balkwill, A., Green, J., Sweetland, S., & Reeves, G. (2008). Gallbladder disease and use of transdermal versus oral hormone replacement therapy in postmenopausal women: prospective cohort study. BMJ, 337. [DOI:10.1136/bmj.a386]
Lobo, R. A. (1987). Absorption and metabolic effects of different types of estrogens and progestogens. Obstetrics and Gynecology Clinics of North America, 14(1), 143–167. [PubMed] [DOI:10.1016/S0889-8545(21)00577-5] [URL]
Lopez, L. M., Ramesh, S., Chen, M., Edelman, A., Otterness, C., Trussell, J., & Helmerhorst, F. M. (2016). Progestin‐only contraceptives: effects on weight. Cochrane Database of Systematic Reviews, (8). [DOI:10.1002/14651858.CD008815.pub4]
Mikkola, A., Aro, J., Rannikko, S., Oksanen, H., Ruutu, M., & Finnprostate Group. (2005). Cardiovascular complications in patients with advanced prostatic cancer treated by means of orchiectomy or polyestradiol phosphate. Scandinavian Journal of Urology and Nephrology, 39(4), 294–300. [DOI:10.1080/00365590510031228]
Misakian, A. L., Ariel, D., Sullivan, E. A., Singh, G., Loeb, D., Strickland, T., … & Hamnvik, O. P. R. (2025). Injectable estradiol monotherapy effectively suppresses testosterone in gender-affirming hormone therapy. Endocrine Practice. [DOI:10.1016/j.eprac.2025.07.002]
Mishra, S. R., Chung, H. F., Waller, M., & Mishra, G. D. (2021). Duration of estrogen exposure during reproductive years, age at menarche and age at Menopause, and risk of cardiovascular disease events, all‐cause and cardiovascular mortality: a systematic review and meta‐analysis. BJOG: An International Journal of Obstetrics & Gynaecology, 128(5), 809–821. [DOI:10.1111/1471-0528.16524]
Morimont, L., Dogné, J. M., & Douxfils, J. (2020). Letter to the Editors-in-Chief in response to the article of Abou-Ismail, et al. entitled “Estrogen and thrombosis: A bench to bedside review” (Thrombosis Research 192 (2020) 40–51). Thrombosis Research, 193, 221–223. [DOI:10.1016/j.thromres.2020.08.006]
Oliver-Williams, C., Glisic, M., Shahzad, S., Brown, E., Pellegrino Baena, C., Chadni, M., Chowdhury, R., Franco, O. H., & Muka, T. (2019). The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Human Reproduction Update, 25(2), 257–271. [DOI:10.1093/humupd/dmy039]
Pappas, I. I., Craig, W. Y., Spratt, L. V., & Spratt, D. I. (2021). Efficacy of Sex Steroid Therapy Without Progestin or GnRH Agonist for Gonadal Suppression in Adult Transgender Patients. The Journal of Clinical Endocrinology & Metabolism, 106(3), e1290–e1300. [DOI:10.1210/clinem/dgaa884]
Perloff, W. H. (1950). Estradiol buccal tablets in the treatment of the menopause. American Journal of Obstetrics and Gynecology, 59(1), 223–225. [DOI:10.1016/0002-9378(50)90390-5]
Pickar, J. H., Bon, C., Amadio, J. M., Mirkin, S., & Bernick, B. (2015). Pharmacokinetics of the first combination 17β-estradiol/progesterone capsule in clinical development for menopausal hormone therapy. Menopause, 22(12), 1308–1316. [DOI:10.1097/GME.0000000000000467]
Pines, A., Averbuch, M., Fisman, E. Z., & Rosano, G. M. (1999). The acute effects of sublingual 17β-estradiol on the cardiovascular system. Maturitas, 33(1), 81–85. [DOI:10.1016/S0378-5122(99)00036-5]
Potts, R. O., & Lobo, R. A. (2005). Transdermal drug delivery: clinical considerations for the obstetrician–gynecologist. Obstetrics & Gynecology, 105(5), 953–961. [DOI:10.1097/01.AOG.0000161958.70059.db]
Price, T. M., Blauer, K. L., Hansen, M., Stanczyk, F., Lobo, R., & Bates, G. W. (1997). Single-dose pharmacokinetics of sublingual versus oral administration of micronized 17β-estradiol. Obstetrics & Gynecology, 89(3), 340–345. [DOI:10.1016/S0029-7844(96)00513-3]
Rosenfield, R. L., Devine, N., Hunold, J. J., Mauras, N., Moshang Jr, T., & Root, A. W. (2005). Salutary effects of combining early very low-dose systemic estradiol with growth hormone therapy in girls with Turner syndrome. The Journal of Clinical Endocrinology & Metabolism, 90(12), 6424–6430. [DOI:10.1210/jc.2005-1081]
Rovinski, D., Ramos, R. B., Fighera, T. M., Casanova, G. K., & Spritzer, P. M. (2018). Risk of venous thromboembolism events in postmenopausal women using oral versus non-oral hormone therapy: a systematic review and meta-analysis. Thrombosis Research, 168, 83–95. [DOI:10.1016/j.thromres.2018.06.014]
Ruggles, T., & Cheung, A. S. (2024). Re:” Sublingual Estradiol Offers No Apparent Advantage over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study” by Yaish et al. Transgender Health, 9(5), 459. [DOI:10.1089/trgh.2024.0048]
Salakphet, T., Mattawanon, N., Manojai, N., Muangmool, T., & Tangpricha, V. (2022). Hormone concentrations in transgender women who self-prescribe gender affirming hormone therapy: a retrospective study. The Journal of Sexual Medicine, 19(5), 864-871. [DOI:10.1016/j.jsxm.2022.02.023]
Safer, J. D. (2022). Are the Pharmacokinetics of Sublingual Estradiol Superior or Inferior to Those of Oral Estradiol? Endocrine Practice, 28(3), 351–352. [DOI:10.1016/j.eprac.2021.12.018]
Sarvaideo, J., Doll, E., & Tangpricha, V. (2022). More Studies Are Needed to Establish the Safety and Efficacy of Sublingual Estradiol in Transgender Women. Endocrine Practice, 28(3), 353–354. [DOI:10.1016/j.eprac.2022.01.004]
Sciarra, A., Gentile, V., Cattarino, S., Gentilucci, A., Alfarone, A., D’Eramo, G., & Salciccia, S. (2015). Oral ethinylestradiol in castration‐resistant prostate cancer: a 10‐year experience. International Journal of Urology, 22(1), 98–103. [DOI:10.1111/iju.12613]
Serhal, P., & Craft, I. (1989). Oocyte donation in 61 patients. The Lancet, 333(8648), 1185–1187. [DOI:10.1016/S0140-6736(89)92762-1]
Shah, S., Forghani, N., Durham, E., & Neely, E. K. (2014). A randomized trial of transdermal and oral estrogen therapy in adolescent girls with hypogonadism. International Journal of Pediatric Endocrinology, 2014(1), 12. [DOI:10.1186/1687-9856-2014-12]
Slack, D. J., Ioschpe, A. D. V., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., … & Safer, J. D. (2025). Examining the influence of the route of administration and dose of estradiol on serum estradiol and testosterone levels in feminizing gender-affirming hormone therapy. Endocrine Practice, 31(1), 19-27. [DOI:10.1016/j.eprac.2024.10.002]
SoRelle, J. A., Jiao, R., Gao, E., Veazey, J., Frame, I., Quinn, A. M., … & Patel, K. (2019). Impact of hormone therapy on laboratory values in transgender patients. Clinical Chemistry, 65(1), 170-179. [DOI:10.1373/clinchem.2018.292730]
Stanczyk, F. Z., Archer, D. F., & Bhavnani, B. R. (2013). Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception, 87(6), 706–727. [DOI:10.1016/j.contraception.2012.12.011]
Stege, R., Gunnarsson, P. O., Johansson, C. J., Olsson, P., Pousette, Å., & Carlström, K. (1996). Pharmacokinetics and testosterone suppression of a single dose of polyestradiol phosphate (Estradurin®) in prostatic cancer patients. The Prostate, 28(5), 307–310. [DOI:10.1002/(SICI)1097-0045(199605)28:5<307::AID-PROS6>3.0.CO;2-8]
Sudhakar, D., Huang, Z., Zietkowski, M., Powell, N., & Fisher, A. R. (2023). Feminizing gender‐affirming hormone therapy for the transgender and gender diverse population: An overview of treatment modality, monitoring, and risks. Neurourology and Urodynamics, 42(5), 903-920. [DOI:10.1002/nau.25097]
Tebbens, M., Heijboer, A. C., T’Sjoen, G., Bisschop, P. H., & den Heijer, M. (2022). The role of estrone in feminizing hormone treatment. The Journal of Clinical Endocrinology & Metabolism, 107(2), 458-466. [DOI:10.1210/clinem/dgab741]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine position statement “assessment and hormonal management in adolescent and adult trans people, with attention for sexual function and satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
Totaro, M., Palazzi, S., Castellini, C., Parisi, A., D’Amato, F., Tienforti, D., … & Barbonetti, A. (2021). Risk of venous thromboembolism in transgender people undergoing hormone feminizing therapy: a prevalence meta-analysis and meta-regression study. Frontiers In Endocrinology, 12, 741866. [DOI:10.3389/fendo.2021.741866]
Thurman, A., Kimble, T., Hall, P., Schwartz, J. L., & Archer, D. F. (2013). Medroxyprogesterone acetate and estradiol cypionate injectable suspension (Cyclofem) monthly contraceptive injection: steady-state pharmacokinetics. Contraception, 87(6), 738–743. [DOI:10.1016/j.contraception.2012.11.010]
Toh, M. R., Teo, V., Kwan, Y. H., Raaj, S., Tan, S. Y. D., & Tan, J. Z. Y. (2014). Association between number of doses per day, number of medications and patient’s non-compliance, and frequency of readmissions in a multi-ethnic Asian population. Preventive Medicine Reports, 1, 43–47. [DOI:10.1016/j.pmedr.2014.10.001]
Tsai, C. C., & Yen, S. S. C. (1971). Acute effects of intravenous infusion of 17β-estradiol on gonadotropin release in pre-and post-menopausal women. The Journal of Clinical Endocrinology & Metabolism, 32(6), 766–771. [DOI:10.1210/jcem-32-6-766]
Verdonk, S. J., Vesper, H. W., Martens, F., Sluss, P. M., Hillebrand, J. J., & Heijboer, A. C. (2019). Estradiol reference intervals in women during the menstrual cycle, postmenopausal women and men using an LC-MS/MS method. Clinica Chimica Acta, 495, 198–204. [DOI:10.1016/j.cca.2019.04.062]
Wiegratz, I., Fink, T., Rohr, U. D., Lang, E., Leukel, P., & Kuhl, H. (2001). Überkreuz-Vergleich der Pharmakokinetik von Estradiol unter der Hormonsubstitution mit Estradiolvalerat oder mikronisiertem Estradiol. [Cross-over comparison of the pharmacokinetics of estradiol during hormone replacement therapy with estradiol valerate or micronized estradiol.] Zentralblatt für Gynäkologie, 123(9), 505–512. [PubMed] [DOI:10.1055/s-2001-18223]
Wisner, K. L., Sit, D. K., Moses-Kolko, E. L., Driscoll, K. E., Prairie, B. A., Stika, C. S., Eng, H. F., Dills, J. L., Luther, J. F., & Wisniewski, S. R. (2015). Transdermal estradiol treatment for postpartum depression: a pilot randomized trial. Journal of Clinical Psychopharmacology, 35(4), 389–395. [DOI:10.1097/JCP.0000000000000351]
Wren, B. G., Day, R. O., McLachlan, A. J., & Williams, K. M. (2003). Pharmacokinetics of estradiol, progesterone, testosterone and dehydroepiandrosterone after transbuccal administration to postmenopausal women. Climacteric, 6(2), 104–111. [DOI:10.1080/cmt.6.2.104.111]
Yaish, I., Gindis, G., Greenman, Y., Shefer, G., Buch, A., Arbiv, M., Moshe, Y., Sofer, Y., & Tordjman, K. M. (2023). Sublingual Estradiol Only, Compared To Combined Oral Estradiol And Cyproterone Acetate,Offers No Apparent Advantage For Gender Affirming Hormone Therapy (GHAT), In Treatment Naïve Transwomen: Results Of A Prospective Pilot Study. Journal of the Endocrine Society, 7(Suppl 1) [ENDO 2023 Abstracts Annual Meeting of the Endocrine Society], A1104–A1105 (abstract no. SAT409/bvad114.2080). [DOI:10.1210/jendso/bvad114.2080] [PDF]
Yaish, I., Gindis, G., Greenman, Y., Moshe, Y., Arbiv, M., Buch, A., Sofer, Y., Shefer, G., & Tordjman, K. (2023). Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study. Transgender Health, 8(6), 485–493. [DOI:10.1089/trgh.2023.0022]
Yaish, I., Greenman, Y., Sofer, Y., & Tordjman, K. M. (2024). Response to Ruggles and Cheung, Re:” Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study”. Transgender Health, 9(5), 460-461. [DOI:10.1089/trgh.2024.0105]
Yaish, I., Buch, A., Gindis, G., Sofer, Y., Arbiv, M., Moshe, Y., … & Tordjman, K. (2025). Early body composition changes in trans women on low-dose estradiol: comparing oral vs sublingual administration using dual energy absorptiometry and bioelectrical impedance analysis. The Journal of Sexual Medicine, 22(4), 625-635. [DOI:10.1093/jsxmed/qdaf005]
\ No newline at end of file
+An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People - Transfeminine Science
An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People
By Sam | First published June 11, 2021 | Last modified August 14, 2025
Abstract / TL;DR
Sublingually-administered estradiol is an alternative route of administration to oral estradiol that has been used by a limited number of gender-affirming care providers internationally. We do not currently know if sublingual estradiol results in better, worse, or similar feminisation as other routes of administration because there is a paucity of clinical data in this area. There may be practical shortcomings associated with the sublingual route, however clinical experience suggest it to be effective and affordable when dosed correctly. Although much more research is clearly needed to properly characterise this route of administration, sublingual estradiol might have some advantageous properties and may be a useful alternative to oral estradiol for some transfeminine people.
Introduction
Although the most common way to administer medication in the form of pills or tablets is by the oral route, oral estradiol formulations can otherwise be taken sublingually or buccally (Kuhl, 2005). Sublingual administration is the administration of an oral pill or tablet by means of placing under the tongue to dissolve and be absorbed into the bloodstream. Buccal administration is similar and refers to placing the medication between the cheek and gums, where it also quickly dissolves and is absorbed (Gass et al., 2004; Bartlett & Maarschalk, 2012).
Many transfeminine people wonder or ask questions on online forums about the sublingual route of administration for estradiol. Some of the most common queries are “What doses of sublingual estradiol should I take?”, “How often should I take sublingual estradiol?”, “Is sublingual estradiol better than oral estradiol?” and so on.
Until very recently, published data about sublingual estradiol in transfeminine people was scarce, with only a very small number of relevant studies having considered it (eg: Jain, Kwan, & Forcier, 2019, Lim et al., 2019). Accordingly, most information about the sublingual route existed only in older studies of cisgender patient populations (Casper & Yen, 1981; Burnier et al., 1981; Price et al., 1997; Wren et al., 2003). In the last few years, there has been a renewed interest in sublingual estradiol within the literature, specifically with an eye towards gender-affirming hormone therapy. Consequently, many high-level publications including recent clinical guidelines and reviews now make reference to the sublingual route (Coleman et al., 2022; Sudhakar et al., 2023; Grock, Reema, & Ahern, 2024).
It is of note that, although the sublingual and buccal administration are distinct routes of administration, they are very similar to each other in how they are performed and in their pharmacology (Perloff, 1950; Chandrasekhara et al., 2002). As such, although the term “sublingual” has been ostensibly used in this literature review, much of the content here is applicable to buccal administration of estradiol as well.
Pharmacology of Sublingual Estradiol
While sublingual estradiol is not as widely used in clinical practice as oral estradiol and other formulations, a number of studies have examined its pharmacology. These studies include both samples of postmenopausal cisgender women and transfeminine people as well as other patient populations (Casper & Yen, 1981; Serhal & Craft, 1989; Cirrincione et al., 2021; Doll et al., 2022; Kariyawasam et al., 2025). Both oral estradiol and oral estradiol valerate tablets can be taken sublingually (Serhal, 1990).
After the administration of oral estradiol, the medication is heavily metabolised and inactivated into estrogen conjugates by the liver (Kuhl, 2005). In turn, these metabolites are gradually converted back into estradiol, which serves to prolong its half life (to approximately 13–20 hours) (Stanczyk, Archer, & Bhavnani, 2013). In contrast to oral estradiol, sublingual estradiol does not pass as extensively through the liver. Therefore, it does not undergo deactivation into clinically insignificant estrogen metabolites. Sublingually administered estradiol is absorbed rapidly into the bloodstream where it directly enters circulation. Consequently, it has greater bioavailability than oral estradiol, meaning that lower doses are needed to achieve similar area-under-the-curve (AUC) estradiol levels (Kuhl, 2005) (Figures 1 and 2). This is an advantage of sublingual estradiol over oral estradiol, as it allows for the use of lower doses. This in turn might reduce medication costs.
Because accidental swallowing of some of the estradiol seems probable, the sublingual route is, most likely, actually a combination of sublingual and oral delivery of estradiol (Lobo, 1987; Kuhl, 2005). A small pharamcokinetics study of transfeminine people reported that a single 1 mg dose of sublingual estradiol caused an average rise in the level of estradiol up to an average of 144 pg/mL (529 pmol/L) within one to two hours of administration. In contrast, a peak concentration of just 35 pg/mL (128 pmol/L) was found with the same dose of 1 mg administered orally (Doll et al., 2022). Thereafter, estradiol levels decreased rapidly. In another study, it was found that mean estradiol levels measured at any given point were 613 pmol/L (167 pg/mL) on sublingual estradiol (Kariyawasam et al., 2025). In this study, a wide range of doses were used and hence it is not possible to ascertain much about dose-specific peak concentrations. Similar findings have been reported in other studies of postmenopausal women, although a wide range of peak concentrations have been observed (Burnier et al., 1981; Price et al., 1997; Wren et al., 2003). Estradiol levels are found to rapidly rise on the order of about five to ten times that of the peak of oral estradiol, then rapidly decline, with an elimination half-life of only a few hours (Kuhl, 2005). Sublingual estradiol is somewhat analogous in this respect to intravenously administered estradiol, which also shows a rapid increase in estradiol levels and a very short elimination half-life (Kuhnz, Gansau, & Mahler, 1993). Another route of administration that is similar in this regard is intranasal administration (Devissaguet et al., 1999). Owing to the short half-life elimination of sublingual estradiol, it does not achieve as stable concentrations as other formulations do. This is a marked difference to other routes, such as oral estradiol, that produce much more stable hormone levels and that do not fluctuate as much over the course of the day. All these differences by themselves do not necessarily mean that sublingual estradiol is superior or inferior to oral estradiol (Safer, 2022; Sarvaideo, Doll, & Tangpricha, 2022). Nevertheless, they should be kept in mind when considering findings from relevant studies.
A range of estimates have been reported for the relative bioavailability of sublingual estradiol. One small randomised study of postmenopausal women found approximately 2.5-fold higher AUC levels of estradiol with sublingual estradiol than with the same doses of oral estradiol (Price et al., 1997). Other studies have reported relative bioavailability estimates for sublingual estradiol of up to five times that of oral estradiol (Pines et al., 1999). A study in marmoset monkeys found that the absolute bioavailability of sublingual estradiol was 10%; approximately twice that of conventional absolute bioavailability estimates of oral estradiol (5%, though with a wide range of 0.1 to 12%) (Kuhnz, Blode, & Zimmermann, 1993). Therefore, with respect to AUC levels of estradiol, the sublingual route appears to have between approximately two and five times higher estradiol levels compared to oral estradiol when given at the same doses. Based on these findings, approximately equivalent doses of sublingual estradiol can be derived (Table 1). It is notable that due to substantial interindividual variation in the metabolism of different forms of estradiol, these relative doses are unlikely to correspond to one another on a person-by-person basis. Measurement of circulating estradiol concentrations should always be used to guide dose titration.
Table 1: Approximately comparable doses of estradiol (E2) and estradiol valerate (EV) administered by the oral and sublingual routes in terms of total estradiol exposure (Price et al., 1997; Pines et al., 1999):
Low Dose
Moderate Dose
High Dose
Very-High Dose
Oral E2
2 mg/day
4 mg/day
8 mg/day
10 mg/day
Sublingual E2a
0.5–1 mg/day
1–2 mg/day
2–4 mg/day
2.5–5 mg/day
Oral EV
3 mg/day
6 mg/day
10 mg/day
12 mg/day
Sublingual EVa
0.75–1.5 mg/day
1.5–3 mg/day
2.5–5 mg/day
3–6 mg/day
a Range calculated by dividing oral doses by two and four to reflect differences in absolute bioavailability and rounding to the nearest 0.25 mg. * Bioidentical estradiol has wide interindividual variation in its pharmacology and the effects of doses are likely to vary significantly between individuals. EV has greater molecular weight and therefore contains less medication for the same amount/dose by weight. It should be noted that estimates for the relative bioavailability of EV are extrapolated from formulations with no valeric ester attached (i.e., E2).
Administration of Multiple Sublingual Doses Per Day
In order to compensate for the short half-life of sublingually administered estradiol, multiple doses of estrogens can be administered in smaller quantities per day to maintain hormone levels that are somewhat more consistent (Ahokas, Kaukoranta, & Aito, 1999; Yaish et al., 2023a; Cortez et al., 2024).
In one study of premenopausal women with high-dose estrogen therapy, 2 mg of sublingual estradiol was administered three or four times per day (a total of 6–8 mg/day), resulting in significantly more stable hormone levels than would be expected with a single dose per day (Serhal & Craft, 1989). This was replicated in another study where estradiol was administered three to eight times per day (Ahokas et al., 2001). Conversely, a third study investigating low-dose buccal estradiol found little difference between the “steady-state” estradiol concentrations with a once-daily and twice-daily 0.25 mg dose of buccal estradiol over a 12 hour observation period (Wren et al., 2003). These findings may indicate that sublingual and buccal estradiol needs to be taken at least thrice per day in order to achieve concentrations of estradiol that are more stable.
It would seem advantageous for transfeminine people using sublingual estradiol that sublingual estradiol is taken in divided doses throughout the day; perhaps ideally at least three or four times per day. For instance, instead of taking a 2 mg dose every 24 hours, it would be better to take four 0.5 mg doses in the space of 24 hours (as evenly spaced as practical). Administering sublingual estradiol multiple times throughout the day might be less convenient, but is likely to provide at least somewhat more balanced estradiol levels. The administration of multiple doses every day could be regarded as optimal for the use of sublingually administered estradiol.
Sublingually Administered Estradiol and Feminisation
The very short half-life of sublingually and buccally administered estradiol relative to other forms raises a few questions relating to its use in feminising hormone therapy. One of the most commonly asked questions on online forums is regarding which gender-affirming hormone therapy regimens might be most “effective” with respect to the feminising effects of estrogens. These include, but are not limited to, outcomes such as breast development and fat distribution.
In contrast to oral and trandermal estradiol, limited data exist describing the extent of feminisation with the sublingual route (Safer, 2022). A non-randomised study found that self-assessed Tanner stage after 6 months of treatment did not appear to be different in users of sublingual estradiol monotherapy as compared to users of oral estradiol plus 10 mg/day cyproterone acetate (Yaish et al., 2023a; Yaish et al., 2023b). However, since breast development itself was not measured objectively, these particular data are low-quality and prevent definitive conclusions either way about the superiority or inferiority of sublingual estradiol. The same study group reported that although there were similar increases in gynoid fat in the two arms, the oral estradiol group did show an increased amount of android fat as compared to the sublingual group (Yaish et al., 2025). On the other hand, a further complication of this study is the possible confounding by lack of concomitant antiandrogen therapy in the sublingual arm (Ruggles & Cheung, 2024; Yaish et al., 2024). Notably, progestogens like cyproterone acetate have been shown to be associated with weight gain (Lopez et al., 2016). This could explain the difference in android fat accumulation.
Oral estradiol and other non-oral forms of estradiol (such as transdermal administration) have not been found to differ in their effects on breast development or other feminising outcomes in transfeminine people or cisgender hypogonadal girls (Rosenfield et al., 2005; Shah et al., 2014; Klaver et al., 2018; de Blok et al., 2021, Tebbens et al., 2022). In consideration of this, differences in efficacy might not be expected for sublingual estradiol either. However, the use of supraphysiological doses of estrogens from the onset of therapy may stunt breast development and reduce final breast size in transfeminine people (Boogers et al., 2025). Because the use of sublingual estradiol results in estradiol concentrations that routinely achieve the supratherapeutic range, it is possible that this could have deleterious effects on breast development.
The fact that several gender clinics have employed sublingual estradiol for some time is encouraging (Deutsch, Bhakri, & Kubicek, 2015; Lim et al., 2019; Cirrincione et al., 2021). Nevertheless, as there is very limited data comparing the feminising efficacy of sublingual estradiol with objective measures, no firm conclusions about any differences in feminisation outcomes between sublingual estradiol and other routes of administration can currently be drawn. Hopefully, studies in the future will shed more light on this.
Testosterone Suppressing Efficacy of Sublingually Administered Estradiol
Another question that might be raised by the short half-life of sublingual estradiol is how it might compare to more conventional routes of administration in terms of its ability to suppress testosterone and other androgens.
Estrogens were first characterised for their use as antigonadotrophic antiandrogens in the 1940s in the form of oral synthetic estrogens, namely diethylstilbestrol (DES), to treat men with prostate cancer (Huggins & Hodges, 1941). Estrogens given in the form of oral ethinylestradiol (EE), long-acting estradiol esters, such as polyestradiol phosphate, and transdermal estradiol patches have been studied. Their efficacy for this indication is well established (Stege et al., 1996; Kohli, 2006; Sciarra et al., 2015). As data are more limited for testosterone suppression with estrogens in transfeminine people, these data are valuable for informing transfeminine hormone therapy. Since sublingual estradiol has never been used to treat prostatic cancer, no such data exist to show the ability of sublingual estradiol in this capacity.
Some studies have found that physiologic levels of estradiol (i.e., 100–200 pg/mL [367–734 pmol/L]) or slightly higher from non-sublingual estradiol alone result in rapid and near complete, if not complete, suppression of testosterone levels to the female range in many transfeminine people (Leinung, Feustel, & Joseph, 2018; Pappas et al., 2020; Misakian et al., 2025). Additionally, the Prostate Adenocarcinoma TransCutaneous Hormones (PATCH) study, a multicentre randomised controlled trial in the United Kingdom, showed that sustained median estradiol levels of between 215 to 250 pg/mL (789–918 pmol/L) from transdermal patches were similarly effective (~95%) to GnRH analogues in reducing testosterone levels to the castrate range (<50 ng/dL [<1.7 nmol/L]) (Langley et al., 2021). However, because sublingual estradiol differs in its pharmacokinetics to other forms of estradiol, it is plausible that this route of administration might result in sub-par suppression at doses with similar concentrations of estradiol.
A few studies have reported the extent of testosterone suppression under sublingual estradiol in transfeminine people. In a randomised controlled trial (RCT) comparing once-daily and twice-daily administration of 2 mg sublingual estradiol to 0.1 mcg/day transdermal estradiol with and without spironolactone, both of the sublingual arms were found to result in inferior testosterone suppression at 1-month and 6-month follow-up (Cortez et al., 2023; Cortez et al., 2024). The authors hypothesised that this could be due to the ability of high concentrations of estrone, which were seen with sublingual estradiol, to inhibit cooperative binding of the estrogen receptor. However, this notion is contradicted by studies comparing oral and transdermal administration of estradiol which have reported no difference in the ability of these formulations to suppress testosterone at equivalent doses (SoRelle et al., 2019; Salakphet et al., 2022; Slack et al., 2025). This is in spite of the large amount of estrone also known to be generated from oral estradiol. Another study of transfeminine people found that sublingual estradiol at a dose of 0.5 mg administered four times daily was able to suppress testosterone as well as oral estradiol in combination with low-dose cypoterone acetate (Yaish et al., 2023a; Yaish et al., 2023b). The use of the four times daily dosing regimen in this study may account for the difference in findings between these two studies in the ability to suppress testosterone. Sublingual estradiol has been studied in transfeminine people in combination with and without the low-dose use of the progestin medroxyprogesterone acetate (MPA) (Jain, Kwan, & Forcier, 2019). In this study, high rates of suppressed testosterone levels (ie: <50 ng/dL [<1.7 nmol/L]) were achieved by the transfeminine people who took sublingual estradiol with medroxyprogesterone acetate, showing that sublingual estradiol taken together with progestogens such as cyproterone acetate is viable for achieving effective testosterone suppression.
A possibility supported by some evidence from pharmacological studies of estradiol is that sustained estradiol levels may be more efficacious with respect to testosterone suppression than the frequent and short-lived peaks in estradiol concentrations that occur with the sublingual route. In some studies of both sublingual and intravenous administration, limited suppression of the gonadotropins (follicle-stimulating hormone and luteinising hormone) have been reported in women despite sufficiently elevated estradiol levels for several hours (Tsai & Yen, 1971; Burnier et al., 1981; Casper & Yen, 1981; Hoon et al., 1993). These studies are low quality and indirect since testosterone suppression itself was not measured and they were performed in cisgender women. Another problem is that all were single dose studies and their findings may not translate to multiple dosing. Nevertheless, these studies might suggest a mechanism by which sublingual estradiol is unable to fully suppress gonadal function in transfeminine people without the use of excessive doses that would lead to greater health risks or the additional use of other antiandrogens.
For the reasons above, transdermal patches, gels and parenteral estradiol esters, such as estradiol valerate, injected intramuscularly or subcutaneously are probably more reliable choices for estradiol monotherapy regimens. If sublingual estradiol is used as a single agent therapy, it would seem reasonable to suggest the use of many divided doses taken throughout the day, as this is probably more likely to be efficacious. Nevertheless, sublingual estradiol appears to be more effective in terms of testosterone suppression when used with concomitant antiandrogens.
Monitoring of Estradiol Levels with Sublingual Administration
A further consideration regarding the rapid changes in estradiol levels that occur with the use of sublingual estradiol is the relevance of monitoring of estradiol levels through bloodwork. Currently, consensus guidelines do not recommend a specific time for monitoring of the blood relative to the time of a last dose (Cheung et al., 2019; T’Sjoen et al., 2020; Coleman et al., 2022). This may be in part due to practical reasons, or because until very recently there were currently no robust data from randomised controlled trials to guide the specifics of dosing in transgender hormone therapy (Haupt et al., 2020). Nevertheless, because estradiol levels vary so significantly with sublingual estradiol, knowledge of how long after the last dose blood was drawn is important to ensure proper interpretation of laboratory results.
For instance, measuring hormone levels just after a dose of sublingual estradiol has been taken might lead to the misinterpretation that levels of estradiol are excessively high and that one’s dosage should be reduced to achieve a more sensible concentration of estradiol in the blood. In reality, this would be a misunderstanding caused by the pharmacology of sublingual estradiol as the point of measurement would be right around the time when estradiol levels are most likely to be at their highest. These estradiol levels would not be indicative of the average amount of exposure, which is the more accurate measure of overall estrogenicity. Similarly, on the opposite end of the scale, drawing blood just before the administration of a new dose might lead to the belief that estrogen levels are too low and, consequently, lead to the use of excessive doses of estrogens. The latter misinterpretation may be more common among people unfamiliar with the pharmacology of sublingual estradiol as levels of estradiol only remain very high in the first few hours after a dose of sublingual estradiol has been taken before falling rapidly.
A possible solution to the problem of rapidly changing hormone levels associated with the sublingual route might simply be to measure when estradiol levels are most likely to be closest to their average. In the case of sublingual estradiol, studies generally find this to be approximately four hours after the administration of a dose, although there is likely to be considerable variation between individuals (Kuhl, 2005). Nevertheless, this approach may give the most representative “snapshot” of overall estrogenic exposure and might help to avoid misleading laboratory data in users of sublingual estradiol.
Safety and Tolerability
Unfortunately, no long term safety data exist for sublingually administered estradiol in the same way that both oral and transdermal estradiol have been rigorously studied in menopausal women (Rovinski et al., 2018; CGHFBC, 2019). The published medical literature concerning the safety and tolerability of this route of administration leaves many questions unanswered.
Adverse Health Effects of Estrogens
With sufficient exposure, owing to their effects in the liver, estrogens are associated with an increased risk of blood clots (Kuhl, 2005). Additionally, under certain circumstances, estrogens can be associated with other cardiovascular complications (Anderson et al., 2004; Mikkola et al., 2005). Although the absolute risk is low in the short-term, these are the most significant health concerns associated with gender-affirming hormone therapy.
A retrospective cohort study in the United States found that the incidence of thromboembolism in transfeminine people with an average dose of 4 mg/day oral estradiol was approximately twice that of cisgender controls not taking hormone therapy after adjusting for confounders (HR 2.0, 95% CI 1.4–2.8 versus reference women) (Getahun et al., 2018). These increases in risks are much lower compared to regimens in transfeminine people in the past that included high doses of synthetic estrogens, however even such increases can significantly increase morbidity and mortality (Morimont, Dogné, & Douxfils, 2020). A 2021 meta-analysis reported an absolute incidence of VTE of 2% in transfeminine people receiving gender-affirming hormone therapy, although with significant between-study heterogeneity (Totaro et al., 2021).
Some studies have assessed the effects of sublingually administered estradiol on the liver (Pines et al., 1999; Lim et al., 2019). These data found similar effects on lipids and cholesterol to other estrogens. One line of evidence that indicates sublingual estradiol has greater hepatic impact than other non-oral forms such as trandermal estradiol is the significantly greater quantities of estrone and estrone sulphate that are generated by this route; a marker of estrogenic exposure in the liver (Burnier et al., 1981; Cirrincione et al., 2021). Intense estrogenic activation in the liver is the mechanism by which non-oral estradiol induces a hypercoagulable state at high doses (Kuhl, 2005). While a large body of research does exist concerning the short and long term health effects of estrogens, none of these studies have investigated sublingual or buccal estradiol (Oliver-Williams et al., 2019; Mishra et al., 2021). Given that oral estradiol has greater risks than non-oral estradiol, and that sublingual administration partially but not fully avoids first-pass metabolism, it may be the case that its own risk would be somewhere between the risk observed with oral estradiol and the risk observed with other conventional non-oral routes (such as transdermal estradiol). However, an ongoing prospective study reported that use of sublingual estradiol alone resulted in less favourable outcomes on some markers of coagulation in the liver as compared to oral estradiol and cyproterone acetate (Bar et al., 2024). These data are indirect, however could suggest that contrary to theoretical expectations, sublingual estradiol might be closer or even less favourable than the risk profile of oral estradiol.
Other adverse effects of estradiol include breast cancer and gallbladder disease. These risks are believed to be dose-dependent (Cummings et al., 1999; Liu et al., 2008). However, as with cardiovascular and thromboembolic complications, no data exist to describe the long-term risk in these other areas with sublingual formulations. In the interest of harm reduction and the balancing of the risks and benefits of gender-affirming hormone therapy, it would be advisable to limit doses of sublingual and buccal estradiol so that they are not excessive (i.e., <6 mg/day) (Jalal & Baldwin, 2023).
Non-compliance
A practical obstacle to the use of sublingual estradiol in transfeminine people is that it may be highly inconvenient to have to administer doses thrice, four times or perhaps even more often throughout the duration of a single day. It has been found in observational studies that, in general, the number of prescribed medications and doses per day are positively associated with patient non-compliance and the number of missed doses (Jin et al., 2008; Toh et al., 2014). These findings are especially of relevance to transfeminine people as, in most cases, we require decades of hormone therapy. While missing one dose from time to time may be of little consequence, missing doses repeatedly could be more problematic. Despite this, sublingual estradiol has been used in studies of transfeminine people where it has been administered up to four times daily (Yaish et al., 2023a).
In contrast to sublingual estradiol, the half-life of oral estradiol and transdermal gel is long enough to enable once-daily administration (Wiegratz et al., 2001; Potts & Lobo, 2005). In the case of transdermal patches and parenteral estradiol, these forms only have to be replenished every few days or after even longer intervals of time (Thurman et al., 2013; Wisner et al., 2015). Therefore, when considering the use of sublingual estradiol versus other forms, whether or not it would be practical or convenient to consistently take medication several times a day should probably also be an important consideration for transfeminine people. If not, then another formulation may be preferable for the person in question. This may be especially true for long term use.
Summary and Conclusions
Sublingual estradiol is different in its pharmacology to other routes. The main difference is that it is associated with a rapid rise and fall in estradiol levels. It has between two and four times the bioavailability of oral estradiol and hence provides the same total estradiol exposure at doses that are two to four times lower. This could be a particular advantage because sublingual estradiol, therefore, is cheaper than oral estradiol.
There is much less research investigating sublingual estradiol than other forms of estrogen. These forms, such as oral and transdermal estrogens, are widely used in the alleviation of the menopause and for other indications. Consequently, they have received much more attention and characterisation than sublingual estradiol has for transfeminine hormone therapy. However, studies are beginning to add our knowledge of sublingual estradiol. Clinical practice guidelines for transgender care, which historically did not make reference to the use of sublingual estradiol, have now begun to discuss it.
The evidence is inconclusive regarding whether sublingual estradiol results in better, worse, or the same feminisation when compared with other routes of administration. However, it is plausible that the supra-physiologic levels of estradiol that frequently occur with sublingual estradiol could be detrimental to breast development. It also seems that sublingual estradiol could result in lesser testosterone suppression when used as a single agent therapy as compared to other routes. Sublingual estradiol has, nonetheless, been shown to be effective with respect to testosterone suppression when paired with other antiandrogens. Care should be taken with sublingual estradiol when monitoring estradiol levels to ensure correct interpretation. In order to help minimise these potential problems, sublingual estradiol can be taken in multiple doses divided throughout the day.
The health risks of sublingual estradiol have not been quantified in large observational or randomised studies. Therefore, although the first pass effect in the liver is partially avoided, the cardiovascular risks associated with long-term sublingual estradiol remain unknown. Sublingual estradiol may also be inconvenient and other formulations can be used instead if preferred, particularly for more long-term therapy.
Taken together, although much more research is clearly needed to properly characterise sublingual estradiol in transfeminine hormone therapy, it might have some advantageous properties and may be a useful alternative to oral estradiol.
References
Ahokas, A., Kaukoranta, J., & Aito, M. (1999). Effect of oestradiol on postpartum depression. Psychopharmacology, 146(1), 108–110. [DOI:10.1007/s002130051095]
Ahokas, A., Kaukoranta, J., Wahlbeck, K., & Aito, M. (2001). Estrogen deficiency in severe postpartum depression: successful treatment with sublingual physiologic 17β-estradiol: a preliminary study. Journal of Clinical Psychiatry, 62(5), 332–336. [DOI:10.4088/jcp.v62n0504]
Anderson, G. L., Limacher, M., Assaf, A. R., Bassford, T., Beresford, S. A., Black, H., Bonds, D., Brunner, R., Brzyski, R., Caan, B., Chlebowski, R., Curb, D., Gass, M., Hays, J., Heiss, G., Hendrix, S., Howard, B. V., Hsia, J., Hubbell, A., Jackson, R., … & Women’s Health Initiative Steering Committee. (2004). Effects of Conjugated Equine Estrogen in Postmenopausal Women With Hysterectomy: The Women’s Health Initiative Randomized Controlled Trial. JAMA, 291(14), 1701–1712. [DOI:10.1001/jama.291.14.1701]
Bar, O. S., Yaish, I., Barzilai, M., Greenman, Y., Gindis, G., Moshe, Y., & Tordjman, K. (2024). Low dose sublingual estradiol decreases protein s, generating a potentially pro-thrombotic state: interim results of a controlled prospective pilot study of treatment-naive trans women. Endocrine Abstracts, 99 [26th European Congress of Endocrinology, Stockholm, Sweden, 11 May 2024 – 14 May 2024], EP592. [DOI:10.1530/endoabs.99.ep592]
Bartlett, J. A., & van der Voort Maarschalk, K. (2012). Understanding the oral mucosal absorption and resulting clinical pharmacokinetics of asenapine. AAPS Pharmscitech, 13(4), 1110–1115. [DOI:10.1208/s12249-012-9839-7]
Boogers, L. S., Sardo Infirri, S. A., Bouchareb, A., Dijkman, B. A. M., Helder, D., de Blok, C. J. M., Liberton, N. P. T. J., den Heijer, M., van Trotsenburg, A. S. P., Dreijerink, K. M. A., Wiepjes, C. M., & Hannema, S. E. (2025). Variations in Volume: Breast Size in Trans Women in Relation to Timing of Testosterone Suppression. The Journal of Clinical Endocrinology & Metabolism, 110(5), 1404–1410. [DOI:10.1210/clinem/dgae573]
Burnier, A. M., Martin, P. L., Yen, S. S., & Brooks, P. (1981). Sublingual absorption of micronized 17β-estradiol. American Journal of Obstetrics and Gynecology, 140(2), 146–150. [DOI:10.1016/0002-9378(81)90101-0]
Casper, R. F., & Yen, S. S. (1981). Rapid absorption of micronized estradiol-17β following sublingual administration. Obstetrics and Gynecology, 57(1), 62–64. [Google Scholar] [PubMed] [URL] [PDF]
Chandrasekhara, D. S., Ali, V., Prost, H. M., & Nader-Estekhari, S. (2002). Buccal estrogen in toothpaste study: systemic absorption of estradiol in postmenopausal or surgically menopausal women when administered as a component in toothpaste. Fertility and Sterility, 78(Suppl 1), S98–S98 (O-258). [DOI:10.1016/S0015-0282(02)03639-7]
Cheung, A. S., Wynne, K., Erasmus, J., Murray, S., & Zajac, J. D. (2019). Position statement on the hormonal management of adult transgender and gender diverse individuals. Medical Journal of Australia, 211(3), 127–133. [DOI:10.5694/mja2.50259]
Cirrincione, L. R., Winston McPherson, G., Rongitsch, J., Sadilkova, K., Drees, J. C., Krasowski, M. D., Dickerson, J. A., & Greene, D. N. (2021). Sublingual Estradiol Is Associated with Higher Estrone Concentrations than Transdermal or Injectable Preparations in Transgender Women and Gender Nonbinary Adults. LGBT Health, 8(2), 125–132. [DOI:10.1089/lgbt.2020.0249]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/S0140-6736(19)31709-X]
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C., Fields, M., Gray, T., Guo, Z., Nicol, G., & Baranski, T. (2023). Effectiveness and Safety of Different Estradiol Regimens in Transgender Women (TREAT Study): Protocol for a Randomized Controlled Trial. JMIR Research Protocols, 12, e53092. [DOI:10.2196/53092]
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C. J., Fields, M. E., Gray, T., Guo, Z., Nicol, G., & Baranski, T. (2024). Effectiveness and safety of different estradiol regimens in transgender females: a randomized controlled trial. Journal of the Endocrine Society, 8(8), bvae108. [DOI:10.1210/jendso/bvae108]
Cummings, S. R., Eckert, S., Krueger, K. A., Grady, D., Powles, T. J., Cauley, J. A., Norton, L., Nickelsen, T., Bjarnason, N. H., Morrow, M., Lippman, M. E., Black, D., Glusman, J. E., Costa, A., & Jordan, V. C. (1999). The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. JAMA, 281(23), 2189–2197. [DOI:10.1001/jama.281.23.2189]
de Blok, C., Dijkman, B., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K., & den Heijer, M. (2021). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
Deutsch, M. B., Bhakri, V., & Kubicek, K. (2015). Effects of cross-sex hormone treatment on transgender women and men. Obstetrics and Gynecology, 125(3), 605–610. [DOI:10.1097/AOG.0000000000000692]
Devissaguet, J. P., Brion, N., Lhote, O., & Deloffre, P. (1999). Pulsed estrogen therapy: pharmacokinetics of intranasal 17-beta-estradiol (S21400) in postmenopausal women and comparison with oral and transdermal formulations. European Journal of Drug Metabolism and Pharmacokinetics, 24(3), 265–271. [DOI:10.1007/BF03190030]
Doll, E. E., Gunsolus, I., Lamberton, N., Tangpricha, V., & Sarvaideo, J. L. (2020). Pharmacokinetics of Sublingual Versus Oral Estradiol in Transgender Women. Journal of the Endocrine Society, 4(Suppl 1), A1128–A1128 (SUN-LB9). [DOI:10.1210/jendso/bvaa046.2237]
Doll, E., Gunsolus, I., Thorgerson, A., Tangpricha, V., Lamberton, N., & Sarvaideo, J. L. (2022). Pharmacokinetics of Sublingual Versus Oral Estradiol in Transgender Women. Endocrine Practice, 28(3), 237–242. [DOI:10.1016/j.eprac.2021.11.081]
Fiet, J., Hermano, M., Witte, J., Villette, J. M., Haimart, M., Gourmel, B., Tabuteau, F., Rouffy, J., & Dreux, C. (1982). Post-menopausal concentrations of plasma oestradiol, oestrone, FSH and LH and of total urinary oestradiol and oestrone after a single sublingual dose of oestradiol-17β. Acta Endocrinologica, 101(1), 93–97. [DOI:10.1530/acta.0.1010093]
Gass, M. S., Rebar, R. W., Cuffie-Jackson, C., Cedars, M. I., Lobo, R. A., Shoupe, D., Judd, H. L., Buyalos, R. P., & Clisham, P. R. (2004). A short study in the treatment of hot flashes with buccal administration of 17-β estradiol. Maturitas, 49(2), 140–147. [DOI:10.1016/j.maturitas.2003.12.004]
Getahun, D., Nash, R., Flanders, W. D., Baird, T. C., Becerra-Culqui, T. A., Cromwell, L., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Silverberg, M. J., Safer, J., Slovis, J., Tangpricha, V., & Goodman, M. (2018). Cross-sex hormones and acute cardiovascular events in transgender persons: a cohort study. Annals of Internal Medicine, 169(4), 205–213. [DOI:10.7326/M17-2785]
Gindis, G., Yaish, I., Greenman, Y., Moshe, Y., Arbiv, M., Buch, A., Sofer, Y., Shefer, G., & Tordjman, K. (May 2023). Sublingual estradiol only, offers no apparent advantage over combined oral estradiol and cyproterone acetate, for Gender Affirming Hormone Therapy (GAHT) of treatment-naive transwomen: Results of a prospective pilot study. Endocrine Abstracts, 90 [25th European Congress of Endocrinology 2023, 13–16 May 2023, Istanbul, Turkey], 274–274 (abstract no. P182). [DOI:10.1530/endoabs.90.p182] [PDF]
Grock, S., Patel, R., & Ahern, S. (2024). Hormone Therapy for Transgender and Gender-Diverse Patients. In Genital Gender Affirming Surgery: An Illustrated Guide and Video Atlas (pp. 33–49). Cham: Springer Nature Switzerland. [DOI:10.1007/978-3-031-69997-9_4]
Haupt, C., Henke, M., Kutschmar, A., Hauser, B., Baldinger, S., Saenz, S. R., & Schreiber, G. (2020). Antiandrogen or estradiol treatment or both during hormone therapy in transitioning transgender women. Cochrane Database of Systematic Reviews, 11(11), CD013138. [DOI:10.1002/14651858.CD013138.pub2]
Hoon, T. J., Dawood, M. Y., Khan‐Dawood, F. S., Ramos, J., & Batenhorst, R. L. (1993). Bioequivalence of a 17β‐Estradiol Hydroxypropyl‐β‐Cyclodextrin Complex in Postmenopausal Women. The Journal of Clinical Pharmacology, 33(11), 1116–1121. [DOI:10.1002/j.1552-4604.1993.tb01949.x]
Huggins, C., & Hodges, C. V. (1941). Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Research, 1(4), 293–297. [URL]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone acetate in Gender-Affirming therapy for Transwomen: results from a retrospective study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Jalal, E., & Baldwin, C. (2023). Supratherapeutic Estrogen Levels in Transgender Women Likely From Sublingual Estradiol. Journal of the Endocrine Society, 7(Suppl 1) [ENDO 2023 Abstracts Annual Meeting of the Endocrine Society], A1095–A1096 (abstract no. SAT391/bvad114.2062). [DOI:10.1210/jendso/bvad114.2062] [PDF]
Jin, J., Sklar, G. E., Oh, V. M. S., & Li, S. C. (2008). Factors affecting therapeutic compliance: A review from the patient’s perspective. Therapeutics and Clinical Risk Management, 4(1), 269–286. [DOI:10.2147/TCRM.S1458]
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2025). Comparison of estrone/estradiol ratio and levels in transfeminine individuals on different routes of estradiol. Transgender Health, 10(3), 261–268. [DOI:10.1089/trgh.2023.0138]
Klaver, M., de Blok, C. J. M., Wiepjes, C. M., Nota, N. M., Dekker, M. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/EJE-17-0496]
Kohli, M. (2006). Phase II study of transdermal estradiol in androgen‐independent prostate carcinoma. Cancer, 106(1), 234–235. [DOI:10.1002/cncr.21528]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhnz, W., Blode, H., & Zimmermann, H. (1993). Pharmacokinetics of exogenous natural and synthetic estrogens and antiestrogens. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 261–322). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_15]
Kuhnz, W., Gansau, C., & Mahler, M. (1993). Pharmacokinetics of estradiol, free and total estrone, in young women following single intravenous and oral administration of 17β-estradiol. Arzneimittelforschung, 43(9), 966–973. [Google Scholar] [PubMed] [PDF]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., … & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/S0140-6736(21)00100-8]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal treatment of transgender women with oral estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Lim, H. H., Jang, Y. H., Choi, G. Y., Lee, J. J., & Lee, E. S. (2019). Gender affirmative care of transgender people: a single center’s experience in Korea. Obstetrics & Gynecology Science, 62(1), 46–55. [DOI:10.5468/ogs.2019.62.1.46]
Liu, B., Beral, V., Balkwill, A., Green, J., Sweetland, S., & Reeves, G. (2008). Gallbladder disease and use of transdermal versus oral hormone replacement therapy in postmenopausal women: prospective cohort study. BMJ, 337. [DOI:10.1136/bmj.a386]
Lobo, R. A. (1987). Absorption and metabolic effects of different types of estrogens and progestogens. Obstetrics and Gynecology Clinics of North America, 14(1), 143–167. [PubMed] [DOI:10.1016/S0889-8545(21)00577-5] [URL]
Lopez, L. M., Ramesh, S., Chen, M., Edelman, A., Otterness, C., Trussell, J., & Helmerhorst, F. M. (2016). Progestin-only contraceptives: effects on weight. Cochrane Database of Systematic Reviews, 2016(8). [DOI:10.1002/14651858.cd008815.pub4]
Mikkola, A., Aro, J., Rannikko, S., Oksanen, H., Ruutu, M., & Finnprostate Group. (2005). Cardiovascular complications in patients with advanced prostatic cancer treated by means of orchiectomy or polyestradiol phosphate. Scandinavian Journal of Urology and Nephrology, 39(4), 294–300. [DOI:10.1080/00365590510031228]
Misakian, A. L., Ariel, D., Sullivan, E. A., Singh, G., Loeb, D., Strickland, T., Iwamoto, S. J., Rothman, M. S., Botzheim, B., Liang, J. W., Kelley, C., & Hamnvik, O. R. (2025). Injectable Estradiol Monotherapy Effectively Suppresses Testosterone in Gender-Affirming Hormone Therapy. Endocrine Practice, 31(11), 1462–1469. [DOI:10.1016/j.eprac.2025.07.002]
Mishra, S. R., Chung, H. F., Waller, M., & Mishra, G. D. (2021). Duration of estrogen exposure during reproductive years, age at menarche and age at Menopause, and risk of cardiovascular disease events, all‐cause and cardiovascular mortality: a systematic review and meta‐analysis. BJOG: An International Journal of Obstetrics & Gynaecology, 128(5), 809–821. [DOI:10.1111/1471-0528.16524]
Morimont, L., Dogné, J. M., & Douxfils, J. (2020). Letter to the Editors-in-Chief in response to the article of Abou-Ismail, et al. entitled “Estrogen and thrombosis: A bench to bedside review” (Thrombosis Research 192 (2020) 40–51). Thrombosis Research, 193, 221–223. [DOI:10.1016/j.thromres.2020.08.006]
Oliver-Williams, C., Glisic, M., Shahzad, S., Brown, E., Pellegrino Baena, C., Chadni, M., Chowdhury, R., Franco, O. H., & Muka, T. (2019). The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Human Reproduction Update, 25(2), 257–271. [DOI:10.1093/humupd/dmy039]
Pappas, I. I., Craig, W. Y., Spratt, L. V., & Spratt, D. I. (2021). Efficacy of Sex Steroid Therapy Without Progestin or GnRH Agonist for Gonadal Suppression in Adult Transgender Patients. The Journal of Clinical Endocrinology & Metabolism, 106(3), e1290–e1300. [DOI:10.1210/clinem/dgaa884]
Perloff, W. H. (1950). Estradiol buccal tablets in the treatment of the menopause. American Journal of Obstetrics and Gynecology, 59(1), 223–225. [DOI:10.1016/0002-9378(50)90390-5]
Pickar, J. H., Bon, C., Amadio, J. M., Mirkin, S., & Bernick, B. (2015). Pharmacokinetics of the first combination 17β-estradiol/progesterone capsule in clinical development for menopausal hormone therapy. Menopause, 22(12), 1308–1316. [DOI:10.1097/GME.0000000000000467]
Pines, A., Averbuch, M., Fisman, E. Z., & Rosano, G. M. (1999). The acute effects of sublingual 17β-estradiol on the cardiovascular system. Maturitas, 33(1), 81–85. [DOI:10.1016/S0378-5122(99)00036-5]
Potts, R. O., & Lobo, R. A. (2005). Transdermal drug delivery: clinical considerations for the obstetrician–gynecologist. Obstetrics & Gynecology, 105(5), 953–961. [DOI:10.1097/01.AOG.0000161958.70059.db]
Price, T. M., Blauer, K. L., Hansen, M., Stanczyk, F., Lobo, R., & Bates, G. W. (1997). Single-dose pharmacokinetics of sublingual versus oral administration of micronized 17β-estradiol. Obstetrics & Gynecology, 89(3), 340–345. [DOI:10.1016/S0029-7844(96)00513-3]
Rosenfield, R. L., Devine, N., Hunold, J. J., Mauras, N., Moshang Jr, T., & Root, A. W. (2005). Salutary effects of combining early very low-dose systemic estradiol with growth hormone therapy in girls with Turner syndrome. The Journal of Clinical Endocrinology & Metabolism, 90(12), 6424–6430. [DOI:10.1210/jc.2005-1081]
Rovinski, D., Ramos, R. B., Fighera, T. M., Casanova, G. K., & Spritzer, P. M. (2018). Risk of venous thromboembolism events in postmenopausal women using oral versus non-oral hormone therapy: a systematic review and meta-analysis. Thrombosis Research, 168, 83–95. [DOI:10.1016/j.thromres.2018.06.014]
Ruggles, T., & Cheung, A. S. (2024). Re:” Sublingual Estradiol Offers No Apparent Advantage over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study” by Yaish et al. Transgender Health, 9(5), 459. [DOI:10.1089/trgh.2024.0048]
Safer, J. D. (2022). Are the Pharmacokinetics of Sublingual Estradiol Superior or Inferior to Those of Oral Estradiol? Endocrine Practice, 28(3), 351–352. [DOI:10.1016/j.eprac.2021.12.018]
Salakphet, T., Mattawanon, N., Manojai, N., Muangmool, T., & Tangpricha, V. (2022). Hormone concentrations in transgender women who self-prescribe gender affirming hormone therapy: a retrospective study. The Journal of Sexual Medicine, 19(5), 864–871. [DOI:10.1016/j.jsxm.2022.02.023]
Sarvaideo, J., Doll, E., & Tangpricha, V. (2022). More Studies Are Needed to Establish the Safety and Efficacy of Sublingual Estradiol in Transgender Women. Endocrine Practice, 28(3), 353–354. [DOI:10.1016/j.eprac.2022.01.004]
Sciarra, A., Gentile, V., Cattarino, S., Gentilucci, A., Alfarone, A., D’Eramo, G., & Salciccia, S. (2015). Oral ethinylestradiol in castration‐resistant prostate cancer: a 10‐year experience. International Journal of Urology, 22(1), 98–103. [DOI:10.1111/iju.12613]
Serhal, P., & Craft, I. (1989). Oocyte donation in 61 patients. The Lancet, 333(8648), 1185–1187. [DOI:10.1016/S0140-6736(89)92762-1]
Shah, S., Forghani, N., Durham, E., & Neely, E. K. (2014). A randomized trial of transdermal and oral estrogen therapy in adolescent girls with hypogonadism. International Journal of Pediatric Endocrinology, 2014(1), 12. [DOI:10.1186/1687-9856-2014-12]
Slack, D. J., Di Via Ioschpe, A., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., Karim, S., & Safer, J. D. (2025). Examining the Influence of the Route of Administration and Dose of Estradiol on Serum Estradiol and Testosterone Levels in Feminizing Gender-Affirming Hormone Therapy. Endocrine Practice, 31(1), 19–27. [DOI:10.1016/j.eprac.2024.10.002]
SoRelle, J. A., Jiao, R., Gao, E., Veazey, J., Frame, I., Quinn, A. M., Day, P., Pagels, P., Gimpel, N., & Patel, K. (2019). Impact of Hormone Therapy on Laboratory Values in Transgender Patients. Clinical Chemistry, 65(1), 170–179. [DOI:10.1373/clinchem.2018.292730]
Stanczyk, F. Z., Archer, D. F., & Bhavnani, B. R. (2013). Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception, 87(6), 706–727. [DOI:10.1016/j.contraception.2012.12.011]
Stege, R., Gunnarsson, P. O., Johansson, C. J., Olsson, P., Pousette, Å., & Carlström, K. (1996). Pharmacokinetics and testosterone suppression of a single dose of polyestradiol phosphate (Estradurin®) in prostatic cancer patients. The Prostate, 28(5), 307–310. [DOI:10.1002/(SICI)1097-0045(199605)28:5<307::AID-PROS6>3.0.CO;2-8]
Sudhakar, D., Huang, Z., Zietkowski, M., Powell, N., & Fisher, A. R. (2023). Feminizing gender‐affirming hormone therapy for the transgender and gender diverse population: An overview of treatment modality, monitoring, and risks. Neurourology and Urodynamics, 42(5), 903–920. [DOI:10.1002/nau.25097]
Tebbens, M., Heijboer, A. C., T’Sjoen, G., Bisschop, P. H., & den Heijer, M. (2022). The role of estrone in feminizing hormone treatment. The Journal of Clinical Endocrinology & Metabolism, 107(2), 458–466. [DOI:10.1210/clinem/dgab741]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine position statement “assessment and hormonal management in adolescent and adult trans people, with attention for sexual function and satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
Thurman, A., Kimble, T., Hall, P., Schwartz, J. L., & Archer, D. F. (2013). Medroxyprogesterone acetate and estradiol cypionate injectable suspension (Cyclofem) monthly contraceptive injection: steady-state pharmacokinetics. Contraception, 87(6), 738–743. [DOI:10.1016/j.contraception.2012.11.010]
Toh, M. R., Teo, V., Kwan, Y. H., Raaj, S., Tan, S. Y. D., & Tan, J. Z. Y. (2014). Association between number of doses per day, number of medications and patient’s non-compliance, and frequency of readmissions in a multi-ethnic Asian population. Preventive Medicine Reports, 1, 43–47. [DOI:10.1016/j.pmedr.2014.10.001]
Totaro, M., Palazzi, S., Castellini, C., Parisi, A., D’Amato, F., Tienforti, D., Baroni, M. G., Francavilla, S., & Barbonetti, A. (2021). Risk of Venous Thromboembolism in Transgender People Undergoing Hormone Feminizing Therapy: A Prevalence Meta-Analysis and Meta-Regression Study. Frontiers in Endocrinology, 12, 741866. [DOI:10.3389/fendo.2021.741866]
Tsai, C. C., & Yen, S. S. C. (1971). Acute effects of intravenous infusion of 17β-estradiol on gonadotropin release in pre-and post-menopausal women. The Journal of Clinical Endocrinology & Metabolism, 32(6), 766–771. [DOI:10.1210/jcem-32-6-766]
Verdonk, S. J., Vesper, H. W., Martens, F., Sluss, P. M., Hillebrand, J. J., & Heijboer, A. C. (2019). Estradiol reference intervals in women during the menstrual cycle, postmenopausal women and men using an LC-MS/MS method. Clinica Chimica Acta, 495, 198–204. [DOI:10.1016/j.cca.2019.04.062]
Wiegratz, I., Fink, T., Rohr, U. D., Lang, E., Leukel, P., & Kuhl, H. (2001). Überkreuz-Vergleich der Pharmakokinetik von Estradiol unter der Hormonsubstitution mit Estradiolvalerat oder mikronisiertem Estradiol. [Cross-over comparison of the pharmacokinetics of estradiol during hormone replacement therapy with estradiol valerate or micronized estradiol.] Zentralblatt für Gynäkologie, 123(9), 505–512. [PubMed] [DOI:10.1055/s-2001-18223]
Wisner, K. L., Sit, D. K., Moses-Kolko, E. L., Driscoll, K. E., Prairie, B. A., Stika, C. S., Eng, H. F., Dills, J. L., Luther, J. F., & Wisniewski, S. R. (2015). Transdermal estradiol treatment for postpartum depression: a pilot randomized trial. Journal of Clinical Psychopharmacology, 35(4), 389–395. [DOI:10.1097/JCP.0000000000000351]
Wren, B. G., Day, R. O., McLachlan, A. J., & Williams, K. M. (2003). Pharmacokinetics of estradiol, progesterone, testosterone and dehydroepiandrosterone after transbuccal administration to postmenopausal women. Climacteric, 6(2), 104–111. [DOI:10.1080/cmt.6.2.104.111]
Yaish, I., Gindis, G., Greenman, Y., Moshe, Y., Arbiv, M., Buch, A., Sofer, Y., Shefer, G., & Tordjman, K. (2023). Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study. Transgender Health, 8(6), 485–493. [DOI:10.1089/trgh.2023.0022]
Yaish, I., Gindis, G., Greenman, Y., Shefer, G., Buch, A., Arbiv, M., Moshe, Y., Sofer, Y., & Tordjman, K. M. (2023). Sublingual Estradiol Only, Compared To Combined Oral Estradiol And Cyproterone Acetate,Offers No Apparent Advantage For Gender Affirming Hormone Therapy (GHAT), In Treatment Naïve Transwomen: Results Of A Prospective Pilot Study. Journal of the Endocrine Society, 7(Suppl 1) [ENDO 2023 Abstracts Annual Meeting of the Endocrine Society], A1104–A1105 (abstract no. SAT409/bvad114.2080). [DOI:10.1210/jendso/bvad114.2080] [PDF]
Yaish, I., Greenman, Y., Sofer, Y., & Tordjman, K. M. (2024). Response to Ruggles and Cheung, Re:” Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study”. Transgender Health, 9(5), 460–461. [DOI:10.1089/trgh.2024.0105]
Yaish, I., Buch, A., Gindis, G., Sofer, Y., Arbiv, M., Moshe, Y., Grenman, Y., & Tordjman, K. (2025). Early body composition changes in trans women on low-dose estradiol: comparing oral vs sublingual administration using dual energy absorptiometry and bioelectrical impedance analysis. The Journal of Sexual Medicine, 22(4), 625–635. [DOI:10.1093/jsxmed/qdaf005]
\ No newline at end of file
diff --git a/transfemscience.org/articles/sublingual-ev/index.html b/transfemscience.org/articles/sublingual-ev/index.html
index 7e22a7d2..14709bb7 100644
--- a/transfemscience.org/articles/sublingual-ev/index.html
+++ b/transfemscience.org/articles/sublingual-ev/index.html
@@ -1 +1 @@
-Sublingual Administration of Oral Estradiol Valerate Tablets for Transfeminine People - Transfeminine Science
Sublingual Administration of Oral Estradiol Valerate Tablets for Transfeminine People
By Aly | First published January 5, 2019 | Last modified November 28, 2022
Abstract / TL;DR
Oral estradiol tablets can be taken sublingually instead of orally and this allows for greater bioavailability and higher estradiol levels than with oral use. For this reason, sublingual estradiol is frequently used in transfeminine people. Oral estradiol valerate tablets also exist in some parts of the world and it’s often inquired by transfeminine people whether this form of estradiol is likewise effective sublingually. Research in this area is limited and no direct comparisons exist, but at least one study in cisgender women has shown that oral estradiol valerate tablets taken sublingually can result in high estradiol levels and gonadal suppression analogously to sublingual estradiol. In addition, at least one gender-affirming hormone therapy clinic has reported use of sublingual estradiol valerate in transfeminine people. Hence, sublingual estradiol valerate appears to be an effective means of estradiol delivery similarly to sublingual estradiol. One difference between these forms of estradiol is that estradiol valerate contains less estradiol than estradiol by weight and hence should be taken at slightly higher doses. While they can be effective, oral estradiol tablets and oral estradiol valerate tablets were not intended or designed for sublingual use, and formulations of these tablets vary. The implications of this in terms of clinical properties, if any, are unknown. However, it does seem apparent that different tablet formulations may require substantially different amounts of time to dissolve when used sublingually. As a result, some formulations of oral estradiol and oral estradiol valerate may be better-suited for sublingual use than others. Additional characteristics of these formulations like micronization and lipophilicity could also differentially influence their pharmacokinetics in ways that have not been studied. It may be advisable to choose sublingual estradiol over sublingual estradiol valerate where possible simply because sublingual estradiol is much better-characterized in comparison and there are fewer unknowns with it. However, sublingual estradiol valerate can be a clearly effective form of estradiol for transfeminine people as well if needed.
Introduction
Oral estradiol tablets (e.g., Estrace and Estrofem among other brand names) are indicated for oral administration (i.e., taken by mouth/swallowed) and this is how they are normally taken. As an alternative to the standard oral route however, these tablets can be taken sublingually (held under the tongue) or buccally (held in the cheek or lips/gums). Sublingual or buccal administration of oral estradiol tablets allows for much greater bioavailability and estradiol levels in comparison to oral administration (Sam, 2021; Wiki; Graphs; Wiki). Transfeminine people often use sublingual estradiol as the estrogen component of hormone therapy. In various countries, for instance many European countries, estradiol is also provided in oral form as estradiol valerate (EV) tablets (e.g., Progynova among other brand names). It is frequently inquired by transfeminine people whether estradiol valerate tablets can be taken sublingually similarly to estradiol tablets and whether there are any differences between these two estradiol forms for this route. This article is intended to explore and shed some light on these questions.
Effectiveness of Sublingual Estradiol Valerate
Estradiol valerate is an estradiol ester and a prodrug of estradiol. Estradiol esters themselves are pharmacologically inactive prior to conversion into estradiol. Estradiol valerate and other related estradiol esters are cleaved into estradiol by various esteraseenzymes. These esterases are widely expressed throughout the body, and the metabolism of estradiol esters into estradiol occurs not only in the liver but has also been shown to take place rapidly in blood and other tissues (Wiki). Hence, estradiol esters like estradiol valerate do not require the first pass through the liver that occurs with oral administration to become pharmacologically active as estrogens. As such, transformation of estradiol valerate into estradiol should not be an impediment in terms of non-oral administration of estradiol valerate, for instance via sublingual administration (as well as in the form of depot injectables of course).
Studies of estradiol tablets administered sublingually instead of orally are limited, and studies on estradiol valerate tablets used sublingually are extremely scarce. No direct comparisons have been made between sublingual use of estradiol tablets versus sublingual use of estradiol valerate tablets. Hence, we currently don’t have reliable data on how sublingual estradiol valerate compares to sublingual estradiol in terms of pharmacokinetics (e.g., bioavailability, estradiol levels, concentration–time curve, etc.).
Only one study seems to have researched and characterized sublingual administration of oral estradiol valerate tablets. This study assessed the use of oral estradiol valerate tablets (brand name Progynova [Schering]) administered sublingually 3 to 4 times per day in a group of premenopausal cisgender women. The researchers reported the findings of their study in the following two publications:
Serhal, P., & Craft, I. (1989). Oocyte donation in 61 patients. The Lancet, 333(8648), 1185–1187. [DOI:10.1016/S0140-6736(89)92762-1]
The results in terms of hormone levels with sublingual estradiol valerate were as follows (top plot is a control menstrual cycle in untreated women, bottom plot is a cycle with sublingual estradiol valerate):
Figure 1: Levels of estradiol (E2), progesterone (P4), luteinizing hormone (LH), and follicle-stimulating hormone (FSH) with 2 mg oral micronized estradiol valerate tablets (Progynova) administered sublingually (SL) 3 or 4 times per day in a group of premenopausal women (bottom). The normal menstrual cycle in a control group of premenopausal women is also shown (top). The time of blood collection following administration was not specified.
As can be seen in the figure, estradiol levels with sublingual estradiol valerate were much higher than in normally cycling control women. In addition, levels of other hormones were suppressed, which is in accordance with the high estradiol levels exhibiting negative feedback on the hypothalamic–pituitary–gonadal axis and suppressing hormone production. These findings indicate that sublingual estradiol valerate is well-absorbed and is able to achieve high estradiol levels analogously to sublingual estradiol. In other words, although we still don’t have direct comparisons between the two, sublingual estradiol valerate appears to be a highly effective means of estradiol delivery similarly to sublingual estradiol.
It’s notable that at least one gender-affirming hormone therapy clinic, located in South Korea, uses sublingual estradiol valerate in transfeminine people. These clinicians have briefly described their experience with sublingual estradiol valerate and their rationale for using it over oral estradiol in a recent publication (Lim et al., 2019). While they don’t provide actual pharmacokinetic data on sublingual estradiol valerate (e.g., estradiol levels), it seems apparent based on their clinical experience that sublingual use is a therapeutically effective route of administration for this form of estradiol.
Formulation of Estradiol and Estradiol Valerate Tablets and Implications for Sublingual Use
Micronization of Oral Estradiol Valerate Tablets
Micronization is a manufacturing process in which solid particles of a substance are reduced to smaller sizes. Micronization can modify the pharmacokinetics of medications by altering their rate and extent of absorption. Oral estradiol tablets were originally non-micronized and had lower estrogenic potency than those used today, but in the 1970s micronized oral estradiol tablets were introduced and replaced the previous formulations (Wiki; Wiki). It appears that micronization of estradiol crystals to a defined particle size range improves the absorption and bioavailability of oral estradiol tablets by several-fold (Wiki). All modern oral estradiol tablets available today are assumed to be micronized.
Questions arise as to what influence micronization has on sublingual administration (rather than oral administration) of hormonal agents like estradiol and estradiol valerate and whether oral estradiol valerate tablets are micronized similarly to oral estradiol tablets. The influence of micronization on the sublingual absorption and pharmacokinetic characteristics of estradiol and its esters, as well as other hormonal agents like progesterone and testosterone, does not seem to have been studied and hence is unknown. It could be assumed that micronization might improve the rate and extent of absorption with sublingual administration similarly to oral administration and hence may be important however, as touched on by the following literature excerpt (Sayeed & Ashraf, 2014):
The drugs that are administered sublingually generally have low solubility. Therefore, to enhance dissolution, it is crucial to reduce and control the particle size of the [active pharmaceutical ingredient]. This attribute is important in the case of all drugs with low solubility. However, a tighter control on particle size of [active pharmaceutical ingredient] is desirable in sublingual drug products to maintain the reproducible quality and performance of the drug product in view of the limited window of dissolution and absorption time.
As to whether oral estradiol valerate tablets are micronized, some formulations of oral estradiol valerate clearly indicate that they are micronized in their packaging or manufacturer information (Photo), whereas other formulations of oral estradiol valerate do not. The oral estradiol valerate tablets used in the study by Serhal and Craft (Progynova [Schering]) as well as certain other publications (e.g., Devroey & Pados, 1998) have been explicitly noted to be micronized. Considering the similar doses and apparently comparable clinical properties of all oral estradiol valerate tablets used today, it may be the case that all oral estradiol valerate formulations are micronized but that this simply isn’t always explicitly stated. Indeed, the original form of oral estradiol valerate introduced in the late 1960s is said to have been micronized (Wiki). An alternative but perhaps less likely possibility is that micronization might not influence the properties of oral estradiol valerate similarly to how it does with oral estradiol.
The University of California, San Francisco (UCSF) transgender care guidelines state that only micronized oral estradiol tablets can be used sublingually and imply that not all oral estradiol tablets are micronized (Deutsch, 2016). These statements may be assumed to also apply to oral estradiol valerate tablets. However, as discussed in this article, support for these notions is lacking at present.
Physicochemical Properties of Estradiol versus Estradiol Valerate
Estradiol valerate is more lipophilic (fat-soluble) than estradiol due to its fatty acid ester moiety (i.e., valeric acid). Lipophilicity is known to modify the sublingual and buccal absorption of medications (Smart, 2005; Batheja, Thakur, & Michniak, 2006). How the differing lipophilicities of estradiol versus estradiol valerate may influence their pharmacokinetics when used sublingually has not been studied. Hence, the therapeutic implications of this physicochemical difference for this route are unknown. It’s also known however that esterases are present in the oral mucosa and saliva and can cleave carboxylic acid esters like estradiol valerate into their unesterified forms (Yamahara & Lee, 1993; Rathbone, Drummond, & Tucker, 1994). This might serve to reduce the importance of the physicochemical differences between estradiol valerate and estradiol in terms of sublingual and buccal administration.
Another difference between estradiol and estradiol valerate is that estradiol valerate has a higher molecular weight than estradiol due to its ester component and hence estradiol valerate contains less estradiol than estradiol for the same dose of substance. The molecular weight of estradiol valerate is about 131% of that of estradiol and hence estradiol valerate contains 76% of the estradiol as an equal amount of estradiol (Table). This difference has been shown to translate to pharmacokinetic studies of oral estradiol versus oral estradiol valerate, with estradiol levels being around 25% lower with oral estradiol valerate compared to oral estradiol at equal doses (Wiki). This is likely also the case for other routes of administration of these forms of estradiol, including sublingual administration. Hence, slightly higher doses (e.g., 2 mg versus 1.5 mg) are likely needed and should be used for estradiol valerate relative to estradiol for equivalent estradiol levels and therapeutic estrogenic effect (Sam, 2021).
Formulation and Dissolution of Oral Estradiol Valerate Tablets
Oral estradiol and estradiol valerate tablets were intended and designed for oral administration and not specifically for sublingual administration. Although many of these tablets do clearly work quite well when used sublingually, different tablet formulations vary in their coating and their compositions and excipients. It is possible that differences between formulations of these tablets may influence their properties when used sublingually, as touched on in the following literature excerpt (Sayeed & Ashraf, 2014):
The conditions prevailing in the oral cavity for disintegration and dissolution of sublingual tablets are markedly different from the tablets that are orally ingested. […] Other specialized [oral] tablets, such as modified-release or enteric-coated tablets, may also partly release the drug in the stomach. In contrast, sublingual tablets are designed to completely disintegrate and dissolve in the oral cavity under the tongue.
One particular issue is that different oral tablets of estradiol and estradiol valerate may dissolve at very different rates. One study of oral estradiol tablets used sublingually reported that they dissolved within 1 or 2 minutes (Burnier, 1981). Anecdotally this has been the case similarly with one brand of generic sugar-coated oral estradiol tablets in the United States (Photo), which dissolve and disappear sublingually within a few minutes at most. Some transfeminine people on social media sites like Reddit however have reported their tablets taking much longer to dissolve, for instance about an hour, when used sublingually or buccally. In any case, dissolution rates of oral estradiol and estradiol valerate tablets may vary depending on the formulation, and some forms of oral estradiol and oral estradiol valerate may be better-suited for sublingual use than others. If tablets take a long time to dissolve when used sublingually, switching to another brand may be considered.
Estradiol or Estradiol Valerate for Sublingual Use?
Sublingual administration of oral estradiol tablets has been much better researched and characterized than sublingual administration of oral estradiol valerate tablets. Because of this, it may be advisable to select oral estradiol tablets with the intention of sublingual use over oral estradiol valerate tablets simply because there are fewer unknowns with them. However, oral estradiol tablets may not always be available in a given market, a person may be prescribed oral estradiol valerate tablets instead of oral estradiol tablets, or other considerations may make oral estradiol tablets a less feasible option. In this regard, it is clear based on available literature that sublingual estradiol valerate can be a highly effective means of delivering estradiol similarly to sublingual estradiol and can be used instead if needed.
References
Batheja, P., Thakur, R., & Michniak, B. (2006). Basic Biopharmaceutics of Buccal and Sublingual Absorption. In Touitou, E., & Barry, B. W. (Eds.). Enhancement in Drug Delivery (pp. 175–202). Boca Raton/London/New York: CRC Press. [Google Scholar] [Google Books] [DOI:10.1201/9781420004816-17]
Burnier, A. M., Martin, P. L., Yen, S. S., & Brooks, P. (1981). Sublingual absorption of micronized 17β-estradiol. American Journal of Obstetrics and Gynecology, 140(2), 146–150. [DOI:10.1016/0002-9378(81)90101-0]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [Google Scholar] [URL] [PDF]
Devroey, P., & Pados, G. (1998). Preparation of endometrium for egg donation. Human Reproduction Update, 4(6), 856–861. [DOI:10.1093/humupd/4.6.856]
Lim, H. H., Jang, Y. H., Choi, G. Y., Lee, J. J., & Lee, E. S. (2019). Gender affirmative care of transgender people: a single center’s experience in Korea. Obstetrics & Gynecology Science, 62(1), 46–55. [DOI:10.5468/ogs.2019.62.1.46]
Rathbone, M. J., Drummond, B. K., & Tucker, I. G. (1994). The oral cavity as a site for systemic drug delivery. Advanced Drug Delivery Reviews, 13(1–2), 1–22. [DOI:10.1016/0169-409X(94)90024-8]
Sayeed, V. A., & Ashraf, M. (2014). Considerations in Developing Sublingual Tablets—An Overview. Pharmaceutical Technology, 38(11), 34–72. [Google Scholar] [URL] [PDF]
Serhal, P., & Craft, I. (1989). Oocyte donation in 61 patients. The Lancet, 333(8648), 1185–1187. [DOI:10.1016/S0140-6736(89)92762-1]
Smart, J. D. (2005). Buccal drug delivery. Expert Opinion on Drug Delivery, 2(3), 507–517. [DOI:10.1517/17425247.2.3.507]
Yamahara, H., & Lee, V. H. (1993). Drug metabolism in the oral cavity. Advanced Drug Delivery Reviews, 12(1–2), 25–39. [DOI:10.1016/0169-409X(93)90039-7]
\ No newline at end of file
+Sublingual Administration of Oral Estradiol Valerate Tablets for Transfeminine People - Transfeminine Science
Sublingual Administration of Oral Estradiol Valerate Tablets for Transfeminine People
By Aly | First published January 5, 2019 | Last modified November 28, 2022
Abstract / TL;DR
Oral estradiol tablets can be taken sublingually instead of orally and this allows for greater bioavailability and higher estradiol levels than with oral use. For this reason, sublingual estradiol is frequently used in transfeminine people. Oral estradiol valerate tablets also exist in some parts of the world and it’s often inquired by transfeminine people whether this form of estradiol is likewise effective sublingually. Research in this area is limited and no direct comparisons exist, but at least one study in cisgender women has shown that oral estradiol valerate tablets taken sublingually can result in high estradiol levels and gonadal suppression analogously to sublingual estradiol. In addition, at least one gender-affirming hormone therapy clinic has reported use of sublingual estradiol valerate in transfeminine people. Hence, sublingual estradiol valerate appears to be an effective means of estradiol delivery similarly to sublingual estradiol. One difference between these forms of estradiol is that estradiol valerate contains less estradiol than estradiol by weight and hence should be taken at slightly higher doses. While they can be effective, oral estradiol tablets and oral estradiol valerate tablets were not intended or designed for sublingual use, and formulations of these tablets vary. The implications of this in terms of clinical properties, if any, are unknown. However, it does seem apparent that different tablet formulations may require substantially different amounts of time to dissolve when used sublingually. As a result, some formulations of oral estradiol and oral estradiol valerate may be better-suited for sublingual use than others. Additional characteristics of these formulations like micronization and lipophilicity could also differentially influence their pharmacokinetics in ways that have not been studied. It may be advisable to choose sublingual estradiol over sublingual estradiol valerate where possible simply because sublingual estradiol is much better-characterized in comparison and there are fewer unknowns with it. However, sublingual estradiol valerate can be a clearly effective form of estradiol for transfeminine people as well if needed.
Introduction
Oral estradiol tablets (e.g., Estrace and Estrofem among other brand names) are indicated for oral administration (i.e., taken by mouth/swallowed) and this is how they are normally taken. As an alternative to the standard oral route however, these tablets can be taken sublingually (held under the tongue) or buccally (held in the cheek or lips/gums). Sublingual or buccal administration of oral estradiol tablets allows for much greater bioavailability and estradiol levels in comparison to oral administration (Sam, 2021; Wiki; Graphs; Wiki). Transfeminine people often use sublingual estradiol as the estrogen component of hormone therapy. In various countries, for instance many European countries, estradiol is also provided in oral form as estradiol valerate (EV) tablets (e.g., Progynova among other brand names). It is frequently inquired by transfeminine people whether estradiol valerate tablets can be taken sublingually similarly to estradiol tablets and whether there are any differences between these two estradiol forms for this route. This article is intended to explore and shed some light on these questions.
Effectiveness of Sublingual Estradiol Valerate
Estradiol valerate is an estradiol ester and a prodrug of estradiol. Estradiol esters themselves are pharmacologically inactive prior to conversion into estradiol. Estradiol valerate and other related estradiol esters are cleaved into estradiol by various esteraseenzymes. These esterases are widely expressed throughout the body, and the metabolism of estradiol esters into estradiol occurs not only in the liver but has also been shown to take place rapidly in blood and other tissues (Wiki). Hence, estradiol esters like estradiol valerate do not require the first pass through the liver that occurs with oral administration to become pharmacologically active as estrogens. As such, transformation of estradiol valerate into estradiol should not be an impediment in terms of non-oral administration of estradiol valerate, for instance via sublingual administration (as well as in the form of depot injectables of course).
Studies of estradiol tablets administered sublingually instead of orally are limited, and studies on estradiol valerate tablets used sublingually are extremely scarce. No direct comparisons have been made between sublingual use of estradiol tablets versus sublingual use of estradiol valerate tablets. Hence, we currently don’t have reliable data on how sublingual estradiol valerate compares to sublingual estradiol in terms of pharmacokinetics (e.g., bioavailability, estradiol levels, concentration–time curve, etc.).
Only one study seems to have researched and characterized sublingual administration of oral estradiol valerate tablets. This study assessed the use of oral estradiol valerate tablets (brand name Progynova [Schering]) administered sublingually 3 to 4 times per day in a group of premenopausal cisgender women. The researchers reported the findings of their study in the following two publications:
Serhal, P., & Craft, I. (1989). Oocyte donation in 61 patients. The Lancet, 333(8648), 1185–1187. [DOI:10.1016/S0140-6736(89)92762-1]
The results in terms of hormone levels with sublingual estradiol valerate were as follows (top plot is a control menstrual cycle in untreated women, bottom plot is a cycle with sublingual estradiol valerate):
Figure 1: Levels of estradiol (E2), progesterone (P4), luteinizing hormone (LH), and follicle-stimulating hormone (FSH) with 2 mg oral micronized estradiol valerate tablets (Progynova) administered sublingually (SL) 3 or 4 times per day in a group of premenopausal women (bottom). The normal menstrual cycle in a control group of premenopausal women is also shown (top). The time of blood collection following administration was not specified.
As can be seen in the figure, estradiol levels with sublingual estradiol valerate were much higher than in normally cycling control women. In addition, levels of other hormones were suppressed, which is in accordance with the high estradiol levels exhibiting negative feedback on the hypothalamic–pituitary–gonadal axis and suppressing hormone production. These findings indicate that sublingual estradiol valerate is well-absorbed and is able to achieve high estradiol levels analogously to sublingual estradiol. In other words, although we still don’t have direct comparisons between the two, sublingual estradiol valerate appears to be a highly effective means of estradiol delivery similarly to sublingual estradiol.
It’s notable that at least one gender-affirming hormone therapy clinic, located in South Korea, uses sublingual estradiol valerate in transfeminine people. These clinicians have briefly described their experience with sublingual estradiol valerate and their rationale for using it over oral estradiol in a recent publication (Lim et al., 2019). While they don’t provide actual pharmacokinetic data on sublingual estradiol valerate (e.g., estradiol levels), it seems apparent based on their clinical experience that sublingual use is a therapeutically effective route of administration for this form of estradiol.
Formulation of Estradiol and Estradiol Valerate Tablets and Implications for Sublingual Use
Micronization of Oral Estradiol Valerate Tablets
Micronization is a manufacturing process in which solid particles of a substance are reduced to smaller sizes. Micronization can modify the pharmacokinetics of medications by altering their rate and extent of absorption. Oral estradiol tablets were originally non-micronized and had lower estrogenic potency than those used today, but in the 1970s micronized oral estradiol tablets were introduced and replaced the previous formulations (Wiki; Wiki). It appears that micronization of estradiol crystals to a defined particle size range improves the absorption and bioavailability of oral estradiol tablets by several-fold (Wiki). All modern oral estradiol tablets available today are assumed to be micronized.
Questions arise as to what influence micronization has on sublingual administration (rather than oral administration) of hormonal agents like estradiol and estradiol valerate and whether oral estradiol valerate tablets are micronized similarly to oral estradiol tablets. The influence of micronization on the sublingual absorption and pharmacokinetic characteristics of estradiol and its esters, as well as other hormonal agents like progesterone and testosterone, does not seem to have been studied and hence is unknown. It could be assumed that micronization might improve the rate and extent of absorption with sublingual administration similarly to oral administration and hence may be important however, as touched on by the following literature excerpt (Sayeed & Ashraf, 2014):
The drugs that are administered sublingually generally have low solubility. Therefore, to enhance dissolution, it is crucial to reduce and control the particle size of the [active pharmaceutical ingredient]. This attribute is important in the case of all drugs with low solubility. However, a tighter control on particle size of [active pharmaceutical ingredient] is desirable in sublingual drug products to maintain the reproducible quality and performance of the drug product in view of the limited window of dissolution and absorption time.
As to whether oral estradiol valerate tablets are micronized, some formulations of oral estradiol valerate clearly indicate that they are micronized in their packaging or manufacturer information (Photo), whereas other formulations of oral estradiol valerate do not. The oral estradiol valerate tablets used in the study by Serhal and Craft (Progynova [Schering]) as well as certain other publications (e.g., Devroey & Pados, 1998) have been explicitly noted to be micronized. Considering the similar doses and apparently comparable clinical properties of all oral estradiol valerate tablets used today, it may be the case that all oral estradiol valerate formulations are micronized but that this simply isn’t always explicitly stated. Indeed, the original form of oral estradiol valerate introduced in the late 1960s is said to have been micronized (Wiki). An alternative but perhaps less likely possibility is that micronization might not influence the properties of oral estradiol valerate similarly to how it does with oral estradiol.
The University of California, San Francisco (UCSF) transgender care guidelines state that only micronized oral estradiol tablets can be used sublingually and imply that not all oral estradiol tablets are micronized (Deutsch, 2016). These statements may be assumed to also apply to oral estradiol valerate tablets. However, as discussed in this article, support for these notions is lacking at present.
Physicochemical Properties of Estradiol versus Estradiol Valerate
Estradiol valerate is more lipophilic (fat-soluble) than estradiol due to its fatty acid ester moiety (i.e., valeric acid). Lipophilicity is known to modify the sublingual and buccal absorption of medications (Smart, 2005; Batheja, Thakur, & Michniak, 2006). How the differing lipophilicities of estradiol versus estradiol valerate may influence their pharmacokinetics when used sublingually has not been studied. Hence, the therapeutic implications of this physicochemical difference for this route are unknown. It’s also known however that esterases are present in the oral mucosa and saliva and can cleave carboxylic acid esters like estradiol valerate into their unesterified forms (Yamahara & Lee, 1993; Rathbone, Drummond, & Tucker, 1994). This might serve to reduce the importance of the physicochemical differences between estradiol valerate and estradiol in terms of sublingual and buccal administration.
Another difference between estradiol and estradiol valerate is that estradiol valerate has a higher molecular weight than estradiol due to its ester component and hence estradiol valerate contains less estradiol than estradiol for the same dose of substance. The molecular weight of estradiol valerate is about 131% of that of estradiol and hence estradiol valerate contains 76% of the estradiol as an equal amount of estradiol (Table). This difference has been shown to translate to pharmacokinetic studies of oral estradiol versus oral estradiol valerate, with estradiol levels being around 25% lower with oral estradiol valerate compared to oral estradiol at equal doses (Wiki). This is likely also the case for other routes of administration of these forms of estradiol, including sublingual administration. Hence, slightly higher doses (e.g., 2 mg versus 1.5 mg) are likely needed and should be used for estradiol valerate relative to estradiol for equivalent estradiol levels and therapeutic estrogenic effect (Sam, 2021).
Formulation and Dissolution of Oral Estradiol Valerate Tablets
Oral estradiol and estradiol valerate tablets were intended and designed for oral administration and not specifically for sublingual administration. Although many of these tablets do clearly work quite well when used sublingually, different tablet formulations vary in their coating and their compositions and excipients. It is possible that differences between formulations of these tablets may influence their properties when used sublingually, as touched on in the following literature excerpt (Sayeed & Ashraf, 2014):
The conditions prevailing in the oral cavity for disintegration and dissolution of sublingual tablets are markedly different from the tablets that are orally ingested. […] Other specialized [oral] tablets, such as modified-release or enteric-coated tablets, may also partly release the drug in the stomach. In contrast, sublingual tablets are designed to completely disintegrate and dissolve in the oral cavity under the tongue.
One particular issue is that different oral tablets of estradiol and estradiol valerate may dissolve at very different rates. One study of oral estradiol tablets used sublingually reported that they dissolved within 1 or 2 minutes (Burnier, 1981). Anecdotally this has been the case similarly with one brand of generic sugar-coated oral estradiol tablets in the United States (Photo), which dissolve and disappear sublingually within a few minutes at most. Some transfeminine people on social media sites like Reddit however have reported their tablets taking much longer to dissolve, for instance about an hour, when used sublingually or buccally. In any case, dissolution rates of oral estradiol and estradiol valerate tablets may vary depending on the formulation, and some forms of oral estradiol and oral estradiol valerate may be better-suited for sublingual use than others. If tablets take a long time to dissolve when used sublingually, switching to another brand may be considered.
Estradiol or Estradiol Valerate for Sublingual Use?
Sublingual administration of oral estradiol tablets has been much better researched and characterized than sublingual administration of oral estradiol valerate tablets. Because of this, it may be advisable to select oral estradiol tablets with the intention of sublingual use over oral estradiol valerate tablets simply because there are fewer unknowns with them. However, oral estradiol tablets may not always be available in a given market, a person may be prescribed oral estradiol valerate tablets instead of oral estradiol tablets, or other considerations may make oral estradiol tablets a less feasible option. In this regard, it is clear based on available literature that sublingual estradiol valerate can be a highly effective means of delivering estradiol similarly to sublingual estradiol and can be used instead if needed.
References
Batheja, P., Thakur, R., & Michniak, B. (2006). Basic Biopharmaceutics of Buccal and Sublingual Absorption. In Touitou, E., & Barry, B. W. (Eds.). Enhancement in Drug Delivery (pp. 175–202). Boca Raton/London/New York: CRC Press. [Google Scholar] [Google Books] [DOI:10.1201/9781420004816-17]
Burnier, A. M., Martin, P. L., Yen, S. S., & Brooks, P. (1981). Sublingual absorption of micronized 17β-estradiol. American Journal of Obstetrics and Gynecology, 140(2), 146–150. [DOI:10.1016/0002-9378(81)90101-0]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [Google Scholar] [URL] [PDF]
Devroey, P., & Pados, G. (1998). Preparation of endometrium for egg donation. Human Reproduction Update, 4(6), 856–861. [DOI:10.1093/humupd/4.6.856]
Lim, H. H., Jang, Y. H., Choi, G. Y., Lee, J. J., & Lee, E. S. (2019). Gender affirmative care of transgender people: a single center’s experience in Korea. Obstetrics & Gynecology Science, 62(1), 46–55. [DOI:10.5468/ogs.2019.62.1.46]
Rathbone, M. J., Drummond, B. K., & Tucker, I. G. (1994). The oral cavity as a site for systemic drug delivery. Advanced Drug Delivery Reviews, 13(1–2), 1–22. [DOI:10.1016/0169-409X(94)90024-8]
Sam. (2021). An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People. Transfeminine Science. [URL]
Sayeed, V. A., & Ashraf, M. (2014). Considerations in Developing Sublingual Tablets—An Overview. Pharmaceutical Technology, 38(11), 34–72. [Google Scholar] [URL] [PDF]
Serhal, P., & Craft, I. (1989). Oocyte donation in 61 patients. The Lancet, 333(8648), 1185–1187. [DOI:10.1016/S0140-6736(89)92762-1]
Smart, J. D. (2005). Buccal drug delivery. Expert Opinion on Drug Delivery, 2(3), 507–517. [DOI:10.1517/17425247.2.3.507]
Yamahara, H., & Lee, V. H. (1993). Drug metabolism in the oral cavity. Advanced Drug Delivery Reviews, 12(1–2), 25–39. [DOI:10.1016/0169-409X(93)90039-7]
\ No newline at end of file
diff --git a/transfemscience.org/articles/transfem-hormone-guidelines/index.html b/transfemscience.org/articles/transfem-hormone-guidelines/index.html
index 5b9e8282..cb2dfdcc 100644
--- a/transfemscience.org/articles/transfem-hormone-guidelines/index.html
+++ b/transfemscience.org/articles/transfem-hormone-guidelines/index.html
@@ -1 +1 @@
-Clinical Guidelines with Information on Transfeminine Hormone Therapy - Transfeminine Science
Clinical Guidelines with Information on Transfeminine Hormone Therapy
By Aly | First published November 20, 2020 | Last modified November 17, 2024
Abstract / TL;DR
This article is a collection of clinical practice guidelines throughout the world with information on transfeminine hormone therapy. Examples of these clinical guidelines include the World Professional Association for Transgender Health (WPATH) Standards of Care for the Health of Transgender and Gender Diverse People, the Endocrine Society guidelines, and the University of California, San Francisco (UCSF) Center of Excellence for Transgender Health guidelines, among many others.
Introduction
Clinicians use clinical practice guidelines (CPGs) to learn about and guide themselves in administering medical care for different indications. Clinical practice guidelines review and summarize the available scientific literature and research in a given medical area. They allow clinicians to competently administer care without necessarily having to delve into and develop their understanding via the primary scientific literature. Literature reviews can serve a similar function. However, clinical practice guidelines are generally more substantial and are more founded in evidence-based medicine. They are also regularly updated. Clinical practice guidelines are developed and maintained by clinical organizations and societies, universities, government agencies, and sometimes even large medical clinics. They may be international/locationless or oftentimes region-specific.
There are many clinical practice guidelines for transgender medicine (for review, Deutsch, Radix, & Reisner, 2016; Radix, 2019; Radix, 2019; UpToDate; Bewley et al., 2021; Dahlen et al., 2021; Ziegler, Carroll, & Charnish, 2021). These guidelines discuss topics such as psychotherapy, hormone therapy, voice therapy, and surgical management of transgender people, among others. In addition to educating and guiding clinicians, transgender clinical practice guidelines are useful materials for transgender people as they can help to inform them about their care.
1st/1979: Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1979 February 13). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [1st Edition/1979 Original Draft]. Palo Alto, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar]
2nd/1980: Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1980 January 20). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [2nd Edition/1980 Revised Draft]. Stanford, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar 1] [Google Scholar 2]
3rd/1981: Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1981 March 9). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [3rd Edition/1981 Revised Draft]. San Francisco, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [URL 1] [URL 2] [DOI:10.1007/BF01541354—1985 reprint in Archives of Sexual Behavior, 14(1), 79–90)]
4th/1990: Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1990 January 25). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [4th Edition/1990 Revised Draft]. Palo Alto/Sonoma, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar 1] [Google Scholar 2] [URL 1] [URL 2] [URL 3] [URL 4] [URL 5] [URL 6]
5th/1998: Levine, S. B., Brown, G., Coleman, E., Cohen-Kettenis, P., Joris Hage, J., Van Maasdam, J., Petersen, M., Pfaefflin, F., & Schaefer, L. C. (June 1998). [The Harry Benjamin International Gender Dysphoria Association’s] The Standards of Care for Gender Identity Disorders [5th Edition]. International Journal of Transgenderism, 2(2). [Consultants: Denny, D., DiCeglie, D., Eicher, W., Green, J., Green, R., Gooren, L., Laub, D., Lawrence, A., Meyer, W., & Wheeler, C.] [URL 1] [URL 2] [URL 3] [PDF]
6th/2001: Meyer, W., Bockting, W. O., Cohen-Kettenis, P., Coleman, E., DiCeglie, D., Devor, H., Gooren, L., Joris Hage, J., Kirk, S., Kuiper, B., Laub, D., Lawrence, A., Menard, Y., Patton, J., Schaefer, L., Webb, A., & Wheeler, C. C. (February 2001). [The Harry Benjamin International Gender Dysphoria Association’s] The Standards of Care for Gender Identity Disorders – Sixth Version. International Journal of Transgenderism, 5(1). [Google Scholar 1] [Google Scholar 2] [Google Scholar 3] [URL 1] [URL 2] [DOI:10.1300/J056v13n01_01—2001/2002 reprint in Journal of Psychology & Human Sexuality, 13(1), 1–30] [PDF 1] [PDF 2]
7th/2012: Coleman, E., Bockting, W., Botzer, M., Cohen-Kettenis, P., DeCuypere, G., Feldman, J., Fraser, L., Green, J., Knudson, G., Meyer, W. J., Monstrey, S., Adler, R. K., Brown, G. R., Devor, A. H., Ehrbar, R., Ettner, R., Eyler, E., Garofalo, R., Karasic, D. H., … & Zucker, K. (2012). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, Version 7. International Journal of Transgenderism, 13(4), 165–232. [DOI:10.1080/15532739.2011.700873] [URL] [PDF]
Fisher et al. / Italian Society of Gender, Identity and Health (SIGIS) / Italian Society of Andrology and Sexual Medicine (SIAMS) / Italian Society of Endocrinology (SIE)
Godano et al. / Società Italiana di Andrologia e Medicina della Sessualità (SIAMS) [Italian Society of Andrology and Sexual Medicine] / Osservatorio Nazionale sull’Identità di Genere (ONIG) [National Observatory of Gender Identity]
Vacharathit et al. / Center of Excellence in Transgender Health / Chulalongkorn University [Thailand]
2021
Online document
Update: New Endocrine Society and UCSF Guidelines
New Endocrine Society and UCSF guidelines are under development as of February 2024 (Christensen, 2024; Endocrine Society, 2024; UCSF, 2024). It is anticipated that the Endocrine Society guidelines will be published in Spring 2026. The new UCSF guidelines were initially slated for publication in 2024, but have been pushed back until the new Endocrine Society guidelines are published. The new Endocrine Society and UCSF guidelines will be the third edition of each of the guidelines.
References
AusPATH. (2022). Australian Informed Consent Standards of Care for Gender Affirming Hormone Therapy. Australia: Australian Professional Association for Trans Health. [URL] [PDF]
Belzer, M. E., Burnett, J., Deutsch, M., Franicevich, J., Gorton, R. N., Hastings, J., Karasic, D., Kohler, L., Vanderleest, J., Van Maasdam, J., Olson, J., Green, J., & DeVries, C. (April 2011). Primary Care Protocol for Transgender Patient Care, 1st Edition. Center of Excellence for Transgender Health, University of California, San Francisco, Department of Family and Community Medicine. [URL] [PDF]
Bewley, S., Dahlen, S., Connolly, D., Arif, I., Junejo, M., & Catherine, M. (2021). International Clinical Practice Guidelines for Gender Minority/Trans People: Systematic Review & Quality Assessment. How Does the Endocrine Society Fare? Journal of the Endocrine Society, 5(Suppl 1), A791–A791. [DOI:10.1210/jendso/bvab048.1609]
Bourns, A. (2019). Guidelines for Gender-Affirming Primary Care with Trans and Non-Binary Patients, 4th Edition. Toronto: Rainbow Health Ontario/Sherbourne Health. [URL] [PDF]
Callen-Lorde Community Health Center. (2018). Protocols for the Provision of Hormone Therapy. New York City: Callen-Lorde Community Health Center. [URL] [PDF]
Carroll, R., Nicholls, R., Carroll, R. W., Bullock, J., Reid, D., Shields, J., Johnson, R., Oliphant, J., McElrea, E., Whitfield, P., & Veale, J. (2023). Primary Care Gender Affirming Hormone Therapy Initiation Guidelines: Aotearoa New Zealand Guidelines for Commencing GAHT for Adults in Primary Care. Otago: University of Otago/PATHA. [URL 1] [URL 2] [PDF 1] [PDF 2]
Cheung, A. S., Wynne, K., Erasmus, J., Murray, S., & Zajac, J. D. (2019). Position Statement on the Hormonal Management of Adult Transgender and Gender Diverse Individuals. Medical Journal of Australia, 211(3), 127–133. [DOI:10.5694/mja2.50259]
Coleman, E., Bockting, W., Botzer, M., Cohen-Kettenis, P., DeCuypere, G., Feldman, J., Fraser, L., Green, J., Knudson, G., Meyer, W. J., Monstrey, S., Adler, R. K., Brown, G. R., Devor, A. H., Ehrbar, R., Ettner, R., Eyler, E., Garofalo, R., Karasic, D. H., … & Zucker, K. (2012). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, Version 7. International Journal of Transgenderism, 13(4), 165–232. [DOI:10.1080/15532739.2011.700873] [URL] [PDF]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Cundill, P., Wong, P., Cheung, A., & Brownhill, A. (2020). Hormone Therapy Prescribing Guide for General Practitioners working with Trans, Gender Diverse and Non-Binary Patients [Version 3.0]. Australia: Equinox Gender Diverse Health Centre/Thorne Harbour Health. [URL] [PDF]
Dahl, M., Feldman, J. L., Goldberg, J., & Jaberi, A. (2015). Endocrine Therapy for Transgender Adults in British Columbia: Suggested Guidelines. Physical Aspects of Transgender Endocrine Therapy. Vancouver: Vancouver Coastal Health. [Google Scholar] [PDF]
Dahlen, S., Connolly, D., Arif, I., Junejo, M. H., Bewley, S., & Meads, C. (2021). International Clinical Practice Guidelines for Gender Minority/Trans People: Systematic Review and Quality Assessment. BMJ Open, 11(4), e048943. [DOI:10.1136/bmjopen-2021-048943]
Davidson, A., Franicevich, J., Freeman, M., Lin, R., Martinez, L., Monihan, M., Porch, M., Samuel, L., Stukalin, R., Vormohr, J., & Zevin, B. (2013). Protocols for Hormonal Reassignment of Gender. San Francisco: San Francisco Department of Public Health/Tom Waddell Health Center. [Google Scholar] [PDF]
Deutsch, M. B. (Ed.). (2016). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition. San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Deutsch, M. B., Radix, A., & Reisner, S. (2016). What’s in a Guideline? Developing Collaborative and Sound Research Designs that Substantiate Best Practice Recommendations for Transgender Health Care. AMA Journal of Ethics, 18(11), 1098–1106. [DOI:10.1001/journalofethics.2016.18.11.stas1-1611]
Feldman, J., & Safer, J. (2009). Hormone Therapy in Adults: Suggested Revisions to the Sixth Version of the Standards of Care. International Journal of Transgenderism, 11(3), 146–182. [DOI:10.1080/15532730903383757]
Fisher, A. D., Senofonte, G., Cocchetti, C., Guercio, G., Lingiardi, V., Meriggiola, M. C., Mosconi, M., Motta, G., Ristori, J., Speranza, A. M., Pierdominici, M., Maggi, M., Corona, G., & Lombardo, F. (2021). SIGIS–SIAMS–SIE position statement of gender affirming hormonal treatment in transgender and non-binary people. Journal of Endocrinological Investigation, 45(3), 657–673. [DOI:10.1007/s40618-021-01694-2]
Godano, A., Maggi, M., Jannini, E., Meriggiola, M. C., Ghigo, E., Todarello, O., Lenzi, A., & Manieri, C. (2009). SIAMS-ONIG Consensus on Hormonal Treatment in Gender Identity Disorders. Journal of Endocrinological Investigation, 32(10), 857–864. [DOI:10.1007/BF03345758]
Gorton, N., Jaffe, J. M., Thompson, J., Menkin, D., Nesteby, A., Dunn, D., Baker, K. K., Harbatkin, D., Do, T., Radix, A., Meacher, P., Goldstein, Z., Carpenter, W., Caine, M., Henn, S., Murayama, R., Feldmann, J., & Zayas, S. (2019). TransLine Gender Affirming Hormone Therapy Prescriber Guidelines. San Francisco: Lyon-Martin Health Services/TransLine. [URL] [PDF]
Health Policy Project, Asia Pacific Transgender Network, United Nations Development Programme. (2015). Blueprint for the Provision of Comprehensive Care for Trans People and Trans Communities. Washington, DC: Futures Group, Health Policy Project. [URL] [PDF]
Hembree, W. C., Cohen-Kettenis, P., Delemarre-Van De Waal, H. A., Gooren, L. J., Meyer III, W. J., Spack, N. P., Tangpricha, V., & Montori, V. M. (2009). Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism, 94(9), 3132–3154. [DOI:10.1210/jc.2009-0345]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
International Planned Parenthood Federation (IPPF). (2015). IMAP Statement on Hormone Therapy for Transgender People. International Medical Advisory Panel/International Planned Parenthood Federation. [URL] [PDF]
Latkin, S., & Coakley, G. (2017). [Transgender Women] Prescribing Guidelines. Doncaster/Bassetlaw: Doncaster and Bassetlaw Teaching Hospitals NHS Foundation Trust. [PDF]
Levine, S. B., Brown, G., Coleman, E., Cohen-Kettenis, P., Joris Hage, J., Van Maasdam, J., Petersen, M., Pfaefflin, F., & Schaefer, L. C. (June 1998). [The Harry Benjamin International Gender Dysphoria Association’s] The Standards of Care for Gender Identity Disorders [5th Edition]. International Journal of Transgenderism, 2(2). [Consultants: Denny, D., DiCeglie, D., Eicher, W., Green, J., Green, R., Gooren, L., Laub, D., Lawrence, A., Meyer, W., & Wheeler, C.] [URL 1] [URL 2] [URL 3] [PDF]
Majumder, A., Chatterjee, S., Maji, D., Roychaudhuri, S., Ghosh, S., Selvan, C., George, B., Kalra, P., Maisnam, I., & Sanyal, D. (2020). IDEA Group Consensus Statement on Medical Management of Adult Gender Incongruent Individuals Seeking Gender Reaffirmation as Female. Indian Journal of Endocrinology and Metabolism, 24(2), 128–135. [DOI:10.4103/ijem.IJEM_593_19]
Meyer, W., Bockting, W. O., Cohen-Kettenis, P., Coleman, E., DiCeglie, D., Devor, H., Gooren, L., Joris Hage, J., Kirk, S., Kuiper, B., Laub, D., Lawrence, A., Menard, Y., Patton, J., Schaefer, L., Webb, A., & Wheeler, C. C. (February 2001). [The Harry Benjamin International Gender Dysphoria Association’s] The Standards of Care for Gender Identity Disorders – Sixth Version. International Journal of Transgenderism, 5(1). [Google Scholar 1] [Google Scholar 2] [Google Scholar 3] [URL 1] [URL 2] [DOI:10.1300/J056v13n01_01—2001/2002 reprint in Journal of Psychology & Human Sexuality, 13(1), 1–30] [PDF 1] [PDF 2]
Oliphant, J., Veale, J., Macdonald, J., Carroll, R., Johnson, R., Harte, M., Stephenson, C. & Bullock, J. (2018). Guidelines for Gender Affirming Healthcare for Gender Diverse and Transgender Children, Young People and Adults in Aotearoa New Zealand. Waikato: Transgender Health Research Lab/University of Waikato. [URL] [PDF]
Pan American Health Organization, John Snow, Inc., World Professional Association for Transgender Health, et al. (2014). Blueprint for the Provision of Comprehensive Care for Trans Persons andTheir Communities in the Caribbean and Other Anglophone Countries. Arlington: John Snow, Inc. [Alt] [PDF]
Radix, A. (2019). Hormone Therapy for Transgender Adults. The Urologic Clinics of North America, 46(4), 467–473. [DOI:10.1016/j.ucl.2019.07.001]
Radix, A. (2019). Primary Care of Transgender Adults. In Poretsky, L., & Hembree, W. C. (Eds.). Transgender Medicine: A Multidisciplinary Approach(Contemporary Endocrinology) (pp. 51–67). Cham: Humana Press. [DOI:10.1007/978-3-030-05683-4_4]
Sappho for Equality. (2019). A Good Practice Guide to Gender-Affirmative Care. Kolkata: Sappho for Equality. [PDF]
Seal, L. J. (2016). Information About Hormonal Treatment for Trans Women. London: West London Mental Health NHS Trust/West London NHS Trust. [PDF]
Seal, L., & Barrett, J. (2017). Shared Care Prescribing Guidance for Treatment of Gender Dysphoria in Transwomen (Male to Female Transsexuals). London: West London Mental Health NHS Trust/West London NHS Trust. [PDF]
Sullivan, C., & Dean, J. (2015). Prescribing Guideline. Pharmacological Treatment of Gender Dysphoria. Devon: Devon Partnership NHS Trust. [PDF]
Tangpricha, V., & Safer, J. D. (2020). Transgender Women: Evaluation and Management. UpToDate. [URL] [PDF]
Telfer, M. M., Tollit, M. A., Pace, C. C., & Pang, K. C. (2020). Australian Standards of Care and Treatment Guidelines for Trans and Gender Diverse Children and Adolescents Version 1.2. Melbourne: The Royal Children’s Hospital. [PDF]
Thomas, C. (2015). Guidelines for the Use of Feminising Hormone Therapy. Information for Primary Care. Sunderland: City Hospitals Sunderland NHS Trust. [PDF]
Thompson, J., Hopwood, R. A., deNormand, S., & Cavanaugh, T. (2021). Medical Care of Trans and Gender Diverse Adults. Boston: Fenway Health. [URL] [PDF]
Tomson, A., McLachlan, C., Wattrus, C., Adams, K., Addinall, R., Bothma, R., Jankelowitz, L., Kotze, E., Luvuno, Z., Madlala, N., Matyila, S., Padavatan, A., Pillay, M., Rakumakoe, M. D., Tomson-Myburgh, M., Venter, W., & de Vries, E. (2021). Southern African HIV Clinicians’ Society gender-affirming healthcare guideline for South Africa. Southern African Journal of HIV Medicine, 22(1), a1299. [DOI:10.4102/sajhivmed.v22i1.1299] [PDF]
Trans Care BC. (2021). Gender-affirming Care for Trans, Two-Spirit, and Gender Diverse Patients in BC: A Primary Care Toolkit. Vancouver: Provincial Health Services Authority/Trans Care BC. [URL] [PDF]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine Position Statement “Assessment and Hormonal Management in Adolescent and Adult Trans People, With Attention for Sexual Function and Satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
UpToDate. (2021). Society Guideline Links: Transgender Health. UpToDate. [URL]
Vacharathit, V., Ratanalert, W., & Samitpol, K. (2021). The Thai Handbook of Transgender Healthcare Services. Thailand: Center of Excellence in Transgender Health/Chulalongkorn University. [URL] [PDF]
Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1979 February 13). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [1st Edition/1979 Original Draft]. Palo Alto, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar]
Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1980 January 20). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [2nd Edition/1980 Revised Draft]. Stanford, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar 1] [Google Scholar 2]
Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1981 March 9). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [3rd Edition/1981 Revised Draft]. San Francisco, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [URL 1] [URL 2] [DOI:10.1007/BF01541354—1985 reprint in Archives of Sexual Behavior, 14(1), 79–90)]
Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1990 January 25). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [4th Edition/1990 Revised Draft]. Palo Alto/Sonoma, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar 1] [Google Scholar 2] [URL 1] [URL 2] [URL 3] [URL 4] [URL 5] [URL 6]
Wilczynski, C., & Emanuele, M. A. (2014). Treating a transgender patient: overview of the guidelines. Postgraduate Medicine, 126(7), 121–128. [DOI:10.3810/pgm.2014.11.2840]
Wylie, K., Barrett, J., Besser, M., Bouman, W. P., Bridgman, M., Clayton, A., Green, R., Hamilton, M., Hines, M., Ivbijaro, G., Khoosal, D., Lawrence, A., Lenihan, P., Loewenthal, D., Ralph, D., Reed, T., Stevens, J., Terry, T., Thom, B., Thornton, J., Walsh, D., & Ward, D. (2014). Good Practice Guidelines for the Assessment and Treatment of Adults with Gender Dysphoria. Sexual and Relationship Therapy, 29(2), 154–214. [DOI:10.1080/14681994.2014.883353]
Ziegler, E., Carroll, B., & Charnish, E. (2021). Review and Analysis of International Transgender Adult Primary Care Guidelines. Transgender Health, 6(3), 139–147. [DOI:10.1089/trgh.2020.0043]
\ No newline at end of file
+Clinical Guidelines with Information on Transfeminine Hormone Therapy - Transfeminine Science
Clinical Guidelines with Information on Transfeminine Hormone Therapy
By Aly | First published November 20, 2020 | Last modified November 17, 2024
Abstract / TL;DR
This article is a collection of clinical practice guidelines throughout the world with information on transfeminine hormone therapy. Examples of these clinical guidelines include the World Professional Association for Transgender Health (WPATH) Standards of Care for the Health of Transgender and Gender Diverse People, the Endocrine Society guidelines, and the University of California, San Francisco (UCSF) Center of Excellence for Transgender Health guidelines, among many others.
Introduction
Clinicians use clinical practice guidelines (CPGs) to learn about and guide themselves in administering medical care for different indications. Clinical practice guidelines review and summarize the available scientific literature and research in a given medical area. They allow clinicians to competently administer care without necessarily having to delve into and develop their understanding via the primary scientific literature. Literature reviews can serve a similar function. However, clinical practice guidelines are generally more substantial and are more founded in evidence-based medicine. They are also regularly updated. Clinical practice guidelines are developed and maintained by clinical organizations and societies, universities, government agencies, and sometimes even large medical clinics. They may be international/locationless or oftentimes region-specific.
There are many clinical practice guidelines for transgender medicine (for review, Deutsch, Radix, & Reisner, 2016; Radix, 2019; Radix, 2019; UpToDate; Bewley et al., 2021; Dahlen et al., 2021; Ziegler, Carroll, & Charnish, 2021). These guidelines discuss topics such as psychotherapy, hormone therapy, voice therapy, and surgical management of transgender people, among others. In addition to educating and guiding clinicians, transgender clinical practice guidelines are useful materials for transgender people as they can help to inform them about their care.
1st/1979: Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1979 February 13). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [1st Edition/1979 Original Draft]. Palo Alto, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar]
2nd/1980: Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1980 January 20). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [2nd Edition/1980 Revised Draft]. Stanford, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar 1] [Google Scholar 2]
3rd/1981: Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1981 March 9). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [3rd Edition/1981 Revised Draft]. San Francisco, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [URL 1] [URL 2] [DOI:10.1007/BF01541354—1985 reprint in Archives of Sexual Behavior, 14(1), 79–90)]
4th/1990: Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1990 January 25). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [4th Edition/1990 Revised Draft]. Palo Alto/Sonoma, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar 1] [Google Scholar 2] [URL 1] [URL 2] [URL 3] [URL 4] [URL 5] [URL 6]
5th/1998: Levine, S. B., Brown, G., Coleman, E., Cohen-Kettenis, P., Joris Hage, J., Van Maasdam, J., Petersen, M., Pfaefflin, F., & Schaefer, L. C. (June 1998). [The Harry Benjamin International Gender Dysphoria Association’s] The Standards of Care for Gender Identity Disorders [5th Edition]. International Journal of Transgenderism, 2(2). [Consultants: Denny, D., DiCeglie, D., Eicher, W., Green, J., Green, R., Gooren, L., Laub, D., Lawrence, A., Meyer, W., & Wheeler, C.] [URL 1] [URL 2] [URL 3] [PDF]
6th/2001: Meyer, W., Bockting, W. O., Cohen-Kettenis, P., Coleman, E., DiCeglie, D., Devor, H., Gooren, L., Joris Hage, J., Kirk, S., Kuiper, B., Laub, D., Lawrence, A., Menard, Y., Patton, J., Schaefer, L., Webb, A., & Wheeler, C. C. (February 2001). [The Harry Benjamin International Gender Dysphoria Association’s] The Standards of Care for Gender Identity Disorders – Sixth Version. International Journal of Transgenderism, 5(1). [Google Scholar 1] [Google Scholar 2] [Google Scholar 3] [URL 1] [URL 2] [DOI:10.1300/J056v13n01_01—2001/2002 reprint in Journal of Psychology & Human Sexuality, 13(1), 1–30] [PDF 1] [PDF 2]
7th/2012: Coleman, E., Bockting, W., Botzer, M., Cohen-Kettenis, P., DeCuypere, G., Feldman, J., Fraser, L., Green, J., Knudson, G., Meyer, W. J., Monstrey, S., Adler, R. K., Brown, G. R., Devor, A. H., Ehrbar, R., Ettner, R., Eyler, E., Garofalo, R., Karasic, D. H., … & Zucker, K. (2012). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, Version 7. International Journal of Transgenderism, 13(4), 165–232. [DOI:10.1080/15532739.2011.700873] [URL] [PDF]
Fisher et al. / Italian Society of Gender, Identity and Health (SIGIS) / Italian Society of Andrology and Sexual Medicine (SIAMS) / Italian Society of Endocrinology (SIE)
Godano et al. / Società Italiana di Andrologia e Medicina della Sessualità (SIAMS) [Italian Society of Andrology and Sexual Medicine] / Osservatorio Nazionale sull’Identità di Genere (ONIG) [National Observatory of Gender Identity]
Vacharathit et al. / Center of Excellence in Transgender Health / Chulalongkorn University [Thailand]
2021
Online document
Update: New Endocrine Society and UCSF Guidelines
New Endocrine Society and UCSF guidelines are under development as of February 2024 (Christensen, 2024; Endocrine Society, 2024; UCSF, 2024). It is anticipated that the Endocrine Society guidelines will be published in Spring 2026. The new UCSF guidelines were initially slated for publication in 2024, but have been pushed back until the new Endocrine Society guidelines are published. The new Endocrine Society and UCSF guidelines will be the third edition of each of the guidelines.
References
AusPATH. (2022). Australian Informed Consent Standards of Care for Gender Affirming Hormone Therapy. Australia: Australian Professional Association for Trans Health. [URL] [PDF]
Belzer, M. E., Burnett, J., Deutsch, M., Franicevich, J., Gorton, R. N., Hastings, J., Karasic, D., Kohler, L., Vanderleest, J., Van Maasdam, J., Olson, J., Green, J., & DeVries, C. (April 2011). Primary Care Protocol for Transgender Patient Care, 1st Edition. Center of Excellence for Transgender Health, University of California, San Francisco, Department of Family and Community Medicine. [URL] [PDF]
Bewley, S., Dahlen, S., Connolly, D., Arif, I., Junejo, M., & Catherine, M. (2021). International Clinical Practice Guidelines for Gender Minority/Trans People: Systematic Review & Quality Assessment. How Does the Endocrine Society Fare? Journal of the Endocrine Society, 5(Suppl 1), A791–A791. [DOI:10.1210/jendso/bvab048.1609]
Bourns, A. (2019). Guidelines for Gender-Affirming Primary Care with Trans and Non-Binary Patients, 4th Edition. Toronto: Rainbow Health Ontario/Sherbourne Health. [URL] [PDF]
Callen-Lorde Community Health Center. (2018). Protocols for the Provision of Hormone Therapy. New York City: Callen-Lorde Community Health Center. [URL] [PDF]
Carroll, R., Nicholls, R., Carroll, R. W., Bullock, J., Reid, D., Shields, J., Johnson, R., Oliphant, J., McElrea, E., Whitfield, P., & Veale, J. (2023). Primary Care Gender Affirming Hormone Therapy Initiation Guidelines: Aotearoa New Zealand Guidelines for Commencing GAHT for Adults in Primary Care. Otago: University of Otago/PATHA. [URL 1] [URL 2] [PDF 1] [PDF 2]
Cheung, A. S., Wynne, K., Erasmus, J., Murray, S., & Zajac, J. D. (2019). Position Statement on the Hormonal Management of Adult Transgender and Gender Diverse Individuals. Medical Journal of Australia, 211(3), 127–133. [DOI:10.5694/mja2.50259]
Coleman, E., Bockting, W., Botzer, M., Cohen-Kettenis, P., DeCuypere, G., Feldman, J., Fraser, L., Green, J., Knudson, G., Meyer, W. J., Monstrey, S., Adler, R. K., Brown, G. R., Devor, A. H., Ehrbar, R., Ettner, R., Eyler, E., Garofalo, R., Karasic, D. H., … & Zucker, K. (2012). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, Version 7. International Journal of Transgenderism, 13(4), 165–232. [DOI:10.1080/15532739.2011.700873] [URL] [PDF]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Cundill, P., Wong, P., Cheung, A., & Brownhill, A. (2020). Hormone Therapy Prescribing Guide for General Practitioners working with Trans, Gender Diverse and Non-Binary Patients [Version 3.0]. Australia: Equinox Gender Diverse Health Centre/Thorne Harbour Health. [URL] [PDF]
Dahl, M., Feldman, J. L., Goldberg, J., & Jaberi, A. (2015). Endocrine Therapy for Transgender Adults in British Columbia: Suggested Guidelines. Physical Aspects of Transgender Endocrine Therapy. Vancouver: Vancouver Coastal Health. [Google Scholar] [PDF]
Dahlen, S., Connolly, D., Arif, I., Junejo, M. H., Bewley, S., & Meads, C. (2021). International Clinical Practice Guidelines for Gender Minority/Trans People: Systematic Review and Quality Assessment. BMJ Open, 11(4), e048943. [DOI:10.1136/bmjopen-2021-048943]
Davidson, A., Franicevich, J., Freeman, M., Lin, R., Martinez, L., Monihan, M., Porch, M., Samuel, L., Stukalin, R., Vormohr, J., & Zevin, B. (2013). Protocols for Hormonal Reassignment of Gender. San Francisco: San Francisco Department of Public Health/Tom Waddell Health Center. [Google Scholar] [PDF]
Deutsch, M. B. (Ed.). (2016). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition. San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Deutsch, M. B., Radix, A., & Reisner, S. (2016). What’s in a Guideline? Developing Collaborative and Sound Research Designs that Substantiate Best Practice Recommendations for Transgender Health Care. AMA Journal of Ethics, 18(11), 1098–1106. [DOI:10.1001/journalofethics.2016.18.11.stas1-1611]
Feldman, J., & Safer, J. (2009). Hormone Therapy in Adults: Suggested Revisions to the Sixth Version of the Standards of Care. International Journal of Transgenderism, 11(3), 146–182. [DOI:10.1080/15532730903383757]
Fisher, A. D., Senofonte, G., Cocchetti, C., Guercio, G., Lingiardi, V., Meriggiola, M. C., Mosconi, M., Motta, G., Ristori, J., Speranza, A. M., Pierdominici, M., Maggi, M., Corona, G., & Lombardo, F. (2021). SIGIS–SIAMS–SIE position statement of gender affirming hormonal treatment in transgender and non-binary people. Journal of Endocrinological Investigation, 45(3), 657–673. [DOI:10.1007/s40618-021-01694-2]
Godano, A., Maggi, M., Jannini, E., Meriggiola, M. C., Ghigo, E., Todarello, O., Lenzi, A., & Manieri, C. (2009). SIAMS-ONIG Consensus on Hormonal Treatment in Gender Identity Disorders. Journal of Endocrinological Investigation, 32(10), 857–864. [DOI:10.1007/BF03345758]
Gorton, N., Jaffe, J. M., Thompson, J., Menkin, D., Nesteby, A., Dunn, D., Baker, K. K., Harbatkin, D., Do, T., Radix, A., Meacher, P., Goldstein, Z., Carpenter, W., Caine, M., Henn, S., Murayama, R., Feldmann, J., & Zayas, S. (2019). TransLine Gender Affirming Hormone Therapy Prescriber Guidelines. San Francisco: Lyon-Martin Health Services/TransLine. [URL] [PDF]
Health Policy Project, Asia Pacific Transgender Network, United Nations Development Programme. (2015). Blueprint for the Provision of Comprehensive Care for Trans People and Trans Communities. Washington, DC: Futures Group, Health Policy Project. [URL] [PDF]
Hembree, W. C., Cohen-Kettenis, P., Delemarre-Van De Waal, H. A., Gooren, L. J., Meyer III, W. J., Spack, N. P., Tangpricha, V., & Montori, V. M. (2009). Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology & Metabolism, 94(9), 3132–3154. [DOI:10.1210/jc.2009-0345]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
International Planned Parenthood Federation (IPPF). (2015). IMAP Statement on Hormone Therapy for Transgender People. International Medical Advisory Panel/International Planned Parenthood Federation. [URL] [PDF]
Latkin, S., & Coakley, G. (2017). [Transgender Women] Prescribing Guidelines. Doncaster/Bassetlaw: Doncaster and Bassetlaw Teaching Hospitals NHS Foundation Trust. [PDF]
Levine, S. B., Brown, G., Coleman, E., Cohen-Kettenis, P., Joris Hage, J., Van Maasdam, J., Petersen, M., Pfaefflin, F., & Schaefer, L. C. (June 1998). [The Harry Benjamin International Gender Dysphoria Association’s] The Standards of Care for Gender Identity Disorders [5th Edition]. International Journal of Transgenderism, 2(2). [Consultants: Denny, D., DiCeglie, D., Eicher, W., Green, J., Green, R., Gooren, L., Laub, D., Lawrence, A., Meyer, W., & Wheeler, C.] [URL 1] [URL 2] [URL 3] [PDF]
Majumder, A., Chatterjee, S., Maji, D., Roychaudhuri, S., Ghosh, S., Selvan, C., George, B., Kalra, P., Maisnam, I., & Sanyal, D. (2020). IDEA Group Consensus Statement on Medical Management of Adult Gender Incongruent Individuals Seeking Gender Reaffirmation as Female. Indian Journal of Endocrinology and Metabolism, 24(2), 128–135. [DOI:10.4103/ijem.IJEM_593_19]
Meyer, W., Bockting, W. O., Cohen-Kettenis, P., Coleman, E., DiCeglie, D., Devor, H., Gooren, L., Joris Hage, J., Kirk, S., Kuiper, B., Laub, D., Lawrence, A., Menard, Y., Patton, J., Schaefer, L., Webb, A., & Wheeler, C. C. (February 2001). [The Harry Benjamin International Gender Dysphoria Association’s] The Standards of Care for Gender Identity Disorders – Sixth Version. International Journal of Transgenderism, 5(1). [Google Scholar 1] [Google Scholar 2] [Google Scholar 3] [URL 1] [URL 2] [DOI:10.1300/J056v13n01_01—2001/2002 reprint in Journal of Psychology & Human Sexuality, 13(1), 1–30] [PDF 1] [PDF 2]
Oliphant, J., Veale, J., Macdonald, J., Carroll, R., Johnson, R., Harte, M., Stephenson, C. & Bullock, J. (2018). Guidelines for Gender Affirming Healthcare for Gender Diverse and Transgender Children, Young People and Adults in Aotearoa New Zealand. Waikato: Transgender Health Research Lab/University of Waikato. [URL] [PDF]
Pan American Health Organization, John Snow, Inc., World Professional Association for Transgender Health, et al. (2014). Blueprint for the Provision of Comprehensive Care for Trans Persons andTheir Communities in the Caribbean and Other Anglophone Countries. Arlington: John Snow, Inc. [Alt] [PDF]
Radix, A. (2019). Hormone Therapy for Transgender Adults. The Urologic Clinics of North America, 46(4), 467–473. [DOI:10.1016/j.ucl.2019.07.001]
Radix, A. (2019). Primary Care of Transgender Adults. In Poretsky, L., & Hembree, W. C. (Eds.). Transgender Medicine: A Multidisciplinary Approach(Contemporary Endocrinology) (pp. 51–67). Cham: Humana Press. [DOI:10.1007/978-3-030-05683-4_4]
Sappho for Equality. (2019). A Good Practice Guide to Gender-Affirmative Care. Kolkata: Sappho for Equality. [PDF]
Seal, L. J. (2016). Information About Hormonal Treatment for Trans Women. London: West London Mental Health NHS Trust/West London NHS Trust. [PDF]
Seal, L., & Barrett, J. (2017). Shared Care Prescribing Guidance for Treatment of Gender Dysphoria in Transwomen (Male to Female Transsexuals). London: West London Mental Health NHS Trust/West London NHS Trust. [PDF]
Sullivan, C., & Dean, J. (2015). Prescribing Guideline. Pharmacological Treatment of Gender Dysphoria. Devon: Devon Partnership NHS Trust. [PDF]
Tangpricha, V., & Safer, J. D. (2020). Transgender Women: Evaluation and Management. UpToDate. [URL] [PDF]
Telfer, M. M., Tollit, M. A., Pace, C. C., & Pang, K. C. (2020). Australian Standards of Care and Treatment Guidelines for Trans and Gender Diverse Children and Adolescents Version 1.2. Melbourne: The Royal Children’s Hospital. [PDF]
Thomas, C. (2015). Guidelines for the Use of Feminising Hormone Therapy. Information for Primary Care. Sunderland: City Hospitals Sunderland NHS Trust. [PDF]
Thompson, J., Hopwood, R. A., deNormand, S., & Cavanaugh, T. (2021). Medical Care of Trans and Gender Diverse Adults. Boston: Fenway Health. [URL] [PDF]
Tomson, A., McLachlan, C., Wattrus, C., Adams, K., Addinall, R., Bothma, R., Jankelowitz, L., Kotze, E., Luvuno, Z., Madlala, N., Matyila, S., Padavatan, A., Pillay, M., Rakumakoe, M. D., Tomson-Myburgh, M., Venter, W., & de Vries, E. (2021). Southern African HIV Clinicians’ Society gender-affirming healthcare guideline for South Africa. Southern African Journal of HIV Medicine, 22(1), a1299. [DOI:10.4102/sajhivmed.v22i1.1299] [PDF]
Trans Care BC. (2021). Gender-affirming Care for Trans, Two-Spirit, and Gender Diverse Patients in BC: A Primary Care Toolkit. Vancouver: Provincial Health Services Authority/Trans Care BC. [URL] [PDF]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine Position Statement “Assessment and Hormonal Management in Adolescent and Adult Trans People, With Attention for Sexual Function and Satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
UpToDate. (2021). Society Guideline Links: Transgender Health. UpToDate. [URL]
Vacharathit, V., Ratanalert, W., & Samitpol, K. (2021). The Thai Handbook of Transgender Healthcare Services. Thailand: Center of Excellence in Transgender Health/Chulalongkorn University. [URL] [PDF]
Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1979 February 13). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [1st Edition/1979 Original Draft]. Palo Alto, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar]
Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1980 January 20). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [2nd Edition/1980 Revised Draft]. Stanford, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar 1] [Google Scholar 2]
Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1981 March 9). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [3rd Edition/1981 Revised Draft]. San Francisco, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [URL 1] [URL 2] [DOI:10.1007/BF01541354—1985 reprint in Archives of Sexual Behavior, 14(1), 79–90)]
Walker, P. A., Berger, J. C., Green, R., Laub, D. R., Reynolds, C. L., & Wollman, L. (1990 January 25). Standards of Care: The Hormonal and Surgical Sex Reassignment of Gender Dysphoric Persons [4th Edition/1990 Revised Draft]. Palo Alto/Sonoma, California: The Harry Benjamin International Gender Dysphoria Association, Inc. [Google Scholar 1] [Google Scholar 2] [URL 1] [URL 2] [URL 3] [URL 4] [URL 5] [URL 6]
Wilczynski, C., & Emanuele, M. A. (2014). Treating a transgender patient: overview of the guidelines. Postgraduate Medicine, 126(7), 121–128. [DOI:10.3810/pgm.2014.11.2840]
Wylie, K., Barrett, J., Besser, M., Bouman, W. P., Bridgman, M., Clayton, A., Green, R., Hamilton, M., Hines, M., Ivbijaro, G., Khoosal, D., Lawrence, A., Lenihan, P., Loewenthal, D., Ralph, D., Reed, T., Stevens, J., Terry, T., Thom, B., Thornton, J., Walsh, D., & Ward, D. (2014). Good Practice Guidelines for the Assessment and Treatment of Adults with Gender Dysphoria. Sexual and Relationship Therapy, 29(2), 154–214. [DOI:10.1080/14681994.2014.883353]
Ziegler, E., Carroll, B., & Charnish, E. (2021). Review and Analysis of International Transgender Adult Primary Care Guidelines. Transgender Health, 6(3), 139–147. [DOI:10.1089/trgh.2020.0043]
\ No newline at end of file
diff --git a/transfemscience.org/articles/transfem-intro/index.html b/transfemscience.org/articles/transfem-intro/index.html
index ff88bb41..49af48a5 100644
--- a/transfemscience.org/articles/transfem-intro/index.html
+++ b/transfemscience.org/articles/transfem-intro/index.html
@@ -1 +1 @@
-An Introduction to Hormone Therapy for Transfeminine People - Transfeminine Science
An Introduction to Hormone Therapy for Transfeminine People
By Aly | First published August 4, 2018 | Last modified December 17, 2025
Abstract / TL;DR
Sex hormones such as estrogen, testosterone, and progesterone are produced by the gonads. The sex hormones mediate the development of the secondary sexual characteristics. Testosterone causes masculinization, while estradiol causes feminization and breast development. Males have high amounts of testosterone, while females have low testosterone but high amounts of estradiol. These hormonal differences are responsible for the physical differences between males and females. Sex hormones and other hormonal medications are used in transfeminine people to shift the hormonal profile from a male-typical one to a female-typical profile. This causes feminization and demasculinization and allows for alleviation of gender dysphoria. The changes caused by transfeminine hormone therapy occur over a period of months to years. There are many different types and forms of hormonal medications, and these medications can be administered by a variety of different routes. Examples include as pills taken by mouth, as patches or gel applied to the skin, and as injections, among others. Different hormonal medications, routes, and doses have differences in efficacy, side effects, risks, costs, convenience, and availability. Hormone therapy should ideally be regularly monitored in transfeminine people with blood tests to ensure effectiveness and safety and to allow for adjustment as necessary.
The Sex Hormones
Types and Effects
The sex hormones include the estrogens (E), progestogens (P), and androgens. A person’s hormonal profile is a product of the type of gonads that they are born with. Natal males have testes while natal females have ovaries. Testes produce large amounts of androgens and small amounts of estrogens whereas ovaries produce high amounts of estrogens and progesterone and low amounts of androgens.
Progestogens have essentially no known role in feminization or pubertal breast development. Rather than acting as mediators of feminization, progestogens have important effects in the female reproductive system and are essential hormones during pregnancy (Wiki). They also oppose the actions of estrogens in certain parts of the body, such as the uterus, vagina, and breasts (Wiki).
In addition to their effects on the body, sex hormones have actions in the brain. These actions influence cognition, emotions, and behavior. For instance, androgens produce pronounced sexual desire and arousal (including spontaneous erections) in men, while estrogens appear to be the major hormones responsible for sexual desire in women (Cappelletti & Wallen, 2016). As another example, testosterone levels have been negatively associated with agreeableness, whereas estrogen levels have been positively associated with this characteristic (Treleaven et al., 2013). Sex hormones also have important effects on health, which can be both positive and negative. For instance, estrogens maintain bone strength and likely protect against heart disease in cisgender women (NAMS, 2022), but also increase the risk of breast cancer (Aly, 2020) and can increase the risk of blood clots (Aly, 2020).
Estrogens, progestogens, and androgens also have antigonadotropic effects. That is, they inhibit the gonadotropin-releasing hormone (GnRH)-induced secretion of the gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), from the pituitary gland in the brain. The gonadotropins signal the gonads to make sex hormones and to supply the sperm and egg cells necessary for fertility. Hence, lower levels of the gonadotropins will result in reduced gonadal sex hormone production and diminished fertility. If gonadotropin levels are sufficiently suppressed, the gonads will no longer make sex hormones at all and fertility will cease. The vast majorities of the quantities of estradiol, testosterone, and progesterone in the body are produced by the gonads. Most of the small remaining amounts of these hormones are produced via the adrenal glands of the kidneys.
Normal Hormone Levels
In cisgender females, the sex hormones are largely absent during childhood, gradually ramp up in production in late childhood and adolescence, are present in a cyclical manner during adulthood, and then largely stop being produced following the menopause. Hormone levels vary substantially but in a predictable manner during the normal menstrual cycle in adult premenopausal women. The menstrual cycle lasts about 28 days on average and consists of the following parts:
Luteal phase—latter half of the cycle or days 14–28
Hormone levels during the menstrual cycle are shown in the following graph:
Figure 1: Median estradiol and progesterone levels throughout the menstrual cycle in premenopausal cisgender women (Stricker et al., 2006; Abbott, 2009). The horizontal dashed lines are the average levels over the spanned periods. Other figures available elsewhere show variation between individuals (Graph; Graph; Graph).
As can be seen in the graph, estradiol levels are relatively low and progesterone levels are very low during the follicular phase; estradiol but not progesterone levels briefly surge to very high levels and trigger ovulation during mid-cycle; and estradiol and progesterone levels both undergo a bump and are relatively high during the luteal phase (though estradiol is not as high as during the mid-cycle peak).
The table below shows the circulating levels and production rates of estradiol, progesterone, and testosterone in women and men and allows for comparison between them.
Table 1: Ranges for circulating levelsa and estimated production ratesb of the major sex hormones:
Mean integrated estradiol levels are around 100 pg/mL (367 pmol/L) in premenopausal women and around 25 pg/mL (92 pmol/L) in men. The 95% range for mean estradiol levels in women is around 50 to 250 pg/mL (180–918 pmol/L) (e.g., Abbott, 2009 (Graph); Verdonk et al., 2019 (Graph)). The average production of estradiol by the ovaries in premenopausal women is about 6 mg over the course of one menstrual cycle (i.e., one month) (Rosenfield et al., 2008). This corresponds to a mean rate of about 200 μg/day. Estradiol levels increase slowly during normal female puberty, when breast development and feminization take place. Mean estradiol levels during the different stages of female puberty are quite low—less than about 50 to 60 pg/mL (180–220 pmol/L) until late puberty (Aly, 2020). In postmenopausal women, whose ovaries no longer produce considerable quantities of estrogens, estradiol levels are generally less than 10 to 20 pg/mL (37–73 pmol/L) (Nakamoto, 2016). Estradiol levels below 50 pg/mL (184 pmol/L) in adults are concentration-dependently associated with menopausal symptoms, including hot flashes, depressive mood changes, defeminization (e.g., breast atrophy, loss of feminine fat distribution), accelerated skin aging, and bone density loss with increased risk of bone fracture.
Mean testosterone levels are around 30 ng/dL (1.0 nmol/L) in women and 600 ng/dL (21 nmol/L) in men. Based on these values, testosterone levels are on average about 20-fold higher in men than in women. In men who have undergone gonadectomy (castration or surgical gonadal removal), testosterone levels are similar to those in women (<50 ng/dL [1.7 nmol/L]) (Nishiyama, 2014; Itty & Getzenberg, 2020). The mean or median levels of testosterone in women with polycystic ovary syndrome (PCOS), who often have clinically significant symptoms of androgen excess (e.g., excessive facial/body hair growth), range from 41 to 75 ng/dL (1.4–2.6 nmol/L) per different studies (Balen et al., 1995; Steinberger et al., 1998; Legro et al., 2010; Loh et al., 2020). Hence, it appears that even testosterone levels that are marginally elevated relative to normal female levels may produce undesirable androgenic effects.
The goal of hormone therapy for transfeminine people, otherwise known as feminizing hormone therapy (FHT) or (more in the past) as male-to-female (MtF) hormone replacement therapy (HRT), is to produce feminization and demasculinization of the body as well as alleviation of gender dysphoria. Medication therapy with sex hormones and other sex-hormonal medications is used to mediate these changes. Transfeminine people are given estrogens, progestogens, and antiandrogens (AAs) to supersede gonadal sex hormone production and shift the hormonal profile from male-typical to female-typical.
Transfeminine hormone therapy aims to achieve estradiol and testosterone levels within the normal female range. Commonly recommended ranges for transfeminine people in the literature are 100 to 200 pg/mL (367–734 pmol/L) for estradiol levels and less than 50 ng/dL (1.7 nmol/L) for testosterone levels (Table). However, higher estradiol levels of more than 200 pg/mL (734 pmol/L) can be useful in transfeminine hormone therapy to help suppress testosterone levels. Lower estradiol levels (≤50–60 pg/mL [≤180–220 pmol/L]) are recommended and more appropriate for pubertal and adolescent transfeminine individuals. Sex hormone levels in the blood can be measured with blood tests, in which blood is drawn from a vein using a needle and then analyzed in a laboratory. This is useful in transfeminine people to ensure that the hormonal profile has been satisfactorily altered in line with therapeutic goals—specifically that hormone levels are within female ranges.
Gonadal Suppression
At sufficiently high exposure, estrogens and androgens are able to completely suppress gonadal sex hormone production, while progestogens by themselves are able to partially but substantially suppress gonadal sex hormone production. More specifically, studies in cisgender men and transfeminine people have found that estradiol levels of around 200 pg/mL (734 pmol/L) suppress testosterone levels by about 90% on average (to ~50 ng/dL [1.7 nmol/L]), while estradiol levels of around 500 pg/mL (1,840 pmol/L) suppress testosterone levels by about 95% on average (to ~20–30 ng/dL [0.7–1.0 nmol/L]) (Gooren et al., 1984 [Graph]; Herndon et al., 2023 [Discussion]; Wiki; Graphs). Estradiol levels of below 200 pg/mL (734 pmol/L) also suppress testosterone levels, although to a reduced extent compared to higher levels (Aly, 2019; Krishnamurthy et al., 2023; Slack et al., 2023). In one large study in transfeminine people, the rates of adequate testosterone suppression (to testosterone levels of <50 ng/dL or <1.7 nmol/L) were 24% of individuals at estradiol levels of <100 pg/mL (367 pmol/L), 58% at 100 to 200 pg/mL (367–734 pmol/L), and 77% at >200 pg/mL (>734 pmol/L) (Krishnamurthy et al., 2023).
Figure 2: Estradiol and testosterone levels after a single injection of 320 mg polyestradiol phosphate (PEP) (a long-acting prodrug of estradiol) in men with prostate cancer (Stege et al., 1996). The maximal decrease in testosterone levels occurred with estradiol levels of greater than 200 pg/mL (734 pmol/L) and was about 90% (to roughly 50 ng/dL [1.7 nmol/L]). This figure demonstrates the ability of estradiol to concentration-dependently suppress gonadal testosterone production and circulating testosterone levels in people with testes.
Progestogens on their own are able to maximally suppress testosterone levels by about 50 to 70% (to ~150–300 ng/dL [5.2–10.4 nmol/L] on average) (Aly, 2019; Wiki). In combination with relatively small amounts of estrogen however, there is synergism in the antigonadotropic effect—the suppression of gonadal testosterone production with maximally effective doses of progestogens becomes complete, and testosterone levels are reduced by about 95% (to ~20–30 ng/dL [0.7–1.0 nmol/L]) (Aly, 2019). Hence, the combination of an estrogen and a progestogen can be used to achieve maximal testosterone suppression at lower doses than would be necessary if an estrogen or progestogen were used alone.
The antigonadotropic effects of estrogens and progestogens are taken advantage of in transfeminine hormone therapy to suppress gonadal testosterone production and attain testosterone levels that are more consistent with those in cisgender women. It should be noted that the preceding numbers on testosterone suppression with estrogens and progestogens are averages and there is significant variation between individuals in terms of testosterone suppression. In other words, some may need more or less in terms of hormonal dosages to achieve the same decrease in testosterone levels.
Effects and Timeline
During normal puberty in both males and females, sex hormone exposure increases slowly over a period of several years (Aly, 2020). In relation to this, sexual maturation occurs gradually during normal puberty. In non-adolescent transgender people, adult or higher amounts of hormones are generally administered right away, and this can result in changes in secondary sex characteristics happening more quickly. Most of the effects of feminizing hormone therapy in transfeminine people onset within 1 to 6 months of commencing treatment and complete within 1 to 3 years. The table below is reproduced from literature sources with slight modification and is commonly cited as a timeline of the effects (Table). It is based on a mixture of anecdotal clinical experience, expert opinion, and available clinical studies of hormone therapy in transfeminine people. Due to limited research characterizing the effects of transfeminine hormone therapy at present, the table may or may not be completely accurate.
Table 2: Effects of hormone therapy at typical doses in adult transfeminine people (Wiki):
Effect
Onseta
Completiona
Permanency
Breast development
2–6 months
2–3 years
Permanent
Reduced and slowed growth of facial and body hair
3–12 months
>3 yearsb
Reversible
Cessation and reversal of scalp hair loss
1–3 months
1–2 years
Reversible
Softening of skin and decreased skin oiliness and acne
3–6 months
Unknown
Reversible
Redistribution of body fat in a feminine pattern
3–6 months
2–5 years
Reversible
Decreased muscle mass and strength
3–6 months
1–2 yearsc
Reversible
Widening and rounding of the pelvisd
Unknown
Unknown
Permanent
Changes in mood, emotionality, and behavior
Immediate
Unknown
Reversible
Decreased sex drive and spontaneous erections
1–3 months
3–6 months
Reversible
Erectile dysfunction and decreased ejaculate volume
1–3 months
Variable
Reversible
Decreased sperm production and infertility
Unknown
>3 years
Mixede
Decreased testicular volume
3–6 months
2–3 years
Unknown
Voice changes (e.g., more feminine pitch/resonance)
Nonef
N/A
N/A
Height changes (e.g., decrease)
Noneg
N/A
N/A
a Effects in general may vary significantly between individuals due to factors like genetics, diet/nutrition, hormone levels, etc. b Hormone therapy usually has little influence on facial hair density in transfeminine people. Complete removal of facial and body hair can be achieved with laser hair removal and electrolysis. Temporary hair removal can be achieved with shaving, epilating, waxing, and other methods. c Reduced muscle mass and strength may vary significantly depending on amount of physical exercise. d Pelvic changes occur only in young individuals who have not yet completed growth plate closure (may not occur at all in post-adolescent people). e Only estrogens, particularly at high doses, seem to have the potential for long-lasting or irreversible infertility; impaired fertility caused by antiandrogens is usually readily reversible with discontinuation. fVoice training can be an effective means of feminizing the voice. g Height attainment may be reduced in adolescents, but height is not meaningfully changed or reduced in adults per clinical data (Gooren & Bunck, 2004; Ingram & Thomas, 2019; Hilton & Lundberg, 2021; Talathi et al., 2025).
Breast Development
Breast development is among the most anticipated effects of hormone therapy in transfeminine people (Masumori et al., 2021; Grock et al., 2024). This relates to the key significance of breasts as a feminine characteristic, component of sexual attractiveness, and signal of sex and gender. Breast growth in transfeminine people usually starts within 1 to 6 months and completes over a period of 1 to 3 years (e.g., de Blok et al., 2021). The developed breasts of transfeminine people are highly variable in terms of size and shape, as with natal women (de Blok et al., 2021). Based on available high-quality clinical studies, transfeminine people tend to have much smaller mature breasts than those of natal women on average, and this appears to be the case regardless of hormonal regimen or age at which hormone therapy is commenced (e.g., de Blok et al., 2021; Boogers et al., 2025). The reasons for this are unknown, but one key possibility, observed in animals, is that prenatal androgen exposure limits subsequent breast growth potential. Despite usually modest breast development, many transfeminine people still express overall satisfaction with their breasts (de Blok et al., 2021; Boogers et al., 2025).
Beyond ensuring adequate testosterone suppression and maintaining sufficient estradiol levels above a specific low threshold, there are currently no known or substantiated methods to permanently enhance or optimize breast development. However, research suggests that avoiding high or excessive doses of estradiol and progestogens may be beneficial. In addition, high levels of estradiol, progesterone, and/or prolactin, as with the normal menstrual cycle and pregnancy, are known to induce temporary and reversible breast tenderness and enlargement, for instance due to local fluid retention and lobuloalveolar maturation (Aly, 2020). However, the breast size increases are modest, and high hormone levels come with health risks (Aly, 2020). Surgical breast augmentation is an option to increase breast size if it is unsatisfactory. Some transfeminine people, for instance many non-binary individuals, may wish to avoid or minimize breast growth, and there are possible therapeutic approaches in this area (Aly, 2019).
Additional review content on breast development in transfeminine people exists on this site (e.g., Aly, 2020; Aly, 2020). Breast growth can be measured and tracked with a variety of methods for individuals who are interested in monitoring their progress (Wiki). Photographs and timelines of breast development and feminization with hormone therapy in transfeminine people are available in communities like r/TransTimelines and r/TransBreastTimelines on the social media website Reddit.
Specific Hormonal Medications
The medications that are used in transfeminine hormone therapy include estrogens, progestogens, and antiandrogens. Estrogens produce feminization and testosterone suppression. Progestogens and antiandrogens do not mediate feminization themselves but further suppress and/or block testosterone. Testosterone suppression causes demasculinization and disinhibition of estrogen-mediated feminization. Androgens are sometimes used at low doses in transfeminine people who have low testosterone levels, although they are not required and benefits are uncertain. There are many different types of these hormonal medications available for transfeminine hormone therapy, with different benefits and risks.
Estrogens, progestogens, and antiandrogens are available in a variety of different formulations and for use by many different routes of administration in transfeminine people. The route of administration influences the absorption, distribution, metabolism, and elimination of the hormone in the body, resulting in significant differences between routes in terms of bioavailability, hormone levels in blood and specific tissues, and patterns of metabolites. These differences can have important therapeutic consequences.
Table 3: Major routes of administration of hormonal medications for transfeminine people:
Insertion via surgical incision into fat under skin
Pellet
Vaginal administration is a major additional route of administration of hormonal medications in cisgender women. While vaginal administration via a natal vagina is of course not possible in transfeminine people, neovaginal administration is a possiblility in those who have undergone vaginoplasty. However, the lining of the neovagina is not the vaginal epithelium of natal females but instead is usually skin or colon—depending on the type of vaginoplasty performed (penile inversion or sigmoid colon vaginoplasty, respectively). For this reason, neovaginal administration in transfeminine people is likely more similar in its properties to transdermal and rectal administration—depending on the type of neovagina—than to vaginal administration in cisgender women. It is noteworthy that the vaginal and rectal routes are said to be similar in their properties for hormonal medications however (Goletiani, Keith, & Gorsky, 2007; Wiki). Moreover, absorption of estradiol via neovaginas constructed from peritoneum (internal abdominal lining)—a less commonly employed vaginoplasty approach in transfeminine people—was reported in one study to be similar to that with vaginal administration of estradiol in cisgender women (Willemsen et al., 1985). As such, neovaginal administration may be an additional possible route for certain transfeminine people depending on the circumstances. However, this route still remains to be more adequately characterized.
An often-encountered question from people who take hormonal medications is whether there is an optimal time of the day to take them (Colonnello et al., 2025). As of present, there is little research in this area, and the answer to the question is essentially unknown (Colonnello et al., 2025). In any case, there is currently no evidence or persuasive theoretical basis to favor specific times of day to take these medications (Colonnello et al., 2025). In all likelihood, it makes little or no difference.
Estrogens
Estradiol, the primary bioidentical form normally found in the human body, is the main estrogen that is used in transfeminine hormone therapy. Estradiol hemihydrate (EH) is another form that is essentially identical to and interchangeable with estradiol. Estradiol esters are also sometimes used in place of estradiol. They are prodrugs of estradiol (i.e., are converted into estradiol in the body) and have essentially identical biological activity to estradiol. However, they have longer durations when used by injection due to slower absorption from the injection site, and this allows them to be administered less often. Some examples of major estradiol esters include estradiol valerate (EV; Progynova, Progynon Depot, Delestrogen) and estradiol cypionate (EC; Depo-Estradiol). Polyestradiol phosphate (PEP; Estradurin) is an injectable estradiol prodrug in the form of a polymer (i.e., linked chain of estradiol molecules) which is metabolized slowly and has a very long duration.
Non-bioidentical estrogens such as ethinylestradiol (EE; found in birth control pills), conjugated estrogens (CEEs; Premarin; used in menopausal hormone therapy), and diethylstilbestrol (DES; widely used previously but now abandoned) are resistant to metabolism in the liver and have disproportionate effects on estrogen-modulated liver synthesis when compared to bioidentical estrogens like estradiol (Aly, 2020). As a result, they have stronger influence on coagulation and greater risk of certain health problems like blood clots and associated cardiovascular issues (Aly, 2020). For this reason, as well as the fact that relatively high doses of estrogens may be needed for testosterone suppression, non-bioidentical estrogens should ideally never be used in transfeminine hormone therapy.
Estradiol dose-dependently suppresses testosterone levels in people with testes. Physiological and guideline-based levels of estradiol (<200 pg/mL or <734 pmol/L) are often not sufficient to suppress testosterone levels into the female range in transfeminine people who have not had their gonads removed (e.g., Liang et al., 2018; Krishnamurthy et al., 2023; Slack et al., 2023). As a result, estradiol is generally used in combination with an antiandrogen or progestogen in transfeminine hormone therapy (Hembree et al., 2017; Coleman et al., 2022; Rose et al., 2023). This results in partial suppression of testosterone levels by estradiol and further suppression or blockade of the remaining testosterone by the antiandrogen or progestogen. While combination therapy can be effective in fully suppressing or blocking testosterone (e.g., Aly, 2019; Aly, 2020), testosterone suppression can also still remain incomplete with antiandrogens and progestogens in certain forms (e.g., Aly, 2018; Jain, Kwan, & Forcier, 2019). In contrast to physiological estradiol levels, supraphysiological levels of estradiol are able to consistently and fully suppress testosterone levels into the normal female range with estradiol alone in transfeminine people (e.g., Gooren et al., 1984 [Graph]; Igo & Visram, 2021; Herndon et al., 2023 [Discussion]). This alternative approach, often referred to as high-dose estradiol monotherapy, has the advantage of avoiding the side effects, risks, and costs of antiandrogens and progestogens. However, it has the disadvantage of exposure to supraphysiological estradiol levels that are above those recommended by guidelines and that may have greater health risks. Physiological estradiol doses and combination therapy are more often used in transfeminine people treated by clinicians, whereas high-dose estradiol monotherapy is more frequently encountered in transfeminine people on DIY hormone therapy.
The feminizing effects of estradiol appear to be maximal at relatively low levels in the absence of androgens. Higher doses of estradiol and supraphysiological estradiol levels, aside from allowing for greater testosterone suppression, are not known to result in better feminization in transfeminine people (Deutsch, 2016; Nolan & Cheung, 2021). In fact, there is some indication that higher estrogen doses early into hormone therapy could actually result in worse breast development. Hence, the therapeutic emphasis in transfeminine hormone therapy is more on testosterone suppression than on achieving a specific estradiol level, at least above a certain low threshold level. Higher doses of estrogens, including of estradiol, also have a greater risk of adverse health effects such as blood clots and cardiovascular problems (Aly, 2020). As such, the use of physiological doses of estradiol is optimal in transfeminine people. At the same time however, high estrogen doses can be useful for improving testosterone suppression when it is inadequate, and the absolute risks, in the case of non-oral bioidentical estradiol, are low and are more important in people with specific risk factors (e.g., older age, physical inactivity, obesity, concomitant progestogen use, smoking, surgery, and rare thrombophilic abnormalities). If more adequate testosterone suppression is necessary, limitedly supraphysiological doses of non-oral estradiol may have a reasonable ratio of benefit to risk in this context, at least in those without relevant risk factors for estrogen-related complications (e.g., many healthy young people) (Aly, 2020).
Estradiol and estradiol esters are usually used orally, sublingually, transdermally, by injection (intramuscularly or subcutaneously), or by implant in transfeminine hormone therapy (Wiki).
Oral Estradiol
Estradiol is used orally in the form of tablets of estradiol (Wiki; Graphs). Alternatively, oral estradiol valerate tablets are used in some countries, for instance in many European countries. The only real difference between these oral estradiol forms is that estradiol valerate contains slightly less estradiol by weight (~76% of that of estradiol) due to its ester component and hence requires somewhat higher doses (~1.3-fold) in comparison for equivalent estradiol levels (Wiki; Table). Oral estradiol has a duration suitable for once-daily administration. Oral administration of estradiol is a very convenient and inexpensive route, which makes it the most popular and widely used form of estradiol in transfeminine people. Oral estradiol has relatively low bioavailability (~5%), and there is substantial variability between people in terms of estradiol levels achieved with the same dose. Hence, in some transfeminine people estradiol levels may be low with oral estradiol, and testosterone suppression may be inadequate depending on the antiandrogen.
A major drawback of oral estradiol is that it results in excessive levels of estradiol in the liver due to the first pass that occurs with oral administration and has a disproportionate impact on estrogen-modulated liver synthesis (Aly, 2020). This in turn increases coagulation and the risk of associated health complications like blood clots and cardiovascular problems (Aly, 2020). These particular health concerns are largely allayed if estradiol is taken non-orally at reasonable and non-excessive doses. Hence non-oral forms of estradiol, like transdermal estradiol, although less convenient and often more expensive than oral estradiol, are preferable in transfeminine hormone therapy. It is recommended that all transfeminine people who are over 40 to 45 years of age use non-oral routes due to the greater risk of blood clots and cardiovascular problems that occurs with age (Aly, 2020; Coleman et al., 2022). Oral estradiol is not a good choice for high-dose estradiol monotherapy in transfeminine people due to the high estradiol levels required and the greater risks than with non-oral routes. In addition to its disproportionate liver impact, oral estradiol results in unphysiological levels of estradiol metabolites like estrone and estrone sulfate when compared to non-oral forms. The clinical implications of this, if any, are unknown. Oral and non-oral estradiol have in any case been found to have similar effectiveness in terms of feminization and breast development in transfeminine people in available studies (Sam, 2020).
Sublingual Estradiol
Oral estradiol tablets can be taken sublingually instead of orally. Sublingual use of estradiol tablets has several-fold higher bioavailability relative to oral administration and hence achieves much higher overall estradiol levels in comparison (Sam, 2021; Wiki; Graphs). Sublingual use of oral estradiol tablets can be employed instead of oral administration to reduce doses and hence medication costs or to produce higher estradiol levels for the purpose of achieving better testosterone suppression when needed. However, sublingual estradiol is very spiky in terms of estradiol levels when compared to oral estradiol and has a short duration of highly elevated estradiol levels. As such, it may be advisable for sublingual estradiol to be used in divided doses multiple times throughout the day in order to maintain at least somewhat steadier estradiol levels. The therapeutic implications for transfeminine people of the spikiness of sublingual estradiol, for instance in terms of testosterone suppression and health risks, have been little-studied and are mostly unknown. In any case, when used as a form of high-dose estradiol monotherapy and taken multiple times per day, strong though still incomplete testosterone suppression has been observed (Yaish et al., 2023). Oral estradiol valerate tablets can be taken sublingually instead of orally similarly to estradiol and are likewise highly effective when used in this way (Aly, 2019; Wiki). Due to partial swallowing of tablets, sublingual estradiol may in practice be a mixture of sublingual and oral administration and may have some of the same health risks of oral estradiol (Wiki). Buccal administration of estradiol appears to have similar properties as sublingual administration but is much less researched in comparison and is not used as often in transfeminine people (Wiki).
Transdermal Estradiol
Transdermal estradiol is available in the form of patches, gel, emulsions, and sprays (Wiki). These forms are usually applied to skin areas such as the arms, abdomen, or buttocks. Gel, emulsions, and sprays are applied and left to dry for a short period, whereas patches are applied and remain adhesed to the skin for a specified amount of time. Due to rate-limited absorption through the skin, there is a depot effect with transdermal estradiol and this route has a long duration with very steady estradiol levels. As a result, estradiol gel, emulsions, and sprays are all suitable for once-daily use. Patches stay applied and continuously deliver estradiol for either 3–4 days or 7 days depending on the patch brand (Table). Transdermal estradiol is more expensive than oral estradiol. Gel, emulsions, and sprays may be less convenient than oral administration, but patches can be more convenient due to their infrequent application. However, patches can sometimes cause application site problems like redness and irritation and can occasionally come off prematurely due to adhesive failure. As with oral estradiol, there is substantial variability in estradiol levels with transdermal estradiol, and some transfeminine people may have poor absorption, low estradiol levels, and inadequate testosterone suppression with this route. Estradiol sprays, such as Lenzetto, have been found to achieve very low estradiol levels that are probably not therapeutically adequate for use in transfeminine hormone therapy (Aly, 2020; Graph).
Transdermal estradiol is the form of estradiol most commonly used in transfeminine people who are over 40 years of age due to its lower health risks relative to oral estradiol. Transdermal estradiol gel is not a favorable option for high-dose estradiol monotherapy as it has difficulty achieving the high estradiol levels needed for adequate testosterone suppression (Aly, 2019). On the other hand, transdermal estradiol patches can be an effective option for high-dose estradiol monotherapy if multiple 100 μg/day patches are used, although this can require the use of many patches and can be expensive (Wiki). Different skin sites absorb transdermal estradiol to different extents (Wiki). Genital application of transdermal estradiol, specifically to the scrotum or neolabia, is particularly better-absorbed than conventional skin sites and can result in much higher estradiol levels than usual (Aly, 2019). This can be useful for reducing doses and hence medication costs or for achieving higher estradiol levels for better testosterone suppression when needed, for instance in the context of high-dose estradiol monotherapy. Transdermal estradiol should not be applied to the breasts as this is not known to result in improved breast development and the potential health consequences of doing so are unknown (e.g., influence on breast cancer risk).
Injectable Estradiol
Injectable estradiol preparations can be administered via either intramuscular or subcutaneous injection (Wiki; Wiki; Graphs). There is a depot effect with injection of estradiol esters such that they are slowly absorbed from the injection site and have a prolonged duration. This ranges from days to months depending on the ester. Commonly used injectable estradiol esters, which all have short to moderate durations, include estradiol valerate (EV), estradiol cypionate (EC), estradiol enanthate (EEn), and estradiol benzoate (EB). Longer-acting injectable estradiol esters, such as estradiol undecylate (EU) and polyestradiol phosphate (PEP), have been discontinued and are no longer pharmaceutically available. In the case of intramuscular injection, common injection sites include the deltoid muscle (upper arm), vastus lateralis and rectus femoris muscles (thigh), and ventrogluteal muscle (buttocks). Subcutaneous injection of estradiol injectables, while less commonly used, has comparable pharmacokinetics to intramuscular injection, and is easier, less painful, and more convenient in comparison (Wiki). However, the maximum volume that can be safely and comfortably injected subcutaneously (1.5–3 mL) is less than that which can be injected intramuscularly (2–5 mL) (Hopkins, & Arias, 2013; Usach et al., 2019). Injectable estradiol tends to be fairly inexpensive, but may be less convenient than other routes due to the need for regular injections. There may also be a risk of internal scar tissue build-up long-term. Estradiol injectables have been discontinued in many parts of the world (e.g., most of Europe), and their availability is limited. In recent years, many transfeminine people have turned to black market homebrewed injectable estradiol preparations to use this route.
Injectable estradiol preparations are typically used at higher doses than other forms of estradiol, and can easily achieve very high levels of estradiol. This can be useful for testosterone suppression, making this form of estradiol likely the best choice for high-dose estradiol monotherapy in transfeminine people. However, the high doses that are possible with injectable estradiol preparations can also easily lead to overdosage and unnecessarily increased risks (e.g., Aly, 2020). Resources are available on this site for guiding selection of appropriate doses and intervals of injectable estradiol esters in transfeminine people. This includes a simulator and informal meta-analysis of estradiol levels with these preparations (Aly, 2021; Aly, 2021) and a table providing approximate equivalent doses between injectable estradiol esters and other estradiol routes and forms (Aly, 2020). It is notable and unfortunate that currently recommended doses and intervals for injectable estradiol esters by transgender care guidelines (e.g., 10–40 mg/2 weeks estradiol valerate) appear to be highly excessive and too widely spaced, and are likely to be therapeutically inadvisable (Aly, 2021). Doses and intervals of injectable estradiol esters recommended by the present author for use as a means of high-dose estradiol monotherapy, targeting mean estradiol levels of around 300 pg/mL (1,100 pmol/L), are provided below (Table 4).
Table 4: Recommended doses and intervals of injectable estradiol esters for high-dose estradiol monotherapy (targeting estradiol levels of around 300 pg/mL [1,100 pmol/L]):
a Injection interval. b Doses and intervals for estradiol undecylate are extrapolated and hypothetical (Aly, 2021).
These doses and intervals should be considered a starting point, and should be fine-tuned as necessary based on blood tests. In terms of injection intervals, the shorter interval, the more stable the estradiol levels, but the more often that injections need to be done. Doses may be increased if estradiol levels are too low and testosterone suppression is inadequate, and doses may be decreased if estradiol levels are too high so long as adequate testosterone suppression is maintained. Doses should be lower (targeting mean estradiol levels of 100–200 pg/mL [367–734 pmol/L]) if combined with an antiandrogen or progestogen as these agents will help with testosterone suppression. Similarly, doses should be lower following surgical gonadal removal as testosterone suppression will no longer be necessary.
Estradiol Pellets
Estradiol implants are pellets of pure crystalline hormone and are surgically placed into subcutaneous fat by a physician (Wiki). They are slowly absorbed by the body following implantation, and new implants are given once every 4 to 6 months. Due to the need for minor surgery, their high cost, and limited availability, estradiol implants are not as commonly used as other estradiol routes. Notably, almost all pharmaceutical estradiol implants throughout the world have been discontinued, and the implants that remain available are almost exclusively compounded products provided by compounding pharmacies. Dosage adjustment with estradiol implants is also more difficult than with other estradiol routes. Despite their various practical limitations however, estradiol implants allow for very steady estradiol levels, and their very long duration can allow for unusual convenience among available estradiol forms.
Additional Notes
Table 5: Available forms and recommended doses of estradiol for adulta transfeminine people:
a Estradiol doses in pubertal adolescent transfeminine people should be lower to mimic estradiol exposure during normal female puberty (Aly, 2020). b May be advisable to use divided doses 2 to 4 times per day (i.e., once every 6 to 12 hours) instead of once per day (Sam, 2021). c This estradiol form achieves very low estradiol levels at typical doses that don’t appear to be well-suited for transfeminine hormone therapy (Aly, 2020; Graph). d Estradiol valerate contains about 75% of the same amount of estradiol as estradiol so doses are about 1.3-fold higher for the same estradiol levels (Aly, 2019; Sam, 2021). e Doses and intervals for estradiol undecylate are extrapolated and hypothetical (Aly, 2021). f A higher initial loading dose of e.g., 240 or 320 mg polyestradiol phosphate can be used for the first one or two injections to reach steady-state estradiol levels more quickly. However, this preparation has recently been discontinued and appears to no longer be available.
Additional informational resources are available in terms of estradiol levels (Wiki; Table) and approximate equivalent doses (Aly, 2020) with different forms, routes, and doses of estradiol.
There is high variability between individuals in the levels of estradiol achieved during estradiol therapy. That is, estradiol levels during treatment with the same dosage of estradiol can differ substantially between individuals. This variability is greatest with oral and transdermal estradiol but is also considerable even with injectable estradiol preparations and other estradiol forms. As such, estradiol doses are not absolute and should be individualized on a case-by-case basis in conjunction with blood work as a guide. It should also be noted that due to fluctuations in estradiol concentrations with certain routes, levels of estradiol can vary considerably from one blood test to another. This is most notable with sublingual estradiol and injectable estradiol. The fluctuations in estradiol levels with these routes are predictable and must be understood when interpreting blood work results. Differences in blood test results can be minimized with informed and consistent timing of blood draws.
If or when the gonads are surgically removed, testosterone suppression is no longer needed in transfeminine people. As a result, estradiol doses, if they are high or supraphysiological, can be lowered to more closely approximate normal physiological levels in cisgender women.
Progestogens
Progestogens include progesterone and progestins. Progestins are synthetic progestogens derived from structural modification of progesterone or testosterone. There are dozens of different progestins and these progestins can be divided into a variety of different structural classes with varying properties (Table). Examples of some major progestins of different classes include the 17α-hydroxyprogesterone derivative medroxyprogesterone acetate (MPA; Provera, Depo-Provera), the 19-nortestosterone derivative norethisterone (NET; many brand names), the retroprogesterone derivative dydrogesterone (Duphaston), and the 17α-spirolactone derivative drospirenone (Slynd, Yasmin). Progestins were developed because they have a more favorable disposition in the body than progesterone for use as medications. Only a few clinically used progestins have been employed in transfeminine hormone therapy. However, progestogens largely produce the same progestogenic effects, with a few exceptions, and theoretically almost any progestogen could be used.
Besides helping with testosterone suppression, progestogens are of no clear or known benefit for feminization or breast development in transfeminine people. While some transfeminine people anecdotally claim to experience improved breast development with progestogens, an involvement of progestogens in improving breast size or shape is controversial and is not supported by theory nor evidence at present (Wiki; Aly, 2020). It is possible that premature introduction of progestogens, particularly at high doses, could actually have an unfavorable influence on breast development (Aly, 2020). Many transfeminine people have also anecdotally claimed that progestogens have a beneficial effect on their sexual desire. However, a review of the literature by the present author found that neither progesterone nor progestins positively influence sexual desire in humans (Aly, 2020). Instead, the available evidence suggests either a neutral influence or an inhibitory effect of progestogens on sexual desire, although the latter may be specific only to high doses of progestogens (Aly, 2020). Claims have been made that progesterone may have beneficial effects on mood in transfeminine people as well, but clinical support for such notions is likewise lacking at this time (Coleman et al., 2022; Nolan et al., 2022). It is notable that progesterone at luteal-phase levels, due to its neurosteroid metabolites like allopregnanolone, actually appears to worsen mood in around 30% of cisgender women, and produces more overt negative reactions, which constitute the diagnoses of premenstrual syndrome (PMS) and premenstrual dysphoric disorder (PMDD), in around 2 to 10% of women (Bäckström et al., 2011; Edler Schiller, Schmidt, & Rubinow, 2014; Sundström-Poromaa et al., 2020). More research is needed to evaluate the possible beneficial effects of progestogens in transfeminine people.
Most clinically used progestogens have off-target activities in addition to their progestogenic activity, and these activities may be desirable or undesirable depending on the action in question (Kuhl, 2005; Stanczyk et al., 2013; Wiki; Table). Progesterone has a variety of neurosteroid as well as other activities that can result in central nervous system effects among others which are not shared by progestins. MPA as well as NET and its derivatives have weak androgenic activity, which is unfavorable in the context of transfeminine hormone therapy. NET and certain related progestins produce ethinylestradiol as a metabolite at high doses and hence can produce ethinylestradiol-like estrogenic effects, including increased risk of blood clots and associated cardiovascular problems. Other off-target actions of progestogens include antiandrogenic, glucocorticoid, and antimineralocorticoid activities. These actions can result in differences in therapeutic effectiveness (e.g., androgen suppression or blockade) as well as side effects and health risks. Some notable progestins without undesirable off-target activities (i.e., androgenic or glucocorticoid activity) include low-dose CPA, drospirenone (DRSP), dienogest, nomegestrol acetate (NOMAC), dydrogesterone, and hydroxyprogesterone caproate (OHPC). However, of these progestins, only CPA has been considerably used and studied in transfeminine people.
The addition of progestogens to estrogen therapy has been associated with a number of unfavorable health effects. These include increased risk of blood clots (Wiki; Aly, 2020), coronary heart disease (Wiki), and breast cancer (Wiki; Aly, 2020). High doses of progestogens are also associated with increased risk of certain non-cancerousbrain tumors including meningiomas and prolactinomas (Wiki; Aly, 2020). The coronary heart disease risk may be due to changes in blood lipids caused by the weak androgenic activity of certain progestogens, but the rest of the aforementioned risks are probably due to their progestogenic activity (Stanczyk et al., 2013; Jiang & Tian, 2017). Aside from health risks, progestogens have also been associated with adverse mood changes (Wiki; Wiki). However, besides the case of progesterone and its neurosteroid metabolites, these effects of progestogens are controversial and are not well-supported by evidence (Wiki; Wiki). Progestogens are otherwise generally well-tolerated and are regarded as producing little in the way of side effects.
In contrast to certain progestins, progesterone has no unfavorable off-target hormonal activities. Due to its lack of androgenic activity, progesterone has no adverse influence on blood lipids and is not expected to raise the risk of coronary heart disease. The addition of oral progesterone to estrogen therapy notably has not been associated with increased risk of blood clots (Wiki). In addition, oral progesterone seems to have less risk of breast cancer than progestins with shorter-term therapy, although this is notably not the case with longer-term exposure (Wiki; Aly, 2020). Consequently, it has been suggested that progesterone, for reasons that have yet to be fully elucidated, may be a safer progestogen than progestins and that it should be the preferred progestogen for hormone therapy in cisgender women and transfeminine people. However, there are also theoretical arguments against such notions. Oral progesterone is known to produce very low progesterone levels and to have only weak progestogenic effects at typical doses (Aly, 2018; Wiki). The seemingly better safety of oral progesterone may simply be an artifact of the low progesterone levels that occur with it, and hence of progestogenic dosage. Non-oral progesterone, at doses resulting in physiological and full progestogenic strength, has never been properly evaluated in terms of health outcomes, and may have similar risks as progestins (Aly, 2018; Wiki).
Due to their lack of known influence on feminization and breast development and their known and possible adverse effects and risks, progestogens are not routinely used in transfeminine hormone therapy at present. Major transgender health guidelines note the limitations of the available evidence on progestogens for transfeminine people and have mixed attitudes on their use, either explictly recommending against their use (Coleman et al., 2022—WPATH SOC8), taking a more neutral stance (Hembree et al., 2017—Endocrine Society guidelines), or being permissive of their use (Deutsch, 2016—UCSF guidelines). There is however a very major exception to the preceding in the form of CPA, an antiandrogen which is widely used in transfeminine hormone therapy to suppress testosterone production and which happens to be a powerful progestogen at the typical doses used in transfeminine people. CPA will be described below in the section on antiandrogens. Although progestogens have various health risks, cisgender women of course have progesterone, and the absolute risks of progestogens are very low in healthy young people. Risks like breast cancer also are exposure-dependent and take many years to develop. The testosterone suppression provided by progestogens can furthermore be very useful in transfeminine people, as is widely taken advantage of with CPA. Given these considerations, a limited duration of progestogen therapy in transfeminine people, for instance a few years to help suppress testosterone levels before surgical gonadal removal, may be considered quite acceptable.
Progesterone can be used in transfeminine people by oral administration, sublingual administration, rectal administration, or by intramuscular or subcutaneous injection (Wiki). Progestins are usually used via oral administration, but certain progestins are also available in injectable formulations (Wiki).
Oral Progesterone
Progesterone is most commonly taken orally. It is used by this route in the form of oil-filled capsules containing 100 or 200 mg micronized progesterone under brand names such as Prometrium, Utrogestan, and Microgest (Wiki). Despite its widespread use, levels of progesterone via oral administration have been found using state-of-the-art assays (LC–MS) to be very low (<2 ng/mL [<6.4 nmol/L] at 100 mg/day) and inadequate for satisfactory progestogenic effects in various areas (Aly, 2018; Wiki). In relation to this, even high doses of oral progesterone (400 mg/day) showed no antigonadotropic effect or testosterone suppression in cisgender men (Aly, 2018; Wiki). This is in major contrast to non-oral forms of progesterone and to progestins, which produce dose-dependent and robust testosterone suppression (Aly, 2019; Wiki). In addition to its low progestogenic potency, oral progesterone is excessively converted into neurosteroid metabolites like allopregnanolone and pregnanolone. These metabolites act as potent GABAA receptor positive allosteric modulators, and can produce undesirable alcohol-like side effects such as sedation, cognitive, memory, and motor impairment, and mood changes (Wiki; Wiki). As such, while inconvenient, non-oral routes are greatly preferable for progesterone.
Sublingual Progesterone
Sublingual progesterone tablets exist and are marketed under the brand name Luteina but today are only available in Poland and Ukraine (Wiki). Oral progesterone could theoretically be taken sublingually, analogously to sublingual use of oral estradiol. However, because oral progesterone is formulated as oil-filled capsules, this makes it difficult and unpleasant to use by sublingual administration. Buccal progesterone, which would be expected to have similar characteristics to those of sublingual progesterone, has been used in medicine in the past, but is no longer marketed today (Wiki).
Rectal Progesterone
Progesterone is approved for use by rectal administration in the form of suppositories under the brand name Cyclogest (Wiki). This product is marketed in only a limited number of countries however, although it is available in the United Kingdom (Wiki). While not approved for use by rectal administration, oral progesterone capsules can be taken rectally instead of orally, and using them in this way may allow for much higher progesterone levels than would be achieved by oral administration due to avoidance of most first-pass metabolism. Rectal administration of oral progesterone capsules has not been formally studied, but oral progesterone capsules have been administered vaginally in cisgender women with success (Miles et al., 1994; Wang et al., 2019), and the vaginal and rectal routes are said to have similar pharmacokinetics in general (Goletiani, Keith, & Gorsky, 2007; Wiki). Hence, there is good theoretical basis for rectal administration of oral progesterone capsules being an effective route of progesterone. Whereas oral progesterone achieves very low levels of progesterone, rectal progesterone can readily achieve normal luteal-phase levels of progesterone (Wiki). Although inconvenient, rectal administration may be the overall best route of administration of progesterone for transfeminine people. A significant subset of transfeminine people on progestogens take progesterone rectally (Chang et al., 2024).
Injectable Progesterone
Progesterone by injection is available as an oil solution for intramuscular injection under brand names such as Proluton, Progestaject, and Gestone (Wiki) and as an aqueous solution for subcutaneous injection under the brand name Prolutex (Wiki). Oil solutions of progesterone for intramuscular injection are widely available, whereas the aqueous solution of progesterone for subcutaneous injection is available only in some European countries (Wiki). Injectable progesterone, regardless of route, has a relatively short duration and must be injected once every one to three days (Wiki; Wiki). This makes it too inconvenient to use for most people. Unlike with estradiol, progesterone esters with longer durations than progesterone itself by injection are not chemically possible as progesterone has no hydroxyl groups available for esterification (Wiki). Injectable aqueous suspensions of microcrystalline progesterone were previously marketed and had a duration of 1 to 2 weeks, but these preparations were associated with pain at the injection site and were eventually discontinued (Aly, 2019; Wiki).
Other Progesterone Routes
Other progesterone routes, such as transdermal progesterone and subcutaneous progesterone pellets, are also known, but are not available as pharmaceutical drugs and are little-used medically (Wiki). This is related to the low potency of progesterone and difficulty achieving progesterone levels high enough for adequate therapeutic effects with these routes (Wiki; Wiki). In addition, progesterone pellets tend to be extruded at high rates (Wiki). In any case, certain compounding pharmacies may make forms of progesterone that could be used by these routes.
Oral and Injectable Progestins
Most progestins are taken orally in the form of solid tablets (Wiki). In contrast to progesterone, progestins, owing to their synthetic nature, are resistant to metabolism in the intestines and liver and have high oral bioavailability. In addition, unlike the case of the estrogen receptors, the progesterone receptors are expressed minimally or not at all in the liver, and there is no known first pass influence of progestogenic activity on liver synthesis (Lax, 1987; Stanczyk, Mathews, & Cortessis, 2017). As a result, there are no apparent problems with oral administration in the case of purely progestogenic progestins. However, some progestins have liver-impacting off-target hormonal actions, such as androgenic, estrogenic, and/or glucocorticoid activity, and this can result in adverse effects like unfavorable lipid changes or procoagulation—which may be augmented by the first pass with oral administration.
A selection of progestins are available in injectable formulations, including for intramuscular or subcutaneous injection (Wiki). Some of the more notable ones include medroxyprogesterone acetate (MPA), norethisterone enanthate (NETE), hydroxyprogesterone caproate (OHPC), and algestone acetophenide (dihydroxyprogesterone acetophenide; DHPA) (Wiki). In addition to being used alone, injectable progestins are used together with estradiol esters in combined injectable contraceptives (Wiki). These preparations are often used as a means of hormone therapy by transfeminine people in Latin America. Whereas injectable progesterone has a duration measured in days, injectable progestins have durations ranging from weeks to months, and can be injected much less often in comparison (Table).
Additional Notes
Table 6: Available forms and recommended doses of progestogens for transfeminine people:
For progesterone levels with different forms, routes, and doses of progesterone, see the table here (only LC–MS and IA + CS assays for oral progesterone) and the graphs here.
As with estradiol, there is high variability between individuals in progesterone levels. Conversely, there is less variability between individuals in the case of progestins.
After removal of the gonads, progestogen doses can be lowered or adjusted to approximate normal female physiological exposure or they can be discontinued entirely.
Antiandrogens
Aside from estrogens and progestogens, there is another class of hormonal medications used in transfeminine hormone therapy known as antiandrogens (AAs). These medications reduce the effects of androgens in the body by either decreasing androgen production and thereby lowering androgen levels or by directly blocking the actions of androgens. They work via a variety of different mechanisms of action, and include androgen receptor antagonists, antigonadotropins, and androgen synthesis inhibitors.
Androgen receptor antagonists act by directly blocking the effects of androgens, including testosterone, DHT, and other androgens, at the level of their biological target. They bind to the androgen receptor without activating it, thereby displacing androgens from the receptor. Due to the nature of their mechanism of action as competitive blockers of androgens, the antiandrogenic efficacy of androgen receptor antagonists is both highly dose-dependent and fundamentally dependent on testosterone levels. They do not act by lowering testosterone levels, although some androgen receptor antagonists may have additional antiandrogenic actions that result in decreased testosterone levels. Because androgen receptor antagonists do not work by lowering testosterone levels, blood work can be less informative for them compared to antiandrogens that suppress testosterone levels. Androgen receptor antagonists include steroidal antiandrogens (SAAs) like spironolactone (Aldactone) and cyproterone acetate (CPA; Androcur) and nonsteroidal antiandrogens (NSAAs) like bicalutamide (Casodex).
Antigonadotropins suppress the gonadal production of androgens by inhibiting the GnRH-mediated secretion of gonadotropins from the pituitary gland. They include estrogens and progestogens. In addition, GnRH agonists such as leuprorelin (Lupron) and GnRH antagonists such as elagolix (Orilissa) act similarly and could likewise be described as antigonadotropins.
Androgen synthesis inhibitors inhibit the enzyme-mediated synthesis of androgens. They include 5α-reductase inhibitors (5α-RIs) like finasteride (Propecia) and dutasteride (Avodart). There are also other types of androgen synthesis inhibitors, for instance potent 17α-hydroxylase/17,20-lyase inhibitors like ketoconazole (Nizoral) and abiraterone acetate (Zytiga). However, these agents have limitations (e.g., toxicity, high cost, and lack of experience) and have not been used in transfeminine hormone therapy.
Although antigonadotropins and androgen synthesis inhibitors have antiandrogenic effects secondary to decreased androgen levels, they are not usually referred to as “antiandrogens”. Instead, this term is most commonly reserved to refer specifically to androgen receptor antagonists. However, antigonadotropins and androgen synthesis inhibitors may nonetheless be described as antiandrogens as well.
After removal of the gonads, antiandrogens can be discontinued. If unwanted androgen-dependent symptoms, such as acne, seborrhea, or scalp hair loss, persist despite full suppression or ablation of gonadal testosterone, then a lower dose of an androgen receptor antagonist, such as 100 to 200 mg/day spironolactone or 12.5 to 25 mg/day bicalutamide, can be continued to treat these symptoms.
Table 7: Available forms and recommended doses of antiandrogens for transfeminine people:
a For CPA, this dose range is specifically one-quarter of a 10-mg tablet to one full 10-mg tablet per day (2.5–10 mg/day) or a quarter of a 50-mg tablet every other day or every 2 to 3 days (4.2–12.5 mg/day). A dosage of 5–10 mg/day or 6.25–12.5 mg/day is likely to ensure maximal testosterone suppression, while lower doses may be less effective (Aly, 2019). b For spironolactone and bicalutamide, it is assumed that testosterone levels are substantially suppressed (≤200 ng/dL [<6.9 nmol/L]). If testosterone levels are not suppressed to this range, then higher doses may be warranted. c Spironolactone and its metabolites have relatively short half-lives, and twice-daily administration in divided doses (e.g., 100–200 mg twice per day) is recommended.
Figure 3: Suppression of gonadal testosterone production and circulating testosterone levels (ng/dL) with estradiol in combination with different antiandrogens over one year of hormone therapy in transfeminine people (Sofer et al., 2020). The estradiol forms included oral tablets 2–8 mg/day, transdermal gel 2.5–5 mg/day, and transdermal patches 50–200 μg/day. The antiandrogens included spironolactone 50–200 mg/day (n=16), cyproterone acetate (n=41), and GnRH agonists (specifically triptorelin 3.75 mg/month or goserelin 3.6 mg/month by injection) (n=10) (Sofer et al., 2020). It should be noted that lower doses of cyproterone acetate (10–12.5 mg/day) show equal testosterone suppression to higher doses (25–100 mg/day) and higher doses should no longer be used (Aly, 2019). The dashed horizontal line corresponds to the upper limit of the normal female range for testosterone levels.
As an antiandrogen, CPA has a dual mechanism of action of suppressing testosterone levels via its progestogenic and hence antigonadotropic activity and of acting as an androgen receptor antagonist (Aly, 2019). The progestogenic activity of CPA is of far greater potency than its androgen receptor antagonism however (Aly, 2019). The dose of CPA used as a progestogen in cisgender women is about 2 mg per day, which produces similar progestogenic effects to those of physiological luteal-phase levels of progesterone (e.g., suppression of gonadotropin secretion, ovulation inhibition, and endometrial transformation and protection) (Aly, 2019). Conversely, much higher doses of CPA of 50 to 300 mg/day have typically been used for androgen-dependent indications (Aly, 2019). These high doses of CPA result in profound progestogenic overdosage and associated side effects and risks (Aly, 2019). In transfeminine people, CPA has historically been used at doses of 50 to 150 mg/day (Aly, 2019). However, CPA doses have dramatically fallen in recent years, and today doses of no more than 10 to 12.5 mg/day are recommended (Aly, 2019; Coleman et al., 2022—WPATH SOC8). These lower doses of CPA still produce strong progestogenic effects, and in combination with estradiol, are equally effective as higher doses in suppressing testosterone levels (Aly, 2019; Meyer et al., 2020; Even Zohar et al., 2021; Kuijpers et al., 2021; Coleman et al., 2022). Even lower doses of CPA, for instance 5 to 6.25 mg/day, are currently being studied, and may still be fully effective (Aly, 2019).
Given by itself without estrogen, CPA typically suppresses testosterone levels in people with testes by about 50 to 70%, down to about 150 to 300 ng/dL (5.2–10.4 nmol/L) (Meriggiola et al., 2002; Toorians et al., 2003; Giltay et al., 2004; T’Sjoen et al., 2005; Tack et al., 2017; Zitzmann et al., 2017; Aly, 2019). Lower doses of CPA alone (e.g., 10 mg/day) show the same degree of testosterone suppression as higher doses of CPA alone (e.g., 50–100 mg/day), indicating that the antigonadotropic effects of CPA are maximal at relatively low therapeutic doses of this medication (Aly, 2019). This is on the order of about 5 to 10 times the ovulation-inhibiting dosage of CPA in cisgender women, a dose–response relationship that has also been observed with a number of other progestogens (Aly, 2019). Per the preceding, CPA alone, regardless of dosage, is unable to reduce testosterone levels into the normal female range (<50 ng/dL [<1.7 nmol/L]). But when CPA is combined with estradiol, even at relatively small doses of estradiol, it consistently suppresses testosterone levels into the normal female range (Aly, 2019; Angus et al., 2019; Gava et al., 2020; Sofer et al., 2020; Collet et al., 2022). However, it appears that a certain minimum level of estradiol, perhaps around 60 pg/mL (220 pmol/L) on average, is required for this to occur (Aly, 2019). Estradiol levels lower than this threshold in those taking CPA, which can occasionally be encountered in transfeminine people due to estradiol being dosed too low, have the potential to compromise full testosterone suppression (Aly, 2019).
In addition to testosterone suppression, CPA can dose-dependently block the androgen receptor (Aly, 2019). However, relatively high doses of CPA are needed to considerably antagonize the androgen receptor (e.g., 50–300 mg/day), and lower doses (e.g., ≤12.5 mg/day) may not be able to do this to a meaningful degree (Aly, 2019). As such, lower doses of CPA may essentially be purely progestogenic, with minimal or no androgen receptor antagonism. In this regard, referring to CPA at such doses as an “antiandrogen”—rather than as a “progestogen”—may be considered somewhat of a misnomer. Higher doses of CPA (>12.5 mg/day) can no longer be considered safe due to the massive progestogenic overdosage that occurs with them, and should no longer be used in transfeminine people. Moreover, as testosterone levels are usually suppressed into the normal female range in transfeminine people taking estradiol plus CPA, there is no actual need for any additional androgen receptor blockade (Aly, 2019).
CPA has been reported to produce various side effects. Some of these side effects include fatigue and a degree of weight gain (Belisle & Love, 1986; Hammerstein, 1990; Martinez-Martin et al., 2022). CPA might be able to produce a magnitude of sexual dysfunction (e.g., reduced sexual desire) beyond that expected with testosterone suppression alone (Wiki; Aly, 2019). It may also have a small risk of depressive mood changes (Wiki). In transfeminine people, CPA has been documented to produce pregnancy-like breast changes (i.e., lobuloalveolar development of the mammary glands) (Kanhai et al., 2000). In relation to this, CPA sometimes causes lactation as a side effect (Dewhurst & Underhill, 1979; Gooren, Harmsen-Louman, & van Kessel, 1985; Schlatterer et al., 1998; Bazarra-Castro, 2009). Concerns have been raised about premature introduction of progestogens—particularly at high doses like with CPA—and possible adverse influence on breast development (Aly, 2020). However, little data exists in humans to substantiate such concerns at present. The side effects of CPA are assumed to be dose-dependent, and using the lowest effective doses is expected to minimize its side effects.
CPA is usually taken orally in the form of tablets (e.g., 10, 50, and 100 mg) (Wiki). Under the brand name Androcur Depot, it is also available as a long-lasting 300 mg depot injectable in some countries (Wiki). However, this formulation is not commonly used in transfeminine people, and happens to correspond to very high doses in terms of CPA exposure. A pill cutter (Amazon) can be used to split CPA tablets and achieve lower doses (e.g., 12.5 mg doses with 50-mg tablets). CPA has a relatively long elimination half-life of about 1.6 to 4.3 days (Wiki; Aly, 2019). As such, it can be taken once daily, or even as infrequently as once every 2 or 3 days, if needed (Aly, 2019). In addition to splitting of CPA tablets, dosing CPA once every 2 or 3 days can also be useful for achieving lower doses (Aly, 2019).
As already described, CPA is a powerful progestogen even at the relatively low doses now used in transfeminine people (e.g., 5–12.5 mg/day). As such, there is no need, nor point, in adding another progestogen, for instance progesterone, in those who are taking CPA—at least if the goal of doing so is to produce progestogenic effects. This is something that is often overlooked in people taking CPA, and can result in increased costs, side effects, and inconvenience without any expected benefit.
Spironolactone
Spironolactone (Aldactone) is an antiandrogen and antimineralocorticoid. It is widely used as an antiandrogen in cisgender women for treatment of androgen-dependent hair and skin conditions like acne, hirsutism (excessive facial/body hair growth), and scalp hair loss, in cisgender women for treatment of hyperandrogenism (high androgen levels) due to polycystic ovary syndrome (PCOS), and in transfeminine people as a component of hormone therapy. Spironolactone is particularly widely used in transfeminine people in the United States, where it is the most commonly used antiandrogen in this population. As an antimineralocorticoid, the original and primary use of spironolactone in medicine, it is used to treat heart failure, high blood pressure, high mineralocorticoid levels, low potassium levels, and conditions of excess fluid retention like nephrotic syndrome and ascites, among others (Wiki). In terms of its antiandrogenic actions, spironolactone is a relatively weak androgen receptor antagonist as well as a weak androgen synthesis inhibitor (Wiki). The androgen synthesis inhibition of spironolactone is mediated specifically via inhibition of 17α-hydroxylase and 17,20-lyase (Wiki). Spironolactone does not appear to have meaningful progestogenic activity, 5α-reductase inhibition, or direct estrogenic activity (Wiki). However, indirect estrogenic effects secondary to its antiandrogenic activity (e.g., breast development and feminization) can occur with it at sufficiently high doses (Wiki).
Spironolactone shows limited and highly inconsistent effects on testosterone levels in clinical studies in cisgender men, cisgender women, and transfeminine people, with most studies finding no change in levels, some studies finding a decrease in levels, and a small number even finding an increase in levels (Aly, 2018). In spite of this, studies commonly find that spironolactone still produces antiandrogenic effects even when androgen levels remain unchanged. Hence, the primary mechanism of action of spironolactone as an antiandrogen appears to be androgen receptor blockade. In relation to this, in transfeminine people taking spironolactone as an antiandrogen, the estrogen component of the regimen is likely to be the main or possibly sole agent suppressing testosterone production. This is in part based on studies in transfeminine people comparing estradiol plus spironolactone to estradiol alone (e.g., Leinung, 2014; Leinung, Feustel, & Joseph, 2018; Angus et al., 2019) and on studies comparing testosterone levels with different doses of spironolactone (e.g., Liang et al., 2018; SoRelle et al., 2019; Allen et al., 2021). Due to the minimal influence of spironolactone on testosterone production, testosterone levels are not usually suppressed into the female range in transfeminine people taking estradiol plus spironolactone, with testosterone levels often remaining well above this range (e.g., 50–450 ng/dL [1.7–15.6 nmol/L] on average) (Leinung, 2014; Leinung, Feustel, & Joseph, 2018; Liang et al., 2018; Angus et al., 2019; Jain, Kwan, & Forcier, 2019; SoRelle et al., 2019; Sofer et al., 2020; Burinkul et al., 2021). However, testosterone levels do tend to decline gradually over time in transfeminine people on this regimen (e.g., Liang et al., 2018; Sofer et al., 2020 (Graph); Allen et al., 2021).
Due to its relatively weak androgen receptor antagonism, spironolactone is likely best-suited for blocking female-range or somewhat-higher testosterone levels (e.g., <100 ng/dL [<3.5 nmol/L]) (Aly, 2018). This is based on clinical dose-ranging studies of spironolactone (typically using 50–200 mg/day) in healthy cisgender women and cisgender women with PCOS (Goodfellow et al., 1984; Lobo et al., 1985; Hammerstein, 1990; James, Jamerson, & Aguh, 2022) as well as comparative studies of spironolactone against the more-potent antiandrogen flutamide (Cusan et al., 1994; Erenus et al., 1994; Shaw, 1996). The clinical antiandrogenic efficacy of spironolactone has been very limitedly assessed in transfeminine people to date, and is largely unknown (Angus et al., 2021). In any case, the antiandrogenic efficacy of spironolactone in cisgender women with androgen-dependent hair and skin conditions is well-established, and the medication thus does appear to be effective so long as testosterone levels are not too high (Brown et al., 2009; van Zuuren & Fedorowicz, 2016; Layton et al., 2017; Barrionuevo et al., 2018; James, Jamerson, & Aguh, 2022). In addition, higher doses of spironolactone (e.g., 300–400 mg/day) may be more useful for blocking higher testosterone levels in transfeminine people, and are allowed for by transgender care guidelines (Aly, 2020).
Consequent to spironolactone’s limited and inconsistent influence on testosterone levels and its relatively weak androgen receptor antagonism, estradiol plus spironolactone regimens will likely not be fully effective in terms of testosterone suppression for many transfeminine people. This is liable to result in suboptimal demasculinization, feminization, and breast development in these individuals. Other antiandrogenic approaches, such as bicalutamide, CPA, GnRH modulators, and high-dose estradiol monotherapy, will likely be more effective in these cases owing either to their ability to more potently block androgens or their capacity to reliably reduce testosterone levels into the female range. If testosterone levels are still too high with estradiol plus spironolactone, a switch to a different antiandrogen, increasing to a higher dosage of estradiol, or addition of a clinically antigonadotropic progestogen (e.g., non-oral progesterone or a progestin) should be considered.
Spironolactone is a strong antimineralocorticoid, or antagonist of the mineralocorticoid receptor, the biological target of the mineralocorticoid steroid hormones aldosterone and 11-deoxycorticosterone. This is an action that spironolactone shares with progesterone, although spironolactone is a much more potent antimineralocorticoid than progesterone. The mineralocorticoid receptor is involved in regulating electrolyte and fluid balances, among other roles. Spironolactone is associated with modestly lowered blood pressure, which may be considered a beneficial effect of its antimineralocorticoid activity (Martinez-Martin et al., 2022). Although spironolactone is usually well-tolerated, it can sometimes produce antimineralocorticoid side effects such as excessively lowered blood pressure, dizziness, fatigue, urinary frequency, and increased cortisol levels, among others (Kellner & Wiedemann, 2008; Kim & Del Rosso, 2012; Zaenglein et al., 2016; Layton et al., 2017; James, Jamerson, & Aguh, 2022). It has been argued by some in the online transgender community that spironolactone, via its antimineralocorticoid activity and increased cortisol levels, may increase visceral fat in transfeminine people (Aly, 2020). However, evidence does not support this hypothetical side effect at present (Aly, 2020). Available data also do not support spironolactone stunting breast development in transfeminine people or producing serious neuropsychiatric side effects, such as prominent depressive mood changes.
In people who are at-risk for hyperkalemia, dietary restriction to limit intake of potassium-rich foods is often recommended (Roscioni et al., 2012; Cupisti et al., 2018). This is often encountered in transgender health as transfeminine people being told “not to eat bananas”, which are said to be high in potassium. However, limiting dietary potassium with spironolactone to avoid hyperkalemia is theoretical and not actually evidence-based, with data so far contradicting its efficacy (St-Jules, Goldfarb, & Sevick, 2016; St-Jules & Fouque, 2021; Babich, Kalantar-Zadeh, & Joshi, 2022; St-Jules & Fouque, 2022). As such, routine restriction of dietary potassium with spironolactone may not be warranted.
Aside from its antimineralocorticoid activity, spironolactone has been reported to increase levels of LDL (“bad”) cholesterol levels and to decrease levels of HDL (“good”) cholesterol in women with PCOS (Nakhjavani et al., 2009). However, findings appear to be conflicting, with other studies not finding unfavorable influences on cholesterol levels with spironolactone (Polyzos et al., 2011). Long-term, adverse effects on cholesterol levels could result in an increase in the risk of coronary heart disease.
Spironolactone is taken orally in the form of tablets (e.g., 25, 50, and 100 mg) (Wiki). It is a prodrug of several active metabolites, including 7α-thiomethylspironolactone, 6β-hydroxy-7α-thiomethylspironolactone, and canrenone (7α-desthioacetyl-δ6-spironolactone) (Wiki). Spironolactone and these active metabolites have elimination half-lives of 1.4 hours, 13.8 hours, 15.0 hours, and 16.5 hours, respectively (Wiki). Due to the relatively short duration of elevated drug levels with spironolactone and its active metabolites (Graph), twice-daily administration of spironolactone in divided doses may be more optimal than once-daily intake and is advised (Reiter et al., 2010).
Bicalutamide
Bicalutamide (Casodex) is a nonsteroidal antiandrogen (NSAA) which acts as a potent and highly selective androgen receptor antagonist (Wiki). It is primarily used in the treatment of prostate cancer in cisgender men. Prostate cancer is an androgen-dependent cancer which antiandrogens can help to slow the progression of, and this use constitutes the vast majority of prescriptions for bicalutamide (Wiki). In addition to prostate cancer, although to a much lesser extent, bicalutamide has been used in the treatment of hirsutism (excessive facial/body hair growth), scalp hair loss, and polycystic ovary syndrome (PCOS) in cisgender women, peripheral or gonadotropin-independent precocious puberty (a rare form of precocious puberty in which antigonadotropins such as GnRH agonists are not effective) in cisgender boys, and priapism in cisgender men (Wiki). Bicalutamide is also becoming increasingly adopted for use as an antiandrogen in transfeminine people (Aly, 2020; Wiki). However, its use in transgender health is still very limited, and well-regarded transgender care guidelines either recommend against its use (Deutsch, 2016—UCSF guidelines; Coleman et al., 2022—WPATH SOC8) or are only cautiously permissive of its use (Thompson et al., 2021—Fenway Health guidelines). This is due to a lack of studies of bicalutamide in transfeminine people and its potential risks. Nonetheless, a small but growing number of clinicians are using bicalutamide in transfeminine people or are willing to prescribe it, with these clinicians located particularly in the United States. A single small clinical study has assessed bicalutamide in transfeminine people so far, specifically as a puberty blocker in 13 transfeminine adolescents who were denied insurance coverage for GnRH agonists (Neyman, Fuqua, & Eugster, 2019). (Update: More studies of bicalutamide in transfeminine people have since been published, see Aly (2020).)
Bicalutamide is a much more potent androgen receptor antagonist than either spironolactone or CPA (Wiki; Neyman, Fuqua, & Eugster, 2019). It is typically used in transfeminine people at a dosage of 25 to 50 mg/day, although this dosage has been arbitrarily selected and is not based on clinical data. Nonetheless, due to its relatively high potency as an androgen receptor antagonist and concomitant suppression of testosterone levels by estradiol, these doses may be adequate for testosterone blockade for many transfeminine people. At higher doses (>50 mg/day), bicalutamide is able to substantially block male-range testosterone levels (>300 ng/dL [>10.4 nmol/L]) based on studies of bicalutamide monotherapy in cisgender men with prostate cancer (Wiki). This is something that spironolactone and CPA are not capable of in the same way. Owing to its selectivity for the androgen receptor, bicalutamide has no off-target hormonal activity and produces almost no side effects in women (Wiki; Erem, 2013; Moretti et al., 2018). The only apparent side effect of bicalutamide in a rigorous clinical trial of the drug for hirsutism in cisgender women was significantly increased total and LDL (“bad”) cholesterol levels (Moretti et al., 2018). Hence, bicalutamide tends to be very well-tolerated. The relative lack of side effects with bicalutamide is in contrast to other antiandrogens like spironolactone and CPA, which are not pure androgen receptor antagonists and have off-target hormonal actions like antimineralocorticoid activity or strong progestogenic activity with consequent side effects and risks.
As a selective androgen receptor antagonist, bicalutamide taken by itself does not decrease testosterone production or levels but rather increases them (Wiki). This is due to a loss of androgen receptor-mediated negative feedback on gonadotropin secretion and a consequent compensatory upregulation of gonadal testosterone production (Wiki). Bicalutamide more than blocks the effects of any increase in testosterone it causes, and in fact fundamentally cannot increase testosterone levels more than it can block them (Wiki). In addition, increases in testosterone levels with bicalutamide will be blunted or abolished if it is combined with an adequate dose of an antigonadotropin such as estradiol (Wiki; Wiki). Since estradiol is made from testosterone in the body, bicalutamide taken alone also preserves and increases estradiol production and levels (Wiki). Because of this, although bicalutamide has no other important intrinsic hormonal activity besides its antiandrogenic activity, it produces robust indirect estrogenic effects including feminization and breast development even when it is not combined with estrogen (Wiki; Wiki; Neyman, Fuqua, & Eugster, 2019). This has important implications for the use of bicalutamide as a puberty blocker in transfeminine adolescents, as bicalutamide does not actually block puberty like conventional puberty blockers (GnRH agonists) but instead has the effect of dose-dependently converting male puberty into female puberty (Wiki; Neyman, Fuqua, & Eugster, 2019).
Bicalutamide has certain health risks, which has been a major reason that it has not been more readily adopted in transfeminine hormone therapy (Aly, 2020). It has a small risk of liver toxicity (Wiki; Aly, 2020) and of lung toxicity (Wiki). Abnormal liver function tests (LFTs), such as elevated liver enzymes and elevated bilirubin, occurred in about 3.4% of men with bicalutamide monotherapy plus standard care versus 1.9% of men with placebo plus standard care in the Early Prostate Trial (EPC) clinical programme after 3.0 years of follow-up (Wiki). In clinical trials, treatment with bicalutamide had to be discontinued in about 0.3 to 1.5% of men due to LFTs that became too highly elevated and could have progressed to serious liver toxicity (Wiki). To date, there are around 10 published case reports of serious liver toxicity, including cases of death, with bicalutamide, all of which have been in men with prostate cancer (Wiki; Table; Aly, 2020). There have also been a few unpublished reports of serious liver toxicity including deaths with bicalutamide in transfeminine people (Aly, 2020). However, these reports have not been confirmed, and they may or may not be reliable. In addition to the preceding reports, hundreds of additional instances of liver complications in people taking bicalutamide exist in the United States FDA Adverse Event Reporting System (FAERS) database (Wiki; FDA). Abnormal LFTs with bicalutamide usually occur within the first 3 to 6 months of treatment (Kolvenbag & Blackledge, 1996; Casodex FDA Label), and all case reports of liver toxicity with bicalutamide have had an onset of less than 6 months (Table). The liver toxicity of bicalutamide is not known to be dose-dependent across its clinically used dose range (Wiki). Abnormal LFTs have occurred with bicalutamide (at rates of 2.9% to 11.4%) even at relatively low doses in cisgender women (e.g., 10–50 mg/day) (de Melo Carvalho, 2022). Due to its risk of liver toxicity, periodic liver monitoring is strongly advised with bicalutamide, especially within the first 6 months of treatment. Possible signs of liver toxicity include nausea, vomiting, abdominal pain, fatigue, appetite loss, flu-like symptoms, dark urine, and jaundice (yellowing of the skin/eyes) (Wiki).
In terms of its lung toxicity risk, bicalutamide has been associated rarely with interstitial pneumonitis, which can lead to pulmonary fibrosis and can be fatal, and also less often with eosinophilic lung disease (Wiki; Table). As of writing, 15 published case reports of interstitial pneumonitis and 2 case reports of eosinophilic lung disease in association with bicalutamide therapy exist, likewise all in men with prostate cancer (Table). As with liver toxicity, hundreds of additional cases of interstitial pneumonitis in people taking bicalutamide exist in the United States FAERS database (Wiki; FDA). It has been estimated that interstitial pneumonitis with bicalutamide occurs at a rate of around 1 in 10,000 people, although this may be an underestimate due to under-reporting (Wiki; Ahmad & Graham, 2003). Asian people may be especially likely to experience lung toxicity with bicalutamide and other NSAAs, as much higher incidences have been observed in this population (Mahler et al., 1996; Wu et al., 2022). There is no laboratory test for routine monitoring of lung changes with bicalutamide. Possible signs of relevant lung toxicity include dyspnea (difficulty breathing or shortness of breath), coughing, and pharyngitis (inflammation of the throat, typically manifesting as sore throat) (Wiki).
Aside from liver and lung toxicity, bicalutamide monotherapy has been found in cisgender men with prostate cancer to increase the risk of death due to causes other than prostate cancer (Iversen et al., 2004; Iversen et al., 2006; Wellington & Keam, 2006; Jia & Spratt, 2022; Wiki). This led to marketing authorization of bicalutamide for treatment of the earliest stage of prostate cancer being revoked and to the drug being abandoned for this use (Wiki). Bicalutamide remains approved and used for treatment of later stages of prostate cancer, as the antiandrogenic benefits of bicalutamide against prostate cancer outweigh any adverse influence it has on non-prostate-cancer mortality in these more severe stages. The mechanisms underlying the increase in risk of death with bicalutamide in men are unknown (Wiki). It is also unclear whether bicalutamide could likewise increase the risk of death in transfeminine people. Limitations of generalizing these studies to transfeminine people include the men in the trials being relatively old and ill, a relatively high dosage of bicalutamide (150 mg/day) being used in the trials for an extended duration (e.g., 5 years), the question of whether the risks were due to androgen deprivation or to specific drug-related toxicity of bicalutamide, and estradiol levels with bicalutamide monotherapy in men with prostate cancer being only about 30 to 50 pg/mL (110–184 pmol/L) (Wiki). The preceding estradiol levels are well above castrate levels and are sufficient for a substantial degree of estrogenic effect, but are nonetheless below those recommended for transfeminine people and potentially needed for full sex-hormone replacement (which are ≥50 pg/mL [≥184 pmol/L]). In any case, as the specific mechanisms underlying the increased mortality risk with bicalutamide seen in men with prostate cancer are uncertain, and as clinical safety data showing that the risk does not generalize do not exist, it remains a possibility that bicalutamide could also increase the risk of death in transfeminine people.
Bicalutamide is taken orally in the form of tablets (e.g., 50 and 150 mg) (Wiki). Due to saturation of absorption in the gastrointestinal tract, the oral bioavailability of bicalutamide progressively starts to decrease above a dosage of about 150 mg/day, and there is no further increase in bicalutamide levels above 300 mg/day (Wiki; Graph). Bicalutamide has a very long elimination half-life of about 6 to 10 days (Wiki; Graphs). As a result, it does not necessarily have to be taken daily, and can be dosed less often (in proportionally higher doses)—for instance twice weekly or even once weekly—if this is more convenient or otherwise desired. Due to its long half-life, bicalutamide requires about 4 to 12 weeks to fully reach steady-state levels (Wiki; Graph; Wiki). However, about 50% of steady state is reached within 1 week of administration of bicalutamide, and about 80 to 90% of steady state is reached after 3 to 4 weeks (Wiki; Graph; Wiki). Loading doses of bicalutamide can be taken to reach steady state more quickly if desired. Animal studies originally suggested that bicalutamide did not cross the blood–brain barrier and hence was peripherally selective (i.e., did not block androgen receptors in the brain) (Wiki). However, subsequent clinical studies found that this was not similarly the case in humans, in whom bicalutamide shows clear and robust centrally mediated antiandrogenic effects (Wiki).
Older NSAAs related to bicalutamide like flutamide (Eulexin) and nilutamide (Anandron, Nilandron) have much greater risks in comparison to bicalutamide and should not be used in transfeminine people. Nilutamide was previously characterized as an antiandrogen in transfeminine people in several studies, but was not further pursued probably due to its very high incidence of lung toxicity and other side effects (Aly, 2020; Wiki; Wiki). Flutamide has been used limitedly as an antiandrogen in transfeminine people in the past, but should no longer be used due to a much higher risk of liver toxicity than bicalutamide as well as other side effects and drawbacks (Aly, 2020; Wiki). Other newer and more-potent NSAAs like enzalutamide (Xtandi), apalutamide (Erleada), and darolutamide (Nubeqa) also have risks and have been studied and used little outside of prostate cancer to date.
5α-Reductase Inhibitors
Testosterone is converted into DHT within certain tissues in the body (Swerdloff et al., 2017). DHT is an androgen metabolite of testosterone with several-fold higher activity than testosterone. The transformation of testosterone into DHT is mediated by the enzyme 5α-reductase. The tissues in which 5α-reductase is present and testosterone is converted into DHT are limited but most importantly include the skin, hair follicles, and prostate gland. Although DHT is more potent than testosterone, it is thought to have minimal biological role as a circulating hormone (Horton, 1992; Swerdloff et al., 2017). Instead, testosterone serves as the main circulating androgen, and the role of DHT is thought to be mainly via local metabolism and potentiation of testosterone into DHT within certain tissues.
5α-Reductase inhibitors (5α-RIs), such as finasteride (Proscar, Propecia) and dutasteride (Avodart), inhibit 5α-reductase and thereby block the conversion of testosterone into DHT. This results in marked decreases in circulating and within-tissue levels of DHT. Due to the primary role of DHT as a mediator in tissues rather than as circulating hormone, the antiandrogenic efficacy of 5α-RIs is limited. This is evidenced by the fact that they are well-tolerated in cisgender men and do not cause notable demasculinization in these individuals (Hirshburg, 2016). The medical use of 5α-RIs is mainly restricted to the treatment of scalp hair loss in men and women, hirsutism (excessive facial/body hair) in women, and prostate enlargement in men. They might also be useful for acne in women, but evidence of this is very limited (Wiki). Due to their specificity, 5α-RIs are inappropriate as general antiandrogens in transfeminine people. Moreover, DHT levels decrease in tandem with testosterone levels with suppression of testosterone production in transfeminine hormone therapy, and routine use of 5α-RIs in transfeminine people with testosterone levels within the female range is of limited usefulness and can be considered unnecessary (Gooren et al., 2016; Irwig, 2020; Prince & Safer, 2020; Glintborg et al., 2021). In any case, 5α-RIs may be useful in transfeminine people on hormone therapy who have persistent body hair growth or scalp hair loss—as they have been shown to be in cisgender women (Barrionuevo et al., 2018; Prince & Safer, 2020). However, it is notable that evidence of effectiveness in cisgender women is better for androgen receptor antagonists for such indications (van Zuuren et al., 2015). This is intuitive as androgen receptor antagonists block both testosterone and DHT whereas 5α-RIs only prevent conversion of testosterone into DHT. Hence, although 5α-RIs strongly reduce or eliminate DHT and their net effect is antiandrogenic, they do not decrease testosterone levels and in fact increase them.
There are three subtypes of 5α-reductase. Dutasteride inhibits all three subtypes of 5α-reductase whereas finasteride only inhibits two of the subtypes. As a result of this, dutasteride is a more complete 5α-RI than finasteride. Dutasteride decreases DHT levels in the blood by up to 98% while finasteride can only decrease them by around 65 to 70%. As nearly all circulating DHT originates from synthesis in peripheral tissues, these decreases indicate parallel reductions in tissue DHT production (Horton, 1992). In accordance with these findings, dutasteride has been found to be more effective than finasteride in the treatment of scalp hair loss in men (Zhou et al., 2018; Dhurat et al., 2020; Wiki). For these reasons, although both finasteride and dutasteride are effective 5α-RIs, dutasteride may be the preferable choice if a 5α-RI is used (Zhou et al., 2018; Dhurat et al., 2020).
A potentially undesirable effect of 5α-RIs in transfeminine people is that they may increase circulating testosterone levels to a degree in those in whom testosterone production isn’t fully suppressed (Leinung, Feustel, & Joseph, 2018; Aly, 2019; Traish et al., 2019; Irwig, 2020; Glintborg et al., 2021). It appears that DHT adds significantly to negative feedback on gonadotropin secretion in the pituitary gland in people with testes who have low testosterone levels relative to the normal male range (Traish et al., 2019). The therapeutic implications of this for transfeminine people, if any, are uncertain.
Clinical dose-ranging studies have found that lower doses of finasteride and dutasteride than are typically used still provide substantial or near-maximal 5α-reductase inhibition (Gormley et al., 1990; Vermeulen et al., 1991; Sudduth & Koronkowski, 1993; Drake et al., 1999; Roberts et al., 1999; Clark et al., 2004; Frye, 2006; Olsen et al., 2006; Harcha et al., 2014; Kuhl & Wiegratz, 2017). In one study with finasteride for instance, DHT levels decreased by 49.5% at 0.05 mg/day, 68.6% at 0.2 mg/day, 71.4% at 1 mg/day, and 72.2% at 5 mg/day (Drake et al., 1999). Parallel reductions in DHT levels were seen locally in the scalp (Drake et al., 1999). In a study with dutasteride, DHT levels were decreased by 52.9% at 0.05 mg/day, 94.7% at 0.5 mg/day, 97.7% at 2.5 mg/day, and 98.4% at 5 mg/day (Clark et al., 2004). Based on these findings, 5α-RIs can potentially be taken at lower doses to help reduce medication costs if needed. Finasteride tablets can be split to achieve smaller doses. Conversely, dutasteride cannot be split as it is formulated as an oil capsule. However, dutasteride has a long half-life, and instead of dividing pills, it can be taken less frequently (e.g., once every few days) as a means of reducing dosage.
5α-Reductase inhibitors are taken orally in the form of tablets and capsules. Compoundedtopical formulations of finasteride also exist (Marks et al., 2020). However, caution is advised with these preparations as they have been found to be excessively dosed and to produce equivalent systemic DHT suppression as oral finasteride formulations (Marks et al., 2020). Lower-concentration formulations of topical finsteride on the other hand may be more locally selective (Marks et al., 2020).
Table 8: Available forms and recommended doses of 5α-reductase inhibitors for transfeminine people:
GnRH agonists and antagonists (GnRHa), also known as GnRH receptor agonists and antagonists or GnRH modulators, are antiandrogens which work by preventing the effects of GnRH in the pituitary gland and thereby suppressing LH and FSH secretion. Receptor agonists normally activate receptors while receptor antagonists block and thereby inhibit the activation of receptors. Due to a physiological quirk however, GnRH agonists and antagonists have the same effects in the pituitary gland. This is because GnRH is secreted in pulses under normal physiological circumstances, and when the GnRH receptor is unnaturally activated in a continuous manner, as with exogenous GnRH agonists, the GnRH receptor in the pituitary gland is strongly desensitized to the point of becoming inactive. Consequently, both GnRH agonists and GnRH antagonists have the effect of abolishing gonadal sex hormone production. This, in turn, reduces testosterone levels into the castrate or normal female range (both <50 ng/dL or <1.7 nmol/L) in people with testes. GnRHa are like a reversible gonadectomy, and for this reason, are also sometimes referred to as “medical castration”. Provided that an estrogen is taken in combination with a GnRHa to prevent sex hormone deficiency, these medications have essentially no known side effects or risks. For these reasons, GnRHa are the ideal antiandrogens for use in transfeminine people.
GnRHa are widely used to suppess puberty in adolescent transgender individuals. Unfortunately however, they are very expensive (e.g., ~US$10,000 per year) and medical insurance does not usually cover them for adult transgender people. Consequently, GnRHa are not commonly used in adult transfeminine people at this time. An exception is in the United Kingdom, where GnRH agonists are covered for all adult transgender people by the National Health Service (NHS). Another exception is buserelin (Suprefact), which has become available very inexpensively in its nasal spray form from certain Eastern European online pharmacies in recent years (Aly, 2018).
GnRH agonists cause a brief flare in testosterone levels at the start of therapy prior to the GnRH receptors in the pituitary gland becoming desensitized (Wiki). Testosterone levels increase by up to about 1.5- to 2-fold for about 1 week and then decrease thereafter (Wiki). Castrate or female-range levels of testosterone are generally reached within 2 to 4 weeks (Wiki). In contrast to GnRH agonists, there is no testosterone flare with GnRH antagonists and testosterone levels start decreasing immediately, reaching castrate levels within a few days (Wiki; Graph). This is because GnRH antagonists work by blocking the GnRH receptor without initially activating it, and hence desensitization of the receptor is not necessary for their action. If desired, the testosterone flare at the initiation of GnRH agonist therapy can be prevented or blunted with the use of antigonadotropins, for instance estrogens and progestogens, as well as with potent androgen receptor antagonists such as bicalutamide (Wiki).
GnRH agonists must be injected subcutaneously or intramuscularly once per day or once every one to six months depending on the formulation employed (buserelin, goserelin, leuprorelin, triptorelin). Alternatively, they can be surgically implanted once a year (histrelin, leuprorelin) or used as a nasal spray two to three times per day (buserelin, nafarelin). The first GnRH antagonists were developed for use by once-monthly intramuscular or subcutaneous injection (abarelix, degarelix). More recently, orally administered GnRH antagonists such as elagolix and relugolix have been introduced for medical use. They are taken in the form of tablets once or twice daily.
Table 9: Available forms and recommended doses of GnRH agonists for transfeminine people:
a 500 μg 3x/day for the first week then 200 μg/day. b 800 μg 3x/day for the first week then 400 μg 3x/day. c 500 μg 2x/day can be used instead of 400 μg 3x/day but is less effective (70% decrease in testosterone levels (to ~180 ng/dL [6.2 nmol/L]) instead of 90% decrease (to ~50 ng/dL [1.7 nmol/L]) per available studies of buserelin in men with prostate cancer) (Aly, 2018; Wiki).
Table 10: Available forms and recommended doses of GnRH antagonists for transfeminine people:
a First month is 240 mg then 80 mg per month thereafter. b 150 mg 1x/day is less effective than 200 mg 2x/day (which provides full gonadal sex-hormone suppression in cisgender women) (Wiki). c 80–120 mg/day for full gonadal sex-hormone suppression and 20–40 mg/day for substantial but partial gonadal sex-hormone suppression (MacLean et al., 2015; DailyMed).
Other Hormonal Medications
Androgens and Anabolic Steroids
In addition to estrogens, progestogens, and antiandrogens, androgens/anabolic steroids (AAS) are sometimes added to transfeminine hormone therapy. This is when testosterone levels are low (e.g., below the female average of 30 ng/dL [1.0 nmol/L]) and androgen replacement is desired. It has been proposed that adequate levels of testosterone may provide benefits such as increased sexual desire, improved mood and energy, positive effects on skin health and cellulite (Avram, 2004), and increased muscle size and strength (Huang & Basaria, 2017). However, there is insufficient clinical evidence to support such benefits at present, and androgens can produce adverse effects in cisgender women and transfeminine people, for instance acne, hirsutism, scalp hair loss, and masculinization (Wiki). For transfeminine people who nonetheless desire androgen replacement therapy, possible options for androgen medications include testosterone and its esters, dehydroepiandrosterone (DHEA; prasterone), and nandroloneesters such as nandrolone decanoate (ND) (Aly, 2020; Table), among others.
Monitoring of Therapy
Transfeminine people on hormone therapy should undergo regular laboratory monitoring in the form of blood work to assess efficacy and monitor for safety. Total estradiol levels and total testosterone levels should be measured to assess the effectiveness of therapy—that is, whether hormone levels are in appropriate ranges for cisgender females—and determine whether medication adjustments may be necessary. Levels of free testosterone, free estradiol, estrone (E1), dihydrotestosterone (DHT), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and sex hormone-binding globulin (SHBG) can also be measured to provide further information although they’re not absolutely necessary. If progesterone is used as a part of hormone therapy, progesterone levels can be measured to provide insight on the degree of progesterone exposure. In addition to hormone blood tests, transfeminine people can monitor their physical changes with hormone therapy, such as breast development and other aspects of feminization, using various physical and digital measurement methods (e.g., Wiki).
Certain therapeutic situations can result in inaccurate lab blood work results. Monitoring of progesterone levels with oral progesterone using immunoassay-based blood tests can result in falsely high readings for progesterone levels due to cross-reactivity with high levels of progesterone metabolites such as allopregnanolone (Aly, 2018; Wiki). Instead of immunoassay-based tests, mass spectrometry-based tests should be used to determine progesterone levels with oral progesterone (Aly, 2018; Wiki). Conversely, either type of test may be used to measure progesterone levels with non-oral progesterone therapy. High-dose biotin (vitamin B7) supplements can interfere with the accuracy of immunoassay-based hormone blood tests, causing falsely low or falsely high readings (Samarasinghe et al., 2017; Avery, 2019; Bowen et al., 2019; FDA, 2019; Luong, Male, & Glennon, 2019). Transdermal estradiol formulations applied to the arm can result in contamination of blood draws taken from the same arm and can result in falsely high readings for estradiol levels (Vihtamäkia, Luukkaala, & Tuimala, 2004).
Certain cancers are known to be hormone-sensitive and their incidence can be influenced by hormone therapy. Screening for breast and prostate cancer is recommended in transfeminine people (Sterling & Garcia, 2020; Iwamoto et al., 2021). The risk of breast cancer appears to be dramatically increased with transfeminine hormone therapy, perhaps especially with progestogens (Aly, 2020). However, the risk still remains lower than in cisgender women (Aly, 2020). The incidence of prostate cancer is greatly decreased with hormone therapy in transfeminine people as a consequence of androgen deprivation, but the risk is not abolished and prostate cancer can still occur (de Nie et al., 2020). The prostate gland is not removed with vaginoplasty, so transfeminine people who have undergone vaginoplasty will also require monitoring for prostate cancer still. Testicular cancer is not known to be a hormone-dependent cancer and its incidence does not appear to be increased with hormone therapy in transfeminine people (Bensley et al., 2021; de Nie et al., 2021; Jacoby et al., 2021).
Ahmad, S. R., & Graham, D. J. (2003). Pneumonitis with Antiandrogens. Annals of Internal Medicine, 139(6), 528–529. [DOI:10.7326/0003-4819-139-6-200309160-00023]
Allen, A. N., Jiao, R., Day, P., Pagels, P., Gimpel, N., & SoRelle, J. A. (2020). Dynamic Impact of Hormone Therapy on Laboratory Values in Transgender Patients over Time. The Journal of Applied Laboratory Medicine, 6(1), 27–40. [DOI:10.1093/jalm/jfaa192]
Angus, L., Leemaqz, S., Ooi, O., Cundill, P., Silberstein, N., Locke, P., Zajac, J. D., & Cheung, A. S. (2019). Cyproterone acetate or spironolactone in lowering testosterone concentrations for transgender individuals receiving oestradiol therapy. Endocrine Connections, 8(7), 935–940. [DOI:10.1530/ec-19-0272]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2020). A systematic review of antiandrogens and feminization in transgender women. Clinical Endocrinology, 94(5), 743–752. [DOI:10.1111/cen.14329]
Antoniou, T., Gomes, T., Mamdani, M. M., Yao, Z., Hellings, C., Garg, A. X., Weir, M. A., & Juurlink, D. N. (2011). Trimethoprim-sulfamethoxazole induced hyperkalaemia in elderly patients receiving spironolactone: nested case-control study. BMJ, 343, d5228. [DOI:10.1136/bmj.d5228]
Antoniou, T., Hollands, S., Macdonald, E. M., Gomes, T., Mamdani, M. M., & Juurlink, D. N. (2015). Trimethoprim–sulfamethoxazole and risk of sudden death among patients taking spironolactone. Canadian Medical Association Journal, 187(4), E138–E143. [DOI:10.1503/cmaj.140816]
Aufrère, M. B., & Benson, H. (1976). Progesterone: An overview and recent advances. Journal of Pharmaceutical Sciences, 65(6), 783–800. [DOI:10.1002/jps.2600650602]
Avery, G. (2019). Biotin interference in immunoassay: a review for the laboratory scientist. Annals of Clinical Biochemistry: International Journal of Laboratory Medicine, 56(4), 424–430. [DOI:10.1177/0004563219842231]
Avram, M. M. (2004). Cellulite: a review of its physiology and treatment. Journal of Cosmetic and Laser Therapy, 6(4), 181–185. [DOI:10.1080/14764170410003057]
Babich, J. S., Kalantar-Zadeh, K., & Joshi, S. (2022). Taking the Kale out of Hyperkalemia: Plant Foods and Serum Potassium in Patients With Kidney Disease. Journal of Renal Nutrition, 32(6), 641–649. [DOI:10.1053/j.jrn.2022.01.013]
Bäckström, T., Haage, D., Löfgren, M., Johansson, I., Strömberg, J., Nyberg, S., Andréen, L., Ossewaarde, L., van Wingen, G., Turkmen, S., & Bengtsson, S. (2011). Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons. Neuroscience, 191, 46–54. [DOI:10.1016/j.neuroscience.2011.03.061]
Barbieri, J. S., Margolis, D. J., & Mostaghimi, A. (2021). Temporal Trends and Clinician Variability in Potassium Monitoring of Healthy Young Women Treated for Acne With Spironolactone. JAMA Dermatology, 157(3), 296–296. [DOI:10.1001/jamadermatol.2020.5468]
Barrionuevo, P., Nabhan, M., Altayar, O., Wang, Z., Erwin, P. J., Asi, N., Martin, K. A., & Murad, M. H. (2018). Treatment Options for Hirsutism: A Systematic Review and Network Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism, 103(4), 1258–1264. [DOI:10.1210/jc.2017-02052]
Bazarra-Castro, M. A. (2009). Etiological aspects, therapy regimes, side effects and treatment satisfaction of transsexual patients. (Doctoral dissertation, Ludwig Maximilian University of Munich.) [DOI:10.5282/edoc.9984] [URN:urn:nbn:de:bvb:19-99840] [PDF]
Belisle, S., & Love, E. J. (1986). Clinical efficacy and safety of cyproterone acetate in severe hirsutism: results of a multicentered Canadian study. Fertility and Sterility, 46(6), 1015–1020. [DOI:10.1016/s0015-0282(16)49873-0]
Ben Salem, C., Badreddine, A., Fathallah, N., Slim, R., & Hmouda, H. (2014). Drug-Induced Hyperkalemia. Drug Safety, 37(9), 677–692. [DOI:10.1007/s40264-014-0196-1]
Bensley, J. G., Cheung, A. S., Grossmann, M., & Papa, N. (2022). Testicular Cancer in Trans People Using Feminising Hormone Therapy–A Brief Review. Urology, 160, 1–4. [DOI:10.1016/j.urology.2021.11.014]
Bessone, F., Lucena, M., Roma, M. G., Stephens, C., Medina-Cáliz, I., Frider, B., Tsariktsian, G., Hernández, N., Bruguera, M., Gualano, G., Fassio, E., Montero, J., Reggiardo, M. V., Ferretti, S., Colombato, L., Tanno, F., Ferrer, J., Zeno, L., Tanno, H., & Andrade, R. J. (2015). Cyproterone acetate induces a wide spectrum of acute liver damage including corticosteroid-responsive hepatitis: report of 22 cases. Liver International, 36(2), 302–310. [DOI:10.1111/liv.12899]
Boogers, L. S., Sardo Infirri, S. A., Bouchareb, A., Dijkman, B. A., Helder, D., de Blok, C. J., Liberton, N. P., den Heijer, M., van Trotsenburg, A. S., Dreijerink, K. M., Wiepjes, C. M., & Hannema, S. E. (2025). Variations in Volume: Breast Size in Trans Women in Relation to Timing of Testosterone Suppression. The Journal of Clinical Endocrinology & Metabolism, 110(5), e1404–e1410. [DOI:10.1210/clinem/dgae573]
Bowen, R., Benavides, R., Colón-Franco, J. M., Katzman, B. M., Muthukumar, A., Sadrzadeh, H., Straseski, J., Klause, U., & Tran, N. (2019). Best practices in mitigating the risk of biotin interference with laboratory testing. Clinical Biochemistry, 74, 1–11. [DOI:10.1016/j.clinbiochem.2019.08.012]
Brown, J., Farquhar, C., Lee, O., Toomath, R., & Jepson, R. G. (2009). Spironolactone versus placebo or in combination with steroids for hirsutism and/or acne. Cochrane Database of Systematic Reviews, 2009(2), CD000194. [DOI:10.1002/14651858.cd000194.pub2]
Burinkul, S., Panyakhamlerd, K., Suwan, A., Tuntiviriyapun, P., & Wainipitapong, S. (2021). Anti-Androgenic Effects Comparison Between Cyproterone Acetate and Spironolactone in Transgender Women: A Randomized Controlled Trial. The Journal of Sexual Medicine, 18(7), 1299–1307. [DOI:10.1016/j.jsxm.2021.05.003]
Callen-Lorde Community Health Center. (2018). Protocols for the Provision of Hormone Therapy. New York City: Callen-Lorde Community Health Center. [URL] [PDF]
Cappelletti, M., & Wallen, K. (2016). Increasing women’s sexual desire: The comparative effectiveness of estrogens and androgens. Hormones and Behavior, 78, 178–193. [DOI:10.1016/j.yhbeh.2015.11.003]
Carmina, E., Stanczyk, F. Z., & Lobo, R. A. (2019). Evaluation of Hormonal Status. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 887–915.e4). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-47912-7.00034-2]
Carvalho, R. d., Santos, L. D., Ramos, P. M., Machado, C. J., Acioly, P., Frattini, S. C., Barcaui, C. B., Donda, A. L., & Melo, D. F. (2022). Bicalutamide and the new perspectives for female pattern hair loss treatment: What dermatologists should know. Journal of Cosmetic Dermatology, 21(10), 4171–4175. [DOI:10.1111/jocd.14773]
Chang, J. J., Tran, N. K., Flentje, A., Lubensky, M. E., Obedin-Maliver, J., Lunn, M. R., & Ariel, D. (2024). 12330 Progestogen Experience And Perception Among Transfeminine Adults - A National Survey. Journal of the Endocrine Society, 8(Suppl 1), bvae163.1657. [DOI:10.1210/jendso/bvae163.1657]
Clark, R. V., Hermann, D. J., Cunningham, G. R., Wilson, T. H., Morrill, B. B., & Hobbs, S. (2004). Marked Suppression of Dihydrotestosterone in Men with Benign Prostatic Hyperplasia by Dutasteride, a Dual 5α-Reductase Inhibitor. The Journal of Clinical Endocrinology & Metabolism, 89(5), 2179–2184. [DOI:10.1210/jc.2003-030330]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/s0140-6736(19)31709-x]
Collet, S., Gieles, N. C., Wiepjes, C. M., Heijboer, A. C., Reyns, T., Fiers, T., Lapauw, B., den Heijer, M., & T’Sjoen, G. (2022). Changes in Serum Testosterone and Adrenal Androgen Levels in Transgender Women With and Without Gonadectomy. The Journal of Clinical Endocrinology & Metabolism, 108(2), 331–338. [DOI:10.1210/clinem/dgac576]
Colonnello, E., Graziani, A., Rossetti, R., Voltan, G., Masi, D., Lubrano, C., Mariani, S., Watanabe, M., Isidori, A. M., Ferlin, A., & Gnessi, L. (2025). The Chronobiology of Hormone Administration: “Doctor, What Time Should I Take My Medication?”. Endocrine Reviews, bnaf013. [DOI:10.1210/endrev/bnaf013]
Connors, J. M., & Middeldorp, S. (2019). Transgender patients and the role of the coagulation clinician. Journal of Thrombosis and Haemostasis, 17(11), 1790–1797. [DOI:10.1111/jth.14626]
Cupisti, A., Kovesdy, C., D’Alessandro, C., & Kalantar-Zadeh, K. (2018). Dietary Approach to Recurrent or Chronic Hyperkalaemia in Patients with Decreased Kidney Function. Nutrients, 10(3), 261–261. [DOI:10.3390/nu10030261]
Cusan, L., Dupont, A., Gomez, J., Tremblay, R. R., & Labrie, F. (1994). Comparison of flutamide and spironolactone in the treatment of hirsutism: a randomized controlled trial. Fertility and Sterility, 61(2), 281–287. [DOI:10.1016/s0015-0282(16)56518-2]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, I. R., & den Heijer, M. (2019). Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in the Netherlands. BMJ, 365, l1652. [DOI:10.1136/bmj.l1652]
de Blok, C. J., Dijkman, B. A., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K. M., & den Heijer, M. (2021). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
de Nie, I., de Blok, C. J., van der Sluis, T. M., Barbé, E., Pigot, G. L., Wiepjes, C. M., Nota, N. M., van Mello, N. M., Valkenburg, N. E., Huirne, J., Gooren, L. J., van Moorselaar, R. J., Dreijerink, K. M., & den Heijer, M. (2020). Prostate Cancer Incidence under Androgen Deprivation: Nationwide Cohort Study in Trans Women Receiving Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 105(9), e3293–e3299. [DOI:10.1210/clinem/dgaa412]
Deng, T., Duan, X., He, Z., Zhao, Z., & Zeng, G. (2020). Association Between 5-Alpha Reductase Inhibitor Use and The Risk of Depression: A Meta-Analysis. Urology Journal, 18(2), 144–150. [DOI:10.22037/uj.v16i7.5866]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Dewhurst, J., & Underhill, R. (1979). The Suppression of Facial Hair Growth in Transsexuals Using Cyproterone Acetate. British Journal of Sexual Medicine, 6, 19–23. [Google Scholar] [PDF]
Dhurat, R., Sharma, A., Rudnicka, L., Kroumpouzos, G., Kassir, M., Galadari, H., Wollina, U., Lotti, T., Golubovic, M., Binic, I., Grabbe, S., & Goldust, M. (2020). 5‐Alpha reductase inhibitors in androgenetic alopecia: Shifting paradigms, current concepts, comparative efficacy, and safety. Dermatologic Therapy, 33(3), e13379. [DOI:10.1111/dth.13379]
Drake, L., Hordinsky, M., Fiedler, V., Swinehart, J., Unger, W. P., Cotterill, P. C., Thiboutot, D. M., Lowe, N., Jacobson, C., Whiting, D., Stieglitz, S., Kraus, S. J., Griffin, E. I., Weiss, D., Carrington, P., Gencheff, C., Cole, G. W., Pariser, D. M., Epstein, E. S., Tanaka, W., Dallob, A., Vandormael, K., Geissler, L., & Waldsteicher, J. (1999). The effects of finasteride on scalp skin and serum androgen levels in men with androgenetic alopecia. Journal of the American Academy of Dermatology, 41(4), 550–554. [DOI:10.1016/s0190-9622(99)80051-6]
Dyson, T. E., Cantrell, M. A., & Lund, B. C. (2020). Lack of Association between 5α-Reductase Inhibitors and Depression. Journal of Urology, 204(4), 793–798. [DOI:10.1097/ju.0000000000001079]
Erem, C. (2013). Update on idiopathic hirsutism: diagnosis and treatment. Acta Clinica Belgica, 68(4), 268–274. [DOI:10.2143/acb.3267]
Erenus, M., Gürbüz, O., Durmuşoğlu, F., Demirçay, Z., & Pekin, S. (1994). Comparison of the efficacy of spironolactone versus flutamide in the treatment of hirsutism. Fertility and Sterility, 61(4), 613–616. [DOI:10.1016/s0015-0282(16)56634-5]
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF]
Even Zohar, N., Sofer, Y., Yaish, I., Serebro, M., Tordjman, K., & Greenman, Y. (2021). Low-Dose Cyproterone Acetate Treatment for Transgender Women. The Journal of Sexual Medicine, 18(7), 1292–1298. [DOI:10.1016/j.jsxm.2021.04.008]
Fertig, R., Shapiro, J., Bergfeld, W., & Tosti, A. (2016). Investigation of the Plausibility of 5-Alpha-Reductase Inhibitor Syndrome. Skin Appendage Disorders, 2(3–4), 120–129. [DOI:10.1159/000450617]
Food and Drug Administration. (2019). UPDATE: The FDA Warns that Biotin May Interfere with Lab Tests: FDA Safety Communication. Food and Drug Administration. [URL]
Fournier, A., Berrino, F., & Clavel-Chapelon, F. (2007). Unequal risks for breast cancer associated with different hormone replacement therapies: results from the E3N cohort study. Breast Cancer Research and Treatment, 107(1), 103–111. [DOI:10.1007/s10549-007-9523-x]
Frye, S. (2006). Discovery and Clinical Development of Dutasteride, a Potent Dual 5α-Reductase Inhibitor. Current Topics in Medicinal Chemistry, 6(5), 405–421. [DOI:10.2174/156802606776743101]
Gava, G., Mancini, I., Alvisi, S., Seracchioli, R., & Meriggiola, M. C. (2020). A comparison of 5-year administration of cyproterone acetate or leuprolide acetate in combination with estradiol in transwomen. European Journal of Endocrinology, 183(6), 561–569. [DOI:10.1530/eje-20-0370]
Getzenberg, R., & Itty, S. (2020). How do we define “castration” in men on androgen deprivation therapy? Asian Journal of Andrology, 22(5), 441–446. [DOI:10.4103/aja.aja_139_19]
Giltay, E. J., Gooren, L. J., Toorians, A. W., Katan, M. B., & Zock, P. L. (2004). Docosahexaenoic acid concentrations are higher in women than in men because of estrogenic effects. The American Journal of Clinical Nutrition, 80(5), 1167–1174. [DOI:10.1093/ajcn/80.5.1167]
Glintborg, D., T’Sjoen, G., Ravn, P., & Andersen, M. S. (2021). MANAGEMENT OF ENDOCRINE DISEASE: Optimal feminizing hormone treatment in transgender people. European Journal of Endocrinology, 185(2), R49–R63. [DOI:10.1530/eje-21-0059]
Goletiani, N. V., Keith, D. R., & Gorsky, S. J. (2007). Progesterone: Review of safety for clinical studies. Experimental and Clinical Psychopharmacology, 15(5), 427–444. [DOI:10.1037/1064-1297.15.5.427]
Goodfellow, A., Alaghband-Zadeh, J., Carter, G., Cream, J., Holland, S., Scully, J., & Wise, P. (1984). Oral spironolactone improves acne vulgaris and reduces sebum excretion. British Journal of Dermatology, 111(2), 209–214. [DOI:10.1111/j.1365-2133.1984.tb04045.x]
Gooren, L. J. (2016). The Endocrinology of Sexual Behavior and Gender Identity. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition, Volume 2 (pp. 2163–2176.e4). Philadelphia: Saunders/Elsevier. [Google Books] [DOI:10.1016/B978-0-323-18907-1.00124-4]
Gooren, L., & Bunck, M. (2004). Transsexuals and competitive sports. European Journal of Endocrinology, 151(4), 425–429. [DOI:10.1530/eje.0.1510425]
Gooren, L. J., Harmsen-Louman, W., & Kessel, H. (1985). Follow-up of prolactin levels in long-term oestrogen-treated male-to-female transsexuals with regard to prolactinoma induction. Clinical Endocrinology, 22(2), 201–207. [DOI:10.1111/j.1365-2265.1985.tb01081.x]
Gooren, L., Rao, B., van Kessel, H., & Harmsen-Louman, W. (1984). Estrogen positive feedback on LH secretion in transsexuality. Psychoneuroendocrinology, 9(3), 249–259. [DOI:10.1016/0306-4530(84)90004-0]
Gormley, G. J., Stoner, E., Rittmaster, R. S., Gregg, H., Thompson, D. L., Lasseter, K. C., Vlasses, P. H., & Stein, E. A. (1990). Effects of Finasteride (MK-906), a 5α-Reductase Inhibitor, on Circulating Androgens in Male Volunteers. The Journal of Clinical Endocrinology & Metabolism, 70(4), 1136–1141. [DOI:10.1210/jcem-70-4-1136]
Grock, S., Weinreb, J., Williams, K. C., Weimer, A., Fadich, S., Patel, R., Geft, A., & Korenman, S. (2024). Priorities for efficacy trials of gender-affirming hormone therapy with estrogen: collaborative design and results of a community survey. Hormones, online ahead of print. [DOI:10.1007/s42000-024-00532-3]
Gubelin Harcha, W., Barboza Martínez, J., Tsai, T., Katsuoka, K., Kawashima, M., Tsuboi, R., Barnes, A., Ferron-Brady, G., & Chetty, D. (2014). A randomized, active- and placebo-controlled study of the efficacy and safety of different doses of dutasteride versus placebo and finasteride in the treatment of male subjects with androgenetic alopecia. Journal of the American Academy of Dermatology, 70(3), 489–498.e3. [DOI:10.1016/j.jaad.2013.10.049]
Gupta, P., Suppakitjanusant, P., Stevenson, M., Goodman, M., & Tangpricha, V. (2022). Potassium Concentrations in Transgender Women Using Spironolactone: A Retrospective Chart Review. Endocrine Practice, 28(11), 1113–1117. [DOI:10.1016/j.eprac.2022.08.007]
Hammerstein, J. (1990). Antiandrogens: Clinical Aspects. In Orfanos, C. E., & Happle, R. (Eds.). Hair and Hair Diseases (pp. 827–886). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-74612-3_35]
Hayes, H., Russell, R., Haugen, A., Nagavally, S., & Sarvaideo, J. (2022). The Utility of Monitoring Potassium in Transgender, Gender Diverse, and Nonbinary Individuals on Spironolactone. Journal of the Endocrine Society, 6(11), bvac133. [DOI:10.1210/jendso/bvac133]
Heinemann, L. A., Will-Shahab, L., van Kesteren, P., Gooren, L. J., & (1997). Safety of Cyproterone Acetate: Report of Active Surveillance. Pharmacoepidemiology and Drug Safety, 6(3), 169–178. [DOI:10.1002/(sici)1099-1557(199705)6:3<169::aid-pds263>3.0.co;2-3]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of the Subcutaneous and Intramuscular Estradiol Regimens as Part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
Hilton, E. N., & Lundberg, T. R. (2020). Transgender Women in the Female Category of Sport: Perspectives on Testosterone Suppression and Performance Advantage. Sports Medicine, 51(2), 199–214. [DOI:10.1007/s40279-020-01389-3]
Hirshburg, J. M., Kelsey, P. A., Therrien, C. A., Gavino, A. C., & Reichenberg, J. S. (2016). Adverse Effects and Safety of 5-alpha Reductase Inhibitors (Finasteride, Dutasteride): A Systematic Review. The Journal of Clinical and Aesthetic Dermatology, 9(7), 56–62. [PubMed] [PubMed Central]
Hopkins, U., & Arias, C. Y. (2013). Large-volume IM injections: a review of best practices. Oncology Nurse Advisor, 4(1), 32–37. [Google Scholar] [URL] [PDF]
Horton, R. (1992). Dihydrotestosterone Is a Peripheral Paracrine Hormone. Journal of Andrology, 13(1), 23–27. [DOI:10.1002/j.1939-4640.1992.tb01621.x]
Huang, G., & Basaria, S. (2017). The Case for Androgens in Menopausal Women: When and How? In Pal, L., & Sayegh, R. A. (Eds.). Essentials of Menopause Management: A Case-Based Approach (pp. 173–196). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-42451-4_10]
Igo, J., & Visram, H. (2021). Testosterone Suppression With Injectable Estrogen Therapy Alone in Male to Female Transgender Patients. Canadian Journal of Diabetes, 45(7 Suppl), S40–S40 (abstract no. 114). [DOI:10.1016/j.jcjd.2021.09.124] [URL]
Ingram, B. J., & Thomas, C. L. (2019). Transgender Policy in Sport, A Review of Current Policy and Commentary of the Challenges of Policy Creation. Current Sports Medicine Reports, 18(6), 239–247. [DOI:10.1249/jsr.0000000000000605]
Irwig, M. S. (2020). Is there a role for 5α‐reductase inhibitors in transgender individuals? Andrology, 9(6), 1729–1731. [DOI:10.1111/andr.12881]
Iversen, P., Johansson, J. E., Lodding, P., Lukkarinen, O., Lundmo, P., Klarskov, P., Tammela, T. L., Tasdemir, I., Morris, T., Carroll, K., & Scandinavian Prostatic Cancer Group. (2004). Bicalutamide (150 mg) versus placebo as immediate therapy alone or as adjuvant to therapy with curative intent for early nonmetastatic prostate cancer: 5.3-year median followup from the Scandinavian Prostate Cancer Group Study Number 6. The Journal of Urology, 172(5 Part 1), 1871–1876. [DOI:10.1097/01.ju.0000139719.99825.54]
Iversen, P., Johansson, J., Lodding, P., Kylmälä, T., Lundmo, P., Klarskov, P., Tammela, T. L., Tasdemir, I., Morris, T., Armstrong, J., & (2006). Bicalutamide 150 mg in addition to standard care for patients with early non-metastatic prostate cancer Updated results from the Scandinavian Prostate Cancer Period Group-6 Study after a median follow-up period of 7.1 years. Scandinavian Journal of Urology and Nephrology, 40(6), 441–452. [DOI:10.1080/00365590601017329]
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 204201881987116. [DOI:10.1177/2042018819871166]
Iwamoto, S. J., Grimstad, F., Irwig, M. S., & Rothman, M. S. (2021). Routine Screening for Transgender and Gender Diverse Adults Taking Gender-Affirming Hormone Therapy: a Narrative Review. Journal of General Internal Medicine, 36(5), 1380–1389. [DOI:10.1007/s11606-021-06634-7]
Jacoby, A., Rifkin, W., Zhao, L. C., & Bluebond-Langner, R. (2020). Incidence of Cancer and Premalignant Lesions in Surgical Specimens of Transgender Patients. Plastic & Reconstructive Surgery, 147(1), 194–198. [DOI:10.1097/prs.0000000000007452]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
James, J. F., Jamerson, T. A., & Aguh, C. (2022). Efficacy and safety profile of oral spironolactone use for androgenic alopecia: A systematic review. Journal of the American Academy of Dermatology, 86(2), 425–429. [DOI:10.1016/j.jaad.2021.07.048]
Jia, A. Y., & Spratt, D. E. (2022). Bicalutamide Monotherapy With Radiation Therapy for Localized Prostate Cancer: A Non-Evidence-Based Alternative. International Journal of Radiation Oncology*Biology*Physics, 113(2), 316–319. [DOI:10.1016/j.ijrobp.2022.01.037]
Jiang, Y., & Tian, W. (2017). The effects of progesterones on blood lipids in hormone replacement therapy. Lipids in Health and Disease, 16(1), 219. [DOI:10.1186/s12944-017-0612-5]
Kanhai, R. C., Hage, J. J., van Diest, P. J., Bloemena, E., & Mulder, J. W. (2000). Short-Term and Long-Term Histologic Effects of Castration and Estrogen Treatment on Breast Tissue of 14 Male-to-Female Transsexuals in Comparison With Two Chemically Castrated Men. The American Journal of Surgical Pathology, 24(1), 74–80. [DOI:10.1097/00000478-200001000-00009]
Kellner, M., & Wiedemann, K. (2008). Mineralocorticoid receptors in brain, in health and disease: Possibilities for new pharmacotherapy. European Journal of Pharmacology, 583(2–3), 372–378. [DOI:10.1016/j.ejphar.2007.07.072]
Kim, G. K., & Del Rosso, J. Q. (2012). Oral Spironolactone in Post-teenage Female Patients with Acne Vulgaris: Practical Considerations for the Clinician Based on Current Data and Clinical Experience. The Journal of Clinical and Aesthetic Dermatology, 5(3), 37–50. [PubMed] [PubMed Central]
King, S. R. (2012). Neurosteroids and the Nervous System. In King, S. R. Neurosteroids and the Nervous System (pp. 1–122). New York: Springer New York. [DOI:10.1007/978-1-4614-5559-2_1]
Krishnamurthy, N., Slack, D., Kyweluk, M., Kirkley, J., Trakhtenberg, E., Contreras-Castro, F., & Safer, J. (2023). Not All Transfeminine Individuals on Estradiol Can Reach Both Target Testosterone and Target Estradiol Levels—Time to Revisit Treatment Guidelines? USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 94–94 (abstract no. SAT-B2-T4). [Symposium Schedule] [PDF] [Full Abstract Book]
Kolvenbag, G. J., & Blackledge, G. R. (1996). Worldwide activity and safety of bicalutamide: a summary review. Urology, 47(1), 70–79. [DOI:10.1016/s0090-4295(96)80012-4]
Kuhl, H. (2003). Östrogene für den Mann? [Estrogen for men?] Blickpunkt der Mann, 1(3), 6–12. [Google Scholar] [URL] [PDF]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H., & Wiegratz, I. (2017). Das Post-Finasterid-Syndrom. [Post Finasteride Syndrome.] Gynäkologische Endokrinologie, 15(2), 153–163. [DOI:10.1007/s10304-017-0126-2]
Kuijpers, S. M., Wiepjes, C. M., Conemans, E. B., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2021). Toward a Lowest Effective Dose of Cyproterone Acetate in Trans Women: Results From the ENIGI Study. The Journal of Clinical Endocrinology & Metabolism, 106(10), e3936–e3945. [DOI:10.1210/clinem/dgab427]
Kumar, P., Reddy, S., Kulkarni, A., Sharma, M., & Rao, P. N. (2021). Cyproterone Acetate–Induced Acute Liver Failure: A Case Report and Review of the Literature. Journal of Clinical and Experimental Hepatology, 11(6), 739–741. [DOI:10.1016/j.jceh.2021.01.003]
Lauritzen, C. (1988). Natürliche und synthetische Sexualhormone – Biologische Grundlagen und Behandlungsprinzipien. [Natural and Synthetic Sexual Hormones – Biological Basis and Medical Treatment Principles.] In Lauritzen, C., Schneider, H. P. G., & Nieschlag, E. (Eds.). Grundlagen und Klinik der Menschlichen Fortpflanzung [Foundations and Clinic of Human Reproduction] (pp. 229–306). Berlin: de Gruyter. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL] [PDF] [Translation]
Lax, E. (1987). Mechanisms of physiological and pharmacological sex hormone action on the mammalian liver. Journal of Steroid Biochemistry, 27(4–6), 1119–1128. [DOI:10.1016/0022-4731(87)90198-1]
Layton, A. M., Eady, E. A., Whitehouse, H., Del Rosso, J. Q., Fedorowicz, Z., & van Zuuren, E. J. (2017). Oral Spironolactone for Acne Vulgaris in Adult Females: A Hybrid Systematic Review. American Journal of Clinical Dermatology, 18(2), 169–191. [DOI:10.1007/s40257-016-0245-x]
Legro, R. S., Schlaff, W. D., Diamond, M. P., Coutifaris, C., Casson, P. R., Brzyski, R. G., Christman, G. M., Trussell, J. C., Krawetz, S. A., Snyder, P. J., Ohl, D., Carson, S. A., Steinkampf, M. P., Carr, B. R., McGovern, P. G., Cataldo, N. A., Gosman, G. G., Nestler, J. E., Myers, E. R., Santoro, N., Eisenberg, E., Zhang, M., & Zhang, H. (2010). Total Testosterone Assays in Women with Polycystic Ovary Syndrome: Precision and Correlation with Hirsutism. The Journal of Clinical Endocrinology & Metabolism, 95(12), 5305–5313. [DOI:10.1210/jc.2010-1123]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Liang, J. J., Jolly, D., Chan, K. J., & Safer, J. D. (2018). Testosterone Levels Achieved by Medically Treated Transgender Women in a United States Endocrinology Clinic Cohort. Endocrine Practice, 24(2), 135–142. [DOI:10.4158/ep-2017-0116]
Lobo, R. A., Shoupe, D., Serafini, P., Brinton, D., & Horton, R. (1985). The effects of two doses of spironolactone on serum androgens and anagen hair in hirsute women. Fertility and Sterility, 43(2), 200–205. [DOI:10.1016/s0015-0282(16)48373-1]
Loh, H. H., Yee, A., Loh, H. S., Kanagasundram, S., Francis, B., & Lim, L. (2020). Sexual dysfunction in polycystic ovary syndrome: a systematic review and meta-analysis. Hormones, 19(3), 413–423. [DOI:10.1007/s42000-020-00210-0]
Luong, J. H., Male, K. B., & Glennon, J. D. (2019). Biotin interference in immunoassays based on biotin-strept(avidin) chemistry: An emerging threat. Biotechnology Advances, 37(5), 634–641. [DOI:10.1016/j.biotechadv.2019.03.007]
MacLean, D. B., Shi, H., Faessel, H. M., & Saad, F. (2015). Medical Castration Using the Investigational Oral GnRH Antagonist TAK-385 (Relugolix): Phase 1 Study in Healthy Males. The Journal of Clinical Endocrinology & Metabolism, 100(12), 4579–4587. [DOI:10.1210/jc.2015-2770]
Mahler, C. (1996). A Review of the Clinical Studies with Nilutamide. In Denis, L. (Ed.). Antiandrogens in Prostate Cancer: A Key to Tailored Endocrine Treatment (ESO Monographs) (pp. 105–111). Berlin/Heidelberg: Springer Berlin Heidelberg. [DOI:10.1007/978-3-642-45745-6_10]
Maksym, R. B., Kajdy, A., & Rabijewski, M. (2019). Post-finasteride syndrome – does it really exist? The Aging Male, 22(4), 250–259. [DOI:10.1080/13685538.2018.1548589]
Marks, D. H., Prasad, S., De Souza, B., Burns, L. J., & Senna, M. M. (2019). Topical Antiandrogen Therapies for Androgenetic Alopecia and Acne Vulgaris. American Journal of Clinical Dermatology, 21(2), 245–254. [DOI:10.1007/s40257-019-00493-z]
Martinez-Martin, F. J., Kuzior, A., Hernandez-Lazaro, A., de Leon-Durango, R. J., Rios-Gomez, C., Santana-Ojeda, B., Perez-Rivero, J. M., Fernandez-Trujillo-Comenge, P. M., Gonzalez-Diaz, P., Arnas-Leon, C., Acosta-Calero, C., Perdomo-Herrera, E., Tocino-Hernandez, A. L., del Sol Sanchez-Bacaicoa, M., & del Pino Perez-Garcia, M. (2022). Incidence of hypertension in young transgender people after a 5-year follow-up: association with gender-affirming hormonal therapy. Hypertension Research, 46(1), 219–225. [DOI:10.1038/s41440-022-01067-z]
Masumori, N., Baba, T., Abe, T., & Niwa, K. (2021). What is the most anticipated change induced by treatment using gender‐affirming hormones in individuals with gender incongruence?. International Journal of Urology, 28(5), 526–529. [DOI:10.1111/iju.14499]
McFarlane, T., Zajac, J. D., & Cheung, A. S. (2018). Gender-affirming hormone therapy and the risk of sex hormone-dependent tumours in transgender individuals—A systematic review. Clinical Endocrinology, 89(6), 700–711. [DOI:10.1111/cen.13835]
Meriggiola, M. C., Bremner, W. J., Costantino, A., Bertaccini, A., Morselli-Labate, A. M., Huebler, D., Kaufmann, G., Oettel, M., & Flamigni, C. (2002). Twenty-One Day Administration of Dienogest Reversibly Suppresses Gonadotropins and Testosterone in Normal Men. The Journal of Clinical Endocrinology & Metabolism, 87(5), 2107–2113. [DOI:10.1210/jcem.87.5.8514]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463]
Miles, R. A., Paulson, R. J., Lobo, R. A., Press, M. F., Dahmoush, L., & Sauer, M. V. (1994). Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: a comparative study. Fertility and Sterility, 62(3), 485–490. [DOI:10.1016/s0015-0282(16)56935-0]
Millington, K., Liu, E., & Chan, Y. (2019). The Utility of Potassium Monitoring in Gender-Diverse Adolescents Taking Spironolactone. Journal of the Endocrine Society, 3(5), 1031–1038. [DOI:10.1210/js.2019-00030]
Millward, C. P., Keshwara, S. M., Islim, A. I., Jenkinson, M. D., Alalade, A. F., & Gilkes, C. E. (2022). Development and Growth of Intracranial Meningiomas in Transgender Women Taking Cyproterone Acetate as Gender-Affirming Progestogen Therapy: A Systematic Review. Transgender Health, 7(6), 473–483. [DOI:10.1089/trgh.2021.0025]
Moretti, C., Guccione, L., Di Giacinto, P., Simonelli, I., Exacoustos, C., Toscano, V., Motta, C., De Leo, V., Petraglia, F., & Lenzi, A. (2017). Combined Oral Contraception and Bicalutamide in Polycystic Ovary Syndrome and Severe Hirsutism: A Double-Blind Randomized Controlled Trial. The Journal of Clinical Endocrinology & Metabolism, 103(3), 824–838. [DOI:10.1210/jc.2017-01186]
Nakamoto, J. (2016). Endocrine Testing. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition (pp. 2655–2688.e1). Philadelphia: Saunders/Elsevier. [DOI:10.1016/B978-0-323-18907-1.00154-2]
Nakhjavani, M., Hamidi, S., Esteghamati, A., Abbasi, M., Nosratian-Jahromi, S., & Pasalar, P. (2009). Short term effects of spironolactone on blood lipid profile: a 3-month study on a cohort of young women with hirsutism. British Journal of Clinical Pharmacology, 68(4), 634–637. [DOI:10.1111/j.1365-2125.2009.03483.x]
Neyman, A., Fuqua, J. S., & Eugster, E. A. (2019). Bicalutamide as an Androgen Blocker With Secondary Effect of Promoting Feminization in Male-to-Female Transgender Adolescents. Journal of Adolescent Health, 64(4), 544–546. [DOI:10.1016/j.jadohealth.2018.10.296]
Nguyen, D., Marchese, M., Cone, E. B., Paciotti, M., Basaria, S., Bhojani, N., & Trinh, Q. (2021). Investigation of Suicidality and Psychological Adverse Events in Patients Treated With Finasteride. JAMA Dermatology, 157(1), 35–42. [DOI:10.1001/jamadermatol.2020.3385]
Nie, I., Wiepjes, C. M., Blok, C. J., Moorselaar, R. J., Pigot, G. L., Sluis, T. M., Barbé, E., Voorn, P., Mello, N. M., Huirne, J., & Heijer, M. (2021). Incidence of testicular cancer in trans women using gender‐affirming hormonal treatment: a nationwide cohort study. BJU International, 129(4), 491–497. [DOI:10.1111/bju.15575]
Nieschlag, E., Zitzmann, M., & Kamischke, A. (2003). Use of progestins in male contraception. Steroids, 68(10–13), 965–972. [DOI:10.1016/s0039-128x(03)00135-1]
Nieschlag, E. (2010). Clinical trials in male hormonal contraception. Contraception, 82(5), 457–470. [DOI:10.1016/j.contraception.2010.03.020]
Nieschlag, E., & Behre, H. M. (2012). The essential role of testosterone in hormonal male contraception. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Testosterone: Action · Deficiency · Substitution, 4th Edition (pp. 470–493). Cambridge/New York: Cambridge University Press. [DOI:10.1017/cbo9781139003353.023]
Nishiyama, T. (2014). Serum testosterone levels after medical or surgical androgen deprivation: A comprehensive review of the literature. Urologic Oncology: Seminars and Original Investigations, 32(1), 38.e17–38.e28. [DOI:10.1016/j.urolonc.2013.03.007]
Nolan, B. J., & Cheung, A. S. (2021). Relationship Between Serum Estradiol Concentrations and Clinical Outcomes in Transgender Individuals Undergoing Feminizing Hormone Therapy: A Narrative Review. Transgender Health, 6(3), 125–131. [DOI:10.1089/trgh.2020.0077]
Nolan, B. J., Frydman, A. S., Leemaqz, S. Y., Carroll, M., Grossmann, M., Zajac, J. D., & Cheung, A. S. (2022). Effects of low-dose oral micronised progesterone on sleep, psychological distress, and breast development in transgender individuals undergoing feminising hormone therapy: a prospective controlled study. Endocrine Connections, 11(5), e220170. [DOI:10.1530/EC-22-0170]
Norman, A. W., & Henry, H. L. (2015). Androgens. In Norman, A. W., & Henry, H. L. Hormones, 3rd Edition (pp. 255–273). London: Academic Press/Elsevier. [DOI:10.1016/b978-0-08-091906-5.00012-4]
Norman, A. W., & Henry, H. L. (2015). Estrogens and Progestins. In Norman, A. W., & Henry, H. L. Hormones, 3rd Edition (pp. 275–296). London: Academic Press/Elsevier. [DOI:10.1016/b978-0-08-091906-5.00013-6]
Nota, N. M., Wiepjes, C. M., de Blok, C. J., Gooren, L. J., Peerdeman, S. M., Kreukels, B. P., & den Heijer, M. (2018). The occurrence of benign brain tumours in transgender individuals during cross-sex hormone treatment. Brain, 141(7), 2047–2054. [DOI:10.1093/brain/awy108]
North American Menopause Society. (2022). The 2022 hormone therapy position statement of The North American Menopause Society. Menopause, 29(7), 767–794. [DOI:10.1097/gme.0000000000002028]
O’Connell, M. B. (1995). Pharmacokinetic and Pharmacologic Variation Between Different Estrogen Products. The Journal of Clinical Pharmacology, 35(9S), 18S–24S. [DOI:10.1002/j.1552-4604.1995.tb04143.x]
Olsen, E. A., Hordinsky, M., Whiting, D., Stough, D., Hobbs, S., Ellis, M. L., Wilson, T., & Rittmaster, R. S. (2006). The importance of dual 5α-reductase inhibition in the treatment of male pattern hair loss: Results of a randomized placebo-controlled study of dutasteride versus finasteride. Journal of the American Academy of Dermatology, 55(6), 1014–1023. [DOI:10.1016/j.jaad.2006.05.007]
Plovanich, M., Weng, Q. Y., & Mostaghimi, A. (2015). Low Usefulness of Potassium Monitoring Among Healthy Young Women Taking Spironolactone for Acne. JAMA Dermatology, 151(9), 941–944. [DOI:10.1001/jamadermatol.2015.34]
Polyzos, S. A., Kountouras, J., Zavos, C., & Deretzi, G. (2011). Spironolactone Revisited. The Journal of Clinical Hypertension, 13(10), 783–784. [DOI:10.1111/j.1751-7176.2011.00484.x]
Powers, M. S., Schenkel, L., Darley, P. E., Good, W. R., Balestra, J. C., & Place, V. A. (1985). Pharmacokinetics and pharmacodynamics of transdermal dosage forms of 17β-estradiol: Comparison with conventional oral estrogens used for hormone replacement. American Journal of Obstetrics and Gynecology, 152(8), 1099–1106. [DOI:10.1016/0002-9378(85)90569-1]
Prince, J. C., & Safer, J. D. (2020). Endocrine treatment of transgender individuals: current guidelines and strategies. Expert Review of Endocrinology & Metabolism, 15(6), 395–403. [DOI:10.1080/17446651.2020.1825075]
Reiter, E. O., Mauras, N., McCormick, K., Kulshreshtha, B., Amrhein, J., De Luca, F., O’Brien, S., Armstrong, J., & Melezinkova, H. (2010). Bicalutamide plus Anastrozole for the Treatment of Gonadotropin-Independent Precocious Puberty in Boys with Testotoxicosis: A Phase II, Open-Label Pilot Study (BATT). Journal of Pediatric Endocrinology and Metabolism, 23(10), 999–1009. [DOI:10.1515/jpem.2010.161]
Rezende, H. D., Dias, M. F. R. G., & Trüeb, R. M. (2018). A Comment on the Post-Finasteride Syndrome. International Journal of Trichology, 10(6), 255–261. [PubMed] [PubMed Central] [DOI:10.4103/ijt.ijt_61_18]
Roberts, J. L., Fiedler, V., Imperato-McGinley, J., Whiting, D., Olsen, E., Shupack, J., Stough, D., DeVillez, R., Rietschel, R., Savin, R., Bergfeld, W., Swinehart, J., Funicella, T., Hordinsky, M., Lowe, N., Katz, I., Lucky, A., Drake, L., Price, V. H., Weiss, D., Whitmore, E., Millikan, L., Muller, S., Gencheff, C., Carrington, P., Binkowitz, B., Kotey, P., He, W., Bruno, K., Jacobsen, C., Terranella, L., Gormley, G. J., & Kaufman, K. D. (1999). Clinical dose ranging studies with finasteride, a type 2 5α-reductase inhibitor, in men with male pattern hair loss. Journal of the American Academy of Dermatology, 41(4), 555–563. [DOI:10.1016/s0190-9622(99)80052-8]
Roscioni, S. S., de Zeeuw, D., Bakker, S. J., & Lambers Heerspink, H. J. (2012). Management of hyperkalaemia consequent to mineralocorticoid-receptor antagonist therapy. Nature Reviews Nephrology, 8(12), 691–699. [DOI:10.1038/nrneph.2012.217]
Rose, A. J., Hughto, J. M., Dunbar, M. S., Quinn, E. K., Deutsch, M., Feldman, J., Radix, A., Safer, J. D., Shipherd, J. C., Thompson, J., & Jasuja, G. K. (2023). Trends in Feminizing Hormone Therapy for Transgender Patients, 2006–2017. Transgender Health, 8(2), 188–194. [DOI:10.1089/trgh.2021.0041]
Rosenfield, R. L., Cooke, D. W., & Radovick, S. (2008). Puberty and its disorders in the female. In Sperling, M. A. (Ed.). Pediatric Endocrinology, 3rd Edition (pp. 530–609). Philadelphia: Saunders. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Rosenfield, R. L., Cooke, D. W., & Radovick, S. (2021). Puberty in the Female and Its Disorders. In Sperling, M. A., Majzoub, J. A., Menon, R. K., & Stratakis, C. A. (Eds.). Sperling Pediatric Endocrinology, 5th Edition (pp. 528–626). Philadelphia: Elsevier. [DOI:10.1016/B978-0-323-62520-3.00016-6]
Samarasinghe, S., Meah, F., Singh, V., Basit, A., Emanuele, N., Emanuele, M. A., Mazhari, A., & Holmes, E. W. (2017). Biotin Interference with Routine Clinical Immunoassays: Understand the Causes and Mitigate the Risks. Endocrine Practice, 23(8), 989–998. [DOI:10.4158/ep171761.ra]
Schiller, C. E., Schmidt, P. J., & Rubinow, D. R. (2014). Allopregnanolone as a mediator of affective switching in reproductive mood disorders. Psychopharmacology, 231(17), 3557–3567. [DOI:10.1007/s00213-014-3599-x]
Schlatterer, K., Yassouridis, A., Werder, K. V., Poland, D., Kemper, J., & Stalla, G. K. (1998). Archives of Sexual Behavior, 27(5), 475–492. [DOI:10.1023/a:1018704630036]
Seaman, H. E., Langley, S. E., Farmer, R. D., & de Vries, C. S. (2007). Venous thromboembolism and cyproterone acetate in men with prostate cancer: a study using the General Practice Research Database. BJU International, 99(6), 1398–1403. [DOI:10.1111/j.1464-410x.2007.06859.x]
Shackleton, C. (2010). Clinical steroid mass spectrometry: A 45-year history culminating in HPLC–MS/MS becoming an essential tool for patient diagnosis. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 481–490. [DOI:10.1016/j.jsbmb.2010.02.017]
Shaw, J. C. (1996). Antiandrogen and hormonal treatment of acne. Dermatologic Clinics, 14(4), 803–811. [DOI:10.1016/s0733-8635(05)70405-8]
Slack, D., Krishnamurthy, N., Contreras-Castro, F., & Safer, J. D. (2023). Achieving Both Target Testosterone And Target Estradiol Levels May Not Be Practical In All Trans Feminine Users Of Estradiol - Which To Prioritize? Journal of the Endocrine Society, 7(Suppl 1), A1102–A1102 (abstract no. SAT404/bvad114.2075). [DOI:10.1210/jendso/bvad114.2075] [PubMed Central]
Sofer, Y., Yaish, I., Yaron, M., Bach, M. Y., Stern, N., & Greenman, Y. (2020). Differential Endocrine and Metabolic Effects of Testosterone Suppressive Agents in Transgender Women. Endocrine Practice, 26(8), 883–890. [DOI:10.4158/ep-2020-0032]
SoRelle, J. A., Jiao, R., Gao, E., Veazey, J., Frame, I., Quinn, A. M., Day, P., Pagels, P., Gimpel, N., & Patel, K. (2019). Impact of Hormone Therapy on Laboratory Values in Transgender Patients. Clinical Chemistry, 65(1), 170–179. [DOI:10.1373/clinchem.2018.292730]
St-Jules, D. E., Goldfarb, D. S., & Sevick, M. A. (2016). Nutrient Non-equivalence: Does Restricting High-Potassium Plant Foods Help to Prevent Hyperkalemia in Hemodialysis Patients? Journal of Renal Nutrition, 26(5), 282–287. [DOI:10.1053/j.jrn.2016.02.005]
St-Jules, D. E., & Fouque, D. (2020). Is it time to abandon the nutrient-based renal diet model? Nephrology Dialysis Transplantation, 36(4), 574–577. [DOI:10.1093/ndt/gfaa257]
St-Jules, D. E., & Fouque, D. (2022). Etiology-based dietary approach for managing hyperkalemia in people with chronic kidney disease. Nutrition Reviews, 80(11), 2198–2205. [DOI:10.1093/nutrit/nuac026]
Stanczyk, F. Z., & Clarke, N. J. (2010). Advantages and challenges of mass spectrometry assays for steroid hormones. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 491–495. [DOI:10.1016/j.jsbmb.2010.05.001]
Stanczyk, F. Z., Hapgood, J. P., Winer, S., & Mishell, D. R. (2012). Progestogens Used in Postmenopausal Hormone Therapy: Differences in Their Pharmacological Properties, Intracellular Actions, and Clinical Effects. Endocrine Reviews, 34(2), 171–208. [DOI:10.1210/er.2012-1008]
Stanczyk, F. Z., Mathews, B. W., & Cortessis, V. K. (2017). Does the type of progestin influence the production of clotting factors? Contraception, 95(2), 113–116. [DOI:10.1016/j.contraception.2016.07.007]
Stege, R., Gunnarsson, P. O., Johansson, C., Olsson, P., Pousette, Å., & Carlström, K. (1996). Pharmacokinetics and testosterone suppression of a single dose of polyestradiol phosphate (Estradurin®) in prostatic cancer patients. The Prostate, 28(5), 307–310. [DOI:10.1002/(sici)1097-0045(199605)28:5<307::aid-pros6>3.0.co;2-8]
Steinberger, E., Ayala, C., Hsi, B., Smith, K. D., Rodriguez-Rigau, L. J., Weidman, E. R., & Reimondo, G. G. (1998). Utilization of Commercial Laboratory Results in Management of Hyperandrogenism in Women. Endocrine Practice, 4(1), 1–10. [DOI:10.4158/ep.4.1.1]
Sterling, J., & Garcia, M. M. (2020). Cancer screening in the transgender population: a review of current guidelines, best practices, and a proposed care model. Translational Andrology and Urology, 9(6), 2771–2785. [DOI:10.21037/tau-20-954]
Strauss, J. F., & FitzGerald, G. A. (2019). Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 75–114.e7). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/b978-0-323-47912-7.00004-4]
Stricker, R., Eberhart, R., Chevailler, M., Quinn, F. A., Bischof, P., & Stricker, R. (2006). Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT® analyzer. Clinical Chemistry and Laboratory Medicine (CCLM), 44(7), 883–887. [DOI:10.1515/cclm.2006.160]
Styne, D. M. (2016). Laboratory Values for Pediatric Endocrinology. In Styne, D. M. Pediatric Endocrinology: A Clinical Handbook (pp. 385–434). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-18371-8_16]
Sudduth, S. L., & Koronkowski, M. J. (1993). Finasteride: The First 5α-Reductase Inhibitor. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 13(4), 309–329. [DOI:10.1002/j.1875-9114.1993.tb02739.x]
Sundström-Poromaa, I., Comasco, E., Sumner, R., & Luders, E. (2020). Progesterone – Friend or foe? Frontiers in Neuroendocrinology, 59, 100856. [DOI:10.1016/j.yfrne.2020.100856]
Swerdloff, R. S., Dudley, R. E., Page, S. T., Wang, C., & Salameh, W. A. (2017). Dihydrotestosterone: Biochemistry, Physiology, and Clinical Implications of Elevated Blood Levels. Endocrine Reviews, 38(3), 220–254. [DOI:10.1210/er.2016-1067]
T’Sjoen, G. G., Beguin, Y., Feyen, E., Rubens, R., Kaufman, J., & Gooren, L. (2005). Influence of exogenous oestrogen or (anti-) androgen administration on soluble transferrin receptor in human plasma. Journal of Endocrinology, 186(1), 61–67. [DOI:10.1677/joe.1.06112]
T’Sjoen, G., Arcelus, J., Gooren, L., Klink, D. T., & Tangpricha, V. (2018). Endocrinology of Transgender Medicine. Endocrine Reviews, 40(1), 97–117. [DOI:10.1210/er.2018-00011]
Tack, L. J., Heyse, R., Craen, M., Dhondt, K., Bossche, H. V., Laridaen, J., & Cools, M. (2017). Consecutive Cyproterone Acetate and Estradiol Treatment in Late-Pubertal Transgender Female Adolescents. The Journal of Sexual Medicine, 14(5), 747–757. [DOI:10.1016/j.jsxm.2017.03.251]
Talathi, R., Juhasz, V., Delgado, M., Quinaglia, T., Ghamari, A., Wang, M., Alhallak, I., Stinebaugh, S., Campbell, S., Stockman, S. L., Ozturk, M. A., Ahmadi, S. M., Looby, S. E., Lee, H., Poteat, T. C., Szczepaniak, L. S., Zanni, M. V., Neilan, T. G., & Toribio, M. (2025). Visceral adipose tissue and liver fat on 17-beta estradiol-dominant gender-affirming hormone therapy: A US-based cohort. The Journal of Clinical Endocrinology and Metabolism, online ahead of print. [DOI:10.1210/clinem/dgaf665]
Thompson, J., Hopwood, R. A., deNormand, S., & Cavanaugh, T. (2021). Medical Care of Trans and Gender Diverse Adults. Boston: Fenway Health. [URL] [PDF]
Toorians, A. W., Thomassen, M. C., Zweegman, S., Magdeleyns, E. J., Tans, G., Gooren, L. J., & Rosing, J. (2003). Venous Thrombosis and Changes of Hemostatic Variables during Cross-Sex Hormone Treatment in Transsexual People. The Journal of Clinical Endocrinology & Metabolism, 88(12), 5723–5729. [DOI:10.1210/jc.2003-030520]
Traish, A. M., Krakowsky, Y., Doros, G., & Morgentaler, A. (2019). Do 5α-Reductase Inhibitors Raise Circulating Serum Testosterone Levels? A Comprehensive Review and Meta-Analysis to Explaining Paradoxical Results. Sexual Medicine Reviews, 7(1), 95–114. [DOI:10.1016/j.sxmr.2018.06.002]
Traish, A. M. (2020). Post-finasteride syndrome: a surmountable challenge for clinicians. Fertility and Sterility, 113(1), 21–50. [DOI:10.1016/j.fertnstert.2019.11.030]
Treleaven, M. M., Jackowich, R. A., Roberts, L., Wassersug, R. J., & Johnson, T. (2013). Castration and personality: Correlation of androgen deprivation and estrogen supplementation with the Big Five factor personality traits of adult males. Journal of Research in Personality, 47(4), 376–379. [DOI:10.1016/j.jrp.2013.03.005]
Usach, I., Martinez, R., Festini, T., & Peris, J. (2019). Subcutaneous Injection of Drugs: Literature Review of Factors Influencing Pain Sensation at the Injection Site. Advances in Therapy, 36(11), 2986–2996. [DOI:10.1007/s12325-019-01101-6]
van Zuuren, E. J., Fedorowicz, Z., Carter, B., & Pandis, N. (2015). Interventions for hirsutism (excluding laser and photoepilation therapy alone). Cochrane Database of Systematic Reviews, 2015, CD010334. [DOI:10.1002/14651858.cd010334.pub2]
Verdonk, S. J., Vesper, H. W., Martens, F., Sluss, P. M., Hillebrand, J. J., & Heijboer, A. C. (2019). Estradiol reference intervals in women during the menstrual cycle, postmenopausal women and men using an LC-MS/MS method. Clinica Chimica Acta, 495, 198–204. [DOI:10.1016/j.cca.2019.04.062]
Vermeulen, A., Giagulli, V., De Schepper, P., & Buntinx, A. (1991). Hormonal Effects of a 5α-Reductase Inhibitor (Finasteride) onHormonal Levels in Normal Men and in Patients withBenign Prostatic Hyperplasia. European Urology, 20(1), 82–86. [DOI:10.1159/000471752]
Vihtamäki, T., Luukkaala, T., & Tuimala, R. (2004). Skin contamination by oestradiol gel—a remarkable source of error in plasma oestradiol measurements during percutaneous hormone replacement therapy. Maturitas, 48(4), 347–353. [DOI:10.1016/s0378-5122(03)00043-4]
Wang, H., Liu, M., Fu, Q., & Deng, C. (2019). Pharmacokinetics of hard micronized progesterone capsules via vaginal or oral route compared with soft micronized capsules in healthy postmenopausal women: a randomized open-label clinical study. Drug Design, Development and Therapy, 13, 2475–2482. [DOI:10.2147/dddt.s204624]
Wang, Y., & Lipner, S. R. (2020). Retrospective analysis of adverse events with spironolactone in females reported to the United States Food and Drug Administration. International Journal of Women’s Dermatology, 6(4), 272–276. [DOI:10.1016/j.ijwd.2020.05.002]
Weill, A., Nguyen, P., Labidi, M., Cadier, B., Passeri, T., Duranteau, L., Bernat, A., Yoldjian, I., Fontanel, S., Froelich, S., & Coste, J. (2021). Use of High Dose Cyproterone Acetate and Risk of Intracranial Meningioma in Women: Cohort Study. BMJ, 372, n37. [DOI:10.1136/bmj.n37]
Welk, B., McArthur, E., Ordon, M., Anderson, K. K., Hayward, J., & Dixon, S. (2017). Association of Suicidality and Depression With 5α-Reductase Inhibitors. JAMA Internal Medicine, 177(5), 683–691. [DOI:10.1001/jamainternmed.2017.0089]
Wellington, K., & Keam, S. J. (2006). Bicalutamide 150mg: A Review of its Use in the Treatment of Locally Advanced Prostate Cancer. Drugs, 66(6), 837–850. [DOI:10.2165/00003495-200666060-00007]
Willemsen, W. N., Mastboom, J. L., Thomas, C. M., & Rolland, R. (1985). Absorption of 17β-estradiol in a neovagina constructed from the peritoneum. European Journal of Obstetrics & Gynecology and Reproductive Biology, 19(4), 247–253. [DOI:10.1016/0028-2243(85)90036-x]
Wilson, L. M., Baker, K. E., Sharma, R., Dukhanin, V., McArthur, K., & Robinson, K. A. (2020). Effects of antiandrogens on prolactin levels among transgender women on estrogen therapy: A systematic review. International Journal of Transgender Health, 21(4), 391–402. [DOI:10.1080/15532739.2020.1819505]
Wu, B., Shen, P., Yin, X., Yu, L., Wu, F., Chen, C., Li, J., & Xu, T. (2022). Analysis of adverse event of interstitial lung disease in men with prostate cancer receiving hormone therapy using the Food and Drug Administration Adverse Event Reporting System. British Journal of Clinical Pharmacology, 89(2), 440–448. [DOI:10.1111/bcp.15336]
Wu, F. C., Balasubramanian, R., Mulders, T. M., & Coelingh-Bennink, H. J. (1999). Oral Progestogen Combined with Testosterone as a Potential Male Contraceptive: Additive Effects between Desogestrel and Testosterone Enanthate in Suppression of Spermatogenesis, Pituitary-Testicular Axis, and Lipid Metabolism. The Journal of Clinical Endocrinology & Metabolism, 84(1), 112–122. [DOI:10.1210/jcem.84.1.5412]
Yaish, I., Gindis, G., Greenman, Y., Moshe, Y., Arbiv, M., Buch, A., Sofer, Y., Shefer, G., & Tordjman, K. (2023). Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study. Transgender Health, 8(6), 485–493. [DOI:10.1089/trgh.2023.0022]
Zaenglein, A. L., Pathy, A. L., Schlosser, B. J., Alikhan, A., Baldwin, H. E., Berson, D. S., Bowe, W. P., Graber, E. M., Harper, J. C., Kang, S., Keri, J. E., Leyden, J. J., Reynolds, R. V., Silverberg, N. B., Stein Gold, L. F., Tollefson, M. M., Weiss, J. S., Dolan, N. C., Sagan, A. A., Stern, M., Boyer, K. M., & Bhushan, R. (2016). Guidelines of care for the management of acne vulgaris. Journal of the American Academy of Dermatology, 74(5), 945–973.e33. [DOI:10.1016/j.jaad.2015.12.037]
Zhang, J., & Stanczyk, F. Z. (2013). LC-MS Bioanalysis of Steroids. In Li, W., Zhang, J., & Tse, F. L. S. (Eds.). Handbook of LC-MS Bioanalysis: Best Practices, Experimental Protocols, and Regulations (pp. 573–590). Hoboken, New Jersey: John Wiley & Sons. [DOI:10.1002/9781118671276.ch45]
Zhou, Z., Song, S., Gao, Z., Wu, J., Ma, J., & Cui, Y. (2019). The efficacy and safety of dutasteride compared with finasteride in treating men with androgenetic alopecia: a systematic review and meta-analysis. Clinical Interventions in Aging, 14, 399–406. [DOI:10.2147/cia.s192435]
Zitzmann, M., Rohayem, J., Raidt, J., Kliesch, S., Kumar, N., Sitruk-Ware, R., & Nieschlag, E. (2017). Impact of various progestins with or without transdermal testosterone on gonadotropin levels for non-invasive hormonal male contraception: a randomized clinical trial. Andrology, 5(3), 516–526. [DOI:10.1111/andr.12328]
Zuuren, E., & Fedorowicz, Z. (2016). Interventions for hirsutism excluding laser and photoepilation therapy alone: abridged Cochrane systematic review including GRADE assessments. British Journal of Dermatology, 175(1), 45–61. [DOI:10.1111/bjd.14486]
\ No newline at end of file
+An Introduction to Hormone Therapy for Transfeminine People - Transfeminine Science
An Introduction to Hormone Therapy for Transfeminine People
By Aly | First published August 4, 2018 | Last modified December 17, 2025
Abstract / TL;DR
Sex hormones such as estrogen, testosterone, and progesterone are produced by the gonads. The sex hormones mediate the development of the secondary sexual characteristics. Testosterone causes masculinization, while estradiol causes feminization and breast development. Males have high amounts of testosterone, while females have low testosterone but high amounts of estradiol. These hormonal differences are responsible for the physical differences between males and females. Sex hormones and other hormonal medications are used in transfeminine people to shift the hormonal profile from a male-typical one to a female-typical profile. This causes feminization and demasculinization and allows for alleviation of gender dysphoria. The changes caused by transfeminine hormone therapy occur over a period of months to years. There are many different types and forms of hormonal medications, and these medications can be administered by a variety of different routes. Examples include as pills taken by mouth, as patches or gel applied to the skin, and as injections, among others. Different hormonal medications, routes, and doses have differences in efficacy, side effects, risks, costs, convenience, and availability. Hormone therapy should ideally be regularly monitored in transfeminine people with blood tests to ensure effectiveness and safety and to allow for adjustment as necessary.
The Sex Hormones
Types and Effects
The sex hormones include the estrogens (E), progestogens (P), and androgens. A person’s hormonal profile is a product of the type of gonads that they are born with. Natal males have testes while natal females have ovaries. Testes produce large amounts of androgens and small amounts of estrogens whereas ovaries produce high amounts of estrogens and progesterone and low amounts of androgens.
Progestogens have essentially no known role in feminization or pubertal breast development. Rather than acting as mediators of feminization, progestogens have important effects in the female reproductive system and are essential hormones during pregnancy (Wiki). They also oppose the actions of estrogens in certain parts of the body, such as the uterus, vagina, and breasts (Wiki).
In addition to their effects on the body, sex hormones have actions in the brain. These actions influence cognition, emotions, and behavior. For instance, androgens produce pronounced sexual desire and arousal (including spontaneous erections) in men, while estrogens appear to be the major hormones responsible for sexual desire in women (Cappelletti & Wallen, 2016). As another example, testosterone levels have been negatively associated with agreeableness, whereas estrogen levels have been positively associated with this characteristic (Treleaven et al., 2013). Sex hormones also have important effects on health, which can be both positive and negative. For instance, estrogens maintain bone strength and likely protect against heart disease in cisgender women (NAMS, 2022), but also increase the risk of breast cancer (Aly, 2020) and can increase the risk of blood clots (Aly, 2020).
Estrogens, progestogens, and androgens also have antigonadotropic effects. That is, they inhibit the gonadotropin-releasing hormone (GnRH)-induced secretion of the gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), from the pituitary gland in the brain. The gonadotropins signal the gonads to make sex hormones and to supply the sperm and egg cells necessary for fertility. Hence, lower levels of the gonadotropins will result in reduced gonadal sex hormone production and diminished fertility. If gonadotropin levels are sufficiently suppressed, the gonads will no longer make sex hormones at all and fertility will cease. The vast majorities of the quantities of estradiol, testosterone, and progesterone in the body are produced by the gonads. Most of the small remaining amounts of these hormones are produced via the adrenal glands of the kidneys.
Normal Hormone Levels
In cisgender females, the sex hormones are largely absent during childhood, gradually ramp up in production in late childhood and adolescence, are present in a cyclical manner during adulthood, and then largely stop being produced following the menopause. Hormone levels vary substantially but in a predictable manner during the normal menstrual cycle in adult premenopausal women. The menstrual cycle lasts about 28 days on average and consists of the following parts:
Luteal phase—latter half of the cycle or days 14–28
Hormone levels during the menstrual cycle are shown in the following graph:
Figure 1: Median estradiol and progesterone levels throughout the menstrual cycle in premenopausal cisgender women (Stricker et al., 2006; Abbott, 2009). The horizontal dashed lines are the average levels over the spanned periods. Other figures available elsewhere show variation between individuals (Graph; Graph; Graph).
As can be seen in the graph, estradiol levels are relatively low and progesterone levels are very low during the follicular phase; estradiol but not progesterone levels briefly surge to very high levels and trigger ovulation during mid-cycle; and estradiol and progesterone levels both undergo a bump and are relatively high during the luteal phase (though estradiol is not as high as during the mid-cycle peak).
The table below shows the circulating levels and production rates of estradiol, progesterone, and testosterone in women and men and allows for comparison between them.
Table 1: Ranges for circulating levelsa and estimated production ratesb of the major sex hormones:
Mean integrated estradiol levels are around 100 pg/mL (367 pmol/L) in premenopausal women and around 25 pg/mL (92 pmol/L) in men. The 95% range for mean estradiol levels in women is around 50 to 250 pg/mL (180–918 pmol/L) (e.g., Abbott, 2009 (Graph); Verdonk et al., 2019 (Graph)). The average production of estradiol by the ovaries in premenopausal women is about 6 mg over the course of one menstrual cycle (i.e., one month) (Rosenfield et al., 2008). This corresponds to a mean rate of about 200 μg/day. Estradiol levels increase slowly during normal female puberty, when breast development and feminization take place. Mean estradiol levels during the different stages of female puberty are quite low—less than about 50 to 60 pg/mL (180–220 pmol/L) until late puberty (Aly, 2020). In postmenopausal women, whose ovaries no longer produce considerable quantities of estrogens, estradiol levels are generally less than 10 to 20 pg/mL (37–73 pmol/L) (Nakamoto, 2016). Estradiol levels below 50 pg/mL (184 pmol/L) in adults are concentration-dependently associated with menopausal symptoms, including hot flashes, depressive mood changes, defeminization (e.g., breast atrophy, loss of feminine fat distribution), accelerated skin aging, and bone density loss with increased risk of bone fracture.
Mean testosterone levels are around 30 ng/dL (1.0 nmol/L) in women and 600 ng/dL (21 nmol/L) in men. Based on these values, testosterone levels are on average about 20-fold higher in men than in women. In men who have undergone gonadectomy (castration or surgical gonadal removal), testosterone levels are similar to those in women (<50 ng/dL [1.7 nmol/L]) (Nishiyama, 2014; Getzenberg & Itty, 2020). The mean or median levels of testosterone in women with polycystic ovary syndrome (PCOS), who often have clinically significant symptoms of androgen excess (e.g., excessive facial/body hair growth), range from 41 to 75 ng/dL (1.4–2.6 nmol/L) per different studies (Balen et al., 1995; Steinberger et al., 1998; Legro et al., 2010; Loh et al., 2020). Hence, it appears that even testosterone levels that are marginally elevated relative to normal female levels may produce undesirable androgenic effects.
The goal of hormone therapy for transfeminine people, otherwise known as feminizing hormone therapy (FHT) or (more in the past) as male-to-female (MtF) hormone replacement therapy (HRT), is to produce feminization and demasculinization of the body as well as alleviation of gender dysphoria. Medication therapy with sex hormones and other sex-hormonal medications is used to mediate these changes. Transfeminine people are given estrogens, progestogens, and antiandrogens (AAs) to supersede gonadal sex hormone production and shift the hormonal profile from male-typical to female-typical.
Transfeminine hormone therapy aims to achieve estradiol and testosterone levels within the normal female range. Commonly recommended ranges for transfeminine people in the literature are 100 to 200 pg/mL (367–734 pmol/L) for estradiol levels and less than 50 ng/dL (1.7 nmol/L) for testosterone levels (Table). However, higher estradiol levels of more than 200 pg/mL (734 pmol/L) can be useful in transfeminine hormone therapy to help suppress testosterone levels. Lower estradiol levels (≤50–60 pg/mL [≤180–220 pmol/L]) are recommended and more appropriate for pubertal and adolescent transfeminine individuals. Sex hormone levels in the blood can be measured with blood tests, in which blood is drawn from a vein using a needle and then analyzed in a laboratory. This is useful in transfeminine people to ensure that the hormonal profile has been satisfactorily altered in line with therapeutic goals—specifically that hormone levels are within female ranges.
Gonadal Suppression
At sufficiently high exposure, estrogens and androgens are able to completely suppress gonadal sex hormone production, while progestogens by themselves are able to partially but substantially suppress gonadal sex hormone production. More specifically, studies in cisgender men and transfeminine people have found that estradiol levels of around 200 pg/mL (734 pmol/L) suppress testosterone levels by about 90% on average (to ~50 ng/dL [1.7 nmol/L]), while estradiol levels of around 500 pg/mL (1,840 pmol/L) suppress testosterone levels by about 95% on average (to ~20–30 ng/dL [0.7–1.0 nmol/L]) (Gooren et al., 1984 [Graph]; Herndon et al., 2023 [Discussion]; Wiki; Graphs). Estradiol levels of below 200 pg/mL (734 pmol/L) also suppress testosterone levels, although to a reduced extent compared to higher levels (Aly, 2019; Krishnamurthy et al., 2023; Slack et al., 2023). In one large study in transfeminine people, the rates of adequate testosterone suppression (to testosterone levels of <50 ng/dL or <1.7 nmol/L) were 24% of individuals at estradiol levels of <100 pg/mL (367 pmol/L), 58% at 100 to 200 pg/mL (367–734 pmol/L), and 77% at >200 pg/mL (>734 pmol/L) (Krishnamurthy et al., 2023).
Figure 2: Estradiol and testosterone levels after a single injection of 320 mg polyestradiol phosphate (PEP) (a long-acting prodrug of estradiol) in men with prostate cancer (Stege et al., 1996). The maximal decrease in testosterone levels occurred with estradiol levels of greater than 200 pg/mL (734 pmol/L) and was about 90% (to roughly 50 ng/dL [1.7 nmol/L]). This figure demonstrates the ability of estradiol to concentration-dependently suppress gonadal testosterone production and circulating testosterone levels in people with testes.
Progestogens on their own are able to maximally suppress testosterone levels by about 50 to 70% (to ~150–300 ng/dL [5.2–10.4 nmol/L] on average) (Aly, 2019; Wiki). In combination with relatively small amounts of estrogen however, there is synergism in the antigonadotropic effect—the suppression of gonadal testosterone production with maximally effective doses of progestogens becomes complete, and testosterone levels are reduced by about 95% (to ~20–30 ng/dL [0.7–1.0 nmol/L]) (Aly, 2019). Hence, the combination of an estrogen and a progestogen can be used to achieve maximal testosterone suppression at lower doses than would be necessary if an estrogen or progestogen were used alone.
The antigonadotropic effects of estrogens and progestogens are taken advantage of in transfeminine hormone therapy to suppress gonadal testosterone production and attain testosterone levels that are more consistent with those in cisgender women. It should be noted that the preceding numbers on testosterone suppression with estrogens and progestogens are averages and there is significant variation between individuals in terms of testosterone suppression. In other words, some may need more or less in terms of hormonal dosages to achieve the same decrease in testosterone levels.
Effects and Timeline
During normal puberty in both males and females, sex hormone exposure increases slowly over a period of several years (Aly, 2020). In relation to this, sexual maturation occurs gradually during normal puberty. In non-adolescent transgender people, adult or higher amounts of hormones are generally administered right away, and this can result in changes in secondary sex characteristics happening more quickly. Most of the effects of feminizing hormone therapy in transfeminine people onset within 1 to 6 months of commencing treatment and complete within 1 to 3 years. The table below is reproduced from literature sources with slight modification and is commonly cited as a timeline of the effects (Table). It is based on a mixture of anecdotal clinical experience, expert opinion, and available clinical studies of hormone therapy in transfeminine people. Due to limited research characterizing the effects of transfeminine hormone therapy at present, the table may or may not be completely accurate.
Table 2: Effects of hormone therapy at typical doses in adult transfeminine people (Wiki):
Effect
Onseta
Completiona
Permanency
Breast development
2–6 months
2–3 years
Permanent
Reduced and slowed growth of facial and body hair
3–12 months
>3 yearsb
Reversible
Cessation and reversal of scalp hair loss
1–3 months
1–2 years
Reversible
Softening of skin and decreased skin oiliness and acne
3–6 months
Unknown
Reversible
Redistribution of body fat in a feminine pattern
3–6 months
2–5 years
Reversible
Decreased muscle mass and strength
3–6 months
1–2 yearsc
Reversible
Widening and rounding of the pelvisd
Unknown
Unknown
Permanent
Changes in mood, emotionality, and behavior
Immediate
Unknown
Reversible
Decreased sex drive and spontaneous erections
1–3 months
3–6 months
Reversible
Erectile dysfunction and decreased ejaculate volume
1–3 months
Variable
Reversible
Decreased sperm production and infertility
Unknown
>3 years
Mixede
Decreased testicular volume
3–6 months
2–3 years
Unknown
Voice changes (e.g., more feminine pitch/resonance)
Nonef
N/A
N/A
Height changes (e.g., decrease)
Noneg
N/A
N/A
a Effects in general may vary significantly between individuals due to factors like genetics, diet/nutrition, hormone levels, etc. b Hormone therapy usually has little influence on facial hair density in transfeminine people. Complete removal of facial and body hair can be achieved with laser hair removal and electrolysis. Temporary hair removal can be achieved with shaving, epilating, waxing, and other methods. c Reduced muscle mass and strength may vary significantly depending on amount of physical exercise. d Pelvic changes occur only in young individuals who have not yet completed growth plate closure (may not occur at all in post-adolescent people). e Only estrogens, particularly at high doses, seem to have the potential for long-lasting or irreversible infertility; impaired fertility caused by antiandrogens is usually readily reversible with discontinuation. fVoice training can be an effective means of feminizing the voice. g Height attainment may be reduced in adolescents, but height is not meaningfully changed or reduced in adults per clinical data (Gooren & Bunck, 2004; Ingram & Thomas, 2019; Hilton & Lundberg, 2021; Talathi et al., 2025).
Breast Development
Breast development is among the most anticipated effects of hormone therapy in transfeminine people (Masumori et al., 2021; Grock et al., 2024). This relates to the key significance of breasts as a feminine characteristic, component of sexual attractiveness, and signal of sex and gender. Breast growth in transfeminine people usually starts within 1 to 6 months and completes over a period of 1 to 3 years (e.g., de Blok et al., 2021). The developed breasts of transfeminine people are highly variable in terms of size and shape, as with natal women (de Blok et al., 2021). Based on available high-quality clinical studies, transfeminine people tend to have much smaller mature breasts than those of natal women on average, and this appears to be the case regardless of hormonal regimen or age at which hormone therapy is commenced (e.g., de Blok et al., 2021; Boogers et al., 2025). The reasons for this are unknown, but one key possibility, observed in animals, is that prenatal androgen exposure limits subsequent breast growth potential. Despite usually modest breast development, many transfeminine people still express overall satisfaction with their breasts (de Blok et al., 2021; Boogers et al., 2025).
Beyond ensuring adequate testosterone suppression and maintaining sufficient estradiol levels above a specific low threshold, there are currently no known or substantiated methods to permanently enhance or optimize breast development. However, research suggests that avoiding high or excessive doses of estradiol and progestogens may be beneficial. In addition, high levels of estradiol, progesterone, and/or prolactin, as with the normal menstrual cycle and pregnancy, are known to induce temporary and reversible breast tenderness and enlargement, for instance due to local fluid retention and lobuloalveolar maturation (Aly, 2020). However, the breast size increases are modest, and high hormone levels come with health risks (Aly, 2020). Surgical breast augmentation is an option to increase breast size if it is unsatisfactory. Some transfeminine people, for instance many non-binary individuals, may wish to avoid or minimize breast growth, and there are possible therapeutic approaches in this area (Aly, 2019).
Additional review content on breast development in transfeminine people exists on this site (e.g., Aly, 2020; Aly, 2020). Breast growth can be measured and tracked with a variety of methods for individuals who are interested in monitoring their progress (Wiki). Photographs and timelines of breast development and feminization with hormone therapy in transfeminine people are available in communities like r/TransTimelines and r/TransBreastTimelines on the social media website Reddit.
Specific Hormonal Medications
The medications that are used in transfeminine hormone therapy include estrogens, progestogens, and antiandrogens. Estrogens produce feminization and testosterone suppression. Progestogens and antiandrogens do not mediate feminization themselves but further suppress and/or block testosterone. Testosterone suppression causes demasculinization and disinhibition of estrogen-mediated feminization. Androgens are sometimes used at low doses in transfeminine people who have low testosterone levels, although they are not required and benefits are uncertain. There are many different types of these hormonal medications available for transfeminine hormone therapy, with different benefits and risks.
Estrogens, progestogens, and antiandrogens are available in a variety of different formulations and for use by many different routes of administration in transfeminine people. The route of administration influences the absorption, distribution, metabolism, and elimination of the hormone in the body, resulting in significant differences between routes in terms of bioavailability, hormone levels in blood and specific tissues, and patterns of metabolites. These differences can have important therapeutic consequences.
Table 3: Major routes of administration of hormonal medications for transfeminine people:
Insertion via surgical incision into fat under skin
Pellet
Vaginal administration is a major additional route of administration of hormonal medications in cisgender women. While vaginal administration via a natal vagina is of course not possible in transfeminine people, neovaginal administration is a possiblility in those who have undergone vaginoplasty. However, the lining of the neovagina is not the vaginal epithelium of natal females but instead is usually skin or colon—depending on the type of vaginoplasty performed (penile inversion or sigmoid colon vaginoplasty, respectively). For this reason, neovaginal administration in transfeminine people is likely more similar in its properties to transdermal and rectal administration—depending on the type of neovagina—than to vaginal administration in cisgender women. It is noteworthy that the vaginal and rectal routes are said to be similar in their properties for hormonal medications however (Goletiani, Keith, & Gorsky, 2007; Wiki). Moreover, absorption of estradiol via neovaginas constructed from peritoneum (internal abdominal lining)—a less commonly employed vaginoplasty approach in transfeminine people—was reported in one study to be similar to that with vaginal administration of estradiol in cisgender women (Willemsen et al., 1985). As such, neovaginal administration may be an additional possible route for certain transfeminine people depending on the circumstances. However, this route still remains to be more adequately characterized.
An often-encountered question from people who take hormonal medications is whether there is an optimal time of the day to take them (Colonnello et al., 2025). As of present, there is little research in this area, and the answer to the question is essentially unknown (Colonnello et al., 2025). In any case, there is currently no evidence or persuasive theoretical basis to favor specific times of day to take these medications (Colonnello et al., 2025). In all likelihood, it makes little or no difference.
Estrogens
Estradiol, the primary bioidentical form normally found in the human body, is the main estrogen that is used in transfeminine hormone therapy. Estradiol hemihydrate (EH) is another form that is essentially identical to and interchangeable with estradiol. Estradiol esters are also sometimes used in place of estradiol. They are prodrugs of estradiol (i.e., are converted into estradiol in the body) and have essentially identical biological activity to estradiol. However, they have longer durations when used by injection due to slower absorption from the injection site, and this allows them to be administered less often. Some examples of major estradiol esters include estradiol valerate (EV; Progynova, Progynon Depot, Delestrogen) and estradiol cypionate (EC; Depo-Estradiol). Polyestradiol phosphate (PEP; Estradurin) is an injectable estradiol prodrug in the form of a polymer (i.e., linked chain of estradiol molecules) which is metabolized slowly and has a very long duration.
Non-bioidentical estrogens such as ethinylestradiol (EE; found in birth control pills), conjugated estrogens (CEEs; Premarin; used in menopausal hormone therapy), and diethylstilbestrol (DES; widely used previously but now abandoned) are resistant to metabolism in the liver and have disproportionate effects on estrogen-modulated liver synthesis when compared to bioidentical estrogens like estradiol (Aly, 2020). As a result, they have stronger influence on coagulation and greater risk of certain health problems like blood clots and associated cardiovascular issues (Aly, 2020). For this reason, as well as the fact that relatively high doses of estrogens may be needed for testosterone suppression, non-bioidentical estrogens should ideally never be used in transfeminine hormone therapy.
Estradiol dose-dependently suppresses testosterone levels in people with testes. Physiological and guideline-based levels of estradiol (<200 pg/mL or <734 pmol/L) are often not sufficient to suppress testosterone levels into the female range in transfeminine people who have not had their gonads removed (e.g., Liang et al., 2018; Krishnamurthy et al., 2023; Slack et al., 2023). As a result, estradiol is generally used in combination with an antiandrogen or progestogen in transfeminine hormone therapy (Hembree et al., 2017; Coleman et al., 2022; Rose et al., 2023). This results in partial suppression of testosterone levels by estradiol and further suppression or blockade of the remaining testosterone by the antiandrogen or progestogen. While combination therapy can be effective in fully suppressing or blocking testosterone (e.g., Aly, 2019; Aly, 2020), testosterone suppression can also still remain incomplete with antiandrogens and progestogens in certain forms (e.g., Aly, 2018; Jain, Kwan, & Forcier, 2019). In contrast to physiological estradiol levels, supraphysiological levels of estradiol are able to consistently and fully suppress testosterone levels into the normal female range with estradiol alone in transfeminine people (e.g., Gooren et al., 1984 [Graph]; Igo & Visram, 2021; Herndon et al., 2023 [Discussion]). This alternative approach, often referred to as high-dose estradiol monotherapy, has the advantage of avoiding the side effects, risks, and costs of antiandrogens and progestogens. However, it has the disadvantage of exposure to supraphysiological estradiol levels that are above those recommended by guidelines and that may have greater health risks. Physiological estradiol doses and combination therapy are more often used in transfeminine people treated by clinicians, whereas high-dose estradiol monotherapy is more frequently encountered in transfeminine people on DIY hormone therapy.
The feminizing effects of estradiol appear to be maximal at relatively low levels in the absence of androgens. Higher doses of estradiol and supraphysiological estradiol levels, aside from allowing for greater testosterone suppression, are not known to result in better feminization in transfeminine people (Deutsch, 2016; Nolan & Cheung, 2021). In fact, there is some indication that higher estrogen doses early into hormone therapy could actually result in worse breast development. Hence, the therapeutic emphasis in transfeminine hormone therapy is more on testosterone suppression than on achieving a specific estradiol level, at least above a certain low threshold level. Higher doses of estrogens, including of estradiol, also have a greater risk of adverse health effects such as blood clots and cardiovascular problems (Aly, 2020). As such, the use of physiological doses of estradiol is optimal in transfeminine people. At the same time however, high estrogen doses can be useful for improving testosterone suppression when it is inadequate, and the absolute risks, in the case of non-oral bioidentical estradiol, are low and are more important in people with specific risk factors (e.g., older age, physical inactivity, obesity, concomitant progestogen use, smoking, surgery, and rare thrombophilic abnormalities). If more adequate testosterone suppression is necessary, limitedly supraphysiological doses of non-oral estradiol may have a reasonable ratio of benefit to risk in this context, at least in those without relevant risk factors for estrogen-related complications (e.g., many healthy young people) (Aly, 2020).
Estradiol and estradiol esters are usually used orally, sublingually, transdermally, by injection (intramuscularly or subcutaneously), or by implant in transfeminine hormone therapy (Wiki).
Oral Estradiol
Estradiol is used orally in the form of tablets of estradiol (Wiki; Graphs). Alternatively, oral estradiol valerate tablets are used in some countries, for instance in many European countries. The only real difference between these oral estradiol forms is that estradiol valerate contains slightly less estradiol by weight (~76% of that of estradiol) due to its ester component and hence requires somewhat higher doses (~1.3-fold) in comparison for equivalent estradiol levels (Wiki; Table). Oral estradiol has a duration suitable for once-daily administration. Oral administration of estradiol is a very convenient and inexpensive route, which makes it the most popular and widely used form of estradiol in transfeminine people. Oral estradiol has relatively low bioavailability (~5%), and there is substantial variability between people in terms of estradiol levels achieved with the same dose. Hence, in some transfeminine people estradiol levels may be low with oral estradiol, and testosterone suppression may be inadequate depending on the antiandrogen.
A major drawback of oral estradiol is that it results in excessive levels of estradiol in the liver due to the first pass that occurs with oral administration and has a disproportionate impact on estrogen-modulated liver synthesis (Aly, 2020). This in turn increases coagulation and the risk of associated health complications like blood clots and cardiovascular problems (Aly, 2020). These particular health concerns are largely allayed if estradiol is taken non-orally at reasonable and non-excessive doses. Hence non-oral forms of estradiol, like transdermal estradiol, although less convenient and often more expensive than oral estradiol, are preferable in transfeminine hormone therapy. It is recommended that all transfeminine people who are over 40 to 45 years of age use non-oral routes due to the greater risk of blood clots and cardiovascular problems that occurs with age (Aly, 2020; Coleman et al., 2022). Oral estradiol is not a good choice for high-dose estradiol monotherapy in transfeminine people due to the high estradiol levels required and the greater risks than with non-oral routes. In addition to its disproportionate liver impact, oral estradiol results in unphysiological levels of estradiol metabolites like estrone and estrone sulfate when compared to non-oral forms. The clinical implications of this, if any, are unknown. Oral and non-oral estradiol have in any case been found to have similar effectiveness in terms of feminization and breast development in transfeminine people in available studies (Sam, 2020).
Sublingual Estradiol
Oral estradiol tablets can be taken sublingually instead of orally. Sublingual use of estradiol tablets has several-fold higher bioavailability relative to oral administration and hence achieves much higher overall estradiol levels in comparison (Sam, 2021; Wiki; Graphs). Sublingual use of oral estradiol tablets can be employed instead of oral administration to reduce doses and hence medication costs or to produce higher estradiol levels for the purpose of achieving better testosterone suppression when needed. However, sublingual estradiol is very spiky in terms of estradiol levels when compared to oral estradiol and has a short duration of highly elevated estradiol levels. As such, it may be advisable for sublingual estradiol to be used in divided doses multiple times throughout the day in order to maintain at least somewhat steadier estradiol levels. The therapeutic implications for transfeminine people of the spikiness of sublingual estradiol, for instance in terms of testosterone suppression and health risks, have been little-studied and are mostly unknown. In any case, when used as a form of high-dose estradiol monotherapy and taken multiple times per day, strong though still incomplete testosterone suppression has been observed (Yaish et al., 2023). Oral estradiol valerate tablets can be taken sublingually instead of orally similarly to estradiol and are likewise highly effective when used in this way (Aly, 2019; Wiki). Due to partial swallowing of tablets, sublingual estradiol may in practice be a mixture of sublingual and oral administration and may have some of the same health risks of oral estradiol (Wiki). Buccal administration of estradiol appears to have similar properties as sublingual administration but is much less researched in comparison and is not used as often in transfeminine people (Wiki).
Transdermal Estradiol
Transdermal estradiol is available in the form of patches, gel, emulsions, and sprays (Wiki). These forms are usually applied to skin areas such as the arms, abdomen, or buttocks. Gel, emulsions, and sprays are applied and left to dry for a short period, whereas patches are applied and remain adhesed to the skin for a specified amount of time. Due to rate-limited absorption through the skin, there is a depot effect with transdermal estradiol and this route has a long duration with very steady estradiol levels. As a result, estradiol gel, emulsions, and sprays are all suitable for once-daily use. Patches stay applied and continuously deliver estradiol for either 3–4 days or 7 days depending on the patch brand (Table). Transdermal estradiol is more expensive than oral estradiol. Gel, emulsions, and sprays may be less convenient than oral administration, but patches can be more convenient due to their infrequent application. However, patches can sometimes cause application site problems like redness and irritation and can occasionally come off prematurely due to adhesive failure. As with oral estradiol, there is substantial variability in estradiol levels with transdermal estradiol, and some transfeminine people may have poor absorption, low estradiol levels, and inadequate testosterone suppression with this route. Estradiol sprays, such as Lenzetto, have been found to achieve very low estradiol levels that are probably not therapeutically adequate for use in transfeminine hormone therapy (Aly, 2020; Graph).
Transdermal estradiol is the form of estradiol most commonly used in transfeminine people who are over 40 years of age due to its lower health risks relative to oral estradiol. Transdermal estradiol gel is not a favorable option for high-dose estradiol monotherapy as it has difficulty achieving the high estradiol levels needed for adequate testosterone suppression (Aly, 2019). On the other hand, transdermal estradiol patches can be an effective option for high-dose estradiol monotherapy if multiple 100 μg/day patches are used, although this can require the use of many patches and can be expensive (Wiki). Different skin sites absorb transdermal estradiol to different extents (Wiki). Genital application of transdermal estradiol, specifically to the scrotum or neolabia, is particularly better-absorbed than conventional skin sites and can result in much higher estradiol levels than usual (Aly, 2019). This can be useful for reducing doses and hence medication costs or for achieving higher estradiol levels for better testosterone suppression when needed, for instance in the context of high-dose estradiol monotherapy. Transdermal estradiol should not be applied to the breasts as this is not known to result in improved breast development and the potential health consequences of doing so are unknown (e.g., influence on breast cancer risk).
Injectable Estradiol
Injectable estradiol preparations can be administered via either intramuscular or subcutaneous injection (Wiki; Wiki; Graphs). There is a depot effect with injection of estradiol esters such that they are slowly absorbed from the injection site and have a prolonged duration. This ranges from days to months depending on the ester. Commonly used injectable estradiol esters, which all have short to moderate durations, include estradiol valerate (EV), estradiol cypionate (EC), estradiol enanthate (EEn), and estradiol benzoate (EB). Longer-acting injectable estradiol esters, such as estradiol undecylate (EU) and polyestradiol phosphate (PEP), have been discontinued and are no longer pharmaceutically available. In the case of intramuscular injection, common injection sites include the deltoid muscle (upper arm), vastus lateralis and rectus femoris muscles (thigh), and ventrogluteal muscle (buttocks). Subcutaneous injection of estradiol injectables, while less commonly used, has comparable pharmacokinetics to intramuscular injection, and is easier, less painful, and more convenient in comparison (Wiki). However, the maximum volume that can be safely and comfortably injected subcutaneously (1.5–3 mL) is less than that which can be injected intramuscularly (2–5 mL) (Hopkins, & Arias, 2013; Usach et al., 2019). Injectable estradiol tends to be fairly inexpensive, but may be less convenient than other routes due to the need for regular injections. There may also be a risk of internal scar tissue build-up long-term. Estradiol injectables have been discontinued in many parts of the world (e.g., most of Europe), and their availability is limited. In recent years, many transfeminine people have turned to black market homebrewed injectable estradiol preparations to use this route.
Injectable estradiol preparations are typically used at higher doses than other forms of estradiol, and can easily achieve very high levels of estradiol. This can be useful for testosterone suppression, making this form of estradiol likely the best choice for high-dose estradiol monotherapy in transfeminine people. However, the high doses that are possible with injectable estradiol preparations can also easily lead to overdosage and unnecessarily increased risks (e.g., Aly, 2020). Resources are available on this site for guiding selection of appropriate doses and intervals of injectable estradiol esters in transfeminine people. This includes a simulator and informal meta-analysis of estradiol levels with these preparations (Aly, 2021; Aly, 2021) and a table providing approximate equivalent doses between injectable estradiol esters and other estradiol routes and forms (Aly, 2020). It is notable and unfortunate that currently recommended doses and intervals for injectable estradiol esters by transgender care guidelines (e.g., 10–40 mg/2 weeks estradiol valerate) appear to be highly excessive and too widely spaced, and are likely to be therapeutically inadvisable (Aly, 2021). Doses and intervals of injectable estradiol esters recommended by the present author for use as a means of high-dose estradiol monotherapy, targeting mean estradiol levels of around 300 pg/mL (1,100 pmol/L), are provided below (Table 4).
Table 4: Recommended doses and intervals of injectable estradiol esters for high-dose estradiol monotherapy (targeting estradiol levels of around 300 pg/mL [1,100 pmol/L]):
a Injection interval. b Doses and intervals for estradiol undecylate are extrapolated and hypothetical (Aly, 2021).
These doses and intervals should be considered a starting point, and should be fine-tuned as necessary based on blood tests. In terms of injection intervals, the shorter interval, the more stable the estradiol levels, but the more often that injections need to be done. Doses may be increased if estradiol levels are too low and testosterone suppression is inadequate, and doses may be decreased if estradiol levels are too high so long as adequate testosterone suppression is maintained. Doses should be lower (targeting mean estradiol levels of 100–200 pg/mL [367–734 pmol/L]) if combined with an antiandrogen or progestogen as these agents will help with testosterone suppression. Similarly, doses should be lower following surgical gonadal removal as testosterone suppression will no longer be necessary.
Estradiol Pellets
Estradiol implants are pellets of pure crystalline hormone and are surgically placed into subcutaneous fat by a physician (Wiki). They are slowly absorbed by the body following implantation, and new implants are given once every 4 to 6 months. Due to the need for minor surgery, their high cost, and limited availability, estradiol implants are not as commonly used as other estradiol routes. Notably, almost all pharmaceutical estradiol implants throughout the world have been discontinued, and the implants that remain available are almost exclusively compounded products provided by compounding pharmacies. Dosage adjustment with estradiol implants is also more difficult than with other estradiol routes. Despite their various practical limitations however, estradiol implants allow for very steady estradiol levels, and their very long duration can allow for unusual convenience among available estradiol forms.
Additional Notes
Table 5: Available forms and recommended doses of estradiol for adulta transfeminine people:
a Estradiol doses in pubertal adolescent transfeminine people should be lower to mimic estradiol exposure during normal female puberty (Aly, 2020). b May be advisable to use divided doses 2 to 4 times per day (i.e., once every 6 to 12 hours) instead of once per day (Sam, 2021). c This estradiol form achieves very low estradiol levels at typical doses that don’t appear to be well-suited for transfeminine hormone therapy (Aly, 2020; Graph). d Estradiol valerate contains about 75% of the same amount of estradiol as estradiol so doses are about 1.3-fold higher for the same estradiol levels (Aly, 2019; Sam, 2021). e Doses and intervals for estradiol undecylate are extrapolated and hypothetical (Aly, 2021). f A higher initial loading dose of e.g., 240 or 320 mg polyestradiol phosphate can be used for the first one or two injections to reach steady-state estradiol levels more quickly. However, this preparation has recently been discontinued and appears to no longer be available.
Additional informational resources are available in terms of estradiol levels (Wiki; Table) and approximate equivalent doses (Aly, 2020) with different forms, routes, and doses of estradiol.
There is high variability between individuals in the levels of estradiol achieved during estradiol therapy. That is, estradiol levels during treatment with the same dosage of estradiol can differ substantially between individuals. This variability is greatest with oral and transdermal estradiol but is also considerable even with injectable estradiol preparations and other estradiol forms. As such, estradiol doses are not absolute and should be individualized on a case-by-case basis in conjunction with blood work as a guide. It should also be noted that due to fluctuations in estradiol concentrations with certain routes, levels of estradiol can vary considerably from one blood test to another. This is most notable with sublingual estradiol and injectable estradiol. The fluctuations in estradiol levels with these routes are predictable and must be understood when interpreting blood work results. Differences in blood test results can be minimized with informed and consistent timing of blood draws.
If or when the gonads are surgically removed, testosterone suppression is no longer needed in transfeminine people. As a result, estradiol doses, if they are high or supraphysiological, can be lowered to more closely approximate normal physiological levels in cisgender women.
Progestogens
Progestogens include progesterone and progestins. Progestins are synthetic progestogens derived from structural modification of progesterone or testosterone. There are dozens of different progestins and these progestins can be divided into a variety of different structural classes with varying properties (Table). Examples of some major progestins of different classes include the 17α-hydroxyprogesterone derivative medroxyprogesterone acetate (MPA; Provera, Depo-Provera), the 19-nortestosterone derivative norethisterone (NET; many brand names), the retroprogesterone derivative dydrogesterone (Duphaston), and the 17α-spirolactone derivative drospirenone (Slynd, Yasmin). Progestins were developed because they have a more favorable disposition in the body than progesterone for use as medications. Only a few clinically used progestins have been employed in transfeminine hormone therapy. However, progestogens largely produce the same progestogenic effects, with a few exceptions, and theoretically almost any progestogen could be used.
Besides helping with testosterone suppression, progestogens are of no clear or known benefit for feminization or breast development in transfeminine people. While some transfeminine people anecdotally claim to experience improved breast development with progestogens, an involvement of progestogens in improving breast size or shape is controversial and is not supported by theory nor evidence at present (Wiki; Aly, 2020). It is possible that premature introduction of progestogens, particularly at high doses, could actually have an unfavorable influence on breast development (Aly, 2020). Many transfeminine people have also anecdotally claimed that progestogens have a beneficial effect on their sexual desire. However, a review of the literature by the present author found that neither progesterone nor progestins positively influence sexual desire in humans (Aly, 2020). Instead, the available evidence suggests either a neutral influence or an inhibitory effect of progestogens on sexual desire, although the latter may be specific only to high doses of progestogens (Aly, 2020). Claims have been made that progesterone may have beneficial effects on mood in transfeminine people as well, but clinical support for such notions is likewise lacking at this time (Coleman et al., 2022; Nolan et al., 2022). It is notable that progesterone at luteal-phase levels, due to its neurosteroid metabolites like allopregnanolone, actually appears to worsen mood in around 30% of cisgender women, and produces more overt negative reactions, which constitute the diagnoses of premenstrual syndrome (PMS) and premenstrual dysphoric disorder (PMDD), in around 2 to 10% of women (Bäckström et al., 2011; Edler Schiller, Schmidt, & Rubinow, 2014; Sundström-Poromaa et al., 2020). More research is needed to evaluate the possible beneficial effects of progestogens in transfeminine people.
Most clinically used progestogens have off-target activities in addition to their progestogenic activity, and these activities may be desirable or undesirable depending on the action in question (Kuhl, 2005; Stanczyk et al., 2013; Wiki; Table). Progesterone has a variety of neurosteroid as well as other activities that can result in central nervous system effects among others which are not shared by progestins. MPA as well as NET and its derivatives have weak androgenic activity, which is unfavorable in the context of transfeminine hormone therapy. NET and certain related progestins produce ethinylestradiol as a metabolite at high doses and hence can produce ethinylestradiol-like estrogenic effects, including increased risk of blood clots and associated cardiovascular problems. Other off-target actions of progestogens include antiandrogenic, glucocorticoid, and antimineralocorticoid activities. These actions can result in differences in therapeutic effectiveness (e.g., androgen suppression or blockade) as well as side effects and health risks. Some notable progestins without undesirable off-target activities (i.e., androgenic or glucocorticoid activity) include low-dose CPA, drospirenone (DRSP), dienogest, nomegestrol acetate (NOMAC), dydrogesterone, and hydroxyprogesterone caproate (OHPC). However, of these progestins, only CPA has been considerably used and studied in transfeminine people.
The addition of progestogens to estrogen therapy has been associated with a number of unfavorable health effects. These include increased risk of blood clots (Wiki; Aly, 2020), coronary heart disease (Wiki), and breast cancer (Wiki; Aly, 2020). High doses of progestogens are also associated with increased risk of certain non-cancerousbrain tumors including meningiomas and prolactinomas (Wiki; Aly, 2020). The coronary heart disease risk may be due to changes in blood lipids caused by the weak androgenic activity of certain progestogens, but the rest of the aforementioned risks are probably due to their progestogenic activity (Stanczyk et al., 2013; Jiang & Tian, 2017). Aside from health risks, progestogens have also been associated with adverse mood changes (Wiki; Wiki). However, besides the case of progesterone and its neurosteroid metabolites, these effects of progestogens are controversial and are not well-supported by evidence (Wiki; Wiki). Progestogens are otherwise generally well-tolerated and are regarded as producing little in the way of side effects.
In contrast to certain progestins, progesterone has no unfavorable off-target hormonal activities. Due to its lack of androgenic activity, progesterone has no adverse influence on blood lipids and is not expected to raise the risk of coronary heart disease. The addition of oral progesterone to estrogen therapy notably has not been associated with increased risk of blood clots (Wiki). In addition, oral progesterone seems to have less risk of breast cancer than progestins with shorter-term therapy, although this is notably not the case with longer-term exposure (Wiki; Aly, 2020). Consequently, it has been suggested that progesterone, for reasons that have yet to be fully elucidated, may be a safer progestogen than progestins and that it should be the preferred progestogen for hormone therapy in cisgender women and transfeminine people. However, there are also theoretical arguments against such notions. Oral progesterone is known to produce very low progesterone levels and to have only weak progestogenic effects at typical doses (Aly, 2018; Wiki). The seemingly better safety of oral progesterone may simply be an artifact of the low progesterone levels that occur with it, and hence of progestogenic dosage. Non-oral progesterone, at doses resulting in physiological and full progestogenic strength, has never been properly evaluated in terms of health outcomes, and may have similar risks as progestins (Aly, 2018; Wiki).
Due to their lack of known influence on feminization and breast development and their known and possible adverse effects and risks, progestogens are not routinely used in transfeminine hormone therapy at present. Major transgender health guidelines note the limitations of the available evidence on progestogens for transfeminine people and have mixed attitudes on their use, either explictly recommending against their use (Coleman et al., 2022—WPATH SOC8), taking a more neutral stance (Hembree et al., 2017—Endocrine Society guidelines), or being permissive of their use (Deutsch, 2016—UCSF guidelines). There is however a very major exception to the preceding in the form of CPA, an antiandrogen which is widely used in transfeminine hormone therapy to suppress testosterone production and which happens to be a powerful progestogen at the typical doses used in transfeminine people. CPA will be described below in the section on antiandrogens. Although progestogens have various health risks, cisgender women of course have progesterone, and the absolute risks of progestogens are very low in healthy young people. Risks like breast cancer also are exposure-dependent and take many years to develop. The testosterone suppression provided by progestogens can furthermore be very useful in transfeminine people, as is widely taken advantage of with CPA. Given these considerations, a limited duration of progestogen therapy in transfeminine people, for instance a few years to help suppress testosterone levels before surgical gonadal removal, may be considered quite acceptable.
Progesterone can be used in transfeminine people by oral administration, sublingual administration, rectal administration, or by intramuscular or subcutaneous injection (Wiki). Progestins are usually used via oral administration, but certain progestins are also available in injectable formulations (Wiki).
Oral Progesterone
Progesterone is most commonly taken orally. It is used by this route in the form of oil-filled capsules containing 100 or 200 mg micronized progesterone under brand names such as Prometrium, Utrogestan, and Microgest (Wiki). Despite its widespread use, levels of progesterone via oral administration have been found using state-of-the-art assays (LC–MS) to be very low (<2 ng/mL [<6.4 nmol/L] at 100 mg/day) and inadequate for satisfactory progestogenic effects in various areas (Aly, 2018; Wiki). In relation to this, even high doses of oral progesterone (400 mg/day) showed no antigonadotropic effect or testosterone suppression in cisgender men (Aly, 2018; Wiki). This is in major contrast to non-oral forms of progesterone and to progestins, which produce dose-dependent and robust testosterone suppression (Aly, 2019; Wiki). In addition to its low progestogenic potency, oral progesterone is excessively converted into neurosteroid metabolites like allopregnanolone and pregnanolone. These metabolites act as potent GABAA receptor positive allosteric modulators, and can produce undesirable alcohol-like side effects such as sedation, cognitive, memory, and motor impairment, and mood changes (Wiki; Wiki). As such, while inconvenient, non-oral routes are greatly preferable for progesterone.
Sublingual Progesterone
Sublingual progesterone tablets exist and are marketed under the brand name Luteina but today are only available in Poland and Ukraine (Wiki). Oral progesterone could theoretically be taken sublingually, analogously to sublingual use of oral estradiol. However, because oral progesterone is formulated as oil-filled capsules, this makes it difficult and unpleasant to use by sublingual administration. Buccal progesterone, which would be expected to have similar characteristics to those of sublingual progesterone, has been used in medicine in the past, but is no longer marketed today (Wiki).
Rectal Progesterone
Progesterone is approved for use by rectal administration in the form of suppositories under the brand name Cyclogest (Wiki). This product is marketed in only a limited number of countries however, although it is available in the United Kingdom (Wiki). While not approved for use by rectal administration, oral progesterone capsules can be taken rectally instead of orally, and using them in this way may allow for much higher progesterone levels than would be achieved by oral administration due to avoidance of most first-pass metabolism. Rectal administration of oral progesterone capsules has not been formally studied, but oral progesterone capsules have been administered vaginally in cisgender women with success (Miles et al., 1994; Wang et al., 2019), and the vaginal and rectal routes are said to have similar pharmacokinetics in general (Goletiani, Keith, & Gorsky, 2007; Wiki). Hence, there is good theoretical basis for rectal administration of oral progesterone capsules being an effective route of progesterone. Whereas oral progesterone achieves very low levels of progesterone, rectal progesterone can readily achieve normal luteal-phase levels of progesterone (Wiki). Although inconvenient, rectal administration may be the overall best route of administration of progesterone for transfeminine people. A significant subset of transfeminine people on progestogens take progesterone rectally (Chang et al., 2024).
Injectable Progesterone
Progesterone by injection is available as an oil solution for intramuscular injection under brand names such as Proluton, Progestaject, and Gestone (Wiki) and as an aqueous solution for subcutaneous injection under the brand name Prolutex (Wiki). Oil solutions of progesterone for intramuscular injection are widely available, whereas the aqueous solution of progesterone for subcutaneous injection is available only in some European countries (Wiki). Injectable progesterone, regardless of route, has a relatively short duration and must be injected once every one to three days (Wiki; Wiki). This makes it too inconvenient to use for most people. Unlike with estradiol, progesterone esters with longer durations than progesterone itself by injection are not chemically possible as progesterone has no hydroxyl groups available for esterification (Wiki). Injectable aqueous suspensions of microcrystalline progesterone were previously marketed and had a duration of 1 to 2 weeks, but these preparations were associated with pain at the injection site and were eventually discontinued (Aly, 2019; Wiki).
Other Progesterone Routes
Other progesterone routes, such as transdermal progesterone and subcutaneous progesterone pellets, are also known, but are not available as pharmaceutical drugs and are little-used medically (Wiki). This is related to the low potency of progesterone and difficulty achieving progesterone levels high enough for adequate therapeutic effects with these routes (Wiki; Wiki). In addition, progesterone pellets tend to be extruded at high rates (Wiki). In any case, certain compounding pharmacies may make forms of progesterone that could be used by these routes.
Oral and Injectable Progestins
Most progestins are taken orally in the form of solid tablets (Wiki). In contrast to progesterone, progestins, owing to their synthetic nature, are resistant to metabolism in the intestines and liver and have high oral bioavailability. In addition, unlike the case of the estrogen receptors, the progesterone receptors are expressed minimally or not at all in the liver, and there is no known first pass influence of progestogenic activity on liver synthesis (Lax, 1987; Stanczyk, Mathews, & Cortessis, 2017). As a result, there are no apparent problems with oral administration in the case of purely progestogenic progestins. However, some progestins have liver-impacting off-target hormonal actions, such as androgenic, estrogenic, and/or glucocorticoid activity, and this can result in adverse effects like unfavorable lipid changes or procoagulation—which may be augmented by the first pass with oral administration.
A selection of progestins are available in injectable formulations, including for intramuscular or subcutaneous injection (Wiki). Some of the more notable ones include medroxyprogesterone acetate (MPA), norethisterone enanthate (NETE), hydroxyprogesterone caproate (OHPC), and algestone acetophenide (dihydroxyprogesterone acetophenide; DHPA) (Wiki). In addition to being used alone, injectable progestins are used together with estradiol esters in combined injectable contraceptives (Wiki). These preparations are often used as a means of hormone therapy by transfeminine people in Latin America. Whereas injectable progesterone has a duration measured in days, injectable progestins have durations ranging from weeks to months, and can be injected much less often in comparison (Table).
Additional Notes
Table 6: Available forms and recommended doses of progestogens for transfeminine people:
For progesterone levels with different forms, routes, and doses of progesterone, see the table here (only LC–MS and IA + CS assays for oral progesterone) and the graphs here.
As with estradiol, there is high variability between individuals in progesterone levels. Conversely, there is less variability between individuals in the case of progestins.
After removal of the gonads, progestogen doses can be lowered or adjusted to approximate normal female physiological exposure or they can be discontinued entirely.
Antiandrogens
Aside from estrogens and progestogens, there is another class of hormonal medications used in transfeminine hormone therapy known as antiandrogens (AAs). These medications reduce the effects of androgens in the body by either decreasing androgen production and thereby lowering androgen levels or by directly blocking the actions of androgens. They work via a variety of different mechanisms of action, and include androgen receptor antagonists, antigonadotropins, and androgen synthesis inhibitors.
Androgen receptor antagonists act by directly blocking the effects of androgens, including testosterone, DHT, and other androgens, at the level of their biological target. They bind to the androgen receptor without activating it, thereby displacing androgens from the receptor. Due to the nature of their mechanism of action as competitive blockers of androgens, the antiandrogenic efficacy of androgen receptor antagonists is both highly dose-dependent and fundamentally dependent on testosterone levels. They do not act by lowering testosterone levels, although some androgen receptor antagonists may have additional antiandrogenic actions that result in decreased testosterone levels. Because androgen receptor antagonists do not work by lowering testosterone levels, blood work can be less informative for them compared to antiandrogens that suppress testosterone levels. Androgen receptor antagonists include steroidal antiandrogens (SAAs) like spironolactone (Aldactone) and cyproterone acetate (CPA; Androcur) and nonsteroidal antiandrogens (NSAAs) like bicalutamide (Casodex).
Antigonadotropins suppress the gonadal production of androgens by inhibiting the GnRH-mediated secretion of gonadotropins from the pituitary gland. They include estrogens and progestogens. In addition, GnRH agonists such as leuprorelin (Lupron) and GnRH antagonists such as elagolix (Orilissa) act similarly and could likewise be described as antigonadotropins.
Androgen synthesis inhibitors inhibit the enzyme-mediated synthesis of androgens. They include 5α-reductase inhibitors (5α-RIs) like finasteride (Propecia) and dutasteride (Avodart). There are also other types of androgen synthesis inhibitors, for instance potent 17α-hydroxylase/17,20-lyase inhibitors like ketoconazole (Nizoral) and abiraterone acetate (Zytiga). However, these agents have limitations (e.g., toxicity, high cost, and lack of experience) and have not been used in transfeminine hormone therapy.
Although antigonadotropins and androgen synthesis inhibitors have antiandrogenic effects secondary to decreased androgen levels, they are not usually referred to as “antiandrogens”. Instead, this term is most commonly reserved to refer specifically to androgen receptor antagonists. However, antigonadotropins and androgen synthesis inhibitors may nonetheless be described as antiandrogens as well.
After removal of the gonads, antiandrogens can be discontinued. If unwanted androgen-dependent symptoms, such as acne, seborrhea, or scalp hair loss, persist despite full suppression or ablation of gonadal testosterone, then a lower dose of an androgen receptor antagonist, such as 100 to 200 mg/day spironolactone or 12.5 to 25 mg/day bicalutamide, can be continued to treat these symptoms.
Table 7: Available forms and recommended doses of antiandrogens for transfeminine people:
a For CPA, this dose range is specifically one-quarter of a 10-mg tablet to one full 10-mg tablet per day (2.5–10 mg/day) or a quarter of a 50-mg tablet every other day or every 2 to 3 days (4.2–12.5 mg/day). A dosage of 5–10 mg/day or 6.25–12.5 mg/day is likely to ensure maximal testosterone suppression, while lower doses may be less effective (Aly, 2019). b For spironolactone and bicalutamide, it is assumed that testosterone levels are substantially suppressed (≤200 ng/dL [<6.9 nmol/L]). If testosterone levels are not suppressed to this range, then higher doses may be warranted. c Spironolactone and its metabolites have relatively short half-lives, and twice-daily administration in divided doses (e.g., 100–200 mg twice per day) is recommended.
Figure 3: Suppression of gonadal testosterone production and circulating testosterone levels (ng/dL) with estradiol in combination with different antiandrogens over one year of hormone therapy in transfeminine people (Sofer et al., 2020). The estradiol forms included oral tablets 2–8 mg/day, transdermal gel 2.5–5 mg/day, and transdermal patches 50–200 μg/day. The antiandrogens included spironolactone 50–200 mg/day (n=16), cyproterone acetate (n=41), and GnRH agonists (specifically triptorelin 3.75 mg/month or goserelin 3.6 mg/month by injection) (n=10) (Sofer et al., 2020). It should be noted that lower doses of cyproterone acetate (10–12.5 mg/day) show equal testosterone suppression to higher doses (25–100 mg/day) and higher doses should no longer be used (Aly, 2019). The dashed horizontal line corresponds to the upper limit of the normal female range for testosterone levels.
As an antiandrogen, CPA has a dual mechanism of action of suppressing testosterone levels via its progestogenic and hence antigonadotropic activity and of acting as an androgen receptor antagonist (Aly, 2019). The progestogenic activity of CPA is of far greater potency than its androgen receptor antagonism however (Aly, 2019). The dose of CPA used as a progestogen in cisgender women is about 2 mg per day, which produces similar progestogenic effects to those of physiological luteal-phase levels of progesterone (e.g., suppression of gonadotropin secretion, ovulation inhibition, and endometrial transformation and protection) (Aly, 2019). Conversely, much higher doses of CPA of 50 to 300 mg/day have typically been used for androgen-dependent indications (Aly, 2019). These high doses of CPA result in profound progestogenic overdosage and associated side effects and risks (Aly, 2019). In transfeminine people, CPA has historically been used at doses of 50 to 150 mg/day (Aly, 2019). However, CPA doses have dramatically fallen in recent years, and today doses of no more than 10 to 12.5 mg/day are recommended (Aly, 2019; Coleman et al., 2022—WPATH SOC8). These lower doses of CPA still produce strong progestogenic effects, and in combination with estradiol, are equally effective as higher doses in suppressing testosterone levels (Aly, 2019; Meyer et al., 2020; Even Zohar et al., 2021; Kuijpers et al., 2021; Coleman et al., 2022). Even lower doses of CPA, for instance 5 to 6.25 mg/day, are currently being studied, and may still be fully effective (Aly, 2019).
Given by itself without estrogen, CPA typically suppresses testosterone levels in people with testes by about 50 to 70%, down to about 150 to 300 ng/dL (5.2–10.4 nmol/L) (Meriggiola et al., 2002; Toorians et al., 2003; Giltay et al., 2004; T’Sjoen et al., 2005; Tack et al., 2017; Zitzmann et al., 2017; Aly, 2019). Lower doses of CPA alone (e.g., 10 mg/day) show the same degree of testosterone suppression as higher doses of CPA alone (e.g., 50–100 mg/day), indicating that the antigonadotropic effects of CPA are maximal at relatively low therapeutic doses of this medication (Aly, 2019). This is on the order of about 5 to 10 times the ovulation-inhibiting dosage of CPA in cisgender women, a dose–response relationship that has also been observed with a number of other progestogens (Aly, 2019). Per the preceding, CPA alone, regardless of dosage, is unable to reduce testosterone levels into the normal female range (<50 ng/dL [<1.7 nmol/L]). But when CPA is combined with estradiol, even at relatively small doses of estradiol, it consistently suppresses testosterone levels into the normal female range (Aly, 2019; Angus et al., 2019; Gava et al., 2020; Sofer et al., 2020; Collet et al., 2022). However, it appears that a certain minimum level of estradiol, perhaps around 60 pg/mL (220 pmol/L) on average, is required for this to occur (Aly, 2019). Estradiol levels lower than this threshold in those taking CPA, which can occasionally be encountered in transfeminine people due to estradiol being dosed too low, have the potential to compromise full testosterone suppression (Aly, 2019).
In addition to testosterone suppression, CPA can dose-dependently block the androgen receptor (Aly, 2019). However, relatively high doses of CPA are needed to considerably antagonize the androgen receptor (e.g., 50–300 mg/day), and lower doses (e.g., ≤12.5 mg/day) may not be able to do this to a meaningful degree (Aly, 2019). As such, lower doses of CPA may essentially be purely progestogenic, with minimal or no androgen receptor antagonism. In this regard, referring to CPA at such doses as an “antiandrogen”—rather than as a “progestogen”—may be considered somewhat of a misnomer. Higher doses of CPA (>12.5 mg/day) can no longer be considered safe due to the massive progestogenic overdosage that occurs with them, and should no longer be used in transfeminine people. Moreover, as testosterone levels are usually suppressed into the normal female range in transfeminine people taking estradiol plus CPA, there is no actual need for any additional androgen receptor blockade (Aly, 2019).
CPA has been reported to produce various side effects. Some of these side effects include fatigue and a degree of weight gain (Belisle & Love, 1986; Hammerstein, 1990; Martinez-Martin et al., 2022). CPA might be able to produce a magnitude of sexual dysfunction (e.g., reduced sexual desire) beyond that expected with testosterone suppression alone (Wiki; Aly, 2019). It may also have a small risk of depressive mood changes (Wiki). In transfeminine people, CPA has been documented to produce pregnancy-like breast changes (i.e., lobuloalveolar development of the mammary glands) (Kanhai et al., 2000). In relation to this, CPA sometimes causes lactation as a side effect (Dewhurst & Underhill, 1979; Gooren, Harmsen-Louman, & van Kessel, 1985; Schlatterer et al., 1998; Bazarra-Castro, 2009). Concerns have been raised about premature introduction of progestogens—particularly at high doses like with CPA—and possible adverse influence on breast development (Aly, 2020). However, little data exists in humans to substantiate such concerns at present. The side effects of CPA are assumed to be dose-dependent, and using the lowest effective doses is expected to minimize its side effects.
CPA is usually taken orally in the form of tablets (e.g., 10, 50, and 100 mg) (Wiki). Under the brand name Androcur Depot, it is also available as a long-lasting 300 mg depot injectable in some countries (Wiki). However, this formulation is not commonly used in transfeminine people, and happens to correspond to very high doses in terms of CPA exposure. A pill cutter (Amazon) can be used to split CPA tablets and achieve lower doses (e.g., 12.5 mg doses with 50-mg tablets). CPA has a relatively long elimination half-life of about 1.6 to 4.3 days (Wiki; Aly, 2019). As such, it can be taken once daily, or even as infrequently as once every 2 or 3 days, if needed (Aly, 2019). In addition to splitting of CPA tablets, dosing CPA once every 2 or 3 days can also be useful for achieving lower doses (Aly, 2019).
As already described, CPA is a powerful progestogen even at the relatively low doses now used in transfeminine people (e.g., 5–12.5 mg/day). As such, there is no need, nor point, in adding another progestogen, for instance progesterone, in those who are taking CPA—at least if the goal of doing so is to produce progestogenic effects. This is something that is often overlooked in people taking CPA, and can result in increased costs, side effects, and inconvenience without any expected benefit.
Spironolactone
Spironolactone (Aldactone) is an antiandrogen and antimineralocorticoid. It is widely used as an antiandrogen in cisgender women for treatment of androgen-dependent hair and skin conditions like acne, hirsutism (excessive facial/body hair growth), and scalp hair loss, in cisgender women for treatment of hyperandrogenism (high androgen levels) due to polycystic ovary syndrome (PCOS), and in transfeminine people as a component of hormone therapy. Spironolactone is particularly widely used in transfeminine people in the United States, where it is the most commonly used antiandrogen in this population. As an antimineralocorticoid, the original and primary use of spironolactone in medicine, it is used to treat heart failure, high blood pressure, high mineralocorticoid levels, low potassium levels, and conditions of excess fluid retention like nephrotic syndrome and ascites, among others (Wiki). In terms of its antiandrogenic actions, spironolactone is a relatively weak androgen receptor antagonist as well as a weak androgen synthesis inhibitor (Wiki). The androgen synthesis inhibition of spironolactone is mediated specifically via inhibition of 17α-hydroxylase and 17,20-lyase (Wiki). Spironolactone does not appear to have meaningful progestogenic activity, 5α-reductase inhibition, or direct estrogenic activity (Wiki). However, indirect estrogenic effects secondary to its antiandrogenic activity (e.g., breast development and feminization) can occur with it at sufficiently high doses (Wiki).
Spironolactone shows limited and highly inconsistent effects on testosterone levels in clinical studies in cisgender men, cisgender women, and transfeminine people, with most studies finding no change in levels, some studies finding a decrease in levels, and a small number even finding an increase in levels (Aly, 2018). In spite of this, studies commonly find that spironolactone still produces antiandrogenic effects even when androgen levels remain unchanged. Hence, the primary mechanism of action of spironolactone as an antiandrogen appears to be androgen receptor blockade. In relation to this, in transfeminine people taking spironolactone as an antiandrogen, the estrogen component of the regimen is likely to be the main or possibly sole agent suppressing testosterone production. This is in part based on studies in transfeminine people comparing estradiol plus spironolactone to estradiol alone (e.g., Leinung, 2014; Leinung, Feustel, & Joseph, 2018; Angus et al., 2019) and on studies comparing testosterone levels with different doses of spironolactone (e.g., Liang et al., 2018; SoRelle et al., 2019; Allen et al., 2021). Due to the minimal influence of spironolactone on testosterone production, testosterone levels are not usually suppressed into the female range in transfeminine people taking estradiol plus spironolactone, with testosterone levels often remaining well above this range (e.g., 50–450 ng/dL [1.7–15.6 nmol/L] on average) (Leinung, 2014; Leinung, Feustel, & Joseph, 2018; Liang et al., 2018; Angus et al., 2019; Jain, Kwan, & Forcier, 2019; SoRelle et al., 2019; Sofer et al., 2020; Burinkul et al., 2021). However, testosterone levels do tend to decline gradually over time in transfeminine people on this regimen (e.g., Liang et al., 2018; Sofer et al., 2020 (Graph); Allen et al., 2021).
Due to its relatively weak androgen receptor antagonism, spironolactone is likely best-suited for blocking female-range or somewhat-higher testosterone levels (e.g., <100 ng/dL [<3.5 nmol/L]) (Aly, 2018). This is based on clinical dose-ranging studies of spironolactone (typically using 50–200 mg/day) in healthy cisgender women and cisgender women with PCOS (Goodfellow et al., 1984; Lobo et al., 1985; Hammerstein, 1990; James, Jamerson, & Aguh, 2022) as well as comparative studies of spironolactone against the more-potent antiandrogen flutamide (Cusan et al., 1994; Erenus et al., 1994; Shaw, 1996). The clinical antiandrogenic efficacy of spironolactone has been very limitedly assessed in transfeminine people to date, and is largely unknown (Angus et al., 2021). In any case, the antiandrogenic efficacy of spironolactone in cisgender women with androgen-dependent hair and skin conditions is well-established, and the medication thus does appear to be effective so long as testosterone levels are not too high (Brown et al., 2009; van Zuuren & Fedorowicz, 2016; Layton et al., 2017; Barrionuevo et al., 2018; James, Jamerson, & Aguh, 2022). In addition, higher doses of spironolactone (e.g., 300–400 mg/day) may be more useful for blocking higher testosterone levels in transfeminine people, and are allowed for by transgender care guidelines (Aly, 2020).
Consequent to spironolactone’s limited and inconsistent influence on testosterone levels and its relatively weak androgen receptor antagonism, estradiol plus spironolactone regimens will likely not be fully effective in terms of testosterone suppression for many transfeminine people. This is liable to result in suboptimal demasculinization, feminization, and breast development in these individuals. Other antiandrogenic approaches, such as bicalutamide, CPA, GnRH modulators, and high-dose estradiol monotherapy, will likely be more effective in these cases owing either to their ability to more potently block androgens or their capacity to reliably reduce testosterone levels into the female range. If testosterone levels are still too high with estradiol plus spironolactone, a switch to a different antiandrogen, increasing to a higher dosage of estradiol, or addition of a clinically antigonadotropic progestogen (e.g., non-oral progesterone or a progestin) should be considered.
Spironolactone is a strong antimineralocorticoid, or antagonist of the mineralocorticoid receptor, the biological target of the mineralocorticoid steroid hormones aldosterone and 11-deoxycorticosterone. This is an action that spironolactone shares with progesterone, although spironolactone is a much more potent antimineralocorticoid than progesterone. The mineralocorticoid receptor is involved in regulating electrolyte and fluid balances, among other roles. Spironolactone is associated with modestly lowered blood pressure, which may be considered a beneficial effect of its antimineralocorticoid activity (Martinez-Martin et al., 2022). Although spironolactone is usually well-tolerated, it can sometimes produce antimineralocorticoid side effects such as excessively lowered blood pressure, dizziness, fatigue, urinary frequency, and increased cortisol levels, among others (Kellner & Wiedemann, 2008; Kim & Del Rosso, 2012; Zaenglein et al., 2016; Layton et al., 2017; James, Jamerson, & Aguh, 2022). It has been argued by some in the online transgender community that spironolactone, via its antimineralocorticoid activity and increased cortisol levels, may increase visceral fat in transfeminine people (Aly, 2020). However, evidence does not support this hypothetical side effect at present (Aly, 2020). Available data also do not support spironolactone stunting breast development in transfeminine people or producing serious neuropsychiatric side effects, such as prominent depressive mood changes.
In people who are at-risk for hyperkalemia, dietary restriction to limit intake of potassium-rich foods is often recommended (Roscioni et al., 2012; Cupisti et al., 2018). This is often encountered in transgender health as transfeminine people being told “not to eat bananas”, which are said to be high in potassium. However, limiting dietary potassium with spironolactone to avoid hyperkalemia is theoretical and not actually evidence-based, with data so far contradicting its efficacy (St-Jules, Goldfarb, & Sevick, 2016; St-Jules & Fouque, 2020; Babich, Kalantar-Zadeh, & Joshi, 2022; St-Jules & Fouque, 2022). As such, routine restriction of dietary potassium with spironolactone may not be warranted.
Aside from its antimineralocorticoid activity, spironolactone has been reported to increase levels of LDL (“bad”) cholesterol levels and to decrease levels of HDL (“good”) cholesterol in women with PCOS (Nakhjavani et al., 2009). However, findings appear to be conflicting, with other studies not finding unfavorable influences on cholesterol levels with spironolactone (Polyzos et al., 2011). Long-term, adverse effects on cholesterol levels could result in an increase in the risk of coronary heart disease.
Spironolactone is taken orally in the form of tablets (e.g., 25, 50, and 100 mg) (Wiki). It is a prodrug of several active metabolites, including 7α-thiomethylspironolactone, 6β-hydroxy-7α-thiomethylspironolactone, and canrenone (7α-desthioacetyl-δ6-spironolactone) (Wiki). Spironolactone and these active metabolites have elimination half-lives of 1.4 hours, 13.8 hours, 15.0 hours, and 16.5 hours, respectively (Wiki). Due to the relatively short duration of elevated drug levels with spironolactone and its active metabolites (Graph), twice-daily administration of spironolactone in divided doses may be more optimal than once-daily intake and is advised (Reiter et al., 2010).
Bicalutamide
Bicalutamide (Casodex) is a nonsteroidal antiandrogen (NSAA) which acts as a potent and highly selective androgen receptor antagonist (Wiki). It is primarily used in the treatment of prostate cancer in cisgender men. Prostate cancer is an androgen-dependent cancer which antiandrogens can help to slow the progression of, and this use constitutes the vast majority of prescriptions for bicalutamide (Wiki). In addition to prostate cancer, although to a much lesser extent, bicalutamide has been used in the treatment of hirsutism (excessive facial/body hair growth), scalp hair loss, and polycystic ovary syndrome (PCOS) in cisgender women, peripheral or gonadotropin-independent precocious puberty (a rare form of precocious puberty in which antigonadotropins such as GnRH agonists are not effective) in cisgender boys, and priapism in cisgender men (Wiki). Bicalutamide is also becoming increasingly adopted for use as an antiandrogen in transfeminine people (Aly, 2020; Wiki). However, its use in transgender health is still very limited, and well-regarded transgender care guidelines either recommend against its use (Deutsch, 2016—UCSF guidelines; Coleman et al., 2022—WPATH SOC8) or are only cautiously permissive of its use (Thompson et al., 2021—Fenway Health guidelines). This is due to a lack of studies of bicalutamide in transfeminine people and its potential risks. Nonetheless, a small but growing number of clinicians are using bicalutamide in transfeminine people or are willing to prescribe it, with these clinicians located particularly in the United States. A single small clinical study has assessed bicalutamide in transfeminine people so far, specifically as a puberty blocker in 13 transfeminine adolescents who were denied insurance coverage for GnRH agonists (Neyman, Fuqua, & Eugster, 2019). (Update: More studies of bicalutamide in transfeminine people have since been published, see Aly (2020).)
Bicalutamide is a much more potent androgen receptor antagonist than either spironolactone or CPA (Wiki; Neyman, Fuqua, & Eugster, 2019). It is typically used in transfeminine people at a dosage of 25 to 50 mg/day, although this dosage has been arbitrarily selected and is not based on clinical data. Nonetheless, due to its relatively high potency as an androgen receptor antagonist and concomitant suppression of testosterone levels by estradiol, these doses may be adequate for testosterone blockade for many transfeminine people. At higher doses (>50 mg/day), bicalutamide is able to substantially block male-range testosterone levels (>300 ng/dL [>10.4 nmol/L]) based on studies of bicalutamide monotherapy in cisgender men with prostate cancer (Wiki). This is something that spironolactone and CPA are not capable of in the same way. Owing to its selectivity for the androgen receptor, bicalutamide has no off-target hormonal activity and produces almost no side effects in women (Wiki; Erem, 2013; Moretti et al., 2018). The only apparent side effect of bicalutamide in a rigorous clinical trial of the drug for hirsutism in cisgender women was significantly increased total and LDL (“bad”) cholesterol levels (Moretti et al., 2018). Hence, bicalutamide tends to be very well-tolerated. The relative lack of side effects with bicalutamide is in contrast to other antiandrogens like spironolactone and CPA, which are not pure androgen receptor antagonists and have off-target hormonal actions like antimineralocorticoid activity or strong progestogenic activity with consequent side effects and risks.
As a selective androgen receptor antagonist, bicalutamide taken by itself does not decrease testosterone production or levels but rather increases them (Wiki). This is due to a loss of androgen receptor-mediated negative feedback on gonadotropin secretion and a consequent compensatory upregulation of gonadal testosterone production (Wiki). Bicalutamide more than blocks the effects of any increase in testosterone it causes, and in fact fundamentally cannot increase testosterone levels more than it can block them (Wiki). In addition, increases in testosterone levels with bicalutamide will be blunted or abolished if it is combined with an adequate dose of an antigonadotropin such as estradiol (Wiki; Wiki). Since estradiol is made from testosterone in the body, bicalutamide taken alone also preserves and increases estradiol production and levels (Wiki). Because of this, although bicalutamide has no other important intrinsic hormonal activity besides its antiandrogenic activity, it produces robust indirect estrogenic effects including feminization and breast development even when it is not combined with estrogen (Wiki; Wiki; Neyman, Fuqua, & Eugster, 2019). This has important implications for the use of bicalutamide as a puberty blocker in transfeminine adolescents, as bicalutamide does not actually block puberty like conventional puberty blockers (GnRH agonists) but instead has the effect of dose-dependently converting male puberty into female puberty (Wiki; Neyman, Fuqua, & Eugster, 2019).
Bicalutamide has certain health risks, which has been a major reason that it has not been more readily adopted in transfeminine hormone therapy (Aly, 2020). It has a small risk of liver toxicity (Wiki; Aly, 2020) and of lung toxicity (Wiki). Abnormal liver function tests (LFTs), such as elevated liver enzymes and elevated bilirubin, occurred in about 3.4% of men with bicalutamide monotherapy plus standard care versus 1.9% of men with placebo plus standard care in the Early Prostate Trial (EPC) clinical programme after 3.0 years of follow-up (Wiki). In clinical trials, treatment with bicalutamide had to be discontinued in about 0.3 to 1.5% of men due to LFTs that became too highly elevated and could have progressed to serious liver toxicity (Wiki). To date, there are around 10 published case reports of serious liver toxicity, including cases of death, with bicalutamide, all of which have been in men with prostate cancer (Wiki; Table; Aly, 2020). There have also been a few unpublished reports of serious liver toxicity including deaths with bicalutamide in transfeminine people (Aly, 2020). However, these reports have not been confirmed, and they may or may not be reliable. In addition to the preceding reports, hundreds of additional instances of liver complications in people taking bicalutamide exist in the United States FDA Adverse Event Reporting System (FAERS) database (Wiki; FDA). Abnormal LFTs with bicalutamide usually occur within the first 3 to 6 months of treatment (Kolvenbag & Blackledge, 1996; Casodex FDA Label), and all case reports of liver toxicity with bicalutamide have had an onset of less than 6 months (Table). The liver toxicity of bicalutamide is not known to be dose-dependent across its clinically used dose range (Wiki). Abnormal LFTs have occurred with bicalutamide (at rates of 2.9% to 11.4%) even at relatively low doses in cisgender women (e.g., 10–50 mg/day) (de Melo Carvalho, 2022). Due to its risk of liver toxicity, periodic liver monitoring is strongly advised with bicalutamide, especially within the first 6 months of treatment. Possible signs of liver toxicity include nausea, vomiting, abdominal pain, fatigue, appetite loss, flu-like symptoms, dark urine, and jaundice (yellowing of the skin/eyes) (Wiki).
In terms of its lung toxicity risk, bicalutamide has been associated rarely with interstitial pneumonitis, which can lead to pulmonary fibrosis and can be fatal, and also less often with eosinophilic lung disease (Wiki; Table). As of writing, 15 published case reports of interstitial pneumonitis and 2 case reports of eosinophilic lung disease in association with bicalutamide therapy exist, likewise all in men with prostate cancer (Table). As with liver toxicity, hundreds of additional cases of interstitial pneumonitis in people taking bicalutamide exist in the United States FAERS database (Wiki; FDA). It has been estimated that interstitial pneumonitis with bicalutamide occurs at a rate of around 1 in 10,000 people, although this may be an underestimate due to under-reporting (Wiki; Ahmad & Graham, 2003). Asian people may be especially likely to experience lung toxicity with bicalutamide and other NSAAs, as much higher incidences have been observed in this population (Mahler et al., 1996; Wu et al., 2022). There is no laboratory test for routine monitoring of lung changes with bicalutamide. Possible signs of relevant lung toxicity include dyspnea (difficulty breathing or shortness of breath), coughing, and pharyngitis (inflammation of the throat, typically manifesting as sore throat) (Wiki).
Aside from liver and lung toxicity, bicalutamide monotherapy has been found in cisgender men with prostate cancer to increase the risk of death due to causes other than prostate cancer (Iversen et al., 2004; Iversen et al., 2006; Wellington & Keam, 2006; Jia & Spratt, 2022; Wiki). This led to marketing authorization of bicalutamide for treatment of the earliest stage of prostate cancer being revoked and to the drug being abandoned for this use (Wiki). Bicalutamide remains approved and used for treatment of later stages of prostate cancer, as the antiandrogenic benefits of bicalutamide against prostate cancer outweigh any adverse influence it has on non-prostate-cancer mortality in these more severe stages. The mechanisms underlying the increase in risk of death with bicalutamide in men are unknown (Wiki). It is also unclear whether bicalutamide could likewise increase the risk of death in transfeminine people. Limitations of generalizing these studies to transfeminine people include the men in the trials being relatively old and ill, a relatively high dosage of bicalutamide (150 mg/day) being used in the trials for an extended duration (e.g., 5 years), the question of whether the risks were due to androgen deprivation or to specific drug-related toxicity of bicalutamide, and estradiol levels with bicalutamide monotherapy in men with prostate cancer being only about 30 to 50 pg/mL (110–184 pmol/L) (Wiki). The preceding estradiol levels are well above castrate levels and are sufficient for a substantial degree of estrogenic effect, but are nonetheless below those recommended for transfeminine people and potentially needed for full sex-hormone replacement (which are ≥50 pg/mL [≥184 pmol/L]). In any case, as the specific mechanisms underlying the increased mortality risk with bicalutamide seen in men with prostate cancer are uncertain, and as clinical safety data showing that the risk does not generalize do not exist, it remains a possibility that bicalutamide could also increase the risk of death in transfeminine people.
Bicalutamide is taken orally in the form of tablets (e.g., 50 and 150 mg) (Wiki). Due to saturation of absorption in the gastrointestinal tract, the oral bioavailability of bicalutamide progressively starts to decrease above a dosage of about 150 mg/day, and there is no further increase in bicalutamide levels above 300 mg/day (Wiki; Graph). Bicalutamide has a very long elimination half-life of about 6 to 10 days (Wiki; Graphs). As a result, it does not necessarily have to be taken daily, and can be dosed less often (in proportionally higher doses)—for instance twice weekly or even once weekly—if this is more convenient or otherwise desired. Due to its long half-life, bicalutamide requires about 4 to 12 weeks to fully reach steady-state levels (Wiki; Graph; Wiki). However, about 50% of steady state is reached within 1 week of administration of bicalutamide, and about 80 to 90% of steady state is reached after 3 to 4 weeks (Wiki; Graph; Wiki). Loading doses of bicalutamide can be taken to reach steady state more quickly if desired. Animal studies originally suggested that bicalutamide did not cross the blood–brain barrier and hence was peripherally selective (i.e., did not block androgen receptors in the brain) (Wiki). However, subsequent clinical studies found that this was not similarly the case in humans, in whom bicalutamide shows clear and robust centrally mediated antiandrogenic effects (Wiki).
Older NSAAs related to bicalutamide like flutamide (Eulexin) and nilutamide (Anandron, Nilandron) have much greater risks in comparison to bicalutamide and should not be used in transfeminine people. Nilutamide was previously characterized as an antiandrogen in transfeminine people in several studies, but was not further pursued probably due to its very high incidence of lung toxicity and other side effects (Aly, 2020; Wiki; Wiki). Flutamide has been used limitedly as an antiandrogen in transfeminine people in the past, but should no longer be used due to a much higher risk of liver toxicity than bicalutamide as well as other side effects and drawbacks (Aly, 2020; Wiki). Other newer and more-potent NSAAs like enzalutamide (Xtandi), apalutamide (Erleada), and darolutamide (Nubeqa) also have risks and have been studied and used little outside of prostate cancer to date.
5α-Reductase Inhibitors
Testosterone is converted into DHT within certain tissues in the body (Swerdloff et al., 2017). DHT is an androgen metabolite of testosterone with several-fold higher activity than testosterone. The transformation of testosterone into DHT is mediated by the enzyme 5α-reductase. The tissues in which 5α-reductase is present and testosterone is converted into DHT are limited but most importantly include the skin, hair follicles, and prostate gland. Although DHT is more potent than testosterone, it is thought to have minimal biological role as a circulating hormone (Horton, 1992; Swerdloff et al., 2017). Instead, testosterone serves as the main circulating androgen, and the role of DHT is thought to be mainly via local metabolism and potentiation of testosterone into DHT within certain tissues.
5α-Reductase inhibitors (5α-RIs), such as finasteride (Proscar, Propecia) and dutasteride (Avodart), inhibit 5α-reductase and thereby block the conversion of testosterone into DHT. This results in marked decreases in circulating and within-tissue levels of DHT. Due to the primary role of DHT as a mediator in tissues rather than as circulating hormone, the antiandrogenic efficacy of 5α-RIs is limited. This is evidenced by the fact that they are well-tolerated in cisgender men and do not cause notable demasculinization in these individuals (Hirshburg, 2016). The medical use of 5α-RIs is mainly restricted to the treatment of scalp hair loss in men and women, hirsutism (excessive facial/body hair) in women, and prostate enlargement in men. They might also be useful for acne in women, but evidence of this is very limited (Wiki). Due to their specificity, 5α-RIs are inappropriate as general antiandrogens in transfeminine people. Moreover, DHT levels decrease in tandem with testosterone levels with suppression of testosterone production in transfeminine hormone therapy, and routine use of 5α-RIs in transfeminine people with testosterone levels within the female range is of limited usefulness and can be considered unnecessary (Gooren et al., 2016; Irwig, 2020; Prince & Safer, 2020; Glintborg et al., 2021). In any case, 5α-RIs may be useful in transfeminine people on hormone therapy who have persistent body hair growth or scalp hair loss—as they have been shown to be in cisgender women (Barrionuevo et al., 2018; Prince & Safer, 2020). However, it is notable that evidence of effectiveness in cisgender women is better for androgen receptor antagonists for such indications (van Zuuren et al., 2015). This is intuitive as androgen receptor antagonists block both testosterone and DHT whereas 5α-RIs only prevent conversion of testosterone into DHT. Hence, although 5α-RIs strongly reduce or eliminate DHT and their net effect is antiandrogenic, they do not decrease testosterone levels and in fact increase them.
There are three subtypes of 5α-reductase. Dutasteride inhibits all three subtypes of 5α-reductase whereas finasteride only inhibits two of the subtypes. As a result of this, dutasteride is a more complete 5α-RI than finasteride. Dutasteride decreases DHT levels in the blood by up to 98% while finasteride can only decrease them by around 65 to 70%. As nearly all circulating DHT originates from synthesis in peripheral tissues, these decreases indicate parallel reductions in tissue DHT production (Horton, 1992). In accordance with these findings, dutasteride has been found to be more effective than finasteride in the treatment of scalp hair loss in men (Zhou et al., 2018; Dhurat et al., 2020; Wiki). For these reasons, although both finasteride and dutasteride are effective 5α-RIs, dutasteride may be the preferable choice if a 5α-RI is used (Zhou et al., 2018; Dhurat et al., 2020).
A potentially undesirable effect of 5α-RIs in transfeminine people is that they may increase circulating testosterone levels to a degree in those in whom testosterone production isn’t fully suppressed (Leinung, Feustel, & Joseph, 2018; Aly, 2019; Traish et al., 2019; Irwig, 2020; Glintborg et al., 2021). It appears that DHT adds significantly to negative feedback on gonadotropin secretion in the pituitary gland in people with testes who have low testosterone levels relative to the normal male range (Traish et al., 2019). The therapeutic implications of this for transfeminine people, if any, are uncertain.
Clinical dose-ranging studies have found that lower doses of finasteride and dutasteride than are typically used still provide substantial or near-maximal 5α-reductase inhibition (Gormley et al., 1990; Vermeulen et al., 1991; Sudduth & Koronkowski, 1993; Drake et al., 1999; Roberts et al., 1999; Clark et al., 2004; Frye, 2006; Olsen et al., 2006; Harcha et al., 2014; Kuhl & Wiegratz, 2017). In one study with finasteride for instance, DHT levels decreased by 49.5% at 0.05 mg/day, 68.6% at 0.2 mg/day, 71.4% at 1 mg/day, and 72.2% at 5 mg/day (Drake et al., 1999). Parallel reductions in DHT levels were seen locally in the scalp (Drake et al., 1999). In a study with dutasteride, DHT levels were decreased by 52.9% at 0.05 mg/day, 94.7% at 0.5 mg/day, 97.7% at 2.5 mg/day, and 98.4% at 5 mg/day (Clark et al., 2004). Based on these findings, 5α-RIs can potentially be taken at lower doses to help reduce medication costs if needed. Finasteride tablets can be split to achieve smaller doses. Conversely, dutasteride cannot be split as it is formulated as an oil capsule. However, dutasteride has a long half-life, and instead of dividing pills, it can be taken less frequently (e.g., once every few days) as a means of reducing dosage.
5α-Reductase inhibitors are taken orally in the form of tablets and capsules. Compoundedtopical formulations of finasteride also exist (Marks et al., 2020). However, caution is advised with these preparations as they have been found to be excessively dosed and to produce equivalent systemic DHT suppression as oral finasteride formulations (Marks et al., 2020). Lower-concentration formulations of topical finsteride on the other hand may be more locally selective (Marks et al., 2020).
Table 8: Available forms and recommended doses of 5α-reductase inhibitors for transfeminine people:
GnRH agonists and antagonists (GnRHa), also known as GnRH receptor agonists and antagonists or GnRH modulators, are antiandrogens which work by preventing the effects of GnRH in the pituitary gland and thereby suppressing LH and FSH secretion. Receptor agonists normally activate receptors while receptor antagonists block and thereby inhibit the activation of receptors. Due to a physiological quirk however, GnRH agonists and antagonists have the same effects in the pituitary gland. This is because GnRH is secreted in pulses under normal physiological circumstances, and when the GnRH receptor is unnaturally activated in a continuous manner, as with exogenous GnRH agonists, the GnRH receptor in the pituitary gland is strongly desensitized to the point of becoming inactive. Consequently, both GnRH agonists and GnRH antagonists have the effect of abolishing gonadal sex hormone production. This, in turn, reduces testosterone levels into the castrate or normal female range (both <50 ng/dL or <1.7 nmol/L) in people with testes. GnRHa are like a reversible gonadectomy, and for this reason, are also sometimes referred to as “medical castration”. Provided that an estrogen is taken in combination with a GnRHa to prevent sex hormone deficiency, these medications have essentially no known side effects or risks. For these reasons, GnRHa are the ideal antiandrogens for use in transfeminine people.
GnRHa are widely used to suppess puberty in adolescent transgender individuals. Unfortunately however, they are very expensive (e.g., ~US$10,000 per year) and medical insurance does not usually cover them for adult transgender people. Consequently, GnRHa are not commonly used in adult transfeminine people at this time. An exception is in the United Kingdom, where GnRH agonists are covered for all adult transgender people by the National Health Service (NHS). Another exception is buserelin (Suprefact), which has become available very inexpensively in its nasal spray form from certain Eastern European online pharmacies in recent years (Aly, 2018).
GnRH agonists cause a brief flare in testosterone levels at the start of therapy prior to the GnRH receptors in the pituitary gland becoming desensitized (Wiki). Testosterone levels increase by up to about 1.5- to 2-fold for about 1 week and then decrease thereafter (Wiki). Castrate or female-range levels of testosterone are generally reached within 2 to 4 weeks (Wiki). In contrast to GnRH agonists, there is no testosterone flare with GnRH antagonists and testosterone levels start decreasing immediately, reaching castrate levels within a few days (Wiki; Graph). This is because GnRH antagonists work by blocking the GnRH receptor without initially activating it, and hence desensitization of the receptor is not necessary for their action. If desired, the testosterone flare at the initiation of GnRH agonist therapy can be prevented or blunted with the use of antigonadotropins, for instance estrogens and progestogens, as well as with potent androgen receptor antagonists such as bicalutamide (Wiki).
GnRH agonists must be injected subcutaneously or intramuscularly once per day or once every one to six months depending on the formulation employed (buserelin, goserelin, leuprorelin, triptorelin). Alternatively, they can be surgically implanted once a year (histrelin, leuprorelin) or used as a nasal spray two to three times per day (buserelin, nafarelin). The first GnRH antagonists were developed for use by once-monthly intramuscular or subcutaneous injection (abarelix, degarelix). More recently, orally administered GnRH antagonists such as elagolix and relugolix have been introduced for medical use. They are taken in the form of tablets once or twice daily.
Table 9: Available forms and recommended doses of GnRH agonists for transfeminine people:
a 500 μg 3x/day for the first week then 200 μg/day. b 800 μg 3x/day for the first week then 400 μg 3x/day. c 500 μg 2x/day can be used instead of 400 μg 3x/day but is less effective (70% decrease in testosterone levels (to ~180 ng/dL [6.2 nmol/L]) instead of 90% decrease (to ~50 ng/dL [1.7 nmol/L]) per available studies of buserelin in men with prostate cancer) (Aly, 2018; Wiki).
Table 10: Available forms and recommended doses of GnRH antagonists for transfeminine people:
a First month is 240 mg then 80 mg per month thereafter. b 150 mg 1x/day is less effective than 200 mg 2x/day (which provides full gonadal sex-hormone suppression in cisgender women) (Wiki). c 80–120 mg/day for full gonadal sex-hormone suppression and 20–40 mg/day for substantial but partial gonadal sex-hormone suppression (MacLean et al., 2015; DailyMed).
Other Hormonal Medications
Androgens and Anabolic Steroids
In addition to estrogens, progestogens, and antiandrogens, androgens/anabolic steroids (AAS) are sometimes added to transfeminine hormone therapy. This is when testosterone levels are low (e.g., below the female average of 30 ng/dL [1.0 nmol/L]) and androgen replacement is desired. It has been proposed that adequate levels of testosterone may provide benefits such as increased sexual desire, improved mood and energy, positive effects on skin health and cellulite (Avram, 2004), and increased muscle size and strength (Huang & Basaria, 2017). However, there is insufficient clinical evidence to support such benefits at present, and androgens can produce adverse effects in cisgender women and transfeminine people, for instance acne, hirsutism, scalp hair loss, and masculinization (Wiki). For transfeminine people who nonetheless desire androgen replacement therapy, possible options for androgen medications include testosterone and its esters, dehydroepiandrosterone (DHEA; prasterone), and nandroloneesters such as nandrolone decanoate (ND) (Aly, 2020; Table), among others.
Monitoring of Therapy
Transfeminine people on hormone therapy should undergo regular laboratory monitoring in the form of blood work to assess efficacy and monitor for safety. Total estradiol levels and total testosterone levels should be measured to assess the effectiveness of therapy—that is, whether hormone levels are in appropriate ranges for cisgender females—and determine whether medication adjustments may be necessary. Levels of free testosterone, free estradiol, estrone (E1), dihydrotestosterone (DHT), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and sex hormone-binding globulin (SHBG) can also be measured to provide further information although they’re not absolutely necessary. If progesterone is used as a part of hormone therapy, progesterone levels can be measured to provide insight on the degree of progesterone exposure. In addition to hormone blood tests, transfeminine people can monitor their physical changes with hormone therapy, such as breast development and other aspects of feminization, using various physical and digital measurement methods (e.g., Wiki).
Certain therapeutic situations can result in inaccurate lab blood work results. Monitoring of progesterone levels with oral progesterone using immunoassay-based blood tests can result in falsely high readings for progesterone levels due to cross-reactivity with high levels of progesterone metabolites such as allopregnanolone (Aly, 2018; Wiki). Instead of immunoassay-based tests, mass spectrometry-based tests should be used to determine progesterone levels with oral progesterone (Aly, 2018; Wiki). Conversely, either type of test may be used to measure progesterone levels with non-oral progesterone therapy. High-dose biotin (vitamin B7) supplements can interfere with the accuracy of immunoassay-based hormone blood tests, causing falsely low or falsely high readings (Samarasinghe et al., 2017; Avery, 2019; Bowen et al., 2019; FDA, 2019; Luong, Male, & Glennon, 2019). Transdermal estradiol formulations applied to the arm can result in contamination of blood draws taken from the same arm and can result in falsely high readings for estradiol levels (Vihtamäkia, Luukkaala, & Tuimala, 2004).
Certain cancers are known to be hormone-sensitive and their incidence can be influenced by hormone therapy. Screening for breast and prostate cancer is recommended in transfeminine people (Sterling & Garcia, 2020; Iwamoto et al., 2021). The risk of breast cancer appears to be dramatically increased with transfeminine hormone therapy, perhaps especially with progestogens (Aly, 2020). However, the risk still remains lower than in cisgender women (Aly, 2020). The incidence of prostate cancer is greatly decreased with hormone therapy in transfeminine people as a consequence of androgen deprivation, but the risk is not abolished and prostate cancer can still occur (de Nie et al., 2020). The prostate gland is not removed with vaginoplasty, so transfeminine people who have undergone vaginoplasty will also require monitoring for prostate cancer still. Testicular cancer is not known to be a hormone-dependent cancer and its incidence does not appear to be increased with hormone therapy in transfeminine people (Bensley et al., 2021; de Nie et al., 2021; Jacoby et al., 2021).
Ahmad, S. R., & Graham, D. J. (2003). Pneumonitis with Antiandrogens. Annals of Internal Medicine, 139(6), 528–529. [DOI:10.7326/0003-4819-139-6-200309160-00023]
Allen, A. N., Jiao, R., Day, P., Pagels, P., Gimpel, N., & SoRelle, J. A. (2020). Dynamic Impact of Hormone Therapy on Laboratory Values in Transgender Patients over Time. The Journal of Applied Laboratory Medicine, 6(1), 27–40. [DOI:10.1093/jalm/jfaa192]
Aly. (2018). A Review of Studies on Spironolactone and Testosterone Suppression in Cisgender Men, Cisgender Women, and Transfeminine People. Transfeminine Science. [URL]
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
Aly. (2018). Buserelin, a Gonadotropin-Releasing Hormone Agonist, is Inexpensively Available as a Nasal Spray From Online Pharmacies. Transfeminine Science. [URL]
Aly. (2018). EC508 (Estradiol Aminosulfonylbenzoylproline), a Unique and Highly Promising Estradiol Ester and Possible Oral Estradiol Form of the Future. Transfeminine Science. [URL]
Aly. (2018). Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects. Transfeminine Science. [URL]
Aly. (2019). A Review of Studies on Estradiol Levels and Testosterone Suppression with High-Dose Transdermal Estradiol Gel and Ointment in Cisgender Men with Prostate Cancer. Transfeminine Science. [URL]
Aly. (2019). An Exploration of Possibilities for Hormone Therapy in Non-Binary Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). Analysis of Estradiol and Testosterone Levels with Oral Estradiol in Transfeminine People Based on Leinung et al. (2018). Transfeminine Science. [URL]
Aly. (2019). Aqueous Suspensions of Sex Hormones and How Depot Injectables Work. Transfeminine Science. [URL]
Aly. (2019). Genital Application via the Scrotum and Neolabia for Greatly Enhanced Absorption of Transdermal Estradiol in Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). Literature on Early Progestogen Exposure and Breast Development. Transfeminine Science. [URL]
Aly. (2019). Low Doses of Cyproterone Acetate Are Maximally Effective for Testosterone Suppression in Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). Published Case Reports of Lactation and/or Breastfeeding in Transfeminine People. Transfeminine Science. [URL]
Aly. (2019). Sublingual Administration of Oral Estradiol Valerate Tablets for Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Aly. (2020). Bicalutamide and its Adoption by the Medical Community for Use in Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Aly. (2020). Breast Cancer Risk with Hormone Therapy in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Clinical Guidelines with Information on Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
Aly. (2020). Hormone Levels During Normal Puberty in Cisgender Girls. Transfeminine Science. [URL]
Aly. (2020). Nandrolone as a Potential Alternative Androgen with Reduced Androgenic Side Effects for Transfeminine and Transmasculine People. Transfeminine Science. [URL]
Aly. (2020). Recent Developments on Cyproterone Acetate and Meningioma Risk Out of France and Implications for Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). Spironolactone and Claims About Increased Visceral Fat in Transfeminine People. Transfeminine Science. [URL]
Aly. (2020). The Influence of Progesterone and Other Progestogens on Sexual Desire and Function. Transfeminine Science. [URL]
Aly. (2020). The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Aly. (2020). The Possible Role of a Second X Chromosome in Breast Cancer Risk. Transfeminine Science. [URL]
Aly. (2021). An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations. Transfeminine Science. [URL]
Aly. (2021). An Interactive Web Simulator for Estradiol Levels with Injectable Estradiol Esters. Transfeminine Science. [URL]
Angus, L., Leemaqz, S., Ooi, O., Cundill, P., Silberstein, N., Locke, P., Zajac, J. D., & Cheung, A. S. (2019). Cyproterone acetate or spironolactone in lowering testosterone concentrations for transgender individuals receiving oestradiol therapy. Endocrine Connections, 8(7), 935–940. [DOI:10.1530/ec-19-0272]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2020). A systematic review of antiandrogens and feminization in transgender women. Clinical Endocrinology, 94(5), 743–752. [DOI:10.1111/cen.14329]
Antoniou, T., Gomes, T., Mamdani, M. M., Yao, Z., Hellings, C., Garg, A. X., Weir, M. A., & Juurlink, D. N. (2011). Trimethoprim-sulfamethoxazole induced hyperkalaemia in elderly patients receiving spironolactone: nested case-control study. BMJ, 343, d5228. [DOI:10.1136/bmj.d5228]
Antoniou, T., Hollands, S., Macdonald, E. M., Gomes, T., Mamdani, M. M., & Juurlink, D. N. (2015). Trimethoprim–sulfamethoxazole and risk of sudden death among patients taking spironolactone. Canadian Medical Association Journal, 187(4), E138–E143. [DOI:10.1503/cmaj.140816]
Aufrère, M. B., & Benson, H. (1976). Progesterone: An overview and recent advances. Journal of Pharmaceutical Sciences, 65(6), 783–800. [DOI:10.1002/jps.2600650602]
Avery, G. (2019). Biotin interference in immunoassay: a review for the laboratory scientist. Annals of Clinical Biochemistry: International Journal of Laboratory Medicine, 56(4), 424–430. [DOI:10.1177/0004563219842231]
Avram, M. M. (2004). Cellulite: a review of its physiology and treatment. Journal of Cosmetic and Laser Therapy, 6(4), 181–185. [DOI:10.1080/14764170410003057]
Babich, J. S., Kalantar-Zadeh, K., & Joshi, S. (2022). Taking the Kale out of Hyperkalemia: Plant Foods and Serum Potassium in Patients With Kidney Disease. Journal of Renal Nutrition, 32(6), 641–649. [DOI:10.1053/j.jrn.2022.01.013]
Bäckström, T., Haage, D., Löfgren, M., Johansson, I., Strömberg, J., Nyberg, S., Andréen, L., Ossewaarde, L., van Wingen, G., Turkmen, S., & Bengtsson, S. (2011). Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons. Neuroscience, 191, 46–54. [DOI:10.1016/j.neuroscience.2011.03.061]
Barbieri, J. S., Margolis, D. J., & Mostaghimi, A. (2021). Temporal Trends and Clinician Variability in Potassium Monitoring of Healthy Young Women Treated for Acne With Spironolactone. JAMA Dermatology, 157(3), 296–296. [DOI:10.1001/jamadermatol.2020.5468]
Balen, A. H., Conway, G. S., Kaltsas, G., Techatraisak, K., Manning, P. J., West, C., & Jacobs, H. S. (1995). Andrology: Polycystic ovary syndrome: the spectrum of the disorder in 1741 patients. Human Reproduction, 10(8), 2107–2111. [DOI:10.1093/oxfordjournals.humrep.a136243]
Barrionuevo, P., Nabhan, M., Altayar, O., Wang, Z., Erwin, P. J., Asi, N., Martin, K. A., & Murad, M. H. (2018). Treatment Options for Hirsutism: A Systematic Review and Network Meta-Analysis. The Journal of Clinical Endocrinology & Metabolism, 103(4), 1258–1264. [DOI:10.1210/jc.2017-02052]
Bazarra-Castro, M. A. (2009). Etiological aspects, therapy regimes, side effects and treatment satisfaction of transsexual patients. (Doctoral dissertation, Ludwig Maximilian University of Munich.) [DOI:10.5282/edoc.9984] [URN:urn:nbn:de:bvb:19-99840] [PDF]
Belisle, S., & Love, E. J. (1986). Clinical efficacy and safety of cyproterone acetate in severe hirsutism: results of a multicentered Canadian study. Fertility and Sterility, 46(6), 1015–1020. [DOI:10.1016/s0015-0282(16)49873-0]
Ben Salem, C., Badreddine, A., Fathallah, N., Slim, R., & Hmouda, H. (2014). Drug-Induced Hyperkalemia. Drug Safety, 37(9), 677–692. [DOI:10.1007/s40264-014-0196-1]
Bensley, J. G., Cheung, A. S., Grossmann, M., & Papa, N. (2022). Testicular Cancer in Trans People Using Feminising Hormone Therapy–A Brief Review. Urology, 160, 1–4. [DOI:10.1016/j.urology.2021.11.014]
Bessone, F., Lucena, M., Roma, M. G., Stephens, C., Medina-Cáliz, I., Frider, B., Tsariktsian, G., Hernández, N., Bruguera, M., Gualano, G., Fassio, E., Montero, J., Reggiardo, M. V., Ferretti, S., Colombato, L., Tanno, F., Ferrer, J., Zeno, L., Tanno, H., & Andrade, R. J. (2015). Cyproterone acetate induces a wide spectrum of acute liver damage including corticosteroid-responsive hepatitis: report of 22 cases. Liver International, 36(2), 302–310. [DOI:10.1111/liv.12899]
Boogers, L. S., Sardo Infirri, S. A., Bouchareb, A., Dijkman, B. A., Helder, D., de Blok, C. J., Liberton, N. P., den Heijer, M., van Trotsenburg, A. S., Dreijerink, K. M., Wiepjes, C. M., & Hannema, S. E. (2025). Variations in Volume: Breast Size in Trans Women in Relation to Timing of Testosterone Suppression. The Journal of Clinical Endocrinology & Metabolism, 110(5), e1404–e1410. [DOI:10.1210/clinem/dgae573]
Bowen, R., Benavides, R., Colón-Franco, J. M., Katzman, B. M., Muthukumar, A., Sadrzadeh, H., Straseski, J., Klause, U., & Tran, N. (2019). Best practices in mitigating the risk of biotin interference with laboratory testing. Clinical Biochemistry, 74, 1–11. [DOI:10.1016/j.clinbiochem.2019.08.012]
Brown, J., Farquhar, C., Lee, O., Toomath, R., & Jepson, R. G. (2009). Spironolactone versus placebo or in combination with steroids for hirsutism and/or acne. Cochrane Database of Systematic Reviews, 2009(2), CD000194. [DOI:10.1002/14651858.cd000194.pub2]
Burinkul, S., Panyakhamlerd, K., Suwan, A., Tuntiviriyapun, P., & Wainipitapong, S. (2021). Anti-Androgenic Effects Comparison Between Cyproterone Acetate and Spironolactone in Transgender Women: A Randomized Controlled Trial. The Journal of Sexual Medicine, 18(7), 1299–1307. [DOI:10.1016/j.jsxm.2021.05.003]
Callen-Lorde Community Health Center. (2018). Protocols for the Provision of Hormone Therapy. New York City: Callen-Lorde Community Health Center. [URL] [PDF]
Cappelletti, M., & Wallen, K. (2016). Increasing women’s sexual desire: The comparative effectiveness of estrogens and androgens. Hormones and Behavior, 78, 178–193. [DOI:10.1016/j.yhbeh.2015.11.003]
Carmina, E., Stanczyk, F. Z., & Lobo, R. A. (2019). Evaluation of Hormonal Status. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 887–915.e4). Philadelphia: Elsevier. [DOI:10.1016/b978-0-323-47912-7.00034-2]
Carvalho, R. d., Santos, L. D., Ramos, P. M., Machado, C. J., Acioly, P., Frattini, S. C., Barcaui, C. B., Donda, A. L., & Melo, D. F. (2022). Bicalutamide and the new perspectives for female pattern hair loss treatment: What dermatologists should know. Journal of Cosmetic Dermatology, 21(10), 4171–4175. [DOI:10.1111/jocd.14773]
Chang, J. J., Tran, N. K., Flentje, A., Lubensky, M. E., Obedin-Maliver, J., Lunn, M. R., & Ariel, D. (2024). 12330 Progestogen Experience And Perception Among Transfeminine Adults - A National Survey. Journal of the Endocrine Society, 8(Suppl 1), bvae163.1657. [DOI:10.1210/jendso/bvae163.1657]
Clark, R. V., Hermann, D. J., Cunningham, G. R., Wilson, T. H., Morrill, B. B., & Hobbs, S. (2004). Marked Suppression of Dihydrotestosterone in Men with Benign Prostatic Hyperplasia by Dutasteride, a Dual 5α-Reductase Inhibitor. The Journal of Clinical Endocrinology & Metabolism, 89(5), 2179–2184. [DOI:10.1210/jc.2003-030330]
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/s0140-6736(19)31709-x]
Collet, S., Gieles, N. C., Wiepjes, C. M., Heijboer, A. C., Reyns, T., Fiers, T., Lapauw, B., den Heijer, M., & T’Sjoen, G. (2022). Changes in Serum Testosterone and Adrenal Androgen Levels in Transgender Women With and Without Gonadectomy. The Journal of Clinical Endocrinology & Metabolism, 108(2), 331–338. [DOI:10.1210/clinem/dgac576]
Colonnello, E., Graziani, A., Rossetti, R., Voltan, G., Masi, D., Lubrano, C., Mariani, S., Watanabe, M., Isidori, A. M., Ferlin, A., & Gnessi, L. (2025). The Chronobiology of Hormone Administration: “Doctor, What Time Should I Take My Medication?”. Endocrine Reviews, bnaf013. [DOI:10.1210/endrev/bnaf013]
Connors, J. M., & Middeldorp, S. (2019). Transgender patients and the role of the coagulation clinician. Journal of Thrombosis and Haemostasis, 17(11), 1790–1797. [DOI:10.1111/jth.14626]
Cupisti, A., Kovesdy, C., D’Alessandro, C., & Kalantar-Zadeh, K. (2018). Dietary Approach to Recurrent or Chronic Hyperkalaemia in Patients with Decreased Kidney Function. Nutrients, 10(3), 261–261. [DOI:10.3390/nu10030261]
Cusan, L., Dupont, A., Gomez, J., Tremblay, R. R., & Labrie, F. (1994). Comparison of flutamide and spironolactone in the treatment of hirsutism: a randomized controlled trial. Fertility and Sterility, 61(2), 281–287. [DOI:10.1016/s0015-0282(16)56518-2]
de Blok, C. J., Wiepjes, C. M., Nota, N. M., van Engelen, K., Adank, M. A., Dreijerink, K. M., Barbé, E., Konings, I. R., & den Heijer, M. (2019). Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in the Netherlands. BMJ, 365, l1652. [DOI:10.1136/bmj.l1652]
de Blok, C. J., Dijkman, B. A., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K. M., & den Heijer, M. (2021). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
de Nie, I., de Blok, C. J., van der Sluis, T. M., Barbé, E., Pigot, G. L., Wiepjes, C. M., Nota, N. M., van Mello, N. M., Valkenburg, N. E., Huirne, J., Gooren, L. J., van Moorselaar, R. J., Dreijerink, K. M., & den Heijer, M. (2020). Prostate Cancer Incidence under Androgen Deprivation: Nationwide Cohort Study in Trans Women Receiving Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 105(9), e3293–e3299. [DOI:10.1210/clinem/dgaa412]
de Nie, I., Wiepjes, C. M., Blok, C. J., Moorselaar, R. J., Pigot, G. L., Sluis, T. M., Barbé, E., Voorn, P., Mello, N. M., Huirne, J., & Heijer, M. (2021). Incidence of testicular cancer in trans women using gender‐affirming hormonal treatment: a nationwide cohort study. BJU International, 129(4), 491–497. [DOI:10.1111/bju.15575]
Deng, T., Duan, X., He, Z., Zhao, Z., & Zeng, G. (2020). Association Between 5-Alpha Reductase Inhibitor Use and The Risk of Depression: A Meta-Analysis. Urology Journal, 18(2), 144–150. [DOI:10.22037/uj.v16i7.5866]
Deutsch, M. B. (2016). Overview of feminizing hormone therapy. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 26–48). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Dewhurst, J., & Underhill, R. (1979). The Suppression of Facial Hair Growth in Transsexuals Using Cyproterone Acetate. British Journal of Sexual Medicine, 6, 19–23. [Google Scholar] [PDF]
Dhurat, R., Sharma, A., Rudnicka, L., Kroumpouzos, G., Kassir, M., Galadari, H., Wollina, U., Lotti, T., Golubovic, M., Binic, I., Grabbe, S., & Goldust, M. (2020). 5‐Alpha reductase inhibitors in androgenetic alopecia: Shifting paradigms, current concepts, comparative efficacy, and safety. Dermatologic Therapy, 33(3), e13379. [DOI:10.1111/dth.13379]
Drake, L., Hordinsky, M., Fiedler, V., Swinehart, J., Unger, W. P., Cotterill, P. C., Thiboutot, D. M., Lowe, N., Jacobson, C., Whiting, D., Stieglitz, S., Kraus, S. J., Griffin, E. I., Weiss, D., Carrington, P., Gencheff, C., Cole, G. W., Pariser, D. M., Epstein, E. S., Tanaka, W., Dallob, A., Vandormael, K., Geissler, L., & Waldsteicher, J. (1999). The effects of finasteride on scalp skin and serum androgen levels in men with androgenetic alopecia. Journal of the American Academy of Dermatology, 41(4), 550–554. [DOI:10.1016/s0190-9622(99)80051-6]
Dyson, T. E., Cantrell, M. A., & Lund, B. C. (2020). Lack of Association between 5α-Reductase Inhibitors and Depression. Journal of Urology, 204(4), 793–798. [DOI:10.1097/ju.0000000000001079]
Erem, C. (2013). Update on idiopathic hirsutism: diagnosis and treatment. Acta Clinica Belgica, 68(4), 268–274. [DOI:10.2143/acb.3267]
Erenus, M., Gürbüz, O., Durmuşoğlu, F., Demirçay, Z., & Pekin, S. (1994). Comparison of the efficacy of spironolactone versus flutamide in the treatment of hirsutism. Fertility and Sterility, 61(4), 613–616. [DOI:10.1016/s0015-0282(16)56634-5]
Esoterix/LabCorp. (2020). Endocrinology Expected Values and S.I. Unit Conversion Tables. LabCorp/Endocrine Sciences. [PDF]
Even Zohar, N., Sofer, Y., Yaish, I., Serebro, M., Tordjman, K., & Greenman, Y. (2021). Low-Dose Cyproterone Acetate Treatment for Transgender Women. The Journal of Sexual Medicine, 18(7), 1292–1298. [DOI:10.1016/j.jsxm.2021.04.008]
Fertig, R., Shapiro, J., Bergfeld, W., & Tosti, A. (2016). Investigation of the Plausibility of 5-Alpha-Reductase Inhibitor Syndrome. Skin Appendage Disorders, 2(3–4), 120–129. [DOI:10.1159/000450617]
Food and Drug Administration. (2019). UPDATE: The FDA Warns that Biotin May Interfere with Lab Tests: FDA Safety Communication. Food and Drug Administration. [URL]
Fournier, A., Berrino, F., & Clavel-Chapelon, F. (2007). Unequal risks for breast cancer associated with different hormone replacement therapies: results from the E3N cohort study. Breast Cancer Research and Treatment, 107(1), 103–111. [DOI:10.1007/s10549-007-9523-x]
Frye, S. (2006). Discovery and Clinical Development of Dutasteride, a Potent Dual 5α-Reductase Inhibitor. Current Topics in Medicinal Chemistry, 6(5), 405–421. [DOI:10.2174/156802606776743101]
Gava, G., Mancini, I., Alvisi, S., Seracchioli, R., & Meriggiola, M. C. (2020). A comparison of 5-year administration of cyproterone acetate or leuprolide acetate in combination with estradiol in transwomen. European Journal of Endocrinology, 183(6), 561–569. [DOI:10.1530/eje-20-0370]
Getzenberg, R., & Itty, S. (2020). How do we define “castration” in men on androgen deprivation therapy? Asian Journal of Andrology, 22(5), 441–446. [DOI:10.4103/aja.aja_139_19]
Giltay, E. J., Gooren, L. J., Toorians, A. W., Katan, M. B., & Zock, P. L. (2004). Docosahexaenoic acid concentrations are higher in women than in men because of estrogenic effects. The American Journal of Clinical Nutrition, 80(5), 1167–1174. [DOI:10.1093/ajcn/80.5.1167]
Glintborg, D., T’Sjoen, G., Ravn, P., & Andersen, M. S. (2021). MANAGEMENT OF ENDOCRINE DISEASE: Optimal feminizing hormone treatment in transgender people. European Journal of Endocrinology, 185(2), R49–R63. [DOI:10.1530/eje-21-0059]
Goletiani, N. V., Keith, D. R., & Gorsky, S. J. (2007). Progesterone: Review of safety for clinical studies. Experimental and Clinical Psychopharmacology, 15(5), 427–444. [DOI:10.1037/1064-1297.15.5.427]
Goodfellow, A., Alaghband-Zadeh, J., Carter, G., Cream, J., Holland, S., Scully, J., & Wise, P. (1984). Oral spironolactone improves acne vulgaris and reduces sebum excretion. British Journal of Dermatology, 111(2), 209–214. [DOI:10.1111/j.1365-2133.1984.tb04045.x]
Gooren, L. J. (2016). The Endocrinology of Sexual Behavior and Gender Identity. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition, Volume 2 (pp. 2163–2176.e4). Philadelphia: Saunders/Elsevier. [Google Books] [DOI:10.1016/B978-0-323-18907-1.00124-4]
Gooren, L., & Bunck, M. (2004). Transsexuals and competitive sports. European Journal of Endocrinology, 151(4), 425–429. [DOI:10.1530/eje.0.1510425]
Gooren, L. J., Harmsen-Louman, W., & Kessel, H. (1985). Follow-up of prolactin levels in long-term oestrogen-treated male-to-female transsexuals with regard to prolactinoma induction. Clinical Endocrinology, 22(2), 201–207. [DOI:10.1111/j.1365-2265.1985.tb01081.x]
Gooren, L., Rao, B., van Kessel, H., & Harmsen-Louman, W. (1984). Estrogen positive feedback on LH secretion in transsexuality. Psychoneuroendocrinology, 9(3), 249–259. [DOI:10.1016/0306-4530(84)90004-0]
Gormley, G. J., Stoner, E., Rittmaster, R. S., Gregg, H., Thompson, D. L., Lasseter, K. C., Vlasses, P. H., & Stein, E. A. (1990). Effects of Finasteride (MK-906), a 5α-Reductase Inhibitor, on Circulating Androgens in Male Volunteers. The Journal of Clinical Endocrinology & Metabolism, 70(4), 1136–1141. [DOI:10.1210/jcem-70-4-1136]
Grock, S., Weinreb, J., Williams, K. C., Weimer, A., Fadich, S., Patel, R., Geft, A., & Korenman, S. (2024). Priorities for efficacy trials of gender-affirming hormone therapy with estrogen: collaborative design and results of a community survey. Hormones, online ahead of print. [DOI:10.1007/s42000-024-00532-3]
Gubelin Harcha, W., Barboza Martínez, J., Tsai, T., Katsuoka, K., Kawashima, M., Tsuboi, R., Barnes, A., Ferron-Brady, G., & Chetty, D. (2014). A randomized, active- and placebo-controlled study of the efficacy and safety of different doses of dutasteride versus placebo and finasteride in the treatment of male subjects with androgenetic alopecia. Journal of the American Academy of Dermatology, 70(3), 489–498.e3. [DOI:10.1016/j.jaad.2013.10.049]
Gupta, P., Suppakitjanusant, P., Stevenson, M., Goodman, M., & Tangpricha, V. (2022). Potassium Concentrations in Transgender Women Using Spironolactone: A Retrospective Chart Review. Endocrine Practice, 28(11), 1113–1117. [DOI:10.1016/j.eprac.2022.08.007]
Hammerstein, J. (1990). Antiandrogens: Clinical Aspects. In Orfanos, C. E., & Happle, R. (Eds.). Hair and Hair Diseases (pp. 827–886). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-74612-3_35]
Hayes, H., Russell, R., Haugen, A., Nagavally, S., & Sarvaideo, J. (2022). The Utility of Monitoring Potassium in Transgender, Gender Diverse, and Nonbinary Individuals on Spironolactone. Journal of the Endocrine Society, 6(11), bvac133. [DOI:10.1210/jendso/bvac133]
Heinemann, L. A., Will-Shahab, L., van Kesteren, P., Gooren, L. J., & (1997). Safety of Cyproterone Acetate: Report of Active Surveillance. Pharmacoepidemiology and Drug Safety, 6(3), 169–178. [DOI:10.1002/(sici)1099-1557(199705)6:3<169::aid-pds263>3.0.co;2-3]
Hembree, W. C., Cohen-Kettenis, P. T., Gooren, L., Hannema, S. E., Meyer, W. J., Murad, M. H., Rosenthal, S. M., Safer, J. D., Tangpricha, V., & T’Sjoen, G. G. (2017). Endocrine Treatment of Gender-Dysphoric/Gender-Incongruent Persons: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology and Metabolism, 102(11), 3869–3903. [DOI:10.1210/jc.2017-01658] [PDF]
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of the Subcutaneous and Intramuscular Estradiol Regimens as Part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
Hilton, E. N., & Lundberg, T. R. (2020). Transgender Women in the Female Category of Sport: Perspectives on Testosterone Suppression and Performance Advantage. Sports Medicine, 51(2), 199–214. [DOI:10.1007/s40279-020-01389-3]
Hirshburg, J. M., Kelsey, P. A., Therrien, C. A., Gavino, A. C., & Reichenberg, J. S. (2016). Adverse Effects and Safety of 5-alpha Reductase Inhibitors (Finasteride, Dutasteride): A Systematic Review. The Journal of Clinical and Aesthetic Dermatology, 9(7), 56–62. [PubMed] [PubMed Central]
Hopkins, U., & Arias, C. Y. (2013). Large-volume IM injections: a review of best practices. Oncology Nurse Advisor, 4(1), 32–37. [Google Scholar] [URL] [PDF]
Horton, R. (1992). Dihydrotestosterone Is a Peripheral Paracrine Hormone. Journal of Andrology, 13(1), 23–27. [DOI:10.1002/j.1939-4640.1992.tb01621.x]
Huang, G., & Basaria, S. (2017). The Case for Androgens in Menopausal Women: When and How? In Pal, L., & Sayegh, R. A. (Eds.). Essentials of Menopause Management: A Case-Based Approach (pp. 173–196). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-42451-4_10]
Igo, J., & Visram, H. (2021). Testosterone Suppression With Injectable Estrogen Therapy Alone in Male to Female Transgender Patients. Canadian Journal of Diabetes, 45(7 Suppl), S40–S40 (abstract no. 114). [DOI:10.1016/j.jcjd.2021.09.124] [URL]
Ingram, B. J., & Thomas, C. L. (2019). Transgender Policy in Sport, A Review of Current Policy and Commentary of the Challenges of Policy Creation. Current Sports Medicine Reports, 18(6), 239–247. [DOI:10.1249/jsr.0000000000000605]
Irwig, M. S. (2020). Is there a role for 5α‐reductase inhibitors in transgender individuals? Andrology, 9(6), 1729–1731. [DOI:10.1111/andr.12881]
Iversen, P., Johansson, J. E., Lodding, P., Lukkarinen, O., Lundmo, P., Klarskov, P., Tammela, T. L., Tasdemir, I., Morris, T., Carroll, K., & Scandinavian Prostatic Cancer Group. (2004). Bicalutamide (150 mg) versus placebo as immediate therapy alone or as adjuvant to therapy with curative intent for early nonmetastatic prostate cancer: 5.3-year median followup from the Scandinavian Prostate Cancer Group Study Number 6. The Journal of Urology, 172(5 Part 1), 1871–1876. [DOI:10.1097/01.ju.0000139719.99825.54]
Iversen, P., Johansson, J., Lodding, P., Kylmälä, T., Lundmo, P., Klarskov, P., Tammela, T. L., Tasdemir, I., Morris, T., Armstrong, J., & (2006). Bicalutamide 150 mg in addition to standard care for patients with early non-metastatic prostate cancer Updated results from the Scandinavian Prostate Cancer Period Group-6 Study after a median follow-up period of 7.1 years. Scandinavian Journal of Urology and Nephrology, 40(6), 441–452. [DOI:10.1080/00365590601017329]
Iwamoto, S. J., Defreyne, J., Rothman, M. S., Van Schuylenbergh, J., Van de Bruaene, L., Motmans, J., & T’Sjoen, G. (2019). Health considerations for transgender women and remaining unknowns: a narrative review. Therapeutic Advances in Endocrinology and Metabolism, 10, 204201881987116. [DOI:10.1177/2042018819871166]
Iwamoto, S. J., Grimstad, F., Irwig, M. S., & Rothman, M. S. (2021). Routine Screening for Transgender and Gender Diverse Adults Taking Gender-Affirming Hormone Therapy: a Narrative Review. Journal of General Internal Medicine, 36(5), 1380–1389. [DOI:10.1007/s11606-021-06634-7]
Jacoby, A., Rifkin, W., Zhao, L. C., & Bluebond-Langner, R. (2020). Incidence of Cancer and Premalignant Lesions in Surgical Specimens of Transgender Patients. Plastic & Reconstructive Surgery, 147(1), 194–198. [DOI:10.1097/prs.0000000000007452]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone Acetate in Gender-Affirming Therapy for Transwomen: Results From a Retrospective Study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
James, J. F., Jamerson, T. A., & Aguh, C. (2022). Efficacy and safety profile of oral spironolactone use for androgenic alopecia: A systematic review. Journal of the American Academy of Dermatology, 86(2), 425–429. [DOI:10.1016/j.jaad.2021.07.048]
Jia, A. Y., & Spratt, D. E. (2022). Bicalutamide Monotherapy With Radiation Therapy for Localized Prostate Cancer: A Non-Evidence-Based Alternative. International Journal of Radiation Oncology*Biology*Physics, 113(2), 316–319. [DOI:10.1016/j.ijrobp.2022.01.037]
Jiang, Y., & Tian, W. (2017). The effects of progesterones on blood lipids in hormone replacement therapy. Lipids in Health and Disease, 16(1), 219. [DOI:10.1186/s12944-017-0612-5]
Kanhai, R. C., Hage, J. J., van Diest, P. J., Bloemena, E., & Mulder, J. W. (2000). Short-Term and Long-Term Histologic Effects of Castration and Estrogen Treatment on Breast Tissue of 14 Male-to-Female Transsexuals in Comparison With Two Chemically Castrated Men. The American Journal of Surgical Pathology, 24(1), 74–80. [DOI:10.1097/00000478-200001000-00009]
Kellner, M., & Wiedemann, K. (2008). Mineralocorticoid receptors in brain, in health and disease: Possibilities for new pharmacotherapy. European Journal of Pharmacology, 583(2–3), 372–378. [DOI:10.1016/j.ejphar.2007.07.072]
Kim, G. K., & Del Rosso, J. Q. (2012). Oral Spironolactone in Post-teenage Female Patients with Acne Vulgaris: Practical Considerations for the Clinician Based on Current Data and Clinical Experience. The Journal of Clinical and Aesthetic Dermatology, 5(3), 37–50. [PubMed] [PubMed Central]
King, S. R. (2012). Neurosteroids and the Nervous System. In King, S. R. Neurosteroids and the Nervous System (pp. 1–122). New York: Springer New York. [DOI:10.1007/978-1-4614-5559-2_1]
Krishnamurthy, N., Slack, D., Kyweluk, M., Kirkley, J., Trakhtenberg, E., Contreras-Castro, F., & Safer, J. (2023). Not All Transfeminine Individuals on Estradiol Can Reach Both Target Testosterone and Target Estradiol Levels—Time to Revisit Treatment Guidelines? USPATH Scientific Symposium, November 1-5, 2023, The Westin Westminster, Westminster, Colorado, Abstract Submissions, 94–94 (abstract no. SAT-B2-T4). [Symposium Schedule] [PDF] [Full Abstract Book]
Kolvenbag, G. J., & Blackledge, G. R. (1996). Worldwide activity and safety of bicalutamide: a summary review. Urology, 47(1), 70–79. [DOI:10.1016/s0090-4295(96)80012-4]
Kuhl, H. (2003). Östrogene für den Mann? [Estrogen for men?] Blickpunkt der Mann, 1(3), 6–12. [Google Scholar] [URL] [PDF]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H., & Wiegratz, I. (2017). Das Post-Finasterid-Syndrom. [Post Finasteride Syndrome.] Gynäkologische Endokrinologie, 15(2), 153–163. [DOI:10.1007/s10304-017-0126-2]
Kuijpers, S. M., Wiepjes, C. M., Conemans, E. B., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2021). Toward a Lowest Effective Dose of Cyproterone Acetate in Trans Women: Results From the ENIGI Study. The Journal of Clinical Endocrinology & Metabolism, 106(10), e3936–e3945. [DOI:10.1210/clinem/dgab427]
Kumar, P., Reddy, S., Kulkarni, A., Sharma, M., & Rao, P. N. (2021). Cyproterone Acetate–Induced Acute Liver Failure: A Case Report and Review of the Literature. Journal of Clinical and Experimental Hepatology, 11(6), 739–741. [DOI:10.1016/j.jceh.2021.01.003]
Lauritzen, C. (1988). Natürliche und synthetische Sexualhormone – Biologische Grundlagen und Behandlungsprinzipien. [Natural and Synthetic Sexual Hormones – Biological Basis and Medical Treatment Principles.] In Lauritzen, C., Schneider, H. P. G., & Nieschlag, E. (Eds.). Grundlagen und Klinik der Menschlichen Fortpflanzung [Foundations and Clinic of Human Reproduction] (pp. 229–306). Berlin: de Gruyter. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [URL] [PDF] [Translation]
Lax, E. (1987). Mechanisms of physiological and pharmacological sex hormone action on the mammalian liver. Journal of Steroid Biochemistry, 27(4–6), 1119–1128. [DOI:10.1016/0022-4731(87)90198-1]
Layton, A. M., Eady, E. A., Whitehouse, H., Del Rosso, J. Q., Fedorowicz, Z., & van Zuuren, E. J. (2017). Oral Spironolactone for Acne Vulgaris in Adult Females: A Hybrid Systematic Review. American Journal of Clinical Dermatology, 18(2), 169–191. [DOI:10.1007/s40257-016-0245-x]
Legro, R. S., Schlaff, W. D., Diamond, M. P., Coutifaris, C., Casson, P. R., Brzyski, R. G., Christman, G. M., Trussell, J. C., Krawetz, S. A., Snyder, P. J., Ohl, D., Carson, S. A., Steinkampf, M. P., Carr, B. R., McGovern, P. G., Cataldo, N. A., Gosman, G. G., Nestler, J. E., Myers, E. R., Santoro, N., Eisenberg, E., Zhang, M., & Zhang, H. (2010). Total Testosterone Assays in Women with Polycystic Ovary Syndrome: Precision and Correlation with Hirsutism. The Journal of Clinical Endocrinology & Metabolism, 95(12), 5305–5313. [DOI:10.1210/jc.2010-1123]
Leinung, M. C., Feustel, P. J., & Joseph, J. (2018). Hormonal Treatment of Transgender Women with Oral Estradiol. Transgender Health, 3(1), 74–81. [DOI:10.1089/trgh.2017.0035]
Liang, J. J., Jolly, D., Chan, K. J., & Safer, J. D. (2018). Testosterone Levels Achieved by Medically Treated Transgender Women in a United States Endocrinology Clinic Cohort. Endocrine Practice, 24(2), 135–142. [DOI:10.4158/ep-2017-0116]
Lobo, R. A., Shoupe, D., Serafini, P., Brinton, D., & Horton, R. (1985). The effects of two doses of spironolactone on serum androgens and anagen hair in hirsute women. Fertility and Sterility, 43(2), 200–205. [DOI:10.1016/s0015-0282(16)48373-1]
Loh, H. H., Yee, A., Loh, H. S., Kanagasundram, S., Francis, B., & Lim, L. (2020). Sexual dysfunction in polycystic ovary syndrome: a systematic review and meta-analysis. Hormones, 19(3), 413–423. [DOI:10.1007/s42000-020-00210-0]
Luong, J. H., Male, K. B., & Glennon, J. D. (2019). Biotin interference in immunoassays based on biotin-strept(avidin) chemistry: An emerging threat. Biotechnology Advances, 37(5), 634–641. [DOI:10.1016/j.biotechadv.2019.03.007]
MacLean, D. B., Shi, H., Faessel, H. M., & Saad, F. (2015). Medical Castration Using the Investigational Oral GnRH Antagonist TAK-385 (Relugolix): Phase 1 Study in Healthy Males. The Journal of Clinical Endocrinology & Metabolism, 100(12), 4579–4587. [DOI:10.1210/jc.2015-2770]
Mahler, C. (1996). A Review of the Clinical Studies with Nilutamide. In Denis, L. (Ed.). Antiandrogens in Prostate Cancer: A Key to Tailored Endocrine Treatment (ESO Monographs) (pp. 105–111). Berlin/Heidelberg: Springer Berlin Heidelberg. [DOI:10.1007/978-3-642-45745-6_10]
Maksym, R. B., Kajdy, A., & Rabijewski, M. (2019). Post-finasteride syndrome – does it really exist? The Aging Male, 22(4), 250–259. [DOI:10.1080/13685538.2018.1548589]
Marks, D. H., Prasad, S., De Souza, B., Burns, L. J., & Senna, M. M. (2019). Topical Antiandrogen Therapies for Androgenetic Alopecia and Acne Vulgaris. American Journal of Clinical Dermatology, 21(2), 245–254. [DOI:10.1007/s40257-019-00493-z]
Martinez-Martin, F. J., Kuzior, A., Hernandez-Lazaro, A., de Leon-Durango, R. J., Rios-Gomez, C., Santana-Ojeda, B., Perez-Rivero, J. M., Fernandez-Trujillo-Comenge, P. M., Gonzalez-Diaz, P., Arnas-Leon, C., Acosta-Calero, C., Perdomo-Herrera, E., Tocino-Hernandez, A. L., del Sol Sanchez-Bacaicoa, M., & del Pino Perez-Garcia, M. (2022). Incidence of hypertension in young transgender people after a 5-year follow-up: association with gender-affirming hormonal therapy. Hypertension Research, 46(1), 219–225. [DOI:10.1038/s41440-022-01067-z]
Masumori, N., Baba, T., Abe, T., & Niwa, K. (2021). What is the most anticipated change induced by treatment using gender‐affirming hormones in individuals with gender incongruence?. International Journal of Urology, 28(5), 526–529. [DOI:10.1111/iju.14499]
McFarlane, T., Zajac, J. D., & Cheung, A. S. (2018). Gender-affirming hormone therapy and the risk of sex hormone-dependent tumours in transgender individuals—A systematic review. Clinical Endocrinology, 89(6), 700–711. [DOI:10.1111/cen.13835]
Meriggiola, M. C., Bremner, W. J., Costantino, A., Bertaccini, A., Morselli-Labate, A. M., Huebler, D., Kaufmann, G., Oettel, M., & Flamigni, C. (2002). Twenty-One Day Administration of Dienogest Reversibly Suppresses Gonadotropins and Testosterone in Normal Men. The Journal of Clinical Endocrinology & Metabolism, 87(5), 2107–2113. [DOI:10.1210/jcem.87.5.8514]
Meyer, G., Mayer, M., Mondorf, A., Flügel, A. K., Herrmann, E., & Bojunga, J. (2020). Safety and rapid efficacy of guideline-based gender-affirming hormone therapy: an analysis of 388 individuals diagnosed with gender dysphoria. European Journal of Endocrinology, 182(2), 149–156. [DOI:10.1530/eje-19-0463]
Miles, R. A., Paulson, R. J., Lobo, R. A., Press, M. F., Dahmoush, L., & Sauer, M. V. (1994). Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: a comparative study. Fertility and Sterility, 62(3), 485–490. [DOI:10.1016/s0015-0282(16)56935-0]
Millington, K., Liu, E., & Chan, Y. (2019). The Utility of Potassium Monitoring in Gender-Diverse Adolescents Taking Spironolactone. Journal of the Endocrine Society, 3(5), 1031–1038. [DOI:10.1210/js.2019-00030]
Millward, C. P., Keshwara, S. M., Islim, A. I., Jenkinson, M. D., Alalade, A. F., & Gilkes, C. E. (2022). Development and Growth of Intracranial Meningiomas in Transgender Women Taking Cyproterone Acetate as Gender-Affirming Progestogen Therapy: A Systematic Review. Transgender Health, 7(6), 473–483. [DOI:10.1089/trgh.2021.0025]
Moretti, C., Guccione, L., Di Giacinto, P., Simonelli, I., Exacoustos, C., Toscano, V., Motta, C., De Leo, V., Petraglia, F., & Lenzi, A. (2017). Combined Oral Contraception and Bicalutamide in Polycystic Ovary Syndrome and Severe Hirsutism: A Double-Blind Randomized Controlled Trial. The Journal of Clinical Endocrinology & Metabolism, 103(3), 824–838. [DOI:10.1210/jc.2017-01186]
Nakamoto, J. (2016). Endocrine Testing. In Jameson, J. L., & De Groot, L. J. (Eds.). Endocrinology: Adult and Pediatric, 7th Edition (pp. 2655–2688.e1). Philadelphia: Saunders/Elsevier. [DOI:10.1016/B978-0-323-18907-1.00154-2]
Nakhjavani, M., Hamidi, S., Esteghamati, A., Abbasi, M., Nosratian-Jahromi, S., & Pasalar, P. (2009). Short term effects of spironolactone on blood lipid profile: a 3-month study on a cohort of young women with hirsutism. British Journal of Clinical Pharmacology, 68(4), 634–637. [DOI:10.1111/j.1365-2125.2009.03483.x]
Neyman, A., Fuqua, J. S., & Eugster, E. A. (2019). Bicalutamide as an Androgen Blocker With Secondary Effect of Promoting Feminization in Male-to-Female Transgender Adolescents. Journal of Adolescent Health, 64(4), 544–546. [DOI:10.1016/j.jadohealth.2018.10.296]
Nguyen, D., Marchese, M., Cone, E. B., Paciotti, M., Basaria, S., Bhojani, N., & Trinh, Q. (2021). Investigation of Suicidality and Psychological Adverse Events in Patients Treated With Finasteride. JAMA Dermatology, 157(1), 35–42. [DOI:10.1001/jamadermatol.2020.3385]
Nieschlag, E., Zitzmann, M., & Kamischke, A. (2003). Use of progestins in male contraception. Steroids, 68(10–13), 965–972. [DOI:10.1016/s0039-128x(03)00135-1]
Nieschlag, E. (2010). Clinical trials in male hormonal contraception. Contraception, 82(5), 457–470. [DOI:10.1016/j.contraception.2010.03.020]
Nieschlag, E., & Behre, H. M. (2012). The essential role of testosterone in hormonal male contraception. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Testosterone: Action · Deficiency · Substitution, 4th Edition (pp. 470–493). Cambridge/New York: Cambridge University Press. [DOI:10.1017/cbo9781139003353.023]
Nishiyama, T. (2014). Serum testosterone levels after medical or surgical androgen deprivation: A comprehensive review of the literature. Urologic Oncology: Seminars and Original Investigations, 32(1), 38.e17–38.e28. [DOI:10.1016/j.urolonc.2013.03.007]
Nolan, B. J., & Cheung, A. S. (2021). Relationship Between Serum Estradiol Concentrations and Clinical Outcomes in Transgender Individuals Undergoing Feminizing Hormone Therapy: A Narrative Review. Transgender Health, 6(3), 125–131. [DOI:10.1089/trgh.2020.0077]
Nolan, B. J., Frydman, A. S., Leemaqz, S. Y., Carroll, M., Grossmann, M., Zajac, J. D., & Cheung, A. S. (2022). Effects of low-dose oral micronised progesterone on sleep, psychological distress, and breast development in transgender individuals undergoing feminising hormone therapy: a prospective controlled study. Endocrine Connections, 11(5), e220170. [DOI:10.1530/EC-22-0170]
Norman, A. W., & Henry, H. L. (2015). Androgens. In Norman, A. W., & Henry, H. L. Hormones, 3rd Edition (pp. 255–273). London: Academic Press/Elsevier. [DOI:10.1016/b978-0-08-091906-5.00012-4]
Norman, A. W., & Henry, H. L. (2015). Estrogens and Progestins. In Norman, A. W., & Henry, H. L. Hormones, 3rd Edition (pp. 275–296). London: Academic Press/Elsevier. [DOI:10.1016/b978-0-08-091906-5.00013-6]
Nota, N. M., Wiepjes, C. M., de Blok, C. J., Gooren, L. J., Peerdeman, S. M., Kreukels, B. P., & den Heijer, M. (2018). The occurrence of benign brain tumours in transgender individuals during cross-sex hormone treatment. Brain, 141(7), 2047–2054. [DOI:10.1093/brain/awy108]
North American Menopause Society. (2022). The 2022 hormone therapy position statement of The North American Menopause Society. Menopause, 29(7), 767–794. [DOI:10.1097/gme.0000000000002028]
O’Connell, M. B. (1995). Pharmacokinetic and Pharmacologic Variation Between Different Estrogen Products. The Journal of Clinical Pharmacology, 35(9S), 18S–24S. [DOI:10.1002/j.1552-4604.1995.tb04143.x]
Olsen, E. A., Hordinsky, M., Whiting, D., Stough, D., Hobbs, S., Ellis, M. L., Wilson, T., & Rittmaster, R. S. (2006). The importance of dual 5α-reductase inhibition in the treatment of male pattern hair loss: Results of a randomized placebo-controlled study of dutasteride versus finasteride. Journal of the American Academy of Dermatology, 55(6), 1014–1023. [DOI:10.1016/j.jaad.2006.05.007]
Plovanich, M., Weng, Q. Y., & Mostaghimi, A. (2015). Low Usefulness of Potassium Monitoring Among Healthy Young Women Taking Spironolactone for Acne. JAMA Dermatology, 151(9), 941–944. [DOI:10.1001/jamadermatol.2015.34]
Polyzos, S. A., Kountouras, J., Zavos, C., & Deretzi, G. (2011). Spironolactone Revisited. The Journal of Clinical Hypertension, 13(10), 783–784. [DOI:10.1111/j.1751-7176.2011.00484.x]
Powers, M. S., Schenkel, L., Darley, P. E., Good, W. R., Balestra, J. C., & Place, V. A. (1985). Pharmacokinetics and pharmacodynamics of transdermal dosage forms of 17β-estradiol: Comparison with conventional oral estrogens used for hormone replacement. American Journal of Obstetrics and Gynecology, 152(8), 1099–1106. [DOI:10.1016/0002-9378(85)90569-1]
Prince, J. C., & Safer, J. D. (2020). Endocrine treatment of transgender individuals: current guidelines and strategies. Expert Review of Endocrinology & Metabolism, 15(6), 395–403. [DOI:10.1080/17446651.2020.1825075]
Reiter, E. O., Mauras, N., McCormick, K., Kulshreshtha, B., Amrhein, J., De Luca, F., O’Brien, S., Armstrong, J., & Melezinkova, H. (2010). Bicalutamide plus Anastrozole for the Treatment of Gonadotropin-Independent Precocious Puberty in Boys with Testotoxicosis: A Phase II, Open-Label Pilot Study (BATT). Journal of Pediatric Endocrinology and Metabolism, 23(10), 999–1009. [DOI:10.1515/jpem.2010.161]
Rezende, H. D., Dias, M. F. R. G., & Trüeb, R. M. (2018). A Comment on the Post-Finasteride Syndrome. International Journal of Trichology, 10(6), 255–261. [PubMed] [PubMed Central] [DOI:10.4103/ijt.ijt_61_18]
Roberts, J. L., Fiedler, V., Imperato-McGinley, J., Whiting, D., Olsen, E., Shupack, J., Stough, D., DeVillez, R., Rietschel, R., Savin, R., Bergfeld, W., Swinehart, J., Funicella, T., Hordinsky, M., Lowe, N., Katz, I., Lucky, A., Drake, L., Price, V. H., Weiss, D., Whitmore, E., Millikan, L., Muller, S., Gencheff, C., Carrington, P., Binkowitz, B., Kotey, P., He, W., Bruno, K., Jacobsen, C., Terranella, L., Gormley, G. J., & Kaufman, K. D. (1999). Clinical dose ranging studies with finasteride, a type 2 5α-reductase inhibitor, in men with male pattern hair loss. Journal of the American Academy of Dermatology, 41(4), 555–563. [DOI:10.1016/s0190-9622(99)80052-8]
Roscioni, S. S., de Zeeuw, D., Bakker, S. J., & Lambers Heerspink, H. J. (2012). Management of hyperkalaemia consequent to mineralocorticoid-receptor antagonist therapy. Nature Reviews Nephrology, 8(12), 691–699. [DOI:10.1038/nrneph.2012.217]
Rose, A. J., Hughto, J. M., Dunbar, M. S., Quinn, E. K., Deutsch, M., Feldman, J., Radix, A., Safer, J. D., Shipherd, J. C., Thompson, J., & Jasuja, G. K. (2023). Trends in Feminizing Hormone Therapy for Transgender Patients, 2006–2017. Transgender Health, 8(2), 188–194. [DOI:10.1089/trgh.2021.0041]
Rosenfield, R. L., Cooke, D. W., & Radovick, S. (2008). Puberty and its disorders in the female. In Sperling, M. A. (Ed.). Pediatric Endocrinology, 3rd Edition (pp. 530–609). Philadelphia: Saunders. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat]
Rosenfield, R. L., Cooke, D. W., & Radovick, S. (2021). Puberty in the Female and Its Disorders. In Sperling, M. A., Majzoub, J. A., Menon, R. K., & Stratakis, C. A. (Eds.). Sperling Pediatric Endocrinology, 5th Edition (pp. 528–626). Philadelphia: Elsevier. [DOI:10.1016/B978-0-323-62520-3.00016-6]
Sam. (2020). A Comparison of Oral and Transdermal Estradiol in Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Sam. (2020). Analysis of Cardiovascular and Thromboembolic Toxicity with High Dose Parenteral Polyestradiol Phosphate in the Treatment of Prostate Cancer. Transfeminine Science. [URL]
Sam. (2020). Excerpts: Perspectives and Opinions of Clinicians and Researchers Concerning the Use of Progestogens in Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Sam. (2020). Progestogens and Breast Cancer in Transgender Women: A Review and Discussion of Risk. Transfeminine Science. [URL]
Sam. (2021). An Exploration of Sublingual Estradiol as an Alternative to Oral Estradiol in Transfeminine People. Transfeminine Science. [URL]
Sam. (2025). A Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People. Transfeminine Science. [URL]
Samarasinghe, S., Meah, F., Singh, V., Basit, A., Emanuele, N., Emanuele, M. A., Mazhari, A., & Holmes, E. W. (2017). Biotin Interference with Routine Clinical Immunoassays: Understand the Causes and Mitigate the Risks. Endocrine Practice, 23(8), 989–998. [DOI:10.4158/ep171761.ra]
Schiller, C. E., Schmidt, P. J., & Rubinow, D. R. (2014). Allopregnanolone as a mediator of affective switching in reproductive mood disorders. Psychopharmacology, 231(17), 3557–3567. [DOI:10.1007/s00213-014-3599-x]
Schlatterer, K., Yassouridis, A., Werder, K. V., Poland, D., Kemper, J., & Stalla, G. K. (1998). Archives of Sexual Behavior, 27(5), 475–492. [DOI:10.1023/a:1018704630036]
Seaman, H. E., Langley, S. E., Farmer, R. D., & de Vries, C. S. (2007). Venous thromboembolism and cyproterone acetate in men with prostate cancer: a study using the General Practice Research Database. BJU International, 99(6), 1398–1403. [DOI:10.1111/j.1464-410x.2007.06859.x]
Shackleton, C. (2010). Clinical steroid mass spectrometry: A 45-year history culminating in HPLC–MS/MS becoming an essential tool for patient diagnosis. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 481–490. [DOI:10.1016/j.jsbmb.2010.02.017]
Shaw, J. C. (1996). Antiandrogen and hormonal treatment of acne. Dermatologic Clinics, 14(4), 803–811. [DOI:10.1016/s0733-8635(05)70405-8]
Slack, D., Krishnamurthy, N., Contreras-Castro, F., & Safer, J. D. (2023). Achieving Both Target Testosterone And Target Estradiol Levels May Not Be Practical In All Trans Feminine Users Of Estradiol - Which To Prioritize? Journal of the Endocrine Society, 7(Suppl 1), A1102–A1102 (abstract no. SAT404/bvad114.2075). [DOI:10.1210/jendso/bvad114.2075] [PubMed Central]
Sofer, Y., Yaish, I., Yaron, M., Bach, M. Y., Stern, N., & Greenman, Y. (2020). Differential Endocrine and Metabolic Effects of Testosterone Suppressive Agents in Transgender Women. Endocrine Practice, 26(8), 883–890. [DOI:10.4158/ep-2020-0032]
SoRelle, J. A., Jiao, R., Gao, E., Veazey, J., Frame, I., Quinn, A. M., Day, P., Pagels, P., Gimpel, N., & Patel, K. (2019). Impact of Hormone Therapy on Laboratory Values in Transgender Patients. Clinical Chemistry, 65(1), 170–179. [DOI:10.1373/clinchem.2018.292730]
St-Jules, D. E., Goldfarb, D. S., & Sevick, M. A. (2016). Nutrient Non-equivalence: Does Restricting High-Potassium Plant Foods Help to Prevent Hyperkalemia in Hemodialysis Patients? Journal of Renal Nutrition, 26(5), 282–287. [DOI:10.1053/j.jrn.2016.02.005]
St-Jules, D. E., & Fouque, D. (2020). Is it time to abandon the nutrient-based renal diet model? Nephrology Dialysis Transplantation, 36(4), 574–577. [DOI:10.1093/ndt/gfaa257]
St-Jules, D. E., & Fouque, D. (2022). Etiology-based dietary approach for managing hyperkalemia in people with chronic kidney disease. Nutrition Reviews, 80(11), 2198–2205. [DOI:10.1093/nutrit/nuac026]
Stanczyk, F. Z., & Clarke, N. J. (2010). Advantages and challenges of mass spectrometry assays for steroid hormones. The Journal of Steroid Biochemistry and Molecular Biology, 121(3–5), 491–495. [DOI:10.1016/j.jsbmb.2010.05.001]
Stanczyk, F. Z., Hapgood, J. P., Winer, S., & Mishell, D. R. (2012). Progestogens Used in Postmenopausal Hormone Therapy: Differences in Their Pharmacological Properties, Intracellular Actions, and Clinical Effects. Endocrine Reviews, 34(2), 171–208. [DOI:10.1210/er.2012-1008]
Stanczyk, F. Z., Mathews, B. W., & Cortessis, V. K. (2017). Does the type of progestin influence the production of clotting factors? Contraception, 95(2), 113–116. [DOI:10.1016/j.contraception.2016.07.007]
Stege, R., Gunnarsson, P. O., Johansson, C., Olsson, P., Pousette, Å., & Carlström, K. (1996). Pharmacokinetics and testosterone suppression of a single dose of polyestradiol phosphate (Estradurin®) in prostatic cancer patients. The Prostate, 28(5), 307–310. [DOI:10.1002/(sici)1097-0045(199605)28:5<307::aid-pros6>3.0.co;2-8]
Steinberger, E., Ayala, C., Hsi, B., Smith, K. D., Rodriguez-Rigau, L. J., Weidman, E. R., & Reimondo, G. G. (1998). Utilization of Commercial Laboratory Results in Management of Hyperandrogenism in Women. Endocrine Practice, 4(1), 1–10. [DOI:10.4158/ep.4.1.1]
Sterling, J., & Garcia, M. M. (2020). Cancer screening in the transgender population: a review of current guidelines, best practices, and a proposed care model. Translational Andrology and Urology, 9(6), 2771–2785. [DOI:10.21037/tau-20-954]
Strauss, J. F., & FitzGerald, G. A. (2019). Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction. In Strauss, J. F., & Barbieri, R. L. (Eds.). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th Edition (pp. 75–114.e7). Philadelphia: Elsevier. [Google Books] [DOI:10.1016/b978-0-323-47912-7.00004-4]
Stricker, R., Eberhart, R., Chevailler, M., Quinn, F. A., Bischof, P., & Stricker, R. (2006). Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT® analyzer. Clinical Chemistry and Laboratory Medicine (CCLM), 44(7), 883–887. [DOI:10.1515/cclm.2006.160]
Styne, D. M. (2016). Laboratory Values for Pediatric Endocrinology. In Styne, D. M. Pediatric Endocrinology: A Clinical Handbook (pp. 385–434). Cham: Springer International Publishing. [DOI:10.1007/978-3-319-18371-8_16]
Sudduth, S. L., & Koronkowski, M. J. (1993). Finasteride: The First 5α-Reductase Inhibitor. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 13(4), 309–329. [DOI:10.1002/j.1875-9114.1993.tb02739.x]
Sundström-Poromaa, I., Comasco, E., Sumner, R., & Luders, E. (2020). Progesterone – Friend or foe? Frontiers in Neuroendocrinology, 59, 100856. [DOI:10.1016/j.yfrne.2020.100856]
Swerdloff, R. S., Dudley, R. E., Page, S. T., Wang, C., & Salameh, W. A. (2017). Dihydrotestosterone: Biochemistry, Physiology, and Clinical Implications of Elevated Blood Levels. Endocrine Reviews, 38(3), 220–254. [DOI:10.1210/er.2016-1067]
T’Sjoen, G. G., Beguin, Y., Feyen, E., Rubens, R., Kaufman, J., & Gooren, L. (2005). Influence of exogenous oestrogen or (anti-) androgen administration on soluble transferrin receptor in human plasma. Journal of Endocrinology, 186(1), 61–67. [DOI:10.1677/joe.1.06112]
T’Sjoen, G., Arcelus, J., Gooren, L., Klink, D. T., & Tangpricha, V. (2018). Endocrinology of Transgender Medicine. Endocrine Reviews, 40(1), 97–117. [DOI:10.1210/er.2018-00011]
Tack, L. J., Heyse, R., Craen, M., Dhondt, K., Bossche, H. V., Laridaen, J., & Cools, M. (2017). Consecutive Cyproterone Acetate and Estradiol Treatment in Late-Pubertal Transgender Female Adolescents. The Journal of Sexual Medicine, 14(5), 747–757. [DOI:10.1016/j.jsxm.2017.03.251]
Talathi, R., Juhasz, V., Delgado, M., Quinaglia, T., Ghamari, A., Wang, M., Alhallak, I., Stinebaugh, S., Campbell, S., Stockman, S. L., Ozturk, M. A., Ahmadi, S. M., Looby, S. E., Lee, H., Poteat, T. C., Szczepaniak, L. S., Zanni, M. V., Neilan, T. G., & Toribio, M. (2025). Visceral adipose tissue and liver fat on 17-beta estradiol-dominant gender-affirming hormone therapy: A US-based cohort. The Journal of Clinical Endocrinology and Metabolism, online ahead of print. [DOI:10.1210/clinem/dgaf665]
Thompson, J., Hopwood, R. A., deNormand, S., & Cavanaugh, T. (2021). Medical Care of Trans and Gender Diverse Adults. Boston: Fenway Health. [URL] [PDF]
Toorians, A. W., Thomassen, M. C., Zweegman, S., Magdeleyns, E. J., Tans, G., Gooren, L. J., & Rosing, J. (2003). Venous Thrombosis and Changes of Hemostatic Variables during Cross-Sex Hormone Treatment in Transsexual People. The Journal of Clinical Endocrinology & Metabolism, 88(12), 5723–5729. [DOI:10.1210/jc.2003-030520]
Traish, A. M., Krakowsky, Y., Doros, G., & Morgentaler, A. (2019). Do 5α-Reductase Inhibitors Raise Circulating Serum Testosterone Levels? A Comprehensive Review and Meta-Analysis to Explaining Paradoxical Results. Sexual Medicine Reviews, 7(1), 95–114. [DOI:10.1016/j.sxmr.2018.06.002]
Traish, A. M. (2020). Post-finasteride syndrome: a surmountable challenge for clinicians. Fertility and Sterility, 113(1), 21–50. [DOI:10.1016/j.fertnstert.2019.11.030]
Treleaven, M. M., Jackowich, R. A., Roberts, L., Wassersug, R. J., & Johnson, T. (2013). Castration and personality: Correlation of androgen deprivation and estrogen supplementation with the Big Five factor personality traits of adult males. Journal of Research in Personality, 47(4), 376–379. [DOI:10.1016/j.jrp.2013.03.005]
Usach, I., Martinez, R., Festini, T., & Peris, J. (2019). Subcutaneous Injection of Drugs: Literature Review of Factors Influencing Pain Sensation at the Injection Site. Advances in Therapy, 36(11), 2986–2996. [DOI:10.1007/s12325-019-01101-6]
van Zuuren, E. J., Fedorowicz, Z., Carter, B., & Pandis, N. (2015). Interventions for hirsutism (excluding laser and photoepilation therapy alone). Cochrane Database of Systematic Reviews, 2015, CD010334. [DOI:10.1002/14651858.cd010334.pub2]
Verdonk, S. J., Vesper, H. W., Martens, F., Sluss, P. M., Hillebrand, J. J., & Heijboer, A. C. (2019). Estradiol reference intervals in women during the menstrual cycle, postmenopausal women and men using an LC-MS/MS method. Clinica Chimica Acta, 495, 198–204. [DOI:10.1016/j.cca.2019.04.062]
Vermeulen, A., Giagulli, V., De Schepper, P., & Buntinx, A. (1991). Hormonal Effects of a 5α-Reductase Inhibitor (Finasteride) onHormonal Levels in Normal Men and in Patients withBenign Prostatic Hyperplasia. European Urology, 20(1), 82–86. [DOI:10.1159/000471752]
Vihtamäki, T., Luukkaala, T., & Tuimala, R. (2004). Skin contamination by oestradiol gel—a remarkable source of error in plasma oestradiol measurements during percutaneous hormone replacement therapy. Maturitas, 48(4), 347–353. [DOI:10.1016/s0378-5122(03)00043-4]
Wang, H., Liu, M., Fu, Q., & Deng, C. (2019). Pharmacokinetics of hard micronized progesterone capsules via vaginal or oral route compared with soft micronized capsules in healthy postmenopausal women: a randomized open-label clinical study. Drug Design, Development and Therapy, 13, 2475–2482. [DOI:10.2147/dddt.s204624]
Wang, Y., & Lipner, S. R. (2020). Retrospective analysis of adverse events with spironolactone in females reported to the United States Food and Drug Administration. International Journal of Women’s Dermatology, 6(4), 272–276. [DOI:10.1016/j.ijwd.2020.05.002]
Weill, A., Nguyen, P., Labidi, M., Cadier, B., Passeri, T., Duranteau, L., Bernat, A., Yoldjian, I., Fontanel, S., Froelich, S., & Coste, J. (2021). Use of High Dose Cyproterone Acetate and Risk of Intracranial Meningioma in Women: Cohort Study. BMJ, 372, n37. [DOI:10.1136/bmj.n37]
Welk, B., McArthur, E., Ordon, M., Anderson, K. K., Hayward, J., & Dixon, S. (2017). Association of Suicidality and Depression With 5α-Reductase Inhibitors. JAMA Internal Medicine, 177(5), 683–691. [DOI:10.1001/jamainternmed.2017.0089]
Wellington, K., & Keam, S. J. (2006). Bicalutamide 150mg: A Review of its Use in the Treatment of Locally Advanced Prostate Cancer. Drugs, 66(6), 837–850. [DOI:10.2165/00003495-200666060-00007]
Willemsen, W. N., Mastboom, J. L., Thomas, C. M., & Rolland, R. (1985). Absorption of 17β-estradiol in a neovagina constructed from the peritoneum. European Journal of Obstetrics & Gynecology and Reproductive Biology, 19(4), 247–253. [DOI:10.1016/0028-2243(85)90036-x]
Wilson, L. M., Baker, K. E., Sharma, R., Dukhanin, V., McArthur, K., & Robinson, K. A. (2020). Effects of antiandrogens on prolactin levels among transgender women on estrogen therapy: A systematic review. International Journal of Transgender Health, 21(4), 391–402. [DOI:10.1080/15532739.2020.1819505]
Wu, B., Shen, P., Yin, X., Yu, L., Wu, F., Chen, C., Li, J., & Xu, T. (2022). Analysis of adverse event of interstitial lung disease in men with prostate cancer receiving hormone therapy using the Food and Drug Administration Adverse Event Reporting System. British Journal of Clinical Pharmacology, 89(2), 440–448. [DOI:10.1111/bcp.15336]
Wu, F. C., Balasubramanian, R., Mulders, T. M., & Coelingh-Bennink, H. J. (1999). Oral Progestogen Combined with Testosterone as a Potential Male Contraceptive: Additive Effects between Desogestrel and Testosterone Enanthate in Suppression of Spermatogenesis, Pituitary-Testicular Axis, and Lipid Metabolism. The Journal of Clinical Endocrinology & Metabolism, 84(1), 112–122. [DOI:10.1210/jcem.84.1.5412]
Yaish, I., Gindis, G., Greenman, Y., Moshe, Y., Arbiv, M., Buch, A., Sofer, Y., Shefer, G., & Tordjman, K. (2023). Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study. Transgender Health, 8(6), 485–493. [DOI:10.1089/trgh.2023.0022]
Zaenglein, A. L., Pathy, A. L., Schlosser, B. J., Alikhan, A., Baldwin, H. E., Berson, D. S., Bowe, W. P., Graber, E. M., Harper, J. C., Kang, S., Keri, J. E., Leyden, J. J., Reynolds, R. V., Silverberg, N. B., Stein Gold, L. F., Tollefson, M. M., Weiss, J. S., Dolan, N. C., Sagan, A. A., Stern, M., Boyer, K. M., & Bhushan, R. (2016). Guidelines of care for the management of acne vulgaris. Journal of the American Academy of Dermatology, 74(5), 945–973.e33. [DOI:10.1016/j.jaad.2015.12.037]
Zhang, J., & Stanczyk, F. Z. (2013). LC-MS Bioanalysis of Steroids. In Li, W., Zhang, J., & Tse, F. L. S. (Eds.). Handbook of LC-MS Bioanalysis: Best Practices, Experimental Protocols, and Regulations (pp. 573–590). Hoboken, New Jersey: John Wiley & Sons. [DOI:10.1002/9781118671276.ch45]
Zhou, Z., Song, S., Gao, Z., Wu, J., Ma, J., & Cui, Y. (2019). The efficacy and safety of dutasteride compared with finasteride in treating men with androgenetic alopecia: a systematic review and meta-analysis. Clinical Interventions in Aging, 14, 399–406. [DOI:10.2147/cia.s192435]
Zitzmann, M., Rohayem, J., Raidt, J., Kliesch, S., Kumar, N., Sitruk-Ware, R., & Nieschlag, E. (2017). Impact of various progestins with or without transdermal testosterone on gonadotropin levels for non-invasive hormonal male contraception: a randomized clinical trial. Andrology, 5(3), 516–526. [DOI:10.1111/andr.12328]
Zuuren, E., & Fedorowicz, Z. (2016). Interventions for hirsutism excluding laser and photoepilation therapy alone: abridged Cochrane systematic review including GRADE assessments. British Journal of Dermatology, 175(1), 45–61. [DOI:10.1111/bjd.14486]
\ No newline at end of file
diff --git a/transfemscience.org/articles/transfem-lactation-literature/index.html b/transfemscience.org/articles/transfem-lactation-literature/index.html
index 740f730c..d20f83cd 100644
--- a/transfemscience.org/articles/transfem-lactation-literature/index.html
+++ b/transfemscience.org/articles/transfem-lactation-literature/index.html
@@ -1 +1 @@
-Published Case Reports of Lactation and/or Breastfeeding in Transfeminine People - Transfeminine Science
Published Case Reports of Lactation and/or Breastfeeding in Transfeminine People
By Aly | First published March 26, 2019 | Last modified August 20, 2025
Abstract / TL;DR
A number of case reports of hormonally induced lactation and breastfeeding in transfeminine people have been published. The earliest report of lactation in a transfeminine person was in the 1950s and the earliest report of breastfeeding was in the 1980s. Starting in 2019, more case reports have been published in the modern scientific literature. Unpublished cases also exist (e.g., that of Dr. Christine McGinn), and lactation has been induced or occurred in cisgender men as well. Lactation may be induced in transfeminine people with the use estrogens, progestogens, and/or prolactin releasers. Reviews discussing lactation induction in transfeminine people have recently been published.
Introduction
Last year, a case report of lactation and breastfeeding in a transgender woman was published:
Reisman, T., & Goldstein, Z. (2018). Case report: Induced lactation in a transgender woman. Transgender Health, 3(1), 24–26. [DOI:10.1089/trgh.2017.0044]
In the paper, the authors state the following:
We believe that this is the first formal report in the medical literature of induced lactation in a transgender woman.
However, this actually wasn’t the first case report of lactation and/or breastfeeding in a transfeminine person in the literature. There are various previous published cases dating back as far as the 1950s. These instances are provided below in the format of sources and excerpts.
Published Case Reports
Foss (1956)
Foss, G. L. (1956). Abnormalities of form and function of the human breast. Journal of Endocrinology, 14(4 Suppl) [Proceedings of the Society for Endocrinology: Fifty-Fourth Meeting. Symposium on Selected Aspects of the Practice of Hormone Administration in Animals and Man], vi–vii. [Google Scholar] [Google Books] [URL] [PDF]:
Based on the theories of lactogenesis and stimulated by the success of Lyons, Li, Johnson & Cole [1955], who succeeded in producing lactation in male rats, an attempt was made to initiate lactogenesis in a male transvestist. Six years ago this patient had been given oestrogens. Both testes and penis were then removed and an artificial vagina was constructed by plastic surgery. The patient was implanted with 500 mg oestradiol in September 1954, and 600 mg in July 1955. The breasts were then developed more intensively with daily injections of oestradiol dipropionate and progesterone for 6 weeks. Immediately following withdrawal of this treatment, prolactin 22·9 mg was injected daily for 3 days without effect. After a second month on oestradiol and progesterone daily, combined injections of prolactin and somatotrophin were given for 4 days and suction was applied by a breast pump—four times daily. On the 4th and 5th days a few drops of colostrum were expressed from the right nipple.
Tindal & McNaught (1958)
Tindal, J. S., & McNaught, M. L. (1958). Hormonal Factors in Breast Development and Milk Secretion. In Gardiner-Hill, H. (Ed.). Modern Trends in Endocrinology, Volume 1 (pp. 188–211) (Modern Trends). London: Butterworth. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]:
Recently, an attempt has been made by Foss (1956) to initiate lactation in a castrated male transvestist. He was given an implant of 500 milligrams of oestradiol, and 10 months later, a further 600 milligrams of oestradiol, followed by daily injections of oestradiol dipropionate and progesterone for 6 weeks. Immediately after withdrawal of this treatment, 22·9 milligrams of prolactin were injected daily for 3 days but without effect. After a second month of treatment with oestradiol and progesterone daily, he was given combined injections of prolactin and somatotrophin for 4 days, suction with a breast-pump being employed 4 times daily. On the fourth and fifth days a few drops of colostrum were expressed from the right nipple. There is a possible application here of modern hormone knowledge to man, and further trials would be of interest.
Experimentally I have been able to induce lactogenesis in a male transvestite whose testes had been removed some years before and whose breasts had been well developed over a long period with stilbestrol and ethisterone.9 In July, 1955, 600 mg. of estradiol was implanted subcutaneously and weekly injections of 50 mg. of progesterone were given for four months. For the next month daily injections of 10 mg. estradiol dipropionate and 50 mg. progesterone were given. These injections were continued for another month, increasing progesterone to 100 mg. daily. Both hormones were then withdrawn, and daily injections of increasing doses of prolactin and somatotropin were given for four days; at the same time, the patient used a breast pump four times daily for 5 minutes on both sides. During this time the mammary veins were visibly enlarged and on the sixth and seventh days 1 to 2 cc. of milky fluid was collected.
Flückiger, Del Pozo, & von Werder (1982)
Flückiger, E., Del Pozo, E., & von Werder, K. (1982). Prolactin: Synthesis, Fate and Actions. In Flückiger, E. W., Del Pozo, E., & von Werder, K. (Eds.). Prolactin: Physiology, Pharmacology, and Clinical Findings (Monographs on Endocrinology, Volume 23) (pp. 1–23). Berlin/Heidelberg: Springer-Verlag. [Google Scholar] [Google Books] [DOI:10.1007/978-3-642-81721-2_1]:
An observation (Wyss and Del Pozo unpublished) in a male transsexual showed that induction of lactation can be similarly achieved in the human male.
Flückiger, E., Del Pozo, E., & von Werder, K. (1982). Nontumoral hyperprolactinemia. In Flückiger, E. W., Del Pozo, E., & von Werder, K. (Eds.). Prolactin: Physiology, Pharmacology, and Clinical Findings (Monographs on Endocrinology, Volume 23) (pp. 102–152). Berlin/Heidelberg: Springer-Verlag. [Google Scholar] [Google Books] [DOI:10.1007/978-3-642-81721-2_4]:
4.3.2 Effect of Hyperprolactinemia in Male Subjects
Although PRL circulates in male blood in appreciable concentrations its physiologic role has not been clarified. The lack of lactational requirements does not preclude that under adequate priming the male mammary gland will respond to a PRL challenge with milk production. Thus, Wyss and del Pozo (unpublished data) found that PRL stimulation with TRH was able to induce milk secretion in a male individual pretreated with estrogens. Certainly, the chronic ingestion of dopamine antagonists or estrogens may lead to sustained hyperprolactinemia, and the same effect can be expected in male subjects on chronic estrogen therapy of prostatic cancer or transsexualism (Frantz 1973; del Pozo, to be published).
Certainly, the prolonged intake of estrogens, in male subjects also, as observed in the treatment of prostatic carcinoma and in transsexuals, can lead to hyperprolactinemia (Frantz 1972b; del Pozo, to be published).
Kozlov, Mel’nichenko, & Golubeva (1985)
Kozlov, G. I., Mel’nichenko, G. A., & Golubeva, I. V. (1985). Случай лактореи у больного мужского пола с транссексуализмом. [Sluchai laktorei u bol’nogo muzhskogo pola s transseksualizmom. / Case of galactorrhea in a transsexual male patient.] Проблемы Эндокринологии [Problemy Èndokrinologii (Moskva) / Problems of Endocrinology (Moscow)], 31(1), 37–38. [ISSN:0375-9660] [Google Scholar 1] [Google Scholar 2] [PubMed] [DOI:10.14341/probl198531137-38] [PDF] [Translation] [Translated]:
The appearance of galactorrhea in men is most often a symptom of pituitary prolactinoma. Combined with gynecomastia and atrophy of the testicles, galactorrhea caused by adenomas of the pituitary gland in men is known as O’Connell syndrome (1).
In recent years, however, cases of galactorrhea have been described in men without radiological or clinical signs of pituitary adenoma (12). Of course, in these cases, the presence of undetected microadenomas of the pituitary gland cannot be excluded, especially since the level of prolactin in these patients is significantly increased (1, 2).
Some medications, especially antipsychotics and estrogen-containing oral contraceptives (7, 10), increase serum levels of prolactin and can lead to the development of galactorrhea.
There is information about the influence of psycho-emotional factors on the lactation process: the possibility of the development (induction) of psychogenic lactation during false pregnancy (3) is known, and, conversely, the possibility of the termination of lactation in nursing mothers after mental stress.
Accumulated clinical observations on the frequent development of depressive states in persistent galactorrhea–amenorrhea syndrome (4), cases of galactorrhea in the mentally ill, even in the absence of neuroleptics (7), as well as experimental observations on the effect of hyperprolactinemia on the behavioral responses of animals (5), require careful study of the relationship of hyperprolactinemia and psycho-emotional factors. In connection with this, we present the following observation.
The patient (P), was born a normal, full-term boy. He remembers well from 6 years. Early development was unremarkable, he did not differ from peers, but loved to play more with girls. He played with dolls and cars. At 10 years of age, there was a desire to wear women’s clothes. From the age of 12 he swam with girls in a shirt and shorts, as he was embarrassed by the lack of breasts. From the age of 14 he changed clothes in his mother’s dress, and only in such clothes “felt like a person”. From the same age in a woman’s dress he went to get acquainted with young men and got pleasure from it. At the age of 15, he came to the firm conviction that he was a girl, began to urinate like a girl, squatting, use lipstick, and put on powdered makeup. He suffered greatly from the presence of “deformities” – male genital organs. At the age of 17, while working as a “nurse” in a hospital, he began to self-inject himself with folliculin (estrogen) and progesterone, which caused the development of the breasts. With pleasure, he did women’s housework, and loved to tinker with children. Having received a passport, he redid it as female, thus resulting in a female civilian gender.
Twice he tried to commit suicide (he took sleeping pills), since he could not bear the duality of his existence. Twice he was treated in psychiatric hospitals about transsexualism, unsuccessfully.
During the examination in IEE and HCG at the age of 20 years, no abnormalities in somatic status were revealed: complex as a man, male genitals, shaved from 17 years of age daily. Erotic dreams were frequent, wherein he played the role of a woman, and denied emissions. The ejaculate was studied (obtained by vibratory massage): volume – 1.4 mL, pH 8.8 (norm 7.6–8.2), sperm count 31 million per 1 mL, mobility 57%, and morphologically normal 69%. Sex chromatin is negative.
At age 22, a course of treatment with cyproterone acetate was conducted at the Institute of Psychiatry of the Ministry of Health of the USSR. Muscle weakness, reduction of sexual hairiness, and appearance of colostrum excretion was noted.
When examined in IEE and HCG at 23 years, the breasts corresponded to the age of 15–16 years (on his own initiative he periodically took estrogens), and colostrum was secreted from the nipples (abundant drops when pressed – galactorrhea (++)). He insisted on castration and amputation of the penis, since, being a “woman”, he was ashamed of not having the appropriate genitals for his sex, which he called “deformities”.
On X-ray of the skull, the shape and size of the sella turcica were normal, but signs of increased intracranial pressure were revealed. On EEG against the background of the general phenomena of irritation, the focus of pathology was recorded in the left parietal lead. Indicators of the functional state of the thyroid gland were in the normal range. In the study of the radioimmunoassay method using standard kits from the Sorin company, some increase in prolactin level of 24 ng/mL was detected in the serum (normal for men is 4–15 ng/mL).
In connection with the repeated suicidal attempts, failure of psychiatric treatment, and in consideration of the fact that the patient has a female civilian sex and performs a female social role, castration and feminizing plastic surgery of the external genitalia were performed for the purpose of social rehabilitation.
Some time after the operation, the patient developed a renewed interest in life. After the surgical and hormonal correction, the patient irresistibly developed maternal instincts. Unmarried, the patient obtained permission for the adoption of a child, simulated pregnancy, and was discharged from the maternity hospital with a son. From the first days after the “birth”, galactorrhea sharply increased, and spontaneous outflow of milk appeared, with galactorrhea (+++). The baby was breastfed up to 6 months of age.
Thus, it can be thought that several factors played a role in the genesis of galactorrhea in this patient:
Increased prolactin levels with estrogen and cyproterone acetate. The hyperprolactic properties of estrogens have long been known; the ability of cyproterone acetate to increase serum prolactin levels was shown by K. Schmidt–Golewizer et al (9).
Increased intracranial pressure, the role of this factor and the genesis of neuroendocrine disorders and, in particular, in the development of galactorrhea was shown by R. Peterson (8).
Our message is the second in the world literature describing galactorrhea in a male patient with transsexualism. The first description of this kind was made in 1983 by R. Flüskiger et al. (6).
This observation demonstrates the independence of the mechanism of lactation development from one’s genetic sex and is alarming with regard to the possibility of drug-induced galactorrhea development in men.
Barber et al. (2004)
Barber, T., Basu, A., Rizvi, K., & Chapman, J. (2004). Normoprolactinaemic galactorrhoea in a male-to-female transsexual. Endocrine Abstracts, 7 [23rd Joint Meeting of the British Endocrine Societies with the European Federation of Endocrine Societies], 271–271. [Google Scholar] [URL]:
Hormonal therapies in the form of oestrogens, anti-androgens and progestogens are often used in the treatment of male-to-female transsexuals. We present the case of a 36 year old phenotypic male with karyotype 46XY who presented with normoprolactinaemic galactorrhoea likely to be related to prior oestrogen administration. He had been self-administering oestrogen and progesterone preparations continuously for 7 years (aged 26 - 33 years) in an attempt to develop female phenotypic characteristics in spite of a heterosexual desire. During this time he developed gynaecomastia with galactorrhoea, increased energy and libido, voice change and an attraction towards both men and women. However due to lack of financial resources to secure a complete gender change, he stopped self-medication with these preparations 3 years ago. Instead he embarked on a regime involving self-administered testosterone in an attempt to reverse the biological changes. After discontinuation of oestrogen the gynaecomastia regressed somewhat, although galactorrhoea continued and worsened with testosterone. Prior to referral he had been treated with dopamine agonists with little improvement in galactorrhoea and gynaecomastia.
Routine biochemistry and haematology are within their reference ranges. Baseline endocrinology is normal: Prolactin 197 milliUnits per litre, LH 2.9 Units per litre, FSH 7.9 Units per litre, free Testosterone 20 nanoMoles per litre, 17 beta-oestradiol less than 110 picoMoles per litre, TSH 0.96 milliUnits per litre and free T4 16.5 picoMoles per litre.
This case illustrates fascinating effects of exogenous oestrogen in the male. The effects of oestrogenic products of aromatised endogenous and briefly also exogenous testosterone acting on oestrogen-primed breast tissue may account for, at least in part, his continuing symptom of normoprolactinaemic galactorrhoea. However two other features do not have any direct explanations: the development of osteopenia during this period, and complete disappearance of vascular migraine, a condition worsened with oestrogens in the female. He is now on Tamoxifen although an opportunity to use the aromatase inhibitor, Anastrozole still remains.
Subsequent Case Reports
Moravek & Pasque (2019)
Moravek, M. B., & Pasque, K. B. (2019). Lactation Can Be Successfully Induced in Transgender Women While Maintaining Gender-Congruent Serum Hormone Levels. Reproductive Sciences, 26(Suppl 1), 136A–136A (abstract no. T-055). [Google Scholar] [DOI:10.1177/1933719119834079]:
Introduction: Transgender women may be interested in breastfeeding their children, but there are no established protocols for lactation induction in this population. The only case report of a lactation induction protocol in a transgender woman significantly lowered her estradiol dose, which would likely result in decreased serum estradiol and increased testosterone levels, with resultant increase in gender dysphoria. Our objective was to induce lactation in a transgender woman without interrupting her gendercongruent hormone profile.
Methods: A 34-year-old transgender woman with a 15-year history of gender-affirming hormone therapy with estradiol and spironolactone presented for lactation induction once her cisgender wife conceived. A modification of the Newman-Goldfarb method for adoptive mothers was used to induce lactation, and serum hormone levels followed.
Results: Baseline labs were obtained (time point 1), then medroxyprogesterone 1.25mg daily was added to her existing hormone regimen of estradiol 6mg daily and spironolactone 100mg twice daily (time point 2). Domperidone 10mg four times daily was initiated 1 month later. Approximately 5 weeks prior to the due date, the patient stopped medroxyprogesterone, decreased estradiol to 2mg daily, and began breast pumping (time point 3). Just prior to the infant’s birth, the patient was pumping 2-3 ounces of breastmilk every 3 hours (time point 4). Spironolactone was decreased to 50mg twice daily. Her son was born at term, via uncomplicated vaginal delivery. The infant was able to breastfeed from both mothers without difficulty, with both mothers pumping when they weren’t actively breastfeeding to maintain supply (time point 5). When the infant was approximately 2 months old, the patient noticed an increase in facial hair growth. Estradiol was increased to 3mg daily and spironolactone increased to 100mg twice daily, with resolution of hair growth and no decrease in milk supply (time point 6). The patient continued to breastfeed on this regimen for >6 months following her son’s birth. Serum hormone levels on the hormone regimens referenced at each time point throughout the patient’s course are displayed in table 1.
Conclusion: Lactation can be successfully induced in transgender women, without a significant decrease in estradiol supplementation. This regimen allows transgender women to breastfeed without developing male secondary sex characteristics incongruent with their gender identity
Table 1 Hormone profile at different time points.
Time Point
Estradiol (pg/mL)
Progesterone (ng/mL)
Testosterone (ng/mL)
Prolactin (ng/mL)
1
114
1.1
0.36
2
130
1.1
0.05
9
3
30
1.3
0.06
152
4
39
5
29
1.4
0.89
184
6
51
0.16
59
Unnithan, Elson, & Shenker (2020)
Unnithan, R., Elson, D. F., & Shenker, Y. (2020). Galactorrhea and Hyperprolactinemia in a Transgender Female. Journal of the Endocrine Society, 4(Suppl 1), A899–A899 (abstract no. SUN-043). [Google Scholar] [PubMed Central] [DOI:10.1210/jendso/bvaa046.1781] [PDF]:
Background: Galactorrhea is a rare manifestation of hyper-prolactinemia in males and post-menopausal females, however the hormonal milieu of the transgender female may increase its incidence
Clinical Case: A 43 year old transgender female presented with three years of bilateral breast discharge. She had chronic, stable headaches and fatigue, but no vision changes or other symptoms. Notably, she had breast augmentation surgery with saline breast implants placed shortly before the galactorrhea commenced. She was on a stable dose of estradiol tablets 1 mg twice daily for six years. On physical exam she had pronounced bilateral breast discharge of a milky quality with nipple compression. Prolactin levels were checked several times and were 40-50 ng/mL, TSH was 2.36 uIU/mL. An MRI showed a left inferior pituitary lesion measuring 6 mm x 3 mm x 5 mm with no mass effect on adjacent structures. Her breast discharge was not bothersome to her, and her pituitary lesion was small. It was unclear whether there was a relationship between her prolactin levels and the lesion seen on MRI, as we expected more pronounced prolactin elevation with a prolactinoma. Instead, given the timing of her symptoms in relation to her breast augmentation surgery, her galactorrhea and hyper-prolactinemia were thought to be the result of nipple irritation related to her breast implants combined with a hyper-estrogenemic state.
Clinical Lessons: In the setting of a prolactin secreting micro-adenoma, galactorrhea in a male is highly unusual. This case highlights the importance of recognizing that the unique medical and surgical characteristics of male to female transgender patients can lead to hyper-prolactinemia and galactorrhea.
Reference: Reisman T, Goldstein Z. Case report: induced lactation in a transgender woman. Transgender Health. 2018;3(1):24-26.
Wamboldt, Shuster, & Sidhu (2021)
Wamboldt, R., Shuster, S., & Sidhu, B. S. (2021). Lactation Induction in a Transgender Woman Wanting to Breastfeed: Case Report. The Journal of Clinical Endocrinology & Metabolism, 106(5), e2047–e2052. [DOI:10.1210/clinem/dgaa976]:
Context: Breastfeeding is known to have many health and wellness benefits to the mother and infant; however, breastfeeding in trans women has been greatly under-researched.
Objective: To review potential methods of lactation induction in trans women wishing to breastfeed and to review the embryological basis for breastfeeding in trans women.
Design: This article summarizes a case of successful lactation in a trans woman, in which milk production was achieved in just over 1 month.
Setting: This patient was followed in an outpatient endocrinology clinic.
Participant: A single trans woman was followed in our endocrinology clinic for a period of 9 months while she took hormone therapy to help with lactation.
Interventions: Readily available lactation induction protocols for nonpuerpural mothers were reviewed and used to guide hormone therapy selection. Daily dose of progesterone was increased from 100 mg to 200 mg daily. The galactogogue domperidone was started at 10 mg 3 times daily and titrated up to effect. She was encouraged to use an electric pump and to increase her frequency of pumping.
Main outcome measure: Lactation induction.
Results: At one month, she had noticed a significant increase in her breast size and fullness. Her milk supply had increased rapidly, and she was producing up to 3 to 5 ounces of milk per day with manual expression alone.
Conclusions: We report the second case in the medical literature to demonstrate successful breastfeeding in a trans woman through use of hormonal augmentation.
Further Case Reports
Delgado, D., Stellwagen, L., McCune, S., Sejane, K., & Bode, L. (2023). Experience of Induced Lactation in a Transgender Woman: Analysis of Human Milk and a Suggested Protocol. Breastfeeding Medicine, 18(11), 888–893. [DOI:10.1089/bfm.2023.0197]
Weimer, A. K. (2023). Lactation induction in a transgender woman: macronutrient analysis and patient perspectives. Journal of Human Lactation, 39(3), 488–494. [DOI:10.1177/08903344231170559]
van Amesfoort, J. E., Van Mello, N. M., & van Genugten, R. (2024). Lactation induction in a transgender woman: case report and recommendations for clinical practice. International Breastfeeding Journal, 19(1), 18. [DOI:10.1186/s13006-024-00624-1]
Trahair, E. D., Kokosa, S., Weinhold, A., Parnell, H., Dotson, A. B., & Kelley, C. E. (2024). Novel Lactation Induction Protocol for a Transgender Woman Wishing to Breastfeed: A Case Report. Breastfeeding Medicine, online ahead of print. [DOI:10.1089/bfm.2024.0012]
Dr. Christine McGinn
Dr. Christine McGinn is a transgender woman and well-known surgeon in Pennsylvania who performs gender-affirming surgeries for transgender people. When she had children with her cisgender female partner, McGinn induced a hormonal pseudopregnancy in herself and her and her partner breastfed their twins together. This was described in the media, including in books and television. McGinn’s case was never formally published as a case report in the scientific literature however.
The Oprah Winfrey Show (2010)
Terry, J. C. (Director), & Winfrey, O. G. (Presenter). (2010 September 29). The Mom Who “Fathered” Her Own Children, Plus the Cast of Modern Family [Television series episode]. The Oprah Winfrey Show (Season 25, Episode 13). Chicago: Harpo Studios. [URL 1] [URL 2] [URL 3]
Trans (2012)
Arnold, C. (Director), Schoen, M. (Producer), RoseWorks (Firm), & Sex Smart Films (Firm). (2012). Trans [DVD] (1:21:32–1:21:55). [WorldCat] [IMDB] [Amazon Prime Video]
Boylan (2014)
Boylan, J. F. (2014). Dr. Christine McGinn. In Boylan, J. F. Stuck in the Middle with You: A Memoir of Parenting in Three Genders (pp. 223–233). New York: Broadway Books. [Google Scholar] [Google Books 1] [Google Books 2] [WorldCat] [PDF]:
Dr. Christine McGinn is a surgeon, a mother of two, a backup flight surgeon for the space shuttle progarm, and a transgender woman. As a man, she saved her sperm before transition; ten years later she used that sperm to have children with her partner Lisa. The two of them are both biological mothers of their son and daughter, and each mother was able to breast-feed the twins. I sat down with Christine at her office in New Hope, Pennsylvania, on a hot summer day in 2011.
CM: […] Then there’s the scientist in me that knows that there is a difference, there is not a binary, but a gender spectrum. There are chemicals that are different in men and women. And when a transgender woman transitions, we are somewhere in the middle. Especialy having gone through a simulated pregnancy, in order to breast-feed, I felt the changes of those hormones. I felt my milk let down when not only my baby would cry, but a baby on TV would cry, and even, ridiculously, when a door would close and make a squeak.
JFB: You had to induce a false pregnancy in order to breast-feed? Tell me how you did that.
CM: As a doctor, I knew it was possible. I followed the protocol that involves simulating pregnancy with hormones. It’s estrogen and progesterone. My simulation pregnancy was over a month before Lisa delivered—with twins, we were expecting them to be born earlier. That entire month I was just pumping nonstop, every two hours. We had a whole freezer full of milk. And you know, the first couple of weeks it was no good, because it had all of the hormones in it. So we only saved, like, the last week or so. But still, it was a freezer full of milk.
Lisa had no idea about the way breast-feeding takes over your life, because this was her first. It was kind of funny that I went through that on my own, first, weeks before she did. And then it took her a couple of days to actually—for her milk to let down.
The children were so small when they were born. They were only five pounds. At first we had to feed them with a syringe. They were breast-feeding as well, but they weren’t latching that great on either of us.
JFB: What was it like when they finally muckled on to you?
CM: Oh, I can’t even put it in words. I really cannot put it in words. It was—I was just—oh.
JFB: Were you amazed? Were you afraid?
CM: It was heaven. I was afraid. I don’t know, it was uncharted territory. Like, I knew the milk was good. Lisa was a little concerned that it would be like skimmed milk, or something, you know. [Laughs] Like—she’s like, “Is it the same stuff?”
JFB: Is it the same milk?
CM: And she was a little dubious about, like, is this really all right? I think that’s totally natural for a mother, to be concerned.
I will just say that there are things snobody thinks about when two women are both breast-feeding. Like, technical stuff that you don’t think about. When you have a mother and a father, the mother decides when the kids get fed. Right? The father doesn’t, really. Right?
But you know, when you have two women who are filled with pregnancy hormones and have that, like, mother-bear attitude about how things should be done… It was really crazy.
JFB: So did that cause serious conflict between you and Lisa?
CM: Totally not serious conflict, because the most important thing are the babies.
Eden finally latched—I breast-fed her more than Luke. Luke was never really good. Lisa hated breast-feeding. Eventually we decided to stop.
I’m putting on my science hat again—when you decide to stop, there are hormonal issues. The strongest emotion a person can feel in their life comes frm oxytocin, which is the love drug.
JFB: Oxytocin?
CM: That’s what’s responsible for babies’ bonding during breastfeeding. So the baby latches on, breast-feeds, your brain just [makes oozing sounds], just like, oozes this gooey love substance, oxytocin. Fathers are proven to have higher oxytocin before the delivery, and just stroking your child’s head. You know, when the baby—when you smell a newborn’s head, it really—that smell, it’s like—
JFB: I just saw a friend’s newborn on Friday, and I was like, [makes sniffing sound]—
CM: My niece said it best. She came in and smell them, and she was five years old at the time, and she’s like, “They smell like cupcakes.” [Laughs] And it’s universal. When you ask me what that’s like, I can’t describe it, you know, and I’m a huge fan of food and cupcakes and chocolate, and so that’s the closest I can come to it—it’s like chocolate. [Laughs]
JFB: So when you stopped breast-feeding, was it a kind of a mourning, a loss?
CM: Yes. Lisa wanted to stop before I did. The problem is, once a baby gets a nipple, a plastic nipple, it gives more milk. And so they don’t have to work as hard.
It’s a unique situation that two breast-feeders in a relationship would experience, but a mother and father would not.
JFB: So did one of you stop breast-feeding before the other?
CM: Yes, Lisa did.
JFB: Lisa stopped. And how much longer did you keep it up?
CM: Not long, because they got the nipple.
They were both so small. We weren’t all that successful at it. We were so worried about their birth weight, and making sure they got enough with the syringes. There were definitely times where, you know, we both would breast-feed and, man, I will never forget that. Like, three ‘clock in the morning, four o’clock in the morning, in the little cocoon, nursing.
The heat of their body, their naked body on your chest. The amazing thing is, it really does kind of hurt when they really get going, you know. And you just… I don’t know how else to describe it. You feel like the life force is just coming out through you. It’s so powerful. It relieves that pain that you have in your breast. It releases that oxytocin, and it’s just—it’s even.
JFB: Did you ever do that thing where you would fall asleep with the children in the bed, and wake up with the children in the bed beside you?
CM: Yeah.
JFB: I loved that. It’s one of my stnogest memories of being a father. Having gotten up in the middle of the night. And they are so small, but such an incredibly powerful feeling, the two of you together surrounding the child. With us, we also had a dog at the bottom of the bed. [Laughs]
CM: And we have two, and that was also very important to me, too. We have miniature pinschers.
JFB: So how many months along did you stop breast-feeding?
CM: Three months. It was really emotionally painful, and I cried a lot. I was really sad.
I was pretty sure we were not going to have any more kids. So I’m like, “This is it.” It was very sad.
JFB: Is there a moment frm the last year and two months where you think, This is what it’s like to be a mother, this is it?
CM: Yes, immediately. It was hot as Hades outside. It was, like, a million degrees. We had just had the kids. It was like, May or June, and my mom was over, and it was, like, we had all this help, initially, because Lisa and I were just not getting any sleep and it was, like, round-the-clock feedings and the kids were small, and Lucas had an apnea monitor that he had to wear all the time, and it was just really hard. And there was a big thunderstorm, and the power went out.
And so, at this point, they weren’t really latching very well, so we both had to pump, and then feed them with the syringes. So Lisa and I are totally, like, engorged with milk. And the power’s out, and the pumps are electric. Right?
JFB: Right.
CM: So there’s no electricity, it’s hot as hell, we’re worried for the kids. Lisa and I are in pain. We’re both leaking. And it was the weirdest, funniest situation. And my mom’s there. She runs out to the store to get batteries, and you know, she’s just beng a mom. She’s getting everything, running around like an angel. And Lisa and I are in pain we’re miserable. When she finally came back, the batteries wouldn’t work on the pumps—something else was wrong. Lisa and I are dying.
And so, here’s the guy part of me… I get the pump that has the backup battery power and the backup car charger. Like, I got all tech on it. [Laughs] I’m out int he car trying to get the car charger to work on the pump in the pouring rain. And it’s ninety-five degrees out. It’s all wet inside, like, the humidity on the windows.
And I’m just trying to get some kind of relief.
And this stupid pump didn’t work that way, either. We come back in and my mom has candles lit.
And then the electricity comes back on. And we all just laugh and pump and breast-feed. And every one of us is in heaven.
Pfeffer (2017)
Pfeffer, C. A. (2017). Trans Partnerships and Families: Historical Traces and Contemporary Representations. In Pfeffer, C. A. Queering Families: The Postmodern Partnerships of Cisgender Women and Transgender Men (pp. 1–34). New York: Oxford University Press. [Google Scholar] [Google Books] [WorldCat] [DOI:10.1093/acprof:oso/9780199908059.003.0001] [Archive.org]:
Just 2 years later, Winfrey would feature another interview that elicited many of the same audience reactions. In this 2010 episode, lesbian partners Dr. Christine McGinn and Lisa Bortz beamed with joy as they held their infant twins. Again, audience members’ jaws dropped when it was revealed that beautiful Christine was a male-to-female transsexual who used to be a handsome military officer Chris, and that Lisa had given birth to the couple’s biological children using sperm Chris banked prior to gender confirmation surgeries.10 And it was Winfrey’s chin that nearly hit the floor as she watched video of Christine breastfeeding the couples’ children (the episode is referred to online as “The Mom Who Fathered Her Own Children”).
Many unpublished reports of lactation and breastfeeding in transfeminine people have been described on the web including at the following pages:
Richards, A. (2003). Lactation and the Transsexual Woman. Second Type Woman. [Updated August 2018] [URL] [PDF]
MacDonald, T. (2013). Trans Women and Breastfeeding: A Personal Interview. Milk Junkies. [URL]
MacDonald, T. (2013). Trans Women and Breastfeeding: The Health Care Provider. Milk Junkies. [URL]
MacDonald, T. (2017). Jenna’s Breastfeeding Journey: Trans Motherhood. Milk Junkies. [URL]
Burns, K. (2018). Yes, Trans Women Can Breastfeed — Here’s How. them. [URL]
Cisgender Men
Induction of lactation has been reported in cisgender men and is noteworthy:
Geschickter (1945)
Geschickter, C. F. (1945). Endocrine Physiology of the Breast. In Geschickter, C. F. Diseases of the Breast: Diagnosis, Pathology, Treatment, 2nd Edition (pp. 42–81). Philadelphia: J.B. Lippincott. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]:
The results obtained indicate that a lactogenic substance in anterior pituitary extracts may cause mammary secretion in nonpregnant women when they have been previously stimulated with estrogenic hormone but true lactation does not occur. Secretion was also obtained in two adult men with gynecomastia after injections of lactogenic hormone.
Huggins (1949)
Huggins, C. (1949). Endocrine substances in the treatment of cancers. Journal of the American Medical Association, 141(11), 750–754. [DOI:10.1001/jama.1949.02910110002002]:
The administration of estrogen in effective amounts causes testicular atrophy and mammary hypertrophy. Growth of the breasts can be so extensive that lactation may be induced, as illustrated in the following case.
W. N., aged 64, had carcinoma of the prostate with osseous metastases, for which he was treated by a permanent suprapubic cystotomy in 1941. Diethylstilbestrol, 20 mg. daily, was given orally for two years beginning September 1942. In September 1944, 25 mg. (500 international units) of prolactin14 was injected daily for five days, and at the end of this time creamy milk could be expressed from both breasts. Orchiectomy and removal of the cystostomy tube were carried out September 6, when administration of estrogen was discontinued; both incisions healed promptly. Since then the patient has been clinically well but has continued to lactate, a large drop of milk being easily expressed from each breast at frequent intervals.
Huggins & Dao (1954)
Huggins, C., & Dao, T. L. (1954). Lactation induced by luteotrophin in women with mammary cancer. Growth of the breast of the human male following estrogenic treatment. Cancer Research, 14(4), 303–306. [Google Scholar] [PubMed] [URL]:
In the observations to be presented luteotrophin [prolactin] was employed as a stimulus for mammary secretion in patients with cancer of the breast, and the results throw new light on the physiology of women bearing this neoplasm. We shall also describe conditions which resulted in the induction of physiologic maturity in the human male, since knowledge of the action of hormones on the human breast is vague.
The effects of luteotrophin on the breast of women post partum has been extensively investigated, but otherwise few observations have been made in the human. Werner (14) administered a crude pituitary extract containing luteotrophin to eight castrate women 21–35 years of age; lactation was not observed, although in one woman “a few drops of colostrum-like fluid” could be expressed from the breasts. Goldzieher (4) treated menstrual disorders in women with luteotrophin, but mammary secretion was not described by him.
PROCEDURE
Luteotrophin,1 dissolved in physiological saline made slight ly alkaline (pH 9) with sodium hydroxide, was injected subcutaneously in daily amounts of 500 International Units; the solutions were freshly prepared, and the injections were continued for 7 days only.
This series comprised 21 female patients who had dis seminated mammary cancer, and all had been subjected to unilateral mastectomy. There were also three men with advanced prostatic carcinoma who had been treated for thera peutic purposes with oral diethylstilbestrol for 20 months, 2, and 6 years, respectively. There were eight persons without mammary or prostatic cancers who served as controls.
In each case of mammary cancer a biopsy of the breast was obtained for histological purposes, the material being stained with Sudan III.3
OBSERVATIONS
Lactation, when it occurred, was never profuse; it varied from a tiny drop to ca. 0.5 cc. from each breast. Clear colostrum was not observed, and the mammary secretion was always milk, as defined above.
Mammary growth in the human male.—Estrogenic substances had been administered to three men in the treatment of disseminated prostate cancer for many months; after luteotrophin injection two lactated and one did not lactate.
W. N. (reported in brief earlier [5]), age 64, had taken diethylstilbestrol, 20 mg/day, orally for 2 years, after which interval sub-areolar button-like masses of mammary tissue could be palpated bilaterally; luteotrophin was then injected for 5 days, and milk was expressed from the breast on the 6th day. Orchiectomy was then performed, and both luteotrophin and estrogenic substances were discontinued. This man continued to lactate for 7 years when the formation of milk gradually ceased.
In the case of A. W., age 62, diethylstilbestrol (5–15 mg/day) had been ingested for 20 months after bilateral orchiectomy; the breasts became slightly enlarged. Luteotrophin was injected, and lactation occurred on the 7th day. A biopsy of the breast showed moderately well developed mammary ducts and alveoli containing milk. In the case of E. G., age 59, diethylstilbestrol (5 mg/day) was ingested almost continuously for 6 years; this resulted in the development of large pendulous breasts, but no lactation occurred after injections of luteotrophin.
Lactation in humans without cancer.—Luteotrophin was administered to two normal males, age 51 and 59, and to four normal females, age 84–59, and none lactated.
DISCUSSION
It must be emphasized that lactation was not copious in any of the humans when it had been induced by luteotrophin; merely small amounts of milk were obtained. It was apparent, however, from the histological studies of the mammary tissue obtained by biopsy that the secretion of milk in any quantity was a criterion of maturity of mammary epithelium.
In the goat and guinea pig it is known that estrogenic substances can induce mammary ma turity without the intervention of exogenous synergistic steroids. In the experiments of Lewis and Turner (9) diethylstilbestrol was implanted in two castrate male goats; one of these animals failed to lactate, while the other produced a small quantity of milk without luteotrophin injections. They obtained small amounts of milk from a male kid similarly treated. Nelson (10) found that estrone induced mammary growth with, later, lactation in the male guinea pig. Our observations demonstrate that diethylstilbestrol ingested for prolonged periods of time can induce maturity of the breast in certain elderly human males. However, the human male differs from the animals just described in that spontaneous lactation was not observed; the injection of luteotrophin was necessary for milk formation.
The duration of lactation induced by luteo trophin was impressive, since milk commonly persisted for many months—and in one male for 7 years. The mechanism whereby this type of lactation is maintained for such long periods of time is at present unknown; we know that milk continues to be secreted both in the presence of the adrenal glands and in the absence of these structures and the gonads as well. Observations (8) have been made on experimental animals which are analogous to the clinical findings; most dogs with spontaneous mammary cancer possess lactation, and this characteristic persists for many months, at least, despite the removal of the adrenal glands and the ovaries.
SUMMARY
The breast of the human male can be induced to grow to a functionally mature state by the administration of estrogenic substances without additional exogenous steroid synergists. Spontaneous lactation was not observed in these men, but it was induced by luteotrophin.
The formation of milk in any amount by the breast is a criterion of functional maturity of the mammary epithelium. Luteotrophin induced the secretion of small amounts of milk in a group of women with mammary cancer and in a number of healthy women as well, and, in addition, in two human males to whom estrogenic substances had been administered for therapeutic purposes. Lactation did not occur in two normal males.
When lactation was induced in human beings, the secretion often persisted for many months; it lasted for 7 years in one man.
HUGGINS,C. Endocrine Substances in the Treatment of Cancers. J.A.M.A., 141:750–54, 1949.
Brodribb, W., & Academy of Breastfeeding Medicine. (2018). ABM Clinical Protocol #9: Use of galactogogues in initiating or augmenting maternal milk production, second revision 2018. Breastfeeding Medicine, 13(5), 307–314. [DOI:10.1089/bfm.2018.29092.wjb]
MacDonald, T. K. (2019). Lactation care for transgender and non-binary patients: Empowering clients and avoiding aversives. Journal of Human Lactation, 35(2), 223–226. [DOI:10.1177/0890334419830989]
Paynter, M. J. (2019). Medication and Facilitation of Transgender Women’s Lactation. Journal of Human Lactation, 35(2), 239–243. [DOI:10.1177/0890334419829729]
Cazorla-Ortiz, G., Obregón-Guitérrez, N., Rozas-Garcia, M. R., & Goberna-Tricas, J. (2020). Methods and Success Factors of Induced Lactation: A Scoping Review. Journal of Human Lactation, 36(4), 739–749. [DOI:10.1177/0890334420950321]
Ferri, R. L., Rosen-Carole, C. B., Jackson, J., Carreno-Rijo, E., Greenberg, K. B., & Academy of Breastfeeding Medicine. (2020). ABM Clinical Protocol #33: Lactation Care for Lesbian, Gay, Bisexual, Transgender, Queer, Questioning, Plus Patients. Breastfeeding Medicine, 15(5), 284–293. [DOI:10.1089/bfm.2020.29152.rlf]
García-Acosta, J. M., Juan-Valdivia, S., María, R., Fernández-Martínez, A. D., Lorenzo-Rocha, N. D., & Castro-Peraza, M. E. (2020). Trans* Pregnancy and Lactation: A Literature Review from a Nursing Perspective. International Journal of Environmental Research and Public Health, 17(1), 44. [DOI:10.3390/ijerph17010044]
LeCain, M., Fraterrigo, G., & Drake, W. M. (2020). Induced Lactation in a Mother Through Surrogacy With Complete Androgen Insensitivity Syndrome (CAIS). Journal of Human Lactation, 36(4), 791–794. [DOI:10.1177/0890334419888752]
Trautner, E., McCool-Myers, M., & Joyner, A. B. (2020). Knowledge and practice of induction of lactation in trans women among professionals working in trans health. International Breastfeeding Journal, 15(1), 63. [DOI:10.1186/s13006-020-00308-6]
References (Inline Citations)
Bazarra-Castro, M. A. (2009). Etiological aspects, therapy regimes, side effects and treatment satisfaction of transsexual patients. (Doctoral dissertation, Ludwig Maximilian University of Munich.) [DOI:10.5282/edoc.9984] [URN:urn:nbn:de:bvb:19-99840] [PDF]
Delgado, D., Stellwagen, L., McCune, S., Sejane, K., & Bode, L. (2023). Experience of Induced Lactation in a Transgender Woman: Analysis of Human Milk and a Suggested Protocol. Breastfeeding Medicine, 18(11), 888–893. [DOI:10.1089/bfm.2023.0197]
Dewhurst, J., & Underhill, R. (1979). The Suppression of Facial Hair Growth in Transsexuals Using Cyproterone Acetate. British Journal of Sexual Medicine, 6, 19–23. [Google Scholar] [PDF]
Futterweit, W. (1980). Endocrine management of transsexual. Hormonal profiles of serum prolactin, testosterone, and estradiol. New York State Journal of Medicine, 80(8), 1260–1264. [Google Scholar] [PubMed] [Archive.org] [PDF]
Gooren, L. J., Harmsen-Louman, W., & Kessel, H. (1985). Follow-up of prolactin levels in long-term oestrogen-treated male-to-female transsexuals with regard to prolactinoma induction. Clinical Endocrinology, 22(2), 201–207. [DOI:10.1111/j.1365-2265.1985.tb01081.x]
Greenblatt, R. B. (1972). Inappropriate lactation in men and women. Medical Aspects of Human Sexuality, 6(6), 25–33. [Google Scholar] [Google Books]
Greenblatt, R. B., & Leng, J. J. (1972). Lactation anormale chez l’homme. [Abnormal lactation in males.] Bordeaux Medical, 5(3), 241–243. [Google Scholar] [PubMed]
Kanhai, R. C., Hage, J. J., van Diest, P. J., Bloemena, E., & Mulder, J. W. (2000). Short-Term and Long-Term Histologic Effects of Castration and Estrogen Treatment on Breast Tissue of 14 Male-to-Female Transsexuals in Comparison With Two Chemically Castrated Men. The American Journal of Surgical Pathology, 24(1), 74–80. [DOI:10.1097/00000478-200001000-00009]
Levy, A., Crown, A., & Reid, R. (2003). Endocrine intervention for transsexuals. Clinical Endocrinology, 59(4), 409–418. [DOI:10.1046/j.1365-2265.2003.01821.x]
Neumann, F. V., von Berswordt-Wallrabe, R., Elger, W., Steinbeck, H., Hahn, J. D., & Kramer, M. (1970). Aspects of Androgen-Dependent Events as Studied by Antiandrogens. In Astwood, E. B. (Ed.). Proceedings of the 1969 Laurentian Hormone Conference (Recent Progress in Hormone Research, Volume 26) (pp. 337–410). New York/London: Academic Press. [DOI:10.1016/B978-0-12-571126-5.50013-3]
Schlatterer, K., Yassouridis, A., Werder, K. V., Poland, D., Kemper, J., & Stalla, G. K. (1998). Archives of Sexual Behavior, 27(5), 475–492. [DOI:10.1023/a:1018704630036]
Trahair, E. D., Kokosa, S., Weinhold, A., Parnell, H., Dotson, A. B., & Kelley, C. E. (2024). Novel Lactation Induction Protocol for a Transgender Woman Wishing to Breastfeed: A Case Report. Breastfeeding Medicine, online ahead of print. [DOI:10.1089/bfm.2024.0012]
van Amesfoort, J. E., Van Mello, N. M., & van Genugten, R. (2024). Lactation induction in a transgender woman: case report and recommendations for clinical practice. International Breastfeeding Journal, 19(1), 18. [DOI:10.1186/s13006-024-00624-1]
Weimer, A. K. (2023). Lactation induction in a transgender woman: macronutrient analysis and patient perspectives. Journal of Human Lactation, 39(3), 488–494. [DOI:10.1177/08903344231170559]
\ No newline at end of file
+Published Case Reports of Lactation and/or Breastfeeding in Transfeminine People - Transfeminine Science
Published Case Reports of Lactation and/or Breastfeeding in Transfeminine People
By Aly | First published March 26, 2019 | Last modified August 20, 2025
Abstract / TL;DR
A number of case reports of hormonally induced lactation and breastfeeding in transfeminine people have been published. The earliest report of lactation in a transfeminine person was in the 1950s and the earliest report of breastfeeding was in the 1980s. Starting in 2019, more case reports have been published in the modern scientific literature. Unpublished cases also exist (e.g., that of Dr. Christine McGinn), and lactation has been induced or occurred in cisgender men as well. Lactation may be induced in transfeminine people with the use estrogens, progestogens, and/or prolactin releasers. Reviews discussing lactation induction in transfeminine people have recently been published.
Introduction
Last year, a case report of lactation and breastfeeding in a transgender woman was published:
Reisman, T., & Goldstein, Z. (2018). Case report: Induced lactation in a transgender woman. Transgender Health, 3(1), 24–26. [DOI:10.1089/trgh.2017.0044]
In the paper, the authors state the following:
We believe that this is the first formal report in the medical literature of induced lactation in a transgender woman.
However, this actually wasn’t the first case report of lactation and/or breastfeeding in a transfeminine person in the literature. There are various previous published cases dating back as far as the 1950s. These instances are provided below in the format of sources and excerpts.
Published Case Reports
Foss (1956)
Foss, G. L. (1956). Abnormalities of form and function of the human breast. Journal of Endocrinology, 14(4 Suppl) [Proceedings of the Society for Endocrinology: Fifty-Fourth Meeting. Symposium on Selected Aspects of the Practice of Hormone Administration in Animals and Man], vi–vii. [Google Scholar] [Google Books] [URL] [PDF]:
Based on the theories of lactogenesis and stimulated by the success of Lyons, Li, Johnson & Cole [1955], who succeeded in producing lactation in male rats, an attempt was made to initiate lactogenesis in a male transvestist. Six years ago this patient had been given oestrogens. Both testes and penis were then removed and an artificial vagina was constructed by plastic surgery. The patient was implanted with 500 mg oestradiol in September 1954, and 600 mg in July 1955. The breasts were then developed more intensively with daily injections of oestradiol dipropionate and progesterone for 6 weeks. Immediately following withdrawal of this treatment, prolactin 22·9 mg was injected daily for 3 days without effect. After a second month on oestradiol and progesterone daily, combined injections of prolactin and somatotrophin were given for 4 days and suction was applied by a breast pump—four times daily. On the 4th and 5th days a few drops of colostrum were expressed from the right nipple.
Tindal & McNaught (1958)
Tindal, J. S., & McNaught, M. L. (1958). Hormonal Factors in Breast Development and Milk Secretion. In Gardiner-Hill, H. (Ed.). Modern Trends in Endocrinology, Volume 1 (pp. 188–211) (Modern Trends). London: Butterworth. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org]:
Recently, an attempt has been made by Foss (1956) to initiate lactation in a castrated male transvestist. He was given an implant of 500 milligrams of oestradiol, and 10 months later, a further 600 milligrams of oestradiol, followed by daily injections of oestradiol dipropionate and progesterone for 6 weeks. Immediately after withdrawal of this treatment, 22·9 milligrams of prolactin were injected daily for 3 days but without effect. After a second month of treatment with oestradiol and progesterone daily, he was given combined injections of prolactin and somatotrophin for 4 days, suction with a breast-pump being employed 4 times daily. On the fourth and fifth days a few drops of colostrum were expressed from the right nipple. There is a possible application here of modern hormone knowledge to man, and further trials would be of interest.
Experimentally I have been able to induce lactogenesis in a male transvestite whose testes had been removed some years before and whose breasts had been well developed over a long period with stilbestrol and ethisterone.9 In July, 1955, 600 mg. of estradiol was implanted subcutaneously and weekly injections of 50 mg. of progesterone were given for four months. For the next month daily injections of 10 mg. estradiol dipropionate and 50 mg. progesterone were given. These injections were continued for another month, increasing progesterone to 100 mg. daily. Both hormones were then withdrawn, and daily injections of increasing doses of prolactin and somatotropin were given for four days; at the same time, the patient used a breast pump four times daily for 5 minutes on both sides. During this time the mammary veins were visibly enlarged and on the sixth and seventh days 1 to 2 cc. of milky fluid was collected.
Flückiger, Del Pozo, & von Werder (1982)
Flückiger, E., Del Pozo, E., & von Werder, K. (1982). Prolactin: Synthesis, Fate and Actions. In Flückiger, E. W., Del Pozo, E., & von Werder, K. (Eds.). Prolactin: Physiology, Pharmacology, and Clinical Findings (Monographs on Endocrinology, Volume 23) (pp. 1–23). Berlin/Heidelberg: Springer-Verlag. [Google Scholar] [Google Books] [DOI:10.1007/978-3-642-81721-2_1]:
An observation (Wyss and Del Pozo unpublished) in a male transsexual showed that induction of lactation can be similarly achieved in the human male.
Flückiger, E., Del Pozo, E., & von Werder, K. (1982). Nontumoral hyperprolactinemia. In Flückiger, E. W., Del Pozo, E., & von Werder, K. (Eds.). Prolactin: Physiology, Pharmacology, and Clinical Findings (Monographs on Endocrinology, Volume 23) (pp. 102–152). Berlin/Heidelberg: Springer-Verlag. [Google Scholar] [Google Books] [DOI:10.1007/978-3-642-81721-2_4]:
4.3.2 Effect of Hyperprolactinemia in Male Subjects
Although PRL circulates in male blood in appreciable concentrations its physiologic role has not been clarified. The lack of lactational requirements does not preclude that under adequate priming the male mammary gland will respond to a PRL challenge with milk production. Thus, Wyss and del Pozo (unpublished data) found that PRL stimulation with TRH was able to induce milk secretion in a male individual pretreated with estrogens. Certainly, the chronic ingestion of dopamine antagonists or estrogens may lead to sustained hyperprolactinemia, and the same effect can be expected in male subjects on chronic estrogen therapy of prostatic cancer or transsexualism (Frantz 1973; del Pozo, to be published).
Certainly, the prolonged intake of estrogens, in male subjects also, as observed in the treatment of prostatic carcinoma and in transsexuals, can lead to hyperprolactinemia (Frantz 1972b; del Pozo, to be published).
Kozlov, Mel’nichenko, & Golubeva (1985)
Kozlov, G. I., Mel’nichenko, G. A., & Golubeva, I. V. (1985). Случай лактореи у больного мужского пола с транссексуализмом. [Sluchai laktorei u bol’nogo muzhskogo pola s transseksualizmom. / Case of galactorrhea in a transsexual male patient.] Проблемы Эндокринологии [Problemy Èndokrinologii (Moskva) / Problems of Endocrinology (Moscow)], 31(1), 37–38. [ISSN:0375-9660] [Google Scholar 1] [Google Scholar 2] [PubMed] [DOI:10.14341/probl198531137-38] [PDF] [Translation] [Translated]:
The appearance of galactorrhea in men is most often a symptom of pituitary prolactinoma. Combined with gynecomastia and atrophy of the testicles, galactorrhea caused by adenomas of the pituitary gland in men is known as O’Connell syndrome (1).
In recent years, however, cases of galactorrhea have been described in men without radiological or clinical signs of pituitary adenoma (12). Of course, in these cases, the presence of undetected microadenomas of the pituitary gland cannot be excluded, especially since the level of prolactin in these patients is significantly increased (1, 2).
Some medications, especially antipsychotics and estrogen-containing oral contraceptives (7, 10), increase serum levels of prolactin and can lead to the development of galactorrhea.
There is information about the influence of psycho-emotional factors on the lactation process: the possibility of the development (induction) of psychogenic lactation during false pregnancy (3) is known, and, conversely, the possibility of the termination of lactation in nursing mothers after mental stress.
Accumulated clinical observations on the frequent development of depressive states in persistent galactorrhea–amenorrhea syndrome (4), cases of galactorrhea in the mentally ill, even in the absence of neuroleptics (7), as well as experimental observations on the effect of hyperprolactinemia on the behavioral responses of animals (5), require careful study of the relationship of hyperprolactinemia and psycho-emotional factors. In connection with this, we present the following observation.
The patient (P), was born a normal, full-term boy. He remembers well from 6 years. Early development was unremarkable, he did not differ from peers, but loved to play more with girls. He played with dolls and cars. At 10 years of age, there was a desire to wear women’s clothes. From the age of 12 he swam with girls in a shirt and shorts, as he was embarrassed by the lack of breasts. From the age of 14 he changed clothes in his mother’s dress, and only in such clothes “felt like a person”. From the same age in a woman’s dress he went to get acquainted with young men and got pleasure from it. At the age of 15, he came to the firm conviction that he was a girl, began to urinate like a girl, squatting, use lipstick, and put on powdered makeup. He suffered greatly from the presence of “deformities” – male genital organs. At the age of 17, while working as a “nurse” in a hospital, he began to self-inject himself with folliculin (estrogen) and progesterone, which caused the development of the breasts. With pleasure, he did women’s housework, and loved to tinker with children. Having received a passport, he redid it as female, thus resulting in a female civilian gender.
Twice he tried to commit suicide (he took sleeping pills), since he could not bear the duality of his existence. Twice he was treated in psychiatric hospitals about transsexualism, unsuccessfully.
During the examination in IEE and HCG at the age of 20 years, no abnormalities in somatic status were revealed: complex as a man, male genitals, shaved from 17 years of age daily. Erotic dreams were frequent, wherein he played the role of a woman, and denied emissions. The ejaculate was studied (obtained by vibratory massage): volume – 1.4 mL, pH 8.8 (norm 7.6–8.2), sperm count 31 million per 1 mL, mobility 57%, and morphologically normal 69%. Sex chromatin is negative.
At age 22, a course of treatment with cyproterone acetate was conducted at the Institute of Psychiatry of the Ministry of Health of the USSR. Muscle weakness, reduction of sexual hairiness, and appearance of colostrum excretion was noted.
When examined in IEE and HCG at 23 years, the breasts corresponded to the age of 15–16 years (on his own initiative he periodically took estrogens), and colostrum was secreted from the nipples (abundant drops when pressed – galactorrhea (++)). He insisted on castration and amputation of the penis, since, being a “woman”, he was ashamed of not having the appropriate genitals for his sex, which he called “deformities”.
On X-ray of the skull, the shape and size of the sella turcica were normal, but signs of increased intracranial pressure were revealed. On EEG against the background of the general phenomena of irritation, the focus of pathology was recorded in the left parietal lead. Indicators of the functional state of the thyroid gland were in the normal range. In the study of the radioimmunoassay method using standard kits from the Sorin company, some increase in prolactin level of 24 ng/mL was detected in the serum (normal for men is 4–15 ng/mL).
In connection with the repeated suicidal attempts, failure of psychiatric treatment, and in consideration of the fact that the patient has a female civilian sex and performs a female social role, castration and feminizing plastic surgery of the external genitalia were performed for the purpose of social rehabilitation.
Some time after the operation, the patient developed a renewed interest in life. After the surgical and hormonal correction, the patient irresistibly developed maternal instincts. Unmarried, the patient obtained permission for the adoption of a child, simulated pregnancy, and was discharged from the maternity hospital with a son. From the first days after the “birth”, galactorrhea sharply increased, and spontaneous outflow of milk appeared, with galactorrhea (+++). The baby was breastfed up to 6 months of age.
Thus, it can be thought that several factors played a role in the genesis of galactorrhea in this patient:
Increased prolactin levels with estrogen and cyproterone acetate. The hyperprolactic properties of estrogens have long been known; the ability of cyproterone acetate to increase serum prolactin levels was shown by K. Schmidt–Golewizer et al (9).
Increased intracranial pressure, the role of this factor and the genesis of neuroendocrine disorders and, in particular, in the development of galactorrhea was shown by R. Peterson (8).
Our message is the second in the world literature describing galactorrhea in a male patient with transsexualism. The first description of this kind was made in 1983 by R. Flüskiger et al. (6).
This observation demonstrates the independence of the mechanism of lactation development from one’s genetic sex and is alarming with regard to the possibility of drug-induced galactorrhea development in men.
Barber et al. (2004)
Barber, T., Basu, A., Rizvi, K., & Chapman, J. (2004). Normoprolactinaemic galactorrhoea in a male-to-female transsexual. Endocrine Abstracts, 7 [23rd Joint Meeting of the British Endocrine Societies with the European Federation of Endocrine Societies], 271–271. [Google Scholar] [URL]:
Hormonal therapies in the form of oestrogens, anti-androgens and progestogens are often used in the treatment of male-to-female transsexuals. We present the case of a 36 year old phenotypic male with karyotype 46XY who presented with normoprolactinaemic galactorrhoea likely to be related to prior oestrogen administration. He had been self-administering oestrogen and progesterone preparations continuously for 7 years (aged 26 - 33 years) in an attempt to develop female phenotypic characteristics in spite of a heterosexual desire. During this time he developed gynaecomastia with galactorrhoea, increased energy and libido, voice change and an attraction towards both men and women. However due to lack of financial resources to secure a complete gender change, he stopped self-medication with these preparations 3 years ago. Instead he embarked on a regime involving self-administered testosterone in an attempt to reverse the biological changes. After discontinuation of oestrogen the gynaecomastia regressed somewhat, although galactorrhoea continued and worsened with testosterone. Prior to referral he had been treated with dopamine agonists with little improvement in galactorrhoea and gynaecomastia.
Routine biochemistry and haematology are within their reference ranges. Baseline endocrinology is normal: Prolactin 197 milliUnits per litre, LH 2.9 Units per litre, FSH 7.9 Units per litre, free Testosterone 20 nanoMoles per litre, 17 beta-oestradiol less than 110 picoMoles per litre, TSH 0.96 milliUnits per litre and free T4 16.5 picoMoles per litre.
This case illustrates fascinating effects of exogenous oestrogen in the male. The effects of oestrogenic products of aromatised endogenous and briefly also exogenous testosterone acting on oestrogen-primed breast tissue may account for, at least in part, his continuing symptom of normoprolactinaemic galactorrhoea. However two other features do not have any direct explanations: the development of osteopenia during this period, and complete disappearance of vascular migraine, a condition worsened with oestrogens in the female. He is now on Tamoxifen although an opportunity to use the aromatase inhibitor, Anastrozole still remains.
Subsequent Case Reports
Moravek & Pasque (2019)
Moravek, M. B., & Pasque, K. B. (2019). Lactation Can Be Successfully Induced in Transgender Women While Maintaining Gender-Congruent Serum Hormone Levels. Reproductive Sciences, 26(Suppl 1), 136A–136A (abstract no. T-055). [Google Scholar] [DOI:10.1177/1933719119834079]:
Introduction: Transgender women may be interested in breastfeeding their children, but there are no established protocols for lactation induction in this population. The only case report of a lactation induction protocol in a transgender woman significantly lowered her estradiol dose, which would likely result in decreased serum estradiol and increased testosterone levels, with resultant increase in gender dysphoria. Our objective was to induce lactation in a transgender woman without interrupting her gendercongruent hormone profile.
Methods: A 34-year-old transgender woman with a 15-year history of gender-affirming hormone therapy with estradiol and spironolactone presented for lactation induction once her cisgender wife conceived. A modification of the Newman-Goldfarb method for adoptive mothers was used to induce lactation, and serum hormone levels followed.
Results: Baseline labs were obtained (time point 1), then medroxyprogesterone 1.25mg daily was added to her existing hormone regimen of estradiol 6mg daily and spironolactone 100mg twice daily (time point 2). Domperidone 10mg four times daily was initiated 1 month later. Approximately 5 weeks prior to the due date, the patient stopped medroxyprogesterone, decreased estradiol to 2mg daily, and began breast pumping (time point 3). Just prior to the infant’s birth, the patient was pumping 2-3 ounces of breastmilk every 3 hours (time point 4). Spironolactone was decreased to 50mg twice daily. Her son was born at term, via uncomplicated vaginal delivery. The infant was able to breastfeed from both mothers without difficulty, with both mothers pumping when they weren’t actively breastfeeding to maintain supply (time point 5). When the infant was approximately 2 months old, the patient noticed an increase in facial hair growth. Estradiol was increased to 3mg daily and spironolactone increased to 100mg twice daily, with resolution of hair growth and no decrease in milk supply (time point 6). The patient continued to breastfeed on this regimen for >6 months following her son’s birth. Serum hormone levels on the hormone regimens referenced at each time point throughout the patient’s course are displayed in table 1.
Conclusion: Lactation can be successfully induced in transgender women, without a significant decrease in estradiol supplementation. This regimen allows transgender women to breastfeed without developing male secondary sex characteristics incongruent with their gender identity
Table 1 Hormone profile at different time points.
Time Point
Estradiol (pg/mL)
Progesterone (ng/mL)
Testosterone (ng/mL)
Prolactin (ng/mL)
1
114
1.1
0.36
2
130
1.1
0.05
9
3
30
1.3
0.06
152
4
39
5
29
1.4
0.89
184
6
51
0.16
59
Unnithan, Elson, & Shenker (2020)
Unnithan, R., Elson, D. F., & Shenker, Y. (2020). Galactorrhea and Hyperprolactinemia in a Transgender Female. Journal of the Endocrine Society, 4(Suppl 1), A899–A899 (abstract no. SUN-043). [Google Scholar] [PubMed Central] [DOI:10.1210/jendso/bvaa046.1781] [PDF]:
Background: Galactorrhea is a rare manifestation of hyper-prolactinemia in males and post-menopausal females, however the hormonal milieu of the transgender female may increase its incidence
Clinical Case: A 43 year old transgender female presented with three years of bilateral breast discharge. She had chronic, stable headaches and fatigue, but no vision changes or other symptoms. Notably, she had breast augmentation surgery with saline breast implants placed shortly before the galactorrhea commenced. She was on a stable dose of estradiol tablets 1 mg twice daily for six years. On physical exam she had pronounced bilateral breast discharge of a milky quality with nipple compression. Prolactin levels were checked several times and were 40-50 ng/mL, TSH was 2.36 uIU/mL. An MRI showed a left inferior pituitary lesion measuring 6 mm x 3 mm x 5 mm with no mass effect on adjacent structures. Her breast discharge was not bothersome to her, and her pituitary lesion was small. It was unclear whether there was a relationship between her prolactin levels and the lesion seen on MRI, as we expected more pronounced prolactin elevation with a prolactinoma. Instead, given the timing of her symptoms in relation to her breast augmentation surgery, her galactorrhea and hyper-prolactinemia were thought to be the result of nipple irritation related to her breast implants combined with a hyper-estrogenemic state.
Clinical Lessons: In the setting of a prolactin secreting micro-adenoma, galactorrhea in a male is highly unusual. This case highlights the importance of recognizing that the unique medical and surgical characteristics of male to female transgender patients can lead to hyper-prolactinemia and galactorrhea.
Reference: Reisman T, Goldstein Z. Case report: induced lactation in a transgender woman. Transgender Health. 2018;3(1):24-26.
Wamboldt, Shuster, & Sidhu (2021)
Wamboldt, R., Shuster, S., & Sidhu, B. S. (2021). Lactation Induction in a Transgender Woman Wanting to Breastfeed: Case Report. The Journal of Clinical Endocrinology & Metabolism, 106(5), e2047–e2052. [DOI:10.1210/clinem/dgaa976]:
Context: Breastfeeding is known to have many health and wellness benefits to the mother and infant; however, breastfeeding in trans women has been greatly under-researched.
Objective: To review potential methods of lactation induction in trans women wishing to breastfeed and to review the embryological basis for breastfeeding in trans women.
Design: This article summarizes a case of successful lactation in a trans woman, in which milk production was achieved in just over 1 month.
Setting: This patient was followed in an outpatient endocrinology clinic.
Participant: A single trans woman was followed in our endocrinology clinic for a period of 9 months while she took hormone therapy to help with lactation.
Interventions: Readily available lactation induction protocols for nonpuerpural mothers were reviewed and used to guide hormone therapy selection. Daily dose of progesterone was increased from 100 mg to 200 mg daily. The galactogogue domperidone was started at 10 mg 3 times daily and titrated up to effect. She was encouraged to use an electric pump and to increase her frequency of pumping.
Main outcome measure: Lactation induction.
Results: At one month, she had noticed a significant increase in her breast size and fullness. Her milk supply had increased rapidly, and she was producing up to 3 to 5 ounces of milk per day with manual expression alone.
Conclusions: We report the second case in the medical literature to demonstrate successful breastfeeding in a trans woman through use of hormonal augmentation.
Further Case Reports
Delgado, D., Stellwagen, L., McCune, S., Sejane, K., & Bode, L. (2023). Experience of Induced Lactation in a Transgender Woman: Analysis of Human Milk and a Suggested Protocol. Breastfeeding Medicine, 18(11), 888–893. [DOI:10.1089/bfm.2023.0197]
Weimer, A. K. (2023). Lactation induction in a transgender woman: macronutrient analysis and patient perspectives. Journal of Human Lactation, 39(3), 488–494. [DOI:10.1177/08903344231170559]
van Amesfoort, J. E., Van Mello, N. M., & van Genugten, R. (2024). Lactation induction in a transgender woman: case report and recommendations for clinical practice. International Breastfeeding Journal, 19(1), 18. [DOI:10.1186/s13006-024-00624-1]
Trahair, E. D., Kokosa, S., Weinhold, A., Parnell, H., Dotson, A. B., & Kelley, C. E. (2024). Novel Lactation Induction Protocol for a Transgender Woman Wishing to Breastfeed: A Case Report. Breastfeeding Medicine, online ahead of print. [DOI:10.1089/bfm.2024.0012]
Dr. Christine McGinn
Dr. Christine McGinn is a transgender woman and well-known surgeon in Pennsylvania who performs gender-affirming surgeries for transgender people. When she had children with her cisgender female partner, McGinn induced a hormonal pseudopregnancy in herself and her and her partner breastfed their twins together. This was described in the media, including in books and television. McGinn’s case was never formally published as a case report in the scientific literature however.
The Oprah Winfrey Show (2010)
Terry, J. C. (Director), & Winfrey, O. G. (Presenter). (2010 September 29). The Mom Who “Fathered” Her Own Children, Plus the Cast of Modern Family [Television series episode]. The Oprah Winfrey Show (Season 25, Episode 13). Chicago: Harpo Studios. [URL 1] [URL 2] [URL 3]
Trans (2012)
Arnold, C. (Director), Schoen, M. (Producer), RoseWorks (Firm), & Sex Smart Films (Firm). (2012). Trans [DVD] (1:21:32–1:21:55). [WorldCat] [IMDB] [Amazon Prime Video]
Boylan (2014)
Boylan, J. F. (2014). Dr. Christine McGinn. In Boylan, J. F. Stuck in the Middle with You: A Memoir of Parenting in Three Genders (pp. 223–233). New York: Broadway Books. [Google Scholar] [Google Books 1] [Google Books 2] [WorldCat] [PDF]:
Dr. Christine McGinn is a surgeon, a mother of two, a backup flight surgeon for the space shuttle progarm, and a transgender woman. As a man, she saved her sperm before transition; ten years later she used that sperm to have children with her partner Lisa. The two of them are both biological mothers of their son and daughter, and each mother was able to breast-feed the twins. I sat down with Christine at her office in New Hope, Pennsylvania, on a hot summer day in 2011.
CM: […] Then there’s the scientist in me that knows that there is a difference, there is not a binary, but a gender spectrum. There are chemicals that are different in men and women. And when a transgender woman transitions, we are somewhere in the middle. Especialy having gone through a simulated pregnancy, in order to breast-feed, I felt the changes of those hormones. I felt my milk let down when not only my baby would cry, but a baby on TV would cry, and even, ridiculously, when a door would close and make a squeak.
JFB: You had to induce a false pregnancy in order to breast-feed? Tell me how you did that.
CM: As a doctor, I knew it was possible. I followed the protocol that involves simulating pregnancy with hormones. It’s estrogen and progesterone. My simulation pregnancy was over a month before Lisa delivered—with twins, we were expecting them to be born earlier. That entire month I was just pumping nonstop, every two hours. We had a whole freezer full of milk. And you know, the first couple of weeks it was no good, because it had all of the hormones in it. So we only saved, like, the last week or so. But still, it was a freezer full of milk.
Lisa had no idea about the way breast-feeding takes over your life, because this was her first. It was kind of funny that I went through that on my own, first, weeks before she did. And then it took her a couple of days to actually—for her milk to let down.
The children were so small when they were born. They were only five pounds. At first we had to feed them with a syringe. They were breast-feeding as well, but they weren’t latching that great on either of us.
JFB: What was it like when they finally muckled on to you?
CM: Oh, I can’t even put it in words. I really cannot put it in words. It was—I was just—oh.
JFB: Were you amazed? Were you afraid?
CM: It was heaven. I was afraid. I don’t know, it was uncharted territory. Like, I knew the milk was good. Lisa was a little concerned that it would be like skimmed milk, or something, you know. [Laughs] Like—she’s like, “Is it the same stuff?”
JFB: Is it the same milk?
CM: And she was a little dubious about, like, is this really all right? I think that’s totally natural for a mother, to be concerned.
I will just say that there are things snobody thinks about when two women are both breast-feeding. Like, technical stuff that you don’t think about. When you have a mother and a father, the mother decides when the kids get fed. Right? The father doesn’t, really. Right?
But you know, when you have two women who are filled with pregnancy hormones and have that, like, mother-bear attitude about how things should be done… It was really crazy.
JFB: So did that cause serious conflict between you and Lisa?
CM: Totally not serious conflict, because the most important thing are the babies.
Eden finally latched—I breast-fed her more than Luke. Luke was never really good. Lisa hated breast-feeding. Eventually we decided to stop.
I’m putting on my science hat again—when you decide to stop, there are hormonal issues. The strongest emotion a person can feel in their life comes frm oxytocin, which is the love drug.
JFB: Oxytocin?
CM: That’s what’s responsible for babies’ bonding during breastfeeding. So the baby latches on, breast-feeds, your brain just [makes oozing sounds], just like, oozes this gooey love substance, oxytocin. Fathers are proven to have higher oxytocin before the delivery, and just stroking your child’s head. You know, when the baby—when you smell a newborn’s head, it really—that smell, it’s like—
JFB: I just saw a friend’s newborn on Friday, and I was like, [makes sniffing sound]—
CM: My niece said it best. She came in and smell them, and she was five years old at the time, and she’s like, “They smell like cupcakes.” [Laughs] And it’s universal. When you ask me what that’s like, I can’t describe it, you know, and I’m a huge fan of food and cupcakes and chocolate, and so that’s the closest I can come to it—it’s like chocolate. [Laughs]
JFB: So when you stopped breast-feeding, was it a kind of a mourning, a loss?
CM: Yes. Lisa wanted to stop before I did. The problem is, once a baby gets a nipple, a plastic nipple, it gives more milk. And so they don’t have to work as hard.
It’s a unique situation that two breast-feeders in a relationship would experience, but a mother and father would not.
JFB: So did one of you stop breast-feeding before the other?
CM: Yes, Lisa did.
JFB: Lisa stopped. And how much longer did you keep it up?
CM: Not long, because they got the nipple.
They were both so small. We weren’t all that successful at it. We were so worried about their birth weight, and making sure they got enough with the syringes. There were definitely times where, you know, we both would breast-feed and, man, I will never forget that. Like, three ‘clock in the morning, four o’clock in the morning, in the little cocoon, nursing.
The heat of their body, their naked body on your chest. The amazing thing is, it really does kind of hurt when they really get going, you know. And you just… I don’t know how else to describe it. You feel like the life force is just coming out through you. It’s so powerful. It relieves that pain that you have in your breast. It releases that oxytocin, and it’s just—it’s even.
JFB: Did you ever do that thing where you would fall asleep with the children in the bed, and wake up with the children in the bed beside you?
CM: Yeah.
JFB: I loved that. It’s one of my stnogest memories of being a father. Having gotten up in the middle of the night. And they are so small, but such an incredibly powerful feeling, the two of you together surrounding the child. With us, we also had a dog at the bottom of the bed. [Laughs]
CM: And we have two, and that was also very important to me, too. We have miniature pinschers.
JFB: So how many months along did you stop breast-feeding?
CM: Three months. It was really emotionally painful, and I cried a lot. I was really sad.
I was pretty sure we were not going to have any more kids. So I’m like, “This is it.” It was very sad.
JFB: Is there a moment frm the last year and two months where you think, This is what it’s like to be a mother, this is it?
CM: Yes, immediately. It was hot as Hades outside. It was, like, a million degrees. We had just had the kids. It was like, May or June, and my mom was over, and it was, like, we had all this help, initially, because Lisa and I were just not getting any sleep and it was, like, round-the-clock feedings and the kids were small, and Lucas had an apnea monitor that he had to wear all the time, and it was just really hard. And there was a big thunderstorm, and the power went out.
And so, at this point, they weren’t really latching very well, so we both had to pump, and then feed them with the syringes. So Lisa and I are totally, like, engorged with milk. And the power’s out, and the pumps are electric. Right?
JFB: Right.
CM: So there’s no electricity, it’s hot as hell, we’re worried for the kids. Lisa and I are in pain. We’re both leaking. And it was the weirdest, funniest situation. And my mom’s there. She runs out to the store to get batteries, and you know, she’s just beng a mom. She’s getting everything, running around like an angel. And Lisa and I are in pain we’re miserable. When she finally came back, the batteries wouldn’t work on the pumps—something else was wrong. Lisa and I are dying.
And so, here’s the guy part of me… I get the pump that has the backup battery power and the backup car charger. Like, I got all tech on it. [Laughs] I’m out int he car trying to get the car charger to work on the pump in the pouring rain. And it’s ninety-five degrees out. It’s all wet inside, like, the humidity on the windows.
And I’m just trying to get some kind of relief.
And this stupid pump didn’t work that way, either. We come back in and my mom has candles lit.
And then the electricity comes back on. And we all just laugh and pump and breast-feed. And every one of us is in heaven.
Pfeffer (2017)
Pfeffer, C. A. (2017). Trans Partnerships and Families: Historical Traces and Contemporary Representations. In Pfeffer, C. A. Queering Families: The Postmodern Partnerships of Cisgender Women and Transgender Men (pp. 1–34). New York: Oxford University Press. [Google Scholar] [Google Books] [WorldCat] [DOI:10.1093/acprof:oso/9780199908059.003.0001] [Archive.org]:
Just 2 years later, Winfrey would feature another interview that elicited many of the same audience reactions. In this 2010 episode, lesbian partners Dr. Christine McGinn and Lisa Bortz beamed with joy as they held their infant twins. Again, audience members’ jaws dropped when it was revealed that beautiful Christine was a male-to-female transsexual who used to be a handsome military officer Chris, and that Lisa had given birth to the couple’s biological children using sperm Chris banked prior to gender confirmation surgeries.10 And it was Winfrey’s chin that nearly hit the floor as she watched video of Christine breastfeeding the couples’ children (the episode is referred to online as “The Mom Who Fathered Her Own Children”).
Many unpublished reports of lactation and breastfeeding in transfeminine people have been described on the web including at the following pages:
Richards, A. (2003). Lactation and the Transsexual Woman. Second Type Woman. [Updated August 2018] [URL] [PDF]
MacDonald, T. (2013). Trans Women and Breastfeeding: A Personal Interview. Milk Junkies. [URL]
MacDonald, T. (2013). Trans Women and Breastfeeding: The Health Care Provider. Milk Junkies. [URL]
MacDonald, T. (2017). Jenna’s Breastfeeding Journey: Trans Motherhood. Milk Junkies. [URL]
Burns, K. (2018). Yes, Trans Women Can Breastfeed — Here’s How. them. [URL]
Cisgender Men
Induction of lactation has been reported in cisgender men and is noteworthy:
Geschickter (1945)
Geschickter, C. F. (1945). Endocrine Physiology of the Breast. In Geschickter, C. F. Diseases of the Breast: Diagnosis, Pathology, Treatment, 2nd Edition (pp. 42–81). Philadelphia: J.B. Lippincott. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [PDF]:
The results obtained indicate that a lactogenic substance in anterior pituitary extracts may cause mammary secretion in nonpregnant women when they have been previously stimulated with estrogenic hormone but true lactation does not occur. Secretion was also obtained in two adult men with gynecomastia after injections of lactogenic hormone.
Huggins (1949)
Huggins, C. (1949). Endocrine substances in the treatment of cancers. Journal of the American Medical Association, 141(11), 750–754. [DOI:10.1001/jama.1949.02910110002002]:
The administration of estrogen in effective amounts causes testicular atrophy and mammary hypertrophy. Growth of the breasts can be so extensive that lactation may be induced, as illustrated in the following case.
W. N., aged 64, had carcinoma of the prostate with osseous metastases, for which he was treated by a permanent suprapubic cystotomy in 1941. Diethylstilbestrol, 20 mg. daily, was given orally for two years beginning September 1942. In September 1944, 25 mg. (500 international units) of prolactin14 was injected daily for five days, and at the end of this time creamy milk could be expressed from both breasts. Orchiectomy and removal of the cystostomy tube were carried out September 6, when administration of estrogen was discontinued; both incisions healed promptly. Since then the patient has been clinically well but has continued to lactate, a large drop of milk being easily expressed from each breast at frequent intervals.
Huggins & Dao (1954)
Huggins, C., & Dao, T. L. (1954). Lactation induced by luteotrophin in women with mammary cancer. Growth of the breast of the human male following estrogenic treatment. Cancer Research, 14(4), 303–306. [Google Scholar] [PubMed] [URL]:
In the observations to be presented luteotrophin [prolactin] was employed as a stimulus for mammary secretion in patients with cancer of the breast, and the results throw new light on the physiology of women bearing this neoplasm. We shall also describe conditions which resulted in the induction of physiologic maturity in the human male, since knowledge of the action of hormones on the human breast is vague.
The effects of luteotrophin on the breast of women post partum has been extensively investigated, but otherwise few observations have been made in the human. Werner (14) administered a crude pituitary extract containing luteotrophin to eight castrate women 21–35 years of age; lactation was not observed, although in one woman “a few drops of colostrum-like fluid” could be expressed from the breasts. Goldzieher (4) treated menstrual disorders in women with luteotrophin, but mammary secretion was not described by him.
PROCEDURE
Luteotrophin,1 dissolved in physiological saline made slight ly alkaline (pH 9) with sodium hydroxide, was injected subcutaneously in daily amounts of 500 International Units; the solutions were freshly prepared, and the injections were continued for 7 days only.
This series comprised 21 female patients who had dis seminated mammary cancer, and all had been subjected to unilateral mastectomy. There were also three men with advanced prostatic carcinoma who had been treated for thera peutic purposes with oral diethylstilbestrol for 20 months, 2, and 6 years, respectively. There were eight persons without mammary or prostatic cancers who served as controls.
In each case of mammary cancer a biopsy of the breast was obtained for histological purposes, the material being stained with Sudan III.3
OBSERVATIONS
Lactation, when it occurred, was never profuse; it varied from a tiny drop to ca. 0.5 cc. from each breast. Clear colostrum was not observed, and the mammary secretion was always milk, as defined above.
Mammary growth in the human male.—Estrogenic substances had been administered to three men in the treatment of disseminated prostate cancer for many months; after luteotrophin injection two lactated and one did not lactate.
W. N. (reported in brief earlier [5]), age 64, had taken diethylstilbestrol, 20 mg/day, orally for 2 years, after which interval sub-areolar button-like masses of mammary tissue could be palpated bilaterally; luteotrophin was then injected for 5 days, and milk was expressed from the breast on the 6th day. Orchiectomy was then performed, and both luteotrophin and estrogenic substances were discontinued. This man continued to lactate for 7 years when the formation of milk gradually ceased.
In the case of A. W., age 62, diethylstilbestrol (5–15 mg/day) had been ingested for 20 months after bilateral orchiectomy; the breasts became slightly enlarged. Luteotrophin was injected, and lactation occurred on the 7th day. A biopsy of the breast showed moderately well developed mammary ducts and alveoli containing milk. In the case of E. G., age 59, diethylstilbestrol (5 mg/day) was ingested almost continuously for 6 years; this resulted in the development of large pendulous breasts, but no lactation occurred after injections of luteotrophin.
Lactation in humans without cancer.—Luteotrophin was administered to two normal males, age 51 and 59, and to four normal females, age 84–59, and none lactated.
DISCUSSION
It must be emphasized that lactation was not copious in any of the humans when it had been induced by luteotrophin; merely small amounts of milk were obtained. It was apparent, however, from the histological studies of the mammary tissue obtained by biopsy that the secretion of milk in any quantity was a criterion of maturity of mammary epithelium.
In the goat and guinea pig it is known that estrogenic substances can induce mammary ma turity without the intervention of exogenous synergistic steroids. In the experiments of Lewis and Turner (9) diethylstilbestrol was implanted in two castrate male goats; one of these animals failed to lactate, while the other produced a small quantity of milk without luteotrophin injections. They obtained small amounts of milk from a male kid similarly treated. Nelson (10) found that estrone induced mammary growth with, later, lactation in the male guinea pig. Our observations demonstrate that diethylstilbestrol ingested for prolonged periods of time can induce maturity of the breast in certain elderly human males. However, the human male differs from the animals just described in that spontaneous lactation was not observed; the injection of luteotrophin was necessary for milk formation.
The duration of lactation induced by luteo trophin was impressive, since milk commonly persisted for many months—and in one male for 7 years. The mechanism whereby this type of lactation is maintained for such long periods of time is at present unknown; we know that milk continues to be secreted both in the presence of the adrenal glands and in the absence of these structures and the gonads as well. Observations (8) have been made on experimental animals which are analogous to the clinical findings; most dogs with spontaneous mammary cancer possess lactation, and this characteristic persists for many months, at least, despite the removal of the adrenal glands and the ovaries.
SUMMARY
The breast of the human male can be induced to grow to a functionally mature state by the administration of estrogenic substances without additional exogenous steroid synergists. Spontaneous lactation was not observed in these men, but it was induced by luteotrophin.
The formation of milk in any amount by the breast is a criterion of functional maturity of the mammary epithelium. Luteotrophin induced the secretion of small amounts of milk in a group of women with mammary cancer and in a number of healthy women as well, and, in addition, in two human males to whom estrogenic substances had been administered for therapeutic purposes. Lactation did not occur in two normal males.
When lactation was induced in human beings, the secretion often persisted for many months; it lasted for 7 years in one man.
HUGGINS,C. Endocrine Substances in the Treatment of Cancers. J.A.M.A., 141:750–54, 1949.
Brodribb, W., & Academy of Breastfeeding Medicine. (2018). ABM Clinical Protocol #9: Use of galactogogues in initiating or augmenting maternal milk production, second revision 2018. Breastfeeding Medicine, 13(5), 307–314. [DOI:10.1089/bfm.2018.29092.wjb]
MacDonald, T. K. (2019). Lactation care for transgender and non-binary patients: Empowering clients and avoiding aversives. Journal of Human Lactation, 35(2), 223–226. [DOI:10.1177/0890334419830989]
Paynter, M. J. (2019). Medication and Facilitation of Transgender Women’s Lactation. Journal of Human Lactation, 35(2), 239–243. [DOI:10.1177/0890334419829729]
Cazorla-Ortiz, G., Obregón-Guitérrez, N., Rozas-Garcia, M. R., & Goberna-Tricas, J. (2020). Methods and Success Factors of Induced Lactation: A Scoping Review. Journal of Human Lactation, 36(4), 739–749. [DOI:10.1177/0890334420950321]
Ferri, R. L., Rosen-Carole, C. B., Jackson, J., Carreno-Rijo, E., Greenberg, K. B., & Academy of Breastfeeding Medicine. (2020). ABM Clinical Protocol #33: Lactation Care for Lesbian, Gay, Bisexual, Transgender, Queer, Questioning, Plus Patients. Breastfeeding Medicine, 15(5), 284–293. [DOI:10.1089/bfm.2020.29152.rlf]
García-Acosta, J. M., Juan-Valdivia, S., María, R., Fernández-Martínez, A. D., Lorenzo-Rocha, N. D., & Castro-Peraza, M. E. (2020). Trans* Pregnancy and Lactation: A Literature Review from a Nursing Perspective. International Journal of Environmental Research and Public Health, 17(1), 44. [DOI:10.3390/ijerph17010044]
LeCain, M., Fraterrigo, G., & Drake, W. M. (2020). Induced Lactation in a Mother Through Surrogacy With Complete Androgen Insensitivity Syndrome (CAIS). Journal of Human Lactation, 36(4), 791–794. [DOI:10.1177/0890334419888752]
Trautner, E., McCool-Myers, M., & Joyner, A. B. (2020). Knowledge and practice of induction of lactation in trans women among professionals working in trans health. International Breastfeeding Journal, 15(1), 63. [DOI:10.1186/s13006-020-00308-6]
References (Inline Citations)
Bazarra-Castro, M. A. (2009). Etiological aspects, therapy regimes, side effects and treatment satisfaction of transsexual patients. (Doctoral dissertation, Ludwig Maximilian University of Munich.) [DOI:10.5282/edoc.9984] [URN:urn:nbn:de:bvb:19-99840] [PDF]
Delgado, D., Stellwagen, L., McCune, S., Sejane, K., & Bode, L. (2023). Experience of Induced Lactation in a Transgender Woman: Analysis of Human Milk and a Suggested Protocol. Breastfeeding Medicine, 18(11), 888–893. [DOI:10.1089/bfm.2023.0197]
Dewhurst, J., & Underhill, R. (1979). The Suppression of Facial Hair Growth in Transsexuals Using Cyproterone Acetate. British Journal of Sexual Medicine, 6, 19–23. [Google Scholar] [PDF]
Futterweit, W. (1980). Endocrine management of transsexual. Hormonal profiles of serum prolactin, testosterone, and estradiol. New York State Journal of Medicine, 80(8), 1260–1264. [Google Scholar] [PubMed] [Archive.org] [PDF]
Gooren, L. J., Harmsen-Louman, W., & Kessel, H. (1985). Follow-up of prolactin levels in long-term oestrogen-treated male-to-female transsexuals with regard to prolactinoma induction. Clinical Endocrinology, 22(2), 201–207. [DOI:10.1111/j.1365-2265.1985.tb01081.x]
Greenblatt, R. B. (1972). Inappropriate lactation in men and women. Medical Aspects of Human Sexuality, 6(6), 25–33. [Google Scholar] [Google Books]
Greenblatt, R. B., & Leng, J. J. (1972). Lactation anormale chez l’homme. [Abnormal lactation in males.] Bordeaux Medical, 5(3), 241–243. [Google Scholar] [PubMed]
Kanhai, R. C., Hage, J. J., van Diest, P. J., Bloemena, E., & Mulder, J. W. (2000). Short-Term and Long-Term Histologic Effects of Castration and Estrogen Treatment on Breast Tissue of 14 Male-to-Female Transsexuals in Comparison With Two Chemically Castrated Men. The American Journal of Surgical Pathology, 24(1), 74–80. [DOI:10.1097/00000478-200001000-00009]
Levy, A., Crown, A., & Reid, R. (2003). Endocrine intervention for transsexuals. Clinical Endocrinology, 59(4), 409–418. [DOI:10.1046/j.1365-2265.2003.01821.x]
Neumann, F. V., von Berswordt-Wallrabe, R., Elger, W., Steinbeck, H., Hahn, J. D., & Kramer, M. (1970). Aspects of Androgen-Dependent Events as Studied by Antiandrogens. In Astwood, E. B. (Ed.). Proceedings of the 1969 Laurentian Hormone Conference (Recent Progress in Hormone Research, Volume 26) (pp. 337–410). New York/London: Academic Press. [DOI:10.1016/B978-0-12-571126-5.50013-3]
Schlatterer, K., Yassouridis, A., Werder, K. V., Poland, D., Kemper, J., & Stalla, G. K. (1998). Archives of Sexual Behavior, 27(5), 475–492. [DOI:10.1023/a:1018704630036]
Trahair, E. D., Kokosa, S., Weinhold, A., Parnell, H., Dotson, A. B., & Kelley, C. E. (2024). Novel Lactation Induction Protocol for a Transgender Woman Wishing to Breastfeed: A Case Report. Breastfeeding Medicine, online ahead of print. [DOI:10.1089/bfm.2024.0012]
van Amesfoort, J. E., Van Mello, N. M., & van Genugten, R. (2024). Lactation induction in a transgender woman: case report and recommendations for clinical practice. International Breastfeeding Journal, 19(1), 18. [DOI:10.1186/s13006-024-00624-1]
Weimer, A. K. (2023). Lactation induction in a transgender woman: macronutrient analysis and patient perspectives. Journal of Human Lactation, 39(3), 488–494. [DOI:10.1177/08903344231170559]
\ No newline at end of file
diff --git a/transfemscience.org/assets/css/hover-refs.css b/transfemscience.org/assets/css/hover-refs.css
new file mode 100644
index 00000000..9e1d1d72
--- /dev/null
+++ b/transfemscience.org/assets/css/hover-refs.css
@@ -0,0 +1,45 @@
+/* Hover References Popup Styling */
+
+.reference-hover-box {
+ margin-top: 5px;
+ position: absolute;
+ z-index: 9999;
+ background-color: var(--background-color, #ffffff);
+ color: var(--font-color, #333333);
+ border: 1px solid var(--code-border-color, #ccc);
+ box-shadow: 0 4px 12px rgba(0, 0, 0, 0.15), 0 8px 24px rgba(0, 0, 0, 0.1);
+ /* Deeper shadow */
+ padding: 8px;
+ border-radius: 4px;
+ max-width: 400px;
+ font-size: 0.9em;
+ line-height: 1.4;
+ pointer-events: auto;
+ /* Allow interaction with the box */
+
+ /* Ensure it handles overflow or long text gracefully */
+ word-wrap: break-word;
+}
+
+/* Dark mode support if variables are set */
+html[data-theme='dark'] .reference-hover-box {
+ background-color: var(--background-color, #222);
+ /* Fallback to dark grey */
+ border-color: var(--code-border-color, #444);
+ box-shadow: 0 4px 12px rgba(0, 0, 0, 0.4), 0 8px 24px rgba(0, 0, 0, 0.3);
+}
+
+/* Match link styles to article content links */
+.reference-hover-box a {
+ color: var(--link-color);
+ text-decoration: none;
+ background-image: linear-gradient(var(--link-underline-color) 0%, var(--link-underline-color) 100%);
+ background-repeat: repeat-x;
+ background-position: 0 100%;
+ background-size: 1px 1px;
+ word-break: break-all;
+}
+
+.reference-hover-box a:hover {
+ background-image: linear-gradient(var(--link-underline-color-hover) 0%, var(--link-underline-color-hover) 100%);
+}
\ No newline at end of file
diff --git a/transfemscience.org/assets/js/hover-refs.js b/transfemscience.org/assets/js/hover-refs.js
new file mode 100644
index 00000000..51622943
--- /dev/null
+++ b/transfemscience.org/assets/js/hover-refs.js
@@ -0,0 +1,373 @@
+/**
+ * Hover References Feature
+ *
+ * Matches inline citation links with their corresponding full reference
+ * from the reference list by comparing URLs (hrefs).
+ */
+
+(function () {
+ document.addEventListener('DOMContentLoaded', function () {
+ initHoverReferences();
+ });
+
+ function initHoverReferences() {
+ // 1. Find the References section and list
+ const referencesHeader = findReferencesHeader();
+ if (!referencesHeader) return;
+
+ // Helper to normalize URLs for matching
+ // 1. Decode URI (handles %5B vs [ mismatch)
+ // 2. Lowercase
+ const normalizeRefUrl = (url) => {
+ let u = "";
+ try {
+ u = decodeURI(url).toLowerCase();
+ } catch (e) {
+ u = url.toLowerCase();
+ }
+ u = u.replace(/^(https?:)?\/\//, '').replace(/^www\./, '');
+
+ // Check for archive.org URL (e.g. web.archive.org/web/20070430161048/http://... or .../20210717084442if_/...)
+ const archiveRegex = /^web\.archive\.org\/web\/\d+(?:if_)?\//;
+ if (archiveRegex.test(u)) {
+ u = u.replace(archiveRegex, '');
+ // Re-strip protocol and www from the target URL
+ u = u.replace(/^(https?:)?\/\//, '').replace(/^www\./, '');
+ }
+
+ return u.split('#')[0].replace(/\/$/, '');
+ };
+
+ // Find all reference lists within the References section
+ // The References section might have subsections, each with their own list
+ // We need to find the container element that encompasses all References content
+
+ // Strategy: Find the parent section/container, or collect all siblings until next same-level header
+ let referenceLists = [];
+ let currentElement = referencesHeader.nextElementSibling;
+ const headerLevel = parseInt(referencesHeader.tagName.charAt(1)) || 2;
+
+ while (currentElement) {
+ // Stop if we hit another header of same or higher level (lower number)
+ if (/^H[1-6]$/.test(currentElement.tagName)) {
+ const currentLevel = parseInt(currentElement.tagName.charAt(1));
+ if (currentLevel <= headerLevel) {
+ break; // End of References section
+ }
+ }
+
+ // Collect all UL and OL elements (including nested ones in subsections)
+ if (currentElement.tagName === 'UL' || currentElement.tagName === 'OL') {
+ referenceLists.push(currentElement);
+ }
+ // Also check for lists inside divs or other container elements
+ const nestedLists = currentElement.querySelectorAll('ul, ol');
+ nestedLists.forEach(list => referenceLists.push(list));
+
+ currentElement = currentElement.nextElementSibling;
+ }
+
+ if (referenceLists.length === 0) return;
+
+ // 2. Parse all reference list items from all lists
+ // Map of "URL" -> HTML content
+ const urlMap = new Map();
+
+ referenceLists.forEach(referenceList => {
+ const listItems = referenceList.querySelectorAll(':scope > li'); // Direct children only to avoid duplicates
+
+ listItems.forEach(item => {
+ // Find all links in the reference item
+ const links = item.querySelectorAll('a');
+ links.forEach(link => {
+ const rawHref = link.getAttribute('href');
+ if (rawHref && !rawHref.startsWith('#')) { // Ignore anchor links if any
+ const href = normalizeRefUrl(rawHref); // Normalized for storage
+ // Store the item HTML for this URL
+ // If multiple refs share a URL (unlikely but possible), the last one wins,
+ // or we could store an array. For citations, usually unique DOI/URL per ref.
+ urlMap.set(href, item.innerHTML);
+ }
+ });
+ });
+ });
+
+ // 3. Find inline citation links
+ // We look for links inside the article body
+ const articleBody = document.getElementById('article');
+ if (!articleBody) return;
+
+ const links = articleBody.querySelectorAll('a');
+
+ // Create the hover box element
+ const hoverBox = document.createElement('div');
+ hoverBox.classList.add('reference-hover-box');
+ hoverBox.style.display = 'none';
+ document.body.appendChild(hoverBox);
+
+ let hideTimeout;
+
+ // Keep box open when hovering over it
+ hoverBox.addEventListener('mouseenter', () => {
+ if (hideTimeout) clearTimeout(hideTimeout);
+ });
+
+ hoverBox.addEventListener('mouseleave', () => {
+ hideTimeout = setTimeout(() => {
+ hoverBox.style.display = 'none';
+ }, 100);
+ });
+
+ links.forEach(link => {
+ const rawHref = link.getAttribute('href');
+
+ // Skip links inside any of the reference lists
+ const isInReferenceList = referenceLists.some(list => list.contains(link));
+ if (isInReferenceList) return;
+
+ // We only care if the link HAS an href and it's in our map
+ if (rawHref && !rawHref.startsWith('#')) {
+ const exactHref = normalizeRefUrl(rawHref);
+ const baseHref = exactHref.split('#')[0];
+
+ let bestMatchHTML = urlMap.get(exactHref);
+ if (!bestMatchHTML) {
+ bestMatchHTML = urlMap.get(baseHref);
+ }
+
+ // Fallback for unmatched links (e.g. Wiki links, or refs without date/year)
+ // User requested to show just the URL, but ONLY if it looks like a ref (in parentheses)
+ if (!bestMatchHTML) {
+ // Check if the link itself contains parentheses with a year (e.g. "(2005)" or "(2005a)" or "(Aly, 2020)")
+ // We allow other text inside the parens, but it MUST contain a year-like number.
+ const yearParensRegex = /\([^)]*\b\d{4}[a-z]?\b[^)]*\)/i;
+ const textHasParens = link.textContent.includes('(') || link.textContent.includes(')');
+ const textMatchingParensYear = yearParensRegex.test(link.textContent);
+
+ // Check if enclosed in parentheses or brackets by walking siblings
+ let isEnclosed = false;
+ // We only scan if:
+ // 1. The text itself doesn't contain matching parens (if it does, we already know if it's valid or not)
+ // OR
+ // 2. The text doesn't contain parens at all (so we look for surrounding ones)
+
+ if (!textMatchingParensYear && !textHasParens) {
+ // If text has parens but didn't match yearParensRegex, it's invalid (e.g. "Kuhl (Citation)")
+ // So we only scan if text does NOT have parens.
+
+ let openParenCount = 0;
+ let openBracketCount = 0;
+ let foundOpen = false;
+
+ let curr = link.previousSibling;
+ let scans = 0;
+ const MAX_SCANS = 100; // Reasonable lookbehind limit
+
+ while (curr && scans < MAX_SCANS) {
+ if (curr.nodeType === 3) { // Text node
+ const txt = curr.textContent;
+ // Count parens from right to left
+ for (let i = txt.length - 1; i >= 0; i--) {
+ const c = txt[i];
+ if (c === ')') openParenCount--;
+ else if (c === '(') openParenCount++;
+ else if (c === ']') openBracketCount--;
+ else if (c === '[') openBracketCount++;
+
+ if (openParenCount > 0 || openBracketCount > 0) {
+ foundOpen = true;
+ break;
+ }
+ }
+ } else if (curr.nodeType === 1) { // Element node
+ const tagName = curr.tagName;
+ // Stop at block boundaries
+ if (/^(DIV|P|BODY|MAIN|SECTION|BLOCKQUOTE|UL|OL|LI|TABLE|BR|HR|H[1-6])$/.test(tagName)) {
+ break;
+ }
+ // Check text content of inline elements
+ const txt = curr.textContent;
+ for (let i = txt.length - 1; i >= 0; i--) {
+ const c = txt[i];
+ if (c === ')') openParenCount--;
+ else if (c === '(') openParenCount++;
+ else if (c === ']') openBracketCount--;
+ else if (c === '[') openBracketCount++;
+
+ if (openParenCount > 0 || openBracketCount > 0) {
+ foundOpen = true;
+ break;
+ }
+ }
+ }
+
+ if (foundOpen) break;
+ curr = curr.previousSibling;
+ scans++;
+ }
+
+ if (foundOpen) {
+ isEnclosed = true;
+ }
+ }
+
+ if (textMatchingParensYear || isEnclosed) {
+ let displayUrl = rawHref;
+ if (rawHref.startsWith('/')) {
+ displayUrl = 'https://transfemscience.org' + rawHref;
+ }
+ bestMatchHTML = `
`;
+ }
+ }
+
+ if (bestMatchHTML) {
+
+ link.classList.add('reference-link');
+
+ link.addEventListener('mouseenter', (e) => {
+ // Clear any pending hide timeout
+ if (hideTimeout) clearTimeout(hideTimeout);
+
+ hoverBox.innerHTML = bestMatchHTML;
+ hoverBox.style.display = 'block';
+
+ // Use getClientRects to handle multi-line links; find the rect under the mouse
+ const rects = link.getClientRects();
+ let rect = rects.length > 0 ? rects[0] : link.getBoundingClientRect();
+
+ // Find the rect that contains the mouse Y position
+ if (rects.length > 1) {
+ let bestRect = rects[0];
+ let minDistance = Infinity;
+
+ for (let i = 0; i < rects.length; i++) {
+ const r = rects[i];
+ // Check if mouse Y is within this rect's vertical bounds
+ if (e.clientY >= r.top && e.clientY <= r.bottom) {
+ bestRect = r;
+ break; // Found exact line match
+ }
+
+ // Fallback: distance to vertical center
+ const centerY = r.top + (r.height / 2);
+ const dist = Math.abs(e.clientY - centerY);
+ if (dist < minDistance) {
+ minDistance = dist;
+ bestRect = r;
+ }
+ }
+ rect = bestRect;
+ }
+
+ // Positioning
+ let top = rect.bottom + window.scrollY; // 0px gap
+ let left = rect.left + window.scrollX;
+
+ // Boundary checks
+ if (left + hoverBox.offsetWidth > window.innerWidth) {
+ left = window.innerWidth - hoverBox.offsetWidth - 10;
+ }
+
+ hoverBox.style.top = `${top}px`;
+ hoverBox.style.left = `${left}px`;
+ });
+
+ link.addEventListener('mouseleave', () => {
+ // Set a timeout to hide the box, giving time to move into it
+ hideTimeout = setTimeout(() => {
+ hoverBox.style.display = 'none';
+ }, 100);
+
+ // Mobile Long Press Support (Touch) AND Desktop Click-and-Hold
+ let longPressTimer;
+ let isLongPress = false;
+
+ const startPress = (e) => {
+ // Only left click for mouse (button 0)
+ if (e.type === 'mousedown' && e.button !== 0) return;
+
+ isLongPress = false;
+ longPressTimer = setTimeout(() => {
+ isLongPress = true;
+
+ // Show hover box
+ if (hideTimeout) clearTimeout(hideTimeout);
+ hoverBox.innerHTML = bestMatchHTML;
+ hoverBox.style.display = 'block';
+
+ hoverBox.style.display = 'block';
+
+ const rects = link.getClientRects();
+ const rect = rects.length > 0 ? rects[0] : link.getBoundingClientRect();
+
+ let top = rect.bottom + window.scrollY;
+ let left = rect.left + window.scrollX;
+
+ if (left + hoverBox.offsetWidth > window.innerWidth) {
+ left = window.innerWidth - hoverBox.offsetWidth - 10;
+ }
+
+ hoverBox.style.top = `${top}px`;
+ hoverBox.style.left = `${left}px`;
+
+ }, 500); // 500ms for long press
+ };
+
+ const cancelPress = () => {
+ clearTimeout(longPressTimer);
+ };
+
+ const endPress = (e) => {
+ clearTimeout(longPressTimer);
+ if (isLongPress) {
+ e.preventDefault(); // Prevent default action (click/navigate)
+ // Note for desktop: 'click' event might still fire after mouseup if we don't prevent it there too
+ }
+ };
+
+ // Touch Listeners
+ link.addEventListener('touchstart', startPress, { passive: true });
+ link.addEventListener('touchend', endPress);
+ link.addEventListener('touchmove', cancelPress);
+
+ // Mouse Listeners (Desktop)
+ link.addEventListener('mousedown', startPress);
+ link.addEventListener('mouseup', endPress);
+ link.addEventListener('mouseleave', cancelPress);
+
+ link.addEventListener('click', (e) => {
+ if (isLongPress) {
+ e.preventDefault();
+ e.stopPropagation();
+ isLongPress = false; // Reset
+ }
+ });
+
+ link.addEventListener('contextmenu', (e) => {
+ if (isLongPress) {
+ e.preventDefault(); // Prevent default context menu
+ isLongPress = false; // Reset
+ }
+ });
+ });
+ }
+ }
+ });
+ }
+
+ function findReferencesHeader() {
+ // Try by ID first
+ let header = document.getElementById('references');
+ if (header) return header;
+
+ // Try by text content
+ const headers = document.querySelectorAll('h1, h2, h3, h4, h5, h6');
+ for (const h of headers) {
+ if (h.textContent.trim().toLowerCase() === 'references') {
+ return h;
+ }
+ }
+ return null;
+ }
+
+})();
diff --git a/transfemscience.org/feed-posts.xml b/transfemscience.org/feed-posts.xml
index 04ea9f25..b2b95e82 100644
--- a/transfemscience.org/feed-posts.xml
+++ b/transfemscience.org/feed-posts.xml
@@ -1 +1 @@
-Jekyll2025-12-20T18:17:02-08:00https://transfemscience.org/feed-posts.xmlTransfeminine ScienceTransfeminine Science is a site for information on hormone therapy for transfeminine people.Transfeminine Science
\ No newline at end of file
+Jekyll2026-02-06T16:23:17-08:00https://transfemscience.org/feed-posts.xmlTransfeminine ScienceTransfeminine Science is a site for information on hormone therapy for transfeminine people.Transfeminine Science
\ No newline at end of file
diff --git a/transfemscience.org/feed.xml b/transfemscience.org/feed.xml
index 488abd86..f7bc6f46 100644
--- a/transfemscience.org/feed.xml
+++ b/transfemscience.org/feed.xml
@@ -1,4 +1,4 @@
-Jekyll2025-12-20T18:17:02-08:00https://transfemscience.org/feed.xmlTransfeminine Science | ArticlesTransfeminine Science is a site for information on hormone therapy for transfeminine people.Transfeminine ScienceA Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People2025-09-08T18:00:00-07:002025-09-09T00:00:00-07:00https://transfemscience.org/articles/hair-lossA Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People
+Jekyll2026-02-06T16:23:17-08:00https://transfemscience.org/feed.xmlTransfeminine Science | ArticlesTransfeminine Science is a site for information on hormone therapy for transfeminine people.Transfeminine ScienceA Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People2025-09-08T18:00:00-07:002025-09-09T00:00:00-07:00https://transfemscience.org/articles/hair-lossA Review of Pharmaceutical Interventions for Scalp Hair Loss and Implications for Transfeminine People
@@ -38,7 +38,7 @@
In the United States, finasteride prescriptions have increased exponentially in recent years, largely driven by its use in the treatment of hair loss (AHLA, 2024). Dutasteride prescriptions are far fewer, estimated in the hundreds of thousands, but also growing due to increased use for AGA. This difference is largely due to finasteride being widely licensed for this indication throughout the world, whilst dutasteride remains off-label in most countries (Altendorf et al., 2025). However, dutasteride is licensed for use in the treatment of AGA in South Korea, Japan and Mexico. Increasing interest is driving further adoption and research into its use.
-
Despite its use for AGA being mostly off-label, dutasteride is now widely regarded as a more efficacious and hence superior 5-ARI than finasteride. Numerous studies have established that dutasteride results in greater suppression of serum DHT concentrations (i.e., about 70% with finasteride vs 90–95% with dutasteride) (Clark et al., 2004; Olsen et al., 2006; Amory et al., 2007; Upreti et al., 2015). Another study directly comparing scalp tissue concentrations found that dutasteride reduced DHT substantially more than finasteride (mean reduction of about 65% with finasteride versus 90% with dutasteride, though with wide interindividual variation). (Hobo et al., 2023). These differences have been primarily attributed to its broader inhibition of the 5α-reductase enzyme. More specifically, finasteride is a selective inhibitor of type II and III 5α-reductase, whereas dutasteride indiscriminately acts on all three isoforms (Gisleskog et al., 1998; Keam & Scott, 2008; Yamana, Labrie, & Luu-The, 2010). Dutasteride has also been theorised to accumulate inside certain tissues, further enhancing its therapeutic effect.
+
Despite its use for AGA being mostly off-label, dutasteride is now widely regarded as a more efficacious and hence superior 5-ARI than finasteride. Numerous studies have established that dutasteride results in greater suppression of serum DHT concentrations (i.e., about 70% with finasteride vs 90–95% with dutasteride) (Clark et al., 2004; Olsen et al., 2006; Amory et al., 2007; Upreti et al., 2015). Another study directly comparing scalp tissue concentrations found that dutasteride reduced DHT substantially more than finasteride (mean reduction of about 65% with finasteride versus 90% with dutasteride, though with wide interindividual variation) (Hobo et al., 2023). These differences have been primarily attributed to its broader inhibition of the 5α-reductase enzyme. More specifically, finasteride is a selective inhibitor of type II and III 5α-reductase, whereas dutasteride indiscriminately acts on all three isoforms (Gisleskog et al., 1998; Keam & Scott, 2008; Yamana, Labrie, & Luu-The, 2010). Dutasteride has also been theorised to accumulate inside certain tissues, further enhancing its therapeutic effect.
In accordance with the above, two large network meta-analysis studies found that oral dutasteride is superior to oral and topical finasteride in the treatment of male AGA in terms of both total hair density and terminal hair density (Gupta et al., 2024a; Gupta et al., 2025a). These studies also found the effects of finasteride and dutasteride to be dose-dependent. On average, there was no difference between treatment groups using oral and topical finasteride. A systematic review found that dutasteride was superior to finasteride in some studies in terms of hair thickness (Almudimeegh et al., 2024). However, in contrast to the above findings in the case of male AGA, a network meta-analysis of studies investigating different interventions for female AGA found that clinical trials assessing the effectiveness of dutasteride do not yet exist (Gupta et al., 2024b). Notably, oral finasteride given at a dose of 1 mg/day was not found to be effective in treating female AGA, yet oral finasteride used at a dose of 5 mg/day outperformed all other single-agent interventions. Because of the dose-dependent effects of 5-ARIs, it could well be that dutasteride might be more efficacious than finasteride in the treatment of female AGA, as in male AGA. Hopefully, future clinical trials will shed light on this.
@@ -92,7 +92,7 @@
A retrospective study of users of oral minoxidil investigated the frequency of adverse effects in both men and women receiving a median dose of 1.63 mg/day (Vañó-Galván et al., 2021). The following were found to occur: hypertrichosis (excessive facial/body hair) in 15.1%, lightheadedness in 1.7%, fluid retention in 1.3%, tachycardia in 0.9%, headache in 0.4%, periorbital edema (temporary swelling around the eyes) in 0.3%, and insomnia in 0.2%. The total frequency of adverse effects was 20.4%, which prompted discontinuation in 1.2% of users, overall. Another study reported an overall hypertrichosis incidence of 24%, with the highest rates being found in the sideburns (81%), temples (73%), arms (63%), and upper lip (51%) (Jimenez-Cauhe et al., 2021). By contrast, topical minoxidil is associated with much lower overall rates of hypertrichosis. Most studies have reported incidence rates of between 0 and 5% (Lucky et al., 2004; Blume-Peytavi et al., 2016; Ramos et al., 2020; Penha et al., 2024; Yang et al., 2024). These findings are consistent with a meta-analysis that reported point estimates of incidence rates for hypertrichosis of 10%, 15%, and 33% for oral minoxidil at 0.25 mg/day, 0.5 mg/day, and 1.25 mg/day, respectively, and 0% and 2% for topical minoxidil at a 2% and 5% concentration, respectively (Wiechert et al., 2025). Despite this, the discontinuation rate across all studies was 0.49%. There also seemed to be no statistically significant difference between the rate of discontinuation for oral and topical formulations, suggesting that hypertrichosis appears to be very well tolerated.
-
A concern associated with the use of oral minoxidil is its potential impact on cardiovascular health (Ibraheim et al., 2023). Since tachycardia can increase myocardial workload and lead to symptoms such as palpitations or chest discomfort, oral minoxidil should be approached cautiously, especially by individuals with underlying cardiovascular issues. Fortunately, the overall risk of severe cardiovascular complications from low-dose oral minoxidil seems to be very low in the general population (Randolph & Tosti, 2021; Vañó-Galván et al., 2021). Meanwhile, skin reactions appear to be relatively common in users of topical minoxidil. This often manifests as scalp eczema and itching, although rates of incidence vary by study (Lucky et al., 2004; Rossi et al., 2012; Penha et al., 2024). The culprit behind this irritating effect appears not to be minoxidil itself, but rather the ingredients in certain formulations such as propylene glycol (Suchonwanit, Thammarucha, & Leerunyakul, 2019). These solvents help deliver minoxidil into the scalp, but are known to cause skin irritation in susceptible individuals. It also appears that, for most people, long-term topical minoxidil therapy may be precluded by non-compliance (Ali Mapar & Omidian, 2007; Shadi, 2023).
+
A concern associated with the use of oral minoxidil is its potential impact on cardiovascular health (Ibraheim et al., 2023). Since tachycardia can increase myocardial workload and lead to symptoms such as palpitations or chest discomfort, oral minoxidil should be approached cautiously, especially by individuals with underlying cardiovascular issues. Fortunately, the overall risk of severe cardiovascular complications from low-dose oral minoxidil seems to be very low in the general population (Randolph & Tosti, 2021; Vañó-Galván et al., 2021). Meanwhile, skin reactions appear to be relatively common in users of topical minoxidil. This often manifests as scalp eczema and itching, although rates of incidence vary by study (Lucky et al., 2004; Rossi et al., 2012; Penha et al., 2024). The culprit behind this irritating effect appears not to be minoxidil itself, but rather the ingredients in certain formulations such as propylene glycol (Suchonwanit, Thammarucha, & Leerunyakul, 2019). These solvents help deliver minoxidil into the scalp, but are known to cause skin irritation in susceptible individuals. It also appears that, for most people, long-term topical minoxidil therapy may be precluded by non-compliance (Ali Mapar & Omidian, 2007; Shadi, 2023).
The increase in overall body hair growth (i.e., hypertrichosis) is arguably the most consequential side effects for transfeminine people found to occur with minoxidil. As noted above, hypertrichosis is much more common with oral minoxidil than with topical minoxidil. This is a result of the differences in pharmacology between these routes and the extensive systemic absorption that occurs in the case of the former (Desai et al., 2024; Wiechert et al., 2025). In transmasculine people, an increase in body hair growth and diameter could be beneficial. However, these effects are usually not desired by transfeminine people. Consequently, some transfeminine people may prefer to use topical minoxidil over oral minoxidil, despite possible benefits to effectiveness from the latter in some individuals.
@@ -161,7 +161,7 @@
Angus, L. M., Hong, Q. V., Cheung, A. S., & Nolan, B. J. (2024). Effect of bicalutamide on serum total testosterone concentration in transgender adults: a case series. Therapeutic Advances in Endocrinology and Metabolism, 15, 20420188241305022. [DOI:10.1177/20420188241305022]
Angus, L. M., Nolan, B. J., Zajac, J. D., & Cheung, A. S. (2021). A systematic review of antiandrogens and feminization in transgender women. Clinical Endocrinology, 94(5), 743–752. [DOI:10.1111/cen.14329]
Asad, N., Naseer, M., & Ghafoor, R. (2024). Efficacy of Topical Finasteride 0.25% With Minoxidil 5% Versus Topical Minoxidil 5% Alone in Treatment of Male Pattern Androgenic Alopecia, Journal of Drugs in Dermatology, 23(11), 1003–1008. [DOI:10.36849/JDD.7826]
-
Barbieri, J. S., Margolis, D. J., & Mostaghimi, A. (2021). Temporal trends and clinician variability in potassium monitoring of healthy young women treated for acne with spironolactone. JAMA Dermatology, 157(3), 296–300. [DOI:10.1001/jamadermatol.2020.5468]
+
Barbieri, J. S., Margolis, D. J., & Mostaghimi, A. (2021). Temporal trends and clinician variability in potassium monitoring of healthy young women treated for acne with spironolactone. JAMA Dermatology, 157(3), 296–300. [DOI:10.1001/jamadermatol.2020.5468]
Basendwh, M. A., Alharbi, A. A., Bukhamsin, S. A., Abdulwahab, R. A., & Alaboud, S. A. (2024). The efficacy of Topical Clascoterone versus systematic spironolactone for treatment of acne vulgaris: A systematic review and network meta-analysis. Plos One, 19(5), e0298155. [DOI:10.1371/journal.pone.0298155]
Blume-Peytavi, U., Lönnfors, S., Hillmann, K., & Bartels, N. G. (2012). A randomized double-blind placebo-controlled pilot study to assess the efficacy of a 24-week topical treatment by latanoprost 0.1% on hair growth and pigmentation in healthy volunteers with androgenetic alopecia. Journal of the American Academy of Dermatology, 66(5), 794–800. [DOI:10.1016/j.jaad.2011.05.026]
Blume-Peytavi, U., Shapiro, J., Messenger, A. G., Hordinsky, M. K., Zhang, P., Quiza, C., Doshi, U., & Olsen, E. A. (2016). Efficacy and Safety of Once-Daily Minoxidil Foam 5% Versus Twice-Daily Minoxidil Solution 2% in Female Pattern Hair Loss: A Phase III, Randomized, Investigator-Blinded Study. Journal of Drugs in Dermatology, 15(7), 883–889. [Google Scholar] [PubMed] [URL]
@@ -202,7 +202,7 @@
Goren, A., & Naccarato, T. (2018). Minoxidil in the treatment of androgenetic alopecia. Dermatologic Therapy, 31(5), e12686. [DOI:10.1111/dth.12686]
Guo, M., Heran, B., Flannigan, R., Kezouh, A., & Etminan, M. (2016). Persistent sexual dysfunction with finasteride 1 mg taken for hair loss. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 36(11), 1180–1184. [DOI:10.1002/phar.1837]
Gupta, A. K., Venkataraman, M., Talukder, M., & Bamimore, M. A. (2022). Relative efficacy of minoxidil and the 5-α reductase inhibitors in androgenetic alopecia treatment of male patients: a network meta-analysis. JAMA Dermatology, 158(3), 266–274. [DOI:10.1001/jamadermatol.2021.5743]
-
Gupta, A. K., Hall, D. C., Talukder, M., & Bamimore, M. A. (2022). There is a positive dose-dependent association between low-dose oral minoxidil and its efficacy for androgenetic alopecia: findings from a systematic review with meta-regression analyses. Skin Appendage Disorders, 8(5), 355–361. [DOI:10.1159/000525137]
+
Gupta, A. K., Hall, D. C., Talukder, M., & Bamimore, M. A. (2022). There is a positive dose-dependent association between low-dose oral minoxidil and its efficacy for androgenetic alopecia: findings from a systematic review with meta-regression analyses. Skin Appendage Disorders, 8(5), 355–361. [DOI:10.1159/000525137]
Gupta, A. K., Talukder, M., Shemar, A., Piraccini, B. M., & Tosti, A. (2023). Low-dose oral minoxidil for alopecia: a comprehensive review. Skin Appendage Disorders, 9(6), 423–437. [DOI:10.1159/000531890]
Gupta, A. K., Bamimore, M. A., Wang, T., & Talukder, M. (2024). The impact of monotherapies for male androgenetic alopecia: A network meta‐analysis study. Journal of Cosmetic Dermatology, 23(9), 2964–2972. [DOI:10.1111/jocd.16362]
Gupta, A. K., Wang, T., Bamimore, M. A., & Talukder, M. (2024). The relative effect of monotherapy with 5‐alpha reductase inhibitors and minoxidil for female pattern hair loss: A network meta‐analysis study. Journal of Cosmetic Dermatology, 23(1), 154–160. [DOI:10.1111/jocd.15910]
@@ -211,6 +211,7 @@
Gupta, M. A., Vujcic, B., & Gupta, A. K. (2020). Finasteride Use Is Associated with Higher Odds of Obstructive Sleep Apnea: Results from the US Food and Drug Administration Adverse Events Reporting System. Skinmed, 18(3), 146–150. [Google Scholar] [PubMed]
Hebert, A., Thiboutot, D., Gold, L. S., Cartwright, M., Gerloni, M., Fragasso, E., & Mazzetti, A. (2020). Efficacy and safety of topical clascoterone cream, 1%, for treatment in patients with facial acne: two phase 3 randomized clinical trials. JAMA Dermatology, 156(6), 621–630. [DOI:10.1001/jamadermatol.2020.0465]
Hirshburg, J. M., Kelsey, P. A., Therrien, C. A., Gavino, A. C., & Reichenberg, J. S. (2016). Adverse effects and safety of 5-alpha reductase inhibitors (finasteride, dutasteride): a systematic review. The Journal of Clinical and Aesthetic Dermatology, 9(7), 56–62. [Google Scholar] [PubMed] [PubMed Central] [PDF]
+
Hobo, Y., Nishikawa, J., Taniguchi Asai, N., Yoneyama, K., Watanabe, Y., Miyashiro, Y., & Fujikata, A. (2023). Evaluation of the therapeutic effects of AGA drugs by measuring finasteride, dutasteride, and dihydrotestosterone in hair. Clinica Chimica Acta, 547, 117456. [DOI:10.1016/j.cca.2023.117456]
Hu, R., Xu, F., Sheng, Y., Qi, S., Han, Y., Miao, Y., Rui, W., & Yang, Q. (2015). Combined treatment with oral finasteride and topical minoxidil in male androgenetic alopecia: a randomized and comparative study in Chinese patients. Dermatologic Therapy, 28(5), 303–308. [DOI:10.1111/dth.12246]
Ibraheim, M. K., Elsensohn, A., Hauschild, C., Hilliard, A., & Dao, H. (2023). Low dose oral minoxidil and the conundrum of cardiovascular complications. Dermatology Online Journal, 29(4). [DOI:10.5070/D329461861]
Imperato-McGinley, J., & Zhu, Y. S. (2002). Androgens and male physiology the syndrome of 5α-reductase-2 deficiency. Molecular and Cellular Endocrinology, 198(1–2), 51–59. [DOI:10.1016/S0303-7207(02)00368-4]
@@ -257,11 +258,11 @@
Pirastu, N., Joshi, P. K., de Vries, P. S., Cornelis, M. C., McKeigue, P. M., Keum, N., Franceschini, N., Colombo, M., Giovannucci, E. L., Spiliopoulou, A., Franke, L., North, K. E., Kraft, P., Morrison, A. C., Esko, T., & Wilson, J. F. (2017). GWAS for male-pattern baldness identifies 71 susceptibility loci explaining 38% of the risk. Nature Communications, 8(1), 1584. [DOI:10.1038/s41467-017-01490-8]
Prince, J. C. J., & Safer, J. D. (2020). Endocrine treatment of transgender individuals: current guidelines and strategies. Expert Review of Endocrinology & Metabolism, 15(6), 395–403. [DOI:10.1080/17446651.2020.1825075]
Randolph, M., & Tosti, A. (2021). Oral minoxidil treatment for hair loss: a review of efficacy and safety. Journal of the American Academy of Dermatology, 84(3), 737–746. [DOI:10.1016/j.jaad.2020.06.1009]
-
Ramos, P. M., Sinclair, R. D., Kasprzak, M., & Miot, H. A. (2020). Minoxidil 1 mg oral versus minoxidil 5% topical solution for the treatment of female-pattern hair loss: a randomized clinical trial. Journal of the American Academy of Dermatology, 82(1), 252–253. [DOI:10.1016/j.jaad.2019.08.060]
-
Rosenthal, A., Conde, G., Greco, J. F., & Gharavi, N. M. (2024). Management of androgenic alopecia: a systematic review of the literature. Journal of Cosmetic and Laser Therapy, 26(1–4), 1–16. [DOI:10.1080/14764172.2024.2362126]
+
Ramos, P. M., Sinclair, R. D., Kasprzak, M., & Miot, H. A. (2020). Minoxidil 1 mg oral versus minoxidil 5% topical solution for the treatment of female-pattern hair loss: a randomized clinical trial. Journal of the American Academy of Dermatology, 82(1), 252–253. [DOI:10.1016/j.jaad.2019.08.060]
+
Rosenthal, A., Conde, G., Greco, J. F., & Gharavi, N. M. (2024). Management of androgenic alopecia: a systematic review of the literature. Journal of Cosmetic and Laser Therapy, 26(1–4), 1–16. [DOI:10.1080/14764172.2024.2362126]
Rossi, A., Cantisani, C., Melis, L., Iorio, A., Scali, E., & Calvieri, S. (2012). Minoxidil use in dermatology, side effects and recent patents. Recent Patents on Inflammation & Allergy Drug Discovery, 6(2), 130–136. [DOI:10.2174/187221312800166859]
Rossi, A., & Caro, G. (2024). Efficacy of the association of topical minoxidil and topical finasteride compared to their use in monotherapy in men with androgenetic alopecia: A prospective, randomized, controlled, assessor blinded, 3‐arm, pilot trial. Journal of Cosmetic Dermatology, 23(2), 502–509. [DOI:10.1111/jocd.15953]
-
Ryu, H. K., Kim, K. M., Yoo, E. A., Sim, W. Y., & Chung, B. C. (2006). Evaluation of androgens in the scalp hair and plasma of patients with male‐pattern baldness before and after finasteride administration. British Journal of Dermatology, 154(4), 730–734. [DOI:10.4103/ijd.ijd_729_23]
+
Ryu, H. K., Kim, K. M., Yoo, E. A., Sim, W. Y., & Chung, B. C. (2006). Evaluation of androgens in the scalp hair and plasma of patients with male‐pattern baldness before and after finasteride administration. British Journal of Dermatology, 154(4), 730–734. [DOI:10.1111/j.1365-2133.2005.07072.x]
Saceda-Corralo, D., Domínguez-Santas, M., Vañó-Galván, S., & Grimalt, R. (2023). What’s new in therapy for male androgenetic alopecia? American Journal of Clinical Dermatology, 24(1), 15–24. [DOI:10.1007/s40257-022-00730-y]
Sadasivam, I. P., Sambandam, R., Kaliyaperumal, D., & Dileep, J. E. (2024). Androgenetic Alopecia in Men: An Update On Genetics. Indian Journal of Dermatology, 69(3), 282. [DOI:10.4103/ijd.ijd_729_23]
Sanabria, B. D., Perdomo, Y. C., Miot, H. A., & Ramos, P. M. (2024). Oral minoxidil 7.5 mg for hair loss increases heart rate with no change in blood pressure in 24 h Holter and 24 h ambulatory blood pressure monitoring. Anais Brasileiros de Dermatologia, 99(5), 734–736. [DOI:10.1016/j.abd.2023.08.016]
@@ -599,6 +600,8 @@ Using the term desistence in this way does not imply anything about the identity
Achille, C., Taggart, T., Eaton, N. R., Osipoff, J., Tafuri, K., Lane, A., & Wilson, T. A. (2020). Longitudinal impact of gender-affirming endocrine intervention on the mental health and well-being of transgender youths: preliminary results. International Journal of Pediatric Endocrinology, 2020, 8. [DOI:10.1186/s13633-020-00078-2]
+
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
+
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
Antoniazzi, F., Zamboni, G., Bertoldo, F., Lauriola, S., Mengarda, F., Pietrobelli, A., & Tatò, L. (2003). Bone mass at final height in precocious puberty after gonadotropin-releasing hormone agonist with and without calcium supplementation. The Journal of Clinical Endocrinology and Metabolism, 88(3), 1096–1101. [DOI:10.1210/jc.2002-021154]
Arnoldussen, M., Steensma, T. D., Popma, A., van der Miesen, A., Twisk, J., & de Vries, A. (2020). Re-evaluation of the Dutch approach: are recently referred transgender youth different compared to earlier referrals? European Child & Adolescent Psychiatry, 29(6), 803–811. [DOI:10.1007/s00787-019-01394-6]
Bakwin, H. (1968). Deviant gender-role behavior in children: relation to homosexuality. Pediatrics, 41(3), 620–629. [PubMed] [DOI:10.1542/peds.41.3.620]
@@ -773,6 +776,8 @@ Using the term desistence in this way does not imply anything about the identity
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
+
Aly. (2021). An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations. Transfeminine Science. [URL]
Behre, H. M., Abshagen, K., Oettel, M., Hubler, D., & Nieschlag, E. (1999). Intramuscular injection of testosterone undecanoate for the treatment of male hypogonadism: phase I studies. European Journal of Endocrinology, 140(5), 414–419. [DOI:10.1530/eje.0.1400414]
]]>
{"first_name"=>"Aly", "last_name"=>"W.", "author-link"=>"/about/#aly", "articles-link"=>"/articles-by-author/aly/"}An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations2021-07-16T12:00:00-07:002025-05-08T00:00:00-07:00https://transfemscience.org/articles/injectable-e2-meta-analysisAn Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations
@@ -790,7 +795,7 @@ Using the term desistence in this way does not imply anything about the identity
Estradiol is the main estrogen used in transfeminine hormone therapy and is available in a variety of different forms for use by different routes of administration. The most commonly employed forms are oral, sublingual, transdermal, and injectable preparations. Injectable estradiol preparations have been discontinued in many countries and hence are unavailable for use in transfeminine hormone therapy in many parts of the world, for instance in most of Europe (Glintborg et al., 2021). However, they are still used by many transfeminine people particularly in the United States and in the do-it-yourself (DIY) community. The most commonly used forms include estradiol valerate, estradiol cypionate, and estradiol enanthate all in oil. Injectable estradiol preparations have certain advantages over other estradiol forms that make them a popular choice for use in transfeminine hormone therapy. These include often lower cost, capacity to easily achieve higher estradiol levels that can be useful for testosterone suppression, less frequent administration, and theoretically reduced health risks relative to oral estradiol at equivalent doses due to the lack of the first pass with this route (Aly, 2020). The higher estradiol levels with injections are particularly useful for estradiol monotherapy, in which an antiandrogen is not used.
-
Clinically used injectable estradiol preparations are formulated not as estradiol but as estradiol esters. When injected into muscle or fat in oil solutions or crystalline aqueous suspensions, these estradiol esters form depots at the injection site from which they are slowly released. Subsequent to release, estradiol esters are rapidly metabolized into estradiol and hence act as prodrugs. When estradiol itself is given by intramuscular injection in an aqueous solution or oil solution, it is rapidly absorbed and has a very short duration. Due to having lipophilic esters, most clinically used injectable estradiol esters are more fat-soluble than estradiol (as measured by oil–water partition coefficient (P)) (Table). When these esters are administered as oil solutions by intramuscular or subcutaneous injection, their increased lipophilicity causes them to be released from the injection-site depot more slowly than estradiol and to therefore have longer durations. In the case of fatty acid esters, the longer the chain length of the ester—as in e.g. estradiol valerate (5 carbons) vs. estradiol enanthate (7 carbons) vs. estradiol undecylate (10 carbons)—the greater the fat solubility, the slower the rate of release from the depot, and the longer the time to peak levels and duration (Edkins, 1959; Sinkula, 1978; Chien, 1981; Kuhl, 2005; Kalicharan, 2017; Vhora et al., 2019). The durations of both injectable oil solutions and aqueous suspensions depend on the ester and its particular physicochemical properties, but the characteristics of these preparations are different and they work in distinct ways to produce their depot effects (Enever et al., 1983; Aly, 2019). The durations of oil solutions are dependent on the lipophilicity of the ester as well as oil vehicle, whereas the durations of aqueous suspensions depend on the properties of the ester crystal lattice as well as crystal sizes (Chien, 1981; Enever et al., 1983; Aly, 2019). The polymeric estradiol ester polyestradiol phosphate is more hydrophilic (water-soluble) than estradiol and works differently than other injectable estradiol preparations. Ιt is composed of many estradiol molecules linked together via phosphate esters (on average 13 molecules of estradiol per one molecule of polyestradiol phosphate) and has a prolonged duration due to slow cleavage into estradiol following injection. Estradiol esters are able to substantially prolong the duration of estradiol when used as injectables and these preparations have durations ranging from days to months depending on the ester and how it is formulated (Table).
+
Clinically used injectable estradiol preparations are formulated not as estradiol but as estradiol esters. When injected into muscle or fat in oil solutions or crystalline aqueous suspensions, these estradiol esters form depots at the injection site from which they are slowly released. Subsequent to release, estradiol esters are rapidly metabolized into estradiol and hence act as prodrugs. When estradiol itself is given by intramuscular injection in an aqueous solution or oil solution, it is rapidly absorbed and has a very short duration. Due to having lipophilic esters, most clinically used injectable estradiol esters are more fat-soluble than estradiol (as measured by oil–water partition coefficient (P)) (Table). When these esters are administered as oil solutions by intramuscular or subcutaneous injection, their increased lipophilicity causes them to be released from the injection-site depot more slowly than estradiol and to therefore have longer durations. In the case of fatty acid esters, the longer the chain length of the ester—as in e.g. estradiol valerate (5 carbons) vs. estradiol enanthate (7 carbons) vs. estradiol undecylate (10 carbons)—the greater the fat solubility, the slower the rate of release from the depot, and the longer the time to peak levels and duration (Edkins, 1959; Sinkula, 1978; Chien, 1981; Kuhl, 2005; Kalicharan, 2017; Vhora et al., 2019). The durations of both injectable oil solutions and aqueous suspensions depend on the ester and its particular physicochemical properties, but the characteristics of these preparations are different and they work in distinct ways to produce their depot effects (Enever et al., 1983; Aly, 2019). The durations of oil solutions are dependent on the lipophilicity of the ester as well as oil vehicle, whereas the durations of aqueous suspensions depend on the properties of the ester crystal lattice as well as crystal sizes (Chien, 1981; Enever et al., 1983; Aly, 2019). The polymeric estradiol ester polyestradiol phosphate is more hydrophilic (water-soluble) than estradiol and works differently than other injectable estradiol preparations. Ιt is composed of many estradiol molecules linked together via phosphate esters (on average 13 molecules of estradiol per one molecule of polyestradiol phosphate) and has a prolonged duration due to slow cleavage into estradiol following injection. Estradiol esters are able to substantially prolong the duration of estradiol when used as injectables and these preparations have durations ranging from days to months depending on the ester and how it is formulated (Table).
@@ -1988,7 +1993,7 @@ Using the term desistence in this way does not imply anything about the identity
Due to scarcity of data for several injectable estradiol preparations, the study selection criteria maximized data inclusion in order to allow for better curve fits at the risk of including potentially less reliable data. As examples, studies were included regardless of the status of the HPG axis of the participants, and Cmax data were included in the fitting if data were very limited. In the case of HPG axis state, studies with cycling women may result in greater error due to more variable levels of endogenous estradiol. Moreover, acute high levels of estradiol can induce a surge in luteinizing hormone levels after several days in gonadally intact women, and this may cause a delayed bump in estradiol levels (Wiki). One of the more overt instances of this can be seen in a study of estradiol benzoate in such women (Shaw, 1978 [Graph]). Many if not most of the included studies with estradiol benzoate involved women with intact HPG axes, whereas studies of this sort were uncommon with the other preparations. In the case of Cmax data, these data when Cmax corresponds to the mean of individual peaks are a different type of data than the peak of the mean curve of all individuals. Cmax levels can differ in both magnitude and timing compared to the mean curve peak (e.g., Oriowo et al., 1980 [Graph]; Rahimy, Ryan, & Hopkins, 1999). This is because for instance not all individuals peak at the same time and this variability in time to peak normally serves to dilute peak levels for the mean curve when compared to individual maximal concentrations. However, Cmax levels are in any case generally in the vicinity of the mean curve peak. While Cmax levels were excluded in the fitting for most injectable estradiol preparations, they were included in the case of estradiol enanthate. This was because the available mean and individual estradiol curve data were very limited for this specific preparation, and inclusion of Cmax data allowed for improved fitting in spite of its limitations. Lastly, some of the included data was once-monthly multi-dose, and research with once-monthly estradiol enanthate-containing combined injectable contraceptives has found that the time to peak levels may shift with repeated long-term use (Schiavon et al., 1988; Garza-Flores, 1994).
-
There was considerable variability between studies in terms of estradiol levels and concentration–time curve shapes with the same injectable estradiol preparation. The reasons for the large variability across studies are not fully clear. In any case, there are many potential factors that may contribute to this variability. These include preparation- and injection-related factors like formulation (e.g., oil vehicle, other components and excipients, concentration, particle size), injection volume, site of injection (e.g., buttocks, thigh, upper arm), injection technique (e.g., force of injection—and resulting depot droplet dimensions), and syringe dead space. They additionally include various subject- and research-related variables like differing blood-testing methodology, differing sample characteristics (e.g., age, weight, gender, ethnicity, physical activity, HPG axis state), and sampling error (Sinkula, 1978; Chien, 1981; Minto et al., 1997; Larsen & Larsen, 2009; Larsen et al., 2009; Florence, 2010; Larsen, Thing, & Larsen, 2012; Kalicharan, 2017). Older studies, which used potentially less accurate blood tests and tended to have smaller numbers of subjects, seemed to particularly add to the variability between studies. These studies may represent less reliable data than more recent research with larger sample sizes. The exclusion criteria helped to remove outliers for the different injectable estradiol preparations however. This meta-analysis does not take into account the potential factors underlying the variability between studies. To do so would be difficult, as in many cases information on these variables is not provided in individual studies and research quantifying their precise influences and relative importances is limited.
+
There was considerable variability between studies in terms of estradiol levels and concentration–time curve shapes with the same injectable estradiol preparation. The reasons for the large variability across studies are not fully clear. In any case, there are many potential factors that may contribute to this variability. These include preparation- and injection-related factors like formulation (e.g., oil vehicle, other components and excipients, concentration, particle size), injection volume, site of injection (e.g., buttocks, thigh, upper arm), injection technique (e.g., force of injection—and resulting depot droplet dimensions), and syringe dead space. They additionally include various subject- and research-related variables like differing blood-testing methodology, differing sample characteristics (e.g., age, weight, gender, ethnicity, physical activity, HPG axis state), and sampling error (Sinkula, 1978; Chien, 1981; Minto et al., 1997; Larsen & Larsen, 2009; Larsen et al., 2009; Florence, 2010; Larsen, Thing, & Larsen, 2012; Kalicharan, 2017). Older studies, which used potentially less accurate blood tests and tended to have smaller numbers of subjects, seemed to particularly add to the variability between studies. These studies may represent less reliable data than more recent research with larger sample sizes. The exclusion criteria helped to remove outliers for the different injectable estradiol preparations however. This meta-analysis does not take into account the potential factors underlying the variability between studies. To do so would be difficult, as in many cases information on these variables is not provided in individual studies and research quantifying their precise influences and relative importances is limited.
It is in any case known from other studies that different oil vehicles are absorbed at different rates from the injection site (Svendsen & Aaes‐Jørgensen, 1979; Schultz et al., 1998; Larsen et al., 2001) and can result in different concentration–time curve shapes (Ballard, 1978 [Excerpt]; Knudsen, Hansen, & Larsen, 1985). This is thought to be due to differences in oil lipophilicity and depot release rates. Viscosity of oils has also been hypothesized to potentially influence rate of depot escape (Schug, Donath, & Blume, 2012). However, research so far has not supported this hypothesis (Larsen & Larsen, 2009; Larsen, Thing, & Larsen, 2012). Oil vehicles can vary with injectable estradiol preparations even for the same estradiol ester. For instance, pharmaceutical estradiol valerate is formulated in sesame oil, castor oil, or sunflower oil depending on the preparation (Table). It is notable however that these three oils have similar lipophilicities (Table). On the other hand, homebrewed injectable estradiol preparations used by DIY transfeminine people often employ medium-chain triglyceride (MCT) oil as the oil vehicle. This oil (in the proprietary form of Viscoleo) has notably been found to be much more rapidly absorbed than conventional oils like sesame oil and castor oil in animals (Svendsen & Aaes‐Jørgensen, 1979; Schultz et al., 1998; Larsen et al., 2001). In addition, although based on very limited data, MCT oil has been found to give spikier and shorter-lasting depot injectable curves in humans (Knudsen, Hansen, & Larsen, 1985). As such, injectable estradiol preparations using MCT oil as the vehicle may have differing and less favorable concentration–time curve shapes than pharmaceutical injectable estradiol products. Other excipients, like benzyl alcohol, as well as factors like injection site and volume, have additionally been found to influence pharmacokinetic properties with depot injectables (Minto et al., 1997; Kalicharan, Schot, & Vromans, 2016). Excipients besides oil vehicle also vary by formulation (Table).
@@ -2585,6 +2590,12 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Aedo, A. R., Landgren, B. M., Johannisson, E., & Diczfalusy, E. (1985). Pharmacokinetic and pharmacodynamic investigations with monthly injectable contraceptive preparations. Contraception, 31(5), 453–469. [DOI:10.1016/0010-7824(85)90081-2]
Aisaka, K., Ando, S., Kokubo, K., Yoshida, K., & Mori, H. (1986). いわゆる潜在性高prolactin血症患者におけるprolactin分泌予備能の検討. [Studies on Prolactin Secreting Capacity in the Ovulatory Infertile Patients with Transient Hyperprolactinemia.] 日本内分泌学会雑誌 / Nihon Naibunpi Gakkai Zasshi / Folia Endocrinologica Japonica, 62(5), 662–671. [DOI:10.1507/endocrine1927.62.5_662]
Akande, E. O. (1974). The effect of oestrogen on plasma levels of luteinizing hormone in euthyroid and thyrotoxic postmenopausal women. The Journal of Obstetrics and Gynaecology of the British Commonwealth / BJOG, 81(10), 795–803. [DOI:10.1111/j.1471-0528.1974.tb00383.x]
+
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
+
Aly. (2019). Injectable Aqueous Suspensions of Sex Hormones and How Depot Injectables Work. Transfeminine Science. [URL]
+
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
+
Aly. (2020). Clinical Guidelines with Information on Transfeminine Hormone Therapy. Transfeminine Science. [URL]
+
Aly. (2020). Estrogens and Their Influences on Coagulation and Risk of Blood Clots. Transfeminine Science. [URL]
+
Aly. (2021). An Interactive Web Simulator for Estradiol Levels with Injectable Estradiol Esters. Transfeminine Science. [URL]
Ballard, B. E. (1978). An Overview of Prolonged Action Drug Dosage Forms. In Robinson, J. R. (Ed.). Sustained and Controlled Release Drug Delivery Systems (pp. 1–69). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books]
Behre, H. M., Oberpenning, F., & Nieschlag, E. (1990). Comparative pharmacokinetics of androgen preparations: application of computer analysis and simulation. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 1st Edition (pp. 115–135). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-662-00814-0_6]
Behre, H. M., & Nieschlag, E. (1998). Comparative pharmacokinetics of testosterone esters. In Nieschlag, E., & Behre, H. M. (Eds.). Testosterone: Action · Deficiency · Substitution, 2nd Edition (pp. 329–348). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-72185-4_11]
@@ -2611,7 +2622,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Dahl, M., Feldman, J. L., Goldberg, J., & Jaberi, A. (2015). Endocrine Therapy for Transgender Adults in British Columbia: Suggested Guidelines. Physical Aspects of Transgender Endocrine Therapy. Vancouver: Vancouver Coastal Health. [Google Scholar] [PDF]
Davidson, A., Franicevich, J., Freeman, M., Lin, R., Martinez, L., Monihan, M., Porch, M., Samuel, L., Stukalin, R., Vormohr, J., & Zevin, B. (2013). Protocols for Hormonal Reassignment of Gender. San Francisco: San Francisco Department of Public Health/Tom Waddell Health Center. [Google Scholar] [PDF]
Depo®-Estradiol Estradiol Cypionate Label. U.S. Food and Drug Administration/Pharmacia & Upjohn (Pfizer). [URL] [PDF]
-
Derra, C. (1981). Hormonprofile unter Östrogen- und Antiandrogentherapie bei Patienten mit Prostatakarzinom: Östradiolundecylat versus Cyproteronacetat. [Hormone Profiles under Estrogen and Antiandrogen Therapy in Patients with Prostate Cancer: Estradiol Undecylate versus Cyproterone Acetate.] (Doctoral dissertation, University of Mainz.) [Google Scholar] [WorldCat] [PDF] [Translation]
+
Derra, C. (1981). Hormonprofile unter Östrogen- und Antiandrogentherapie bei Patienten mit Prostatakarzinom: Östradiolundecylat versus Cyproteronacetat. [Hormone Profiles under Estrogen and Antiandrogen Therapy in Patients with Prostate Cancer: Estradiol Undecylate versus Cyproterone Acetate.] (Doctoral dissertation, University of Mainz.) [Google Scholar] [WorldCat] [PDF] [Translation]
Deutsch, M. (2014). Medical Transition. In Erickson-Schroth, L. (Ed.). Trans Bodies, Trans Selves: A Resource for the Transgender Community, 1st Edition (pp. 241–264). Oxford: Oxford University Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [PDF]
Deutsch, M. B., Bhakri, V., & Kubicek, K. (2015). Effects of Cross-Sex Hormone Treatment on Transgender Women and Men. Obstetrics & Gynecology, 125(3), 605–610. [DOI:10.1097/AOG.0000000000000692]
Deutsch, M. B. (Ed.). (2016). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition. San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
@@ -2625,7 +2636,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Espino y Sosa, S., Cortés Fuentes, M., Gómez Rico, J. A., & Cortés Bonilla, M. (2019). Non-polymeric Microspheres for the Therapeutic Use of Estrogens: An Innovative Technology. In Khan, W. A. (Ed.). Estrogen. London: IntechOpen. [DOI:10.5772/intechopen.82553]
Fisher, D., & Shafer, S. (2007). Fisher/Shafer NONMEM Workshop Pharmacokinetic and Pharmacodynamic Analysis with NONMEM. Basic Concepts. [PDF]
-
Florence, A. T. (2010). Looking at Formulations. In Florence, A. T. An Introduction to Clinical Pharmaceutics (pp. 69–100). London/Chicago: Pharmaceutical Press. [Google Scholar] [Google Books]
+
Florence, A. T. (2010). Looking at Formulations. In Florence, A. T. An Introduction to Clinical Pharmaceutics (pp. 69–100). London/Chicago: Pharmaceutical Press. [Google Scholar] [Google Books]
Fotherby, K., Benagiano, G., Toppozada, H. K., Abdel-Rahman, A., Navaroli, F., Arce, B., Ramos-Cordero, R., Gual, C., Landgren, B. M., & Johannisson, E. (1982). A preliminary pharmacological trial of the monthly injectable contraceptive Cycloprovera. Contraception, 25(3), 261–272. [DOI:10.1016/0010-7824(82)90049-X]
Futterweit, W., Gabrilove, J., & Smith, H. (1984). Testicular steroidogenic response to human chorionic gonadotropin of fifteen male transsexuals on chronic estrogen treatment. Metabolism, 33(10), 936–942. [DOI:10.1016/0026-0495(84)90248-8] [Figure]
Garner, P. R., & Armstrong, D. T. (1977). The effect of human chorionic gonadotropin and estradiol-17β on the maintenance of the human corpus luteum of early pregnancy. American Journal of Obstetrics and Gynecology, 128(5), 469–475. [DOI:10.1016/0002-9378(77)90026-6]
@@ -2649,8 +2660,8 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Henriksson, P., Carlström, K., Pousette, A., Gunnarsson, P. O., Johansson, C. J., Eriksson, B., Altersgård-Brorsson, A. K., Nordle, O., & Stege, R. (1999). Time for revival of estrogens in the treatment of advanced prostatic carcinoma? Pharmacokinetics, and endocrine and clinical effects, of a parenteral estrogen regimen. The Prostate, 40(2), 76–82. [DOI:10.1002/(SICI)1097-0045(19990701)40:2<76::AID-PROS2>3.0.CO;2-Q]
Herndon, J. S., Maheshwari, A. K., Nippoldt, T. B., Carlson, S. J., Davidge-Pitts, C. J., & Chang, A. Y. (2023). Comparison of Subcutaneous and Intramuscular Estradiol Regimens as part of Gender-Affirming Hormone Therapy. Endocrine Practice, 29(5), 356–361. [DOI:10.1016/j.eprac.2023.02.006]
Hughes, J. H., Woo, K. H., Keizer, R. J., & Goswami, S. (2022). Clinical Decision Support for Precision Dosing: Opportunities for Enhanced Equity and Inclusion in Health Care. Clinical Pharmacology & Therapeutics, 113(3), 565–574. [DOI:10.1002/cpt.2799]:
-
Ibrahim, S. (1996/1998). Pharmakokinetische Untersuchungen mit Östradiolvalerat und Hydroxyprogesteroncaproat in Depotform nach einmaliger Applikation bei 24 postmenopausalen Frauen. [Pharmacokinetic studies with estradiol valerate and hydroxyprogesterone caproate in depot form after a single application in 24 postmenopausal women.] (Doctoral dissertation, Dresden University of Technology.) [Google Scholar] [WorldCat] [Partial PDF]
-
Ittrich, G., & Pots, P. (1965). Östrogenbestimmungen in Blut und Urin nach Verabreichung von Östrogenen. [Estrogen determinations in blood and urine after administration of estrogens.] In Kraatz, H. (Ed.). International Symposium der Gynäkologischen Endokrinologie vom 15. bis 18. Mai 1963. / Abhandlungen der Deutschen Akademie der Wissenschaften zu Berlin, Klasse für Medizin, 1965(1), 53–56. [ISSN:0568-4250] [WorldCat 1] [WorldCat 2] [WorldCat 3] [PDF] [Translation]
+
Ibrahim, S. (1996/1998). Pharmakokinetische Untersuchungen mit Östradiolvalerat und Hydroxyprogesteroncaproat in Depotform nach einmaliger Applikation bei 24 postmenopausalen Frauen. [Pharmacokinetic studies with estradiol valerate and hydroxyprogesterone caproate in depot form after a single application in 24 postmenopausal women.] (Doctoral dissertation, Dresden University of Technology.) [Google Scholar] [WorldCat] [Partial PDF]
+
Ittrich, G., & Pots, P. (1965). Östrogenbestimmungen in Blut und Urin nach Verabreichung von Östrogenen. [Estrogen determinations in blood and urine after administration of estrogens.] In Kraatz, H. (Ed.). International Symposium der Gynäkologischen Endokrinologie vom 15. bis 18. Mai 1963. / Abhandlungen der Deutschen Akademie der Wissenschaften zu Berlin, Klasse für Medizin, 1965(1), 53–56. [ISSN:0568-4250] [WorldCat 1] [WorldCat 2] [WorldCat 3] [PDF] [Translation]
Jaafar, S., Torres-Leguizamon, M., Duplessy, C., & Stambolis-Ruhstorfer, M. (2022). Hormonothérapie injectable et réduction des risques: pratiques, difficultés, santé des personnes trans en France. [Hormone replacement therapy injections and harm reduction: practices, difficulties, and transgender people’s health in France.] Sante Publique, 34(HS2), 109–122. [Google Scholar] [PubMed] [DOI:10.3917/spub.hs2.0109]
Jacobi, G. H., & Altwein, J. E. (1979). Bromocriptin als Palliativtherapie beim fortgeschrittenen Prostatakarzinom. [Bromocriptine for palliation of advanced prostatic carcinoma. Experimental and clinical profile of a drug.] Urologia Internationalis, 34(4), 266–290. [DOI:10.1159/000280272]
Jacobi, G. H., Altwein, J. E., Kurth, K. H., Basting, R., & Hohenfellner, R. (1980). Treatment of advanced prostatic cancer with parenteral cyproterone acetate: a phase III randomised trial. British Journal of Urology, 52(3), 208–215. [DOI:10.1111/j.1464-410X.1980.tb02961.x]
@@ -2698,7 +2709,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Nelson, M. D., Szczepaniak, L. S., Wei, J., Szczepaniak, E., Sánchez, F. J., Vilain, E., Stern, J. H., Bergman, R. N., Bairey Merz, C. N., & Clegg, D. J. (2016). Transwomen and the Metabolic Syndrome: Is Orchiectomy Protective? Transgender Health, 1(1), 165–171. [DOI:10.1089/trgh.2016.0016] [Table]
Nieschlag, E., & Behre, H. M. (2010). Testosterone therapy. In Nieschlag, E., Behre, H. M., & Nieschlag, S. (Eds.). Andrology (pp. 437–455). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-540-78355-8_21] [PDF]
Norlén, B. J., Fritjofsson, Å., Grönquist, L., Gunnarsson, P. O., Johansson, S. Å., & Plym-Forshell, G. (1987). Plasma concentrations of estradiol and testosterone in single-drug polyestradiol phosphate therapy for prostatic cancer. European Urology, 13, 193–197. [DOI:10.1159/000472772]
-
Olson-Kennedy, J., Rosenthal, S. M., Hastings, J., & Wesp, L. (2016). Health considerations for gender non-conforming children and transgender adolescents. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 186–199). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
+
Olson-Kennedy, J., Rosenthal, S. M., Hastings, J., & Wesp, L. (2016). Health considerations for gender non-conforming children and transgender adolescents. In Deutsch, M. B. (Ed.). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition (pp. 186–199). San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
Oriowo, M. A., Landgren, B. M., Stenström, B., & Diczfalusy, E. (1980). A comparison of the pharmacokinetic properties of three estradiol esters. Contraception, 21(4), 415–424. [DOI:10.1016/S0010-7824(80)80018-7]
Parkes, A. S. (1937). Relative duration of action of various esters of oestrone, oestradiol and oestriol. Biochemical Journal, 31(4), 579–585. [DOI:10.1042/bj0310579]
Patel, R., Korenman, S., Weimer, A., & Grock, S. (2024). A Call for Updates to Hormone Therapy Guidelines for Gender-Diverse Adults Assigned Male at Birth. Cureus, 16(6), e62262. [DOI:10.7759/cureus.62262] [PDF]
@@ -2734,7 +2745,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Sierra-Ramírez, J. A., Lara-Ricalde, R., Lujan, M., Velázquez-Ramírez, N., Godínez-Victoria, M., Hernádez-Munguía, I. A., Padilla, A., & Garza-Flores, J. (2011). Comparative pharmacokinetics and pharmacodynamics after subcutaneous and intramuscular administration of medroxyprogesterone acetate (25 mg) and estradiol cypionate (5 mg). Contraception, 84(6), 565–570. [DOI:10.1016/j.contraception.2011.03.014]
Sinkula, A. A. (1978). Methods to Achieve Sustained Drug Delivery. The Chemical Approach. In Robinson, J. R. (Ed.). Sustained and Controlled Release Drug Delivery Systems (pp. 411–555). New York/Basel: Marcel Dekker. [Google Scholar] [Google Books] [PDF]
Slack, D. J., Di Via Ioschpe, A., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., Karim, S., & Safer, J. D. (2025). Examining the Influence of the Route of Administration and Dose of Estradiol on Serum Estradiol and Testosterone Levels in Feminizing Gender-Affirming Hormone Therapy. Endocrine Practice, 31(1), 19–27. [DOI:10.1016/j.eprac.2024.10.002]
-
Somerville, B. W. (1971). The Role of Oestradiol in Menstrual Migraine. In Somerville, B. W. The Influence of Progesterone and Oestradiol on Migraine (Doctoral dissertation, University of New South Wales) (pp. 93–104). [Google Scholar] [URL] [WorldCat] [PDF]
+
Somerville, B. W. (1971). The Role of Oestradiol in Menstrual Migraine. In Somerville, B. W. The Influence of Progesterone and Oestradiol on Migraine (Doctoral dissertation, University of New South Wales) (pp. 93–104). [Google Scholar] [URL] [WorldCat] [PDF]
Somerville, B. W. (1972). The Role of Estradiol Withdrawal in the Etiology of Menstrual Migraine. Neurology, 22(4), 355–365. [DOI:10.1212/WNL.22.4.355]
Somerville, B. W. (1972). The influence of progesterone and estradiol upon migraine. Headache: The Journal of Head and Face Pain, 12(3), 93–102. [DOI:10.1111/j.1526-4610.1972.hed1203093.x]
Somerville, B. W. (1972). The Influence of Hormones Upon Migraine in Women. Medical Journal of Australia, 2(S2), 6–11. [DOI:10.5694/j.1326-5377.1972.tb93039.x]
@@ -2759,7 +2770,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Valle Alvarez, D. C. (2011). Efecto de una Dosis de 50 mg de Enantato de Noretisterona y 5 mg de Valerato de Estradiol en los Niveles de Testosterona Total en Hombres Mexicanos Sanos. [Effect of a Dose of 50 mg of Norethisterone Enanthate and 5 mg of Estradiol Valerate on Total Testosterone Levels in Healthy Mexican Men.] (Masters thesis, National Polytechnic Institute of Mexico.) [Google Scholar] [URL] [PDF] [Translation]
Varangot, J., & Cedard, L. (1957). Modifications des Œstrogènes Sanguins Après Administration Intramusculaire de Benzoate d’Œstradiol. [Changes in Serum Estrogens After Intramuscular Administration of Estradiol Benzoate.] Comptes Rendus des Séances de la Société de Biologie et de ses Filiales, 151(10), 1707–1712. [Google Scholar 1] [Google Scholar 2] [PubMed] [PDF] [Translation]
Vermeulen, A. (1977). Transport and Distribution of Androgens at Different Ages. In Martini, L., & Motta, M. (Eds.). Androgens and Antiandrogens (pp. 53–65). New York: Raven Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [Excerpt]
+
Vermeulen, A. (1977). Transport and Distribution of Androgens at Different Ages. In Martini, L., & Motta, M. (Eds.). Androgens and Antiandrogens (pp. 53–65). New York: Raven Press. [Google Scholar] [Google Books] [OpenLibrary] [WorldCat] [Archive.org] [Excerpt]
Vhora, I., Bardoliwala, D., Ranamalla, S. R., & Javia, A. (2019). Parenteral controlled and prolonged drug delivery systems: therapeutic needs and formulation strategies. In Misra A., & Shahiwala, A. (Eds.). Novel Drug Delivery Technologies (pp. 183–260). Singapore: Springer. [DOI:10.1007/978-981-13-3642-3_7]
Vizziello, G., D’Amato, G., Trentadue, R., & Fanizza, G. (1993). Studio dinamico del blocco ipofisario indotto dalla triptorelina, mediante test all’estradiolo benzoato. [Estradiol benzoate test in the study of pituitary block induced by triptorelin.] Minerva Ginecologica, 45(4), 185–189. [Google Scholar] [PubMed] [PDF] [Translation]
Wagner, J. G. (1993). Pharmacokinetics for the Pharmaceutical Scientist. Lancaster/Basel: Technomic. [DOI:10.1201/9780203743652] [Google Books]
@@ -2876,7 +2887,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
The very short half-life of sublingually and buccally administered estradiol relative to other forms raises a few questions relating to its use in feminising hormone therapy. One of the most commonly asked questions on online forums is regarding which gender-affirming hormone therapy regimens might be most “effective” with respect to the feminising effects of estrogens. These include, but are not limited to, outcomes such as breast development and fat distribution.
-
In contrast to oral and trandermal estradiol, limited data exist describing the extent of feminisation with the sublingual route (Safer, 2022). A non-randomised study found that self-assessed Tanner stage after 6 months of treatment did not appear to be different in users of sublingual estradiol monotherapy as compared to users of oral estradiol plus 10 mg/day cyproterone acetate (Yaish et al., 2023a; Yaish et al., 2023b). However, since breast development itself was not measured objectively, these particular data are low-quality and prevent definitive conclusions either way about the superiority or inferiority of sublingual estradiol. The same study group reported that although there were similar increases in gynoid fat in the two arms, the oral estradiol group did show an increased amount of android fat as compared to the sublingual group (Yaish et al., 2025). On the other hand, a further complication of this study is the possible confounding by lack of concomitant antiandrogen therapy in the sublingual arm (Ruggles & Cheung, 2024; Yaish et al., 2024). Notably, progestogens like cyproterone acetate have been shown to be associated with weight gain (Lopez et al., 2016). This could explain the difference in android fat accumulation.
+
In contrast to oral and trandermal estradiol, limited data exist describing the extent of feminisation with the sublingual route (Safer, 2022). A non-randomised study found that self-assessed Tanner stage after 6 months of treatment did not appear to be different in users of sublingual estradiol monotherapy as compared to users of oral estradiol plus 10 mg/day cyproterone acetate (Yaish et al., 2023a; Yaish et al., 2023b). However, since breast development itself was not measured objectively, these particular data are low-quality and prevent definitive conclusions either way about the superiority or inferiority of sublingual estradiol. The same study group reported that although there were similar increases in gynoid fat in the two arms, the oral estradiol group did show an increased amount of android fat as compared to the sublingual group (Yaish et al., 2025). On the other hand, a further complication of this study is the possible confounding by lack of concomitant antiandrogen therapy in the sublingual arm (Ruggles & Cheung, 2024; Yaish et al., 2024). Notably, progestogens like cyproterone acetate have been shown to be associated with weight gain (Lopez et al., 2016). This could explain the difference in android fat accumulation.
Oral estradiol and other non-oral forms of estradiol (such as transdermal administration) have not been found to differ in their effects on breast development or other feminising outcomes in transfeminine people or cisgender hypogonadal girls (Rosenfield et al., 2005; Shah et al., 2014; Klaver et al., 2018; de Blok et al., 2021, Tebbens et al., 2022). In consideration of this, differences in efficacy might not be expected for sublingual estradiol either. However, the use of supraphysiological doses of estrogens from the onset of therapy may stunt breast development and reduce final breast size in transfeminine people (Boogers et al., 2025). Because the use of sublingual estradiol results in estradiol concentrations that routinely achieve the supratherapeutic range, it is possible that this could have deleterious effects on breast development.
@@ -2886,7 +2897,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Another question that might be raised by the short half-life of sublingual estradiol is how it might compare to more conventional routes of administration in terms of its ability to suppress testosterone and other androgens.
-
Estrogens were first characterised for their use as antigonadotrophic antiandrogens in the 1940s in the form of oral synthetic estrogens, namely diethylstilbestrol (DES), to treat men with prostate cancer (Huggins & Hodges, 1941). Estrogens given in the form of oral ethinylestradiol (EE), long-acting estradiol esters, such as polyestradiol phosphate, and transdermal estradiol patches have been studied. Their efficacy for this indication is well established (Stege et al., 1996; Kohli, 2006; Sciarra et al., 2015). As data are more limited for testosterone suppression with estrogens in transfeminine people, these data are valuable for informing transfeminine hormone therapy. Since sublingual estradiol has never been used to treat prostatic cancer, no such data exist to show the ability of sublingual estradiol in this capacity.
+
Estrogens were first characterised for their use as antigonadotrophic antiandrogens in the 1940s in the form of oral synthetic estrogens, namely diethylstilbestrol (DES), to treat men with prostate cancer (Huggins & Hodges, 1941). Estrogens given in the form of oral ethinylestradiol (EE), long-acting estradiol esters, such as polyestradiol phosphate, and transdermal estradiol patches have been studied. Their efficacy for this indication is well established (Stege et al., 1996; Kohli, 2006; Sciarra et al., 2015). As data are more limited for testosterone suppression with estrogens in transfeminine people, these data are valuable for informing transfeminine hormone therapy. Since sublingual estradiol has never been used to treat prostatic cancer, no such data exist to show the ability of sublingual estradiol in this capacity.
Some studies have found that physiologic levels of estradiol (i.e., 100–200 pg/mL [367–734 pmol/L]) or slightly higher from non-sublingual estradiol alone result in rapid and near complete, if not complete, suppression of testosterone levels to the female range in many transfeminine people (Leinung, Feustel, & Joseph, 2018; Pappas et al., 2020; Misakian et al., 2025). Additionally, the Prostate Adenocarcinoma TransCutaneous Hormones (PATCH) study, a multicentre randomised controlled trial in the United Kingdom, showed that sustained median estradiol levels of between 215 to 250 pg/mL (789–918 pmol/L) from transdermal patches were similarly effective (~95%) to GnRH analogues in reducing testosterone levels to the castrate range (<50 ng/dL [<1.7 nmol/L]) (Langley et al., 2021). However, because sublingual estradiol differs in its pharmacokinetics to other forms of estradiol, it is plausible that this route of administration might result in sub-par suppression at doses with similar concentrations of estradiol.
@@ -2942,19 +2953,19 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Ahokas, A., Kaukoranta, J., & Aito, M. (1999). Effect of oestradiol on postpartum depression. Psychopharmacology, 146(1), 108–110. [DOI:10.1007/s002130051095]
Ahokas, A., Kaukoranta, J., Wahlbeck, K., & Aito, M. (2001). Estrogen deficiency in severe postpartum depression: successful treatment with sublingual physiologic 17β-estradiol: a preliminary study. Journal of Clinical Psychiatry, 62(5), 332–336. [DOI:10.4088/jcp.v62n0504]
Anderson, G. L., Limacher, M., Assaf, A. R., Bassford, T., Beresford, S. A., Black, H., Bonds, D., Brunner, R., Brzyski, R., Caan, B., Chlebowski, R., Curb, D., Gass, M., Hays, J., Heiss, G., Hendrix, S., Howard, B. V., Hsia, J., Hubbell, A., Jackson, R., … & Women’s Health Initiative Steering Committee. (2004). Effects of Conjugated Equine Estrogen in Postmenopausal Women With Hysterectomy: The Women’s Health Initiative Randomized Controlled Trial. JAMA, 291(14), 1701–1712. [DOI:10.1001/jama.291.14.1701]
-
Bar, O. S., Yaish, I., Barzilai, M., Greenman, Y., Gindis, G., Moshe, Y., & Tordjman, K. (2024, May). Low dose sublingual estradiol decreases protein s, generating a potentially pro-thrombotic state: interim results of a controlled prospective pilot study of treatment-naive trans women. In Endocrine Abstracts (Vol. 99). Bioscientifica. [DOI:10.1089/trgh.2024.0105]
+
Bar, O. S., Yaish, I., Barzilai, M., Greenman, Y., Gindis, G., Moshe, Y., & Tordjman, K. (2024). Low dose sublingual estradiol decreases protein s, generating a potentially pro-thrombotic state: interim results of a controlled prospective pilot study of treatment-naive trans women. Endocrine Abstracts, 99 [26th European Congress of Endocrinology, Stockholm, Sweden, 11 May 2024 – 14 May 2024], EP592. [DOI:10.1530/endoabs.99.ep592]
Bartlett, J. A., & van der Voort Maarschalk, K. (2012). Understanding the oral mucosal absorption and resulting clinical pharmacokinetics of asenapine. AAPS Pharmscitech, 13(4), 1110–1115. [DOI:10.1208/s12249-012-9839-7]
-
Boogers, L. S., Sardo Infirri, S. A., Bouchareb, A., Dijkman, B. A., Helder, D., de Blok, C. J., … & Hannema, S. E. (2025). Variations in Volume: Breast Size in Trans Women in Relation to Timing of Testosterone Suppression. The Journal of Clinical Endocrinology & Metabolism, 110(5), 1404-1410. [DOI:10.1210/clinem/dgae573]
+
Boogers, L. S., Sardo Infirri, S. A., Bouchareb, A., Dijkman, B. A. M., Helder, D., de Blok, C. J. M., Liberton, N. P. T. J., den Heijer, M., van Trotsenburg, A. S. P., Dreijerink, K. M. A., Wiepjes, C. M., & Hannema, S. E. (2025). Variations in Volume: Breast Size in Trans Women in Relation to Timing of Testosterone Suppression. The Journal of Clinical Endocrinology & Metabolism, 110(5), 1404–1410. [DOI:10.1210/clinem/dgae573]
Burnier, A. M., Martin, P. L., Yen, S. S., & Brooks, P. (1981). Sublingual absorption of micronized 17β-estradiol. American Journal of Obstetrics and Gynecology, 140(2), 146–150. [DOI:10.1016/0002-9378(81)90101-0]
-
Casper, R. F., & Yen, S. S. (1981). Rapid absorption of micronized estradiol-17β following sublingual administration. Obstetrics and Gynecology, 57(1), 62–64. [Google Scholar] [PubMed] [URL] [PDF]
+
Casper, R. F., & Yen, S. S. (1981). Rapid absorption of micronized estradiol-17β following sublingual administration. Obstetrics and Gynecology, 57(1), 62–64. [Google Scholar] [PubMed] [URL] [PDF]
Chandrasekhara, D. S., Ali, V., Prost, H. M., & Nader-Estekhari, S. (2002). Buccal estrogen in toothpaste study: systemic absorption of estradiol in postmenopausal or surgically menopausal women when administered as a component in toothpaste. Fertility and Sterility, 78(Suppl 1), S98–S98 (O-258). [DOI:10.1016/S0015-0282(02)03639-7]
Cheung, A. S., Wynne, K., Erasmus, J., Murray, S., & Zajac, J. D. (2019). Position statement on the hormonal management of adult transgender and gender diverse individuals. Medical Journal of Australia, 211(3), 127–133. [DOI:10.5694/mja2.50259]
Cirrincione, L. R., Winston McPherson, G., Rongitsch, J., Sadilkova, K., Drees, J. C., Krasowski, M. D., Dickerson, J. A., & Greene, D. N. (2021). Sublingual Estradiol Is Associated with Higher Estrone Concentrations than Transdermal or Injectable Preparations in Transgender Women and Gender Nonbinary Adults. LGBT Health, 8(2), 125–132. [DOI:10.1089/lgbt.2020.0249]
-
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., De Vries, A. L., Deutsch, M. B., … & Arcelus, J. (2022). Standards of care for the health of transgender and gender diverse people, version 8. International Journal of Transgender Health, 23(sup1), S1-S259. [DOI:10.1080/26895269.2022.2100644]
+
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Collaborative Group on Hormonal Factors in Breast Cancer. (2019). Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. The Lancet, 394(10204), 1159–1168. [DOI:10.1016/S0140-6736(19)31709-X]
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C., Fields, M., Gray, T., Guo, Z., Nicol, G., & Baranski, T. (2023). Effectiveness and Safety of Different Estradiol Regimens in Transgender Women (TREAT Study): Protocol for a Randomized Controlled Trial. JMIR Research Protocols, 12, e53092. [DOI:10.2196/53092]
-
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C. J., Fields, M. E., … & Baranski, T. (2024). Effectiveness and safety of different estradiol regimens in transgender females: a randomized controlled trial. Journal of the Endocrine Society, 8(8), bvae108. [DOI: 10.1210/jendso/bvae108]
-
Cummings, S. R., Eckert, S., Krueger, K. A., Grady, D., Powles, T. J., Cauley, J. A., … & Jordan, V. C. (1999). The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. JAMA, 281(23), 2189-2197. [DOI:10.1001/jama.281.23.2189]
+
Cortez, S., Moog, D., Lewis, C., Williams, K., Herrick, C. J., Fields, M. E., Gray, T., Guo, Z., Nicol, G., & Baranski, T. (2024). Effectiveness and safety of different estradiol regimens in transgender females: a randomized controlled trial. Journal of the Endocrine Society, 8(8), bvae108. [DOI:10.1210/jendso/bvae108]
+
Cummings, S. R., Eckert, S., Krueger, K. A., Grady, D., Powles, T. J., Cauley, J. A., Norton, L., Nickelsen, T., Bjarnason, N. H., Morrow, M., Lippman, M. E., Black, D., Glusman, J. E., Costa, A., & Jordan, V. C. (1999). The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. JAMA, 281(23), 2189–2197. [DOI:10.1001/jama.281.23.2189]
de Blok, C., Dijkman, B., Wiepjes, C. M., Staphorsius, A. S., Timmermans, F. W., Smit, J. M., Dreijerink, K., & den Heijer, M. (2021). Sustained Breast Development and Breast Anthropometric Changes in 3 Years of Gender-Affirming Hormone Treatment. The Journal of Clinical Endocrinology & Metabolism, 106(2), e782–e790. [DOI:10.1210/clinem/dgaa841]
Deutsch, M. B., Bhakri, V., & Kubicek, K. (2015). Effects of cross-sex hormone treatment on transgender women and men. Obstetrics and Gynecology, 125(3), 605–610. [DOI:10.1097/AOG.0000000000000692]
Devissaguet, J. P., Brion, N., Lhote, O., & Deloffre, P. (1999). Pulsed estrogen therapy: pharmacokinetics of intranasal 17-beta-estradiol (S21400) in postmenopausal women and comparison with oral and transdermal formulations. European Journal of Drug Metabolism and Pharmacokinetics, 24(3), 265–271. [DOI:10.1007/BF03190030]
@@ -2964,14 +2975,14 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Gass, M. S., Rebar, R. W., Cuffie-Jackson, C., Cedars, M. I., Lobo, R. A., Shoupe, D., Judd, H. L., Buyalos, R. P., & Clisham, P. R. (2004). A short study in the treatment of hot flashes with buccal administration of 17-β estradiol. Maturitas, 49(2), 140–147. [DOI:10.1016/j.maturitas.2003.12.004]
Getahun, D., Nash, R., Flanders, W. D., Baird, T. C., Becerra-Culqui, T. A., Cromwell, L., Hunkeler, E., Lash, T. L., Millman, A., Quinn, V. P., Robinson, B., Roblin, D., Silverberg, M. J., Safer, J., Slovis, J., Tangpricha, V., & Goodman, M. (2018). Cross-sex hormones and acute cardiovascular events in transgender persons: a cohort study. Annals of Internal Medicine, 169(4), 205–213. [DOI:10.7326/M17-2785]
Gindis, G., Yaish, I., Greenman, Y., Moshe, Y., Arbiv, M., Buch, A., Sofer, Y., Shefer, G., & Tordjman, K. (May 2023). Sublingual estradiol only, offers no apparent advantage over combined oral estradiol and cyproterone acetate, for Gender Affirming Hormone Therapy (GAHT) of treatment-naive transwomen: Results of a prospective pilot study. Endocrine Abstracts, 90 [25th European Congress of Endocrinology 2023, 13–16 May 2023, Istanbul, Turkey], 274–274 (abstract no. P182). [DOI:10.1530/endoabs.90.p182] [PDF]
-
Grock, S., Patel, R., & Ahern, S. (2024). Hormone Therapy for Transgender and Gender-Diverse Patients. In Genital Gender Affirming Surgery: An Illustrated Guide and Video Atlas (pp. 33-49). Cham: Springer Nature Switzerland. [DOI:10.1007/978-3-031-69997-9_4]
+
Grock, S., Patel, R., & Ahern, S. (2024). Hormone Therapy for Transgender and Gender-Diverse Patients. In Genital Gender Affirming Surgery: An Illustrated Guide and Video Atlas (pp. 33–49). Cham: Springer Nature Switzerland. [DOI:10.1007/978-3-031-69997-9_4]
Haupt, C., Henke, M., Kutschmar, A., Hauser, B., Baldinger, S., Saenz, S. R., & Schreiber, G. (2020). Antiandrogen or estradiol treatment or both during hormone therapy in transitioning transgender women. Cochrane Database of Systematic Reviews, 11(11), CD013138. [DOI:10.1002/14651858.CD013138.pub2]
Hoon, T. J., Dawood, M. Y., Khan‐Dawood, F. S., Ramos, J., & Batenhorst, R. L. (1993). Bioequivalence of a 17β‐Estradiol Hydroxypropyl‐β‐Cyclodextrin Complex in Postmenopausal Women. The Journal of Clinical Pharmacology, 33(11), 1116–1121. [DOI:10.1002/j.1552-4604.1993.tb01949.x]
-
Huggins, C., & Hodges, C. V. (1941). Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Research, 1(4), 293–297. [DOI:10.3322/canjclin.22.4.232]
+
Huggins, C., & Hodges, C. V. (1941). Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Research, 1(4), 293–297. [URL]
Jain, J., Kwan, D., & Forcier, M. (2019). Medroxyprogesterone acetate in Gender-Affirming therapy for Transwomen: results from a retrospective study. The Journal of Clinical Endocrinology & Metabolism, 104(11), 5148–5156. [DOI:10.1210/jc.2018-02253]
Jalal, E., & Baldwin, C. (2023). Supratherapeutic Estrogen Levels in Transgender Women Likely From Sublingual Estradiol. Journal of the Endocrine Society, 7(Suppl 1) [ENDO 2023 Abstracts Annual Meeting of the Endocrine Society], A1095–A1096 (abstract no. SAT391/bvad114.2062). [DOI:10.1210/jendso/bvad114.2062] [PDF]
Jin, J., Sklar, G. E., Oh, V. M. S., & Li, S. C. (2008). Factors affecting therapeutic compliance: A review from the patient’s perspective. Therapeutics and Clinical Risk Management, 4(1), 269–286. [DOI:10.2147/TCRM.S1458]
-
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2025). Comparison of estrone/estradiol ratio and levels in transfeminine individuals on different routes of estradiol. Transgender Health, 10(3), 261-268. [DOI:10.1089/trgh.2023.0138]
+
Kariyawasam, N. M., Ahmad, T., Sarma, S., & Fung, R. (2025). Comparison of estrone/estradiol ratio and levels in transfeminine individuals on different routes of estradiol. Transgender Health, 10(3), 261–268. [DOI:10.1089/trgh.2023.0138]
Klaver, M., de Blok, C. J. M., Wiepjes, C. M., Nota, N. M., Dekker, M. J., de Mutsert, R., Schreiner, T., Fisher, A. D., T’Sjoen, G., & den Heijer, M. (2018). Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. European Journal of Endocrinology, 178(2), 163–171. [DOI:10.1530/EJE-17-0496]
Kohli, M. (2006). Phase II study of transdermal estradiol in androgen‐independent prostate carcinoma. Cancer, 106(1), 234–235. [DOI:10.1002/cncr.21528]
Kuhl, H. (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
@@ -2982,9 +2993,9 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Lim, H. H., Jang, Y. H., Choi, G. Y., Lee, J. J., & Lee, E. S. (2019). Gender affirmative care of transgender people: a single center’s experience in Korea. Obstetrics & Gynecology Science, 62(1), 46–55. [DOI:10.5468/ogs.2019.62.1.46]
Liu, B., Beral, V., Balkwill, A., Green, J., Sweetland, S., & Reeves, G. (2008). Gallbladder disease and use of transdermal versus oral hormone replacement therapy in postmenopausal women: prospective cohort study. BMJ, 337. [DOI:10.1136/bmj.a386]
Lobo, R. A. (1987). Absorption and metabolic effects of different types of estrogens and progestogens. Obstetrics and Gynecology Clinics of North America, 14(1), 143–167. [PubMed] [DOI:10.1016/S0889-8545(21)00577-5] [URL]
-
Lopez, L. M., Ramesh, S., Chen, M., Edelman, A., Otterness, C., Trussell, J., & Helmerhorst, F. M. (2016). Progestin‐only contraceptives: effects on weight. Cochrane Database of Systematic Reviews, (8). [DOI:10.1002/14651858.CD008815.pub4]
+
Lopez, L. M., Ramesh, S., Chen, M., Edelman, A., Otterness, C., Trussell, J., & Helmerhorst, F. M. (2016). Progestin-only contraceptives: effects on weight. Cochrane Database of Systematic Reviews, 2016(8). [DOI:10.1002/14651858.cd008815.pub4]
Mikkola, A., Aro, J., Rannikko, S., Oksanen, H., Ruutu, M., & Finnprostate Group. (2005). Cardiovascular complications in patients with advanced prostatic cancer treated by means of orchiectomy or polyestradiol phosphate. Scandinavian Journal of Urology and Nephrology, 39(4), 294–300. [DOI:10.1080/00365590510031228]
-
Misakian, A. L., Ariel, D., Sullivan, E. A., Singh, G., Loeb, D., Strickland, T., … & Hamnvik, O. P. R. (2025). Injectable estradiol monotherapy effectively suppresses testosterone in gender-affirming hormone therapy. Endocrine Practice. [DOI:10.1016/j.eprac.2025.07.002]
+
Misakian, A. L., Ariel, D., Sullivan, E. A., Singh, G., Loeb, D., Strickland, T., Iwamoto, S. J., Rothman, M. S., Botzheim, B., Liang, J. W., Kelley, C., & Hamnvik, O. R. (2025). Injectable Estradiol Monotherapy Effectively Suppresses Testosterone in Gender-Affirming Hormone Therapy. Endocrine Practice, 31(11), 1462–1469. [DOI:10.1016/j.eprac.2025.07.002]
Mishra, S. R., Chung, H. F., Waller, M., & Mishra, G. D. (2021). Duration of estrogen exposure during reproductive years, age at menarche and age at Menopause, and risk of cardiovascular disease events, all‐cause and cardiovascular mortality: a systematic review and meta‐analysis. BJOG: An International Journal of Obstetrics & Gynaecology, 128(5), 809–821. [DOI:10.1111/1471-0528.16524]
Morimont, L., Dogné, J. M., & Douxfils, J. (2020). Letter to the Editors-in-Chief in response to the article of Abou-Ismail, et al. entitled “Estrogen and thrombosis: A bench to bedside review” (Thrombosis Research 192 (2020) 40–51). Thrombosis Research, 193, 221–223. [DOI:10.1016/j.thromres.2020.08.006]
Oliver-Williams, C., Glisic, M., Shahzad, S., Brown, E., Pellegrino Baena, C., Chadni, M., Chowdhury, R., Franco, O. H., & Muka, T. (2019). The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Human Reproduction Update, 25(2), 257–271. [DOI:10.1093/humupd/dmy039]
@@ -2997,32 +3008,32 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Rosenfield, R. L., Devine, N., Hunold, J. J., Mauras, N., Moshang Jr, T., & Root, A. W. (2005). Salutary effects of combining early very low-dose systemic estradiol with growth hormone therapy in girls with Turner syndrome. The Journal of Clinical Endocrinology & Metabolism, 90(12), 6424–6430. [DOI:10.1210/jc.2005-1081]
Rovinski, D., Ramos, R. B., Fighera, T. M., Casanova, G. K., & Spritzer, P. M. (2018). Risk of venous thromboembolism events in postmenopausal women using oral versus non-oral hormone therapy: a systematic review and meta-analysis. Thrombosis Research, 168, 83–95. [DOI:10.1016/j.thromres.2018.06.014]
Ruggles, T., & Cheung, A. S. (2024). Re:” Sublingual Estradiol Offers No Apparent Advantage over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study” by Yaish et al. Transgender Health, 9(5), 459. [DOI:10.1089/trgh.2024.0048]
-
Salakphet, T., Mattawanon, N., Manojai, N., Muangmool, T., & Tangpricha, V. (2022). Hormone concentrations in transgender women who self-prescribe gender affirming hormone therapy: a retrospective study. The Journal of Sexual Medicine, 19(5), 864-871. [DOI:10.1016/j.jsxm.2022.02.023]
Safer, J. D. (2022). Are the Pharmacokinetics of Sublingual Estradiol Superior or Inferior to Those of Oral Estradiol? Endocrine Practice, 28(3), 351–352. [DOI:10.1016/j.eprac.2021.12.018]
+
Salakphet, T., Mattawanon, N., Manojai, N., Muangmool, T., & Tangpricha, V. (2022). Hormone concentrations in transgender women who self-prescribe gender affirming hormone therapy: a retrospective study. The Journal of Sexual Medicine, 19(5), 864–871. [DOI:10.1016/j.jsxm.2022.02.023]
Sarvaideo, J., Doll, E., & Tangpricha, V. (2022). More Studies Are Needed to Establish the Safety and Efficacy of Sublingual Estradiol in Transgender Women. Endocrine Practice, 28(3), 353–354. [DOI:10.1016/j.eprac.2022.01.004]
Sciarra, A., Gentile, V., Cattarino, S., Gentilucci, A., Alfarone, A., D’Eramo, G., & Salciccia, S. (2015). Oral ethinylestradiol in castration‐resistant prostate cancer: a 10‐year experience. International Journal of Urology, 22(1), 98–103. [DOI:10.1111/iju.12613]
Serhal, P., & Craft, I. (1989). Oocyte donation in 61 patients. The Lancet, 333(8648), 1185–1187. [DOI:10.1016/S0140-6736(89)92762-1]
Shah, S., Forghani, N., Durham, E., & Neely, E. K. (2014). A randomized trial of transdermal and oral estrogen therapy in adolescent girls with hypogonadism. International Journal of Pediatric Endocrinology, 2014(1), 12. [DOI:10.1186/1687-9856-2014-12]
-
Slack, D. J., Ioschpe, A. D. V., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., … & Safer, J. D. (2025). Examining the influence of the route of administration and dose of estradiol on serum estradiol and testosterone levels in feminizing gender-affirming hormone therapy. Endocrine Practice, 31(1), 19-27. [DOI:10.1016/j.eprac.2024.10.002]
-
SoRelle, J. A., Jiao, R., Gao, E., Veazey, J., Frame, I., Quinn, A. M., … & Patel, K. (2019). Impact of hormone therapy on laboratory values in transgender patients. Clinical Chemistry, 65(1), 170-179. [DOI:10.1373/clinchem.2018.292730]
+
Slack, D. J., Di Via Ioschpe, A., Saturno, M., Kihuwa-Mani, S., Amakiri, U. O., Guerra, D., Karim, S., & Safer, J. D. (2025). Examining the Influence of the Route of Administration and Dose of Estradiol on Serum Estradiol and Testosterone Levels in Feminizing Gender-Affirming Hormone Therapy. Endocrine Practice, 31(1), 19–27. [DOI:10.1016/j.eprac.2024.10.002]
+
SoRelle, J. A., Jiao, R., Gao, E., Veazey, J., Frame, I., Quinn, A. M., Day, P., Pagels, P., Gimpel, N., & Patel, K. (2019). Impact of Hormone Therapy on Laboratory Values in Transgender Patients. Clinical Chemistry, 65(1), 170–179. [DOI:10.1373/clinchem.2018.292730]
Stanczyk, F. Z., Archer, D. F., & Bhavnani, B. R. (2013). Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception, 87(6), 706–727. [DOI:10.1016/j.contraception.2012.12.011]
Stege, R., Gunnarsson, P. O., Johansson, C. J., Olsson, P., Pousette, Å., & Carlström, K. (1996). Pharmacokinetics and testosterone suppression of a single dose of polyestradiol phosphate (Estradurin®) in prostatic cancer patients. The Prostate, 28(5), 307–310. [DOI:10.1002/(SICI)1097-0045(199605)28:5<307::AID-PROS6>3.0.CO;2-8]
-
Sudhakar, D., Huang, Z., Zietkowski, M., Powell, N., & Fisher, A. R. (2023). Feminizing gender‐affirming hormone therapy for the transgender and gender diverse population: An overview of treatment modality, monitoring, and risks. Neurourology and Urodynamics, 42(5), 903-920. [DOI:10.1002/nau.25097]
-
Tebbens, M., Heijboer, A. C., T’Sjoen, G., Bisschop, P. H., & den Heijer, M. (2022). The role of estrone in feminizing hormone treatment. The Journal of Clinical Endocrinology & Metabolism, 107(2), 458-466. [DOI:10.1210/clinem/dgab741]
+
Sudhakar, D., Huang, Z., Zietkowski, M., Powell, N., & Fisher, A. R. (2023). Feminizing gender‐affirming hormone therapy for the transgender and gender diverse population: An overview of treatment modality, monitoring, and risks. Neurourology and Urodynamics, 42(5), 903–920. [DOI:10.1002/nau.25097]
+
Tebbens, M., Heijboer, A. C., T’Sjoen, G., Bisschop, P. H., & den Heijer, M. (2022). The role of estrone in feminizing hormone treatment. The Journal of Clinical Endocrinology & Metabolism, 107(2), 458–466. [DOI:10.1210/clinem/dgab741]
T’Sjoen, G., Arcelus, J., De Vries, A. L., Fisher, A. D., Nieder, T. O., Özer, M., & Motmans, J. (2020). European Society for Sexual Medicine position statement “assessment and hormonal management in adolescent and adult trans people, with attention for sexual function and satisfaction”. The Journal of Sexual Medicine, 17(4), 570–584. [DOI:10.1016/j.jsxm.2020.01.012]
-
Totaro, M., Palazzi, S., Castellini, C., Parisi, A., D’Amato, F., Tienforti, D., … & Barbonetti, A. (2021). Risk of venous thromboembolism in transgender people undergoing hormone feminizing therapy: a prevalence meta-analysis and meta-regression study. Frontiers In Endocrinology, 12, 741866. [DOI:10.3389/fendo.2021.741866]
Thurman, A., Kimble, T., Hall, P., Schwartz, J. L., & Archer, D. F. (2013). Medroxyprogesterone acetate and estradiol cypionate injectable suspension (Cyclofem) monthly contraceptive injection: steady-state pharmacokinetics. Contraception, 87(6), 738–743. [DOI:10.1016/j.contraception.2012.11.010]
Toh, M. R., Teo, V., Kwan, Y. H., Raaj, S., Tan, S. Y. D., & Tan, J. Z. Y. (2014). Association between number of doses per day, number of medications and patient’s non-compliance, and frequency of readmissions in a multi-ethnic Asian population. Preventive Medicine Reports, 1, 43–47. [DOI:10.1016/j.pmedr.2014.10.001]
+
Totaro, M., Palazzi, S., Castellini, C., Parisi, A., D’Amato, F., Tienforti, D., Baroni, M. G., Francavilla, S., & Barbonetti, A. (2021). Risk of Venous Thromboembolism in Transgender People Undergoing Hormone Feminizing Therapy: A Prevalence Meta-Analysis and Meta-Regression Study. Frontiers in Endocrinology, 12, 741866. [DOI:10.3389/fendo.2021.741866]
Tsai, C. C., & Yen, S. S. C. (1971). Acute effects of intravenous infusion of 17β-estradiol on gonadotropin release in pre-and post-menopausal women. The Journal of Clinical Endocrinology & Metabolism, 32(6), 766–771. [DOI:10.1210/jcem-32-6-766]
Verdonk, S. J., Vesper, H. W., Martens, F., Sluss, P. M., Hillebrand, J. J., & Heijboer, A. C. (2019). Estradiol reference intervals in women during the menstrual cycle, postmenopausal women and men using an LC-MS/MS method. Clinica Chimica Acta, 495, 198–204. [DOI:10.1016/j.cca.2019.04.062]
Wiegratz, I., Fink, T., Rohr, U. D., Lang, E., Leukel, P., & Kuhl, H. (2001). Überkreuz-Vergleich der Pharmakokinetik von Estradiol unter der Hormonsubstitution mit Estradiolvalerat oder mikronisiertem Estradiol. [Cross-over comparison of the pharmacokinetics of estradiol during hormone replacement therapy with estradiol valerate or micronized estradiol.] Zentralblatt für Gynäkologie, 123(9), 505–512. [PubMed] [DOI:10.1055/s-2001-18223]
Wisner, K. L., Sit, D. K., Moses-Kolko, E. L., Driscoll, K. E., Prairie, B. A., Stika, C. S., Eng, H. F., Dills, J. L., Luther, J. F., & Wisniewski, S. R. (2015). Transdermal estradiol treatment for postpartum depression: a pilot randomized trial. Journal of Clinical Psychopharmacology, 35(4), 389–395. [DOI:10.1097/JCP.0000000000000351]
Wren, B. G., Day, R. O., McLachlan, A. J., & Williams, K. M. (2003). Pharmacokinetics of estradiol, progesterone, testosterone and dehydroepiandrosterone after transbuccal administration to postmenopausal women. Climacteric, 6(2), 104–111. [DOI:10.1080/cmt.6.2.104.111]
-
Yaish, I., Gindis, G., Greenman, Y., Shefer, G., Buch, A., Arbiv, M., Moshe, Y., Sofer, Y., & Tordjman, K. M. (2023). Sublingual Estradiol Only, Compared To Combined Oral Estradiol And Cyproterone Acetate,Offers No Apparent Advantage For Gender Affirming Hormone Therapy (GHAT), In Treatment Naïve Transwomen: Results Of A Prospective Pilot Study. Journal of the Endocrine Society, 7(Suppl 1) [ENDO 2023 Abstracts Annual Meeting of the Endocrine Society], A1104–A1105 (abstract no. SAT409/bvad114.2080). [DOI:10.1210/jendso/bvad114.2080] [PDF]
Yaish, I., Gindis, G., Greenman, Y., Moshe, Y., Arbiv, M., Buch, A., Sofer, Y., Shefer, G., & Tordjman, K. (2023). Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study. Transgender Health, 8(6), 485–493. [DOI:10.1089/trgh.2023.0022]
-
Yaish, I., Greenman, Y., Sofer, Y., & Tordjman, K. M. (2024). Response to Ruggles and Cheung, Re:” Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study”. Transgender Health, 9(5), 460-461. [DOI:10.1089/trgh.2024.0105]
-
Yaish, I., Buch, A., Gindis, G., Sofer, Y., Arbiv, M., Moshe, Y., … & Tordjman, K. (2025). Early body composition changes in trans women on low-dose estradiol: comparing oral vs sublingual administration using dual energy absorptiometry and bioelectrical impedance analysis. The Journal of Sexual Medicine, 22(4), 625-635. [DOI:10.1093/jsxmed/qdaf005]
+
Yaish, I., Gindis, G., Greenman, Y., Shefer, G., Buch, A., Arbiv, M., Moshe, Y., Sofer, Y., & Tordjman, K. M. (2023). Sublingual Estradiol Only, Compared To Combined Oral Estradiol And Cyproterone Acetate,Offers No Apparent Advantage For Gender Affirming Hormone Therapy (GHAT), In Treatment Naïve Transwomen: Results Of A Prospective Pilot Study. Journal of the Endocrine Society, 7(Suppl 1) [ENDO 2023 Abstracts Annual Meeting of the Endocrine Society], A1104–A1105 (abstract no. SAT409/bvad114.2080). [DOI:10.1210/jendso/bvad114.2080] [PDF]
+
Yaish, I., Greenman, Y., Sofer, Y., & Tordjman, K. M. (2024). Response to Ruggles and Cheung, Re:” Sublingual Estradiol Offers No Apparent Advantage Over Combined Oral Estradiol and Cyproterone Acetate for Gender-Affirming Hormone Therapy of Treatment-Naive Trans Women: Results of a Prospective Pilot Study”. Transgender Health, 9(5), 460–461. [DOI:10.1089/trgh.2024.0105]
+
Yaish, I., Buch, A., Gindis, G., Sofer, Y., Arbiv, M., Moshe, Y., Grenman, Y., & Tordjman, K. (2025). Early body composition changes in trans women on low-dose estradiol: comparing oral vs sublingual administration using dual energy absorptiometry and bioelectrical impedance analysis. The Journal of Sexual Medicine, 22(4), 625–635. [DOI:10.1093/jsxmed/qdaf005]
]]>{"first_name"=>"Sam", "last_name"=>"S.", "author-link"=>"/about/#sam", "articles-link"=>"/articles-by-author/sam/"}Clinical Guidelines with Information on Transfeminine Hormone Therapy2020-11-20T10:00:00-08:002024-11-17T00:00:00-08:00https://transfemscience.org/articles/transfem-hormone-guidelinesClinical Guidelines with Information on Transfeminine Hormone Therapy
@@ -3039,7 +3050,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Clinicians use clinical practice guidelines (CPGs) to learn about and guide themselves in administering medical care for different indications. Clinical practice guidelines review and summarize the available scientific literature and research in a given medical area. They allow clinicians to competently administer care without necessarily having to delve into and develop their understanding via the primary scientific literature. Literature reviews can serve a similar function. However, clinical practice guidelines are generally more substantial and are more founded in evidence-based medicine. They are also regularly updated. Clinical practice guidelines are developed and maintained by clinical organizations and societies, universities, government agencies, and sometimes even large medical clinics. They may be international/locationless or oftentimes region-specific.
-
There are many clinical practice guidelines for transgender medicine (for review, Deutsch, Radix, & Reisner, 2016; Radix, 2019; Radix, 2019; UpToDate; Bewley et al., 2021; Dahlen et al., 2021; Ziegler, Carroll, & Charnish, 2021). These guidelines discuss topics such as psychotherapy, hormone therapy, voice therapy, and surgical management of transgender people, among others. In addition to educating and guiding clinicians, transgender clinical practice guidelines are useful materials for transgender people as they can help to inform them about their care.
+
There are many clinical practice guidelines for transgender medicine (for review, Deutsch, Radix, & Reisner, 2016; Radix, 2019; Radix, 2019; UpToDate; Bewley et al., 2021; Dahlen et al., 2021; Ziegler, Carroll, & Charnish, 2021). These guidelines discuss topics such as psychotherapy, hormone therapy, voice therapy, and surgical management of transgender people, among others. In addition to educating and guiding clinicians, transgender clinical practice guidelines are useful materials for transgender people as they can help to inform them about their care.
Majumder et al. / Integrated Diabetes and Endocrine Academy (IDEA) [India]
2020
Published article
@@ -3403,10 +3414,10 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Coleman, E., Radix, A. E., Bouman, W. P., Brown, G. R., de Vries, A. L., Deutsch, M. B., Ettner, R., Fraser, L., Goodman, M., Green, J., Hancock, A. B., Johnson, T. W., Karasic, D. H., Knudson, G. A., Leibowitz, S. F., Meyer-Bahlburg, H. F., Monstrey, S. J., Motmans, J., Nahata, L., … & Arcelus, J. (2022). [World Professional Association for Transgender Health (WPATH)] Standards of Care for the Health of Transgender and Gender Diverse People, Version 8. International Journal of Transgender Health, 23(Suppl 1), S1–S259. [DOI:10.1080/26895269.2022.2100644] [URL] [PDF]
Cundill, P., Wong, P., Cheung, A., & Brownhill, A. (2020). Hormone Therapy Prescribing Guide for General Practitioners working with Trans, Gender Diverse and Non-Binary Patients [Version 3.0]. Australia: Equinox Gender Diverse Health Centre/Thorne Harbour Health. [URL] [PDF]
Dahl, M., Feldman, J. L., Goldberg, J., & Jaberi, A. (2015). Endocrine Therapy for Transgender Adults in British Columbia: Suggested Guidelines. Physical Aspects of Transgender Endocrine Therapy. Vancouver: Vancouver Coastal Health. [Google Scholar] [PDF]
-
Dahlen, S., Connolly, D., Arif, I., Junejo, M. H., Bewley, S., & Meads, C. (2021). International Clinical Practice Guidelines for Gender Minority/Trans People: Systematic Review and Quality Assessment. BMJ Open, 11(4), e048943. [DOI:10.1136/bmjopen-2021-048943]
+
Dahlen, S., Connolly, D., Arif, I., Junejo, M. H., Bewley, S., & Meads, C. (2021). International Clinical Practice Guidelines for Gender Minority/Trans People: Systematic Review and Quality Assessment. BMJ Open, 11(4), e048943. [DOI:10.1136/bmjopen-2021-048943]
Davidson, A., Franicevich, J., Freeman, M., Lin, R., Martinez, L., Monihan, M., Porch, M., Samuel, L., Stukalin, R., Vormohr, J., & Zevin, B. (2013). Protocols for Hormonal Reassignment of Gender. San Francisco: San Francisco Department of Public Health/Tom Waddell Health Center. [Google Scholar] [PDF]
Deutsch, M. B. (Ed.). (2016). Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People, 2nd Edition. San Francisco: University of California, San Francisco/UCSF Transgender Care. [URL] [PDF]
-
Deutsch, M. B., Radix, A., & Reisner, S. (2016). What’s in a Guideline? Developing Collaborative and Sound Research Designs that Substantiate Best Practice Recommendations for Transgender Health Care. AMA Journal of Ethics, 18(11), 1098–1106. [DOI:10.1001/journalofethics.2016.18.11.stas1-1611]
+
Deutsch, M. B., Radix, A., & Reisner, S. (2016). What’s in a Guideline? Developing Collaborative and Sound Research Designs that Substantiate Best Practice Recommendations for Transgender Health Care. AMA Journal of Ethics, 18(11), 1098–1106. [DOI:10.1001/journalofethics.2016.18.11.stas1-1611]
Feldman, J., & Safer, J. (2009). Hormone Therapy in Adults: Suggested Revisions to the Sixth Version of the Standards of Care. International Journal of Transgenderism, 11(3), 146–182. [DOI:10.1080/15532730903383757]
Fisher, A. D., Senofonte, G., Cocchetti, C., Guercio, G., Lingiardi, V., Meriggiola, M. C., Mosconi, M., Motta, G., Ristori, J., Speranza, A. M., Pierdominici, M., Maggi, M., Corona, G., & Lombardo, F. (2021). SIGIS–SIAMS–SIE position statement of gender affirming hormonal treatment in transgender and non-binary people. Journal of Endocrinological Investigation, 45(3), 657–673. [DOI:10.1007/s40618-021-01694-2]
Godano, A., Maggi, M., Jannini, E., Meriggiola, M. C., Ghigo, E., Todarello, O., Lenzi, A., & Manieri, C. (2009). SIAMS-ONIG Consensus on Hormonal Treatment in Gender Identity Disorders. Journal of Endocrinological Investigation, 32(10), 857–864. [DOI:10.1007/BF03345758]
@@ -4243,6 +4254,13 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Abdul Sultan, A., West, J., Stephansson, O., Grainge, M. J., Tata, L. J., Fleming, K. M., Humes, D., & Ludvigsson, J. F. (2015). Defining venous thromboembolism and measuring its incidence using Swedish health registries: a nationwide pregnancy cohort study. BMJ Open, 5(11), e008864. [DOI:10.1136/bmjopen-2015-008864]
Abou-Ismail, M. Y., Citla Sridhar, D., & Nayak, L. (2020). Estrogen and thrombosis: A bench to bedside review. Thrombosis Research, 192, 40–51. [DOI:10.1016/j.thromres.2020.05.008]
+
Aly. (2018). An Introduction to Hormone Therapy for Transfeminine People. Transfeminine Science. [URL]
+
Aly. (2018). Oral Progesterone Achieves Very Low Levels of Progesterone and Has Only Weak Progestogenic Effects. Transfeminine Science. [URL]
+
Aly. (2019). A Review of Studies on Estradiol Levels and Testosterone Suppression with High-Dose Transdermal Estradiol Gel and Ointment in Cisgender Men with Prostate Cancer. Transfeminine Science. [URL]
+
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
+
Aly. (2020). Clinical Guidelines with Information on Transfeminine Hormone Therapy. Transfeminine Science. [URL]
+
Aly. (2020). The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
+
Aly. (2021). An Informal Meta-Analysis of Estradiol Curves with Injectable Estradiol Preparations. Transfeminine Science. [URL]
Amitrano, L., Guardascione, M. A., Brancaccio, V., & Balzano, A. (2002). Coagulation Disorders in Liver Disease. Seminars in Liver Disease, 22(1), 83–96. [DOI:10.1055/s-2002-23205]
Anderson, G. L., Limacher, M., Assaf, A. R., Bassford, T., Beresford, S. A., Black, H., Bonds, D., Brunner, R., Brzyski, R., Caan, B., Chlebowski, R., Curb, D., Gass, M., Hays, J., Heiss, G., Hendrix, S., Howard, B. V., Hsia, J., Hubbell, A., Jackson, R., … & Women’s Health Initiative Steering Committee. (2004). Effects of Conjugated Equine Estrogen in Postmenopausal Women With Hysterectomy: The Women’s Health Initiative Randomized Controlled Trial. JAMA, 291(14), 1701–1712. [DOI:10.1001/jama.291.14.1701]
Arnold, J. D., Sarkodie, E. P., Coleman, M. E., & Goldstein, D. A. (2016). Incidence of Venous Thromboembolism in Transgender Women Receiving Oral Estradiol. The Journal of Sexual Medicine, 13(11), 1773–1777. [DOI:10.1016/j.jsxm.2016.09.001]
@@ -4347,6 +4365,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Kuhl, H. (1999). Hormonal contraception. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 363–407). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_18]
Kuhl, H. (2005). Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric, 8(Suppl 1), 3–63. [DOI:10.1080/13697130500148875] [PDF]
Kuhl, H., & Stevenson, J. (2006). The effect of medroxyprogesterone acetate on estrogen-dependent risks and benefits – an attempt to interpret the Women’s Health Initiative results. Gynecological Endocrinology, 22(6), 303–317. [DOI:10.1080/09513590600717368]
+
Lain. (2019). A Review of Selective Estrogen Receptor Modulators and their Potential for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Langley, R. E., Cafferty, F. H., Alhasso, A. A., Rosen, S. D., Sundaram, S. K., Freeman, S. C., Pollock, P., Jinks, R. C., Godsland, I. F., Kockelbergh, R., Clarke, N. W., Kynaston, H. G., Parmar, M. K., & Abel, P. D. (2013). Cardiovascular outcomes in patients with locally advanced and metastatic prostate cancer treated with luteinising-hormone-releasing-hormone agonists or transdermal oestrogen: the randomised, phase 2 MRC PATCH trial (PR09). The Lancet Oncology, 14(4), 306–316. [DOI:10.1016/s1470-2045(13)70025-1]
Langley, R. E., Gilbert, D. C., Duong, T., Clarke, N. W., Nankivell, M., Rosen, S. D., Mangar, S., Macnair, A., Sundaram, S. K., Laniado, M. E., Dixit, S., Madaan, S., Manetta, C., Pope, A., Scrase, C. D., Mckay, S., Muazzam, I. A., Collins, G. N., Worlding, J., Williams, S. T., Paez, E., Robinson, A., McFarlane, J., Deighan, J. V., Marshall, J., Forcat, S., Weiss, M., Kockelbergh, R., Alhasso, A., Kynaston, H., & Parmar, M. (2021). Transdermal oestradiol for androgen suppression in prostate cancer: long-term cardiovascular outcomes from the randomised Prostate Adenocarcinoma Transcutaneous Hormone (PATCH) trial programme. The Lancet, 397(10274), 581–591. [DOI:10.1016/s0140-6736(21)00100-8]
Lax, E. (1987). Mechanisms of physiological and pharmacological sex hormone action on the mammalian liver. Journal of Steroid Biochemistry, 27(4–6), 1119–1128. [DOI:10.1016/0022-4731(87)90198-1]
@@ -4419,6 +4438,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Ruiz Garcia, V., López-Briz, E., Carbonell Sanchis, R., Gonzalvez Perales, J. L., & Bort-Martí, S. (2013). Megestrol acetate for treatment of anorexia-cachexia syndrome. Cochrane Database of Systematic Reviews, 2019(3), CD004310. [DOI:10.1002/14651858.cd004310.pub3]
Russell, N., Cheung, A., & Grossmann, M. (2017). Estradiol for the mitigation of adverse effects of androgen deprivation therapy. Endocrine-Related Cancer, 24(8), R297–R313. [DOI:10.1530/erc-17-0153]
Sahlin, L., & Schoultz, B. V. (1999). Liver Inclusive Protein, Lipid and Carbohydrate Metabolism. In Oettel, M., & Schillinger, E. (Eds.). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen (Handbook of Experimental Pharmacology, Volume 135, Part 2) (pp. 163–178). Berlin/Heidelberg: Springer. [DOI:10.1007/978-3-642-60107-1_8]
+
Sam. (2020). Analysis of Cardiovascular and Thromboembolic Toxicity with High Dose Parenteral Polyestradiol Phosphate in the Treatment of Prostate Cancer. Transfeminine Science. [URL]
Scarabin, P. (2018). Progestogens and venous thromboembolism in menopausal women: an updated oral versus transdermal estrogen meta-analysis. Climacteric, 21(4), 341–345. [DOI:10.1080/13697137.2018.1446931]
Scarabin, P., Canonico, M., Plu-Bureau, G., & Oger, E. (2020). Menopause and hormone therapy in the 21st century: why promote transdermal estradiol and progesterone? Heart, 106(16), 1278–1278. [DOI:10.1136/heartjnl-2020-316907]
Scheres, L. J., van Hylckama Vlieg, A., Ballieux, B. E., Fauser, B. C., Rosendaal, F. R., Middeldorp, S., & Cannegieter, S. C. (2019). Endogenous sex hormones and risk of venous thromboembolism in young women. Journal of Thrombosis and Haemostasis, 17(8), 1297–1304. [DOI:10.1111/jth.14474]
@@ -4990,6 +5010,10 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Adamcová, K., Kolátorová, L., Škodová, T., Šimková, M., Pařízek, A., Stárka, L., & Dušková, M. (2018). Steroid hormone levels in the peripartum period – differences caused by fetal sex and delivery type. Physiological Research, 67(Suppl 3), S489–S497. [DOI:10.33549/physiolres.934019]
+
Aly. (2020). A Comprehensive Review of the Potential of Progestogens for Enhancing Breast Development in Transfeminine People. Transfeminine Science. [URL]
+
Aly. (2020). Approximate Comparable Dosages of Estradiol by Different Routes. Transfeminine Science. [URL]
+
Aly. (2020). Hormone Levels During Normal Puberty in Cisgender Girls. Transfeminine Science. [URL]
+
Aly. (2020). Supplement: The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Anderson, P. J., Hancock, K. W., & Oakey, R. E. (1985). Non-protein-bound oestradiol and progesterone in human peripheral plasma before labour and delivery. Journal of Endocrinology, 104(1), 7–15. [DOI:10.1677/joe.0.1040007]
Barini, A., Liberale, I., & Menini, E. (1993). Simultaneous Determination of Free Testosterone and Testosterone Bound to Non-Sex-Hormone-Binding Globulin by Equilibrium Dialysis. Clinical Chemistry, 39(6), 936–941. [DOI:10.1093/clinchem/39.6.936]
@@ -5057,6 +5081,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
Rothman, M. S., Carlson, N. E., Xu, M., Wang, C., Swerdloff, R., Lee, P., Goh, V. H., Ridgway, E. C., & Wierman, M. E. (2011). Reexamination of testosterone, dihydrotestosterone, estradiol and estrone levels across the menstrual cycle and in postmenopausal women measured by liquid chromatography–tandem mass spectrometry. Steroids, 76(1–2), 177–182. [DOI:10.1016/j.steroids.2010.10.010]
Rubinow, D. R., Schmidt, P. J., Roca, C. A., & Daly, R. C. (2002). Gonadal Hormones and Behavior in Women: Concentrations versus Context. In Pfaff, D. W., Arnold, A. P., Etgen, A. M., Fahrbach, S. E., & Rubin, R. T. (Eds.). Hormones, Brain and Behavior, Volume 5 (pp. 37–73). Amsterdam: Academic Press. [Google Books] [DOI:10.1016/B978-012532104-4/50086-X]
Ruokonen, A., Alén, M., Bolton, N., & Vihko, R. (1985). Response of serum testosterone and its precursor steroids, SHBG and CBG to anabolic steroid and testosterone self-administration in man. Journal of Steroid Biochemistry, 23(1), 33–38. [DOI:10.1016/0022-4731(85)90257-2]
+
Sam. (2020). A Comparison of Oral and Transdermal Estradiol in Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Schijf, C. P., van der Mooren, M. J., Doesburg, W. H., Thomas, C. M., & Rolland, R. (1993). Differences in serum lipids, lipoproteins, sex hormone binding globulin and testosterone between the follicular and the luteal phase of the menstrual cycle. Acta Endocrinologica, 129(2), 130–133. [DOI:10.1530/acta.0.1290130]
Schuijt, M. P., Sweep, C. G., van der Steen, R., Olthaar, A. J., Stikkelbroeck, N. M., Ross, H. A., & van Herwaarden, A. E. (2019). Validity of free testosterone calculation in pregnant women. Endocrine Connections, 8(6), 672–679. [DOI:10.1530/ec-19-0110]
Shifren, J. L., Desindes, S., McIlwain, M., Doros, G., & Mazer, N. A. (2007). A randomized, open-label, crossover study comparing the effects of oral versus transdermal estrogen therapy on serum androgens, thyroid hormones, and adrenal hormones in naturally menopausal women. Menopause, 14(6), 985–994. [DOI:10.1097/gme.0b013e31803867a]
@@ -5140,6 +5165,7 @@ Figure 5. Meta-analysis of estradiol concentration-time data from cisgender wome
References
+
Aly. (2020). The Interactions of Sex Hormones with Sex Hormone-Binding Globulin and Relevance for Transfeminine Hormone Therapy. Transfeminine Science. [URL]
Ben-Rafael, Z., Mastroianni, L., Meloni, F., Strauss, J. F., & Flickinger, G. L. (1986). Changes in serum sex hormone-binding globulin, free estradiol, and testosterone during gonadotropin treatment. Fertility and Sterility, 46(4), 593–598. [DOI:10.1016/s0015-0282(16)49633-0]
Carlström, K., Pschera, H., & Lunell, N. (1988). Serum levels of oestrogens, progesterone, follicle-stimulating hormone and sex-hormone-binding globulin during simultaneous vaginal administration of 17β-oestradiol and progesterone in the pre- and post-menopause. Maturitas, 10(4), 307–316. [DOI:10.1016/0378-5122(88)90066-7]
Elaut, E., De Cuypere, G., De Sutter, P., Gijs, L., Van Trotsenburg, M., Heylens, G., Kaufman, J., Rubens, R., & T’Sjoen, G. (2008). Hypoactive sexual desire in transsexual women: prevalence and association with testosterone levels. European Journal of Endocrinology, 158(3), 393–399. [DOI:10.1530/eje-07-0511]
Transfeminine Science was created by Aly and contains articles by different writers on transfeminine hormone therapy. Our authors and other information are listed on our About page. The content on this site is written by transgender people, for transgender people, as well as for medical providers and academics in transgender health. A categorized listing of articles on Transfeminine Science can be found on the Articles page, while a listing of articles by date can be found on the Latest page. The Misc page contains other content like useful tools, notable publications, and links. The articles on this site are wiki-esque living documents, and hence may be updated, expanded, and improved over time.
\ No newline at end of file
+Home - Transfeminine Science
Transfeminine Science was created by Aly and contains articles by different writers on transfeminine hormone therapy. Our authors and other information are listed on our About page. The content on this site is written by transgender people, for transgender people, as well as for medical providers and academics in transgender health. A categorized listing of articles on Transfeminine Science can be found on the Articles page, while a listing of articles by date can be found on the Latest page. The Misc page contains other content like useful tools, notable publications, and links. The articles on this site are wiki-esque living documents, and hence may be updated, expanded, and improved over time.
\ No newline at end of file
diff --git a/transfemscience.org/misc/hormone-conc-unit-conv/index.html b/transfemscience.org/misc/hormone-conc-unit-conv/index.html
index c04386f0..720f7199 100644
--- a/transfemscience.org/misc/hormone-conc-unit-conv/index.html
+++ b/transfemscience.org/misc/hormone-conc-unit-conv/index.html
@@ -1 +1 @@
-Hormone Concentration Unit Conversion - Transfeminine Science
\ No newline at end of file
diff --git a/transfemscience.org/misc/injectable-dose-vol-conc-conv/index.html b/transfemscience.org/misc/injectable-dose-vol-conc-conv/index.html
index 1a827822..e64ca7e6 100644
--- a/transfemscience.org/misc/injectable-dose-vol-conc-conv/index.html
+++ b/transfemscience.org/misc/injectable-dose-vol-conc-conv/index.html
@@ -1 +1 @@
-Dose, Volume, and Concentration Conversion for Injectables - Transfeminine Science
Dose, Volume, and Concentration Conversion for Injectables
Dose and Concentration to Volume
Volume and Concentration to Dose
Notes
The following should be remembered when discussing and using injectables:
Volume is meaningless without concentration (in terms of understanding dose).
What is being used should generally be stated in terms of dose rather than volume. This is because concentration is also required for volume to be meaningful (#1 above) and because dose is much more readily interpretable to other people.
\ No newline at end of file
+Dose, Volume, and Concentration Conversion for Injectables - Transfeminine Science
Dose, Volume, and Concentration Conversion for Injectables
Dose and Concentration to Volume
Volume and Concentration to Dose
Notes
The following should be remembered when discussing and using injectables:
Volume is meaningless without concentration (in terms of understanding dose).
What is being used should generally be stated in terms of dose rather than volume. This is because concentration is also required for volume to be meaningful (#1 above) and because dose is much more readily interpretable to other people.
\ No newline at end of file
diff --git a/transfemscience.org/misc/injectable-e2-simulator-advanced/index.html b/transfemscience.org/misc/injectable-e2-simulator-advanced/index.html
index db1c2599..c571d98d 100644
--- a/transfemscience.org/misc/injectable-e2-simulator-advanced/index.html
+++ b/transfemscience.org/misc/injectable-e2-simulator-advanced/index.html
@@ -1 +1 @@
-Injectable Estradiol Simulator Advanced - Transfeminine Science
Note: WPATH was renamed from the Harry Benjamin International Gender Dysphoria Association (HBIGDA) to the World Professional Association for Transgender Health (WPATH) in 2007.
a ANZPATH (Australian and New Zealand Professional Association for Transgender Health) was formed in 2009 at the 21st WPATH conference in Oslo, Norway. ANZPATH split into AusPATH and PATHA or ANZPATH was renamed to AusPATH in 2019. b Conference was postponed from August 2021 to May 2022 due to COVID-19 pandemic. c AusPATH and PATHA joint conference.
Note: WPATH was renamed from the Harry Benjamin International Gender Dysphoria Association (HBIGDA) to the World Professional Association for Transgender Health (WPATH) in 2007.
a ANZPATH (Australian and New Zealand Professional Association for Transgender Health) was formed in 2009 at the 21st WPATH conference in Oslo, Norway. ANZPATH split into AusPATH and PATHA or ANZPATH was renamed to AusPATH in 2019. b Conference was postponed from August 2021 to May 2022 due to COVID-19 pandemic. c AusPATH and PATHA joint conference.
Transfeminine Science has been cited, mentioned, and/or recognized in the published scientific literature by various academics. This page is a partial list of these instances, with citations and excerpts. We express gratitude to these authors for their comments and that they found our work valuable.
Hughes et al. (2022)
Hughes, J. H., Woo, K. H., Keizer, R. J., & Goswami, S. (2022). Clinical Decision Support for Precision Dosing: Opportunities for Enhanced Equity and Inclusion in Health Care. Clinical Pharmacology & Therapeutics, 113(3), 565–574. [DOI:10.1002/cpt.2799]:
Lastly, we recommend that developers of [clinical decision support software (CDSS)] for dosing take an iterative and participatory approach to designing systems. By involving stakeholders in the design process, they will develop solutions that best suit users’ needs and are more likely to be adopted and used correctly. This participatory approach should involve interviews and usability testing with clinicians. Formal usability testing and analysis with real end users can improve the quality and usefulness of a system.88 Though patients themselves are not typically the end users of CDSS, their expertise (especially that of marginalized groups and organized patient advocacy organizations) can also inform CDSS developers. As an example, transgender people have compiled their own resources to understanding dosing regimens in the absence of clear clinical guidelines.89 Developers of CDSS could provide a great deal of value to these patient populations, and improve their software’s utility, by working with them to understand their needs from a dosing tool.
89. Aly, W. An interactive web simulator for estradiol levels with injectable estradiol esters. Transfeminine Science <https://transfemscience.org/articles/injectable-e2-simulator-release/> (2021) Accessed November 1, 2022.
Linet (2023)
Linet, T. (2023). Prise en charge endocrinologique d’une personne trans. [Endocrinological care of a trans person.] In Faucher, P., Hassoun, D., & Linet, T. (Eds.). Santé sexuelle et reproductive des personnes LGBT [Sexual and Reproductive Health of LGBT People] (pp. 109–124). Issy-les-Moulineaux, France: Elsevier Masson. [Google Books] [URL] [WorldCat] [Excerpt] [Translated]:
It may happen in consultation that the person does not wish to use the prescribed estrogens and wishes to continue the self-prescription of injectable estrogens. It is then possible to evaluate with them the most suitable dosage using the Transfem Science Injection Simulator (https://transfemscience.org/misc/injectable-e2-simulator/).
Barksdale (2024)
Barksdale, A. (2024). Care and Freedom From Below: Experiments in Trans Autonomy Through DIY. (Doctoral dissertation, The University of Arizona.) Tucson: The University of Arizona. [Google Scholar] [URL 1] [URL 2] [PDF]:
Transfeminine Science
Transfeminine Science (TS) is an online resource written by a collective of trans women and non-binary transfeminine people who review and interpret studies on hormonal medicines. TS is not explicitly geared towards people doing DIY, but the DIY hormone therapy community is acknowledged as part of their audience. They state, “Wherever possible, decisions about medical care should be made in partnership with a health care professional. We recognize that many transfeminine people are on do-it-yourself (DIY) hormone therapy however, and we aim to help inform this critical and underserved community of individuals as well” (Transfeminine Science 2023). The primary author of the articles on TS, Aly, has been involved with the DIY hormone therapy community, serving as a moderator for one DIY forum.12 Another author, Mitzi, “is an outspoken critic of her country’s [the United Kingdom] transgender healthcare system, and has self-medicated for the duration of her own transition” (Transfeminine Science 2023). The information and tools produced by TS are well regarded and circulated within the DIY hormone therapy community. Some articles have even been translated by community members into Chinese and Vietnamese.13
12. TS contributors are listed by a singular given name e.g. Aly or Lain. These names are possibly pen names solely for the purpose of TS. No personal profiles are linked on the site.
13. Project Trans has translated some articles into Simplified Chinese. The Vietnamese translations have been produced by Trans Girl VN.
I highlight the work of TS for two primary reasons. Firstly, they demonstrate how knowledge production is put to the use of making hormone use safer and more effective at meeting the needs of trans(feminine) people. Secondly, the level of knowledge available through TS exceeds the health literacy available through institutions. TS is often at the forefront of hormone therapy, able to educate on risks and potentials of drugs in ways that slow-moving institutions are not. Furthermore, just as feminist scientists ask different questions (D. Roy 2008), I argue as a trans project, TS pursues questions and problems not tackled by cis-dominated medical research.
The main activity of TS is reviewing literature on hormone use relevant to transfeminine people. Literature reviews are an important part of scientific knowledge production. While focused on gender-affirming hormone therapy, TS surveys literature on all uses of estrogens, antiandrogens, progestogens, and GnRH modulators. TS offers well-cited and up-to-date reviews of topics in scientific and medical literature, with the information interpreted and articulated for a transfeminine audience. However, in commitment to the rigor of evidence-based medicine and the scientific process, they state in a disclaimer, “The content on this site has not been formally published nor scholarly peer-reviewed. Readers should not take the content on Transfeminine Science as authoritative, but only as a supplementary resource to the information contained in transgender care guidelines and the medical literature in general” (Transfeminine Science 2023). TS is an example of autonomous knowledge production with particular relevance to the DIY community. While DIY hormone therapy guides provide practical knowledge, including protocols for hormone use, TS provides a deeper level of understanding around hormonal medications and their actions within the trans body.14
14. TS too provides immediately practical information and tools, such as dosage equivalencies between routes, concentration and dosage calculators, and the estradiol levels simulator discussed below.
The articles focus on safety and efficacy for transfeminine hormone therapy, highlighting risk and benefit profiles for medications and their delivery routes. The bulk of articles focus on estradiol, as the main component of transfeminine hormone regimens, and the authors review the literature on oral, sublingual/buccal, transdermal, and parenteral administration, including the pharmacokinetics, metabolism pathways, and potential risks. Blood clots and cardiovascular problems are one main risk of using estrogens, particularly long-term. These risks are much lower today, since bioidentical estradiol has largely superseded non-bioidentical estrogens, such as ethinylestradiol and conjugated estrogens. Nevertheless, estradiol still carries risks and the route of administration, which affects how it is metabolized, plays a mediating role. TS highlights that non-oral routes are preferable, because they demonstrate fewer risks compared to oral, suggesting the use of injection or transdermal estradiol.15 Injection and transdermal routes also have the benefit of higher bioavailability of the estradiol.
15. Non-oral routes are also preferable for those with pre-existing conditions, such as HIV.
Beyond estradiol, TS also surveys the use of antiandrogens and highlights issues with the most common drugs used in accordance with standard transgender medicine protocols. In general, guidelines for transfeminine hormone therapy recommend starting with both estradiol and an antiandrogen (Hembree et al. 2017). Antiandrogens are taken to suppress testosterone levels, which may be inadequately suppressed with estradiol alone.16 In the US, the main antiandrogen used for transfeminine hormone therapy is spironolactone, often referred to as “spiro.” As Beverly Cosgrove notes, “Spiro began being prescribed to trans women in the early 90s, when the dangers of taking estrogens in the form of Ethinyl Estradiol or Premarin [conjugated estrogens] became known” (Cosgrove 2018). The side effects of spiro, which for some are unacceptable, in conjunction with its inconsistent efficacy impel critics, including Cosgrove, to question its common use as a standard regimen. Aly argues that the widespread use of spiro is due to flawed studies and misunderstandings about the drug (Aly 2018a). While Aly calls for more and higher quality research on spiro, she concludes “spironolactone is likely to be a limitedly effective antiandrogen in transfeminine people” (Aly 2018a).
16. Though for many, adequate doses of estradiol can suppress testosterone in the desired range.
The primary antiandrogen for transfeminine hormone therapy prescribed in Europe and unavailable in the US is cyproterone acetate, commonly known as cypro. While more effective at suppressing testosterone than spiro, cypro also presents health risks. A 2018 epidemiological study by the French government found “a strong and dose-dependent increase in the risk of meningiomas, a type of hormone-sensitive brain tumor, with typical high doses of cyproterone acetate” (Aly 2020c). Following this study and earlier research on other dose-dependent side effects, such as liver toxicity, TS advocates using as low a dose as is effective. Low-dose cypro for transfeminine hormone therapy remains feasible, because low doses are still effective for testosterone suppression. Aly provides minimum and maximum recommended doses based on which cypro tablets are available. While scholarly literature has suggested using low-doses of cypro for transfeminine hormone therapy for years, medical guidelines are just now catching up.17 This shows the value of the approach of TS, in reviewing the literature and making that knowledge actionable, the collective provides better guidance than institutional guides.
17. Yet the 2017 Endocrine Society Clinical Practice Guideline (Hembree et al. 2017) recommends 25–50 mg/day. The more recently updated WPATH SOC 8 (Coleman et al. 2022), now recommends 10 mg/day.
In addition to reviewing current drugs and regimens for transfeminine hormone therapy, TS considers the use of other drugs that have not yet been used for this purpose. For example, Aly highlights EC508 (Estradiol Aminosulfonylbenzoylproline) as a potential new form of oral estradiol. EC508 was under development as a menopausal hormone therapy and as a hormonal birth control. Unlike other oral forms of estradiol, EC508 “has a pharmacological profile … that is much more similar to that of non-oral estradiol forms” (Aly 2018c). This means EC508 has the potential to be a safer and more effective drug than existing oral estradiols.18 Aly also suggests nandrolone (19-nortestosterone) might be a useful androgen for both transfeminine and transmasculine uses due to its “favorable profile relative to testosterone” (Aly 2020b). By reviewing the literature on hormonal medications beyond gender-affirming hormone therapy, TS suggests new potential avenues for treatment.
18. Unfortunately, as Aly points out in an update to her article, development of the drug seems to have stalled with little information about why.
Of particular significance in the realm of hypothetical treatments is the development of regimens and guidelines for non-binary gender-affirming hormone therapy. Currently, there are no established guidelines for non-binary hormone therapy and non-binary people are poorly served by existing standards and norms for trans medicine (Vincent 2020). For the first time in version 8, the WPATH SOC acknowledges and affirms non-binary transition (Coleman et al. 2022, chap. 8). However, the SOC contains no guidelines for tailoring hormonal therapies for non-binary transition goals. Rather, the WPATH suggests counseling non-binary care-seekers on the supposed impossibility of some common non-binary goals, such as feminization with minimal or no breast development ( S83). In their article on hormone therapy for non-binary transfeminine people, Aly notes, “There is currently a discordance between the number of people who desire non-conventional hormonal transition and the clinical establishment of such therapy. Consequently, an exploration of the possibilities from a theoretical standpoint would be of value and is the aim of this review” (Aly 2019a). She surveys potential ways of achieving demasculinization or partial feminization through hormonal means. This includes selective estrogen receptor modulators (SERMs), which “act like estrogens in some tissues and block estrogens in other tissues” (Lain 2019). Another TS contributor, Lain, published a more comprehensive review focused on SERMs (2019). SERMs do present challenges, including health risks, but are worthy of clinical investigation. The availability of SERMs on DIY hormone therapy source aggregators, also shows that DIYers are likely experimenting with these drugs already and thus information about their safety and efficacy should be available. TS is a valuable source of that information, which is lacking from institutional sources.
Beyond reviews of medical literature, TS has also published practical tools that are useful for hormone therapy users. Some are simple, such as calculators that convert between concentration, volume, and dose for injectables or between different units used to measure hormone concentrations in blood tests. The most impressive tool is the ‘‘Injectable Estradiol Simulator’’ (Aly and Luna 2021). The ‘‘Simulator’’ models blood estradiol levels over time based on the form of estradiol used (e.g. estradiol valerate), the dosage, whether the dose is repeated and how often (see Figure 2.1). The models used are based on an informal meta-analysis performed by Aly (2021).19 The ‘‘Simulator’’ is useful for modeling a dosage regimen to achieve stable estradiol concentration at a desired level (e.g. 200 pg/mL or 735 pmol/L) using the estradiol form one has available. This helps to avoid estradiol levels that are too low, and thus not effective, or too high, and unnecessarily risky. A more advanced version of the simulator including more functionality was coded by computer-scientist Luna based on Aly’s previous work. This tool puts expert knowledge in the hands of DIYers and enables them to experiment in new ways, guided by both blood tests and empirical models.
19. This tool has been recognized in published literature (Hughes et al. 2023; Jaafar et al. 2023), attesting to its value in modeling hormone regimens.
[…] Figure 2.1: Advanced Estradiol Injection Simulator on Transfeminine Science website showing different dosage regimens for injected estradiol valerate and the simulated estradiol levels over time.
Transfeminine Science is a valuable, evidence-based source of information, providing advanced health literacy for transfeminine hormone therapy users, regardless of whether they are doing DIY or not. For DIYers, TS helps to educate on risks that may be absent or downplayed in institutional guides. Impelled by the goal of making hormone therapy safer, more effective, and more responsive to the diverse needs of the transfeminine community, TS is often at the forefront of hormone knowledge and practice.
References […]
Datta (2024)
Datta, S. (2024). Beyond anxiety: Autonomy and harm reduction approaches to DIY Hormone Replacement Therapy. Indian Journal of Medical Ethics, IX(4), 265–270. [PubMed] [DOI:10.20529/IJME.2024.065] [PDF]:
Community-driven initiatives that are led, conceived and executed by transgender persons must be encouraged and supported. One such community-driven initiative seeking to disseminate knowledge on HRT is transfemscience.org. Popular among transfeminine people (ie, people assigned male at birth whose gender identity is predominantly feminine), including those seeking to transition DIY, the online platform features content on HRT written by transgender people with other transgender people, medical providers, and those studying transgender health as its intended audience [39]. All articles on the platform have thorough discussions on efficacy, safety, tolerability, and pharmacology of the drug under review, and are heavily referenced with peer-reviewed medical literature.
Davin, A. (2024). Femboys in the Factory. TSQ [Transgender Studies Quarterly], 11(2), 287–317. [DOI:10.1215/23289252-11215509]:
Communal knowledge recommends the use of bicalutamide and SERMs as hormone options that achieve a significant feminization without breast development, and DIY resources reflect this kind of systematization of femboy transitions as communally recognized and supported, outside of the pathways of coherent gendering demanded by present medical practice (Transfeminine Science [2019] 2022).
Transfeminine Science. (2019) 2022. “An Exploration of Possibilities for Hormone Therapy in Non-binary Transfeminine People.” Transfeminine Science, June. https://transfemscience.org/articles/nonbinary-transfem-overview/.
Rothman et al. (2024)
Rothman, M. S., Ariel, D., Kelley, C., Hamnvik, O. R., Abramowitz, J., Irwig, M. S., Soe, K., Davidge-Pitts, C., Misakian, A. L., Safer, J. D., & Iwamoto, S. J. (2024). The Use of Injectable Estradiol in Transgender and Gender Diverse Adults: A Scoping Review of Dose and Serum Estradiol Levels. Endocrine Practice, 30(9), 870–878. [DOI:10.1016/j.eprac.2024.05.008]:
In recent years, we have noted trends in our clinical practices with TGD adults requesting injectable estradiol, particularly in the United States. The reasons given can vary; it may be due to ease of weekly or every two weeks administration, fatigue of taking daily oral medications and skin reactions to or cost of transdermal preparations. There have been discussions as to the roles of estrone/estradiol ratios in feminization and whether injectable estradiol might lead to more favorable results, however research has not supported a role for estrone in optimizing feminizing outcomes [13]. There is also a belief that higher levels can be attained with injections and may lead to faster and more complete feminization; however, there is a lack of data in the literature to support these conclusions. Such conversations occurring on reddit.com and even some hormone provider websites, are perhaps related to the historical use of high dose injectable estradiol noted above [14]. However, there is a paucity of data to guide clinicians on what dose, type and at what interval estradiol esters should be injected and when levels should be measured to ensure physiologic range estradiol levels. In fact, recent reports and clinical observations have raised concerns that the dosing suggested in guidelines may result in supraphysiological estradiol levels and that higher doses and levels may put patients at elevated risk of thromboembolic events [15-18]. This scoping review examines the available data on levels achieved with various dosages of estradiol injections in TGD adults. We also report on testosterone suppression, route (i.e., SC vs. IM), and type of estradiol ester as well as timing of blood draw relative to dose, where available.
Acknowledgment
[…] [We] thank Aly from Transfemscience for community representation and correspondence.
Toffoli Ribeiro, C., Gois, Í., da Rosa Borges, M., Ferreira, L. G. A., Brandão Vasco, M., Ferreira, J. G., Maia, T. C., & Dias-da-Silva, M. R. (2024). Assessment of parenteral estradiol and dihydroxyprogesterone use among other feminizing regimens for transgender women: insights on satisfaction with breast development from community-based healthcare services. Annals of Medicine, 56(1), 2406458. [DOI:10.1080/07853890.2024.2406458]:
Utilizing a previously published meta-analysis method of estradiol concentration-time data from publicly available information on cisgender women who had used EEn or EEn/DHPA [17], we reanalyzed and integrated data from various studies. […]
[…] The V3C Fitter and Desmos tools, accessible online at https://alyw234237.github.io/injectable-e2-simulator/v3c-fitter/ and https://www.desmos.com/calculator/ndgvp2avhj?lang=pt-BR respectively, were utilized for fitting the three-compartment pharmacokinetic model. […]
Pharmacokinetics of injectable estradiol enanthate
[…] The boxplot graph (Figure 5) illustrates that the median estradiol levels in trans women using EEn/DHPA fell within this population’s expected average range values (100–200pg/mL).
Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. […]
References
[17] Aly. 2021. An informal meta-analysis of estradiol curves with injectable estradiol preparations. Transfeminine Sci. https:// transfemscience.org/articles/injectable-e2-meta-analysis/
\ No newline at end of file
+Literature Recognition - Transfeminine Science
Transfeminine Science has been cited, mentioned, and/or recognized in the published scientific literature by various academics. This page is a partial list of these instances, with citations and excerpts. We express gratitude to these authors for their comments and that they found our work valuable.
Hughes et al. (2022)
Hughes, J. H., Woo, K. H., Keizer, R. J., & Goswami, S. (2022). Clinical Decision Support for Precision Dosing: Opportunities for Enhanced Equity and Inclusion in Health Care. Clinical Pharmacology & Therapeutics, 113(3), 565–574. [DOI:10.1002/cpt.2799]:
Lastly, we recommend that developers of [clinical decision support software (CDSS)] for dosing take an iterative and participatory approach to designing systems. By involving stakeholders in the design process, they will develop solutions that best suit users’ needs and are more likely to be adopted and used correctly. This participatory approach should involve interviews and usability testing with clinicians. Formal usability testing and analysis with real end users can improve the quality and usefulness of a system.88 Though patients themselves are not typically the end users of CDSS, their expertise (especially that of marginalized groups and organized patient advocacy organizations) can also inform CDSS developers. As an example, transgender people have compiled their own resources to understanding dosing regimens in the absence of clear clinical guidelines.89 Developers of CDSS could provide a great deal of value to these patient populations, and improve their software’s utility, by working with them to understand their needs from a dosing tool.
89. Aly, W. An interactive web simulator for estradiol levels with injectable estradiol esters. Transfeminine Science <https://transfemscience.org/articles/injectable-e2-simulator-release/> (2021) Accessed November 1, 2022.
Linet (2023)
Linet, T. (2023). Prise en charge endocrinologique d’une personne trans. [Endocrinological care of a trans person.] In Faucher, P., Hassoun, D., & Linet, T. (Eds.). Santé sexuelle et reproductive des personnes LGBT [Sexual and Reproductive Health of LGBT People] (pp. 109–124). Issy-les-Moulineaux, France: Elsevier Masson. [Google Books] [URL] [WorldCat] [Excerpt] [Translated]:
It may happen in consultation that the person does not wish to use the prescribed estrogens and wishes to continue the self-prescription of injectable estrogens. It is then possible to evaluate with them the most suitable dosage using the Transfem Science Injection Simulator (https://transfemscience.org/misc/injectable-e2-simulator/).
Barksdale (2024)
Barksdale, A. (2024). Care and Freedom From Below: Experiments in Trans Autonomy Through DIY. (Doctoral dissertation, The University of Arizona.) Tucson: The University of Arizona. [Google Scholar] [URL 1] [URL 2] [PDF]:
Transfeminine Science
Transfeminine Science (TS) is an online resource written by a collective of trans women and non-binary transfeminine people who review and interpret studies on hormonal medicines. TS is not explicitly geared towards people doing DIY, but the DIY hormone therapy community is acknowledged as part of their audience. They state, “Wherever possible, decisions about medical care should be made in partnership with a health care professional. We recognize that many transfeminine people are on do-it-yourself (DIY) hormone therapy however, and we aim to help inform this critical and underserved community of individuals as well” (Transfeminine Science 2023). The primary author of the articles on TS, Aly, has been involved with the DIY hormone therapy community, serving as a moderator for one DIY forum.12 Another author, Mitzi, “is an outspoken critic of her country’s [the United Kingdom] transgender healthcare system, and has self-medicated for the duration of her own transition” (Transfeminine Science 2023). The information and tools produced by TS are well regarded and circulated within the DIY hormone therapy community. Some articles have even been translated by community members into Chinese and Vietnamese.13
12. TS contributors are listed by a singular given name e.g. Aly or Lain. These names are possibly pen names solely for the purpose of TS. No personal profiles are linked on the site.
13. Project Trans has translated some articles into Simplified Chinese. The Vietnamese translations have been produced by Trans Girl VN.
I highlight the work of TS for two primary reasons. Firstly, they demonstrate how knowledge production is put to the use of making hormone use safer and more effective at meeting the needs of trans(feminine) people. Secondly, the level of knowledge available through TS exceeds the health literacy available through institutions. TS is often at the forefront of hormone therapy, able to educate on risks and potentials of drugs in ways that slow-moving institutions are not. Furthermore, just as feminist scientists ask different questions (D. Roy 2008), I argue as a trans project, TS pursues questions and problems not tackled by cis-dominated medical research.
The main activity of TS is reviewing literature on hormone use relevant to transfeminine people. Literature reviews are an important part of scientific knowledge production. While focused on gender-affirming hormone therapy, TS surveys literature on all uses of estrogens, antiandrogens, progestogens, and GnRH modulators. TS offers well-cited and up-to-date reviews of topics in scientific and medical literature, with the information interpreted and articulated for a transfeminine audience. However, in commitment to the rigor of evidence-based medicine and the scientific process, they state in a disclaimer, “The content on this site has not been formally published nor scholarly peer-reviewed. Readers should not take the content on Transfeminine Science as authoritative, but only as a supplementary resource to the information contained in transgender care guidelines and the medical literature in general” (Transfeminine Science 2023). TS is an example of autonomous knowledge production with particular relevance to the DIY community. While DIY hormone therapy guides provide practical knowledge, including protocols for hormone use, TS provides a deeper level of understanding around hormonal medications and their actions within the trans body.14
14. TS too provides immediately practical information and tools, such as dosage equivalencies between routes, concentration and dosage calculators, and the estradiol levels simulator discussed below.
The articles focus on safety and efficacy for transfeminine hormone therapy, highlighting risk and benefit profiles for medications and their delivery routes. The bulk of articles focus on estradiol, as the main component of transfeminine hormone regimens, and the authors review the literature on oral, sublingual/buccal, transdermal, and parenteral administration, including the pharmacokinetics, metabolism pathways, and potential risks. Blood clots and cardiovascular problems are one main risk of using estrogens, particularly long-term. These risks are much lower today, since bioidentical estradiol has largely superseded non-bioidentical estrogens, such as ethinylestradiol and conjugated estrogens. Nevertheless, estradiol still carries risks and the route of administration, which affects how it is metabolized, plays a mediating role. TS highlights that non-oral routes are preferable, because they demonstrate fewer risks compared to oral, suggesting the use of injection or transdermal estradiol.15 Injection and transdermal routes also have the benefit of higher bioavailability of the estradiol.
15. Non-oral routes are also preferable for those with pre-existing conditions, such as HIV.
Beyond estradiol, TS also surveys the use of antiandrogens and highlights issues with the most common drugs used in accordance with standard transgender medicine protocols. In general, guidelines for transfeminine hormone therapy recommend starting with both estradiol and an antiandrogen (Hembree et al. 2017). Antiandrogens are taken to suppress testosterone levels, which may be inadequately suppressed with estradiol alone.16 In the US, the main antiandrogen used for transfeminine hormone therapy is spironolactone, often referred to as “spiro.” As Beverly Cosgrove notes, “Spiro began being prescribed to trans women in the early 90s, when the dangers of taking estrogens in the form of Ethinyl Estradiol or Premarin [conjugated estrogens] became known” (Cosgrove 2018). The side effects of spiro, which for some are unacceptable, in conjunction with its inconsistent efficacy impel critics, including Cosgrove, to question its common use as a standard regimen. Aly argues that the widespread use of spiro is due to flawed studies and misunderstandings about the drug (Aly 2018a). While Aly calls for more and higher quality research on spiro, she concludes “spironolactone is likely to be a limitedly effective antiandrogen in transfeminine people” (Aly 2018a).
16. Though for many, adequate doses of estradiol can suppress testosterone in the desired range.
The primary antiandrogen for transfeminine hormone therapy prescribed in Europe and unavailable in the US is cyproterone acetate, commonly known as cypro. While more effective at suppressing testosterone than spiro, cypro also presents health risks. A 2018 epidemiological study by the French government found “a strong and dose-dependent increase in the risk of meningiomas, a type of hormone-sensitive brain tumor, with typical high doses of cyproterone acetate” (Aly 2020c). Following this study and earlier research on other dose-dependent side effects, such as liver toxicity, TS advocates using as low a dose as is effective. Low-dose cypro for transfeminine hormone therapy remains feasible, because low doses are still effective for testosterone suppression. Aly provides minimum and maximum recommended doses based on which cypro tablets are available. While scholarly literature has suggested using low-doses of cypro for transfeminine hormone therapy for years, medical guidelines are just now catching up.17 This shows the value of the approach of TS, in reviewing the literature and making that knowledge actionable, the collective provides better guidance than institutional guides.
17. Yet the 2017 Endocrine Society Clinical Practice Guideline (Hembree et al. 2017) recommends 25–50 mg/day. The more recently updated WPATH SOC 8 (Coleman et al. 2022), now recommends 10 mg/day.
In addition to reviewing current drugs and regimens for transfeminine hormone therapy, TS considers the use of other drugs that have not yet been used for this purpose. For example, Aly highlights EC508 (Estradiol Aminosulfonylbenzoylproline) as a potential new form of oral estradiol. EC508 was under development as a menopausal hormone therapy and as a hormonal birth control. Unlike other oral forms of estradiol, EC508 “has a pharmacological profile … that is much more similar to that of non-oral estradiol forms” (Aly 2018c). This means EC508 has the potential to be a safer and more effective drug than existing oral estradiols.18 Aly also suggests nandrolone (19-nortestosterone) might be a useful androgen for both transfeminine and transmasculine uses due to its “favorable profile relative to testosterone” (Aly 2020b). By reviewing the literature on hormonal medications beyond gender-affirming hormone therapy, TS suggests new potential avenues for treatment.
18. Unfortunately, as Aly points out in an update to her article, development of the drug seems to have stalled with little information about why.
Of particular significance in the realm of hypothetical treatments is the development of regimens and guidelines for non-binary gender-affirming hormone therapy. Currently, there are no established guidelines for non-binary hormone therapy and non-binary people are poorly served by existing standards and norms for trans medicine (Vincent 2020). For the first time in version 8, the WPATH SOC acknowledges and affirms non-binary transition (Coleman et al. 2022, chap. 8). However, the SOC contains no guidelines for tailoring hormonal therapies for non-binary transition goals. Rather, the WPATH suggests counseling non-binary care-seekers on the supposed impossibility of some common non-binary goals, such as feminization with minimal or no breast development ( S83). In their article on hormone therapy for non-binary transfeminine people, Aly notes, “There is currently a discordance between the number of people who desire non-conventional hormonal transition and the clinical establishment of such therapy. Consequently, an exploration of the possibilities from a theoretical standpoint would be of value and is the aim of this review” (Aly 2019a). She surveys potential ways of achieving demasculinization or partial feminization through hormonal means. This includes selective estrogen receptor modulators (SERMs), which “act like estrogens in some tissues and block estrogens in other tissues” (Lain 2019). Another TS contributor, Lain, published a more comprehensive review focused on SERMs (2019). SERMs do present challenges, including health risks, but are worthy of clinical investigation. The availability of SERMs on DIY hormone therapy source aggregators, also shows that DIYers are likely experimenting with these drugs already and thus information about their safety and efficacy should be available. TS is a valuable source of that information, which is lacking from institutional sources.
Beyond reviews of medical literature, TS has also published practical tools that are useful for hormone therapy users. Some are simple, such as calculators that convert between concentration, volume, and dose for injectables or between different units used to measure hormone concentrations in blood tests. The most impressive tool is the ‘‘Injectable Estradiol Simulator’’ (Aly and Luna 2021). The ‘‘Simulator’’ models blood estradiol levels over time based on the form of estradiol used (e.g. estradiol valerate), the dosage, whether the dose is repeated and how often (see Figure 2.1). The models used are based on an informal meta-analysis performed by Aly (2021).19 The ‘‘Simulator’’ is useful for modeling a dosage regimen to achieve stable estradiol concentration at a desired level (e.g. 200 pg/mL or 735 pmol/L) using the estradiol form one has available. This helps to avoid estradiol levels that are too low, and thus not effective, or too high, and unnecessarily risky. A more advanced version of the simulator including more functionality was coded by computer-scientist Luna based on Aly’s previous work. This tool puts expert knowledge in the hands of DIYers and enables them to experiment in new ways, guided by both blood tests and empirical models.
19. This tool has been recognized in published literature (Hughes et al. 2023; Jaafar et al. 2023), attesting to its value in modeling hormone regimens.
[…] Figure 2.1: Advanced Estradiol Injection Simulator on Transfeminine Science website showing different dosage regimens for injected estradiol valerate and the simulated estradiol levels over time.
Transfeminine Science is a valuable, evidence-based source of information, providing advanced health literacy for transfeminine hormone therapy users, regardless of whether they are doing DIY or not. For DIYers, TS helps to educate on risks that may be absent or downplayed in institutional guides. Impelled by the goal of making hormone therapy safer, more effective, and more responsive to the diverse needs of the transfeminine community, TS is often at the forefront of hormone knowledge and practice.
References […]
Datta (2024)
Datta, S. (2024). Beyond anxiety: Autonomy and harm reduction approaches to DIY Hormone Replacement Therapy. Indian Journal of Medical Ethics, IX(4), 265–270. [PubMed] [DOI:10.20529/IJME.2024.065] [PDF]:
Community-driven initiatives that are led, conceived and executed by transgender persons must be encouraged and supported. One such community-driven initiative seeking to disseminate knowledge on HRT is transfemscience.org. Popular among transfeminine people (ie, people assigned male at birth whose gender identity is predominantly feminine), including those seeking to transition DIY, the online platform features content on HRT written by transgender people with other transgender people, medical providers, and those studying transgender health as its intended audience [39]. All articles on the platform have thorough discussions on efficacy, safety, tolerability, and pharmacology of the drug under review, and are heavily referenced with peer-reviewed medical literature.
Davin, A. (2024). Femboys in the Factory. TSQ [Transgender Studies Quarterly], 11(2), 287–317. [DOI:10.1215/23289252-11215509]:
Communal knowledge recommends the use of bicalutamide and SERMs as hormone options that achieve a significant feminization without breast development, and DIY resources reflect this kind of systematization of femboy transitions as communally recognized and supported, outside of the pathways of coherent gendering demanded by present medical practice (Transfeminine Science [2019] 2022).
Transfeminine Science. (2019) 2022. “An Exploration of Possibilities for Hormone Therapy in Non-binary Transfeminine People.” Transfeminine Science, June. https://transfemscience.org/articles/nonbinary-transfem-overview/.
Rothman et al. (2024)
Rothman, M. S., Ariel, D., Kelley, C., Hamnvik, O. R., Abramowitz, J., Irwig, M. S., Soe, K., Davidge-Pitts, C., Misakian, A. L., Safer, J. D., & Iwamoto, S. J. (2024). The Use of Injectable Estradiol in Transgender and Gender Diverse Adults: A Scoping Review of Dose and Serum Estradiol Levels. Endocrine Practice, 30(9), 870–878. [DOI:10.1016/j.eprac.2024.05.008]:
In recent years, we have noted trends in our clinical practices with TGD adults requesting injectable estradiol, particularly in the United States. The reasons given can vary; it may be due to ease of weekly or every two weeks administration, fatigue of taking daily oral medications and skin reactions to or cost of transdermal preparations. There have been discussions as to the roles of estrone/estradiol ratios in feminization and whether injectable estradiol might lead to more favorable results, however research has not supported a role for estrone in optimizing feminizing outcomes [13]. There is also a belief that higher levels can be attained with injections and may lead to faster and more complete feminization; however, there is a lack of data in the literature to support these conclusions. Such conversations occurring on reddit.com and even some hormone provider websites, are perhaps related to the historical use of high dose injectable estradiol noted above [14]. However, there is a paucity of data to guide clinicians on what dose, type and at what interval estradiol esters should be injected and when levels should be measured to ensure physiologic range estradiol levels. In fact, recent reports and clinical observations have raised concerns that the dosing suggested in guidelines may result in supraphysiological estradiol levels and that higher doses and levels may put patients at elevated risk of thromboembolic events [15-18]. This scoping review examines the available data on levels achieved with various dosages of estradiol injections in TGD adults. We also report on testosterone suppression, route (i.e., SC vs. IM), and type of estradiol ester as well as timing of blood draw relative to dose, where available.
Acknowledgment
[…] [We] thank Aly from Transfemscience for community representation and correspondence.
Toffoli Ribeiro, C., Gois, Í., da Rosa Borges, M., Ferreira, L. G. A., Brandão Vasco, M., Ferreira, J. G., Maia, T. C., & Dias-da-Silva, M. R. (2024). Assessment of parenteral estradiol and dihydroxyprogesterone use among other feminizing regimens for transgender women: insights on satisfaction with breast development from community-based healthcare services. Annals of Medicine, 56(1), 2406458. [DOI:10.1080/07853890.2024.2406458]:
Utilizing a previously published meta-analysis method of estradiol concentration-time data from publicly available information on cisgender women who had used EEn or EEn/DHPA [17], we reanalyzed and integrated data from various studies. […]
[…] The V3C Fitter and Desmos tools, accessible online at https://alyw234237.github.io/injectable-e2-simulator/v3c-fitter/ and https://www.desmos.com/calculator/ndgvp2avhj?lang=pt-BR respectively, were utilized for fitting the three-compartment pharmacokinetic model. […]
Pharmacokinetics of injectable estradiol enanthate
[…] The boxplot graph (Figure 5) illustrates that the median estradiol levels in trans women using EEn/DHPA fell within this population’s expected average range values (100–200pg/mL).
Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. […]
References
[17] Aly. 2021. An informal meta-analysis of estradiol curves with injectable estradiol preparations. Transfeminine Sci. https:// transfemscience.org/articles/injectable-e2-meta-analysis/

































 Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. […]
Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. […]





























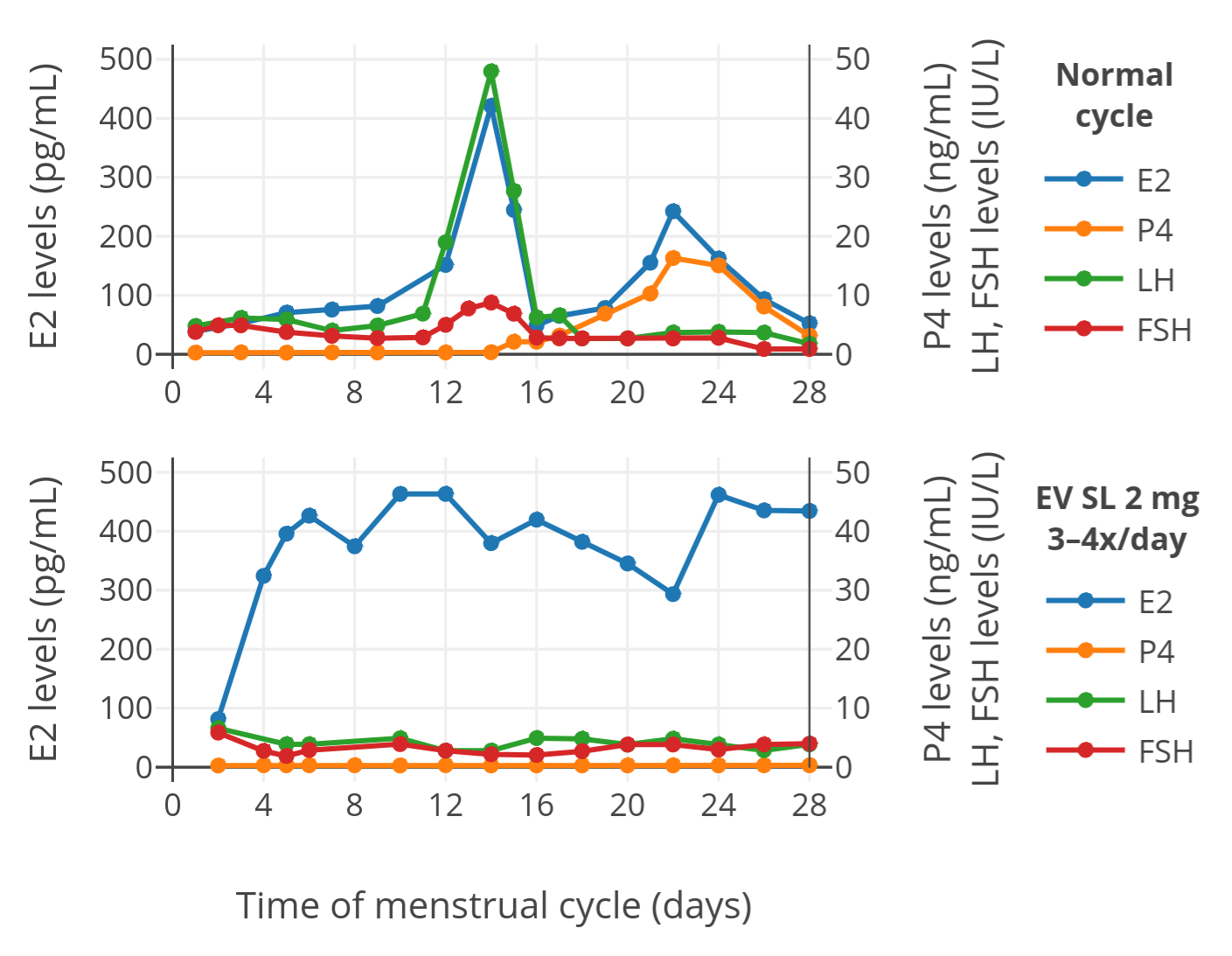

 Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. […]
Figure 5. Meta-analysis of estradiol concentration-time data from cisgender women under EEn alone or EEn/DHPA. Fitted data curves from various studies individually and combined into a single-dose curve over 30 days were generated based on an informal meta-analysis of published estradiol concentration-time data from cisgender women under EEn or EEn/DHPA [17]. […]{kind=link}
{kind=link}
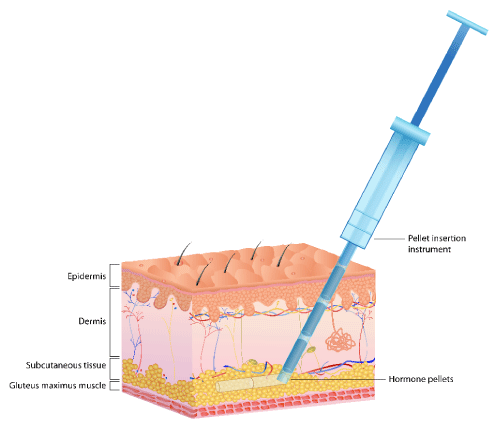{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
_in_postmenopausal_women.png){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}